

Abstract
Interactions between steroid hormone receptors and signal transducer and activator of transcription (Stat)-mediated signaling pathways have already been described. In the present study, we explored the capacity of progestins to modulate Stat3 transcriptional activation in an experimental model of hormonal carcinogenesis in which the synthetic progestin medroxyprogesterone acetate (MPA) induced mammary adenocarcinomas in BALB/c mice and in the human breast cancer cell line T47D. We found that C4HD epithelial cells, from the MPA-induced mammary tumor model, expressed Stat3 and that MPA treatment of C4HD cells up-regulated Stat3 protein expression. In addition, MPA induced rapid, nongenomic Stat3, Jak1, and Jak2 tyrosine phosphorylation in C4HD and T47D cells. MPA treatment of C4HD cells also resulted in rapid c-Src tyrosine phosphorylation. These effects were completely abolished by the progestin antagonist RU486. Abrogation of Jak1 and Jak2 activity by transient transfection of C4HD cells with dominant negative (DN) Jak1 or DN Jak2 vectors, or inhibition of Src activity by preincubation of cells with the Src family kinase inhibitor PP2, blocked the capacity of MPA to induce Stat3 phosphorylation. Treatment of C4HD cells with MPA induced Stat3 binding to DNA. In addition, MPA promoted strong Stat3 transcriptional activation in C4HD and T47D cells that was inhibited by RU486 and by blockage of Jak1, Jak2, and Src activities. To investigate the correlation between MPA-induced Stat3 activation and cell growth, C4HD cells were transiently transfected with a DN Stat3 expression vector, Stat3Y705-F, or with a constitutively activated Stat3 mutant, Stat3-C. While expression of Stat3Y705-F mutant had an inhibitory effect on MPA-induced growth of C4HD cells, transfection with the constitutively activated Stat3-C vector resulted in MPA-independent proliferation. Finally, we addressed the effect of targeting Stat3 in in vivo growth of C4HD breast tumors. Blockage of Stat3 activation by transfection of C4HD cells with the DN Stat3Y705-F expression vector significantly inhibited these cells' ability to form tumors in syngeneic mice. Our results have for the first time demonstrated that progestins are able to induce Stat3 transcriptional activation, which is in turn an obligatory requirement for progestin stimulation of both in vitro and in vivo breast cancer growth.
Progesterone regulates diverse biological effects in a broad range of tissues, mostly by interaction with the classical progesterone receptor (PR), a member of the nuclear receptor superfamily of ligand-dependent transcription factors. Particularly in the mammary gland, progesterone plays a key role in the control of cell proliferation and differentiation (31 and references within). Accumulated evidence also indicates that progestins are involved in controlling mammary gland tumorigenesis, both in women and in animal models (6, 8, 15, 21, 24, 26, 31, 32, 48). Although the mechanisms by which progestins stimulate growth of breast cancer cells have not been completely deciphered, several lines of evidence (6, 26, 33), including our own work (3, 4, 25, 44), have shown that convergence between progestins and growth factor (GF) signaling pathways mediates proliferative effects of progestins in mammary tumor cells.
In addition to their direct transcriptional effects, rapid or nongenomic biological effects of progestins have been described in several species, including fish, amphibian, and mammalian (27). Whether the recently cloned membrane PR from humans and other vertebrates (59) is involved in mediating nongenomic progestin effects in mammalians remains to be elucidated. Nongenomic effects of progestins in breast cancer cells have been unraveled by startling reports from Auricchio and coworkers (5, 13, 33), who demonstrated that progestin treatment of human breast cancer T47D cells activates the signal-transducing c-Src/p21ras/MAPK pathway, which results in cell proliferation (5, 13, 33). Progestin activation of the c-Src/p21ras/MAPK pathway has also been described by Edwards and coworkers (6), who further explored mechanisms involved in progestin modulation of c-Src activity.
Over the last years, a unique family of proteins, the signal transducers and activators of transcription (Stats), was found to be involved in cross talks with both steroid hormones and GF signaling pathways. There are seven Stat genes in mammals, Stat1, Stat2, Stat3, Stat4, Stat5a, Stat5b, and Stat6 (9). They have dual functions: as signaling molecules in the cytoplasm and as transcription factors following nuclear translocation (9, 55). Cytoplasmic Stats were found to be recruited to and activated either by tyrosine kinase receptors (RTKs), after binding of GFs (38, 42, 50), or by soluble tyrosine kinases of the Janus (JAK) and Src kinase families, after binding of cytokines to their receptors (16, 52). Tyrosine-phosphorylated Stats then dimerize and translocate to the nucleus, where they bind DNA and regulate gene transcription.
Interaction between steroid hormone receptors and Stat-mediated pathways has a bidirectional nature, where steroid hormone receptors regulate Stat-dependent transcription and where, conversely, Stats are able to modulate steroid hormone-mediated transcription (18, 51, 58). Functional interactions between progestins and Stats were demonstrated by Lange et al. (26) and Richer et al. (43), who found that treatment of T47D human breast cancer cells with the synthetic progestin R5020 resulted in up-regulation of Stat3 and Stat5 protein levels. In addition, these authors described constitutive association between Stat5 and PRB in HeLa cells transfected with the B isoform of PR (43). Recently, progesterone was found to induce Stat5a expression in MDA-MB-231 breast cancer cells transfected with PR (29). Progesterone also enhanced Stat3 cytoplasmic stores in the decidualized mesometrium during pregnancy in rats (30), an effect which was inhibited by the progestin antagonist RU486, showing that Stat3 is a progesterone-dependent protein. On the other hand, involvement of different RTKs in the activation of Stat proteins has been reported. The best elucidated are the roles of the epidermal growth factor receptor (EGF-R/ErbB-1) (38, 42) and the platelet-derived growth factor receptor (50). Interestingly, constitutive activation of Stats, especially of Stat3 and Stat5, has been found in transformed cells and in a variety of human tumor types, supporting the concept that Stats play a role in malignant transformation (55).
We have already exhaustively characterized mechanisms of progestin-induced growth in an experimental model of hormonal carcinogenesis in which the synthetic progestin medroxyprogesterone acetate (MPA) induced mammary adenocarcinomas in female BALB/c mice (3, 4, 25, 44). In the present study, we addressed the question of whether Stats might play a role in the scenario of progestin-induced growth of breast cancer. We focused our present study on Stat3 not only because accumulated evidence indicates that its activation is associated with cell transformation (9, 11, 55) but also because tumor-derived cell lines or samples from human breast cancer frequently contain activated forms of Stat3 (12, 20, 28, 53). Our findings have for the first time demonstrated that progestins are able to induce Stat3 transcriptional activation in both mouse and human mammary tumor cells by a mechanism requiring Jak and c-Src activities. We also found that the presence of activated Stat3 is a requisite for progestin stimulation of in vitro and in vivo breast cancer growth. Moreover, in the course of our studies of the molecular mechanisms involved in progestin-induced Stat3 activation, we unfolded several rapid, nongenomic progestin effects. Thus, we have here for the first time described the ability of progestin to induce rapid Jak1, Jak2, and Stat3 tyrosine phosphorylation.
MATERIALS AND METHODS
Animals and tumors.
Experiments were carried out with virgin female BALB/c mice raised at the Institute of Biology and Experimental Medicine of Buenos Aires. All animal studies were conducted in accordance with the highest standards of animal care as outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institute of Biology and Experimental Medicine Animal Research Committee. Hormone-dependent ductal tumor line C4HD originated in mice treated with 40 mg MPA every 3 months for 1 year and has been maintained by serial transplantation in animals treated with a 40-mg subcutaneous (s.c.) MPA depot in the flank opposite the tumor inoculum (3, 4, 25, 44). The C4HD tumor line is of ductal origin, expresses PR and estrogen receptor (3, 4, 25, 44), and lacks glucocorticoid receptor and EGF-R/ErbB-1 expression (4, 25).
Cell culture and treatments.
Primary cultures of epithelial cells from C4HD tumors growing in MPA-treated mice were performed as previously described (3, 4, 25, 44). Cells were incubated in Dulbecco modified Eagle medium (DMEM)-F12 (without phenol red, with 100 U/ml penicillin, and with 100 μ/ml streptomycin) with 0.1% charcoal-stripped fetal calf serum (ChFCS) in the presence or absence of 10 nM MPA and MPA-10 nM RU486. To block c-Src activation, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) (10 μM, dissolved in 1:2,000 dimethyl sulfoxide) was added to cells 90 min before incubation with MPA. Controls were performed in order to verify that dimethyl sulfoxide (1:2,000) did not modify MPA-modulated c-Src tyrosine phosphorylation. Proliferation of C4HD cells was evaluated by propidium iodide (PI) staining and flow cytometry analysis, as described below. T47D cells were obtained from the American Type Culture Collection. T47D-Y cells were a generous gift from K. Horwitz (University of Colorado Health Sciences Center, Denver). LM3 cells were cultured as previously described (40). In experiments assessing the effect of MPA on Stat3 activity, T47D, T47D-Y, and LM3 cells were cultured in DMEM (without phenol red) supplemented with 0.1% ChFCS and subjected to the treatments described above for C4HD cells.
Transient transfections.
In experiments assessing the roles of Jak1 and Jak2 in MPA-induced effects on Stat3, C4HD and T47D cells were transiently transfected with 2 μg of a dominant negative (DN) Jak1 vector or with 2 μg of a DN Jak2 vector (38, 39), kindly provided by O. Silvennoinen (Tampere University Hospital, Finland) via N. Hynes (Friedrich Miescher Institute, Basel, Switzerland). C4HD cells were transfected for 48 h in DMEM-F12 supplemented with 10 nM MPA and 2.5% ChFCS, and T47D cells were transfected for 48 h in DMEM with 10% ChFCS without antibiotics. The Fugene 6 transfection reagent technique (Roche Biochemicals) was used in accordance with the manufacturer's instructions. After transfection, cells were washed and cultured for 24 h in 0.1% ChFCS before treatment with MPA for the indicated times. Total cell lysates were then prepared as described below for use in Western blot assays. To study the role of Stat3 in proliferation, C4HD cells were transiently transfected, as described above, for 48 h with 2 μg of a DN Stat3 expression vector, Stat3Y705-F (10, 23, 28), which carries a tyrosine-to-phenylalanine substitution at codon 705 that reduces phosphorylation on tyrosine of the wild-type Stat3 protein, therefore inhibiting both dimerization and DNA binding of Stat3. C4HD cells were also transfected with 2 μg of a constitutively activated Stat3 mutant, Stat3-C, which dimerizes spontaneously, binds to DNA, and activates transcription (11). Both were kindly provided by J. Darnell (New York, NY). As a control, cells were transfected with 2 μg of the empty pRc/CMV vector. After transfection, cells were washed and cultured for 24 h in 0.1% ChFCS before treatment with MPA for 48 h. Cells were then harvested for flow cytometry. Transfection efficiency in C4HD cells was evaluated using the pEGFP-N1 vector (BD Biosciences Clontech, Palo Alto, CA) and determined by the percentage of cells that exhibited green fluorescence 24 h after transfection. Green fluorescent protein was visualized by direct fluorescence imaging using a Nikon Eclipse E800 confocal laser microscopy system (Nikon Instruments, Inc., Melville, NY). In C4HD cells, fluorescence intensities were also measured by fluorescence-activated cell sorter. To investigate the capacity of MPA to induce the transcriptional activation of Stat3, C4HD and T47D cells were transiently transfected with 2 μg of a luciferase reporter plasmid containing four copies of the m67 high-affinity binding site (11, 56) and 1 μg of a β-galactosidase expression plasmid, CMV-βgal (Clontech, Palo Alto, CA), used to correct variations in transfection efficiency. As a control, cells were also transfected with a pTATA-Luc reporter lacking the m67 insertion (11, 56). In experiments assessing the role of Jaks in Stat3 transcriptional activation, cells were cotransfected with 2 μg DN Jak1 or 2 μg DN Jak2 expression vector. The total amount of transfected DNA was standardized by adding the pTATA-Luc reporter lacking the m67 insertion. Cells were then treated with MPA or MPA-RU486 or preincubated with the c-Src inhibitor PP2 for 90 min and then treated with MPA for 48 h or were left untreated growing in ChFCS. Cells were washed once with phosphate-buffered saline without calcium and magnesium and then harvested and lysed in 1× reporter buffer (Promega, Madison, Wis.) using one freeze-thaw cycle. Protein concentrations were determined using a Bio-Rad kit (Bio-Rad, Hercules, CA). Luciferase results were calculated as the ratio of luciferase activity per unit of β-galactosidase activity. Duplicate samples were analyzed for each datum point.
