

Abstract
Many recent studies have demonstrated recruitment of chromatin-modifying enzymes to double-strand breaks. Instead, we wanted to examine chromatin modifications during the repair of these double-strand breaks. We show that homologous recombination triggers the acetylation of N-terminal lysines on histones H3 and H4 flanking a double-strand break, followed by deacetylation of H3 and H4. Consistent with a requirement for acetylation and deacetylation during homologous recombination, Saccharomyces cerevisiae with substitutions of the acetylatable lysines of histone H4, deleted for the N-terminal tail of histone H3 or H4, deleted for the histone acetyltransferase GCN5 gene or the histone deacetylase RPD3 gene, shows inviability following induction of an HO lesion that is repaired primarily by homologous recombination. Furthermore, the histone acetyltransferases Gcn5 and Esa1 and the histone deacetylases Rpd3, Sir2, and Hst1 are recruited to the HO lesion during homologous recombinational repair. We have also observed a distinct pattern of histone deacetylation at the donor locus during homologous recombination. Our results demonstrate that dynamic changes in histone acetylation accompany homologous recombination and that the ability to modulate histone acetylation is essential for viability following homologous recombination.
The repair of DNA damage is essential for the prevention of cell death and carcinogenesis. Lethal DNA damage occurs when both strands of DNA are broken, which can lead to the loss of chromosome arms during mitosis (45). Cells employ two major pathways to repair a double-strand break (DSB), nonhomologous end joining (NHEJ) and homologous recombination. Homologous recombination is mediated by the Rad52 epistasis group of proteins, where Rad52 is the only member essential for homologous recombination (26). Homologous recombination requires significant sequence homology to allow the lesion to be “patched” by copying the homologous donor sequences. The single defined HO site at the Saccharomyces cerevisiae MAT locus has allowed the events of DSB repair to be dissected (12). The HO lesion is primarily repaired by homologous recombination with donor sequences from HMLα or HMRa (21). We chose to use the HO system to investigate whether homologous recombination is regulated by chromatin modifications, as is the case for transcriptional regulation.
Chromatin comprises a basic repeating unit called the nucleosome. The nucleosome is made up of a histone octamer (two molecules each of H2A, H2B, H3, and H4) which has approximately 147 base pairs of DNA wrapped around it (25). There are many types of histone modifications that alter chromatin structure and function, including acetylation, methylation, phosphorylation, ubiquitylation, ADP-ribosylation, and sumoylation. Most of these types of modifications have been found to play a fundamental role in regulating gene expression by modulating the accessibility of the DNA to binding factors and by acting as specific binding sites for factors that regulate transcription (10, 16). These modifications on histone proteins have been dubbed the histone code (8, 33, 42).
Until recently, the evidence for a DNA damage-induced histone code was limited to the phosphorylation of the C-terminal tail of the histone variant H2AX (or H2A in S. cerevisiae) (29, 33). This is a very early response to double-strand DNA breaks, and recent studies in yeast indicate that H2A phosphorylation is required for the recruitment of chromatin-modifying complexes to the vicinity of DNA damage. Phosphorylated H2A in yeast leads to the recruitment of the chromatin-remodeling complex INO80.com to a double-strand DNA break (32, 46), which in turn is required for efficient processing of the double-strand break into single-stranded DNA (46). Similarly, phosphorylation of H2A leads to the recruitment of the Swr1 chromatin-remodeling complex (7). In addition, phosphorylation of H2A in yeast is required for the recruitment of the NuA4 histone acetyltransferase complex to a region proximal to a double-strand break and is accompanied by localized acetylation of histone H4 on lysine 8 (7). Methylated and ubiquitinated histones also appear to play an important role in the signaling of double-strand DNA damage to the cell (11, 15, 40).
There is a growing body of evidence supporting a role for histone acetylation in double-strand DNA repair. The N terminus of histone H3 has five reversibly acetylatable lysine residues at positions 9, 14, 18, 23, and 27, while the N terminus of histone H4 has four reversibly acetylatable lysine residues at positions 5, 8, 12, and 16. Mutation of all four acetylatable lysines of histone H4 causes sensitivity to double-strand DNA-damaging agents (9). Mutation of both lysines 14 and 23 on histone H3 increases sensitivity to expression of the HO endonuclease (35). The enzymes that alter histone acetylation have also been implicated in the repair of DSBs. For example, mutation of a histone acetyltransferase, Gcn5, causes sensitivity to double-strand DNA-damaging agents (6). A major caveat with these studies linking DNA damage sensitivity to histone acetylation is that it may be a result of indirect effects (for example, transcriptional dysregulation of repair factors). In addition, it is not clear whether the repair is by homologous recombination and/or NHEJ.
There is evidence to suggest that changes in histone acetylation accompany NHEJ, but it is unknown whether histone acetylation changes during homologous recombinational repair. For example, the histone acetyltransferase Esa1 is required for efficient NHEJ (4). The human counterpart of the Gcn5 histone acetyltransferase is phosphorylated and inactivated by DNA-dependent protein kinase, which is a central component of the NHEJ machinery (2). There is also clear evidence to suggest that the opposite reaction, histone deacetylation, is important for NHEJ. The histone deacetylase Rpd3 and its binding partner Sin3 are required for efficient NHEJ (18). Sir3, which is part of a complex containing the histone deacetylase Sir2, is known to relocalize to the site of a DSB in yeast that lacks homologous donor sequences for repair by homologous recombination, but the reason for this is unclear (28, 31). Similarly, Esa1 localizes to the HO break in yeast that lacks homologous donor sequences for repair by homologous recombination (7). Two recent reports have shown for the first time that histone acetylation levels actually change around a DSB. Acetylation on H4 K16 is reduced 40 to 50% following induction of a DSB that can be repaired only by NHEJ (18), and acetylation of lysine H4 K8 transiently increases in chromatin flanking the DSB that can only be repaired by NHEJ (7). However, we do not know whether histone acetylation changes during homologous recombination.
Since virtually nothing was known about histone acetylation during homologous recombinational repair, we set out to perform a comprehensive analysis of acetylation levels on all nine acetylatable residues of the N-terminal tails of histones H3 and H4 before, during, and after homologous recombinational repair of an HO lesion. We find that localized histone acetylation, followed by histone deacetylation, occurs in a manner dependent on homologous recombinational repair. These modifications are important for viability following DNA repair and are likely to be mediated by the recruitment of the histone acetylases Gcn5 and Esa1 and the histone deacetylases Sir2, Hst1, and Rpd3 during homologous recombination.
