

Summary
Several X-linked genes escape from X chromosome inactivation (XCI), while differences in escape across cell types and tissues are still poorly characterized. Here, we developed scLinaX for directly quantifying relative gene expression from the inactivated X chromosome with droplet-based single-cell RNA sequencing (scRNA-seq) data. The scLinaX and differentially expressed gene analyses with large-scale blood scRNA-seq datasets consistently identified the stronger escape in lymphocytes than in myeloid cells. An extension of scLinaX to a 10x multiome dataset (scLinaX-multi) suggested a stronger escape in lymphocytes than in myeloid cells at the chromatin-accessibility level. The scLinaX analysis of human multiple-organ scRNA-seq datasets also identified the relatively strong degree of escape from XCI in lymphoid tissues and lymphocytes. Finally, effect size comparisons of genome-wide association studies between sexes suggested the underlying impact of escape on the genotype-phenotype association. Overall, scLinaX and the quantified escape catalog identified the heterogeneity of escape across cell types and tissues.
Keywords: X chromosome, sex differences, single-cell omics
Graphical abstract
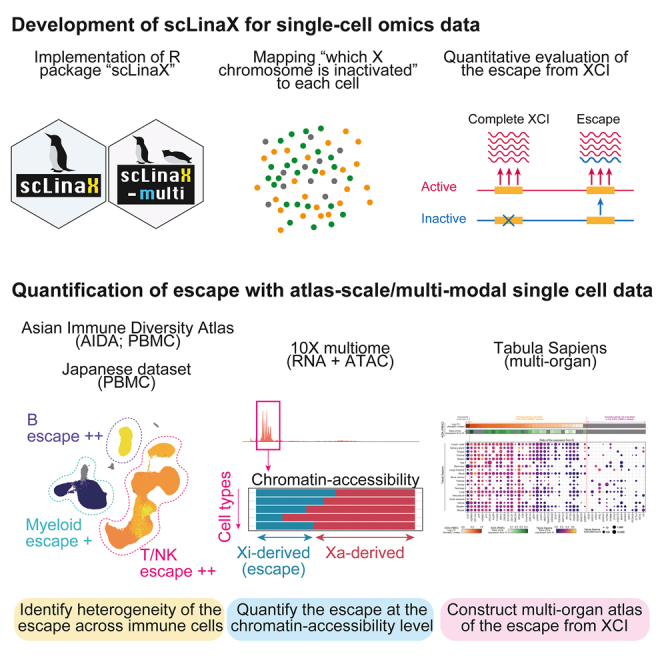
Highlights
-
•
Development of scLinaX software that quantifies escape from XCI with scRNA-seq data
-
•
Lymphocytes showed stronger escape from XCI than myeloid cells
-
•
Extension of scLinaX to multiome can quantify escape at chromatin-accessibility level
-
•
Escape can affect the sex difference of the genotype-phenotype associations
Tomofuji et al. developed scLinaX, a software to quantify escape from X chromosome inactivation (XCI). Their analyses identified the heterogeneity of escape across cell types, namely a stronger escape from XCI in lymphocytes than myeloid cells. scLinaX would be a useful tool for understanding the sex differences in gene regulation.
Introduction
One of the two X chromosomes of females is epigenetically silenced through X chromosome inactivation (XCI) to compensate for the difference in the dosage between sexes. XCI is established on the randomly determined X chromosome in each cell during early embryonic development. Multiple biological processes are involved in XCI, such as upregulation of the non-coding RNA XIST, changes in the histone modifications, and DNA methylation.1 However, several X-linked genes (∼23% of the X-linked genes2) escape from XCI and are then expressed from both active (Xa) and inactive (Xi) X chromosomes.