Cell cycle analysis.
Primary cultures of C4HD cells subjected to the different treatments described in Results were harvested for flow cytometric analysis and fixed in 70% ethanol for 24 h at 4°C. They were washed twice with phosphate-buffered saline, followed by RNA digestion (RNase A at 50 U/ml) and PI (20 μg/ml) staining for 30 min at room temperature in the dark. Cell cycle analysis was performed using a FACScalibur flow cytometer (Becton-Dickinson, Mountain View, CA) and Modfit LT software.
Apoptosis assays.
C4HD cells were transiently transfected with the DN Stat3 expression vector Stat3Y705-F or with the empty pRc/CMV vector as described above. Apoptosis was investigated through cell surface binding of fluorescent Annexin V by using the fluorescein isothiocyanate-annexin V binding assay (Immunotech).
Western blot analysis and immunoprecipitation assays.
Lysates were prepared from C4HD or T47D cells subjected to the different treatments described in each experiment. Cells were lysed in buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 0.5% Nonidet P-40, 1 mM Mg2Cl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 μg/ml leupeptin, 5 μg/ml pepstatin, 5 μg/ml aprotinin, 1 mM sodium orthovanadate, 5 mM NaF, 20 mM sodium molybdate, and 5 mM sodium pyrophosphate. Lysates were centrifuged at 40,000 × g for 40 min at 4°C, and protein content in the supernatant was determined using a Bio-Rad kit (Bio-Rad, Richmond, CA). Proteins were solubilized in sample buffer (60 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate [SDS], 10% glycerol, 0.01% bromophenol blue) and subjected to SDS-polyacrylamide gel electrophoresis (PAGE). Proteins were electroblotted onto nitrocellulose. Membranes were immunoblotted with the following antibodies: phosphotyrosine Stat3 (B-7), total Stat3 (C-20), phosphotyrosine Stat1 (A-2), total Stat1 (E-23), c-Src (N-16), phosphotyrosine Jak1 (Tyr1022/1023), total Jak1 (HR-785), the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI-3K) (Z-8), and Bcl-xL (S-18), all from Santa Cruz Biotechnology (Santa Cruz, CA), phosphotyrosine Src (Tyr416) and phosphotyrosine Jak2 (Tyr1007/1008) from Cell Signaling (Beverly, MA), hPR Ab-7 (clone 7), and actin (clone ACTN05) from Neomarkers (Freemont, CA), and FLAG-M2 monoclonal antibody from Sigma (St. Louis, MO). Association between PR and Stat3 was studied by performing coimmunoprecipitation experiments. Total cell lysates (500 μg protein) from C4HD cells, treated and untreated with MPA, were immunoprecipitated using the PR mouse monoclonal antibody hPR Ab-7 (clone 7; Neomarkers). Immunoprecipitations were rocked for 2 h at 4°C, and the immunocomplexes were then captured by adding protein A-agarose and rocked for an additional 2 h. Beads were washed three times with lysis buffer and then boiled for 10 min in sample buffer and subjected to SDS-PAGE. Proteins were electroblotted onto nitrocellulose and probed with an anti-Stat3 antibody (C-20; Santa Cruz). When the immunoprecipitation-immunoblotting procedure was reversed, lysates were immunoprecipitated with the anti-Stat3 antibody, and Western blotting was performed with the anti-PR antibody (Ab-7; Neomarkers). As a negative control, preimmune rabbit serum was used.
Preparation of nuclear and cytosolic extracts.
C4HD and T47D cells were lysed by scraping at 4°C in buffer A, which contains 10 mM Tris (pH 7.4), 1.5 mM EDTA, 10% glycerol, 0.5 mM dithiothreitol, 1 mM PMSF, 10 μg/ml leupeptin, 5 μg/ml pepstatin, 5 μg/ml aprotinin, and 5 mM NaF. The homogenate was centrifuged for 10 min at 1,000 × g, and the supernatant fraction (cytosol) was then collected. The crude nuclear pellet was washed once with buffer A containing 0.01% NP-40 and centrifuged again at 1,000 × g, and the supernatant was added to the cytosol, which was then centrifuged at 105,000 × g for 30 min. The supernatant yielded the cytoplasmic fraction. The washed crude nuclear pellet was resuspended in buffer A containing 0.4 M KCl and was later incubated for 30 min at 4°C with resuspension every 10 min. The suspension was centrifuged at 105,000 × g (30 min) and diluted with the same volume of buffer A to reduce the salt concentration.
EMSA.
The DNA-binding activity of Stat3 in nuclear cell extracts of C4HD cells was analyzed by electrophoretic mobility shift assay (EMSA). C4HD cells were treated for 15 min at 37°C with MPA or MPA-RU486 or were left untreated growing in ChFCS. Double-stranded DNA containing the high-affinity mutant of the sis-inducible element (SIE) from the human c-fos promoter (5′GTGCATTTCCCGTAAATCTTGTCTACA3′) (m67) was used as probe. M67 oligonucleotide was end labeled with [γ-32P]ATP by T4 polynucleotide kinase to a specific activity of approximately 30,000 to 50,000 cpm/0.1 ng. Twenty micrograms of protein from nuclear extracts was incubated for 20 min at room temperature in a total volume of 30 μl with 1 ng of the [32P]m67. The DNA-binding reaction mixture also consisted of 2.5 μg poly(dI-dC), as nonspecific competitor DNA, in a binding buffer containing 10 mM Tris-HCl (pH 7.4), 50 mM NaCl, 10% glycerol, 1 mM dithiothreitol, 5 mM MgCl2, 5 μg/μl gelatin, 1 mM PMSF, 10 μg/ml leupeptin, 5 μg/ml pepstatin, and 5 μg/ml aprotinin. The specificity of Stat3-DNA complexes was studied by competition with a 25- and 100-fold mass excess of unlabeled m67 oligonucleotide and by the lack of competition with a 100-fold mass excess of a mutant m67 with a TTC-to-CCA substitution in the DNA-binding region (5′GTGCATCCACCGTAAATCTTGTCTACA3′) (Santa Cruz). An anti-Stat3α antibody (clone C-20) or an anti-Stat1 antibody (clone E-23), each from Santa Cruz, was used in the supershift assays at a concentration of 4 μg/assay. Preimmune rabbit serum was used as a control. Samples were subjected to electrophoresis on nondenaturing 5.5% acrylamide gels (30:1) in low-ionic-strength TBE buffer (10 mM Tris-borate [pH 7.5], 0.025 mM EDTA). Gels were preelectrophoresed at 10 mA for 60 min, and samples were run at 20 mA/gel for 120 min at room temperature. To prevent warming of the gels, water was recirculated through the gel apparatus. Gels were dried without fixation under vacuum and were autoradiographed by exposure to Kodak X-OMAT film at −70°C.
In vivo blockage of Stat3 expression.
C4HD cells growing in 10 nM MPA were transiently transfected as already described above with the DN Stat3Y705-F expression vector and with the empty pRc/CMV vector or remained untreated. After 48 h of transfection, 106 cells from each experimental group were inoculated s.c. into animals treated with a 40-mg MPA depot in the flank opposite the cell inoculum. Tumor growth was measured three times a week with a vernier caliper. Tumor volume (mm3) was calculated as (L × W2)/2, where L = length (mm) and W = width (mm). Tumor growth rates were determined as the slopes of growth curves. Percentage of tumor growth inhibition was calculated by dividing the mean tumor volume of the animal group injected with C4HD cells transiently transfected with the DN Stat3Y705-F expression vector, by the mean tumor volume of control groups, subtracting the resulting value from 1, and multiplying it by 100. Tumor growth delay was evaluated as T − C, where T and C are the median times for treated and control tumors, respectively, to reach the same volume. At day 21, animals from each group were killed and tumors were removed. Tissues for molecular studies were stored at −80°C, and tissues for histopathological analysis were fixed in 10% buffered formalin. Samples of liver, lung, heart, and pancreas were also fixed for histological examination. Comparison of tumor volumes between the different groups for specific times was done by analysis of variance followed by Tukey's t test among groups. Linear regression analysis was performed on tumor growth curves, and the slopes were compared using analysis of variance followed by a parallelism test to evaluate the statistical significance of differences.
RESULTS
MPA induces Stat3 tyrosine phosphorylation acting through the classical PR and through a mechanism requiring Jak1, Jak2, and Src activation.
We performed the present study in an experimental model of hormonal carcinogenesis in which the synthetic progestin MPA induced mammary adenocarcinomas in female BALB/c mice (3, 4, 25, 44). Primary cultures of the C4HD murine mammary tumor line (3, 4, 25, 44) were used. As we have already described in detail (19), C4HD epithelial cells derive from a ductal, progestin-dependent mammary tumor line. C4HD cells require MPA administration to proliferate and express high levels of PR (19). We first investigated Stat3 expression and tyrosine phosphorylation in C4HD cells. As shown in Fig. 1, C4HD cells express Stat3 at the protein level. MPA treatment of C4HD cells for 48 h induced a two- to threefold up-regulation of Stat3 protein expression (Fig. 1, first panel), as reported in human breast cancer T47D cells (26, 43). The progestin antagonist RU486 completely inhibited the stimulatory effect of MPA on Stat3 expression (Fig. 1, first panel). C4HD cells also express Stat1 (Fig. 1, third panel). However, MPA not only had no effect on Stat1 expression, but in some experiments it even reduced Stat1 expression slightly (Fig. 1, third panel). We also found that MPA treatment of C4HD cells for 5 to 10 min induced phosphorylation of Stat3 on tyrosine residue 705 by performing Western blot assays with the specific antiphosphotyrosine Stat3 antibody (Fig. 2A, first part). This effect was completely abolished by RU486 (Fig. 2A, first part). On the other hand, Stat1 showed basal levels of phosphorylation on tyrosine 701, which remained unaffected by MPA treatment (Fig. 2A, third part). The involvement of members of the Jak tyrosine kinase family in cytokine-mediated Stat phosphorylation has been well acknowledged (1). However, the role of Jaks in progestin-induced Stat3 activation has never been explored. Here, we found that both Jak1 and Jak2 were expressed in C4HD cells (Fig. 2A, sixth and eighth parts). MPA treatment resulted in strong tyrosine phosphorylation of Jak1 and Jak2, observed as early as 5 min after treatment, which was inhibited by antiprogestin RU486 (Fig. 2A, fifth and seventh parts, respectively). Similar experiments were conducted with T47D cells. As shown in Fig. 2B, MPA was able to induce Stat3 tyrosine phosphorylation in T47D cells, and this effect was abolished by RU486. MPA treatment of T47D cells also resulted in Jak1 and Jak2 tyrosine phosphorylation, which was blocked by RU486 (Fig. 2B). Interestingly, although Stat3 tyrosine phosphorylation was very transient, Jak activation persisted for at least 15 min and returned to basal levels at 30 min of MPA treatment in both the C4HD (Fig. 2A) and T47D (Fig. 2B) cell lines. To further demonstrate that conventional PR is mediating nongenomic MPA-induced Stat3 tyrosine phosphorylation, we used our well-characterized mouse metastatic mammary tumor line LM3, which lacks PR expression (40). MPA was not able to induce Stat3 phosphorylation in wild-type LM3 cells (Fig. 2C). However, as we previously demonstrated (40), treatment of LM3 cells with the type I RTK ligand heregulin strongly induced Stat3 tyrosine phosphorylation (Fig. 2C). On the other hand, when we transiently transfected LM3 cells with a human PRB expression vector, MPA treatment resulted in strong induction of Stat3 tyrosine phosphorylation, which was abolished by RU486 (Fig. 2C). We used a similar strategy with human PR null T47D-Y cells (26). As shown in Fig. 2D, MPA treatment of T47D-Y cells did not induce Stat3 tyrosine phosphorylation. However, when these cells were transfected with PRB, strong Stat3 tyrosine phosphorylation in response to MPA was detected (Fig. 2D). RU486 inhibited MPA-induced Stat3 phosphorylation (Fig. 2D).
