

Abstract
Gene editing technologies hold promise for enabling the next generation of adoptive cellular therapies. In conventional gene editing platforms that rely on nuclease activity, such as clustered regularly interspaced short palindromic repeats CRISPR-associated protein 9 (CRISPR-Cas9), allow efficient introduction of genetic modifications; however, these modifications occur via the generation of DNA double-strand breaks (DSBs) and can lead to unwanted genomic alterations and genotoxicity. Here, we apply a novel modular RNA aptamer-mediated Pin-point base editing platform to simultaneously introduce multiple gene knockouts and site-specific integration of a transgene in human primary T cells. We demonstrate high editing efficiency and purity at all target sites and significantly reduced frequency of chromosomal translocations compared with the conventional CRISPR-Cas9 system. Site-specific knockin of a chimeric antigen receptor and multiplex gene knockout are achieved within a single intervention and without the requirement for additional sequence-targeting components. The ability to perform complex genome editing efficiently and precisely highlights the potential of the Pin-point platform for application in a range of advanced cell therapies.
Keywords: base editing, gene editing, CRISPR, allogeneic cell therapy, multiple base editing, multiple gene knockout, transgene integration, knockin, knockout, advanced genome editing
Graphical abstract
Porreca and colleagues report on the use of the modular aptamer-mediated Pin-point base editing platform for genome modification. Owing to its modularity, the Pin-point platform enables the introduction of multiple discrete modifications simultaneously by utilizing a single targeting element and, therefore, facilitates the generation of complex engineered cell therapies.
Introduction
Gene editing technologies have entered the clinic and show significant potential for advancing next-generation therapies, particularly in the development of more efficient chimeric antigen receptor (CAR)-T cell therapies to address hematological malignancies.1,2,3 To overcome the logistical and infrastructure-related challenges and product variability barriers of the autologous cell therapy paradigm, recent focus has shifted to realizing the potential of allogeneic cell therapies. The manufacture of allogeneic cell products requires multiple edits to prevent both graft-versus-host disease and immune rejection by the host, which would otherwise limit efficacy, persistence, and safety of the cell product. To expand the scope of these innovative off-the-shelf therapies to solid tumors, further edits will also be required to ensure therapeutic cells retain their efficacy in the refractory and heterogeneous tumor microenvironment.4 These factors, together with the need to provide new functions to the cells to make effective and safe therapies that offer wider patient accessibility and therapy deployment, ultimately demand increasingly refined genome editing strategies.
Gene editing technologies such as zinc-finger nucleases, transcription activator-like effector nucleases and CRISPR-Cas9 have all been employed to successfully perform targeted editing at genomic loci for effective knockout and knockin applications. However, the generation of double-strand breaks (DSBs) inherent to their mechanism of conferring a DNA edit can lead to chromosomal loss or structural variation.5,6,7,8,9,10,11 The occurrence of chromosomal aberrations is enhanced in the context of multi-gene editing as more concurrent DSBs are generated, and the extent of this damage is expanded if DNA breaks also occur at off-target sites. Although many structural aberrations in a cell may not be viable, it has been reported that some rearrangements could be stable and persist over time.1,9 While it is uncertain whether these rearrangements increase tumorigenicity risk or compromise the safety and efficacy of engineered cell therapy products, such aberrations remain an important point of concern.
Base editing with its ability to induce genetic modifications without relying on DSB formation12,13 has emerged as a strong contender in the development of advanced cell therapies, particularly in the context of multi-gene editing strategies. The two main categories of base editors, cytosine base editors and adenine base editors mediate efficient C to T and A to G base changes, respectively.12,13 Due to their capacity for programmable introduction of a single point mutation, base editors have been employed to facilitate gene disruption via generation of a premature termination codon (PTC)14,15 or by mutation of splice acceptor (SA) or splice donor (SD) sites 16,17 with high precision and efficiency while generating minimal undesired editing outcomes compared with standard nucleases. Rapid technological developments to increase the precision, efficiency and targeting scope of base editors, alongside an improved safety profile,18,19,20,21,22,23,24 have enabled fast-tracked and successful progression to the clinic.25,26 Base editing combined with lentiviral delivery of the CAR transgene has been applied to introduce multiple gene knockout for the generation of enhanced allogeneic CAR-T therapies,16,19,26,27,28 however, such approaches come with many limitations, including the risk of insertional mutagenesis, variable transgene expression and gene silencing. To overcome these limitations, a targeted transgene integration approach facilitated by CRISPR-Cas technologies has become increasingly popular.29 However, to date, simultaneous site-specific knockin alongside base editing at other loci has only been achieved by combining two Cas homologs (Cas9 for base editing and Cas12 for knockin) to avoid cross utilization of sgRNAs.28,30
We have previously described the modular RNA aptamer-mediated Pin-point base editing system and demonstrated that this technology can edit targeted cytosines with high efficiency in human immortalized cells.31 The specific version of the Pin-point technology utilized in this study (Figure 1A) relies on a CRISPR-Cas module and a recruiting RNA aptamer derived from the operator stem-loop of bacteriophage MS2 (MS2), fused to the guide RNA (gRNA) to recruit the effector module. The effector module is composed of a deaminase (e.g., rAPOBEC1) fused to the MS2 coat protein (MCP), which binds to the aptamer. The recruitment of the deaminase to the target site results in editing of specific residues on the unpaired DNA strand within the CRISPR R-loop. Owing to its modularity the Pin-point platform has the potential to introduce multiple discrete modifications simultaneously by utilizing one individual targeting element and to facilitate the generation of complex edited therapies.
Figure 1.

The Pin-point platform is a highly efficient technology for multiplex editing in T cells
(A) Schematic of the configuration of the Pin-point base editing technology used in this manuscript. An SpCas9 nickase (nCas9-UGI-UGI) binds to the gRNA, the recruiting RNA aptamer (MS2) fused to the gRNA recruits the effector module. The effector module is composed of a cytidine deaminase (rAPOBEC1) fused to the aptamer binding protein (MCP). The recruitment of the deaminase to the target site forms an active complex capable of editing target cytosine residues on the unpaired DNA strand within the CRISPR R-loop. (B) Representative electropherograms from edited and control samples for the four targets following co-delivery of Pin-point mRNAs and four target sgRNAs as analyzed by Sanger sequencing 7 days post electroporation. The target C is highlighted by blue shading. (C) Levels of C to T conversion of the target C at B2M, CD52, TRAC, and PDCD1 loci following co-delivery of Pin-point mRNAs and four target sgRNAs, as analyzed by NGS 7 days post electroporation. Data represented as mean (SEM), n = 3 independent biological T cell donors. (D) Alignment plots showing the top 10 most abundant reads for each of the four targets. The target C is shown in red and underlined. The target splice site is shown in red. (E) Levels of C to G or A conversion of the target C at B2M, CD52, TRAC, and PDCD1 loci following co-delivery of Pin-point mRNAs and four target sgRNAs, as analyzed by NGS. (F) Insertion (INS) and deletion (DEL) frequency at the target C at B2M, CD52, TRAC, and PDCD1 loci following co-delivery of Pin-point mRNAs and four target sgRNAs, as analyzed by NGS. In (E) and (F), data represented as mean (SEM), n = 3 independent biological T cell donors.
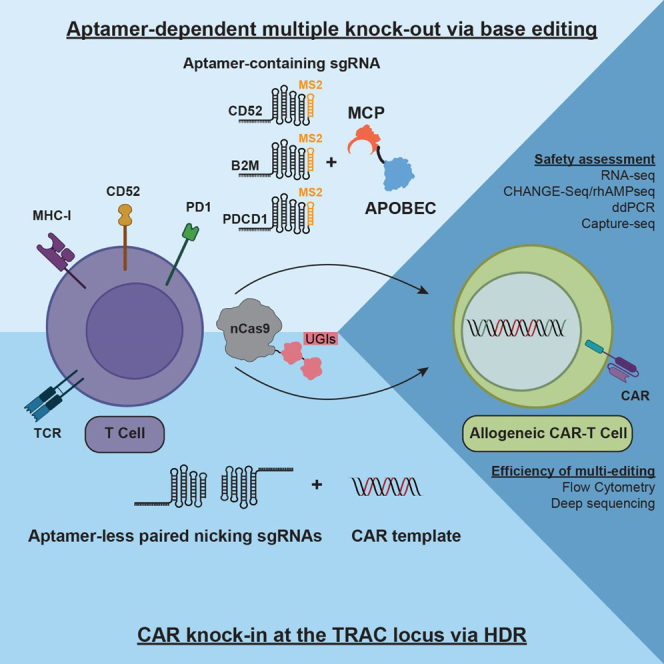
We demonstrate the adaptation of the plasmid-based Pin-point system into a safe and efficient fully synthetic system which can be readily adopted for manufacturing engineered cell therapies by combining mRNAs encoding the requisite Cas and deaminase modules with Pin-point gRNAs. We utilize a version of the Pin-point base editor composed of rAPOBEC1 and Cas9 nickase (nCas9) for the generation of allogeneic human CAR-T cells. Initially, we performed a screen to identify highly functional aptamer-containing gRNAs targeting four well-established genes capable of enhancing CAR-T cell function: beta-2-microglobulin (B2M) to eliminate HLA class I proteins and evade host immune responses, T cell receptor alpha constant (TRAC) to reduce the potential to cause graft versus host disease, CD52 molecule (CD52) to enable the use of the anti-CD52 antibody alemtuzumab to be employed as part of the lymphodepletion regimen prior to the cell infusion, and programmed cell death protein 1 (PDCD1) to reduce exhaustion of the infused CAR-T cells. We demonstrate efficient and specific multi-gene editing with minimal differences in editing efficiencies whether editing a single locus or multiple loci and with undetectable incidence of chromosomal translocations. In addition to being compatible with conventional lentiviral CAR transgene delivery technologies, the Pin-point base editing platform can be employed to perform multiplex genome engineering operations, enabling simultaneous target transgene knockin and multi-target knockout. We demonstrate the utility of this approach by combining aptamer-containing and aptamer-less gRNAs to generate functional engineered CAR-T cells via simultaneous knockout of multiple targets alongside targeted CD19-CAR insertion at the endogenous TRAC locus. The Pin-point platform thus enables complex genetic modifications of T cells using a single DNA-targeting nuclease via a novel single-step process.