MATERIALS AND METHODS
S. cerevisiae strains.
S. cerevisiae strains used in this study are shown in Table 1.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Reference |
|---|---|---|
| BAT009 | W303 MATaade2-1 can1-100 his3-11 leu2-3,112 trp1-1 ura3-1 GAL ade3::Gal10::HO | This study |
| BAT019 | W303 MATalys2 leu2-3,112 his3-11 trp1-1 ura3-1 bar1::LEU2 gcn5::KAN | This study |
| BAT020 | W303 MATalys2 leu2-3,112 his3-11 trp1-1 ura3-1 bar1::LEU2 rpd3::KAN | This study |
| BAT021 | W303 MATaade2-1 can1-100 his3-11 leu2-3,112 trp1-1 ura3-1 GAL | This study |
| BAT022 | W303 MATatrp1-1 ura3-1 can1-100 ADE bar::LEU2 his3-11 GAL rad52::TRP1 | This study |
| BAT035 | W303 MATaade2-1 can1-100 his3-11 leu2-3,112 trp1-1 ura3-1 GAL ade3::Gal10::HO SIR2-3HA::KAN | This study |
| BAT038 | W303 MATaade2-101 his3-D200 lys2-801 trp1-D901 ura3-52 hht1 hhf1::LEU2 hht2,hhf2::HIS3 rad52Δ::Hygro [pRM200 (CNE4 ARS1 TRP1 HHT2 HHF2)] | This study |
| BAT039 | MATaade2-101 his3-D200 lys2-801 trp1-D901 ura3-52 lys2-801::dam+::LYS2 hht1,hhf1::LEU2 hht2,hhf2::HIS4 [pGF17-D4-19]; MATa version of GFY34419 (9) | This study |
| FLY722 | MATaade2-101 leu2-3,112 lys2-801 trp1-D901 ura3-52 hhf1::HIS3 hhf2::TRP1-HHF2-K5,8,12,16R | 23 |
| FLY821 | MATaade2-101 leu2-3,112 lys2-801 trp1-D901 ura3-52 hhf1::HIS3 hhf2::TRP1-HHF2-K5,8,12R | 23 |
| JKM179 | MATα ΔHO::Ade1 Δhml::ADE1 Δhmr::ade1 ade1 leu2-3,112 ADE can1-100 | 22 |
| RMY200 | MATaade2-101 his3-D200 lys2-801 trp1-D901 ura3-52 hht1,hhf1::LEU2 hht2,hhf2::HIS3 [pRM200 (CNE4 ARS1 TRP1 HHT2 HHF2)] | 27 |
| RMY430 | MATaade2-101 his3-D200 lys2-801 trp1-D901 ura3-52 hht1,hhf1::LEU2 hht2,hhf2::HIS3 [pRM430 (CEN4 ARS1 TRP1 HHT1-D4-30)] | 27 |
| Z1256 | W303 MATaade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+psi+ | 37 |
| Z1346 | W303 MATaade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+psi+ESA1::18myc::TRP1 | 37 |
| Z1347 | W303 MATaade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+psi+RPD3::18myc::TRP1 | 37 |
| Z1410 | W303 MATaade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+psi+HST1::9myc::TRP1 | 37 |
| Z1466 | W303 MATaade2-1 trp1-1 can1-100 leu2-3,112 his3-11,15 ura3 GAL+psi+GCN5::9myc::TRP1 | 37 |
HO endonuclease sensitivity assays.
Yeast strains were grown overnight in rich media, media with no uracil but with 2% raffinose, or media with no adenosine but with 2% raffinose. Cells were diluted and grown to an optical density at 600 nm (OD600) of ∼0.75 and concentrated to an OD600 of 1.0 or higher to plate in serial dilutions onto rich media, media with no uracil but with 2% raffinose, or media with no adenosine but with 2% raffinose with increasing amounts of galactose.
Viability assays.
Yeast strains were grown overnight in medium without uracil or adenosine but with 2% raffinose diluted to an OD600 of 0.2, and grown to an OD600 of 0.5 before adding galactose to 2%. Samples were taken for viability analysis before adding galactose (t = 0) and each consecutive hour afterwards for 3 h. Samples were sonicated, counted, and diluted in order to plate 200 cells onto yeast extract-peptone-dextrose (YEPD) plates in triplicate. Plates were incubated for 2 or 3 days until the colonies were large enough to count.
DNA damage and repair quantitation.
Primers flanking the HO site in the MAT locus were used to determine the degree of cutting and repair of mating type by PCR amplification (primer sequences are available upon request). Cultures were grown overnight in raffinose-containing media. Galactose and then glucose were added to 2% at the times indicated in the figure legends. Primers to the RAD27 gene were included in the multiplex PCR as an internal control. The number of PCR cycles to produce amplification in the linear range was determined empirically. The ratio of the MAT product to the control product was quantified using Labworks (GelPro4.0; Media Cybernetics, LP).
ChIP analysis.
Cultures were grown overnight in raffinose-containing media, diluted, and grown for 2 to 3 h until the cells reached an OD600 of approximately 0.5. Galactose and then glucose were added to 2% at the times indicated in the figure legends. Samples were taken for chromatin immunoprecipitation (ChIP) analysis at the time points indicated in the figure legends and were processed as described previously (20). Residue-specific acetyl lysine antibodies (Upstate) were used for ChIP exactly as described previously (44). The specificity of these antibodies had been rigorously established previously (44). Histone levels were determined by ChIP using an antiserum to the C terminus of histone H3 (Abcam). A 1:400 dilution of the anti-mouse hemagglutinin (HA) antibody (Covance) was used to immunoprecipitate Sir2-HA and a 1:266 dilution of anti-myc antibody (9E11) to immunoprecipitate Gcn5-MYC, Rpd3-MYC, Esa1-MYC, and Hst1-MYC. Samples were analyzed using quantitative real-time PCR in a multiplex reaction with primers and probes designed 0.6 kb away from the HO lesion and to an unrelated intergenic region within the SMC2 gene. All experiments were performed at least two independent times.
Quantitative real-time PCR.
Real-time PCR was used to quantitate amounts of DNA fragments in the immunoprecipitated (IP) and input samples from the ChIP analyses, using the ABI 7700 sequence detector and Taqman PCR Master Mix protocol (ABI). Each PCR was performed in triplicate with cycling conditions as follows: 50°C for 2 min, 95°C for 10 min, and then 40 cycles, with 1 cycle consisting of 95°C for 15 s and 60°C for 1 min. The primers used were designed using the ABI Primer Express software (ABI) and are available upon request. Each PCR was carried out in triplicate and was multiplexed with a primer set specific to either 0.6 kb or 2 kb from the HO lesion, a region 0.4 kb from the HO site in the HML region, and a primer set to the internal control SMC2 gene using 6-carboxyfluorescine and VIC probes designed using the ABI Primer Express software (Applied Biosystems). The cycle threshold (CT) value was set so that the fluorescence signal was above the baseline noise and as low as possible in the exponential amplification phase. The amount of change compared to the SMC2 control was calculated for each input and IP, and the ratio of the IP to the input was calculated as the relative change (n-fold) over input using the following equation: [log (base 2) SMC2 primer pair CT from IP (1:100) − HO primer pair CT from IP (1:100)]/[log (base 2) SMC2 primer pair CT from input (1:5,000) − HO primer pair CT from input (1:5,000)]. A change in the CT value of 1 unit was determined empirically for each primer pair to be very close to 2, indicating a change of approximately twofold in amplification per cycle over the range of template amounts used in these analyses. The concentrations of primers, probes, and template DNA that yielded the optimal amplification in the linear range in real time was empirically determined. P values were determined using the unpaired Student t test using Prism software (GraphPad Software, Inc.).