Expression from Xi due to escape can contribute to sex differences in gene expression and diseases, such as cancer3 and autoimmune diseases.4,5,6 Furthermore, escape can introduce changes in the effective allele dosage of females in the context of genotype-phenotype association analyses7,8,9 (e.g., genome-wide association study [GWAS] and expression quantitative trait locus [eQTL] mapping). This effect has contributed to the technical difficulties in X chromosome analyses, resulting in the exclusion of the X chromosome from GWAS and eQTL analyses, which is one of the current limitations of genetic studies. Therefore, understanding XCI escape is important for elucidating biological sex differences and resolving the current limitation of genetic analysis.10
Whether an X-linked gene escapes XCI has historically been determined by evaluating the heterogeneity of metabolic capacity of female cell lines harboring loss-of-function mutations of X-linked genes encoding metabolic enzymes on one allele.11,12 Subsequently, escape was evaluated for hundreds of genes by analyses of female-derived cell lines with skewed XCI13 (i.e., preferential inactivation of a specific X chromosome) and hybridomas from the human and mouse cells.14 However, concerns remained regarding the generalizability of the findings to physiological conditions within the human body. Although several methods had utilized incomplete XCI skew of the tissue samples for evaluating escape,15,16,17 they were often not sensitive and, moreover, were only compatible with samples showing XCI skew.
Differentially expressed gene (DEG) analysis between sexes was also utilized to investigate escape. For example, DEG analysis of Genotype-Tissue Expression (GTEx) project datasets enabled a comprehensive exploration of escape in a tissue/gene-wide manner.2 Although DEG analysis could identify escape in a physiological condition, it did not directly evaluate escape and it was difficult to separately evaluate the effects of escape and other factors, such as sex-hormonal influences. In addition, previous studies had utilized bulk RNA sequencing (RNA-seq) datasets, so heterogeneity of escape across cell types had not been evaluated.
Recently, the single-cell RNA-seq (scRNA-seq) technology has been utilized to analyze XCI escape through inference of the Xi and in silico generation of the nearly completely skewed XCI condition.2,18,19 Although scRNA-seq analyses enabled direct observation of escape under physiological conditions, current computational methods require high per-cell read depth and are compatible only with plate-based scRNA-seq data (e.g., smart-seq). Due to the plate-based method’s relatively limited throughput, analyses have often been performed with a limited number of samples and cells, and the heterogeneity of escape across different cell types has remained unexplored. Given that the droplet-based approach (e.g., 10x Genomics) is high throughput and currently the most widely used method, the development of a computation method compatible with the 10x dataset is necessary to fully utilize the growing number of publicly available datasets and expand the knowledge of escape across multiple cell types.
Here, we investigated escape across immune cell types utilizing the ∼1,000,000 cell-scale 10x peripheral blood mononuclear cells (PBMCs) scRNA-seq datasets. We performed pseudobulk and single-cell-level DEG analysis to evaluate escape across cell types. To directly and quantitatively evaluate escape, we developed a method, single-cell-level inactivated X chromosome mapping (scLinaX), which identified heterogeneity of escape across cell types. We also developed an extension for the multiome (RNA + assay for transposase-accessible chromatin [ATAC]) dataset, scLinaX-multi, to evaluate escape at the chromatin-accessibility level. Our scLinaX analysis with a multi-organ dataset, Tabula Sapiens,20 identified the heterogeneity of escape across tissues and cell types. Finally, utilizing the quantitative estimates of escape, we evaluated the effect sizes of sex-stratified eQTL and GWAS analysis to understand how escape would affect the results of the genotype-phenotype association analyses. scLinaX and scLinaX-multi are publicly available as an R package (https://github.com/ytomofuji/scLinaX).
Results
Pseudobulk and single-cell-level DEG analysis from the scRNA-seq data of PBMCs
To investigate escape in immune cells, we generated scRNA-seq data of PBMCs derived from healthy Asian subjects as a part of the Asian Immune Diversity Atlas (AIDA) project (Figure 1A; Table S1; 498 individuals, 896,511 cells; AIDA).21 We also utilized previously published PBMC scRNA-seq data (Figure S1A; Table S1; 147 individuals, 865,238 cells) derived from COVID-19 patients and healthy subjects of Japanese ancestry.22,23
Figure 1.

Pseudobulk and single-cell-level differentially expressed gene analyses suggested escape from XCI across immune cells
(A) The scRNA-seq datasets used in this study.
(B) The DEG analysis methods used in this study (STAR Methods, Data S1).