Results
Multiplex editing in human T cells with the Pin-point base editing system
To determine the optimal Pin-point platform (Figure 1A) configuration with rAPOBEC1 as the effector module we assessed the impact of aptamer copy number and position within the gRNA on editing efficiencies in mammalian cells. The configuration with one copy of the MS2 aptamer located at the 3′ end of the tracrRNA resulted in optimal base editing (Figures S1A and S1B) and was adopted as the basis for the synthetic gRNA designs employed in this study.
Using fully synthetic RNA reagents, as is conventional in engineered adoptive T cell therapy manufacturing, we screened a panel of crRNAs to identify the best performing gRNAs for knockout of B2M, TRAC, PDCD1, and CD52 with the Pin-point base editing system. The crRNAs were designed to result in the introduction of a PTC or mutation at either the SA or SD sites in each target gene (Table S1). Individual crRNAs were delivered into human primary T cells by electroporation in combination with the aptamer-containing tracrRNA, an mRNA encoding nCas9 fused to uracil glycosylase inhibitor (UGI) and an mRNA encoding rAPOBEC1-MCP. Base conversion was assessed by amplicon sequencing and target protein expression was evaluated by flow cytometry (Figures S1C and S1D). crRNAs exhibiting the highest level of target C to T conversion and associated protein knockout for each gene (SD disruption in exon 1 for B2M, SD disruption in exon 1 for CD52, SA disruption in exon 3 for TRAC, and SD disruption in exon 1 for PDCD1) were selected for synthesis as one-part single gRNAs (sgRNA) for simultaneous multi-gene editing experiments. Delivery of sgRNAs in multiplex achieved high levels of C to T conversion at the target C for each of the four genes (∼76%–90%) (Figures 1B–1D) with efficiencies comparable with that observed for individual sgRNA delivery (Figure S1E). We observed minimal undesired C to A or C to G conversion (Figures 1B, 1D, and 1E) or indel mutations (Figures 1F and S1F) at each of the four target loci. Thus, the Pin-point system configuration consisting of nCas9-UGI, rAPOBEC1-MCP, and an sgRNA containing an MS2 aptamer is capable of simultaneously generating C to T edits with high efficiency and purity at multiple target loci when delivered to human T cells by synthetic RNA reagents.
Characterization of multi-gene knockout T cells
To determine the extent of multiplex target protein knockout in individual cells we performed multi-color flow cytometry analysis (Figure S2). Non-activated T cells express B2M, CD52, and TCRa/b, all of which showed ∼75%–85% reduction in protein expression following multi-editing with either the Pin-point platform or with SpCas9 with sgRNAs designed for optimal indel formation (Figures 2A and 2B). By contrast, PDCD1 induction requires activation of T cells for appreciable expression. PD1 protein levels were therefore quantified in T cells cultured in the presence of phorbol-12-myristate 13-acetate (PMA) and ionomycin32 immediately before analysis (Figures 2C and 2D), which again demonstrated the expected ∼80% reduction in protein expression following multi-editing with either the Pin-point platform or with SpCas9. Thus all four targets exhibited the expected high efficiency of protein knockout consistent with the high base editing efficiency we observed at the genomic level, both in non-activated and activated T cells.
Figure 2.

Quantification of T cell target knockout in individual cells
(A–C) Flow cytometry histograms to show protein levels for CD52, TCRa/b, PD1, and B2M following co-delivery of Pin-point or SpCas9 mRNAs and their compatible four target sgRNAs in (A) non-activated and (C) activated T cells, analyzed 7 days post electroporation. Non-edited mock electroporated cells are used as control. In this comparison, optimal gRNAs for SpCas9 have been used and these differ in their spacer sequence from the optimal Pin-point gRNAs (further details in the materials and methods). Quantification of protein knockout reported as the percentage of (B) non-activated and (D) activated T cells expressing undetectable levels of the protein, normalized to the frequency of expression in mock electroporated cells. Data in (B) and (D) represented as mean (SEM), n = 4 independent biological T cell donors. Protein expression phenotypes within populations of (E) non-activated and (F) activated T cells either mock electroporated as controls or edited with either SpCas9 or the Pin-point platform. Columns are single cells classified as positive (colored) or negative (white) for the four target proteins with respect to the gates shown in (A) and (C). Color bar shows clusters of cells with similar patterns of expression of the protein targets (rows). k-means clustering, k = 16. Quantification of single, double, triple, and quadruple negative target protein expression in (G) non-activated and (H) activated T cells following co-delivery of Pin-point or SpCas9 mRNAs and four target gRNAs, analyzed by flow cytometry 7 days post electroporation. Control is mock electroporated T cells without RNA. Single cells are classified as either positive or negative for the four target proteins according to the gates shown in Figure S2. Data in (G) and (H) represented as mean (SEM), n = 4 independent biological T cell donors. (I) Fold expansion of T cells as measured by cell counts 3 days post co-delivery of Pin-point or SpCas9 mRNAs and three or four target gRNAs. Data represented as mean (SEM), n = 4 independent biological T cell donors. A paired student t-test was applied.
We next sought to determine the degree of simultaneous target knockout. Due to differences in the expression of PDCD1 and TRAC between non-activated and activated states (Figures 2E and 2F), we performed analysis of T cells cultured both in the absence and presence of PMA and ionomycin. Approximately 80% of unedited, non-activated T cells were triple positive for B2M, CD52, and TCRa/b, whereas PD1 was undetectable in the majority of the population (Figures 2E and 2G). Following editing by either the Pin-point platform or by SpCas9, triple-positive cells were almost undetectable; instead 80% of the population were negative for the three markers TCRa/b, B2M, and CD52, while the remaining 20% were double-negative (Figures 2E and 2G). PMA-ionomycin-activated T cells exhibited a more heterogeneous phenotype than unstimulated T cells due to both non-uniform upregulation of PD1 and the downregulation of TCRa/b compared with non-activated cells, with ∼50% of mock electroporated controls expressing all four markers and ∼35% expressing three of the four markers (Figures 2F and 2H). Nonetheless, ∼50% of T cells edited by the Pin-point platform and ∼60% of T cells edited by SpCas9 were negative for the four targets, with an additional 30% negative for three of the four targets (Figures 2F and 2H). This indicates a high degree of simultaneous knockout, consistent with expectations based on individual target knockout efficiencies.
It is well known that nuclease-dependent gene editing technologies have the potential to impair cell fitness and proliferative capacity due to the activation of DNA damage responses, which is exacerbated when introducing multiple DSBs.33,34 Consistent with the DSB-independent mechanism of base editing, we observed that simultaneous editing at three or four loci with SpCas9 adversely impacts T cell proliferation compared with the Pin-point platform (Figure 2I). Thus, the Pin-point base editing technology enables efficient knockout of multiple genes in T cells and preserves their proliferative capacity.
Assessment of gRNA-specific off-target editing
The potential of gene editing technologies to generate off-target edits is an important consideration for clinical risk assessment of engineered cell therapies. To experimentally identify candidate off-target editing sites for each of the four gRNAs, we performed “circularization for high-throughput analysis of nuclease genome-wide effects by sequencing” (CHANGE-seq) using SpCas9 on genomic DNA (gDNA) isolated from T cells.35 To validate the method, we used a well-characterized sgRNA against VEGFA as a positive control.36 All off-targets previously identified for the VEGFA sgRNA37 were identified in the CHANGE-seq experiment (Table S2). For the four targets, we identified variable numbers of candidate off-target sites ranging from 223 to 481 per sgRNA (Table S2). The top 100 CHANGE-seq nominated off-target candidate sites for each gRNA, based on the hierarchical selection strategy described in the materials and methods (Figure S3A; Table S2), were subsequently analyzed by rhAmpSeq in human T cells edited at the four target loci using either SpCas9 or the Pin-point platform (Table S3). We additionally analyzed all the candidate off-targets having up to four mismatches to the target site (Table S4). In the validation rhAmpSeq experiments, we detected high levels of indel formation at all four on-target sites in samples edited with SpCas9 and at low levels at three of the four targets in samples edited with the Pin-point platform (1.2%–2.3%) (Figures 3A and S3B; Table S3), consistent with our previous observations (Figure 1F). Of the total of 456 CHANGE-seq candidate sites that were analyzed, we detected low levels of off-target base editing at four sites in samples edited with the Pin-point platform (0.7%–2.2%) (Figure 3B, and S3B; Table S3), and off-target indel formation at two sites in samples edited with SpCas9 (1.2%–20.2%) (Figures 3A, and S3B; Table S3). Both systems shared a common PDCD1 gRNA off-target site; however, editing levels were substantially lower in samples edited with the Pin-point platform, consistent with the more specific sequence requirements for base editing than cutting with SpCas9. Moreover, we obtained no evidence for indel formation at any off-target sites in samples edited with the Pin-point platform (Tables S3 and S4).
Figure 3.

Assessment of DNA off-target editing
(A and B) On-/off-target activity of sgRNAs targeting B2M, PDCD1, TRAC, or CD52 genes, determined by rhAmpSeq NGS profiling of on-target, 100 nominated off-target sites, and of all the candidates having up to four mismatches to the target site per sgRNA identified by CHANGE-seq. The on-/off-target activity of each sgRNA was profiled in human T cells edited with either the Pin-point base editor or SpCas9 5 days post electroporation and the percent editing (% base editing events in A or % indels events in B) determined in each case. Each dot depicts the maximal percentage editing at a given site in one human donor for control (mock electroporation, x axis) versus edit (edited sample, y axis) with an average coverage per panel of >35,000 reads. Top left quadrant indicates events with more than 0.5% editing in edited samples and less than 0.5% editing in control sample. Blue dots highlight on-target editing, red dots highlight validated off-target activity occurring in at least 0.5% of corrected reads (dotted lines) and in both human donors profiled. Shown on base 10 logarithmic scale.
Assessment of chromosomal translocations
Multiplex editing with DSB-dependent technologies can lead to the generation of chromosomal translocations between the edited loci,1,2 whereas cleavage of only a single DNA strand by the nCas9 variant38,39 used in the Pin-point platform is expected to substantially reduce this risk. As indel formation was minimized across on- and off-target sites in samples edited with the Pin-point platform this suggested to us that translocations between DSBs would indeed be substantially reduced compared with SpCas9.
