

Abstract
The gastrointestinal glutathione peroxidase (GI-GPx, GPx2) is a selenoprotein that was suggested to act as barrier against hydroperoxide absorption but has also been implicated in the control of inflammation and malignant growth. In CaCo-2 cells, GI-GPx was induced by t-butyl hydroquinone (tBHQ) and sulforaphane (SFN), i.e., “antioxidants” known to activate the “antioxidant response element” (ARE) via electrophilic thiol modification of Keap1 in the Nrf2/Keap1 system. The functional significance of a putative ARE in the GI-GPx promoter was validated by transcriptional activation of reporter gene constructs upon exposure to electrophiles (tBHQ, SFN, and curcumin) or overexpression of Nrf2 and by reversal of these effects by mutation of the ARE in the promoter and by overexpressed Keap1. Binding of Nrf2 to the ARE sequence in authentic gpx2 was corroborated by chromatin immunoprecipitation. Thus, the presumed natural antioxidants sulforaphane and curcumin may exert their anti-inflammatory and anticarcinogenic effects not only by induction of phase 2 enzymes but also by the up-regulation of the selenoprotein GI-GPx.
The gastrointestinal glutathione peroxidase (GI-GPx, GPx2) belongs to the family of selenium-dependent glutathione peroxidases, which comprises five members (38). GI-GPx was first described in 1993 (13) as a novel isoenzyme exclusively expressed in the gastrointestinal tract. Like all other glutathione peroxidases, it reduces fatty acid hydroperoxides (8) and, due to its expression in the intestinal epithelium, has been suggested to function as a barrier against hydroperoxide absorption. In a CaCo-2 cell model, selenium deficiency and the resulting decrease in glutathione peroxidase activity indeed led to a higher release of hydroperoxides to the basolateral side. This was paralleled by damage to the cell membrane and could be prevented by selenium concentrations sufficient to restore GI-GPx expression but not the classical GPx (cGPx, GPx1) (66).
The barrier function, however, might not be the only biological role of GI-GPx. This view is supported by some unique features of GI-GPx. (i) GI-GPx mRNA is extremely stable in selenium deficiency, which ranks GI-GPx high in the hierarchy of selenoproteins and, thus, points to more vital functions than those the low-ranking selenoproteins such as cGPx may have (45, 65). (ii) In accordance with the high mRNA stability, GI-GPx protein is synthesized first upon selenium resupplementation, while cGPx follows with a delay of at least 24 h, as has been demonstrated in vitro and in vivo (10, 66). (iii) Unlike cGPx, GI-GPx is not uniformly expressed along the crypt-to-villus axis but is preferentially located in the crypts (22). In the small intestine, its concentration is especially high in Paneth cells (25), which play a major role in mucosal immunity: e.g., by secreting microbicidal defensins upon exposure to bacteria (2). (iv) GI-GPx is up-regulated in human colorectal adenomas (25, 39, 43), in Barrett's esophageal mucosa (44), and during neoplastic transformation of squamous epithelial cells (55). (v) While a single knockout (KO) of cGPx remains largely asymptomatic (31), a double KO of GPx1 and GPx2 (GPx1/2 KO) results in inflammatory bowel disease and increased intestinal cancer incidence (14), making a role for GI-GPx in preventing carcinogenesis likely.
Cruciferous vegetables, such as broccoli, Brussel sprouts, cabbage, and cauliflower, appear to be most effective in reducing the risk of colorectal cancer (61). They contain glucosinolates which are cleaved to chemopreventive isothiocyanates, like sulforaphane (SFN), by myrosinase released from ruptured plant cells. Further effective chemopreventive compounds are, for example, flavonoids, or curcuminoids (19, 47). These compounds, like the synthetic phenolic antioxidant tert-butyl hydroquinone (tBHQ), have been shown to induce multiple antioxidant and/or detoxication enzymes: e.g., γ-glutamylcysteine synthetase, heme oxygenase 1, and NAD(P)H:quinone oxidoreductase 1. Their promoters contain a cis-acting sequence, referred to as the “antioxidant response element” (ARE), which regulates both basal expression and inducible expression (reviewed in references 29 and 47). The core ARE consensus sequence was defined by mutational analysis and comprises the sequence 5′-TGACnnnGC-3′, where “n” can be any nucleotide (47, 53). Apart from the core sequence, also the 5′ and 3′ flanking regions influence the efficacy of ARE. Hence, the extended ARE consensus sequence was described as TA/CAnnA/GTGAC/TnnnGCA/GA/TA/TA/T (63).
The most effective transcription factor that acts through ARE is the NF-E2-related factor 2 (Nrf2), a member of the NF-E2 family of basic leucine zipper transcription factors (33). Nrf2-activated genes typically are phase 2 enzymes. The crucial role of Nrf2 is evidenced from Nrf2-deficient mice that display reduced expression levels of phase 2 enzymes and accordingly an increased susceptibility to carcinogens (52). The molecular link between the chemopreventive agents and Nrf2 is Kelch-like ECH-associated protein-1 (Keap1), a cysteine-rich actin-associated protein that keeps Nrf2 complexed in the cytosol (34, 36). Modification of specific SH groups in Keap1 by oxidation, alkylation, or arylation results in the dissociation of the Keap1/Nrf2 complex and translocation of Nrf2 to the nucleus, where it heterodimerizes with other basic leucine zipper proteins, such as small Maf, and then can bind to ARE (62). The common denominator of the realm of structurally diverse Nrf2 inducers is that they act as electrophiles or can react as Michael addition acceptors and thus can modify Keap1 SH groups (18, 19).
Glutathione peroxidases are generally believed to be up-regulated by oxidants, and, as expected also GI-GPx was found increased in a mouse model of hyperoxia or bleomycin-induced pulmonary fibrosis. Surprisingly, this GI-GPx induction was, however, no longer detected in Nrf2 KO mice (11, 12). These observations suggested that the expression of the antioxidant enzyme GI-GPx might also be up-regulated by the Keap1/Nrf2/ARE system, which canonically responds to antioxidants. Intrigued by this corollary, we analyzed whether transcriptional activation of gpx2 might be regulated by the Nrf2/Keap1 system and indeed could verify the functionality of a putative ARE element in gpx2 and thus classify the GI-GPx gene as an unorthodox target for Nrf2.
MATERIALS AND METHODS
Cell culture.
HepG2 cells (human liver carcinoma cells; ATCC HB8065) were cultured in RPMI 1640 with 2 mM l-alanyl-l-glutamine. CaCo-2 cells (human colon adenocarcinoma cells; DSMZ ACC 169) were grown in Dulbecco's modified Eagle's medium (high glucose) with 1% nonessential amino acids. All media were further supplemented with 10% heat-inactivated fetal calf serum (FCS; Biochrom, Berlin, Germany) and 5 mM HEPES, 100 U/ml penicillin, and 100 μg/ml streptomycin (all from Gibco, Karlsruhe, Germany). Viability of cells in response to treatment with tBHQ (Sigma, Taufkirchen, Germany), dl-sulforaphane (SFN; Alexis, Grünberg, Germany), or curcumin (CUR; Sigma) was confirmed with MTT {3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide}(Sigma). In case of selenium-supplemented cells, media contained 50 nM sodium selenite.
Plasmid constructions.
A 2,125-bp fragment of the human GI-GPx promoter was isolated via PCR from genomic DNA of HepG2 cells by using the primers pGIfor1 and pGIrev (Table 1) and cloned into the plasmid pCR-II-Topo (Invitrogen, Karlsruhe, Germany). The resulting construct was digested with KpnI and MluI and cloned into the luciferase reporter vector pGL3-basic (Promega, Mannheim, Germany), yielding the construct GI-prom-1 containing the promoter fragment of GI-GPx (−2111/+1 [+1 meaning the translation start point]). Deletion constructs GI-prom-2 to -6 (see Fig. 5) were generated by standard PCR using the GI-prom-1-construct as template. For cloning into pGL3-basic, primers were designed to incorporate a KpnI restriction site at the 5′ end and a MluI site at the 3′ end of the respective constructs (Table 1). All clones to be used were sequenced (MWG, Ebersberg, Germany).
TABLE 1.