FIG. 1.

MPA up-regulates Stat3 protein expression. Primary cultures of C4HD cells were treated for 48 h in medium with ChFCS supplemented with 10 nM MPA or MPA-10 nM RU486. Fifty micrograms of protein from cell lysates was electrophoresed and immunoblotted for Stat3 and Stat1. A Western blot assay using an antiactin antibody was carried out using identical protein lysates as a control for the specificity of the effect of MPA. This is a representative experiment of a total of four in which the standard error of the mean was within 10%. W, Western blot assay.
FIG. 2.

MPA induces tyrosine phosphorylation by Stat3, Jak1, and Jak2. Cultures of C4HD (A) and T47D (B) cells were treated with 10 nM MPA or MPA-10 nM RU486 for the indicated times. Fifty micrograms of protein from cell lysates was electrophoresed, and Western blot assays were performed with antiphosphotyrosine 705 Stat3, antiphosphotyrosine 701 Stat1, antiphosphotyrosine 1022/1023 Jak1, and antiphosphotyrosine 1007/1008 Jak2 antibodies. Membranes were then stripped and hybridized with anti-Stat3, -Stat1, -Jak1, and -Jak2 antibodies. This experiment was repeated six times for C4HD cells and three times for T47D cells with similar results. LM3 (C) or T47D-Y (D) cells were transfected with PRB or with the empty pSG5 plasmidor remained untreated. Cells were then stimulated for 5 min with MPA or pretreated with RU486 before MPA stimulation. LM3 cells were also treated with heregulin for 10 min. Fifty micrograms of protein from cell lysates was electrophoresed, and Western blot assays were performed with antiphosphotyrosine 705 Stat3 (upper parts of panels C and D). Membranes were then stripped and hybridized with anti-Stat3 (middle panes of parts C and D) and anti-PR (lower parts of panels C and D) antibodies. This experiment was repeated three times with similar results. W, Western blot assay.
We then investigated the participation of Jaks in MPA-induced Stat3 phosphorylation. For this purpose, C4HD cells were transiently transfected with 2 μg DN Jak1 or DN Jak2 vector (38, 39) and then treated with MPA. Transfection efficiency in C4HD cells, evaluated using the pEGFP-N1 vector, varied from 65 to 75%, as determined by the percentage of cells that exhibited green fluorescence 24 h after transfection. Green fluorescent protein was visualized by direct fluorescence imaging using confocal laser microscopy. High efficiency of transfection in our cell cultures induced high levels of DN Jak expression, shown by an increase in total Jak1 and Jak2 expression in cells transfected with DN Jak expression vectors, compared with nontransfected cells (Fig. 3A and B, respectively). As shown in Fig. 3A and B, when DN Jak1 and Jak2 were expressed in C4HD cells, they were able to work dominant negatively against MPA-induced tyrosine phosphorylation of wild-type Jaks present in C4HD cells. Experiments done transfecting C4HD cells with increasing amounts of DN Jak vectors showed that inhibition of wild-type Jak tyrosine phosphorylation was dose dependent (data not shown). Specificity of DN Jak action was demonstrated by lack of effect of the DN Jak2 vector on MPA-induced tyrosine phosphorylation of endogenous Jak1 (Fig. 3A) and of the DN Jak1 vector on MPA-induced phosphorylation of endogenous Jak2 (Fig. 3B). Abolishment of Jak1 and Jak2 activity resulted in inhibition of the capacity of MPA to induce Stat3 tyrosine phosphorylation (Fig. 3C, upper parts), clearly showing that both kinases are involved in the effect of MPA.
FIG. 3.

Jak1 and Jak2 are involved in MPA-induced Stat3 phosphorylation. C4HD cells were transiently transfected with 2 μg of DN Jak1 or DN Jak2 vector and then treated with MPA for 5 min or left untreated. Fifty micrograms of protein from cell lysates was electrophoresed, and Western blot assays were performed with antiphosphotyrosine Jak1 (A, upper part) or antiphosphotyrosine Jak2 (B, upper part) antibodies. Membranes were then stripped and hybridized with anti-Jak1 (A, lower part) and anti-Jak2 (B, lower part) antibodies. (C) Fifty micrograms of protein from cells transfected with the DN Jak1 (left part) or the DN Jak2 (right part) vector and subsequentlytreated with MPA for 5 min or left untreated was electrophoresed, and Western blot assays were performed with antiphosphotyrosine Stat3 (upper parts). Membranes were then stripped and hybridized with anti-Stat3 (lower parts) antibodies. (D) C4HD cells were treated with MPA for the indicated times or preincubated with the selective Src family kinase inhibitor PP2 or RU486 for 90 min and then treated with MPA. Fifty micrograms of protein from cell lysates was electrophoresed and immunoblotted with an antiphosphotyrosine c-Src antibody (upper part). Membrane was then stripped and hybridized with anti-c-Src antibody (lower part). (E) C4HD cells were preincubated with the selective Src family kinase inhibitor PP2 for 90 min and then treated with MPA for 5 min. Fifty micrograms of protein from cell lysates was electrophoresed and immunoblotted with antiphosphotyrosine Stat3, antiphosphotyrosine Jak1, and antiphosphotyrosine Jak2 antibodies. Membranes were then stripped and hybridized with anti-Stat3, anti-Jak1, and anti-Jak2 antibodies, respectively. These experiments were repeated three times with similar results. W, Western blot assay.
The requirement of Src activity for Stat3 activation in breast cancer cells has already been acknowledged (7, 20, 53, 55, 57). In addition, accumulating data have shown the ability of progestin to induce c-Src phosphorylation by a nongenomic mechanism in mammary tumor cells (5, 6, 13, 33). Therefore, we here assessed the effect of MPA on c-Src activity in C4HD cells. MPA treatment of C4HD cells for 2 to 10 min induced strong c-Src tyrosine phosphorylation, which was significantly inhibited by the selective Src family kinase inhibitor PP2 (Fig. 3D). We then explored the role of c-Src in MPA-induced Stat3 tyrosine phosphorylation. As shown in Fig. 3E, inhibition of Src activity by preincubation of cells with PP2 blocked the capacity of MPA to induce Stat3 phosphorylation. The above results show that both Jaks and Src are able to activate Stat3. To gain further insight into the molecular mechanism through which these kinases participate in Stat3 tyrosine phosphorylation, we examined the tyrosine phosphorylation state of Jak1 and Jak2 in C4HD cells pretreated with the Src inhibitor PP2. We found that Jak1 and Jak2 tyrosine phosphorylation was effectively inhibited by PP2 (Fig. 3E). These findings indicate that Jaks are phosphorylated by c-Src in C4HD cells.
MPA induces association between Stat3 and PR.
Stats have been found to be physically associated with several members of the steroid receptor superfamily, including PR (30, 43). In this work, we investigated the capacity of MPA to induce association between Stat3 and PR by performing coimmunoprecipitation experiments. Protein extracts from C4HD cells were immunoprecipitated with an anti-PR antibody and immunoblotted with an anti-Stat3 antibody. As shown in Fig. 4A, association between PR and Stat3 was detected in C4HD cells growing in 0.1% ChFCS and was dramatically increased by MPA treatment. Coimmunoprecipitation of Stat3 and PR was also detected when the immunoprecipitation-immunoblotting procedure was reversed. Thus, when cell extracts were immunoprecipitated with an anti-Stat3 antibody and Western blotting was performed with an anti-PR a clear MPA-induced association between Stat3 and PR was found (Fig. 4B). Since we found that Src activity is an absolute requirement for MPA-induced Jak and Stat3 tyrosine phosphorylation, we explored Src involvement in the association between PR and Stat3. As shown in Fig. 4A and B, abolishment of Src activity resulted in blockage of the capacity of MPA to induce a physical association between PR and Stat3.
FIG. 4.

MPA induces association of Stat3 with PR. (A) C4HD cells were treated with 10 nM MPA for the indicated times or preincubated with PP2 before MPA treatment for 5 min, and PR was immunoprecipitated from 500 μg of protein extracts. As a control, lysates were also immunoprecipitated with normal mouse serum (NMS). Immunocomplexes were subjected to SDS-PAGE and analyzed by Western blotting with an anti-Stat3 antibody (upper part). Twenty micrograms of protein from cell extracts was directly immunoblotted with the Stat3 antibody (last lane, upper part). Identical aliquots of each immunoprecipitate were subjected to immunoblot analysis with anti-PR antibody to verify that nearly equal amounts of immunoprecipitated proteins were loaded (lower part). (B) Protein lysates (500 μg) from cells treated as indicated in panel A were immunoprecipitated with an anti-Stat3 antibody or with normal rabbit serum (NRS). Immunocomplexes were subjected to SDS-PAGE and analyzed by Western blotting with an anti-PR antibody (upper part). Twenty micrograms of protein from cell extracts was directly immunoblotted with the PR antibody (last lane, upper part). Identical aliquots of each immunoprecipitate were subjected to immunoblot analysis with anti-Stat3 antibody to verify that nearly equal amounts of immunoprecipitated proteins were loaded (lower part). This is a representative experiment out of a total of three. W, Western blot assay; IP, immunoprecipitation.
MPA induces Stat3 nuclear translocation and binding to DNA.
Upon tyrosine phosphorylation, Stats dimerize and translocate to the nucleus, where they bind DNA and regulate gene transcription. In order to investigate the effects of MPA on these events, nuclear and cytoplasmic extracts from C4HD cells were prepared and Western blot assay was performed with an anti-Stat3 antibody. While Stat3 was only detected in the cytoplasmic fraction in untreated cells growing in 0.1% ChFCS, MPA treatment induced an extensive nuclear translocation of Stat3 (Fig. 5, upper panel). In addition, we found that inhibition of Src activity by PP2 inhibited MPA-induced Stat3 nuclear translocation (Fig. 5, upper panel). Cellular fractionation was controlled by immunoblotting with either an anti-PI-3K (Fig. 5, middle panel) or an anti-retinoblastoma (Rb) (Fig. 5, lower panel) antibody.