To test this hypothesis, we performed targeted DNA capture to enrich for genomic regions around the four gRNA target sequences (Figure 4A), followed by paired-end sequencing to an average depth of 4,000× (Capture-seq) (Table S5). Identification and quantification of translocations for each target was performed using the DRAGEN Structural Variant (SV) Caller,40,41 which identified translocations involving untargeted genomic regions in addition to all expected translocation products between pairs of captured gRNA target loci in SpCas9-edited samples (Figures 4A–4C; Tables S5–S7). To validate the Capture-seq method, translocations between the four sgRNA targets were quantified by orthogonal droplet digital polymerase chain reaction (ddPCR) analysis using probes spanning the expected translocation breakpoints. Translocations were identified at comparable frequencies using the two methods and were stable over time for samples edited with the Pin-point platform or SpCas9 (Figures 4D, 4E, and S4A). We consistently detected all expected on-target to on-target translocation events with frequencies ranging between 0.2% and 1.6%, and translocations between gRNA target regions and the PDCD1-associated off-target site on chromosome 15 identified by CHANGE-seq in the SpCas9 multi-edited samples (Figure S4B; Table S6), further confirming it as a contributor to CRISPR-Cas9-mediated genome instability. In summary, the aggregation of translocation frequencies quantified from T cells edited at four loci with SpCas9 (Table S6) indicates that up to 1 in 17–25 diploid cells will potentially carry a translocation event, while these types of chromosomal abnormalities are unlikely to occur in cells edited with the Pin-point platform (see materials and methods). Thus, multi-gene editing with the Pin-point platform greatly reduces the adverse effects on genome stability associated with SpCas9.
Figure 4.

Assessment of translocations following multi-gene editing
(A) gRNA binding sites (orange) and genomic regions spanning the locations of SpCas9-induced translocation breakpoints identified by the DRAGEN Structural Variant (SV) Caller (blue bars) at each target gene locus. Genomic regions include confidence intervals around each breakpoint. (B) Circos plot of interchromosomal translocations involving gRNA target sites identified by MANTA in the SpCas9 multi-edited samples. Thickness of lines joining genomic loci is proportional to the translocation frequency. (C) Percentage of Capture-seq reads marked as translocations mapping to each sgRNA target site. (D and E) Average frequencies of the two outcomes of each predicted reciprocal translocation quantified by ddPCR 3 or 7 days post editing with either (D) the Pin-point platform or (E) SpCas9. For (B)–(E) mRNAs encoding either the Pin-point platform or SpCas9 were delivered with four targeting sgRNAs. Control is mock electroporated T cells without RNA. Samples were analyzed 3 days post electroporation unless specified otherwise. n = 2 independent T cell donors.
Molecular assessment of RNA deamination
As promiscuous activity of the deaminase component of base editors has the potential to deaminate RNA22,42 similarly to the activity of endogenous cellular deaminases,43 we assessed the impact on cytidine deamination of RNA by performing transcriptome-wide messenger RNA sequencing (mRNA-seq). Previous findings have highlighted that thousands of C to U transitions occur throughout the transcriptome when base editors are delivered in plasmid format.22,42,44 To evaluate the impact on RNA deamination of the more therapeutically relevant RNA-based transient expression of the Pin-point platform in human primary T cells, we performed an mRNA-seq time course (days 1, 3, and 7 post electroporation) and observed a low-level, transient, gRNA-independent increase in RNA deamination events compared with nCas9-UGI-UGI alone (approximately 60 additional C to U transitions observed exclusively at day 1 post electroporation) (Figures 5A and 5B). Consistent with these observations, the levels of the mRNAs encoding the different components of the Pin-point base editing platform rapidly decline, becoming undetectable by day 7 in culture (Figure S5A).
Figure 5.

Effect of the Pin-point platform on RNA editing and transcription
(A and B) RNA C to U editing assessed by transcriptome sequencing in primary human T cells that were electroporated with Pin-point (nCas9-UGI-UGI and rAPOBEC1-MCP) or nCas9-UGI-UGI only mRNAs and the four targeting sgRNAs against B2M, CD52, TRAC, and PDCD1 genes (A) or a scrambled non-targeting sgRNA (B). Each dot represents one editing event. The total number of editing events is indicated above each replicate per condition. (C–H) Scatterplots of gene expression levels (log2 transformed TPM +1, TPM with a pseudocount of one added before log transformation) in primary human T cells electroporated with Pin-point mRNAs and either a scramble non-targeting sgRNA (C, D, and E) or the four targeting sgRNAs (TRAC, B2M, CD52, PDCD1) (F, G, and H) compared with control cells that received the pulse electroporation only (x axis). DESeq2 analysis was performed on total mRNA collected at days 1 (C and F), 3 (D and G), and 7 (E and H) post electroporation and was used to identify upregulated and downregulated genes. Upregulated or downregulated genes (p < 0.05) with absolute log2 fold change ≥1.5 in gene expression (represented as log2 transformed TPM +1) marked red and blue, respectively. r indicates the Pearson correlation coefficient, calculated for log-transformed values on all genes.
Phenotypic analysis of edited T cells
We investigated whether the transient mRNA deamination associated with base editing with the Pin-point platform had any major effects on the gene expression profile of T cells by performing differential gene expression analysis on the mRNA-seq time course dataset. Global gene expression was minimally affected by the delivery of Pin-point mRNAs and a non-targeting sgRNA (175, 142, and 2 transcripts were deregulated at day 1, 3, and 7, respectively) (Figures 5C–5E; Table S8). We observed a similar effect on the transcriptome when the four gene-specific sgRNAs were delivered (123, 350, and 66 transcripts were deregulated at days 1, 3, and 7, respectively) (Figures 5F–5H; Table S8), indicating that the major component of the effect on gene expression profile is gRNA sequence independent.
In line with expectations, five transcripts encoding the sgRNA target region B2M, PDCD1, CD52, a miRNA (MIR10393) associated with the B2M gene, and a non-coding and uncharacterized gene associated with the PDCD1 gene (LOC105373977) were stably downregulated in the samples edited with the four gene specific sgRNAs (Figure S5B; Table S8), whereas none of the differentially expressed transcripts were stably deregulated across the time course of samples edited with the non-targeting sgRNA. Of the deregulated genes, 41 were deregulated in both targeted and untargeted conditions one day after electroporation and 56 were deregulated in both conditions at day 3 (Figure S5B). These transient gene expression changes likely reflect an immediate cellular response to the delivery of exogenous RNAs or occurred as a consequence of the transient RNA deamination events described above. In conclusion, we observed low level and transient RNA deamination following base editing using the Pin-point platform that did not result in a significant long-term perturbation of T cell transcriptional identity.
Multi-gene editing of CAR-T cells with the Pin-point platform
Having established that synthetic RNA-based delivery of the Pin-point platform presented minimal detrimental effects on human primary T cells, we next sought to address whether multi-gene base editing had an adverse impact on the functionality of CAR-T cells.
Human primary T cells were first transduced with a lentivirus to deliver the CD19-CAR and then edited at the four target genes by electroporation of mRNA encoding either the Pin-point platform or the appropriate targeting gRNAs for B2M, CD52, PDCD1, and TRAC. High-efficiency protein depletion for all four targets (60%–80%) was achieved (Figure 6A) without interfering with CD19-CAR expression (Figure 6B). Edited CAR-T cells retained the ability to kill antigen-positive cancer cells in vitro (Figure 6C) and to produce the effector cytokines TNF alpha and IFN gamma (Figure 6D) with efficiency comparable with unedited controls, demonstrating that the Pin-point platform is a suitable system for engineering functional multi-gene edited CAR-T cells.
Figure 6.

Generation of allogeneic CAR-T cells by multi-gene editing with the Pin-point platform and lentiviral delivery of the CAR
CAR-T cells were generated by lentivirus delivery of the CD19-CAR and subsequently edited by the Pin-point base editor. (A) Frequency of CD19-CAR-positive cells in the transduced T cell population after delivery of Pin-point reagents and in unedited cells. Control cells are T cells that have been mock transduced. (B) Frequency of CD52, TCRa/b, PD1, and B2M protein knockout following co-delivery of Pin-point mRNAs and four target sgRNAs, as analyzed by flow cytometry 7 days post electroporation in CAR-T cells. (C) Raji cells killing measured by flow cytometry after co-culture with CAR-T cells unedited or multi-edited with the Pin-point platform at 1:1 or 3:1 T cell:target cell ratios. Control cells are T cells that have been mock transduced. (D) Levels of TNF alpha and INF gamma measured in the media of the co-culture at the 1:1 T cell:target cell ratio. Data represented as mean (SEM), n = 2 independent biological T cell donors.
Generation of allogeneic CAR-T cells by multi-gene editing and simultaneous site-specific knockin of the CAR with the Pin-point platform
In contrast to lentiviral delivery, targeted insertion of a CAR transgene can result in a more homogeneous cell therapy with improved functionality45 and reduced insertional oncogenesis risk. We therefore developed site-specific knockin using the Pin-point platform by exploiting the aptamer-dependent recruitment of the deaminase, to achieve simultaneous multiplex gene knockout and CD19-CAR knockin in a single event (Figure 7A). The sgRNA-containing aptamers recruit the entire Pin-point machinery to base editing target sites (Figure 7A, left), while the use of two adjacent aptamer-less sgRNAs enables the recruitment of two nCas9 molecules at the knockin site independently of the deaminase module (Figure 7A, right) allowing to direct discreet functions to specific loci.
Figure 7.

Generation of multiplex edited CAR-T cells by simultaneous multiplex base editing knockout and locus-specific knockin with the Pin-point system
(A) Schematic showing the recruitment of the entire Pin-point platform machinery by aptamer-containing gRNAs on the site where the desired outcome is base editing (left) and of the nCas9 alone by aptamer-less gRNAs on the knockin site (right). CAR-T cells were generated by knockin of the CD19-CAR in the TRAC locus. Pin-point mRNAs have been co-delivered with aptamer-containing sgRNAs directed to base edit B2M, CD52, and PDCD1 and 2 aptamer-less sgRNAs designed to target the exon1 of TRAC locus. Cells electroporated with SpCas9 mRNA received optimal gRNAs to knock out B2M, CD52, and PDCD1 by indel formation and one of the two gRNA designed to target the exon 1 of TRAC locus. Shortly after electroporation, cells were transduced with AAV6 carrying the CD19-CAR transgene flanked by the homology arms to the TRAC locus. (B) Frequency of CD52, TCRa/b, PD1, and B2M protein knockout following co-delivery of Pin-point or SpCas9 reagents and transduction with the AAV6-CAR as analyzed by flow cytometry 7 days post electroporation/transduction. (C) Frequency of CD19-CAR-positive cells in the T cell population after delivery of either Pin-point or SpCas9 reagents and transduction with the AAV6-CAR compared with non-transduced cells. (D) Raji cell killing measured by calcein assay after co-culture with T cells unedited or multi-edited with the Pin-point platform or with SpCas9 and transduced with AAV6-CAR compared with non-transduced cells at 1:1, 3:1, or 5:1 T cell:target cell ratios. Control cells are non-transduced cells. (E) Levels of TNF alpha and INF gamma measured in the media of the co-culture at the 1:1 T cell:target cell ratio. Data represented as mean ± SD, n = 2 independent biological T cell donors.