(C) A boxplot represents log2 fold changes in the gene expression between sexes. Genes are grouped according to the XCI status annotated in the previous study.2
(D) A heatmap represents differential gene expression between sexes. The colors of the tiles represent log2 fold changes in the gene expression between sexes. Only genes that satisfied Bonferroni-corrected significance thresholds at least in one cell type are shown. ∗p < 0.05. ∗∗Per-cell-type false discovery ratio (FDR) <0.05. ∗∗∗Bonferroni-corrected p < 0.05.
(E) A boxplot represents log2 fold changes of the escapee gene expression between sexes across cell types.
(F) Scatterplots represent pairwise comparisons of the log2 fold changes of the escapee gene expression between sexes. The y axes represent the log2 fold changes in monocytes and the x axes represent the log2 fold changes in lymphocytes. The dashed lines represent x = 0, x = y, and y = 0.
(G and H) UMAPs represent the per-cell effect sizes of the sex in the single-cell-level DEG analysis calculated as a sum of the effect sizes of sex and sex × batch-corrected PCs (STAR Methods, top) and gene expression (bottom). Genes that show a stronger degree of escape in lymphocytes than monocytes (G) and other patterns of heterogeneity of effect sizes (H) are indicated. The p values for the interaction between sex and batch-corrected PCs were <1 × 10−200 (G) and 1.5 × 10−12 (H). DEG, differentially expressed genes; PC, principal component; PAR, pseudoautosomal region; PBMC, peripheral blood mononuclear cells; scRNA-seq, single-cell RNA-seq; UMAP, uniform manifold approximation and projection; XCI, X chromosome inactivation.
To evaluate escape from XCI across immune cell types, we performed DEG analysis between sexes for each cell type (Figure 1B). Cell types with a large number of cells tended to have a large number of significant DEGs (Figure S1B; Table S2). X-linked genes were enriched among the significant DEGs (pFisher < 0.05/11 and pFisher < 0.05/8 across cell types, respectively, for the two datasets; Figure S1C). The results of the DEG analyses were consistent across the two datasets (Figures S1D and S1E). We compared the effect sizes of the X-linked genes in the DEG analysis across the XCI status defined in the previous study2 and confirmed that known escapee genes tended to have larger effect sizes than other classes of X-linked genes (Figures 1C, S1F, and S1G). Consistent with the previous study,2 the DEG profile of the X-linked genes is often shared across immune cells (Figure 1D). However, lymphocytes tended to show larger effect sizes than myeloid cells, suggesting differences in the degree of escape from XCI among immune cells (Figures 1E, 1F, S1H, and S1I).
To further elucidate the heterogeneity of the female-biased expression of escapee genes among immune cells, we performed single-cell-level DEG analysis. We used batch-corrected PCs as proxies for continuous cell state and evaluated the interaction between the sex and cell state using a negative binomial model (Figure 1B; STAR Methods). Significant cell-state-interacting sex-biased expression was frequently observed for escapee genes (Figure S2A). The negative binomial model was well calibrated and the results were consistent across the two datasets (Figures S2B–S2D). Larger effect sizes were observed for the lymphocytes in comparison to the myeloid cells for the representative escapee genes (Figure 1G). On the other hand, some of escapee genes, such as the protein kinase, X-linked (PRKX) gene, showed different patterns of heterogeneity of the effect sizes (Figure 1H). Overall, heterogeneity of escape across immune cell types, namely the relatively strong degree of escape in lymphocytes, was suggested from the DEG analysis.
scLinaX can directly evaluate escape from 10x scRNA-seq data
To directly validate the evidence of the heterogeneity of escape, which was indirectly suggested by the DEG analysis, it would be advantageous to directly quantify escape from XCI, namely gene expression from Xi. 10x scRNA-seq information could be useful for the analysis of escape because single-cell-level information enabled us to treat cells with different inactivated X chromosomes separately, while such a method had not been implemented previously due to the sparse nature of 10x scRNA-seq data. Therefore, we developed a method, scLinaX, which is compatible with the 10x scRNA-seq data (Figure 2A; Data S1; STAR Methods). In scLinaX analysis, samples derived from different individuals are processed separately. First, pseudobulk allele-specific expression (ASE) profiles are generated for cells expressing each candidate reference single-nucleotide polymorphism (SNP). Then, alleles of the reference SNPs on the same X chromosome are listed by correlation analysis of the pseudobulk ASE profiles. Finally, scLinaX assigns which X chromosome is inactivated to each cell based on the allelic expression of the reference SNPs and generates a nearly complete XCI skewed condition in silico and the estimates for the ratio of the expression from Xi.