FIG. 5.

MPA induces Stat3 nuclear translocation. C4HD cells were treated with 10 nM MPA for the time indicated or were pretreated with PP2 before MPA stimulation. Nuclear (nuc) and cytosolic (cyt) fractions were prepared, and 30 μg of protein from cell extracts was analyzed by Western blot assay for Stat3 expression level. Membranes were then stripped and hybridized with an anti-p85 PI-3K subunit antibody (middle) or an anti-retinoblastoma (Rb) antibody (bottom) in order to control cellular fractionation efficiency. W, Western blot assay.
We then investigated the capacity of MPA to induce Stat3 binding to DNA, using as a labeled probe a high-affinity mutant of the SIE from the human c-fos promoter (m67), which binds both Stat3 and Stat1 (11, 56). EMSAs using nuclear extracts from C4HD cells treated with MPA for 15 min demonstrated that MPA was able to promote formation of DNA-binding complexes (Fig. 6, left panel). As has been acknowledged in numerous studies (11, 56), three major complexes were found in C4HD cells corresponding to Stat3 homodimers, Stat3-Stat1 heterodimers, and Stat1 homodimers (Fig. 6, left panel). Specificity of the Stats-DNA complexes was demonstrated by competition with excess unlabeled m67 oligonucleotide and by lack of competition with a mutated m67 probe (Fig. 6, left panel). Stat1 binding to DNA was observed in cells growing in 0.1% ChFCS, while no increase in the levels of Stat1-DNA complexes was observed after MPA treatment (Fig. 6, left panel), in accordance with our finding of basal Stat1 tyrosine phosphorylation, which remained unaffected by MPA treatment (Fig. 2). On the contrary, MPA strongly induced the formation of Stat3-DNA complexes, which was completely abolished by preincubation with RU486 (Fig. 6, left panel). To confirm the identity of the DNA-binding complexes, either anti-Stat3 or anti-Stat1 antibodies were included in the EMSA reaction mixture (Fig. 6, right panel). The use of an anti-Stat3α antibody (clone C-20) resulted in complete abolishment of the MPA-induced Stat3 homodimer-DNA complex and dramatically decreased the Stat3-Stat1 heterodimer-DNA complex abundance (Fig. 6, right panel). This antibody also induced the formation of a complex with decreased mobility (Fig. 6, right panel). The Stat1 antibody (clone E-23) was raised against a Stat1 epitope corresponding to an amino acid sequence within the carboxy-terminal SH2 domain sequence present in both Stat1α and Stat1β. The inclusion of this antibody in the EMSA reaction mixture eliminated Stat1 homodimer-DNA complexes (Fig. 6, right panel) and induced a highly significant decrease in Stat3-Stat1-DNA heterodimer complex abundance (Fig. 6, right panel). As previously reported for this antibody (Fig. 6, right panel) (46, 47), no supershift was detected. An equivalent amount of the preimmune rabbit serum used as a control had no effect (Fig. 6, right panel, NRS). These data clearly show the presence of Stat3 in MPA-induced DNA-protein complexes. We found similar results in the EMSAs using the α2-macroglobulin probe, which contains the GAS element in the α2-macroglobulin enhancer (49, 56), as has been previously reported in HEPG2 cell types (49).
FIG. 6.

MPA induces Stat3 binding to the high-affinity mutant of the SIE from the human c-fos promoter. C4HD cells were treated for 15 min at 37°C with MPA or MPA-RU486 or were left untreated growing in ChFCS. Twenty micrograms of protein from nuclear extracts was incubated for 20 min at room temperature with 1 ng of 32P-labeled double-stranded DNA containing the high-affinity mutant of the SIE from the human c-fos promoter (5′GTGCATTTCCCGTAAATCTTGTCTACA3′) (m67) used as a probe and analyzed by EMSA. The specificity of the Stat3-DNA complexes is shown by competition with 25- and 100-fold mass excesses unlabeled m67 oligonucleotide and by the lack of competition with a 100-fold mass excess of mutant m67 (100xm67 mut). The right panel shows a supershift analysis that was performed by including either anti-Stat3 or anti-Stat1 antibodies. An equivalent amount of preimmune rabbit serum was used as a control in the EMSA reaction mixture (NRS [normal rabbit serum]). This experiment was repeated six times with similar results. wt, wild type.
MPA induces Stat3 transcriptional activation through a Jak1-, Jak2-, and Src-dependent pathway.
To investigate the capacity of MPA to induce the transcriptional activation of Stat3, C4HD cells were transiently transfected with a luciferase reporter plasmid containing four copies of the m67 high-affinity binding site (11, 56) and a β-galactosidase expression vector as an internal control. Treatment of C4HD cells with MPA induced Stat3 transcriptional activation that was completely inhibited by preincubation with RU486 (Fig. 7A). Blockage of Jak1 and Jak2 activities by the use of a DN Jak1 or Jak2 expression vector and of Src activity with PP2 also inhibited the capacity of MPA to activate the m67-Luc reporter plasmid (Fig. 7A). Comparable results were obtained with T47D cells, in which MPA also induced Stat3 transcriptional activation. RU486 pretreatment, transfection with a DN Jak1 or Jak2 expression vector, and PP2 preincubation all abolished the capacity of MPA to activate the m67-Luc reporter in T47D cells (Fig. 7B). Similar results with both C4HD and T47D cells were obtained by using a luciferase reporter plasmid containing three copies of the Ly6E Stat1 and Stat3 binding site (10) in the transient-transfection assays (data not shown).
FIG. 7.

MPA induces Stat3 transcriptional activation. C4HD (A) and T47D (B) cells were transiently transfected with 2 μg/well of a luciferase reporter plasmid containing four copies of the m67 high-affinity binding site and with 1 μg/well of a CMV-βgal expression vector as an internal control. In the indicated lanes, C4HD cells were cotransfected with the DN Jak1 and DN Jak2 expression vectors or pretreated with PP2. Cells were also transfected with a pTATA-Luc reporter lacking the m67 insertion. The total amount of transfected DNA was standardized by adding the empty vector. After transfection, cells were treated with MPA and MPA-RU486 at 37°C for 48 h or left untreated growing in ChFCS. C4HD cells were then harvested and lysed. Luciferase and β-galactosidase activities were measured as described in Materials and Methods. Results are presented as n-fold induction of luciferase activity with respect to cells growing in ChFCS. The data shown represent the mean of six independent experiments ± the standard error of the mean. For b versus a and c versus b, P < 0.001.
Stat3 activity is a requisite in MPA-induced proliferation of C4HD cells.
To investigate the correlation between MPA-induced Stat3 activation and cell growth, C4HD cells were transiently transfected with a DN Stat3 expression vector, Stat3Y705-F (10, 23, 28), which carries a tyrosine-to-phenylalanine substitution at codon 705 that reduces phosphorylation on tyrosine of the wild-type Stat3 protein, therefore inhibiting both dimerization and DNA binding of Stat3. C4HD cells were also transfected with a constitutively activated Stat3 mutant, Stat3-C, which dimerizes spontaneously, binds to DNA, and activates transcription (11). Proliferation of transfected C4HD cells was evaluated by PI staining and flow cytometry analysis. As shown in Fig. 8A (middle parts, bottom), expression of the Stat3Y705-F mutant had an inhibitory effect on MPA-induced growth of C4HD cells, compared with MPA-stimulated C4HD cells transfected with the empty vector (middle parts, top). On the other hand, transfection with the constitutively activated Stat3-C vector (Fig. 8A, right part) resulted in MPA-independent proliferation with a percentage of cells in the S and G2/M phases comparable to the one in MPA-treated cells, transfected with the empty vector (Fig. 8A, middle parts, top). Interestingly, profiles of PI staining showed a significant subdiploid peak in C4HD cells transfected with the Stat3Y705-F mutant vector (Fig. 8A, middle parts, bottom), compared with C4HD cells transfected with the empty vector (Fig. 8A, middle parts top), indicating a higher percentage of apoptotic cells. To further explore this finding, apoptosis was investigated through cell surface binding of fluorescent Annexin V, as an early indicator of programmed cell death. As shown in Fig. 8B, the level of apoptosis in Stat3Y705-F-transfected cells was found to increase significantly (right parts, bottom), compared with empty-vector-transfected cells (right parts, top). As a whole, our results indicate that Stat3Y705-F-mediated growth inhibition of C4HD cells involves both cell cycle arrest and apoptosis.
FIG. 8.
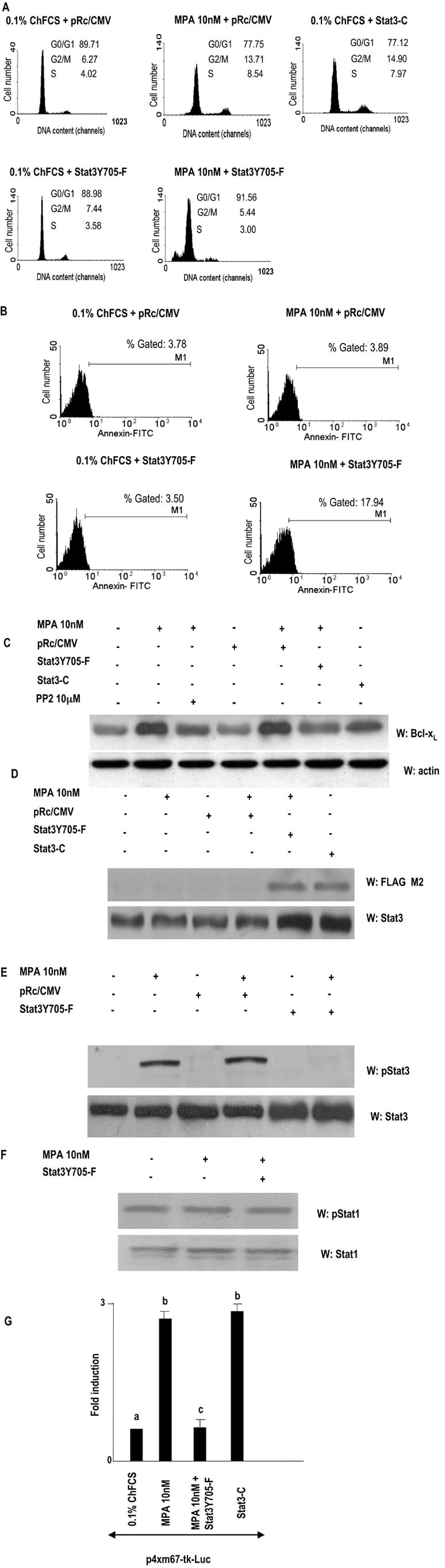
Stat3 is involved in MPA-induced proliferation of C4HD cells. (A) C4HD cells were transiently transfected with 2 μg DN Stat3 expression vector, Stat3Y705-F, with 2 μg constitutively activated Stat3 mutant, Stat3-C, or with 2 μg empty pRc/CMV vector, as a control, for 48 h. Cells were treated with MPA for another 48 h or remained untreated and were then stained with PI and analyzed for cell cycle distribution by flow cytometry. The percentages of total cells in the cell cycle phases are indicated. (B) C4HD cells were transiently transfected with 2 μg DN Stat3 expression vector or with 2 μg empty pRc/CMV vector, as a control, for 48 h. Cells were treated as described for panel A, and cell surface Annexin V binding was measured by flow cytometry. (C) Fifty micrograms of protein from lysates of cells transfected with Stat3Y705-F, Stat3-C, and empty pRc/CMV plasmids and from nontransfected cells, treated with MPA for 48 h or left untreated, was electrophoresed, and Western blot assays were performed with an anti Bcl-xL antibody (upper part). Membrane was then stripped and hybridized with an antiactin antibody (lower part). (D) Fifty micrograms of protein from lysates of cells treated as described for panel C and stimulated or not with MPA for 5 min was electrophoresed, and Western blot assays were performed with an anti-FLAG M2 antibody (upper part). Membrane was then stripped and hybridized with an anti-Stat3 antibody (lower part). (E) Fifty micrograms of protein from C4HD cells transfected with 2 μg Stat3Y705-F vector or with empty pRc/CMV plasmid and subsequently left untreated treated or with MPA for 5 min was electrophoresed, and Western blot assays were performed with antiphosphotyrosine Stat3 antibody (upper part). Membranes were then stripped and hybridized with anti-Stat3 antibodies (lower part). (F) Fifty micrograms of protein from C4HD cells transfected with 2 μg Stat3Y705-F vector and then treated with MPA for 5 min or left untreated was electrophoresed, and Western blot assays were performed with antiphosphotyrosine 701 Stat1 antibody (upper part). Membrane was then stripped and hybridized with anti-Stat1 antibody (lower part). Experiments described in panels A to F were repeated three times with similar results. W, Western blot assay. (G) C4HD cells were transiently transfected with 2 μg/well of the m67-Luc reporter plasmid and with 1 μg/well of a CMV-βgal expression vector as an internal control. In the indicated lanes, C4HD cells were cotransfected with either Stat3Y705-F or Stat3-C plasmid. The total amount of transfected DNA was standardized by adding the empty vector. After transfection, cells were treated when indicated with MPA for 48 h and were then harvested and lysed. Luciferase and β-galactosidase activities were measured as described in Materials and Methods. Results are presented as n-fold induction of luciferase activity with respect to cells growing in ChFCS. Data shown represent the mean of two independent experiments ± the standard error of the mean. For b versus a and c versus b, P < 0.001. FITC, fluorescein isothiocyanate.