RESULTS
The H3 and H4 N-terminal tails and the acetylatable lysines of histone H4 are required for viability following exposure to HO endonuclease. It has previously been shown that S. cerevisiae strains lacking the N-terminal tail or acetylatable lysines of histone H3 and H4 are sensitive to double-strand DNA-damaging agents (4, 30). However, sensitivity to these drugs can be a consequence of a role for the N-terminal tails in either NHEJ or homologous recombination. To begin to address whether histone acetylation modulates DNA repair via homologous recombination, we analyzed whether yeast strains with histone H3 or H4 lacking their N-terminal tail (H3 tailΔ and H4 tailΔ) or with their reversibly acetylatable lysines mutated to arginine have reduced viability when exposed to HO endonuclease. Failure to repair the HO lesion is lethal in yeast (3). The H3 tailΔ, the H4 tailΔ, and the H4 lysine mutant strains showed increasing sensitivity to increasing amounts of galactose, where the HO endonuclease was under the control of a galactose-inducible promoter (pGAL-HO) (Fig. 1A). Strikingly, the HO sensitivity of the H3 tailΔ strain was comparable to that of a rad52Δ strain that is unable to perform any homologous recombination (Fig. 1A). The H4 tailΔ strain was not as sensitive as the H3 tailΔ strain, presumably because yeast strains deleted for their H4 N-terminal tail have 10- to 20-fold decreased induction of the GAL10 promoter (47). Consistent with a role for the H3 N-terminal tail in homologous recombination, it has previously been shown that mutation of lysines 14 and 23 of histone H3 lead to increased sensitivity to an HO lesion that is repaired by homologous recombination (35). To strengthen our argument that the H4 N-terminal tail plays a role in homologous recombination, we examined strains that still had their H4 N-terminal tails but lacked the acetylatable lysines. Yeast strains with substitutions of lysines 5, 8, 12, and 16 or lysines 5, 8, and 12 (strains H4K5,8,12,16R and H4K5,8,12R, respectively) were sensitive to induction of the HO endonuclease (Fig. 1A). Other combinations of H4 lysine substitutions did not show sensitivity to HO endonuclease (data not shown). The inviability on galactose plates was a response to the HO lesion, rather than a general sensitivity to galactose, because the H3 tailΔ and H4 tailΔ strains without pGAL-HO were insensitive to galactose (data not shown). Furthermore, H3 tailΔ and H4 tailΔ strains lacking the HO site at MAT were insensitive to induction of HO endonuclease (data not shown), indicating that the sensitivity seen in the H3 tailΔ and H4 tailΔ strains is a result of cleavage of the HO site at MAT. Taken together, these results indicate that the acetylatable N-terminal tails and lysines of H3 and H4 are required for viability following exposure of yeast to a single DSB that is repaired by homologous recombination.
FIG. 1.

The N-terminal tails of histones H3 and H4 and the acetylatable lysines of histone H4 confer resistance to the HO endonuclease. A. Sensitivity to prolonged exposure to HO endonuclease. S. cerevisiae strains RMY200 (wild type [WT]), RMY430 (H3 tailΔ), BAT039 (H4 tailΔ), FLY722 (H4K5,8,12,16R), FLY821 (H4K5,8,12R), and BAT038 (rad52Δ) containing the pGAL-HO-ADE2 plasmid were serially diluted onto plates containing increasing amounts of galactose (GAL) in order to induce increasing amounts of the HO endonuclease. B. Sensitivity to limited exposure to HO endonuclease. The viability of yeast strains used in panel A was determined after induction of the HO endonuclease for 0, 1, 2, or 3 h. The average viability ± standard deviation (error bar) of three independent ChIP experiments is shown. Viability at 0 h is normalized to 100 percent.
To further investigate the roles of the N-terminal tails of histones H3 and H4 in homologous recombination, we measured sensitivity to a transient exposure to HO endonuclease. To do this, we induced the HO endonuclease for 1, 2, or 3 h and then plated the cells onto glucose-containing media to repress HO endonuclease and to allow DSB repair. While the wild-type cells were able to repair the DNA lesion even after 3 hours of exposure to HO endonuclease, the H4 tailΔ and H3 tailΔ strains showed decreased viability even after 1 hour of exposure to HO endonuclease (Fig. 1B). After 3 hours of exposure to the HO endonuclease, the wild-type strain exhibited 100% viability, the H3 tailΔ strain exhibited 66% viability, the H4 tailΔ strain exhibited 68% viability, and the rad52Δ strain exhibited 65% viability (Fig. 1B). These results are consistent with a role for the N-terminal tails of histones H3 and H4 in DSB repair via homologous recombination.
Histone acetylation and deacetylation occur during homologous recombination.
We predicted that the requirement of the N-terminal tails of histones H3 and H4 and the acetylatable lysines of histone H4 for viability following homologous recombinational repair of an HO lesion is a consequence of their reversible acetylation. However, first we needed to determine the degree of cutting and repair at the HO site. Therefore, we developed a quantitative PCR-based analysis of the MAT region to follow the cutting and repair of the HO lesion (Fig. 2A and B). There are five regions classified within the mating-type loci; from left to right, they are W (723 bp), X (704 bp), Y (a = 642 bp and α = 747 bp), and Z comprising Z1 (239 bp) and Z2 (89 bp) (29) (Fig. 2A). The HO endonuclease cleaves the DNA near the Y/Z junction at MAT, and up to 1.4 kb can be exchanged from HMRa or HMLα during DNA repair (primarily in the X and Y regions) (29, 36). The yeast MAT locus expresses the information for either the a or α mating type. In general, MATa cells are repaired using the information at HMLα to yield MATα yeast, while MATα cells are repaired using the information at HMRa to yield MATa yeast (Fig. 2A) (48). Therefore, we designed a primer pair that would produce a product unique to MAT that would differ by 0.1 kb between MATa and MATα cells (Fig. 2A). A MAT locus with an HO lesion will yield no MAT PCR product. This PCR-based approach generated the same results as Southern analysis (data not shown) but was more convenient and easier to control internally. To analyze HO cleavage and repair, HO endonuclease was induced at 0 h by the addition of galactose, followed by the addition of glucose at 2 h to repress HO endonuclease. HO cleavage and repair were apparent in the wild-type strain (Fig. 2B). Both mating types are generated during repair, because immediate recleavage and repair occur on some templates during the 2-h induction of HO endonuclease.
FIG. 2.

Levels of histone acetylation before, during, and after repair of a double-strand break by homologous recombination. A. Schematic of the mating-type loci in yeast. The locations of PCR primers used to quantify HO cleavage and repair are shown, where the reverse primer is specific to the MAT locus and the forward primers recognize the X sequence. If the mating type is a, then the PCR product is 1.0 kb, and if it is α, then the product is 1.1 kb. Also shown are the positions of primer pairs used to assay the amount of acetylation 0.6 kb and 2 kb away from the HO endonuclease site. Chrom.III, chromosome III. B. Cutting and repair of the HO lesion in wild-type yeast. HO endonuclease was induced at time zero in strain BAT009 (wild type) containing the plasmid pGAL-HO-URA3 by the addition of galactose and was repressed at 2 h by the addition of glucose. Samples were analyzed throughout the time course with the primers shown in panel A and control primers. The MATa and MATα products were quantified from the gel and were normalized to the control product. The amount of MAT product at time zero was normalized to 1. C. ChIP analysis of acetylated lysines on histones H4 0.6 kb and 2.0 kb away from the HO site. HO endonuclease was induced at time zero in strain BAT009 containing the plasmid pGAL-HO-URA3 by the addition of galactose and was repressed at 2 h by the addition of glucose as described above for panel B. Relative percent immunoprecipitation (IP) was quantitated by taking the ratio of the MAT product to the SMC2 control product. To obtain the IP/input values (y axis), the amount of immunoprecipitated DNA was divided by the amount of the input DNA. Values were normalized so that the IP/input ratio was equal to 1 at time zero. The averages and standard errors of the means (error bars) are plotted for at least six independent quantitative real-time PCR experiments from two independent ChIPs. The asterisks represent a significant P value where one asterisk represents a P value of 0.05 to 0.01, two asterisks 0.009 to 0.001, and three asterisks ≤0.0009. Black asterisks indicate a significant change from the previous time point, and small grey circles indicate a significant change from the value before HO induction at time zero. H4AcK5, H4 with lysine 5 acetylated; H3 C-term, H3 C terminus. D. ChIP analysis of histone H3 levels and acetylated lysines on histone H3, as described for panel C.