Oligonucleotides and plasmids used in this study
| Oligonucleotide or plasmida | Sequence (5′→3′) or descriptionb | Accession no. |
|---|---|---|
| Oligonucleotides | ||
| pGlfor1 | CCA CTG AAT TGG AAT CAC TGG AGG | AL139022 |
| pGlfor2 | GCC CAC TAT CGG TAC CAA GTC CTT G | AF199441 |
| pGlfor3 | TCC CAA TGG AGG TAC CCC TGT GAA | AF199441 |
| pGlfor4 | AGA GGA AGG TAC CTG CAG TTC ATC C | AF199441 |
| pGlfor5 | GAC CTT TAG GGT ACC GGT TTC CTG T | AF199441 |
| pGlfor6 | CAA CCT CCT GGT ACC AAC AGC ACT | AF199441 |
| pGlrev | TTG GCA ATG AAA CGC GTG GTG AAG C | AF199441 |
| HO1-ARE fwd | GGA ATT CTG TTT TCG CTG AGT CAT GGT C | Z82244 |
| HO1-ARE rev | GAA CCA TGA CTC AGC GAA AAC AGA ATC C | Z82244 |
| GI-ARE-1 fwd | GAA TTC AGA CCC GCC CCT ACA GAC AA | AF199441 |
| GI-ARE-1 rev | TTG TAC ATG TGA GAG GGC AGG GTC TGA ATT CC | AF199441 |
| GI-ARE-2 fwd | GGA ATT CTG TTT TGC TAA GTC ATC CTG GGG A | AF199441 |
| GI-ARE-2 rev | TCC CCA GGA TGA CTT AGC AAA AAC AGA ATT CC | AF199441 |
| GI-prom Mut up | GGA CCT GTT TTT TAT AAG TCA TCC TGG | AF199441 |
| GI-prom Mut do | CCA GGA TGA CTT ATA AAA AAC AGG TCC | AF199441 |
| GI-GPx up | GGC TTT CAT TGC CAA GTC CTT C | X53463 |
| GI-GPx do | CTA TAT GGC AAC TTT AAG GAG GCG C | X53463 |
| β-Actin fwd | CAA GAG ATG GCC ACG GCT GCT | X00351 |
| β-Actin rev | TCC TTC TGC ATC CTG TCG GCA | X00351 |
| GI-GPx ChIP fwd | CAA CCT CCT TGT TCA AAC AGC ACT | AF199441 |
| GI-GPx ChIP rev | TTG GCA ATG AAA GCC ATG GTG AAG C | AF199441 |
| HO-1 ChIP fwd | CCA TCT GGC GCC GCT CTG C | Z82244 |
| HO-1 ChIP rev | GAG CAG CTG GAA CTC TGA GGA | Z82244 |
| Plasmids | ||
| pGL3-basic | Empty luciferase reporter gene plasmid lacking any promoter or enhancer | |
| GI-prom-1 | 2,111 bp of GI-GPx promoter in pGL3-basic | |
| GI-prom-2 | 1,589 bp of GI-GPx promoter in pGL3-basic | |
| GI-prom-3 | 1,258 bp of GI-GPx promoter in pGL3-basic | |
| GI-prom-4 | 879 bp of GI-GPx promoter in pGL3-basic | |
| GI-prom-5 | 302 bp of GI-GPx promoter in pGL3-basic | |
| GI-prom-6 | 172 bp of GI-GPx promoter in pGL3-basic | |
| GI-prom-1-mut | GI-prom-1 that carries a point mutation within GI-ARE-2 | |
| pGL3-promoter | Empty luciferase reporter gene plasmid with minimal SV40 promoter | |
| HO-ARE-pGL | pGL3-promoter containing 1 copy of the heme oxygenase ARE | |
| GI-ARE-1-pGL | pGL3-promoter containing 1 copy of the GI-GPx ARE-1 | |
| GI-ARE-2-pGL | pGL3-promoter containing 1 copy of the GI-GPx ARE-2 | |
| pcDNA3 | Empty expression plasmid | |
| pcDNA3-mNrf2 | pcDNA3 containing cDNA for murine Nrf2 | |
| pcDNA3-mKeap1 | pcDNA3 containing cDNA for murine Keap1 | |
| pSV-β-Gal | β-Galactosidase control plasmid for transfection normalization |
fwd, forward; rev, reverse; do, down.
Mutations are in boldface and underlined.
FIG. 5.

Nrf2-dependent induction of GI-GPx promoter activity is reversed by Keap1. (A) The GI-GPx promoter constructs GI-prom-1 to -6 and GI-prom-1-mut were cotransfected in HepG2 cells together with pcDNA3-mNrf2 or with empty pcDNA3. The factor of induction by Nrf2 is shown at the right side. Relative luciferase activity of GI-prom-1 pcDNA3 was set as 1. Potential AREs are indicated by boxes (mutated ARE in black). Values are means of three experiments measured in triplicate ± standard deviation. #, P < 0.05 versus pcDNA3. (B) The GI-GPx promoter construct GI-prom-1 was cotransfected with 10 ng pcDNA3-mKeap1 or pcDNA3 plasmid. Twenty-four hours after transfection, cells were exposed to tBHQ (20 μM), SFN (5 μM), or CUR (25 μM) for 24 h. Relative luciferase activity of the untreated construct without Keap1 was set as 1. Values are means of three experiments measured in triplicate ± standard deviation. #, P ≤ 0.05 versus respective control.
HO-ARE-pGL, GI-ARE-1-pGL, and GI-ARE-2-pGL were generated by cloning oligonucleotides (Table 1) containing the ARE of the murine heme oxygenase 1 (HO-1) promoter (accession no. U70472) or the putative AREs of the human GI-GPx-promoter into the EcoRI and HincII sites of pBluescript II KS(+) (Stratagene, La Jolla, Calif.). The resulting constructs were ligated via SmaI and XhoI into pGL3 promoter (Promega, Mannheim, Germany). Expression plasmids for murine Nrf2 (pcDNA3-mNrf2) and Keap1 (pcDNA3-mKeap1) were kindly provided by M. Yamamoto (University of Tsukuba, Japan). Empty pcDNA3 (Invitrogen, Karlsruhe, Germany) served as a control. pSV-β-galactosidase (Promega, Mannheim, Germany) was used for normalization of luciferase activity. Plasmids used for transfection experiments are listed in Table 1.
Site-directed mutagenesis.
Computer-based analysis of the GI-GPx promoter was performed by using the MatInspector program (51). The GI-ARE-2 site within the GI-GPx-promoter construct 1 (GI-prom-1) was changed from GCTAAGTCA into TATAAGTCA by “overlap-extension-PCR” (30). For the primers used, see Table 1. The correctness of GI-prom-1-mut was verified by sequencing.
Transfection and reporter gene assays.
A total of 2 × 105 (HepG2) or 2.5 × 105 (CaCo-2) cells were seeded onto 24-well plates and 24 h later transfected with 0.5 μg pSV-β-galactosidase, 0.15 μg luciferase reporter plasmid, and various amounts (maximum, 0.15 μg) of expression plasmids using Tfx-20 (Promega, Mannheim, Germany) according to the manufacturer's protocol. Stimulation with tBHQ (20 μM), SFN (5 μM), or CUR (25 μM) in serum-free medium was started 24 h after transfection for 24 h. In experiments without stimulation, cells were harvested 48 h after transfection. Cell lysis and determination of luciferase and β-galactosidase activity were performed as described previously (3). Relative luciferase activity was calculated by dividing luciferase activity by β-galactosidase activity. Reporter gene activity of the respective empty luciferase plasmids (pGL3-basic, pGL3-promoter) served as a control.
Nuclear extracts and EMSAs.
Cells were grown for 3 days before tBHQ (20 or 200 μM) or SFN (5 or 15 μM) was added for up to 16 h in serum-free medium. Cells were lysed at a density of 1 × 107 cells in 1.2 ml of lysis buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol [DTT], 0.5 mM phenylmethylsulfonyl fluoride [PMSF], pH 7.9) containing 0.1% Nonidet P-40 (Sigma, Taufkirchen, Germany) for 7 min at 4°C. Nuclei were pelleted by centrifugation and lysed for 30 min on ice in 100 μl of lysis buffer (40 mM HEPES, 400 mM KCl, 10% glycerol, 1 mM DTT, 0.1 mM PMSF, pH 7.9). Prior to use, 6.25 μl of 5 M NaCl was added to 100 μl lysis buffer. Nuclear extracts were centrifuged (20,800 × g, 30 min, 4°C) and subjected to protein determination. DNA fragments containing the consensus element HO-ARE, GI-ARE-1, or GI-ARE-2 (same as for the respective reporter plasmids) were labeled with [γ-32P]ATP. Binding reactions were performed for 30 min at room temperature with 5 μg nuclear protein, 50 fmol of labeled DNA, and 1 μg poly(dI-dC) (Amersham Biosciences, Freiburg, Germany) in 15 mM HEPES, 1 mM EDTA, 1 mM DTT, 10% (wt/vol) glycerol in a total volume of 10 μl. For the electrophoretic mobility shift assays (EMSAs), electrophoresis was carried out on native 4% polyacrylamide gels in 0.25× Tris-borate-EDTA at 200 V. Competition experiments were performed by adding 100-fold excess of unlabeled specific or unspecific oligonucleotide. For supershift analysis, 1 μg Nrf2-sc-722 antibody (Santa Cruz Biotechnology, Santa Cruz, Calif.) per sample was used.