Previous reports (11, 14) have identified members of the Bcl-2 family of antiapoptotic regulatory proteins as being encoded by Stat3-regulated genes. Here, we investigated Bcl-xL gene expression in C4HD transfected with either Stat3Y705-F or Stat3-C plasmids. MPA treatment of C4HD cells resulted in up-regulation of Bcl-xL protein expression (Fig. 8C), in accordance with previous results demonstrating progesterone induction of Bcl-xL expression in breast cancer cells (34). As shown in Fig. 8C, transfection with Stat3Y705-F resulted in significant inhibition of MPA-induced Bcl-xL expression. Inhibition of Src activity by preincubation of cells with PP2 also induced a decrease in MPA-induced Bcl-xL protein (Fig. 8C). On the other hand, the constitutively activated Stat3-C mutant induced an increase in Bcl-xL protein levels, comparable to the effect exerted by MPA (Fig. 8C).
Expression and function of Stat3Y705-F and Stat3-C plasmids under the conditions described above, in which they modulate C4HD cell growth, were then assessed. Expression of either Stat3Y705-F or Stat3-C in transfected C4HD cells was studied by Western blot assay with an anti-FLAG M2 antibody since both Stat3Y705-F and Stat3-C are tagged at the carboxyl-terminal site with the FLAG epitope (Fig. 8D, upper part). Compared with the nontransfected cells, the levels of Stat3 were markedly higher after transfection with both plasmids (Fig. 8D, lower part). As a control of Stat3Y705-F function, Fig. 8E shows that when expressed in C4HD cells, Stat3Y705-F was able to work dominant negatively against MPA-induced tyrosine phosphorylation of Stat3. Specificity of Stat3Y705-F action was demonstrated by its lack of effect on Stat1 constitutive phosphorylation on tyrosine 701 (Fig. 8F). In addition, the capacity of MPA to induce transcriptional activation of endogenous Stat3 was completely inhibited by Stat3Y705-F expression, as shown in the transient-transfection assay with the m67-Luc reporter plasmid (Fig. 8G). In contrast, Stat3-C expression resulted in activated transcription of the reporter gene in cells untreated with MPA (Fig. 8G). As happened with DN Jak transfection, experiments done by transfecting C4HD cells with increasing amounts of Stat3Y705-F showed that inhibition of MPA-induced tyrosine phosphorylation of Stat3 and of the capacity of MPA to induce transcriptional activation of endogenous Stat3 was dose dependent (data not shown).
In vivo blockage of Stat3 expression results in C4HD tumor growth inhibition.
We have already demonstrated that proliferation in our model system is driven by a complex cross talk between progestins and growth factors pathways (3, 4, 25, 44). In the present work, we found that Stat3 is yet one more player in this scenario, since MPA-induced Stat3 transcriptional activation (Fig. 7) is a requisite for MPA stimulation of C4HD cell growth (Fig. 8). Therefore, we here addressed the effect of targeting Stat3 in in vivo growth of C4HD breast tumors. For this purpose, C4HD cells growing in 10 nM MPA were transiently transfected with the DN Stat3Y705-F expression vector or with the empty pRc/CMV vector or were left untreated. After 48 h of transfection, 106 cells from each experimental group were inoculated s.c. into animals treated with a 40-mg MPA depot in the flank opposite the cell inoculum, and tumor width and length were measured three times a week in order to calculate volume. All mice (n = 9) injected with C4HD cells transfected with the pRc/CMV vector or injected with wild-type C4HD cells (n = 9) developed tumors which became palpable after 8 days of inoculation. On the contrary, only four out of nine mice injected with C4HD cells transfected with the DN Stat3Y705-F expression vector developed tumors, with a delay of 3 days in tumor latency compared with tumors from control groups. The mean volume of tumors developed from DN Stat3Y705-F-transfected C4HD cells was significantly lower than that of tumors from both control groups (Fig. 9). Tumor growth rates, determined as the slopes of growth curves, were significantly lower in tumors developed from DN Stat3Y705-F-transfected C4HD cells than those of control groups (Table 1). At day 21, animals in each group were killed and tumors were excised. Results are summarized in Table 1. Both tumor growth and growth rates were found to decrease significantly in tumors developed from DN Stat3Y705-F-transfected C4HD cells (Table 1). At day 21, a delay of 7 days in tumor growth was observed in mice injected with DN Stat3Y705-F-treated cells with respect to tumors developed in mice injected with wild-type C4HD cells and of 6 days with respect to tumors growing in mice injected with pRc/CMV vector-transfected cells. No statistically significant differences were found either in tumor growth, in growth rates, or in growth delay between tumors growing in mice injected with wild-type C4HD cells and tumors growing in mice injected with pRc/CMV vector-transfected cells (Table 1). Histopathological analysis was performed by hematoxylin-and-eosin staining of histological sections obtained from tumors excised at day 21. Tumors from mice receiving DN Stat3Y705-F-transfected C4HD cells showed a significantly lower number of mitoses (0 to 5 mitoses/10 high-power fields) compared to tumors from animals receiving wild-type C4HD cells or pRc/CMV vector-transfected C4HD cells, both of which showed over 10 mitoses/10 high-power fields. To gain further insight into the molecular mechanisms involved in inhibition of tumor growth by Stat3Y705-F transfection, we explored levels of Stat3 tyrosine phosphorylation and of antiapoptotic Bcl-xL gene expression in tumor samples. As shown in Fig. 9B, significantly lower levels of Stat3 tyrosine phosphorylation were found in tumors developed in mice injected with DN Stat3Y705-F-transfected cells than in tumors of mice injected with either pRc/CMV vector-transfected C4HD cells or wild-type C4HD cells. Furthermore, Bcl-xL expression was dramatically lower in tumors from DN Stat3Y705-F-transfected cells than that of both control groups (Fig. 9C).
FIG. 9.

In vivo blockage of Stat3 expression. (A) C4HD cells growing in 10 nM MPA were transiently transfected with the DN Stat3Y705-F expression vector (▾) or the empty pRc/CMV vector (▪) or remained untreated (▴), as described in the legend to Fig. 8. After 48 h of transfection, 106 cells from each experimental group were inoculated s.c. into animals treated with a 40-mg MPA depot in the flank opposite the cell inoculum, and tumor width and length were measured three times a week in order to calculate volume as described in Materials and Methods. Each point represents the mean volume ± the standard error of nine independent tumors for each control group and of four tumors that developed in mice injected with Stat3Y705-F-transfected cells. (B) One hundred micrograms of protein from tumor lysates was electrophoresed and immunoblotted with an anti-phospho-Stat3 antibody (upper part). Shown are two representative samples of mice injected with C4HD wild-type cells (lanes 1 and 2), C4HD cells transfected with the empty pRc/CMV vector (lanes 3 and 4), and C4HD cells transfected with the DN Stat3Y705-F expression vector (lanes 5 and 6). Membrane was stripped and hybridized with an anti-Stat3 antibody (lower part). This is a representative experiment out of a total of three. Densitometric analysis of Stat3 phosphorylated bands from the four tumors that developed in mice injected with C4HD cells transfected with the DN Stat3Y705-F vector and from multiple C4HD tumors that developed in mice injected with either wild-type C4HD cells or with C4HD cells transfected with the empty pRc/CMV vector showed a significant decrease in Stat3 tyrosine phosphorylation in tumors from mice injected with cells transfected with the DN Stat3Y705-F vector, with respect to tumors growing in control animals (P < 0.001). W, Western blot assay. (C) One hundred micrograms of protein from tumor lysates was electrophoresed and immunoblotted with an anti-Bcl-xL antibody (upper part). Shown are two representative samples of mice injected with C4HD wild-type cells (lanes 1 and 2), with C4HD cells transfected with the empty pRc/CMV vector (lanes 3 and 4), andwith C4HD cells transfected with the DN Stat3Y705-F expression vector (lanes 5 and 6). Membrane was then stripped and hybridized with an antiactin antibody (lower part), as a control for the specificity of the DN Stat3Y705-F effect. Densitometric analysis of the Bcl-xL band from tumors that developed in mice injected with C4HD cells transfected with the DN Stat3Y705-F, expressed as a percentage of the control values (i.e., tumors growing in control groups), ranged between 25 and 40% for tumors growing in mice injected with DN Stat3Y705-F-transfected cells. There was significant inhibition of Bcl-xL expression in mice injected with DN Stat3Y705-F-transfected cells with respect to mice injected with empty pRc/CMV vector-transfected cells or with wild-type C4HD cells (P < 0.001).
TABLE 1.
Tumor growth rates on day 21a
| Treatment | Mean tumor vol (mm3) ± SE | Growth rate (mm3/day) | % Growth inhibition |
|---|---|---|---|
| Stat3Y705-F | 101.1 ± 39.09* | 7.55 ± 1.15* | 57.14,b 63.84c |
| pRc/CMV | 235.9 ± 52.55† | 16.52 ± 1.287† | |
| Wild-type C4HD | 279.6 ± 49.15† | 18.92 ± 1.597† |
All experimental protocols were performed with mice treated with a 40-mg s.c. MPA depot in the flank opposite the cell inoculum. Mice were inoculated with C4HD cells transfected with either the DN Stat3Y705-F expression vector or the empty pRc/CMV vector and with wild-type C4HD cells. While control groups contained nine mice each, only four of the nine mice injected with cells transfected with the DN Stat3Y705-F vector developed tumors. Growth rate was calculated as the slopes of growth curves. At day 21, tumor volume and percentage of growth inhibition in tumors from mice injected with cells transfected with the DN Stat3Y705-F expression vector with respect to mice injected with the empty pRc/CMV vector or with wild-type C4HD cells were calculated as described in Materials and Methods. * versus †, P < 0.001.