Firstly, we verified that the deaminase expression did not affect the efficiency of site-specific knockin of a GFP reporter or of CD19-CAR at the TRAC locus by the nCas9 component of the Pin-point base editing system (Figures S6A and S6B, respectively). We next investigated whether inclusion of the adjacent aptamer-less sgRNAs resulted in translocations when combined with the aptamer-encoding base editing sgRNAs using Capture-seq. Primary human T cells were electroporated with mRNAs encoding nCas9 and rAPOBEC1-MCP, aptamer-encoding gRNAs directed to base edit B2M, CD52, and PDCD1, and two aptamer-less gRNAs designed to target nCas9 alone to exon 1 of the TRAC locus to enable homology-directed repair (HDR)-driven integration of the CD19-CAR. Whereas translocations at all four target loci were detectable at similar levels in samples edited with SpCas9, they remained undetectable in T cells edited with the Pin-point platform (Figure S6C; Tables S9–S11).
Having confirmed the safety of combining aptamer-less and aptamer-containing sgRNAs, we investigated the efficiency of simultaneous site-specific knockin of CD19-CAR at the TRAC locus with multiplex base editing of B2M, CD52, and PDCD1 using the Pin-point platform. A CD19-CAR transgene lacking a promoter flanked by sequences homologous to the TRAC locus was delivered by AAV6 particles to T cells electroporated with either SpCas9 or the Pin-point platform in combination with the previously validated aptamer-containing and aptamer-less sgRNA combination. We achieved high levels (60%–90%) of protein depletion for both the base editing targets (B2M, CD52, and PD1) and the integration target (TRAC) (Figure 7B). The level of site-specific knockin, as evaluated by CD19-CAR expression from the endogenous TRAC locus, and achieved using the simultaneous knockin knockout application of the Pin-point platform, was comparable with the results achieved with SpCas9 (∼20%) (Figure 7C). Additionally, there was no detectable AAV integration at the other sgRNA targeted sites associated with multi-gene editing with SpCas946 (Figure S6D; Tables S12 and S13). Moreover, CAR-T cells generated by simultaneous knockin and knockout using the Pin-point platform were functional, showing comparable ability to kill antigen-positive target cells in vitro (Figure 7D) and produce the effector cytokines TNF alpha and IFN gamma (Figure 7E) as SpCas9-engineered controls.
These data demonstrate that the Pin-point platform is a promising technology for simultaneous multiplex gene editing and targeted gene insertion applications, while limiting the deleterious effects of nuclease-dependent gene editing. Furthermore, the ability to efficiently base edit multiple sites while allowing targeted integration without the requirement of additional orthogonal targeting enzymes in a single editing procedure is unique to the Pin-point platform and has large potential in the development of complex, engineered cell and gene therapy products.
Discussion
We present the first proof of functionality of the Pin-point platform in primary human T cells, demonstrating that the technology can be employed to simultaneously introduce base edits at multiple loci at high efficiency in combination with site-specific transgene integration in a single intervention. When applied to the generation of engineered CAR-T cells using fully synthetic RNA components, the Pin-point platform exhibits a favorable safety profile compared with DSB-dependent CRISPR-Cas9 technology. Unbiased identification of candidate gRNA-dependent Cas9 off-target editing sites35 revealed the Pin-point platform to be highly specific with only 4 out of 456 analyzed sites showing low level base editing. When editing 4 targets simultaneously with SpCas9 we detected translocations at frequencies where 1 in every 17–25 cells of the final product would likely contain a translocation. Translocations were undetectable with the Pin-point technology, as it has been previously reported using other base editors.16,27,28 Multi-gene editing with the Pin-point platform also improved proliferative capacity of engineered T cells compared with SpCas9, presenting an advantage for the manufacturing of both autologous and allogeneic therapies, ultimately reducing costs and opening up the potential to make them available at a lower price and broadening access.
While the generation of multiple gene knockouts in immune cells by base editing has already been shown,16,19,27,28 more complex genetic modifications, such as targeted transgene integration alongside base editing knockout at other loci, have only been demonstrated by combining two orthogonal Cas enzymes.28,30 The aptamer-dependent design of the Pin-point platform overcomes this requirement for the delivery of multiple large Cas enzymes by independently controlling which active modules are recruited at each of multiple target loci. Whereas aptamer-containing gRNAs recruit the complete base editing machinery consisting of the nCas9 and the deaminase modules to loci intended for gene knockout via base conversion, aptamer-less gRNAs recruit only the nCas9 module to loci intended for transgene insertion but avoid recruiting the deaminase function, which could otherwise induce deamination at the integration site.
Although the aptamer-dependent design of the Pin-point base editing system has the advantage of increased flexibility, the untethered deaminase could in principle increase the risk of spurious deamination. We addressed concerns about rAPOBEC1-mediated deamination22,44 and determined that delivery of the Pin-point base editing machinery in the form of synthetic reagents into human primary T cells resulted in transient deamination of a minor fraction of expressed mRNAs. Nonetheless, all off-target alterations to the transcriptome rapidly dissipated and would therefore not affect the phenotype of T cells at the point of infusion of the allogeneic product. Taken together with the marked improvements in genome stability and proliferative capacity of multi-gene edited T cell we propose that the Pin-point platform represents a substantive advance in the toolkit available for safely engineering complex adoptive cellular therapies.
Due to its inherent flexibility, we anticipate that the Pin-point platform could be configured to simultaneously perform a suite of independent operations at multiple genomic loci by recruiting the desired effector modules via distinct RNA aptamers. For example, by combining deaminase and epigenetic modulation modules it should be possible to rewire gene regulatory networks to confer novel T cell responses to stimuli by simultaneously modifying the sequence of cis-regulatory elements and the chromatin organization at specific loci in combination with the site-specific incorporation of synthetic signaling receptors. Similarly, it should be possible to rewire metabolic networks to overcome challenges such as T cell exhaustion by rationally engineering the activity of key enzymes in situ by base editing while simultaneously inducing or reducing expression of additional endogenous metabolic enzymes using transcriptional activator or repressor modules. Beyond its application in the creation of next-generation adoptive T cell therapies, we anticipate the Pin-point platform will offer similar opportunities for the engineering of a wide range of allogeneic cell therapies with increasingly advanced safety and functionality profiles.
Materials and methods
gRNA design
gRNAs for base editing have been designed by using an internal design tool for PTCs generation or by manual design for the splice site disruption. The internal tool searches for NGG PAM within exons and 20 bp protospacer sequences that include a C in positions 2–18 that when converted to T introduce a STOP codon. For splice site disruption, the approach was based on editing the conserved SA (intron-AG|exon) or SD (exon|GT-intron) motif to disrupt the functional transcript. This was done by finding an NGG PAM site near the splice junction and 20 bp protospacer that included the SA or donor site to edit. Guides that targeted more than a single location within the genome were removed from consideration. gRNA information for base editing is reported in Table S1. Information regarding gRNAs utilized with SpCas9 for optimal indels formation45,47,48 are reported in Table S14.
For the knockin strategy we designed two gRNAs (Table S15) with PAM-out configuration to target opposite stands in the first exon of the TRAC gene. Both, non-homologous end joining and integration of the CAR by HDR at this locus has been proven to efficiently disrupt the TCR complex.45
Editing reagents
Pin-point platform (nCas9-UGU-UGI and rAPOBEC1-MCP) and SpCas9 mRNAs were produced commercially (Trilink Biotechnologies and Revvity). Sequences are available in the supplemental information (Figures S7 and S8). gRNA reagents (crRNAs, tracrRNA, and sgRNAs) were synthesized at Revvity or at Agilent Technologies (Figures S9–S11).
Primary human T cell isolation and culture
Primary human T cells (CD3+) were either purchased (Hemacare, CA) or isolated in-house from fresh whole peripheral blood (CPD blood bags, Cambridge Bioscience, UK) from healthy donors in accordance with Human Tissue Act (HTA) regulations under license number 12638. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation with Lymphoprep (STEMCELL Technologies, Germany) in SepMate-50 (STEMCELL Technologies) tubes. T cells were subsequently isolated from the PBMC population by immunomagnetic negative selective with the EasySep Human T cell Isolation kit (STEMCELL Technologies, Canada). Isolated T cells with >95% viability and >95% purity were either cryopreserved or directly cultured for subsequent experiments. T cells were cultured at ∼1–2 × 106 mL in ImmunoCult-XF T cell expansion medium (STEMCELL Technologies) supplemented with penicillin-streptomycin (Gibco, NY) and IL-2 (100 IU/mL; Miltenyi Biotech). Cells were activated with Dynabeads Human T-Activator CD3/CD28 (Gibco, Vilnius, Lithuania) at a 1:1 bead:cell ratio for 48 h before electroporation.
T cell electroporation
After activation, Dynabeads were magnetically removed and the cells were washed with Dulbecco’s PBS (Gibco, Paisley, UK) prior to resuspension in the electroporation Buffer R. Activated T cells (2.5 × 105 per reaction) were electroporated with sgRNAs at 2 μM or with tracrRNA/crRNA at 6 μM and either 1 μg of SpCas9 mRNA or 1.6 μg of Pin-point nCas9-UGI-UGI and 0.22 μg of Pin-point rApobec1 using the Neon Transfection System (Invitrogen, South Korea) with 10 μL tips and under the following conditions: 1,600 V, pulse width of 10 ms, 3 pulses. After electroporation, T cells were transferred directly to prewarmed antibiotic-free ImmunoCult-XV T cell expansion medium supplemented with IL-2 (100 IU/mL), IL-7 (100 IU/mL; Peprotech, NJ), and IL-15 (100 IU/mL; Peprotech) and incubated at 37°C, 5% CO2 for 3–7 days. Electroporations were performed in duplicate or triplicate for each condition.