ChIP.
Chromatin immunoprecipitation (ChIP) was performed as follows. Cells were grown in 15-cm dishes for 4 days before tBHQ (200 μM) or SFN (5 or 15 μM) was added for up to 16 h in serum-free medium. Cells were fixed in 1% formaldehyde at 37°C for 10 min. Cross-linking was stopped by the addition of 125 mM glycine in phosphate-buffered saline (PBS) for 10 min. Cells were washed with PBS and harvested by scraping. After centrifugation (18,000 × g, 1 min, 4°C) the pellet was resuspended in 1.5 ml cold lysis buffer (10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EDTA, 0.35 M sucrose, 0.5 mM DTT, pH 7.9) containing 1 mM PMSF and 1 μg/ml pepstatin A to inhibit proteases. After 10 min of incubation on ice, cells were centrifuged for 15 min at 4,000 × g and 4°C. The nuclear pellet was resuspended in 1 ml of cold lysis buffer (20 mM HEPES, 420 mM NaCl, 1.5 mM MgCl2, 0.1 mM EDTA, 10% glycerol, pH 7.9). For fragmentation of DNA to an average length of 200 to 1,000 bp, the resuspended pellet was sonicated on ice (2 × 30 s, 100% amplitude). After centrifugation (18,000 × g, 10 min, 4°C), a 50-μl aliquot was removed as input DNA. The remaining chromatin solution was diluted 10-fold in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, 150 mM NaCl, 0.25 mM EDTA, 1.0% Triton X-100, 0.1% sodium deoxycholate, pH 8.1) and precleared for 2 h at 4°C with a protein G-Sepharose slurry (Amersham Biosciences, Freiburg, Germany) preblocked with sonicated salmon sperm DNA and bovine serum albumin. Half of each sample was incubated overnight at 4°C with 3 μg of the specific Nrf2-sc-13032 antibody (Santa Cruz Biotechnology, Santa Cruz, Calif.) or with 3 μg of a nonspecific immunoglobulin G (IgG) (IκB-α-sc-203; Santa Cruz Biotechnology), respectively. Immune complexes were precipitated by adding 25 μl of the protein G-Sepharose slurry for 2 h at 4°C. Sepharose beads were washed twice with RIPA buffer and twice with PBS. Elution was performed twice in 150 μl of 0.1 M NaHCO3, 1% sodium dodecyl sulfate (SDS) at room temperature for 30 min each. After addition of NaCl to a final concentration of 0.3 M and 20 μg RNase A, cross-linking was reversed at 65°C for 4 h. Proteins were then digested with 100 μg proteinase K in 40 mM Tris-HCl, 10 mM EDTA for 1 h at 55°C. The remaining proteins and proteinase K were removed by extraction with phenol-chloroform and DNA precipitated with ethanol in the presence of 20 μg glycogen and redissolved in 30 μl water. PCR was performed with primers for a 200-bp region of the GI-GPx promoter containing GI-ARE-2 or with primers for the HO-1 promoter spanning a 200-bp region containing three AREs (for primers see Table 1). The PCR protocol was the same as described below. Cycle numbers were 37 for GI-GPx and 38 for HO-1, and the annealing temperatures were set to 64°C (GI-GPx) and 66°C (HO-1), respectively. PCR products were separated on 1.5% agarose gels.
Western blot analysis.
For GI-GPx detection, cells were grown until confluence for 3 days in medium containing 10% fetal calf serum (FCS) that was supplemented with sodium selenite (50 nM) before tBHQ (20 μM) or SFN (5 μM) was added for 48 h in serum-free, selenite-supplemented medium. Cells were lysed for 15 min on ice in RIPA buffer (50 mM Tris, 150 mM NaCl, 2 mM EGTA, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40) supplemented with protease inhibitors. Cellular debris was removed by centrifugation. For Nrf2 detection, nuclear extracts (see above) were used. SDS-polyacrylamide gel electrophoresis and Western blotting were performed as described previously (6). GI-GPx was detected by rabbit anti-human GI-GPx (5) and Nrf2 by Nrf2-sc-722 (Santa Cruz Biotechnology, Santa Cruz, Calif.). Peroxidase-conjugated goat anti-rabbit IgG (Chemicon, Hofheim, Germany) was used as a secondary antibody.
Reverse transcription-PCR.
Total RNA was isolated using the Invisorb Spin Cell RNA Mini kit (Invitek, Berlin, Germany). Reverse transcription was performed with 3 μg of RNA, oligo(dT)15 primers, and a Moloney murine leukemia virus RNase H− reverse transcriptase (Promega, Mannheim, Germany). PCR was performed in 25-μl reaction mixtures containing 25 pmol of each primer (Table 1), 200 μM deoxynucleoside triphosphate (dNTP), 0.625 U Taq DNA polymerase (Promega, Mannheim, Germany), 2.5 μl 10× reaction buffer, 0.5 mM (GI-GPx) or 1 mM (β-actin) MgCl2, and 1 μl cDNA. Initial denaturation (4 min, 94°C) was followed by 23 cycles (GI-GPx) or 20 cycles (β-actin) of 40 s at 94°C, 30 s at 63°C (GI-GPx) or 60°C (β-actin), and 2 min at 72°C. PCR was completed by 8 min at 72°C. Intensity of PCR bands of ethidium bromide-stained gels was quantified densitometrically (Gel Doc 2000; Bio-Rad, Munich, Germany). The amount of GI-GPx was normalized for β-actin.
RESULTS
The GI-GPx promoter contains one ARE that binds Nrf2.
First, the GI-GPx promoter was isolated by PCR using the primers pGIfor1 and pGIrev (Table 1). The promoter sequence was analyzed for potential transcription factor binding sites by using the MatInspector program (51). Two putative AREs were identified: one at bp −442 to −450 (GI-ARE-1) and one at bp −133 to −141 (GI-ARE-2). Whereas GI-ARE-2 covered a perfect ARE core sequence (47), 5′-TGACnnnGC-3′, in the reverse direction, GI-ARE-1 contains one mismatch (Fig. 1). Both AREs contain an embedded AP-1 recognition sequence. In general, AREs display a close sequence similarity to the AP-1-responsive element; however, for functioning as an ARE more features are required (i.e., the sequence −GC- at the 3′ end of the ARE core sequence, which is fulfilled by both GI-ARE-1 and −2). Comparison with the murine GI-GPx promoter sequence (accession number U62658) showed that GI-ARE-2 is also present in the mouse promoter, whereas GI-ARE-1 is not conserved among species.
FIG. 1.

Localization of potential AREs within the GI-GPx promoter. The promoter of human GI-GPx contains two putative AREs. The element GI-ARE-1 differs in 1 base (underlined and bold) from the consensus sequence, whereas GI-ARE-2 matches the consensus sequence completely. Numbers indicate the position starting from the ATG.
To check whether the AREs found in the GI-GPx promoter were able to bind Nrf2, oligonucleotides were synthesized containing GI-ARE-1, GI-ARE-2, or the ARE of HO-1. HO-1 is a well-characterized Nrf2 target gene (1) and was therefore used as a positive control. The oligonucleotides were tested for Nrf2 binding in EMSAs with nuclear extracts from HepG2 cells stimulated with tBHQ, a typical activator of Nrf2. Treatment with tBHQ led to enhanced DNA binding to HO-ARE (Fig. 2A, lanes 1 to 3) and GI-ARE-2 (Fig. 2A, lanes 11 to 13; Fig. 2B, lanes 1 to 3). Specificity was demonstrated by the disappearance of Nrf2 complexes by competition with an excess of the respective unlabeled oligonucleotide but not with an unrelated NF-κB oligonucleotide (Fig. 2A, lanes 4, 5, 14, and 15). With GI-ARE-1, bands running at different positions were obtained which were not further stimulated by tBHQ but which, however, disappeared in the presence of an excess of the unlabeled specific oligonucleotide (Fig. 2A, lanes 6 to 10). Thus, only GI-ARE-2 behaved like the positive control HO-ARE in respect to Nrf2 binding, whereas GI-ARE-1 appeared to interact with proteins rather nonspecifically. Treatment of HepG2 cells with a second Nrf2 activator, SFN, also resulted in enhanced binding to GI-ARE-2 (Fig. 2C, lanes 1 to 3). The specificity was further confirmed by supershift with an Nrf2 antibody (Fig. 2B, lane 4; Fig. 2C, lane 4). The activation of Nrf2 by tBHQ or SFN could also be demonstrated by the translocation of Nrf2 into the nucleus at a protein level, as demonstrated by Western blotting (Fig. 2D). Besides the expected band at 68 kDa, an additional band around 100 kDa, the amount of which was also increased upon tBHQ or SFN exposure, was detected in all samples. The occurrence of such a high-molecular-mass product of Nrf2 has already been described by others and likely indicates an Nrf2-actin complex (35).