Figure 2.

scLinaX, a method to quantify escape from XCI using droplet-based scRNA-seq data
(A) A schematic illustration of scLinaX.
(B) A boxplot represents the estimated ratio of the expression from Xi. Genes are grouped according to the XCI status annotated in the previous study.2
(C) A plot represents the concordance of the ratio of the expression from Xi between the AIDA dataset (x axis) and the Japanese dataset (y axis). Genes that are annotated as escapee genes and the SEPTIN6 gene are included. The black line indicates x = y. Pearson’s correlation = 0.92 with a 95% confidence interval (CI) of 0.82–0.97.
(D) A plot represents the relationship between the log2 fold changes in the DEG analysis (x axis) and the ratio of the expression from Xi (y axis). Genes that are annotated as escapee genes and the SEPTIN6 gene are included. The curved line indicates the theoretical relationship under the assumption that total gene expression in males and Xa-derived gene expression in females are at the same level. Pearson’s correlation = 0.94 with a 95% CI of 0.87–0.97.
(E) A plot representing the ratio of the expression from Xa and Xi at an individual level for the DDX3X gene. The dashed black horizontal line represents the mean ratio of the expression from Xi across samples.
(F and G) Forest plots represent the log2 fold changes in the DEG analysis for each cell type (left) and plots on the right represent the ratio of the expression from Xa and Xi at an individual level. The error bars indicate 95% CI. The colors of the dots represent the log-scaled mean normalized count calculated by DEseq2 (baseMean). ∗p < 0.05. ∗∗Per-cell-type FDR <0.05. The dashed black horizontal line represents the mean ratio of the expression from Xi across samples. AIDA, Asian Immune Diversity Atlas; ALT, alternative allele; ASE, allele-specific expression; REF, reference allele; SNP, single-nucleotide polymorphism; Xa, active X chromosome; Xi, inactive X chromosome.
We applied scLinaX to the PBMC scRNA-seq data and SNP array data and found that previously identified escapee genes tended to show a higher ratio of the expression from Xi than other classes of genes, suggesting that scLinaX had worked successfully (Figures 2B and S3A–S3G; Tables S3, S4, S5, and S6). We also performed the analysis based on the SNP data called from scRNA-seq data and the results were almost consistent with the results based on the SNP array data (Figures S3A–S3J), suggesting that scLinaX would also be useful when germline genotype data were not available. While genotype calls from scRNA-seq data were generally accurate, utilization of the SNP array is expected to yield more accurate and conservative results (Figures S3K and S3L; Data S2). Therefore, we prioritized analyses using both SNP array data and scRNA-seq data whenever SNP array data were available. There was no association between the gene expression level and scLinaX estimates for the escapee genes (Figure S3M). The scLinaX estimates were consistent between the two datasets, suggesting the robustness of the scLinaX analysis (Figure 2C and S3N). In the scLinaX analysis with down-sampling, the number of cells that were mapped with the inactivated X chromosome and the number of the genes that could be included in the analysis increased as the cell number and unique molecular identifier (UMI) count per cell increased (Figures S4A–S4D). Also, the higher the cell number and UMI count per cell were, the higher the observed correlation with the full dataset, while the correlations were overall high in all conditions (Figures S4E–S4G). We observed agreement of phase information inferred from scLinaX and derived from the imputed SNP array data when the distance between SNPs was not so far as to cause switch errors, suggesting the high accuracy of the phase information obtained through scLinaX analysis (Figures S4H and S4I). We also observed agreement between the phase information from scLinaX and PacBio HiFi long-read sequencing (mean coverage = 16.0×), again suggesting the high accuracy of the scLinaX-based phasing (concordant for 83/83 [100%] pairs of SNPs; Figure S4J).