Lentiviral transduction
Lentivirus was generated in HEK293T cells using Lipofectamine 3000 Transfection Reagent (Invitrogen), the ViraSafe Lentiviral Packaging System (Cell Biolabs) and an expression plasmid to deliver the 1928z CAR used in clinical trials (CD19-CAR)49 under the control of EF1a promoter. Viral particles were harvested from the culture, concentrated using 100 kDa Amicon Ultra-15 Centrifugal Filter Units (Merck) and cryopreserved at −80°C. Functional viral titer was estimated by titrating the viral particles on Jurkat cells. Prior to lentiviral transduction, T cells were cultured in ImmunoCult-XV T cell expansion medium supplemented with human serum (10%; Sigma), penicillin-streptomycin, and IL-2 (100 IU/mL) and activated for 24 h in the presence of plate-bound anti-CD3 antibody (2.5 μg/mL; BioLegend) and soluble anti-CD28 antibody (2.5 μg/mL; BioLegend). Cells were transduced on RetroNectin (100 μg/mL; Takara)-coated plates at an MOI of 5 and the transduced population was enriched by puromycin (3 μg/mL; Gibco, China) selection for 5 days. Cells were then reactivated using Dynabeads Human T-Activator CD3/CD28 before electroporation with editing reagents as reported above.
AAV transduction
The HDR donor sequence is similar to that described by Eyquem et al.45 In more detail, it consists of 1.8 kb of genomic TRAC flanking the left and the right gRNA targeting sequences, a self-cleaving P2A peptide in frame with the first exon of TRAC, and by the 1928z CAR used in clinical trials.49 The left and right homology arms, each of 900 nucleotides, start near the position of the nick from the 5′ sgRNA (the left homology arm 3 nucleotides upstream and the right homology arm 9 nucleotides downstream). A silent point mutation has been introduced in the right homology arm to destroy the PAM site for the 3′ gRNA. The HDR sequence was cloned by GenScript in a pAAV background, and the resulting plasmid utilized to generate recombinant AAV6 donor vector by Vigene Bio. Sequence of the CAR constructs with homology arms is reported in supplemental information (Figure S12).
For the locus-specific knockin experiment, activated T cells were electroporated with the gRNAs and Pin-point or SpCas9 mRNAs and immediately after electroporation transduced with the recombinant AAV6 donor vector at multiplicity of infection of 5 × 105. Subsequently, T cells were cultured in antibiotic-free ImmunoCult-XV T cell expansion medium supplemented with IL-2 (100 IU/mL), IL-7 (100 IU/mL; Peprotech), and IL-15 (100 IU/mL; Peprotech) at 37°C, 5% CO2, and culture medium was completely replaced after 24 h.
Flow cytometry
Prior to flow cytometry, T cells edited only at the PDCD1 locus were re-stimulated using Dynabeads Human T-Activator CD3/CD28 for 48 h as described above to induce the expression of PD1. In experiments where the 4 targets were knocked out, cells were activated with PMA (50 ng/mL; Sigma-Aldrich) and ionomycin (250 ng/mL; Millipore) for 48 h prior to flow cytometry analysis to induce the expression of PD1. For flow cytometry, cells were stained with fluorophore-conjugated antibodies against human B2M (BioLegend, no. 316304), CD52 (BD BioSciences, no. 562945), TCR a/b (BioLegend, no. 306742), PD1 (BioLegend, no. 329908), and CD19-CAR (AcroBiosystems, anti-FMC63 scFv). Cell viability was assessed using DAPI (80 ng/mL). Cells were acquired on an IntelliCyte IQue PLUS or Sartorius iQue3 flow cytometer using iQue ForeCyt Enterprise Client Edition 9.0 (R3) software for both acquisition and data analysis. The gating strategy for simultaneous quantification of viability, B2M, CD52, TCR a/b, and PD1 expression was as follows (Figure S2). Within the live population, B2M expression versus CD52 expression was assessed using quadrant gating, then within each of the 4 subpopulations TCR a/b and PD1 expression was assessed using quadrant gating. Each of these 16 populations represents a different expression profile of the 4 targets, and cell counts within each population were used to calculate the frequency of cells which had lost each target or combination of targets. To correct for the proportion of cells in non-edited samples that resulted negative to the marker, level of protein knockout in edited samples was normalized on expression in non-edited samples by applying the following: frequency of negative cells in test sample = 1 − (frequency of positive cells in test sample/frequency of negative cells in control sample).
For fold expansion calculation, CountBright Absolute Counting Beads (Invitrogen) were added to flow cytometry samples to allow counting of the absolute number of live (DAPI negative). A flow cytometry count was performed 2 h after editing (baseline), and a second one 3 days after editing. Fold expansion was calculated by dividing the live cell count for each sample by its own baseline count.
Amplicon sequencing of genomic DNA samples
Locus-specific primers with Illumina universal adapter were designed to amplify a 250–350 bp site surrounding the genomic region of interest (Table S16). For gDNA preparation, T cells were lysed using DirectPCR (cell) (Viagen Biotech, LA) lysis buffer supplemented with proteinase K (10 μg/mL; Sigma-Aldrich) and heated at 55°C for 30 min, then 95°C for 30 min. The crude lysate was then used for the first PCR with locus-specific primers containing Illumina adapters. Products from the first PCR were then amplified using Illumina barcoding primers. Following barcoding, PCR samples were pooled and purified using AMPure XP beads (Beckman Coulter). DNA was sequenced by SourceBioScience on Illumina MiSeq 2 × 300 bp runs (Illumina, San Diego, CA). Raw FASTQ files were analyzed against a reference sequence and sgRNA protospacer sequence using a custom pipeline that was used to count nucleotide substitutions in the base editor window (both expected C:G to T:A conversions and other substitutions) and indels overlapping the spacer sequence as described previously.31
CHANGE-seq off-target discovery
CHANGE-seq was performed as previously described by Lazzarotto et al.35 with minimal modifications on gDNA extracted using the Gentra Puregene Cell Kit (QIAGEN) from two independent human T cell (CD3+) donors, following the manufacturer’s instructions. Size analysis of resultant high-molecular-weight gDNA was assessed in the Fragment Analyzer (Agilent), and subjected to tagmentation with a customized transposome composed of oCRL225/oCRL226 adaptors and the Hyperactive Tn5 transposase (Diagenode). DNA tagmentation was performed in batches of 4 μg, utilizing 17.5 μL of the assembled transposome in a final volume of 200 μL, and incubated for 6 min at 55°C. Reaction was quenched by the addition of 200 μL of SDS 0.4%, and resultant fragments were assessed on the Fragment analyzer and quantified using the Qubit dsDNA BR Assay kit (Thermo Fisher Scientific). After gap repair with KAPA Hi-Fi HotStart Uracil+ DNA Polymerase (KAPA Biosystems) and Taq DNA Ligase (NEB) and treatment with USER enzyme (NEB) and T4 polynucleotide kinase (NEB), the tagmented DNA was circularized with T4 DNA Ligase (NEB) and treated with a cocktail of exonucleases containing Plasmid-Safe ATP-dependent DNase (Lucigen), Lambda exonuclease (NEB), and Exonuclease I (NEB) to degrade residual linear DNA carryover. Circularized material was then in vitro cleaved by SpCas9 RNP in combination with the tested sgRNAs. A validated sgRNA against VEGFA with known off-target profile (GACCCCCTCCACCCCGCCTC) was used to control the method and data are reported in Table S2.36,37 Illumina Universal Adaptor (NEB) was ligated to the blunted end after adenylation, enzymatically treated with USER enzyme (NEB), and amplified with NEBNext Multiplex Oligos for Illumina for 20 amplification cycles. The quality of the amplified and bead-cleaned-up libraries was determined using a 5,300 fragment analyzer with the standard sensitivity NGS kit (Agilent). Libraries were then pooled, diluted, and denatured according to Illumina’s recommendations and sequenced on a NextSeq550 300 cycles kit with a paired-end 2 × 150 configuration (Illumina). Bioinformatic analysis was performed as described by Tsai et al.36 with a minor modification: reads with mapping quality equal to zero were included in the analysis, alongside those passing the MAPQ threshold defined in the pipeline parameters, to nominate putative off-targets located in non-uniquely mappable regions. The pipeline was run with the following parameters: read_threshold: 4, window_size: 3, mapq_threshold: 50, start_threshold: 1, gap_threshold: 3, mismatch_threshold: 6, search_radius: 30.
rhAmpSeq off-target validation
rhAmpSeq panel design
rhAmpSeq panels were designed for each gene target composing 100 sites with a high degree of overlap between technical and biological replicates from CHANGE-seq results. Hierarchical site selection strategy was employed to pick the most likely off-target sites: (1) sites present in both donors and all replicates; (2) sites present in all replicates of one donor; (3) sites present in at least two replicates of either donor; and (4) sites present in at least one replicate from one donor. In cases where we had more than 100 sites we prioritized based on the nuclease-read count (Figure S3A). All the candidate off-target sites having up to four mismatches to the target sites were included in the rhAmpSeq analysis irrespective of their position based on the above selection criteria. The genomic coordinates for on- and off-targets were then entered into IDT’s rhAmpSeq CRISPR analysis portal for assay design and ordering.
rhAmpSeq library preparation
gDNA from non-edited T cells (CD3+) and T cells (CD3+) treated with either the Pin-point platform or SpCas9 was extracted 5 days post electroporation using the Gentra Puregene Cell Kit (QIAGEN) following the manufacturer’s instructions. rhAmpSeq NGS libraries were then generated as per IDT’s rhAmpSeq library preparation protocol. Primary pools, secondary pools and single amplicon rhAmpSeq reactions were then applied on the extracted gDNA. In target rhAmp PCR 1, the 4× rhAmpSeq library mix was mixed with ∼50–80 ng of gDNA (equivalent to ∼15,000–25,000 haploid genomes) and amplified using the following thermocycling conditions: 95°C for 10 min; [95°C for 15 s; 61°C for 8 min] × 14 cycles; 99.5°C for 15 min; 4°C hold. The PCR 1 product was purified using Agencourt AMPure XP beads (Beckman Coulter) and immediately proceeded to the rhAmp PCR 2. In PCR 2, dually indexed Illumina sequencing libraries were generated using PCR 1 product, mixed with 4× rhAmpSeq library mix 2 and unique i5 and i7 primers (IDT), and amplified using the following thermocycling conditions: 95°C for 3 min; [95°C for 15 s; 60°C for 30 s; 72°C for 30 s] × 24 cycles; 72°C for 1 min; 4°C hold. The final libraries were purified using Agencourt AMPure XP (Beckman Coulter), quantified using Qubit 1× dsDNA HS Assay Kit (Thermo Fisher Scientific) and quality was checked by qPCR and on a Tapestation 4200 (Agilent). Paired-end, 151-bp reads were sequenced using the mid-output 300 cycles kit on the Illumina’s NextSeq 550 platform (Illumina). Libraries from the candidate sites having up to four mismatches to the target sites were generated and sequenced in a second round.