With respect to pRc/CMV treatment.
With respect to wild-type C4HD treatment.
There were no signs of overt toxicity in mice injected with DN Stat3Y705-F-transfected C4HD cells. In addition, we observed no weight loss in mice receiving DN Stat3Y705-F-transfected cells, which shows good tolerance by animals. Histological examination of the liver, lung, heart, and pancreas did not reveal any pathological changes (data not shown).
DISCUSSION
In the present study, we have demonstrated that MPA up-regulates Stat3 protein expression in C4HD mammary tumor cells, our experimental model of progestin-induced mammary carcinogenesis. This effect was completely inhibited by the progestin antagonist RU486, indicating involvement of classical PR. The ability of progestin to modulate Stat expression in breast cancer cells has been previously found by Lange et al. (26) and by Richer et al. (43), who demonstrated that treatment of T47D cells with the synthetic progestin R5020 resulted in up-regulation of Stat3, Stat5a, and Stat5b protein levels. These authors also found a constitutive association between Stat5 and PRB in HeLa cells transfected with the B isoform of PR (43). Recently, progesterone was found to induce Stat5a expression in MDA-MB-231 breast cancer cells transfected with PR (29). Functional interaction between progestins and Stat3 was also found in decidual cells (30). Thus, progesterone enhanced cytoplasmic Stat3 stores in the decidualized mesometrium during pregnancy in rats (30), and this effect was inhibited by RU486, showing that Stat3 is a progesterone-dependent protein. Association between PR and Stat3 in decidualized mesometrium has also been found (30). Regarding Stat1, our results showing that MPA not only had no effect on Stat1 expression but even reduced it slightly are in accordance with those previously reported for T47D cells (43). Since MPA exerts a strong proliferative stimulus in C4HD cells, our present findings add further support to accumulating evidence favoring a growth inhibitory role of Stat1 (7, 9, 55).
Here, we have for the first time demonstrated that progestins induce rapid Stat3, Jak1, and Jak2 tyrosine phosphorylation. Moreover, MPA stimulates association between PR and Stat3, Stat3 nuclear translocation, binding to DNA, and transcriptional activation. All these effects were abrogated by RU486, indicating involvement of the classical intracellular PR. Our studies with PR-null LM3 and T47D-Y cells, in which MPA was not able to induce Stat3 tyrosine phosphorylation, and the finding that transfection of these cells with PRB restored the effects of MPA further showed the involvement of conventional PR in progestin-induced Stat3 tyrosine phosphorylation We found these results particularly interesting because of two reasons. First, because they provide support to accumulating evidence showing Stat3 activation in breast cancer cells. In fact, constitutive Stat3 tyrosine phosphorylation and DNA-binding activity have been found in breast cancer cell lines and in breast tumor samples (12, 20, 28, 41, 53). Interestingly, a high frequency of constitutive Stat3 activation has been reported in human breast cancer cell lines possessing elevated levels of EGF-R (45). Second, because we have for the first time uncovered several rapid or nongenomic effects of progestin, i.e., the capacity of MPA to induce Stat3, Jak1, and Jak2 tyrosine phosphorylation in both mouse and human breast cancer cells. Evidence of the capacity of progestins to promote Jak2 tyrosine phosphorylation was provided in pivotal studies by Lange et al. (26) and Richer et al. (43). However, in these studies, short-term treatment of T47Dco cells failed to induce Jak2 tyrosine phosphorylation, which instead became readily detectable after 48 h of R5020 stimulation. Differences between the T47D cell clones used could account for the discrepancy. Nevertheless, it is worth pointing out that either through transcriptional effects (26, 43) or through the rapid, nongenomic mechanism we have described here, progestins are able to modulate Jak2 phosphorylation in breast cancer cells. From our studies with DN Jak1 and Jak2 expression vectors, it is clear that progestins induce Stat3 tyrosine phosphorylation by a Jak-dependent pathway. It should be noted that while MPA-induced Stat3 tyrosine phosphorylation is very transient, Jak activation persists for at least 15 min and returns to basal levels after 30 min of MPA treatment in both C4HD and T47D cells. Prolonged Jak activation may reflect involvement of Jaks in other signaling pathways, activated by MPA by a nongenomic mechanism, besides induction of Stat3 tyrosine phosphorylation.
In the present work, we have also assessed the role of c-Src in progestin-induced Stat3 activation in C4HD cells. We were interested in c-Src because a series of previous reports had shown the requirement of Src activity for Stat3 activation in breast cancer cells (7, 20, 53, 55, 57). In addition, accumulating data showed the ability of progestin to induce Src phosphorylation by a nongenomic mechanism in mammary tumor cells (5, 6, 13, 33). Interestingly, we here found that 2 min of MPA treatment of C4HD cells induced strong c-Src tyrosine phosphorylation in C4HD cells. Blockage of Src activity inhibited both the capacity of MPA to induce Stat3 phosphorylation and transcriptional activation in C4HD and T47D cells. Notably, we also found that abolishment of Src activity results in inhibition of MPA-induced Jak1 and Jak2 tyrosine phosphorylation. Therefore, our findings indicate that progestins induce Stat3 tyrosine phosphorylation via Jak- and Src-dependent pathways. The molecular mechanism through which Jaks and Src participate in Stat3 phosphorylation remains to be elucidated. Jaks and Src can be hypothesized to cooperate in the activation of Stat3 in C4HD cells, as described in other breast cancer cells (20). Our findings led us to hypothesize a mechanism in which Src, activated by progestin binding to the classical nuclear PR, acts as the upstream kinase for phosphorylation of Jak1 and Jak2. We hereby propose two possible molecular mechanisms downstream to this event, consistent with results in our model system. First, both Src and Jaks might act as kinases for Stat3. Second, activated Jaks might serve to recruit Stat3 to Src, which in turn could directly phosphorylate Stat3. A mechanism like the latter has already been proposed in v-Src-transformed NIH 3T3 cells (57). Interestingly, we also unraveled several events in the molecular mechanism of MPA-induced Stat3 activation. Thus, we found that MPA-mediated Src activation, which in turn induces Stat3 tyrosine phosphorylation, is a requirement for Stat3 nuclear translocation and association with PR.
Here have here provided the first demonstration that progestins are able to induce transcriptional activation of Stat3 in both mouse and human breast cancer cells. As expected from results with Stat3 tyrosine phosphorylation, assessment of the molecular mechanism involved in MPA-stimulated Stat3 transcriptional activity in C4HD and T47D cells showed that it is mediated by the classical nuclear PR and that it requires Jak1, Jak2, and Src activities. Similarly, Src and the Jak family of tyrosine kinases have been found to cooperate in mediating constitutive Stat3 activation in several human breast cancer cell lines (20).
We have already demonstrated that proliferation in our model system is driven by a bidirectional interaction between progestins and growth factors pathways (3, 4, 25, 44). In the present work, we found that Stat3 is yet another player in this scenario, since MPA-induced Stat3 transcriptional activation (Fig. 7) is a requisite for MPA stimulation of C4HD cell growth (Fig. 8). Thus, transfection of C4HD cells with the DN Stat3 expression vector Stat3Y705-F (10, 23, 28) strongly inhibited MPA-induced proliferation by inducing cell cycle arrest and apoptosis. These findings, providing the first evidence that Stat3 participates in progestin-induced breast cancer growth, are in line with accumulating evidence showing involvement of Stat3 in the proliferation of breast cancer cells. Thus, in several breast cancer cell lines possessing activated Stat3, pharmacological inhibitors of Src and Jaks, whose activation is a requirement for Stat3 phosphorylation and DNA-binding capacity, resulted in abrogation of cell growth (12, 20), with a significant increase in the percentage of cells undergoing apoptosis (12, 20). In addition, induction of apoptosis and inhibition of cell growth were observed by transfection of breast tumor cells, displaying constitutive Stat3 activity, with a naturally occurring DN Stat3 form, Stat3β, or with DN Stat3Y705-F and DN Stat33E/V expression vectors (12, 20, 28). Very recently, a dramatic increase in Stat3 tyrosine phosphorylation with confluence was found in normal mammary cells, as well as in breast cancer cells (53). Confluence-dependent Stat3 activation was dependent on cell-to-cell contact and on Jak activity but did not require functional Src (53). On the other hand, we have demonstrated that transfection with a constitutively activated Stat3 form, Stat3-C (11), induced progestin-independent proliferation of breast cancer cells. This is the first evidence of a direct link between hormone-independent growth of breast tumors and Stat3 activity. Our results agree with previous studies with the constitutively activated Stat3-C mutant that proved that Stat3 has an intrinsic capacity to induce cell transformation. Thus, rodent fibroblasts expressing Stat3-C form colonies in soft agar and induce tumors in nude mice (11). Moreover, it has recently been shown that Stat3-C transforms immortalized human mammary epithelial cells which then acquire the capacity to develop tumors in irradiated nonobese diabetic-severe combined immunodeficient mice (17).
Members of the Bcl-2 family of antiapoptotic regulatory proteins have been identified as being encoded by Stat3-regulated genes, under conditions in which Stat3 modulates cell growth and survival (11, 14). Thus, constitutive Stat3 activation results in Bcl-xL expression in a range of tumor cells (55) that directly correlates with induction of proliferative responses by Stat3 (reviewed in references 41 and 55). Conversely, blockage of Stat3 activation or transfection with DN Stat3 variants resulted in induction of apoptosis, inhibition of tumor growth, and down-regulation of Bcl-xL (2). In our present work, we found that MPA-induced up-regulation of Bcl-xL protein expression in C4HD cells was inhibited by transfection with Stat3Y705-F. In addition, blockage of Src activity resulted also in significant inhibition of MPA-induced Bcl-xL expression, further demonstrating the involvement of Stat3 in progestin-induced Bcl-xL expression. On the other hand, transfection of C4HD cells with constitutively activated Sta3t-C mutant induced an increase in Bcl-xL protein levels, comparable to the effect exerted by MPA. These findings are consistent with a mechanism of malignant transformation in which persistent MPA-induced Stat3 activation might contribute to tumor growth by preventing apoptosis of C4HD cells. Blockage of Stat3 activity by transfection of DN Stat3Y705-F would therefore result in inhibition of MPA-induced growth by sensitizing tumor cells to apoptosis.