The relationship between the effect sizes of the DEG analysis and the ratio of the expression from Xi estimated by the scLinaX was compatible with the assumption that differential gene expression between sexes is due to the expression from Xi (Figures 2D and S5A; the ratio of the expression from Xi [y axis] = 1 − 1/2log2 fold change [x axis]). In the scLinaX analysis, SEPTIN6 was not annotated as an escapee gene in the previous study2; it showed a relatively high ratio of expression from Xi and female-biased expression, suggesting that SEPTIN6 was thought to actually be an escapee gene as recently reported.17,24 Also, there existed genes that showed female-biased expression in the DEG analysis but had a low ratio of expression from Xi. For example, the CD40 ligand (CD40LG) gene was a female-biased DEG in the PBMC analysis but its ratio of the expression from Xi was low compared to escapee genes such as the DDX3X (Figures 2E, 2F, and S5B). CD40LG was highly expressed by CD4 T cells, but it was not a DEG in the pseudobulk analysis of CD4 T cells, suggesting that it was detected as a DEG due to the confounding effect of the relative subset composition of CD4 T cells, not escape (Figures 2F and S5C). The ITM2A gene was also detected as a significant female-biased DEG in the PBMC analysis while the ratio of the expression from Xi was low (Figures 2G, S5B, and S5C). Since ITM2A showed significant female-biased expression in the per-cell-type DEG analysis, it might be that female-biased ITM2A expression was due to other factors, such as sex-hormonal effects. Considering these examples, scLinaX would be useful to directly evaluate escape and complement the limitation of the DEG analysis.
Quantification of escape across cell types by scLinaX
Next, we evaluated escape by scLinaX as a ratio of the expression from Xi for each cell type (Figure S6A; Tables S4, S5, and S6). Consistent with the results of the DEG analysis, lymphocytes tended to have a higher ratio of expression of the escapee genes from Xi than monocytes (Figures 3A, 3B, and S6B–S6D). When per-cell-type estimates from scLinaX were projected onto the uniform manifold approximation and projection (UMAP), the gradients of the ratio of expression from Xi showed the same pattern as those from the single-cell-level DEG analysis (Figures 1G, 3C, 3D, S6E, and S6F). Although cell or organ specificity of escape for a few genes had been suggested,2,6 consistent differences in the strength of escape across several escapee genes, namely stronger escape in lymphocytes than in monocytes, have not previously been reported. In addition, the PRKX gene, which showed an atypical pattern of the heterogeneity of the effect sizes in the DEG analysis, also showed gradients of the ratio of the expression from Xi with the same pattern as those from the single-cell-level DEG analysis (Figures 1H, 3D, S6G, and S6H). Considering the clear relationship between the results of DEG and scLinaX analyses in the bulk PBMC analysis (Figure 2D), these findings suggested that the inter-cell-type heterogeneity of escape quantified by scLinaX contributed to the heterogeneity of sex differences in gene expression across cell types. We also evaluated the effects of genetic variants on the degree of escape (escape quantitative trait locus [QTL] analysis) but could not find significant associations (Figures S6I and S6J), although future analyses with larger sample sizes may find escape QTLs.
Figure 3.

The scLinaX-based quantification of escape from XCI across immune cell types
(A) A boxplot represents the estimated ratio of the expression from Xi for escapee genes across cell types.
(B) Scatterplots represent pairwise comparisons of the ratio of the expression from Xi for escapee genes. The dashed lines represent x = 0, x = y, and y = 0.
(C) UMAPs colored according to the ratio of the expression from Xi estimated for each cell type. Representative genes that showed a higher ratio of expression from Xi in lymphocytes than monocytes, the DDX3X and EIF2S3 genes, are indicated. Cell types whose ratio of the expression from Xi could not be estimated are colored gray.
(D) The ratio of the expression from Xa and Xi at an individual level for the DDX3X and EIF2S3 genes. The dashed horizontal line represents the mean ratio of the expression from Xi across samples for each cell type. Since the definition of alleles derived from Xa and Xi is consistent within the same individual, the ratio of expression from Xi may exceed 0.5 in some cell types.