FIG. 2.

Nrf2 translocates to the nucleus and binds to GI-ARE-2 in response to tBHQ or SFN exposure. Nuclear extracts were prepared from HepG2 cells treated with tBHQ (20, 200 μM; 16 h) or SFN (5, 15 μM; 4 h). EMSAs were performed as described in Materials and Methods. (A) HO-ARE (lanes 1 to 5), GI-ARE-1 (lanes 6 to 10), and GI-ARE-2 (lanes 11 to 15) probes were incubated with nuclear extracts from stimulated cells as indicated. For control, a 100-fold molar excess of the respective unlabeled specific (lanes 4, 9, 14) or unspecific (κB; lanes 5, 10, 15) oligonucleotide was added during the binding procedure. (B and C) GI-ARE-2 was incubated with nuclear extracts of tBHQ-treated (B) or SFN-treated (C) cells. For supershift, nuclear extracts were incubated with anti-Nrf2 (lane 4). Results are representative of three independent experiments. (D) HepG2 cells were stimulated with tBHQ (200 μM, 16 h) or SFN (15 μM, 4 h). Nuclear extracts were analyzed for Nrf2 by Western blotting as described in Materials and Methods. Results are representative of three independent experiments.
Finally, the direct binding of Nrf2 to the endogenous GI-GPx promoter had to be tested. To this end, chromatin immunoprecipitation was performed in HepG2 cells using an Nrf2-specific antibody and PCR primers covering the ARE-2, but not the ARE-1, site in the GI-GPx promoter. In accordance with the basal expression of GI-GPx in HepG2 cells, specific PCR signals were already obtained in controls and they were distinctly enhanced by treatment with either tBHQ or sulforaphane (Fig. 3A). This clearly demonstrates that Nrf2 indeed binds to the GI-GPx promoter at the ARE-2 site. Controls without Nrf2-specific antibodies (IgG) did not show PCR signals. Again, the HO-1 promoter was used as positive control. Corresponding ChIP analyses revealed the expected increase of Nrf2 binding to the HO-1 promoter by tBHQ and sulforaphane (Fig. 3B).
FIG. 3.
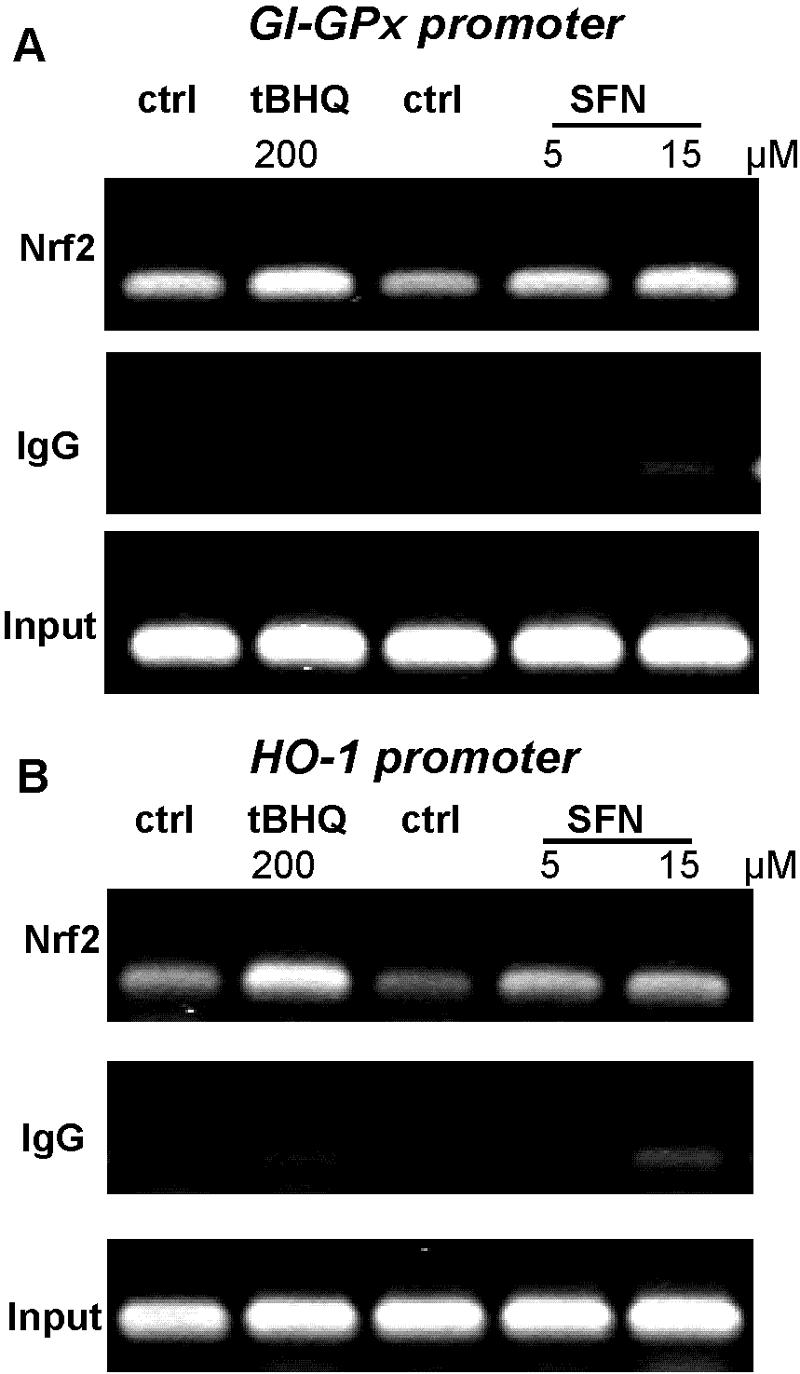
Nrf2 binds to the GI-ARE-2 in the GI-GPx promoter. Chromatin immunoprecipitation was carried out in HepG2 cells, as described in Materials and Methods. Protein-DNA complexes of cells treated with either tBHQ (200 μM, 16 h) or SFN (5 to 15 μM, 4 h) and of untreated cells were cross-linked with formaldehyde. Sheared complexes were precipitated with an antibody against Nrf2 or with a nonspecific immunoglobulin (IgG), and the thus coprecipitated fragmented genomic DNA was analyzed by PCR with primers specific for a fragment of the GI-GPx promoter (A) or the HO-1 promoter (B) containing the specific Nrf2 binding site(s). For control of equal sample amounts, input DNA (sheared DNA prior to immunoprecipitation) was PCR amplified. ctrl, control. Results are representative of three independent experiments.
Taken together, the GI-GPx promoter contains two putative ARE sequences. One, GI-ARE-2, can bind Nrf2 present in the nucleus from control and, to a higher extent, from activated HepG2 cells.
GI-ARE-2 is transcriptionally active.
To verify the functionality of the GI-ARE-2, HepG2 cells were transiently transfected with ARE-driven luciferase reporter plasmids (HO-ARE-pGL, GI-ARE-1-pGL, and GI-ARE-2-pGL) and the expression plasmid pcDNA3-mNrf2. While HO-ARE and GI-ARE-2 were significantly activated by Nrf2, GI-ARE-1 did not respond to Nrf2 at all (Fig. 4A) and was, therefore, excluded from further analysis. The effect of Nrf2 could be mimicked, though to a lesser extent, by Nrf2-activating compounds (tBHQ, SFN, and curcumin) (Fig. 4B).
FIG. 4.