To determine directly whether histone acetylation levels change during homologous recombination, we evaluated the state of histone acetylation before, during, and after DNA repair. We performed ChIP analysis using antibodies specific to each acetylated lysine on histones H3 (lysines 9, 14, 18, 23, and 27) and H4 (lysines 5, 8, 12, and 16) over the identical time course of damage and repair used in Fig. 2B, followed by quantitative real-time PCR analysis (44). Primer pairs were used 0.6 kb and 2.0 kb away from the HO cleavage site at MAT to analyze the enrichment or depletion of acetylation flanking the HO site (Fig. 2A). All PCRs were multiplexed with a primer pair to a control region whose acetylation did not change during the course of the experiment, and all acetylation flanking the HO lesion was normalized to this internal control. Although the exact time point and extent of the maximal change in acetylation flanking the HO lesion was somewhat residue specific, several generalizations can be made. Following HO endonuclease induction, when the amount of HO cutting reached 90 percent (2 h; Fig. 2B), acetylation was increased on all nine acetylatable H4 and H3 residues at 0.6 kb from the HO lesion (Fig. 2C and D). Following repression of the HO endonuclease (after 2 h), acetylation levels decreased on all nine acetylatable H3 and H4 residues at a distance 0.6 kb away from the HO site (Fig. 2C and D). At 2 kb away from the HO site, acetylation also increased on most histone H3 and H4 lysine residues during DNA repair, but not to the same extent seen at 0.6 kb from the HO site (Fig. 2C and D). These results indicate that levels of histone acetylation initially increase in chromatin flanking a DSB and then decrease during homologous recombination.
To determine whether the changes in histone acetylation flanking the HO site were due to histone removal and replacement during repair, we determined whether histone levels changed flanking the DSB during DNA damage and repair. We performed ChIP using an antibody raised to the C terminus of histone H3 that is not influenced by histone acetylation. Because histones H3 and H4 are always heterodimers on DNA (19), any changes in the amount of histone H3 or H4 present will be apparent using this antibody. We found no significant changes in histone H3 levels during DNA damage or repair (Fig. 2D), indicating that the changes in acetylation that we observed are not due to histone loss or gain. Importantly, the changes in acetylation during homologous recombination were not a consequence of the switch from MATa to MATα or changes in the sugar source that occurred during the course of the experiment, because no acetylation changes occurred when we performed the identical analysis described above in MATa and MATα strains that lacked the pGAL-HO plasmid (data not shown). As expected, the relative acetylation levels at 0.6 kb from the HO site in MATα and MATa strains were similar (data not shown). Taken together, our data indicate that the increases and decreases in acetylation that occur flanking the HO site are a direct and dynamic response to DNA damage and repair, presumably due to the action of histone acetylases and deacetylases.
Gcn5 and Rpd3 mutants are sensitive to HO endonuclease.
In order to gain further evidence for a role of histone acetylation and deacetylation during homologous recombination, we examined yeast strains deleted for some of the enzymes that remove or add acetyl groups to histones. We found that yeast strains deleted for the histone deacetylase RPD3 gene (rpd3Δ) were as sensitive to constant exposure to the HO endonuclease as a strain that cannot perform homologous recombination (rad52Δ) (Fig. 3A). Similarly, yeast strains deleted for the histone acetyltransferase GCN5 gene (gcn5Δ) were also sensitive, albeit less so than rpd3Δ cells, to constant exposure to the HO endonuclease (Fig. 3A). We verified that the sensitivity of yeast lacking Gcn5 and Rpd3 to galactose was a consequence of induction of the HO endonuclease and cleavage at the HO site by determining that gcn5Δ and rpd3Δ yeast strains lacking the pGAL-HO plasmid or lacking the HO site are insensitive to galactose (data not shown). These results indicate that the sensitivity of strains lacking Rpd3 and Gcn5 to galactose is a consequence of HO endonuclease induction and the persistence of a DNA double-strand break at MAT.
FIG. 3.

Gcn5, Esa1, Sir2, Hst1, and Rpd3 may act to modify histones during homologous recombination. A. Sensitivity to prolonged HO induction. Serial dilution analysis of strains BAT021 (wild type [WT]), BAT019 (gcn5Δ), BAT020 (rpd3Δ), and BAT022 (rad52Δ), containing plasmid pGAL-HO-URA3 onto plates with and without galactose (GAL). B. Sensitivity to transient induction of HO endonuclease. Viability analysis of the strains used in panel A following transient exposure to HO endonuclease performed as described in the legend to Fig. 1B. C. Gcn5 and Esa1 are recruited to the HO lesion. ChIP analysis of strains Z1466 (Gcn5-MYC), Z1346 (Esa1-Myc), and Z1256 (no tag) containing a pGAL-HO-URA3 plasmid where galactose was added after time zero and glucose was added after 2 h to induce and repress the HO endonuclease. Real-time PCR analysis and quantification was performed as described in the legend to Fig. 2 using primers 0.6 kb to the right of the HO site and an SMC2 internal primer pair for normalization. D. Sir2, Hst1, and Rpd3 are recruited to the HO lesion. ChIP analysis of strains BAT035 (Sir2-HA) and BAT009 (No tag), Z1346 (Hst1-MYC) and Z1256 (No tag), and Z1347 (Rpd3-MYC) and Z1256 (No tag) containing a pGAL-HO-URA3 plasmid as described for panel C.
To further analyze the roles of Gcn5 and Rpd3 during homologous recombination, we examined the sensitivities of the gcn5Δ and rpd3Δ strains to transient exposure to the HO endonuclease. We found that both mutants had reduced viabilities at longer times of HO induction (Fig. 3B). For example, after a 3-hour induction of the HO endonuclease, 86% of wild-type cells survived compared to 55% for gcn5Δ cells, 15% for rpd3Δ cells, and 4% for rad52Δ cells. These data are consistent with a role for Gcn5 and Rpd3 in repair of the chromatin structure at the MAT HO site via homologous recombination. These results suggest that Gcn5 and Rpd3 may acetylate and deacetylate histones, respectively, before, during, and/or after homologous recombination.
Gcn5, Esa1, Sir2, Rpd3, and Hst1 are recruited to the HO lesion during homologous recombination.
To further analyze the roles of histone acetylases and deacetylases in homologous recombination, we tested whether these enzymes are specifically recruited to the HO site during homologous recombination. Using ChIP analysis, we reproducibly found significant enrichment of Gcn5 proximal to the HO site after induction of DNA damage compared to a no-tag control (Fig. 3C). The kinetics of cutting and repair of the HO site were identical to those of the wild type in all the tagged histone acetylase and deacetylase strains that we used (data not shown). The enrichment of Gcn5 at the HO lesion is concurrent with the observed increase in histone acetylation (Fig. 2C and D). Esa1, a histone acetyltransferase specific to H4 lysine residues (17), is also recruited to the HO lesion after 2 hours of exposure to the HO endonuclease, consistent with the time in which we see increases in acetylation on H4 lysine residues (Fig. 3C and Fig. 2C). These data indicate that localization of Gcn5 and Esa1 may directly contribute to the increase in acetylation that occurs flanking a DSB during DNA damage and repair by homologous recombination.
The dramatic increase and subsequent decrease in H4 lysine 16 acetylation during DNA damage and repair by homologous recombination led us to look for recruitment of Sir2, a histone deacetylase that deacetylates H4 lysine 16. We once again used ChIP analysis to measure Sir2 levels 0.6 kb away from the HO site during the DNA damage and repair time course (Fig. 3D). Using two different tagged Sir2 strains (HA and myc epitopes), we saw a dramatic increase in Sir2 recruitment compared to a no-tag control at the time point concurrent with histone deacetylation seen at 3 h and during DNA repair (Fig. 3D and data not shown). Hst1, another NAD+-dependent histone deacetylase named for its homology to Sir2 (named Hst for homolog of Sir two) deacetylates histones H3 and H4 (14, 38, 39) and is also recruited to the HO lesion concurrent with the time points in which repair occurs, presumably to deacetylate histones (Fig. 3D). Rpd3, a global histone deacetylase, is also recruited to the HO lesion during the time we observed histone deacetylation (Fig. 2C and D and Fig. 3D). These data indicate that the Sir2, Hst1, and Rpd3 histone deacetylases may act to deacetylate histones during homologous recombination.