(E) A UMAP colored according to the ratio of the expression from Xi estimated for each cell type. The PRKX gene, which shows a unique pattern of heterogeneity of escape across cell types, is indicated.
(F) The ratio of the expression from Xa and Xi at an individual level for the PRKX gene.
Evaluation of the differential escape in disease conditions
It was reported that some autoimmune-disease-associated genes, e.g., in systemic lupus erythematosus (SLE), were escapee and that escape of such genes could be enhanced in patients with SLE.4,5,6,25 Despite the potential association between escape and diseases, X chromosome-wide evaluation of escape in diseased individuals had not been performed. We analyzed the changes in escape in two diseases, COVID-1922 and SLE,26 based on the scLinaX estimates. After multiple-test correction, we could not detect a significant association, possibly because of the lack of power, suggesting the need for future larger cohort analyses (Figures S7A and S7B; Table S7; Data S3). We also evaluated escape in a male sample with an XXY karyotype and the escape status was almost consistent with that of healthy females (Figure S7C; Table S8).
scLinaX-multi can evaluate escape at the chromatin-accessibility level
XCI escape, which we had observed at the transcription level, was closely linked to gene regulation at the chromatin level. XCI induces chromatin-level transcriptional repression on Xi, while a transcriptionally active chromatin state on Xi can be observed under escape from XCI. Although previous studies had demonstrated escape at the chromatin level through the comparative analyses between sexes27 and allele-specific epigenetic investigations using cell lines,28 the chromatin-level escape had not been directly quantified under physiological conditions. To directly quantify the chromatin-level escape, we developed an extension of scLinaX for multi-modal single-cell data (RNA + ATAC), scLinaX for multi-modal data (scLinaX-multi; Figure 4A; Data S4; STAR Methods). In multi-modal single-cell data, each cell has both RNA and ATAC information. scLinaX-multi utilizes allelic RNA expression information to estimate which X chromosome is inactivated for each cell, as is done in the scLinaX analysis. For the cells in which the inactivated X chromosome has been successfully identified based on the RNA information, allelic ATAC information is utilized to calculate the ratio of the accessible chromatin derived from Xi, namely escape at the chromatin-accessibility level.
Figure 4.

scLinaX-multi, a method to estimate the chromatin accessibility of Xi from multi-modal single-cell omics data
(A) A schematic illustration of the scLinaX-multi (Data S4; STAR Methods).
(B) Boxplots represent the estimated ratio of the accessible chromatin derived from Xi for peaks within 2 kbp of TSS (left) and ≥2 kbp distant from TSS (right). Peaks are grouped according to the XCI status of the nearest gene.
(C) A plot representing the relationship between the ratio of the expression from Xi (RNA level, x axis) and the ratio of the accessible chromatin derived from Xi (y axis) for each peak-nearest gene pair. Genes that are annotated as escape genes or showed evidence of escape in the scLinaX analysis (ratio of the expression from Xi > 0.15) are indicated. The black line indicates x = y. When a single gene has multiple peaks, the average across the peaks for the ratio of the Xi-derived accessible chromatin is used for the calculation of Pearson’s correlation.
(D) Scatterplots represent pairwise comparisons of the accessible chromatin derived from Xi for peaks whose nearest genes are escapee genes. The y axes represent the ratio of the expression from Xi in monocytes and the x axes represent the ratio of the expression from Xi in lymphocytes. The dashed lines represent x = 0, x = y, and y = 0. The p values are calculated by the Wilcoxon signed-rank test.
(E–G) The results of the scLinaX-multi for the representative peaks around escapee genes, namely DDX3X (E), USP9X (F), and ZRSR2 (G). Normalized tag counts across cell types are indicated with peak information (top). The ratio of the accessible chromatin derived from Xa and Xi across cell types is indicated as bar plots (bottom) with information on which SNPs are used for the analysis. Since the definition of alleles derived from Xa and Xi is consistent within the same individual, the ratio of expression from Xi may exceed 0.5 in some cell types. ATAC, assay for transposase-accessible chromatin; TSS, transcription start site.