GI-ARE-2 is responsive to Nrf2, tBHQ, SFN, and CUR. (A) HepG2 cells were transfected with the reporter gene construct HO-ARE-pGL, GI-ARE-1-pGL, GI-ARE-2-pGL, or pGL3-promoter in combination with either pcDNA3-mNrf2 or empty pcDNA3. Cells were harvested 48 h after transfection, and luciferase and β-galactosidase activities were analyzed. Relative luciferase activities (luciferase activity divided by β-galactosidase activity) were normalized to that obtained with the plasmid pGL3-promoter without responsive elements. The respective pcDNA3-transfected sample was set as 1. Values are means of three experiments measured in triplicate ± standard deviation. #, P < 0.05 versus pcDNA3. (B) HepG2 cells were transfected with the reporter gene construct HO-ARE-pGL, GI-ARE-2-pGL, or pGL3-promoter. Twenty-four hours after transfection, cells were exposed to tBHQ (20 μM), SFN (5 μM), or CUR (25 μM) for 24 h. Relative luciferase activity was normalized to the activity of the empty plasmid pGL3-promoter. The respective values obtained in untreated cells (control) were set to 1. Values are means of three experiments measured in triplicate ± standard deviation. #, P < 0.05 versus the respective control.
Having identified the transcriptionally active GI-ARE-2, we next tested whether it also functions within the authentic GI-GPx promoter. For this, six deletion constructs (GI-prom-1 to -6) of the GI-GPx promoter with a maximum length of 2,111 bp were generated and cloned into the reporter plasmid pGL3-basic (Fig. 5A). Constructs 1 to 4 still contain both putative AREs, whereas constructs 5 and 6 only contain GI-ARE-2. In addition, a construct identical to GI-prom-1 but containing a 2-base-pair mutation within the GI-ARE-2 was generated (GI-prom-1-mut). Relative luciferase activity of all constructs was induced in HepG2 cells by cotransfection of pcDNA3-mNrf2 (Fig. 5A). The basal promoter activity was lowest for the shortest fragment (GI-prom-6). The factor of Nrf2-induction, however, was highest for this construct. Mutation of the GI-ARE-2 within the construct GI-prom-1 resulted in an almost complete loss of basal promoter activity as well as of responsiveness to Nrf2 (Fig. 5A). The mutation was designed to only disrupt the Nrf2 recognition sequence but not the potential AP-1 binding site adjacent to it. This demonstrates that GI-ARE-2 not only might be an essential responsive element for the induction of GI-GPx but also is crucial for basal promoter activity. Furthermore, GI-ARE-1 can obviously not compensate for the missing GI-ARE-2, leaving the function of GI-ARE-1 unexplained.
The GI-GPx promoter is activated by typical Nrf2 activators.
Since all GI-GPx-promoter constructs were shown to be responsive to Nrf2, only the longest fragment, GI-prom-1, was used for further experiments. In accordance to the GI-ARE-2-pGL construct, the GI-GPx promoter itself also responded to the Nrf2-activating compounds. Incubation of GI-prom-1-transfected HepG2 cells with tBHQ, SFN, or CUR resulted in a low but significant induction of reporter gene activity, which was strongest in SFN-treated cells (Fig. 5B). Basal promoter activity was inhibited by cotransfection with small amounts of pcDNA3-mKeap1 but could nevertheless be reactivated at an even higher factor of induction by tBHQ, SFN, and CUR, indicating that the transfected Keap1 reacted with these substances in the expected manner (Fig. 5B).
The GI-GPx promoter is regulated by the Nrf2/Keap1 system dose dependently.
Nrf2 activation requires its release from Keap1, which sequesters Nrf2 in the cytoplasm (34). Excess of Keap1 should therefore prevent Nrf2 activation. Such a relationship is here demonstrated for the GI-GPx promoter. Cotransfection of GI-prom-1 with pcDNA3-mNrf2 resulted in the already described sevenfold induction of reporter gene activity (Fig. 6A). The effect of Nrf2 on the promoter activity was dose-dependently reversed by increasing amounts of cotransfected pcDNA3-mKeap1 (Fig. 6A). Vice versa, basal promoter activity was inhibited by overexpression of Keap1, an effect which could be reversed by simultaneous overexpression of Nrf2 in a dose-dependent manner (Fig. 6B).
FIG. 6.

Dose-dependent regulation of GI-GPx-promoter activity by Nrf2 and Keap1. Repression of promoter activity by Keap1 (A) and reversal of the Keap1 effect by Nrf2 (B). HepG2 cells were transfected with the reporter construct GI-prom-1 and either 150 ng pcDNA3-mNrf2 or 150 ng pcDNA3-mKeap1 and mutually transfected with increasing amounts of Keap1 or Nrf2 expression plasmid, respectively. Empty pcDNA3 was used to equalize the amount of cotransfected expression plasmid. Relative luciferase activity in cells transfected with GI-prom-1 and pcDNA3 was set as 1. Values are means of two experiments measured in triplicate ± standard deviation.
Regulation of the GI-GPx promoter by the Nrf2/Keap1 system is also working in CaCo-2 cells.
HepG2 cells usually express fairly large amounts of GI-GPx. To test whether the observed effects of Nrf2 and Nrf2-activating compounds are also prevalent in a cellular system with a generally lower GI-GPx expression level, the most crucial transfection experiments were repeated in CaCo-2 cells. In accordance with the data from HepG2 cells, the activity of the promoter construct GI-prom-1 was strongly enhanced (sevenfold) in CaCo-2 cells by overexpression of Nrf2 and was inhibited by Keap1. Also, Nrf2 activation by tBHQ, SFN, and CUR in CaCo-2 cells was comparable to that observed in HepG2 cells (Fig. 7A). Thus, the regulation of the GI-GPx promoter by the Nrf2/Keap1 system is not restricted to liver cells nor does it depend on the endogenous GI-GPx expression level.
FIG. 7.

Induction of endogenous GI-GPx in CaCo-2 cells. (A) GI-GPx promoter activation. CaCo-2 cells were transfected with the GI-GPx promoter construct GI-prom-1 together with 150 ng pcDNA3-mNrf2, pcDNA3-mKeap1, or empty pcDNA3. Twenty-four hours after transfection, cells were incubated with tBHQ (20 μM), SFN (5 μM), or CUR (25 μM) for 24 h. Relative luciferase activity of the untreated pcDNA3-construct was set as 1. Values are means of three experiments measured in triplicate ± standard deviation. #, P < 0.05 versus control. (B) GI-GPx mRNA expression in CaCo-2 cells. Selenium-supplemented (50 nM sodium selenite) CaCo-2 cells were grown to confluence for 3 days and stimulated with tBHQ (20 μM) or SFN (5 μM) for 8 h. RNA was extracted, reverse transcribed, and amplified by PCR. β-Actin was taken as reference and was not influenced by Nrf2 activators. PCR products were separated on agarose gels and quantified densitometrically. Values are means of three experiments measured in duplicate ± standard deviation. #, P < 0.05 versus control. (C) Expression of endogenous GI-GPx. CaCo-2 cells were grown as in panel B and stimulated with either tBHQ (20 μM) or SFN (5 μM) for 48 h. Cell lysates (75 μg protein per lane) were analyzed for GI-GPx by Western blotting. Results are representative of three independent experiments.
Induction of endogenous GI-GPx by Nrf2 activators.
The expression of GI-GPx as a selenoprotein is primarily regulated at the translational level by the availability of selenocysteyl-tRNA and, thus, by the selenium supply (27). Therefore, an increase in promoter activity might not necessarily be reflected in an increase in the protein content. In HepG2 cells, the levels of endogenous GI-GPx were accordingly higher, if the medium was supplemented with sodium selenite. Surprisingly, however, these cells did not respond to Nrf2 activators with an increase of GI-GPx production irrespective of their selenium status. In contrast to reporter gene-transfected HepG2 cells, the endogenous GI-GPx mRNA did not respond to the Nrf2 activators either. In this cell line, which is characterized by a high basal level of gpx2 expression, the Nrf2-mediated regulation of gpx2 is evidently obscured by dominant regulatory phenomena that may be specific for this type of cancer cell, which, however, remains to be elucidated. In contrast, in CaCo-2 cells, which express a comparatively low level of endogenous GI-GPx, the GI-GPx promoter responded, as predicted from the reporter gene experiments: In selenite-supplemented (50 nM) CaCo-2 cells, GI-GPx mRNA (Fig. 7B) and protein (Fig. 7C) were clearly enhanced by tBHQ and SFN, whereas in selenium-deficient CaCo-2 cells, GI-GPx protein remained below the detection level despite high mRNA levels (not shown). This shows that tBHQ and SFN not only activate transfected reporter genes but indeed induce endogenous GI-GPx. The activation appears to be particularly effective when the basal expression of GI-GPx is low.