Histone acetylation flanking a double-strand break is dependent on homologous recombination.
To investigate whether the changes in histone acetylation were a result of the DSB itself or a consequence of DNA repair, we examined histone acetylation in strains that are unable to initiate homologous recombination. The approach was to perform ChIP analysis with antibodies directed against the acetyl lysine residues on the N termini of histones H3 and H4 and also the C terminus of histone H3 in a strain lacking the homologous donor sequences at the HML and HMR loci (HML/HMRΔ) or lacking the Rad52 protein (rad52Δ), which is essential for homologous recombination, and to quantify the DNA 0.6 kb away from the HO site using quantitative real-time PCR (Fig. 4C and 5B). The inability to repair the DNA lesion in the HML/HMRΔ and rad52Δ yeast strains is shown in Fig. 4A and 5A by the disappearance of the MAT product. Significant histone acetylation does not occur following DNA damage in the HML/HMRΔ strain in contrast to the wild-type strain (Fig. 4C); however, there was still significant histone deacetylation in this strain. In the rad52Δ strain there was also significantly less histone acetylation and histone deacetylation following DNA damage compared to the wild-type strain (Fig. 5B).
FIG. 4.

Changes in histone H3 and H4 acetylation are triggered by homologous recombination. A. Cutting and repair of the HO site were measured when the HO endonuclease was induced at 0 h and repressed at 2 h in strain JKM179 (hml/hmrΔ) containing plasmid pGAL-HO-URA3. B. An H3 C-terminal (H3C-term) antibody was used to measure the relative levels of histone proteins 0.6 kb away from the HO site by ChIP, real-time PCR, and statistical analyses as described in the legend to Fig. 2. C. Levels of histone acetylation in strain JKM179 (hml/hmrΔ) containing plasmid pGAL-HO-URA3 at 0.6 kb from the HO site were measured as described for panel B and in the legend to Fig. 2. Significant changes are noted by small light grey circles as described in the legend to Fig. 2.
FIG. 5.
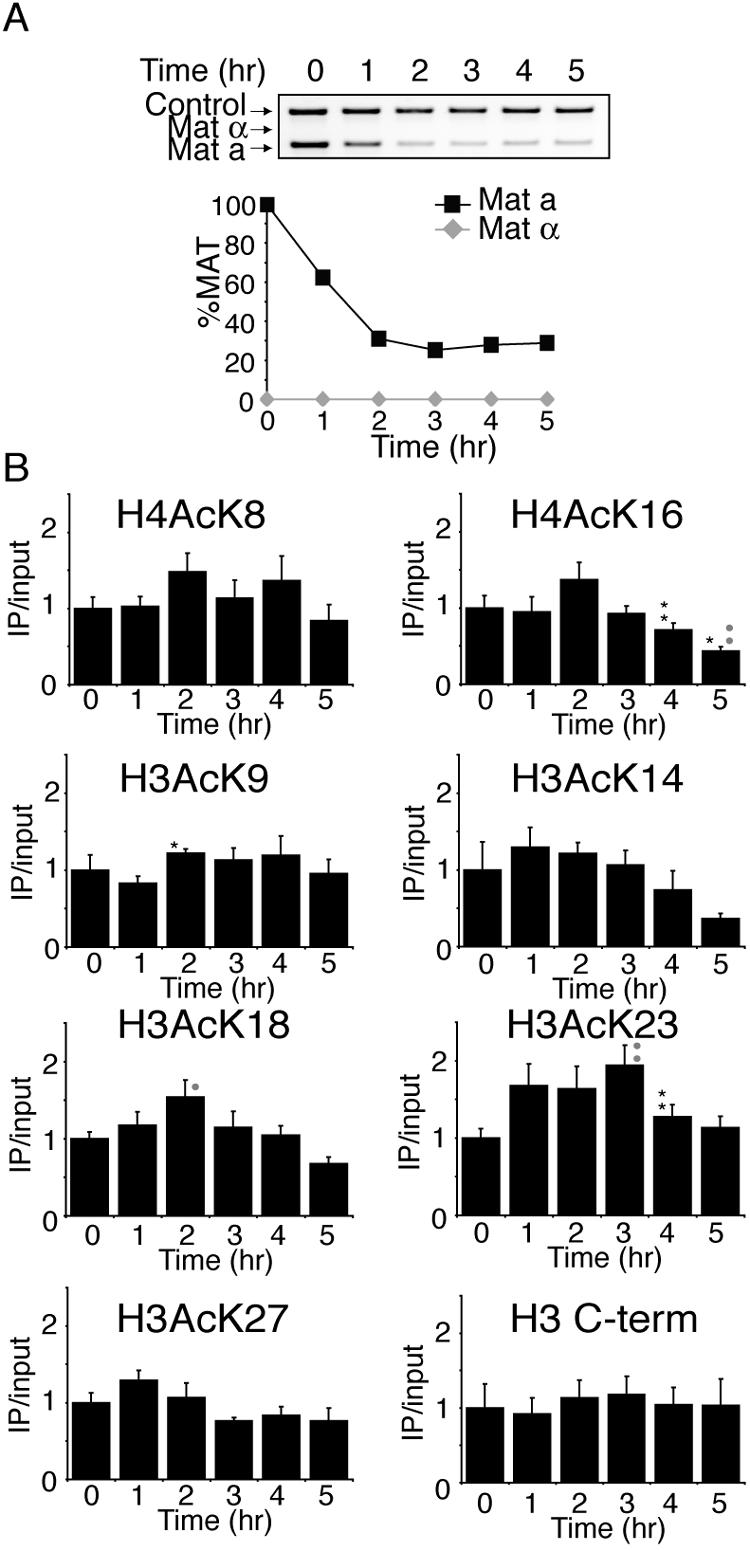
Changes in histone H3 and H4 acetylation are triggered by the Rad52 protein. A. Cutting and repair of the HO site in strain BAT022 (rad52Δ) containing plasmid pGAL-HO-URA3. B. Histone acetylation levels were measured in the same strain used in panel A as described in the legend to Fig. 2. Significant changes are noted by asterisks and small grey circles as described in the legend to Fig. 2.
To demonstrate that the reduced histone acetylation following DNA damage in strains that are unable to repair the HO lesion was not due to histone loss as a consequence of DNA resection, we also analyzed histone H3 levels. We found no significant loss of histone H3 in the HML/HMRΔ strain or in the rad52Δ strain in chromatin flanking the HO lesion (Fig. 4B and 5B), but this is an artifact of the manner in which we analyzed our data. Due to the fact that we normalized all of our data to the input signal for each sample, we would be unable to see a loss of double-stranded DNA and therefore a loss of histones. When the ChIP acetylation signals were not normalized to the input DNA, there was a significant decrease in the amount of DNA flanking the HO lesion due to DNA resection, while the amount of DNA from the control region remained the same (data not shown). As such, histones are removed flanking the DNA break in our strains that are unable to repair the HO lesion, but this is negated in our data by normalizing to the amount of input DNA. Because we analyzed the H3 levels and acetylation levels in the same manner, the reduction in histone acetylation in the HML/HMRΔ strain reflects histone deacetylation and not histone removal. Taken together, our results demonstrate that histone acetylation and deacetylation are dependent on the homologous recombinational repair machinery to differing degrees.
Histone acetylation changes at the homologous donor sequence.