Finally, our results demonstrated that blockage of Stat3 activation by the DN Stat3Y705-F expression vector resulted in inhibition of in vivo breast tumor growth in our model of immunocompetent mice. We have used an experimental strategy that relied on modification of cells ex vivo before mice inoculation. Thus, C4HD cells were transiently transfected with the DNStat3Y705-F expression vector and were then inoculated s.c. into syngeneic mice. No tumors developed in a significantly high percentage of mice injected with DNStat3Y705-F-transfected C4HD cells. In addition, the low percentage of tumors that grew in animals injected with DNStat3Y705-F-transfected C4HD cells showed a greater delay in tumor formation and significantly lower tumor growth and growth rates. A likely explanation for the lack of tumor growth in five out of nine mice injected with DNStat3Y705-F-transfected C4HD cells is that DNStat3Y705-F induced massive apoptosis of C4HD cells (Fig. 8B), which absolutely prevented them from growing at the site of injection. On the other hand, assessment of the mechanisms involved in reduced proliferation of the low percentage of tumors that grew in mice injected with DNStat3Y705-F-transfected C4HD cells revealed a significantly lower Stat3 tyrosine phosphorylation compared to control groups. Lack of Stat3 activation was accompanied by a dramatic reduction in Bcl-xL protein expression. To our knowledge, there is only one more study assessing the effect of inhibition of Stat3 activity in breast tumor growth, in which G-quartet oligodeoxynucleotides (GQ-ODNs) were developed to inhibit Stat3 activation by abolishment of the capacity of Stat3 to bind DNA (22). Systemic administration of these GQ-ODNs blocked the growth of breast and prostate tumor xenografts in nude mice (22). Inhibitory mechanisms of tumor growth by GQ-ODNs were strikingly similar to those we have reported in our experimental strategy. Thus, levels of Stat3 activation and of Bcl-2 and Bcl-xL protein expression were found to be markedly inhibited and apoptosis was found to be much higher in tumors from mice receiving GQ-ODNs compared with tumors from placebo-treated cells (22). We could not define significantly higher levels of apoptosis in tumors from DNStat3Y705-F-transfected C4HD cells than those in control groups, using the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling method (data not shown). Although to some extent this was an unexpected result, a probable explanation is that the rapidity of the death process makes it difficult to visualize and to quantify apoptosis. Our samples were tumors that developed in mice for 21 days. It could be hypothesized that the mechanism of tumor growth inhibition in our in vivo experimental approach relies on blockage of MPA-induced Stat3 activation that results in the abrogation of expression of antiapoptotic proteins, such as Bcl-xL, which in turn triggers apoptosis of breast cancer cells.
In addition to direct regulation of gene expression by transfection with DN variants of Stat3, which might account for the growth inhibitory properties of DN Stat3, a strong bystander effect mediated by DN Stat3 expression has been reported. Thus, a series of previous studies demonstrated that gene therapy by electroinjection of the DN Stat3β expression vector into preexisting murine melanoma B16 tumors resulted in inhibition of tumor growth and tumor regression (35, 36). Interestingly, although only 10 to 15% of the tumor cells were transfected in vivo in these studies, the antitumor effect of Stat3β was associated with massive apoptosis of B16 cells, suggesting a bystander effect (36). Stat3β-transfected B16 cells released soluble factors capable of inducing apoptosis and cell cycle arrest of nontransfected B16 cells, which suggests that the bystander effect could be mediated in part by soluble factors (36). It was very recently demonstrated that blockage of Stat3 in tumor cells increased expression of proinflammatory cytokines and chemokines that activate innate immune cells to produce immunological danger signals such as nitric oxide and tumor necrosis factor alpha (54). Inflammatory mediators synthesized as a consequence of the abrogation of Stat3 function in tumor cells also activate dendritic cells, resulting in tumor-specific T-cell responses (54). Therefore, antitumor immune responses might be involved in the bystander effect. Finally, evidence has been provided demonstrating that inhibition of angiogenic signals could also mediate the bystander effect of abrogation of Stat3 signaling (37). Remarkably, all of the mechanisms mentioned above as mediators of the bystander effect of abrogation of Stat3 signaling might be able to inhibit DNStat3Y705-F-transfected C4HD cells in vivo growth. Particularly interesting is the concept of involvement of a host immune response in the antitumor effect, since we have worked with a model of immunocompetent mice, in contrast to the aforementioned study in which systemic administration of these GQ-ODNs inhibited the growth of breast tumor xenografts in nude mice (22). An intriguing question is the mechanism mediating the inhibition of Stat3 tyrosine phosphorylation in tumors arising from DNStat3Y705-F-transfected C4HD cells. Our in vitro studies showed both a significant increase in Stat3 expression after transfection of C4HD cells with the DNStat3Y705-F vector (Fig. 8D) and a direct correlation between the increase in Stat3 expression after DNStat3Y705-F transfection and the decrease in the levels of Stat3 tyrosine phosphorylation (Fig. 8E). It remains to be elucidated whether the levels of expression of the DNStat3Y705-F vector could be high enough to account for decreased levels of Stat3 phosphorylation after 21 days of in vivo tumor growth. Another interesting possibility to consider is that soluble factors released by cells expressing DNStat3Y705-F might result in abrogation of MPA-induced Stat3 tyrosine phosphorylation.
Here, we have not only unraveled a novel mechanism of progestin-Stat3 interaction but have also demonstrated novel nongenomic progestin effects in breast cancer cells directly associated with proliferation. Our findings have for the first time demonstrated that progestins are able to induce Stat3 transcriptional activation, which is in turn an obligatory requirement for progestin stimulation of both in vitro and in vivo breast cancer growth. Accumulated evidence, including our own results, has shown that breast cancer proliferation is driven by bidirectional cross talk between steroid hormones and growth factors. In the present study, we have demonstrated that Stat3 is yet another player in this scenario. Taking into account the role of Stat3 as a transcription factor, it could interact on chromatin with other transcription factors, such as PR, to regulate gene transcription both in normal proliferation and in the aberrant growth of malignant cells. On the other hand, proliferative effects of GFs are mediated by signaling pathways that ultimately result in activation of transcription factors. In this context, what makes Stats so attractive is that they could constitute a convergence point between steroid hormones and GF signaling pathways so that targeting of Stat3 might in turn be an alternative therapy for effective treatment of breast cancer.
Acknowledgments
This work was supported by grants IDB 201/OC-AR and PICT 2002 05-11055 from the National Agency of Scientific Promotion of Argentina and Oncomed-Reno CONICET 1819/03 from the Henry Moore Institute of Argentina.
We thank E. Arzt and A Carbia-Nagashima for expert assistance with the Stat3 transient-transfection assays, N. Lope for assistance with animal care, G. Rabinovich for assistance with the Annexin V binding assay, C. Lanari for providing the MPA-induced mammary tumor model, and the Gador Laboratory of Argentina for kindly providing the MPA.
REFERENCES
- 1.Aaronson, D. S., and C. M. Horvath. 2002. A road map for those who don't know JAK-STAT. Science 296:1653-1655. [DOI] [PubMed] [Google Scholar]
- 2.Amin, H. M., T. J. McDonnell, Y. Ma, Q. Lin, Y. Fujio, K. Kunisada, V. Leventaki, P. Das, G. Z. Rassidakis, C. Cutler, L. J. Medeiros, and R. Lai. 2004. Selective inhibition of STAT3 induces apoptosis and G1 cell cycle arrest in ALK-positive anaplastic large cell lymphoma. Oncogene 23:5426-5434. [DOI] [PubMed] [Google Scholar]
- 3.Balana, M. E., L. Labriola, M. Salatino, F. Movsichoff, G. Peters, E. H. Charreau, and P. V. Elizalde. 2001. Activation of ErbB-2 via a hierarchical interaction between ErbB-2 and type I insulin-like growth factor receptor in mammary tumor cells. Oncogene 20:34-47. [DOI] [PubMed] [Google Scholar]
- 4.Balana, M. E., R. Lupu, L. Labriola, E. H. Charreau, and P. V. Elizalde. 1999. Interactions between progestins and heregulin (HRG) signaling pathways: HRG acts as mediator of progestins proliferative effects in mouse mammary adenocarcinomas. Oncogene 18:6370-6379. [DOI] [PubMed] [Google Scholar]
- 5.Ballare, C., M. Uhrig, T. Bechtold, E. Sancho, M. Di Domenico, A. Migliaccio, F. Auricchio, and M. Beato. 2003. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol. Cell. Biol. 23:1994-2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boonyaratanakornkit, V., M. P. Scott, V. Ribon, L. Sherman, S. M. Anderson, J. L. Maller, W. T. Miller, and D. P. Edwards. 2001. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol. Cell 8:269-280. [DOI] [PubMed] [Google Scholar]
- 7.Bowman, T., R. Garcia, J. Turkson, and R. Jove. 2000. STATs in oncogenesis. Oncogene 19:2474-2488. [DOI] [PubMed] [Google Scholar]
- 8.Braunsberg, H., N. G. Coldham, R. E. Leake, S. K. Cowan, and W. Wong. 1987. Actions of a progestogen on human breast cancer cells: mechanisms of growth stimulation and inhibition. Eur. J. Cancer Clin. Oncol. 23:563-571. [DOI] [PubMed] [Google Scholar]
- 9.Bromberg, J., and J. E. Darnell, Jr. 2000. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 19:2468-2473. [DOI] [PubMed] [Google Scholar]
- 10.Bromberg, J. F., C. M. Horvath, D. Besser, W. W. Lathem, and J. E. Darnell, Jr. 1998. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 18:2553-2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bromberg, J. F., M. H. Wrzeszczynska, G. Devgan, Y. Zhao, R. G. Pestell, C. Albanese, and J. E. Darnell, Jr. 1999. Stat3 as an oncogene. Cell 98:295-303. [DOI] [PubMed] [Google Scholar]
- 12.Burke, W. M., X. Jin, H. J. Lin, M. Huang, R. Liu, R. K. Reynolds, and J. Lin. 2001. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene 20:7925-7934. [DOI] [PubMed] [Google Scholar]
- 13.Castoria, G., M. V. Barone, M. Di Domenico, A. Bilancio, D. Ametrano, A. Migliaccio, and F. Auricchio. 1999. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. EMBO J. 18:2500-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catlett-Falcone, R., T. H. Landowski, M. M. Oshiro, J. Turkson, A. Levitzki, R. Savino, G. Ciliberto, L. Moscinski, J. L. Fernandez-Luna, G. Nunez, W. S. Dalton, and R. Jove. 1999. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 10:105-115. [DOI] [PubMed] [Google Scholar]
- 15.Clarke, C. L., and R. L. Sutherland. 1990. Progestin regulation of cellular proliferation. Endocr. Rev. 11:266-301. [DOI] [PubMed] [Google Scholar]
- 16.Darnell, J. E., Jr., I. M. Kerr, and G. R. Stark. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415-1421. [DOI] [PubMed] [Google Scholar]