Bioinformatic processing of rhAmpSeq data
To deal with non-specific PCR products, we first aligned merged reads (usingfast lLength adjustment of short reads50) to the intended reference sequences for each target (using bwa mem) and used the alignments produced to identify the variants occurring in each reference (minimum 10 reads and allele frequency 0.01%). From these variants an extended set of reference sequences was constructed comprising the original reference plus putative variant sequences containing the different combinations of variants. From this extended set of sequences the ones that differed from the reference in global pairwise alignment by over 20 (Python Bio.pairwise2.align.globalms with scoring 1 for a match, −1 for a mismatch, 1 gap open, −0.5 gap extend) were considered sufficiently different from constitute non-specific PCR products. This set was clustered based on pairwise alignment scores within 20, and one representative from each cluster formed a “decoy” sequence to add to the targets passed to CRISPResso2 (v.2.1.1) in pooled mode with base editing parameters (-w 20 -wc 1 -be). Following alignment, CRISPResso2 outputs were processed to identify the base position with the highest Insertion, Deletion, or Base Editing event within the windows of gRNA target site +/− 10 bp per sample per amplicon. Off-targets were reported based on the following criteria as previously defined51: (1) off-target editing detected in greater than 0.5% of edited samples and less than 0.5% of mock control samples; (2) off-targets were detected in both human donor replicates; and (3) a minimum of 5,000 reads, with an average coverage per off-target pool of >35,000 reads. Scripts are available on request.
To apply a statistical analysis to the data, a beta regression was fitted using the brms package in R.52,53,54 The model was fitted using a weak informative prior (normal[0,1]) (https://github.com/stan-dev/stan/wiki/Prior-Choice-Recommendations). We compared the editing percentage in the edited cells with the editing in the control cells and tested the hypothesis of whether the editing in edited cells was higher than in the control cells. We removed sites where the editing in the control cells was higher than in edited cells, and sites that were only present in one donor or where the background corrected editing was not higher than 0 in both donors. Based on the ground truth of known on-target sites we set the posterior probability at >0.8 to classify sites that had been edited. We also considered the evidence ratio when assessing these sites. The evidence ratio can be interpreted like this: 1–3, anecdotal evidence; 3–10, substantial evidence; 10–20, strong evidence; 20–100, very strong evidence, and >100 decisive evidence. We used Bayesian statistics instead of frequentist statistics as sample sizes were small and not suitable to test the assumptions of frequentist analyses.
To further confirm true off-targets we have manually assessed the CRISPResso allele plots for all sites with posterior probability >0.8, or with above 0.1% background corrected editing or where high background (>0.5%) was observed and annotated the results in the Tables S3 and S4.
Capture-seq
Library preparation
gDNA samples were prepared from T cells (CD3+) (Hemacare) using DNeasy Blood and Tissue Kit (QIAGEN). gDNA (500–750 ng) were used to prepare sequencing libraries using KAPA HyperPlus kit (Roche). In brief, gDNA was purified using HyperPure beads (Roche), fragmented for 20 min at 37°C, and analyzed using Tapestation to confirm consistent fragmentation. Following end repair and A-tailing, KAPA universal stubby adapters were ligated to gDNA fragments, products were cleaned and size selected using 0.8× HyperPure beads. KAPA Unique Dual Indexed primers were incorporated by PCR (4–6 cycles) using KAPA HiFi Hotstart ReadyMix. Following clean-up with HyperPure beads, libraries were quantified by Qubit and analyzed on a Tapestation 4200 (Agilent).
Hybridization capture probe design
DNA capture probes (120 bp long) complementary to sequences within 200 bp 5′ and 3′ of the PAM sites of gene editing targets were designed using “Oligo” tool (https://github.com/jbkerry/oligo) in OffTarget configuration. Oligo outputs were manually curated to remove probes with stretches of homology >30 bp and 4 probes per target were chosen (Table S17). 5′-Biotinylated DNA probes were synthesized by IDT as xGen Custom Hybridization Capture Panels. For the experiment assessing chromosomal translocations generated by four target multi-gene editing, probes were designed to capture the on-target sites of the four sgRNAs used. For the experiments assessing chromosomal translocations generated by three target base editing with simultaneous dual nicking, or untargeted integration of the AAV HDR vector into Cas9-induced DSBs, probes were designed to capture the off-target sites identified during the course of the study in addition to on-target sites of the five sgRNAs used.
Hybridization capture and sequencing
Libraries were enriched for genomic regions flanking gene editing targets using xGen Hybridization Capture (IDT) workflow. In brief, 1 μg of each indexed library were pooled and ethanol precipitated with COT DNA, resuspended in xGen Hybridisation Buffer containing xGen Universal Blockers TS Mix and 5′-biotinylated custom DNA probes (IDT), heated to 95°C (1 min), and hybridized overnight at 65°C. Streptavidin-conjugated magnetic beads (IDT) were incubated for 45 min at 65°C with the hybridized libraries, immobilized on a magnetic stand, and stringently washed at 65°C. Following washing, beads were immobilized and captured library fragments were eluted in H2O. Post-capture PCR (12 cycles) was performed using KAPA HiFi Hotstart ReadyMix (Roche) and xGen Library Amplification Primer Mix (IDT). Following clean-up with HyperPure beads, enriched libraries were quantified by Qubit, analyzed by Bioanalyser to determine fragment size distribution, and sequenced using 2 × 300 bp paired-end reads on Illumina MiSeq by Source Bioscience.
Read alignment and structural variant identification
Sequencing reads were trimmed to the first 75 bp using a custom Python script to enable unique mapping of each paired read and then processed through the Illumina DRAGEN Structural Variant (SV) Caller55 (v.3.8.4) to identify structural variants, which extends the MANTA56 structural variation pipeline. Each sample was run through the pipeline as an unpaired tumor sample. An example command is as follows:
/opt/edico/bin/dragen -f --ref-dir/ephemeral/ucsc.hg38.3.8.4/--tumor-fastq1 s3://aws_bucket/Sample1_R1_001.paired.75bp.fastq.gz --tumor-fastq2 s3://aws_bucket/Sample1_R2_001.paired.75bp.fastq.gz --output-directory/ephemeral/DRAGEN_Sample1/--output-file-prefix Sample1 --enable-duplicate-marking true --enable-map-align true --enable-map-align-output true --enable-sv true --RGID-tumor Sample1 --RGSM-tumor Sample1 --sv-exome true --remove-duplicates true.
Quantification of translocations
As only translocations with at least one break end mapping to a captured target can be analyzed by Capture-seq we removed read pairs derived from library fragments captured non-specifically during hybridization by filtering the “∗.candidateSV.vcf” output from MANTA to include only clusters with break ends within sequences mapping to genomic regions +/− 1,000 bp either side of gRNA targets with supporting read evidence in the .bam file. Similar translocations with breakpoints within +/− 1,000 bp of the same gRNA target were grouped and quantified by normalizing the sum of all reads supporting a given group of variants (BND_PAIR_COUNT) by the total number of reads mapping to +/− 1,000 bp genomic regions centered on the gRNA target on the captured chromosome. Where both genomic regions involved in a translocation contained sequences targeted by capture probes the average number of reads across these regions was used for normalization. To quantify translocation frequencies at each gRNA target site the total count of all variants with breakpoints mapping within +/− 1,000 bp were normalized to the total number of reads mapping to the same interval.
Estimating the probability of translocations in individual cells
Based on the assumption that translocations occur independently in individual cells, the relative frequencies per haploid genome of all detected translocations within the population were aggregated and adjusted for two genome copies per nucleus to estimate the probability of at least one event occurring per cell.
Quantification of AAV ITR integration
To quantify untargeted AAV integration at edited genomic loci, a modified version of the Capture-seq workflow for assessment of chromosomal translocations generated by three target base editing with simultaneous dual nicking was followed. Reads were mapped against a custom human genome build composed of the hg38.3.8.4 reference genome with an additional patch including the sequence of the AAV ITR. To remove reads derived from TRAC sequences located on unintegrated AAV genomes the “∗.candidateSV.vcf” output from MANTA was filtered to remove clusters with break ends mapping to the TRAC locus. AAV integration frequency was calculated by normalizing the number of reads assigned to clusters supporting translocations between breakpoints within +/− 1,000 bp of the captured gRNA target sequences and the AAV ITR patch by the total number of reads mapping to +/− 1,000 bp of the captured gRNA targets.
Translocation quantification by ddPCR
gDNA samples were prepared from T cells (CD3+) (Hemacare) using DNeasy Blood and Tissue Kit (QIAGEN). qPCR assays were designed to amplify predicted translocation products composed of sequences flanking each gRNA target site using the Integrated DNA Technologies (IDT) PrimeTime qPCR probe design tool. Primers and probe information for ddPCR analysis are reported in Table S18. ddPCR Supermix for Probes (no dUTP) (Bio-Rad) was used for PCR reactions, each containing 40–100 ng EcoR1-digested gDNA, an internal reference primer pair targeting the PPIA gene + HEX labeled probe (IDT), and a translocation targeting primer pair + FAM labeled probe (IDT). Droplets were generated and analyzed using the QX200 Droplet-digital PCR system (Bio-Rad) according to manufacturer’s instructions. Translocation frequency per haploid genome was calculated from two technical replicates per sample as the fraction of translocation events detected relative to the reference sequence using QuantaSoft software (version 1.7.4) (Bio-Rad).
RNA purification and sequencing
Total RNA was isolated from unedited and edited T cells using the RNeasy Mini Kit (QIAGEN) and quality was determined using a BioAnalyser (Agilent) and a NanoDrop Spectrophotometer. Samples were quantified using the RNA assays on the Qubit Fluorometer. Total RNA was subjected to mRNA isolation and strand-specific RNA sequencing library preparation with the Illumina Stranded mRNA Prep Ligation kit according to manufacturer’s instructions. The libraries were validated on the Agilent BioAnalyzer 2100 to check the size distribution of the libraries and on the Qubit to check the concentration. The RNA-seq libraries were sequenced on an Illumina HiSeq X instrument, for an average of minimum 30M 150 bp paired end reads per sample (Source Bioscience).