DISCUSSION
The GI-GPx promoter proved to contain a functionally active ARE that is regulated by the Nrf2/Keap1 system, as is demonstrated by its activity in reporter gene constructs, be it inserted as isolated enhancer or left within a >2,000-bp promoter sequence. Binding of Nrf2 to the ARE was demonstrated by EMSA, and binding of Nrf2 to the endogenous GI-GPx promoter was verified by ChIP analyses. Also, typical Nrf2 activators and overexpressed Nrf2 itself induced the ARE-driven reporter gene and equally induced endogenous GI-GPx, and this induction could be antagonized by overexpression of Keap1. We thus demonstrate that the GI-GPx gene is a novel target for Nrf2.
Up-regulation of a peroxidase that is considered to be part of the antioxidant defense system via ARE is indeed intriguing. Rather an up-regulation by oxidants had to be anticipated. In line with this expectation, cGPx is evidently induced by oxidative stress (reviewed in reference 24). Two oxygen-responsive elements were detected in the cGPx promoter, whose activity is increased upon oxygen exposure (17). However, the regulation of this key player in hydroperoxide detoxification is complex: cGPx promoter activity was shown to be activated in human osteogenic sarcoma (Saos-2) cells by the tumor suppressor p53 (57) and in neutrophils by PU.1 (58). Gender-specific expression levels of cGPx have been known for decades (49) but remained mechanistically unexplored. A regulation of cGPx by the Nrf2 system can almost be excluded. Analysis of the 1,001 base pairs of the published cGPx promoter sequence (accession number AF029317) and of a 2,590-bp region of the human chromosome 3 containing the 5′-untranslated region of cGPx (accession number AC121247) did not reveal any motif reminiscent of ARE, and tBHQ or sulforaphane did not enhance cGPx mRNA in HepG2 or in CaCo-2 cells (our unpublished observations). The extracellular GPx (GPx3) appears to be regulated inversely to cGPx. Its promoter contains a responsive element for the hypoxia-inducible factor-1 (HIF-1) and is activated under hypoxic conditions (4). The same authors describe a putative ARE; however, its functionality has not been investigated yet (4). Little is known about the regulation of PHGPx. Its gene is composed of eight exons. It is transcribed into three different mRNAs, leading to three isoenzymes differing in their N-terminal extensions, a cytosolic, a mitochondrial, and a nuclear PHGPx. The first exon contains two ATGs, the more 5′ one encoding the start for the mitochondrial enzyme (50) and the second one encoding the start for the cytosolic enzyme (9). The promoter regulating both forms of PHGPx contains a functional NF-Y site (32, 59) and SP1/SP3 and members of the SMAD family binding sites (59). An alternative promoter lies in the first intron of the PHGPx gene, which guides the expression of the nuclear form of PHGPx (40, 42). It contains binding sites for EGR1 and SREBP1 (7). Intriguing tissue-specific expression patterns were reported for PHGPx (41), which, however, could not yet be explained by any particular mechanism. Needless to state, the transcriptional regulation of GI-GPx is also complex. Its expression could, e.g., be induced by retinoic acid in MCF-7 breast cancer cells (16), for the first time linking GI-GPx expression to cancer. Taken together, neither the promoters nor the expression patterns of the individual GPx types display any obvious similarities. This observation suggests that it might be naïve to deduce a similar biological role from the common ability of these enzymes to reduce hydroperoxides. Instead, the, in part, opposite regulatory phenomena can be taken as evidence for their diversified functions that are amply discussed elsewhere (8, 23, 60).
Before discussing the potential consequences of ARE-mediated GI-GPx up-regulation, it may be recalled that the term “electrophilic responsive element (EpRE)” (26) more adequately describes the biological role of ARE. Many compounds activating ARE/EpRE have been mislabeled as antioxidants simply because they can scavenge radicals in vitro. In the biological context under consideration, however, they clearly act as SH-modifying electrophiles. This implies that sulforaphane, curcumin, and tBHQ do not likely induce GI-GPx because they counteract oxidative stress, but rather mimic an oxidation of protein thiols, as is most sensitively recognized by Keap1. Viewed this way, up-regulation of GI-GPx by “antioxidants” via Nrf2/Keap1 is in line with the general pattern of ARE-mediated regulation of detoxifying enzymes.
Whereas in cell culture systems used here, GI-GPx could unequivocally be identified as a target for Nrf2, the verification that this also holds true in vivo awaits further investigations. The lack of GI-GPx induction by hyperoxia or bleomycin in Nrf2 KO mice, however, points into this direction (11, 12). Moreover, basal expression of GI-GPx mRNA in the intestine of male and female Nrf2 KO mice was distinctly decreased (T. Suzuki and M. Yamamoto, personal communication), providing further evidence for the relevance of an Nrf2-regulated GI-GPx expression in vivo.
The phenotype of GI-GPx KO clearly points to a relevant role of the enzyme in gastrointestinal hydroperoxide metabolism. Like cGPx KO mice (31), GI-GPx KO mice remain asymptomatic (21), and even the double-KO mice do not display any obvious phenotype if grown under germfree conditions. Upon colonization with an intestinal flora that is tolerated in wild-type mice, they, however, first develop a kind of inflammatory bowel disease (20) and finally develop multiple and different malignant tumors in the lower intestine (14). Prevention of food-borne peroxides by itself cannot explain this phenomenon, since the germfree double-KO mice should have been similarly affected. More likely, the two synergistic peroxidases counteract the synthesis of proinflammatory mediators, such as prostaglandins and leukotrienes, which are formed in response to bacterial toxins. Silencing of cyclooxygenases and lipoxygenases appears to be a potential common to glutathione peroxidases (28, 54, 56, 64), since the cyclooxygenases and lipoxygenases require a certain peroxide tone for activity. A second ability common to glutathione peroxidases might be equally important: They inhibit apoptosis (37, 48; reviewed in reference 46). In the gastrointestinal system, a gradient of GI-GPx decreasing from the ground of the crypts to the top of the villi is superimposed onto an equal level of cGPx (22). This phenomenon may be related to the balance of proliferation and apoptosis of the gastrointestinal epithelium. Acceleration of the physiological apoptosis toward the tip of the villi due to GI-GPx deficiency might facilitate invasion of opportunistic bacteria and thereby inflammatory responses. In GPx1/2-KO mice, apoptotic cells are increased in ileal crypts, pointing to the role of GI-GPx in regulating apoptosis (14). The role of GI-GPx in inflammation and cancer has been comprehensively reviewed and discussed by Chu et al. (15), with the conclusion that an increased expression of GI-GPx prevents cancer by inhibiting preceding inflammation rather than being involved in the development of cancer itself. The experience with the GPx1/2-KO mice tends to forecast beneficial effects of compounds that guarantee optimum enzyme levels or induce these enzymes beyond baseline levels; they will dampen the tendency to develop inflammatory bowel disease, which, if chronic, predisposes to malignant transformation. The still most important “optimizer” of cGPx is the alimentary selenium supply, while GI-GPx, because of its high rank in the hierarchy of selenoproteins, cannot be expected to respond to moderate variations in dietary selenium. By Nrf2 activators, however, the basal GI-GPx levels can easily be doubled in cells of the gastrointestinal system and, interestingly, the natural anticarcinogens sulforaphane and curcumin appear to be particularly efficient in preventing tumors of the gastrointestinal tract. It therefore appears justified to add the induction of GI-GPx via Nrf2/Keap1 to the working hypotheses on how these compounds decrease the risk of developing gastrointestinal cancers.
Acknowledgments
We are grateful to M. Yamamoto, A. Kobayashi, and T. Suzuki (Center for TARA, University of Tsukuba, Japan) for kindly providing the plasmids pcDNA3-mNrf2 and pcDNA3-mKeap1 and communicating unpublished data. We further thank J.-U. Bittner and E. Krohn for excellent technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft, DFG, grant Br 778/5-3.