To investigate whether the changes in histone acetylation that occur during homologous recombination flanking the DSB are a consequence of the donor locus coming into the vicinity of the DNA lesion during synapsis, we measured acetylation levels at the donor locus during homologous recombination. To determine directly whether histones are acetylated or deacetylated at the homologous donor sequence during DNA repair, we designed a primer and probe set for quantitative real-time PCR that was 400 base pairs away from the HO consensus sequence in the homologous donor sequence, the HMLα locus (primer pairs closer than this would have also detected the same sequences at MAT and HMR). Using the same DNA samples obtained from the DNA damage/repair time course in Fig. 2, we detected a significant decrease in acetylation on H4 lysine residues 12 and 16 at the donor locus during DNA damage and then a significant increase in their acetylation back to time zero levels during repair (Fig. 6). In addition, acetylation of H4 lysines 5 and 8 and H3 lysines 9 and 18 decreases significantly at the 3-h time point, after glucose is added for an hour and all cells are allowed to repair, and then increases back to time zero levels at later times (Fig. 6). These results indicate that residue-specific and time-specific changes in acetylation occur at the donor locus during DNA damage and repair. Importantly, the pattern of changes in acetylation at the donor locus is quite distinct from those flanking the DNA lesion, indicating that the acetylation changes at the DNA lesion are not an artifact of synapsis but reflect real changes in acetylation on the chromatin flanking the DNA break.
FIG. 6.

Levels of histone acetylation before, during, and after repair of a double-strand break by homologous recombination at the homologous donor sequence. DNA from the ChIP analysis in Fig. 2 was reexamined using primers adjacent to the HO consensus sequence in the HMLα gene to measure histone acetylation levels. Significant changes are noted by asterisks and small light grey circles as described in the legend to Fig. 2.
DISCUSSION
We present a comprehensive identification of the histone H3 and H4 lysine residues that are acetylated and deacetylated flanking a double-strand DNA lesion and the homologous donor sequences during DNA damage and subsequent repair by homologous recombination. Using deletions and substitutions of the acetylatable regions of the histones and deletions of the enzymes responsible for histone acetylation and deacetylation, we have further demonstrated that acetylation and deacetylation during repair are important for viability following DSB repair. In addition, we have shown that the histone acetyltransferases Gcn5 and Esa1 and the histone deacetylases Rpd3, Sir2, and Hst1 localize to the double-strand break during DNA repair and that localized changes in histone acetylation are dependent on homologous recombinational repair of the DNA lesion.
Histone acetylation flanking a double-strand break is dependent on homologous recombinational repair.
Recent studies have analyzed the changes in histone acetylation on two histone H4 residues at an HO lesion (7, 18) and the recruitment of chromatin modifiers to an HO lesion (7, 32, 46). It is important to note that all of these studies used S. cerevisiae strains that lack the HML and HMR donor sequences. Repair in a donorless strain cannot occur by homologous recombination, and even repair by NHEJ is undetectable at the molecular level in our assay (Fig. 4A). As such, these studies are likely to be showing changes in histone acetylation or recruitment of chromatin-modifying enzymes in response to a double-strand break, rather than during repair. Our study is the first work that has examined changes in histone acetylation or the recruitment of histone acetylases or deacetylases during homologous recombinational repair. During homologous recombination, the increase in acetylation on all nine acetylatable lysines in the N termini of histones H3 and H4 was greatest 0.6 kb from the break and less so 2 kb away from the break, especially on histone H4. We predict that changes in acetylation during homologous recombination may even be greater closer to the DNA lesion, but due to the exchange of the “a” and “α” genetic information during HO repair and the homology between the MAT. HMR, and HML loci, we were unable to analyze acetylation changes closer to the break site. This localized increase in acetylation relative to the HO break is quite distinct from the distribution of H2A phosphorylation, which spreads to 50 kb each side of the HO lesion (41).
The peak of histone acetylation that we observed flanking a DSB is a consequence of DNA repair rather than DNA damage per se. Histone acetylation appears to be triggered by an event during homologous recombination, because the increase in acetylation flanking the HO lesion was greatly reduced when either the homologous donor sequences or the gene for the essential component of homologous recombination, Rad52, were deleted (Fig. 4 and 5). Unlike the transient histone acetylation that occurs on H4 lysine 8 in response to an HO lesion in yeast that lacks the homologous donor sequences (7), the general peak of acetylation that we observe during homologous recombination does not occur in yeast strains that are unable to perform homologous recombination (Fig. 4C and 5B). The small amount of acetylation that did occur in our strains that were unable to perform homologous recombination may reflect histone acetylation occurring in response to a double-strand break or during NHEJ (4), which is the pathway used to repair a double-strand break if homologous recombination is prevented.
Histone deacetylation in the vicinity of a double-strand break is triggered by DNA double-strand break repair.
Histone deacetylation during homologous recombination was not dependent on the prior increase in acetylation that occurs during DNA repair. This was apparent because we were able to uncouple the two processes in the strain that lacked the HMR and HML donor sequences where deacetylation occurred in the absence of prior acetylation (Fig. 4). Histone deacetylation during homologous recombination appears to be triggered by an early event during homologous recombination. In a strain lacking the homologous recombinational repair protein Rad52, histone deacetylation flanking the HO site was greatly reduced (Fig. 5). Yet deacetylation still occurred in a strain lacking the HMR and HML donor sequences (Fig. 4). This is consistent with the decreased acetylation of H4 K16 and H4 K8 observed recently at an HO lesion in a strain that lacked the HMR and HML donor sequences (7, 18). One of these studies (7) also detected a transient increase in H4 lysine 8 acetylation at 30 min after HO induction in a strain that lacked the HMR and HML donor sequences that was gone by 60 min and therefore would have been missed in our experiments. The homologous recombinational repair proteins Rad51, Rad54, Rad55, Rdh4, and Rad59 are not recruited to an HO lesion in a rad52Δ strain (24), while Rad51 (and therefore presumably Rad52) is still recruited to the HO lesion in a strain lacking the HMR and HML donor sequences (24, 43). Due to the correlation between the recruitment of Rad51 and Rad52 and histone deacetylation flanking the HO lesion in the strain lacking the HMR and HML donors but little deacetylation and no Rad51 or Rad52 recruitment in the strain lacking RAD52, it is tempting to speculate that recruitment of Rad51 and/or Rad52 is required to trigger histone deacetylation during homologous recombination.
Distinct acetylation changes at the DNA lesion and the donor locus during homologous recombination.
In contrast to the general increase and subsequent decrease in histone acetylation flanking the lesion during homologous recombinational repair, deacetylation occurred in a residue- and time-specific manner at the donor locus during homologous recombination. Clearly then, the changes in histone acetylation flanking the HO lesion are not a consequence of synapsis leading to cross-linking of the histones at the donor locus to the DNA lesion. The histone acetylation and deacetylation that occurs flanking the HO site appear less like a histone code and more like a global general acetylation that would result first in opening and then closing of the chromatin structure. In contrast, the histone deacetylation at the donor locus is residue specific, more like a histone code. The reason for histone deacetylation at the donor locus is unknown. Perhaps the deacetylation of lysines 12 and 16 of histone H4 immediately after induction of the HO endonuclease helps to prevent cutting of the HO sequence at the donor locus. The deacetylation of H4 lysines 5 and 8 and H3 lysines 9 and 18 during repair may help dissociate chromatin-remodeling factors that may be required to facilitate strand invasion.
Dynamic recruitment of histone acetyltransferases and deacetylases during homologous recombination.
The changes in histone acetylation around a DSB during homologous recombination are not a consequence of histone removal or gain and are therefore likely to be caused by an active local process mediated by histone deacetylases and histone acetyltransferases. Consistent with this idea, Gcn5 and Esa1 are likely to directly contribute to histone acetylation during homologous recombination, because they were specifically recruited to the HO lesion. Similarly, we have observed dynamic recruitment of the histone deacetylases Sir2, Rpd3, and Hst1 to the HO lesion during homologous recombinational repair, and these are presumably mediating the deacetylation that occurs during homologous recombination. Histone acetylation of lysine 8 and deacetylation of lysine 16 have recently been observed during NHEJ (7, 18). Similarly, recruitment of Esa1 to the HO lesion has been observed during NHEJ (7). As such, the dynamic recruitment of enzymes that modify the acetylation status of chromatin structure appears to be common to both NHEJ and homologous recombination.