RNA deamination analysis
RNA-seq variant calling and quality control was performed as described by Grünewald et al.42 In short, Illumina paired-end FASTQ sequences were processed through the GATK best practices for RNA-seq variant calling,57 which produced analysis-ready BAM files aligned against human hg38 reference genome. RNA variants were called using GATK HaplotypeCaller58 targeting single-nucleotide variants (SNVs) across chromosomes 1–22, X and Y. Bam-readcount (https://github.com/genome/bam-readcount) was used to quantify per-base nucleotide abundances per variant.
Variant loci in the experimental samples (nCas9-UGI-UGI alone or Pin-point base editor electroporated cells) were filtered to exclude sites without high confidence reference genotype calls in the control samples. For a given SNV the read coverage in the control samples (electroporation control) was set to be above the 90th percentile of the read coverage across all SNVs in the corresponding experimental samples. Only loci having at least 99% of reads containing the reference allele in the control samples were kept. RNA edits in the experimental samples were filtered to include only loci with 10 or more reads and with greater than 0% reads containing alternate allele. Base edits labeled as C-to-U comprise C-to-U edits called on the positive strand as well as G-to-A edits sourced from the negative strand.
Differential gene expression analysis
Sequences were processed through the Illumina DRAGEN RNA Pipeline v.3.7.5 to quantify transcripts per million and read counts. Differential expression analysis was then performed using DESeq2 v.1.26.0.59
Cytotoxicity assay
CD19-expressing Raji cells (InvivoGen #raji-null) were used as target cells in the cytotoxicity assay. Killing of the target cells was measured by flow cytometry assay or by calcein assay. For the flow cytometry assay, Raji cells were seeded in 96-well plate (5 × 104/well) and co-cultured with T cells stained with CellTrace Violet (Invitrogen) at the indicated E:T ratios. After 3 days of co-culture, cells were stained with LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Invitrogen) and acquired by flow cytometry. With T cells positive for the CellTrace Violet, the Raji cells and T cells were gated into distinct populations prior to live-dead analysis. The percent of viable Raji cells (Rlive) was used to calculate the percent of T cell-mediated (TCM) killing as follows: TCM killing = 100 – Rlive.
For the calcein assay, Raji cells were loaded with Calcein AM Dye (Invitrogen) following the manufacturer’s instructions and co-cultured in a 384-well plate (1 × 104/well) with T cells at the indicated E:T ratios. Target cells without effectors served as a negative control and target cells incubated with 2% Triton X-100 (Merck-Sigma) served as positive control (maximum killing). After 6 h of co-culture, culture supernatant was analyzed at the Envision plate reader (Revvity) with excitation 494 nm and emission 517 nm settings. TCM killing is calculated as follow: TCM killing = (test condition − negative control condition)/(positive control condition − negative control condition) × 100.
Cytokine profiling
The MultiCyt QBeads PlexScreen Secreted Protein Assay Kit (Sartorius) was used to quantify the level of tumor necrosis factor alpha (TNF-α) and interferon gamma (INF-γ) secretion during the T cell cytotoxicity assays. Protocol D (reduced background, with standard curve) in the manufacturer’s handbook was followed. The IntelliCyte IQue PLUS or Sartorius iQue3 flow cytometer using iQue ForeCyt Enterprise Client Edition 9.0 (R3) Software was used for both acquisition and data analysis, including plotting the standard curves and calculating the absolute value of each sample. For samples that were diluted before analysis, analyte concentration was multiplied by the dilution factor. Background analyte concentration (Raji alone) was subtracted from all values and the data plotted using Graph Prism v.9.4.1 Software.
Data and code availability
The NGS datasets reported in this manuscript are available on Sequence Read Archive with BioProject IDs PRJNA1090029 (http://www.ncbi.nlm.nih.gov/bioproject/1090029) and PRJNA1121376 (http://www.ncbi.nlm.nih.gov/bioproject/1121376).
Acknowledgments
S.J. is supported by NJCCR grant COCR23PRG005 and DOD grant MD200088. We thank former Revvity employee Andrea Frapporti for creating the Pin-point platform schematics used in the article and current employee Robin Loesch for creating the graphic abstract. We acknowledge the support from AstraZeneca employee Natalie Van Zuydam for the statistical analysis performed on the rhAMPSeq data. Finally, we thank current and former Revvity colleagues Toby Gould, Sven Hofmann, Susana Vega, Michael Anbar, Steve Lenger, John McGonigle, and Matthew Perkett for their helpful discussion and support.
Author contributions
I.P. and J.J.L. conceived, led the project, and interpreted the data. I.P. and R.B. designed, performed experiments, analyzed data, and wrote the manuscript with contribution from all the authors. J.H., B.J., O.M., and K.H. designed, performed experiments, and analyzed data. J. Stombaugh performed bioinformatic analysis. J. Sumner, D.P., J.L.T., M. Francescatto, M. Firth, and B.T. led, designed, performed, and analyzed the CHANGE-seq and rhAMpSeq-off-target validation. Z.S., C.M.W., and A.v.B.S. supervised and provided critical suggestions. J.C.C., S.J., and T.S. contributed to the ideation of the project. J.C.C. and S.J. provided critical reagents.
Declaration of interests
M. Francescatto, M. Firth, J.L.T., D.P., J. Sumner, and B.T. are all current or past (while engaged in the research project) employees of AstraZeneca. I.P., R.B., J.H., B.J., O.M., J. Stombaugh, K.H., T.S., Z.S., C.W., A.v.B.S., and J.J.L. are current or past (while engaged in the research project) employees at Revvity. Revvity has an exclusive license from Rutgers University to certain base editing patents. Rutgers University and Horizon Discovery Limited have filed patent applications on this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.06.033.
Supplemental information
The average and individual nuclease read counts (replicates 1–3) for each amplified site are reported for two human donors evaluated (identified as D71 and D73). Sites selected for rhAmpSeq validation are identified in column L. These sites have been selected based on hierarchical selection criteria (column M) as defined in the table and in the methods section. Genomic coordinates and sequence for each amplified site are reported. Mismatches to the on-target site are reported in lowercase.
Percentage editing in edited samples (raw editing), control samples (control), and corrected editing calculated by subtracting the value of the control sample from the edited sample (background corrected editing), together with matched read depth at each interrogated site. Data are representative of either base editing (BE) or Insertions/Deletions (INDEL) events in cells edited with the Pin-point system (PnP) or SpCas9 for the indicated on-target (TRAC/B2M/ PDCD-1/CD52) and predicted off-target sites. Bayesian statistics has been performed on the dataset, citing the evidence ratio, posterior probability and ∗ as measures of statistical significance. On-targets are highlighted in green and validated off-targets in red.
Percentage editing in edited samples (raw editing), control samples (control), and corrected editing calculated by subtracting the value of the control sample from the edited sample (background corrected editing), together with matched read depth at each interrogated site. Data are representative of either base editing (BE) or Insertions/Deletions (INDEL) events in cells edited with the Pin-point system (PnP) or SpCas9 for the indicated on-target (TRAC/B2M/ PDCD-1/CD52) and predicted off-target sites. Bayesian statistics has been performed on the dataset, citing the evidence ratio, posterior probability, and ∗ as measures of statistical significance. On-targets are highlighted in green and validated off-targets in red.