REFERENCES
- 1.Alam, J., and J. L. Cook. 2003. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr. Pharm. Des. 9:2499-2511. [DOI] [PubMed] [Google Scholar]
- 2.Ayabe, T., D. P. Satchell, C. L. Wilson, W. C. Parks, M. E. Selsted, and A. J. Ouellette. 2000. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 1:113-118. [DOI] [PubMed] [Google Scholar]
- 3.Banning, A., K. Schnurr, G. F. Böl, D. Kupper, K. Müller-Schmehl, H. Viita, S. Yla-Herttuala, and R. Brigelius-Flohé. 2004. Inhibition of basal and interleukin-1-induced VCAM-1 expression by phospholipid hydroperoxide glutathione peroxidase and 15-lipoxygenase in rabbit aortic smooth muscle cells. Free Radic. Biol. Med. 36:135-144. [DOI] [PubMed] [Google Scholar]
- 4.Bierl, C., B. Voetsch, R. C. Jin, D. E. Handy, and J. Loscalzo. 2004. Determinants of human plasma glutathione peroxidase (GPx-3) expression. J. Biol. Chem. 279:26839-26845. [DOI] [PubMed] [Google Scholar]
- 5.Böcher, M., T. Böldicke, M. Kiess, and U. Bilitewski. 1997. Synthesis of mono- and bifunctional peptide-dextran conjugates for the immobilization of peptide antigens on ELISA plates: properties and application. J. Immunol. Methods 208:191-202. [DOI] [PubMed] [Google Scholar]
- 6.Böl, G. F., N. Jurrmann, and R. Brigelius-Flohé. 2003. Recruitment of the interleukin-1 receptor (IL-1RI)-associated kinase IRAK to the IL-1RI is redox regulated. Biol. Chem. 384:609-617. [DOI] [PubMed] [Google Scholar]
- 7.Borchert, A., N. E. Savaskan, and H. Kühn. 2003. Regulation of expression of the phospholipid hydroperoxide/sperm nucleus glutathione peroxidase gene. Tissue-specific expression pattern and identification of functional cis- and trans-regulatory elements. J. Biol. Chem. 278:2571-2580. [DOI] [PubMed] [Google Scholar]
- 8.Brigelius-Flohé, R. 1999. Tissue-specific functions of individual glutathione peroxidases. Free Radic. Biol. Med. 27:951-965. [DOI] [PubMed] [Google Scholar]
- 9.Brigelius-Flohé, R., K. D. Aumann, H. Blocker, G. Gross, M. Kiess, K. D. Kloppel, M. Maiorino, A. Roveri, R. Schuckelt, F. Ursini, E. Wingender, and L. Flohé. 1994. Phospholipid-hydroperoxide glutathione peroxidase. Genomic DNA, cDNA, and deduced amino acid sequence. J. Biol. Chem. 269:7342-7348. [PubMed] [Google Scholar]
- 10.Brigelius-Flohé, R., C. Müller, J. Menard, S. Florian, K. Schmehl, and K. Wingler. 2001. Functions of GI-GPx: lessons from selenium-dependent expression and intracellular localization. Biofactors 14:101-106. [DOI] [PubMed] [Google Scholar]
- 11.Cho, H. Y., A. E. Jedlicka, S. P. Reddy, T. W. Kensler, M. Yamamoto, L. Y. Zhang, and S. R. Kleeberger. 2002. Role of Nrf2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell Mol. Biol. 26:175-182. [DOI] [PubMed] [Google Scholar]
- 12.Cho, H. Y., S. P. Reddy, M. Yamamoto, and S. R. Kleeberger. 2004. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J. 18:1258-1260. [DOI] [PubMed] [Google Scholar]
- 13.Chu, F. F., J. H. Doroshow, and R. S. Esworthy. 1993. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J. Biol. Chem. 268:2571-2576. [PubMed] [Google Scholar]
- 14.Chu, F. F., R. S. Esworthy, P. G. Chu, J. A. Longmate, M. M. Huycke, S. Wilczynski, and J. H. Doroshow. 2004. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 64:962-968. [DOI] [PubMed] [Google Scholar]
- 15.Chu, F. F., R. S. Esworthy, and J. H. Doroshow. 2004. Role of Se-dependent glutathione peroxidases in gastrointestinal inflammation and cancer. Free Radic. Biol. Med. 36:1481-1495. [DOI] [PubMed] [Google Scholar]
- 16.Chu, F. F., R. S. Esworthy, L. Lee, and S. Wilczynski. 1999. Retinoic acid induces Gpx2 gene expression in MCF-7 human breast cancer cells. J. Nutr. 129:1846-1854. [DOI] [PubMed] [Google Scholar]
- 17.Cowan, D. B., R. D. Weisel, W. G. Williams, and D. A. Mickle. 1993. Identification of oxygen responsive elements in the 5′-flanking region of the human glutathione peroxidase gene. J. Biol. Chem. 268:26904-26910. [PubMed] [Google Scholar]
- 18.Dinkova-Kostova, A. T., W. D. Holtzclaw, R. N. Cole, K. Itoh, N. Wakabayashi, Y. Katoh, M. Yamamoto, and P. Talalay. 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 99:11908-11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dinkova-Kostova, A. T., M. A. Massiah, R. E. Bozak, R. J. Hicks, and P. Talalay. 2001. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc. Natl. Acad. Sci. USA 98:3404-3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esworthy, R. S., R. Aranda, M. G. Martin, J. H. Doroshow, S. W. Binder, and F. F. Chu. 2001. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 281:G848-G855. [DOI] [PubMed] [Google Scholar]
- 21.Esworthy, R. S., J. R. Mann, M. Sam, and F. F. Chu. 2000. Low glutathione peroxidase activity in Gpx1 knockout mice protects jejunum crypts from gamma-irradiation damage. Am. J. Physiol. Gastrointest. Liver Physiol. 279:G426-G436. [DOI] [PubMed] [Google Scholar]
- 22.Esworthy, R. S., K. M. Swiderek, Y. S. Ho, and F. F. Chu. 1998. Selenium-dependent glutathione peroxidase-GI is a major glutathione peroxidase activity in the mucosal epithelium of rodent intestine. Biochim. Biophys. Acta 1381:213-226. [DOI] [PubMed] [Google Scholar]
- 23.Flohé, L., and R. Brigelius-Flohé. 2001. Selenoproteins of the glutathione system, p. 157-178. In D. L. Hatfield (ed.), Selenium: its molecular biology and role in human health. Kluwer Academic Publishers, Boston, Mass.
- 24.Flohé, L., E. Wingender, and R. Brigelius-Flohé. 1997. The regulation of glutathione peroxidases, p. 415-435. In H. Forman and E. Cadenas (ed.), Oxidative stress and signal transduction. Chapman & Hall, New York, N.Y.