We applied scLinaX-multi to the publicly available PBMC multiome datasets from a female and found that peaks whose nearest genes were escapee genes tended to show a higher ratio of the accessible chromatin derived from Xi than other classes of peaks, suggesting that scLinaX-multi had worked successfully (Figures 4B and S8A–S8E; Table S9). The correlation between the ratio of the accessible chromatin derived from Xi (ATAC) and the ratio of the expression from Xi (RNA) for peak-nearest gene pairs, while strongly positive, was not significant for the escapee genes in PBMCs (Figures 4C and S8F; Pearson’s correlation = 0.57 and p = 0.066 in AIDA RNA vs. 10x multiome ATAC; Pearson’s correlation = 0.62 and p = 0.055 in 10x multiome RNA vs. 10x multiome ATAC). The ratio of the accessible chromatin derived from Xi was nominally higher in lymphocytes than in monocytes (Figure 4D, pWilcoxon-signed < 0.05 in CD4+ T cells vs. monocytes and CD8+ T cells vs. monocytes). For example, peaks at the transcription start sites (TSSs) of the escapee genes DDX3X, USP9X, and ZRSR2 showed a higher ratio of accessible chromatin derived from Xi in lymphocytes than in monocytes (Figures 4E–4G). In addition, we found chromatin-level escape at the myeloid cell-specific enhancer in the ZRSR2 gene locus, which was also defined as a cis-regulatory element (cCRE) in the Encyclopedia of DNA Elements (ENCODE) project (EH38E3926410).29 We could not observe such signs of escape at the chromatin level within peaks around the non-escapee genes (Figures S8G–S8I). In summary, scLinaX-multi could be useful in identifying chromatin-level escape and its heterogeneity across cell types.
Direct quantification of escape across multi-organs with scLinaX
To evaluate the heterogeneity of escape beyond blood cells, we applied scLinaX to Tabula Sapiens,20 the current largest publicly available human multi-organ scRNA-seq dataset in terms of number of cells and organs20 (https://tabula-sapiens-portal.ds.czbiohub.org). Although the Tabula Sapiens dataset did not contain genotype data, scLinaX could be applied to datasets without genotype data (Figures S3A–S3L). Data from six females were included in the analysis, and known escapee genes showed relatively high scLinaX estimates across the organs (Figures 5A and S9A–S9G; Table S10), consistent with the previous study.2 To evaluate the heterogeneity of escape across organs, we performed pairwise comparisons of the ratio of the expression from Xi and found that lymphoid tissues, such as lymph node, thymus, and spleen, had a relatively high ratio of the expression from Xi (Figures 5B and 5C).
Figure 5.

Quantitative evaluation of escape from XCI with a human multi-organ atlas of single-cell transcriptome data
(A) The ratio of the expression from Xi across organs from the Tabula Sapiens dataset (y axis) for escapee genes (x axis). (The XIST gene is the exception, showing the expression from Xa.) The color and size of the dots represent the ratio of the expression from Xi and the total allele count. Heatmaps above the dot plot represent the log2 fold change of gene expression between sexes (orange) and the ratio of the expression from Xi (green) calculated from the AIDA dataset. The heatmap on the right of the dot plot represents the number of cells used for the scLinaX analysis across organs and samples.
(B) The results of the pairwise comparison of the ratio of the expression from Xi across organs. The color of the dots represents the ratio of the genes whose ratio of the expression from Xi is higher in organ 1 (y axis) than in organ 2 (x axis). The size of the dots represents the number of genes used for each comparison. The bar plots on the right of the dot plot represent the numbers and types of the cells that were used for the scLinaX analysis.
(C) The pairwise comparisons of the ratio of the expression from Xi for escapee genes. The y- and x axes represent the ratio of the expression from Xi in lymphoid tissues and organs with a relatively weak degree of escape, respectively. Since these organs are commonly evaluated in a sample TSP2, data from TSP2 are presented. The dashed line represents x = y. The numbers in each plot indicate the number of genes that are located in the x > y (lower right, blue) and x < y (upper left, red).
(D) The results of the pairwise comparison of the ratio of the expression from Xi across cell types.