- 17.Dechow, T. N., L. Pedranzini, A. Leitch, K. Leslie, W. L. Gerald, I. Linkov, and J. F. Bromberg. 2004. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by Stat3-C. Proc. Natl. Acad. Sci. USA 101:10602-10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Miguel, F., S. O. Lee, S. A. Onate, and A. C. Gao. 2003. Stat3 enhances transactivation of steroid hormone receptors. Nucl. Recept. 1:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dran, G., I. A. Luthy, A. A. Molinolo, F. Montecchia, E. H. Charreau, C. D. Pasqualini, and C. Lanari. 1995. Effect of medroxyprogesterone acetate (MPA) and serum factors on cell proliferation in primary cultures of an MPA-induced mammary adenocarcinoma. Breast Cancer Res. Treat. 35:173-186. [DOI] [PubMed] [Google Scholar]
- 20.Garcia, R., T. L. Bowman, G. Niu, H. Yu, S. Minton, C. A. Muro-Cacho, C. E. Cox, R. Falcone, R. Fairclough, S. Parsons, A. Laudano, A. Gazit, A. Levitzki, A. Kraker, and R. Jove. 2001. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 20:2499-2513. [DOI] [PubMed] [Google Scholar]
- 21.Groshong, S. D., G. I. Owen, B. Grimison, I. E. Schauer, M. C. Todd, T. A. Langan, R. A. Sclafani, C. A. Lange, and K. B. Horwitz. 1997. Biphasic regulation of breast cancer cell growth by progesterone: role of the cyclin-dependent kinase inhibitors, p21 and p27(Kip1). Mol. Endocrinol. 11:1593-1607. [DOI] [PubMed] [Google Scholar]
- 22.Jing, N., Y. Li, W. Xiong, W. Sha, L. Jing, and D. J. Tweardy. 2004. G-quartet oligonucleotides: a new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 64:6603-6609. [DOI] [PubMed] [Google Scholar]
- 23.Kaptein, A., V. Paillard, and M. Saunders. 1996. Dominant negative stat3 mutant inhibits interleukin-6-induced Jak-STAT signal transduction. J. Biol. Chem. 271:5961-5964. [DOI] [PubMed] [Google Scholar]
- 24.Kiss, R., R. J. Paridaens, J. C. Heuson, and A. J. Danguy. 1986. Effect of progesterone on cell proliferation in the MXT mouse hormone-sensitive mammary neoplasm. J. Natl. Cancer Inst. 77:173-178. [PubMed] [Google Scholar]
- 25.Labriola, L., M. Salatino, C. J. Proietti, A. Pecci, O. A. Coso, A. R. Kornblihtt, E. H. Charreau, and P. V. Elizalde. 2003. Heregulin induces transcriptional activation of the progesterone receptor by a mechanism that requires functional ErbB-2 and mitogen-activated protein kinase activation in breast cancer cells. Mol. Cell. Biol. 23:1095-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lange, C. A., J. K. Richer, T. Shen, and K. B. Horwitz. 1998. Convergence of progesterone and epidermal growth factor signaling in breast cancer. Potentiation of mitogen-activated protein kinase pathways. J. Biol. Chem. 273:31308-31316. [DOI] [PubMed] [Google Scholar]
- 27.Leonhardt, S. A., V. Boonyaratanakornkit, and D. P. Edwards. 2003. Progesterone receptor transcription and non-transcription signaling mechanisms. Steroids 68:761-770. [DOI] [PubMed] [Google Scholar]
- 28.Li, L., and P. E. Shaw. 2002. Autocrine-mediated activation of STAT3 correlates with cell proliferation in breast carcinoma lines. J. Biol. Chem. 277:17397-17405. [DOI] [PubMed] [Google Scholar]
- 29.Lin, V. C., R. Jin, P. H. Tan, S. E. Aw, C. T. Woon, and B. H. Bay. 2003. Progesterone induces cellular differentiation in MDA-MB-231 breast cancer cells transfected with progesterone receptor complementary DNA. Am. J. Pathol. 162:1781-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu, T., and T. F. Ogle. 2002. Signal transducer and activator of transcription 3 is expressed in the decidualized mesometrium of pregnancy and associates with the progesterone receptor through protein-protein interactions. Biol. Reprod. 67:114-118. [DOI] [PubMed] [Google Scholar]
- 31.Lydon, J. P., L. Sivaraman, and O. M. Conneely. 2000. A reappraisal of progesterone action in the mammary gland. J. Mammary Gland Biol. Neoplasia 5:325-338. [DOI] [PubMed] [Google Scholar]
- 32.Manni, A., B. Badger, C. Wright, S. R. Ahmed, and L. M. Demers. 1987. Effects of progestins on growth of experimental breast cancer in culture: interaction with estradiol and prolactin and involvement of the polyamine pathway. Cancer Res. 47:3066-3071. [PubMed] [Google Scholar]
- 33.Migliaccio, A., D. Piccolo, G. Castoria, M. Di Domenico, A. Bilancio, M. Lombardi, W. Gong, M. Beato, and F. Auricchio. 1998. Activation of the Src/p21ras/Erk pathway by progesterone receptor via cross-talk with estrogen receptor. EMBO J. 17:2008-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore, M. R., J. L. Conover, and K. M. Franks. 2000. Progestin effects on long-term growth, death, and Bcl-xL in breast cancer cells. Biochem. Biophys. Res. Commun. 277:650-654. [DOI] [PubMed] [Google Scholar]
- 35.Niu, G., R. Heller, R. Catlett-Falcone, D. Coppola, M. Jaroszeski, W. Dalton, R. Jove, and H. Yu. 1999. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 59:5059-5063. [PubMed] [Google Scholar]
- 36.Niu, G., K. H. Shain, M. Huang, R. Ravi, A. Bedi, W. S. Dalton, R. Jove, and H. Yu. 2001. Overexpression of a dominant-negative signal transducer and activator of transcription 3 variant in tumor cells leads to production of soluble factors that induce apoptosis and cell cycle arrest. Cancer Res. 61:3276-3280. [PubMed] [Google Scholar]
- 37.Niu, G., K. L. Wright, M. Huang, L. Song, E. Haura, J. Turkson, S. Zhang, T. Wang, D. Sinibaldi, D. Coppola, R. Heller, L. M. Ellis, J. Karras, J. Bromberg, D. Pardoll, R. Jove, and H. Yu. 2002. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21:2000-2008. [DOI] [PubMed] [Google Scholar]
- 38.Olayioye, M. A., I. Beuvink, K. Horsch, J. M. Daly, and N. E. Hynes. 1999. ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem. 274:17209-17218. [DOI] [PubMed] [Google Scholar]
- 39.Paukku, K., S. Valgeirsdottir, P. Saharinen, M. Bergman, C. H. Heldin, and O. Silvennoinen. 2000. Platelet-derived growth factor (PDGF)-induced activation of signal transducer and activator of transcription (Stat) 5 is mediated by PDGF beta-receptor and is not dependent on c-src, fyn, jak1 or jak2 kinases. Biochem. J. 345(Pt. 3):759-766. [PMC free article] [PubMed] [Google Scholar]
- 40.Puricelli, L., C. J. Proiettii, L. Labriola, M. Salatino, M. E. Balana, G. J. Aguirre, R. Lupu, O. P. Pignataro, E. H. Charreau, J. E. Bal de Kier, and P. V. Elizalde. 2002. Heregulin inhibits proliferation via ERKs and phosphatidyl-inositol 3-kinase activation but regulates urokinase plasminogen activator independently of these pathways in metastatic mammary tumor cells. Int. J. Cancer 100:642-653. [DOI] [PubMed] [Google Scholar]
- 41.Real, P. J., A. Sierra, A. De Juan, J. C. Segovia, J. M. Lopez-Vega, and J. L. Fernandez-Luna. 2002. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 21:7611-7618. [DOI] [PubMed] [Google Scholar]
- 42.Ren, Z., and T. S. Schaefer. 2002. ErbB-2 activates Stat3 alpha in a Src- and JAK2-dependent manner. J. Biol. Chem. 277:38486-38493. [DOI] [PubMed] [Google Scholar]
- 43.Richer, J. K., C. A. Lange, N. G. Manning, G. Owen, R. Powell, and K. B. Horwitz. 1998. Convergence of progesterone with growth factor and cytokine signaling in breast cancer. Progesterone receptors regulate signal transducers and activators of transcription expression and activity. J. Biol. Chem. 273:31317-31326. [DOI] [PubMed] [Google Scholar]
- 44.Salatino, M., R. Schillaci, C. J. Proietti, R. Carnevale, I. Frahm, A. A. Molinolo, A. Iribarren, E. H. Charreau, and P. V. Elizalde. 2004. Inhibition of in vivo breast cancer growth by antisense oligodeoxynucleotides to type I insulin-like growth factor receptor mRNA involves inactivation of ErbBs, PI-3K/Akt and p42/p44 MAPK signaling pathways but not modulation of progesterone receptor activity. Oncogene 23:5161-5174. [DOI] [PubMed] [Google Scholar]
- 45.Sartor, C. I., M. L. Dziubinski, C. L. Yu, R. Jove, and S. P. Ethier. 1997. Role of epidermal growth factor receptor and STAT-3 activation in autonomous proliferation of SUM-102PT human breast cancer cells. Cancer Res. 57:978-987. [PubMed] [Google Scholar]
- 46.Schaefer, L. K., S. Wang, and T. S. Schaefer. 2000. Oncostatin M activates stat DNA binding and transcriptional activity in primary human fetal astrocytes: low- and high-passage cells have distinct patterns of stat activation. Cytokine 12:1647-1655. [DOI] [PubMed] [Google Scholar]
- 47.Schulze, H., M. Ballmaier, K. Welte, and M. Germeshausen. 2000. Thrombopoietin induces the generation of distinct Stat1, Stat3, Stat5a and Stat5b homo- and heterodimeric complexes with different kinetics in human platelets. Exp. Hematol. 28:294-304. [DOI] [PubMed] [Google Scholar]
- 48.Shen, T., K. B. Horwitz, and C. A. Lange. 2001. Transcriptional hyperactivity of human progesterone receptors is coupled to their ligand-dependent down-regulation by mitogen-activated protein kinase-dependent phosphorylation of serine 294. Mol. Cell. Biol. 21:6122-6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherman, C. T., and A. R. Brasier. 2001. Role of signal transducers and activators of transcription 1 and 3 in inducible regulation of the human angiotensinogen gene by interleukin-6. Mol. Endocrinol. 15:441-457. [DOI] [PubMed] [Google Scholar]
- 50.Silvennoinen, O., C. Schindler, J. Schlessinger, and D. E. Levy. 1993. Ras-independent growth factor signaling by transcription factor tyrosine phosphorylation. Science 261:1736-1739. [DOI] [PubMed] [Google Scholar]
- 51.Stoecklin, E., M. Wissler, D. Schaetzle, E. Pfitzner, and B. Groner. 1999. Interactions in the transcriptional regulation exerted by Stat5 and by members of the steroid hormone receptor family. J. Steroid Biochem. Mol. Biol. 69:195-204. [DOI] [PubMed] [Google Scholar]
- 52.Taga, T., and T. Kishimoto. 1997. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 15:797-819. [DOI] [PubMed] [Google Scholar]
- 53.Vultur, A., J. Cao, R. Arulanandam, J. Turkson, R. Jove, P. Greer, A. Craig, B. Elliott, and L. Raptis. 2004. Cell-to-cell adhesion modulates Stat3 activity in normal and breast carcinoma cells. Oncogene 23:2600-2616. [DOI] [PubMed] [Google Scholar]
- 54.Wang, T., G. Niu, M. Kortylewski, L. Burdelya, K. Shain, S. Zhang, R. Bhattacharya, D. Gabrilovich, R. Heller, D. Coppola, W. Dalton, R. Jove, D. Pardoll, and H. Yu. 2004. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10:48-54. [DOI] [PubMed] [Google Scholar]
- 55.Yu, H., and R. Jove. 2004. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer 4:97-105. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, X., M. H. Wrzeszczynska, C. M. Horvath, and J. E. Darnell, Jr. 1999. Interacting regions in Stat3 and c-Jun that participate in cooperative transcriptional activation. Mol. Cell. Biol. 19:7138-7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang, Y., J. Turkson, C. Carter-Su, T. Smithgall, A. Levitzki, A. Kraker, J. J. Krolewski, P. Medveczky, and R. Jove. 2000. Activation of Stat3 in v-Src-transformed fibroblasts requires cooperation of Jak1 kinase activity. J. Biol. Chem. 275:24935-24944. [DOI] [PubMed] [Google Scholar]
- 58.Zhang, Z., S. Jones, J. S. Hagood, N. L. Fuentes, and G. M. Fuller. 1997. STAT3 acts as a co-activator of glucocorticoid receptor signaling. J. Biol. Chem. 272:30607-30610. [DOI] [PubMed] [Google Scholar]
- 59.Zhu, Y., J. Bond, and P. Thomas. 2003. Identification, classification, and partial characterization of genes in humans and other vertebrates homologous to a fish membrane progestin receptor. Proc. Natl. Acad. Sci. USA 100:2237-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]