- 25.Florian, S., K. Wingler, K. Schmehl, G. Jacobasch, O. J. Kreuzer, W. Meyerhof, and R. Brigelius-Flohé. 2001. Cellular and subcellular localization of gastrointestinal glutathione peroxidase in normal and malignant human intestinal tissue. Free Radic. Res. 35:655-663. [DOI] [PubMed] [Google Scholar]
- 26.Friling, R. S., A. Bensimon, Y. Tichauer, and V. Daniel. 1990. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proc. Natl. Acad. Sci. USA 87:6258-6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hatfield, D. L., and V. N. Gladyshev. 2002. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 22:3565-3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haurand, M., and L. Flohé. 1988. Kinetic studies on arachidonate 5-lipoxygenase from rat basophilic leukemia cells. Biol. Chem. Hoppe-Seyler 369:133-142. [DOI] [PubMed] [Google Scholar]
- 29.Hayes, J. D., and M. McMahon. 2001. Molecular basis for the contribution of the antioxidant responsive element to cancer chemoprevention. Cancer Lett. 174:103-113. [DOI] [PubMed] [Google Scholar]
- 30.Ho, S. N., H. D. Hunt, R. M. Horton, J. K. Pullen, and L. R. Pease. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51-59. [DOI] [PubMed] [Google Scholar]
- 31.Ho, Y. S., J. L. Magnenat, R. T. Bronson, J. Cao, M. Gargano, M. Sugawara, and C. D. Funk. 1997. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 272:16644-16651. [DOI] [PubMed] [Google Scholar]
- 32.Huang, H. S., C. J. Chen, and W. C. Chang. 1999. The CCAAT-box binding factor NF-Y is required for the expression of phospholipid hydroperoxide glutathione peroxidase in human epidermoid carcinoma A431 cells. FEBS Lett. 455:111-116. [DOI] [PubMed] [Google Scholar]
- 33.Itoh, K., T. Chiba, S. Takahashi, T. Ishii, K. Igarashi, Y. Katoh, T. Oyake, N. Hayashi, K. Satoh, I. Hatayama, M. Yamamoto, and Y. Nabeshima. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 236:313-322. [DOI] [PubMed] [Google Scholar]
- 34.Itoh, K., N. Wakabayashi, Y. Katoh, T. Ishii, K. Igarashi, J. D. Engel, and M. Yamamoto. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13:76-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang, K. W., S. J. Lee, J. W. Park, and S. G. Kim. 2002. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol. Pharmacol. 62:1001-1010. [DOI] [PubMed] [Google Scholar]
- 36.Kang, M. I., A. Kobayashi, N. Wakabayashi, S. G. Kim, and M. Yamamoto. 2004. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 101:2046-2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kayanoki, Y., J. Fujii, K. N. Islam, K. Suzuki, S. Kawata, Y. Matsuzawa, and N. Taniguchi. 1996. The protective role of glutathione peroxidase in apoptosis induced by reactive oxygen species. J. Biochem. (Tokyo) 119:817-822. [DOI] [PubMed] [Google Scholar]
- 38.Kryukov, G. V., S. Castellano, S. V. Novoselov, A. V. Lobanov, O. Zehtab, R. Guigo, and V. N. Gladyshev. 2003. Characterization of mammalian selenoproteomes. Science 300:1439-1443. [DOI] [PubMed] [Google Scholar]
- 39.Lin, Y. M., Y. Furukawa, T. Tsunoda, C. T. Yue, K. C. Yang, and Y. Nakamura. 2002. Molecular diagnosis of colorectal tumors by expression profiles of 50 genes expressed differentially in adenomas and carcinomas. Oncogene 21:4120-4128. [DOI] [PubMed] [Google Scholar]
- 40.Maiorino, M., M. Scapin, F. Ursini, M. Biasolo, V. Bosello, and L. Flohé. 2003. Distinct promoters determine alternative transcription of gpx-4 into phospholipid-hydroperoxide glutathione peroxidase variants. J. Biol. Chem. 278:34286-34290. [DOI] [PubMed] [Google Scholar]
- 41.Maiorino, M., J. B. Wissing, R. Brigelius-Flohé, F. Calabrese, A. Roveri, P. Steinert, F. Ursini, and L. Flohé. 1998. Testosterone mediates expression of the selenoprotein PHGPx by induction of spermatogenesis and not by direct transcriptional gene activation. FASEB J. 12:1359-1370. [DOI] [PubMed] [Google Scholar]
- 42.Moreno, S. G., G. Laux, M. Brielmeier, G. W. Bornkamm, and M. Conrad. 2003. Testis-specific expression of the nuclear form of phospholipid hydroperoxide glutathione peroxidase (PHGPx). Biol. Chem. 384:635-643. [DOI] [PubMed] [Google Scholar]
- 43.Mörk, H., O. H. Al-Taie, K. Bahr, A. Zierer, C. Beck, M. Scheurlen, F. Jakob, and J. Köhrle. 2000. Inverse mRNA expression of the selenocysteine-containing proteins GI-GPx and SeP in colorectal adenomas compared with adjacent normal mucosa. Nutr. Cancer 37:108-116. [DOI] [PubMed] [Google Scholar]
- 44.Mörk, H., M. Scheurlen, O. Al-Taie, A. Zierer, M. Kraus, K. Schottker, F. Jakob, and J. Köhrle. 2003. Glutathione peroxidase isoforms as part of the local antioxidative defense system in normal and Barrett's esophagus. Int. J. Cancer 105:300-304. [DOI] [PubMed] [Google Scholar]
- 45.Müller, C., K. Wingler, and R. Brigelius-Flohé. 2003. 3′UTRs of glutathione peroxidases differentially affect selenium-dependent mRNA stability and selenocysteine incorporation efficiency. Biol. Chem. 384:11-18. [DOI] [PubMed] [Google Scholar]
- 46.Nakagawa, Y. 2004. Role of mitochondrial phospholipid hydroperoxide glutathione peroxidase (PHGPx) as a antiapoptotic factor. Biol. Pharm. Bull. 27:956-960. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen, T., P. J. Sherratt, and C. B. Pickett. 2003. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43:233-260. [DOI] [PubMed] [Google Scholar]
- 48.Nomura, K., H. Imai, T. Koumura, M. Arai, and Y. Nakagawa. 1999. Mitochondrial phospholipid hydroperoxide glutathione peroxidase suppresses apoptosis mediated by a mitochondrial death pathway. J. Biol. Chem. 274:29294-29302. [DOI] [PubMed] [Google Scholar]
- 49.Pinto, R. E., and W. Bartley. 1969. The nature of the sex-linked differences in glutathione peroxidase activity and aerobic oxidation of glutathione in male and female rat liver. Biochem. J. 115:449-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pushpa-Rekha, T. R., A. L. Burdsall, L. M. Oleksa, G. M. Chisolm, and D. M. Driscoll. 1995. Rat phospholipid-hydroperoxide glutathione peroxidase.cDNA cloning and identification of multiple transcription and translation start sites. J. Biol. Chem. 270:26993-26999. [DOI] [PubMed] [Google Scholar]
- 51.Quandt, K., K. Frech, H. Karas, E. Wingender, and T. Werner. 1995. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 23:4878-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramos-Gomez, M., M. K. Kwak, P. M. Dolan, K. Itoh, M. Yamamoto, P. Talalay, and T. W. Kensler. 2001. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl. Acad. Sci. USA 98:3410-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rushmore, T. H., M. R. Morton, and C. B. Pickett. 1991. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem. 266:11632-11639. [PubMed] [Google Scholar]
- 54.Schnurr, K., J. Belkner, F. Ursini, T. Schewe, and H. Kühn. 1996. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase controls the activity of the 15-lipoxygenase with complex substrates and preserves the specificity of the oxygenation products. J. Biol. Chem. 271:4653-4658. [DOI] [PubMed] [Google Scholar]
- 55.Serewko, M. M., C. Popa, A. L. Dahler, L. Smith, G. M. Strutton, W. Coman, A. J. Dicker, and N. A. Saunders. 2002. Alterations in gene expression and activity during squamous cell carcinoma development. Cancer Res. 62:3759-3765. [PubMed] [Google Scholar]
- 56.Smith, W. L., and W. E. Lands. 1972. Oxygenation of polyunsaturated fatty acids during prostaglandin biosynthesis by sheep vesicular gland. Biochemistry 11:3276-3285. [DOI] [PubMed] [Google Scholar]
- 57.Tan, M., S. Li, M. Swaroop, K. Guan, L. W. Oberley, and Y. Sun. 1999. Transcriptional activation of the human glutathione peroxidase promoter by p53. J. Biol. Chem. 274:12061-12066. [DOI] [PubMed] [Google Scholar]
- 58.Throm, S. L., and M. J. Klemsz. 2003. PU.1 regulates glutathione peroxidase expression in neutrophils. J. Leukoc. Biol. 74:111-117. [DOI] [PubMed] [Google Scholar]
- 59.Ufer, C., A. Borchert, and H. Kühn. 2003. Functional characterization of cis- and trans-regulatory elements involved in expression of phospholipid hydroperoxide glutathione peroxidase. Nucleic Acids Res. 31:4293-4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ursini, F., M. Maiorino, R. Brigelius-Flohé, K. D. Aumann, A. Roveri, D. Schomburg, and L. Flohé. 1995. Diversity of glutathione peroxidases. Methods Enzymol. 252:38-53. [DOI] [PubMed] [Google Scholar]
- 61.Verhoeven, D. T., H. Verhagen, R. A. Goldbohm, P. A. van den Brandt, and G. van Poppel. 1997. A review of mechanisms underlying anticarcinogenicity by brassica vegetables. Chem. Biol. Interact. 103:79-129. [DOI] [PubMed] [Google Scholar]
- 62.Wakabayashi, N., A. T. Dinkova-Kostova, W. D. Holtzclaw, M. I. Kang, A. Kobayashi, M. Yamamoto, T. W. Kensler, and P. Talalay. 2004. Protection against electrophile and oxidant stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc. Natl. Acad. Sci. USA 101:2040-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wasserman, W. W., and W. E. Fahl. 1997. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 94:5361-5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weitzel, F., and A. Wendel. 1993. Selenoenzymes regulate the activity of leukocyte 5-lipoxygenase via the peroxide tone. J. Biol. Chem. 268:6288-6292. [PubMed] [Google Scholar]
- 65.Wingler, K., M. Böcher, L. Flohé, H. Kollmus, and R. Brigelius-Flohé. 1999. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 259:149-157. [DOI] [PubMed] [Google Scholar]
- 66.Wingler, K., C. Müller, K. Schmehl, S. Florian, and R. Brigelius-Flohé. 2000. Gastrointestinal glutathione peroxidase prevents transport of lipid hydroperoxides in CaCo-2 cells. Gastroenterology 119:420-430. [DOI] [PubMed] [Google Scholar]