The requirement for histone acetylation and deacetylation during double-strand DNA repair is likely to be conserved in all eukaryotes. Chromatin structure and the DNA repair processes are highly conserved through evolution, and furthermore, recent evidence indicates that the Caenorhabditis elegans homologs of the Rpd3 and Gcn5 proteins may be involved in DNA repair. For example, Hda3 was shown to be required for survival after gamma irradiation in C. elegans (5, 34). C. elegans Hda1 and Hda2 and a protein containing a Gcn5 histone acetyltransferase domain were also identified in RNA interference screens for genes that protect against mutation (34).
What role do changes in histone acetylation play during homologous recombination?
Clearly, histone acetylation and deacetylation play an essential role in maintaining viability after homologous recombinational repair. For example, yeast strains with substitutions that prevent acetylation of H3 (35) or H4 (Fig. 1) or yeast strains deleted for the histone acetylase GCN5 or deacetylase RPD3 genes suffered inviability following exposure to an HO lesion that is repaired by homologous recombination (Fig. 3A and B) and to double-strand DNA-damaging agents (4, 30). Why is histone acetylation essential for viability following homologous recombination? Histone acetylation is well-known to influence factor accessibility to DNA (1). Studies of the repair of UV-irradiated DNA suggest that histone acetylation may be important for increasing the accessibility of the repair machinery during single-strand DNA repair (13). However, it appears unlikely that histone acetylation influences the accessibility of the homologous recombinational repair machinery because yeasts that are not able to undergo histone acetylation in response to an HO lesion show no defect in religation of the DNA break during homologous recombination (4). This is in contrast to the apparent requirement for histone acetylation for plasmid rejoining via NHEJ (4). There is evidence that histone acetylation that occurs in response to an HO lesion may be important for recruiting other chromatin remodelers. For example, recruitment of Rvb1-containing chromatin remodelers, such as Swr1 and INO80.com, to an HO lesion is reduced in cells with mutant versions of the histone acetyltransferase Esa1 (7). The consequence of failing to recruit INO80.com is a <50% reduction in processing of double-stranded DNA into single-stranded DNA (46). Whether this decrease in DNA processing that may result from reduced histone acetylation is sufficient to influence the rate of DNA repair or resulting viability is unknown. Furthermore, it should be noted that the acetylation by Esa1 that occurs in response to the HO lesion (7) is distinct from the peak of acetylation that we observe that is dependent on homologous recombination. Whether the homologous recombination-dependent acetylation is also recruiting chromatin remodelers remains to be determined.
We propose that the changes in histone acetylation levels during homologous recombination themselves are important to the cell, rather than contributing to the repair of the DNA lesion. It is possible that histone acetylation and subsequent deacetylation during DNA repair may be important signals to the cell to indicate that chromosomal repair is complete and may be required to turn off a DNA damage or chromatin structure checkpoint. This proposed inability to recover from a cell cycle checkpoint may be a result of a signal or lack thereof from the chromatin structure that would stop cells from ever proceeding through the cell cycle, causing inviability. Consistent with this idea, deletion of the N-terminal tail of histone H4 or substitution of the acetylatable lysines of histone H4 activates the DNA damage checkpoint (30), presumably in response to endogenous DNA damage that occurs during DNA replication and is repaired by homologous recombination. It is not yet technically possible to distinguish between whether it is the changes in histone acetylation at the donor locus or around the HO lesion or both that are important for viability following homologous recombinational repair. As such, it is formally possible that the failure to change acetylation levels at the donor sequence, not the DNA lesion, may be the cause of inviability.
In summary, we have discovered dynamic changes in histone acetylation flanking a DNA lesion that are dependent on the homologous recombinational repair of a double-strand break. We have also mapped quite distinct, but equally dynamic, changes in histone acetylation at the donor locus during homologous recombination. These modifications, which are likely to be in part mediated by Rpd3, Sir2, Hst1, Esa1, and Gcn5, are important for the repair of chromosomal damage. Future studies will determine the precise nature of the essential roles of histone acetylation and deacetylation during double-strand DNA repair in eukaryotes.
Acknowledgments
We thank Melissa Adkins, Susan Zabaronick, and Jeff Linger for critical reading of the manuscript; Michael Grunstein and Francois Robert for yeast strains; and Michael Grunstein for antibodies for our initial experiments. We are also grateful to Jeff Linger for designing the PCR analysis of mating-type switching and the UCCC real-time PCR core facility.
This work was supported by an NIH award (CA95641-01) to J.K.T. and NIH minority supplement to B.A.T. J.K.T. is a scholar of the Leukemia and Lymphoma Society.
REFERENCES
- 1.Anderson, J. D., P. T. Lowary, and J. Widom. 2001. Effects of histone acetylation on the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol. 307:977-985. [DOI] [PubMed] [Google Scholar]
- 2.Barlev, N. A., V. Poltoratsky, T. Owen-Hughes, C. Ying, L. Liu, J. L. Workman, and S. L. Berger. 1998. Repression of GCN5 histone acetyltransferase activity via bromodomain-mediated binding and phosphorylation by the Ku-DNA-dependent protein kinase complex. Mol. Cell. Biol. 18:1349-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett, C. B., A. L. Lewis, K. K. Baldwin, and M. A. Resnick. 1993. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 90:5613-5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird, A. W., D. Y. Yu, M. G. Pray-Grant, Q. Qiu, K. E. Harmon, P. C. Megee, P. A. Grant, M. M. Smith, and M. F. Christman. 2002. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 419:411-415. [DOI] [PubMed] [Google Scholar]
- 5.Boulton, S. J., A. Gartner, J. Reboul, P. Vaglio, N. Dyson, D. E. Hill, and M. Vidal. 2002. Combined functional genomic maps of the C. elegans DNA damage response. Science 295:127-131. [DOI] [PubMed] [Google Scholar]
- 6.Choy, J. S., and S. J. Kron. 2002. NuA4 subunit Yng2 function in intra-S-phase DNA damage response. Mol. Cell. Biol. 22:8215-8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Downs, J. A., S. Allard, O. Jobin-Robitaille, A. Javaheri, A. Auger, N. Bouchard, S. J. Kron, S. P. Jackson, and J. Cote. 2004. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol. Cell 16:979-990. [DOI] [PubMed] [Google Scholar]
- 8.Downs, J. A., N. F. Lowndes, and S. P. Jackson. 2000. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408:1001-1004. [DOI] [PubMed] [Google Scholar]
- 9.Fisher-Adams, G., and M. Grunstein. 1995. Yeast histone H4 and H3 N-termini have different effects on the chromatin structure of the GAL1 promoter. EMBO J. 14:1468-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freiman, R. N., and R. Tjian. 2003. Regulating the regulators: lysine modifications make their mark. Cell 112:11-17. [DOI] [PubMed] [Google Scholar]
- 11.Giannattasio, M., F. Lazzaro, P. Plevani, and M. Muzi-Falconi. 2005. The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J. Biol. Chem. 280:9879-9886. [DOI] [PubMed] [Google Scholar]