References
- 1.Stadtmauer E.A., Fraietta J.A., Davis M.M., Cohen A.D., Weber K.L., Lancaster E., Mangan P.A., Kulikovskaya I., Gupta M., Chen F., et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367 doi: 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ottaviano G., Georgiadis C., Gkazi S.A., Syed F., Zhan H., Etuk A., Preece R., Chu J., Kubat A., Adams S., et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci. Transl. Med. 2022;14 doi: 10.1126/scitranslmed.abq3010. [DOI] [PubMed] [Google Scholar]
- 3.Benjamin R., Graham C., Yallop D., Jozwik A., Mirci-Danicar O.C., Lucchini G., Pinner D., Jain N., Kantarjian H., Boissel N., et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. The Lancet. 2020;396:1885–1894. doi: 10.1016/S0140-6736(20)32334-5. [DOI] [PubMed] [Google Scholar]
- 4.Dimitri A., Herbst F., Fraietta J.A. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol. Cancer. 2022;21:78. doi: 10.1186/s12943-022-01559-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papathanasiou S., Markoulaki S., Blaine L.J., Leibowitz M.L., Zhang C.-Z., Jaenisch R., Pellman D. Whole chromosome loss and genomic instability in mouse embryos after CRISPR-Cas9 genome editing. Nat. Commun. 2021;12:5855. doi: 10.1038/s41467-021-26097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alanis-Lobato G., Zohren J., McCarthy A., Fogarty N.M.E., Kubikova N., Hardman E., Greco M., Wells D., Turner J.M.A., Niakan K.K. Frequent loss of heterozygosity in CRISPR-Cas9-edited early human embryos. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2004832117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zuccaro M.V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E., et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell. 2020;183:1650–1664.e15. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- 8.Leibowitz M.L., Papathanasiou S., Doerfler P.A., Blaine L.J., Sun L., Yao Y., Zhang C.-Z., Weiss M.J., Pellman D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021;53:895–905. doi: 10.1038/s41588-021-00838-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nahmad A.D., Reuveni E., Goldschmidt E., Tenne T., Liberman M., Horovitz-Fried M., Khosravi R., Kobo H., Reinstein E., Madi A., et al. Frequent Aneuploidy in Primary Human T Cells after CRISPR-Cas9 cleavage. Nat. Biotechnol. 2022;40:1807–1813. doi: 10.1038/s41587-022-01377-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boutin J., Rosier J., Cappellen D., Prat F., Toutain J., Pennamen P., Bouron J., Rooryck C., Merlio J.P., Lamrissi-Garcia I., et al. CRISPR-Cas9 globin editing can induce megabase-scale copy-neutral losses of heterozygosity in hematopoietic cells. Nat. Commun. 2021;12:4922. doi: 10.1038/s41467-021-25190-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 12.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A⋅T to G⋅C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Billon P., Bryant E.E., Joseph S.A., Nambiar T.S., Hayward S.B., Rothstein R., Ciccia A. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol. Cell. 2017;67:1068–1079.e4. doi: 10.1016/j.molcel.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuscu C., Parlak M., Tufan T., Yang J., Szlachta K., Wei X., Mammadov R., Adli M. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat. Methods. 2017;14:710–712. doi: 10.1038/nmeth.4327. [DOI] [PubMed] [Google Scholar]
- 16.Webber B.R., Lonetree C.L., Kluesner M.G., Johnson M.J., Pomeroy E.J., Diers M.D., Lahr W.S., Draper G.M., Slipek N.J., Smeester B.A., et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019;10:5222. doi: 10.1038/s41467-019-13007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kluesner M.G., Lahr W.S., Lonetree C.L., Smeester B.A., Qiu X., Slipek N.J., Claudio Vázquez P.N., Pitzen S.P., Pomeroy E.J., Vignes M.J., et al. CRISPR-Cas9 cytidine and adenosine base editing of splice-sites mediates highly-efficient disruption of proteins in primary and immortalized cells. Nat. Commun. 2021;12:2437. doi: 10.1038/s41467-021-22009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.-J., Liquori A.J., et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 20.Tan J., Zhang F., Karcher D., Bock R. Expanding the genome-targeting scope and the site selectivity of high-precision base editors. Nat. Commun. 2020;11:629. doi: 10.1038/s41467-020-14465-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gehrke J.M., Cervantes O., Clement M.K., Wu Y., Zeng J., Bauer D.E., Pinello L., Joung J.K. An APOBEC3A-Cas9 base editor with minimized bystander and off-target activities. Nat. Biotechnol. 2018;36:977–982. doi: 10.1038/nbt.4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grünewald J., Zhou R., Iyer S., Lareau C.A., Garcia S.P., Aryee M.J., Joung J.K. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 2019;37:1041–1048. doi: 10.1038/s41587-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doman J.L., Raguram A., Newby G.A., Liu D.R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020;38:620–628. doi: 10.1038/s41587-020-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam D.K., Feliciano P.R., Arif A., Bohnuud T., Fernandez T.P., Gehrke J.M., Grayson P., Lee K.D., Ortega M.A., Sawyer C., et al. Improved cytosine base editors generated from TadA variants. Nat. Biotechnol. 2023;41:686–697. doi: 10.1038/s41587-022-01611-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kingwell K. Base editors hit the clinic. Nat. Rev. Drug Discov. 2022;21:545–547. doi: 10.1038/d41573-022-00124-z. [DOI] [PubMed] [Google Scholar]
- 26.Chiesa R., Georgiadis C., Syed F., Zhan H., Etuk A., Gkazi S.A., Preece R., Ottaviano G., Braybrook T., Chu J., et al. Base-Edited CAR7 T Cells for Relapsed T-Cell Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2023;389:899–910. doi: 10.1056/NEJMoa2300709. [DOI] [PubMed] [Google Scholar]
- 27.Georgiadis C., Rasaiyaah J., Gkazi S.A., Preece R., Etuk A., Christi A., Qasim W. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia. 2021;35:3466–3481. doi: 10.1038/s41375-021-01282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Diorio C., Murray R., Naniong M., Barrera L., Camblin A., Chukinas J., Coholan L., Edwards A., Fuller T., Gonzales C., et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140:619–629. doi: 10.1182/blood.2022015825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roth T.L., Li P.J., Blaeschke F., Nies J.F., Apathy R., Mowery C., Yu R., Nguyen M.L.T., Lee Y., Truong A., et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell. 2020;181:728–744.e21. doi: 10.1016/j.cell.2020.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glaser V., Flugel C., Kath J., Du W., Drosdek V., Franke C., Stein M., Pruß A., Schmueck-Henneresse M., Volk H.-D., et al. Combining different CRISPR nucleases for simultaneous knock-in and base editing prevents translocations in multiplex-edited CAR T cells. Genome Biol. 2023;24:89. doi: 10.1186/s13059-023-02928-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collantes J.C., Tan V.M., Xu H., Ruiz-Urigüen M., Alasadi A., Guo J., Tao H., Su C., Tyc K.M., Selmi T., et al. Development and Characterization of a Modular CRISPR and RNA Aptamer Mediated Base Editing System. CRISPR J. 2021;4:58–68. doi: 10.1089/crispr.2020.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agata Y., Kawasaki A., Nishimura H., Ishida Y., Tsubata T., Yagita H., Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996;8:765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 33.van den Berg J., G Manjón A., Kielbassa K., Feringa F.M., Freire R., Medema R.H. A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res. 2018;46:10132–10144. doi: 10.1093/nar/gky786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friskes A., Koob L., Krenning L., Severson T.M., Koeleman E.S., Vergara X., Schubert M., van den Berg J., Evers B., Manjón A.G., et al. Double-strand break toxicity is chromatin context independent. Nucleic Acids Res. 2022;50:9930–9947. doi: 10.1093/nar/gkac758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazzarotto C.R., Malinin N.L., Li Y., Zhang R., Yang Y., Lee G., Cowley E., He Y., Lan X., Jividen K., et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat. Biotechnol. 2020;38:1317–1327. doi: 10.1038/s41587-020-0555-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller N.A., Farrow E.G., Gibson M., Willig L.K., Twist G., Yoo B., Marrs T., Corder S., Krivohlavek L., Walter A., et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 2015;7:100. doi: 10.1186/s13073-015-0221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Illumina DRAGEN Bio-IT Platform | Variant calling & genomics software https://www.illumina.com/products/by-type/informatics-products/dragen-bio-it-platform.html.
- 42.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lerner T., Papavasiliou F.N., Pecori R. RNA Editors, Cofactors, and mRNA Targets: An Overview of the C-to-U RNA Editing Machinery and Its Implication in Human Disease. Genes (Basel) 2018;10:13. doi: 10.3390/genes10010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R., et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 45.Eyquem J., Mansilla-Soto J., Giavridis T., van der Stegen S.J.C., Hamieh M., Cunanan K.M., Odak A., Gönen M., Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E., et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barkal A.A., Weiskopf K., Kao K.S., Gordon S.R., Rosental B., Yiu Y.Y., George B.M., Markovic M., Ring N.G., Tsai J.M., et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat. Immunol. 2018;19:76–84. doi: 10.1038/s41590-017-0004-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kamali E., Rahbarizadeh F., Hojati Z., Frödin M. CRISPR/Cas9-mediated knockout of clinically relevant alloantigenes in human primary T cells. BMC Biotechnol. 2021;21:9. doi: 10.1186/s12896-020-00665-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brentjens R.J., Davila M.L., Riviere I., Park J., Wang X., Cowell L.G., Bartido S., Stefanski J., Taylor C., Olszewska M., et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013;5 doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Magoč T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cromer M.K., Majeti K.R., Rettig G.R., Murugan K., Kurgan G.L., Bode N.M., Hampton J.P., Vakulskas C.A., Behlke M.A., Porteus M.H. Comparative analysis of CRISPR off-target discovery tools following ex vivo editing of CD34+ hematopoietic stem and progenitor cells. Mol. Ther. 2023;31:1074–1087. doi: 10.1016/j.ymthe.2023.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bürkner P.-C. brms: An R Package for Bayesian Multilevel Models Using Stan. J. Stat. Softw. 2017;80:1–28. doi: 10.18637/jss.v080.i01. [DOI] [Google Scholar]
- 53.Bürkner P.-C. Advanced Bayesian Multilevel Modeling with the R Package brms. R. J. 2018;10:395–411. [Google Scholar]
- 54.Bürkner P.-C. Bayesian Item Response Modeling in R with brms and Stan. J. Stat. Softw. 2021;100:1–54. doi: 10.18637/jss.v100.i05. [DOI] [Google Scholar]
- 55.Miller N.A., Farrow E.G., Gibson M., Willig L.K., Twist G., Yoo B., Marrs T., Corder S., Krivohlavek L., Walter A., et al. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 2015;7:100–116. doi: 10.1186/S13073-015-0221-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen X., Schulz-Trieglaff O., Shaw R., Barnes B., Schlesinger F., Källberg M., Cox A.J., Kruglyak S., Saunders C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32:1220–1222. doi: 10.1093/BIOINFORMATICS/BTV710. [DOI] [PubMed] [Google Scholar]
- 57.Genomics in the cloud : using Docker, GATK, and WDL in Terra - University of Wolverhampton https://librarysearch.wlv.ac.uk/discovery/fulldisplay/alma991002848267404901/44UOWO_INST:MAIN.
- 58.Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., Van der Auwera G.A., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D., et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv. 2018 doi: 10.1101/201178. Preprint at. [DOI] [Google Scholar]
- 59.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The average and individual nuclease read counts (replicates 1–3) for each amplified site are reported for two human donors evaluated (identified as D71 and D73). Sites selected for rhAmpSeq validation are identified in column L. These sites have been selected based on hierarchical selection criteria (column M) as defined in the table and in the methods section. Genomic coordinates and sequence for each amplified site are reported. Mismatches to the on-target site are reported in lowercase.
Percentage editing in edited samples (raw editing), control samples (control), and corrected editing calculated by subtracting the value of the control sample from the edited sample (background corrected editing), together with matched read depth at each interrogated site. Data are representative of either base editing (BE) or Insertions/Deletions (INDEL) events in cells edited with the Pin-point system (PnP) or SpCas9 for the indicated on-target (TRAC/B2M/ PDCD-1/CD52) and predicted off-target sites. Bayesian statistics has been performed on the dataset, citing the evidence ratio, posterior probability and ∗ as measures of statistical significance. On-targets are highlighted in green and validated off-targets in red.
Percentage editing in edited samples (raw editing), control samples (control), and corrected editing calculated by subtracting the value of the control sample from the edited sample (background corrected editing), together with matched read depth at each interrogated site. Data are representative of either base editing (BE) or Insertions/Deletions (INDEL) events in cells edited with the Pin-point system (PnP) or SpCas9 for the indicated on-target (TRAC/B2M/ PDCD-1/CD52) and predicted off-target sites. Bayesian statistics has been performed on the dataset, citing the evidence ratio, posterior probability, and ∗ as measures of statistical significance. On-targets are highlighted in green and validated off-targets in red.
Data Availability Statement
The NGS datasets reported in this manuscript are available on Sequence Read Archive with BioProject IDs PRJNA1090029 (http://www.ncbi.nlm.nih.gov/bioproject/1090029) and PRJNA1121376 (http://www.ncbi.nlm.nih.gov/bioproject/1121376).