- 12.Haber, J. E. 2000. Lucky breaks: analysis of recombination in Saccharomyces. Mutat. Res. 451:53-69. [DOI] [PubMed] [Google Scholar]
- 13.Hasan, S., and M. O. Hottiger. 2002. Histone acetyl transferases: a role in DNA repair and DNA replication. J. Mol. Med. 80:463-474. [DOI] [PubMed] [Google Scholar]
- 14.Hoppe, G. J., J. C. Tanny, A. D. Rudner, S. A. Gerber, S. Danaie, S. P. Gygi, and D. Moazed. 2002. Steps in assembly of silent chromatin in yeast: Sir3-independent binding of a Sir2/Sir4 complex to silencers and role for Sir2-dependent deacetylation. Mol. Cell. Biol. 22:4167-4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huyen, Y., O. Zgheib, R. A. Ditullio, Jr., V. G. Gorgoulis, P. Zacharatos, T. J. Petty, E. A. Sheston, H. S. Mellert, E. S. Stavridi, and T. D. Halazonetis. 2004. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 432:406-411. [DOI] [PubMed] [Google Scholar]
- 16.Iizuka, M., and M. M. Smith. 2003. Functional consequences of histone modifications. Curr. Opin. Genet. Dev. 13:154-160. [DOI] [PubMed] [Google Scholar]
- 17.Jacobson, S. J., P. M. Laurenson, and L. Pillus. 2004. Functional analyses of chromatin modifications in yeast. Methods Enzymol. 377:3-55. [DOI] [PubMed] [Google Scholar]
- 18.Jazayeri, A., A. D. McAinsh, and S. P. Jackson. 2004. Saccharomyces cerevisiae Sin3p facilitates DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 101:1644-1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kornberg, R. D., and J. O. Thomas. 1974. Chromatin structure: oligomers of the histones. Science 184:865-868. [DOI] [PubMed] [Google Scholar]
- 20.Kuo, M. H., and C. D. Allis. 1999. In vivo cross-linking and immunoprecipitation for studying dynamic protein:DNA associations in a chromatin environment. Methods 19:425-433. [DOI] [PubMed] [Google Scholar]
- 21.Lee, S. E., F. Paques, J. Sylvan, and J. E. Haber. 1999. Role of yeast SIR genes and mating type in directing DNA double-strand breaks to homologous and non-homologous repair paths. Curr. Biol. 9:767-770. [DOI] [PubMed] [Google Scholar]
- 22.Lee, S. E., A. Pellicioli, M. B. Vaze, N. Sugawara, A. Malkova, M. Foiani, and J. E. Haber. 2003. Yeast Rad52 and Rad51 recombination proteins define a second pathway of DNA damage assessment in response to a single double-strand break. Mol. Cell. Biol. 23:8913-8923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lenfant, F., R. K. Mann, B. Thomsen, X. Ling, and M. Grunstein. 1996. All four core histone N-termini contain sequences required for the repression of basal transcription in yeast. EMBO J. 15:3974-3985. [PMC free article] [PubMed] [Google Scholar]
- 24.Lisby, M., J. H. Barlow, R. C. Burgess, and R. Rothstein. 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118:699-713. [DOI] [PubMed] [Google Scholar]
- 25.Luger, K., T. J. Rechsteiner, and T. J. Richmond. 1999. Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 119:1-16. [DOI] [PubMed] [Google Scholar]
- 26.Malone, R. E., and R. E. Esposito. 1980. The RAD52 gene is required for homothallic interconversion of mating types and spontaneous mitotic recombination in yeast. Proc. Natl. Acad. Sci. USA 77:503-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mann, R. K., and M. Grunstein. 1992. Histone H3 N-terminal mutations allow hyperactivation of the yeast GAL1 gene in vivo. EMBO J. 11:3297-3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin, S. G., T. Laroche, N. Suka, M. Grunstein, and S. M. Gasser. 1999. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97:621-633. [DOI] [PubMed] [Google Scholar]
- 29.McGill, C., B. Shafer, and J. Strathern. 1989. Coconversion of flanking sequences with homothallic switching. Cell 57:459-467. [DOI] [PubMed] [Google Scholar]
- 30.Megee, P. C., B. A. Morgan, and M. M. Smith. 1995. Histone H4 and the maintenance of genome integrity. Genes Dev. 9:1716-1727. [DOI] [PubMed] [Google Scholar]
- 31.Mills, K. D., D. A. Sinclair, and L. Guarente. 1999. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell 97:609-620. [DOI] [PubMed] [Google Scholar]
- 32.Morrison, A. J., J. Highland, N. J. Krogan, A. Arbel-Eden, J. F. Greenblatt, J. E. Haber, and X. Shen. 2004. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 119:767-775. [DOI] [PubMed] [Google Scholar]
- 33.Paull, T. T., E. P. Rogakou, V. Yamazaki, C. U. Kirchgessner, M. Gellert, and W. M. Bonner. 2000. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10:886-895. [DOI] [PubMed] [Google Scholar]
- 34.Pothof, J., G. van Haaften, K. Thijssen, R. S. Kamath, A. G. Fraser, J. Ahringer, R. H. Plasterk, and M. Tijsterman. 2003. Identification of genes that protect the C. elegans genome against mutations by genome-wide RNAi. Genes Dev. 17:443-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin, S., and M. R. Parthun. 2002. Histone H3 and the histone acetyltransferase Hat1p contribute to DNA double-strand break repair. Mol. Cell. Biol. 22:8353-8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raveh, D., S. H. Hughes, B. K. Shafer, and J. N. Strathern. 1989. Analysis of the HO-cleaved MAT DNA intermediate generated during the mating type switch in the yeast Saccharomyces cerevisiae. Mol. Gen. Genet. 220:33-42. [PubMed] [Google Scholar]
- 37.Robert, F., D. K. Pokholok, N. M. Hannett, N. J. Rinaldi, M. Chandy, A. Rolfe, J. L. Workman, D. K. Gifford, and R. A. Young. 2004. Global position and recruitment of HATs and HDACs in the yeast genome. Mol. Cell 16:199-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robyr, D., Y. Suka, I. Xenarios, S. K. Kurdistani, A. Wang, N. Suka, and M. Grunstein. 2002. Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109:437-446. [DOI] [PubMed] [Google Scholar]
- 39.Rusche, L. N., and J. Rine. 2001. Conversion of a gene-specific repressor to a regional silencer. Genes Dev. 15:955-967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanders, S. L., M. Portoso, J. Mata, J. Bahler, R. C. Allshire, and T. Kouzarides. 2004. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell 119:603-614. [DOI] [PubMed] [Google Scholar]
- 41.Shroff, R., A. Arbel-Eden, D. Pilch, G. Ira, W. M. Bonner, J. H. Petrini, J. E. Haber, and M. Lichten. 2004. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 14:1703-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strahl, B. D., and C. D. Allis. 2000. The language of covalent histone modifications. Nature 403:41-45. [DOI] [PubMed] [Google Scholar]
- 43.Sugawara, N., X. Wang, and J. E. Haber. 2003. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol. Cell 12:209-219. [DOI] [PubMed] [Google Scholar]
- 44.Suka, N., Y. Suka, A. A. Carmen, J. Wu, and M. Grunstein. 2001. Highly specific antibodies determine histone acetylation site usage in yeast heterochromatin and euchromatin. Mol. Cell 8:473-479. [DOI] [PubMed] [Google Scholar]
- 45.Sung, P., K. M. Trujillo, and S. Van Komen. 2000. Recombination factors of Saccharomyces cerevisiae. Mutat. Res. 451:257-275. [DOI] [PubMed] [Google Scholar]
- 46.van Attikum, H., O. Fritsch, B. Hohn, and S. M. Gasser. 2004. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 119:777-788. [DOI] [PubMed] [Google Scholar]
- 47.Wan, J. S., R. K. Mann, and M. Grunstein. 1995. Yeast histone H3 and H4 N termini function through different GAL1 regulatory elements to repress and activate transcription. Proc. Natl. Acad. Sci. USA 92:5664-5668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu, X., J. K. Moore, and J. E. Haber. 1996. Mechanism of MATα donor preference during mating-type switching of Saccharomyces cerevisiae. Mol. Cell. Biol. 16:657-668. [DOI] [PMC free article] [PubMed] [Google Scholar]