(E) The pairwise comparisons of the ratio of the expression from Xi for each escapee gene and individual. The y axes represent the ratio of the expression from Xi in immune cell types (top, lymphoid; bottom, myeloid) and the x axes represent the ratio of the expression from Xi in other cell types. The color of the points represents each sample.
In our analyses of PBMCs, we found that lymphocytes showed relatively strong escape compared to myeloid cells. Therefore, we hypothesized that the relatively high ratio of the expression from Xi observed in lymphoid tissues was due to their high lymphocyte content. Consistent with the hypothesis, a higher ratio of the expression from Xi was observed for the lymphocytes in the pairwise comparisons of the ratio of the expression from Xi across cell types in the Tabula Sapiens dataset (Figures 5D and 5E; Table S11). In summary, scLinaX analysis suggested a tissue-level escape heterogeneity linked to cell-type-level escape heterogeneity.
A difference in the genetic effects on the complex traits was observed at the escapee gene loci
Although genetic association studies such as GWAS and eQTL mapping have successfully identified the genetic backgrounds of human traits, the sex-associated difference is one of the remaining unresolved issues. Specifically, the X chromosome has often been excluded from these analyses due to technical difficulties, despite its apparent importance in the context of sex-associated differences.10 One of these difficulties is the potential need to adjust the dosage differences between males and females dependent on the degree of escape for obtaining the per-allele estimate of the GWAS effect sizes. For example, previous literature suggested that the effective dosage of the alleles should be 0/2 for males and 0/1/2 for females under the complete XCI and 0/1 for males and 0/1/2 for females under the complete escape.8 On the other hand, a previous study showed that the inter-sex differences in the eQTL effects of escape genes were consistent with complete XCI rather than escape in most cases.7 Therefore, we evaluated the effects of escape on the sex differences of the genotype-phenotype association analyses with the quantified catalog of escape.
First, to evaluate the effects of escape on the eQTL analysis, we performed eQTL mapping with all samples from the AIDA dataset (allele dosages of the males and females were 0/2 and 0/1/2, respectively) and found 202 significant eQTL signals across 10 cell types (Table S12; p < 5 × 10−8). These eQTL signals were highly reproducible in the analysis with the Japanese dataset (Figure S10A; Table S13). Then, we performed eQTL mapping separately for males and females and compared the effect sizes of the significant eQTLs on the X chromosome between sexes. We did not observe apparent female-biased effect sizes across all the XCI statuses including escapees (Figures 6A and S10B). In addition, there was no clear relationship between the sex-associated differences in effect sizes and the degree of escape quantified by the DEG and scLinaX analyses (Figures 6B and S10C). These results are consistent with a previous eQTL study7 but inconsistent with other studies utilizing ASE or DEG analyses2,13 and with the results of the DEG and scLinaX analyses in this study. We speculate that the sex differences in effective allele dosage caused by escape do not cause sex differences in the eQTL effect because of the transformation of the expression data, such as log transformation, which stabilizes variance and resolves heteroskedasticity (Figure S10D).
Figure 6.

Detection of differential effect sizes between sexes in the genotype-phenotype association analysis
(A) The effect sizes of the significant eQTL signals (p < 5 × 10−8) in the female-only (x axis) and male-only (y axis) analyses, separately for each XCI status. The error bars indicate standard errors. The color of the plots indicates the cell type in which the eQTL signals are identified. The oblique lines correspond to the female/male effect size ratios described in the plots. The bar plots in the lower right of each plot indicate the number of eQTL signals that have larger effect sizes in females (left) and males (right).
(B) Scatterplots for escapee genes (A, upper left) are colored according to the estimated female/male effect size ratio based on the DEG analysis (top) and scLinaX analysis (bottom). Genes that were not evaluated in the scLinaX analyses are colored gray.
(C) The association between PRKX gene loci and lymphocyte counts in the BBJ analysis, the UKB analysis, and the BBJ + UKB meta-analysis. The rs6641874 (top variant in the BBJ + UKB meta-analysis and T cells eQTL analysis) and rs6641601 (top variant in the monocytes eQTL analysis) are colored purple and green, respectively. Genes located around the PRKX gene region are indicated at the bottom of the plots.
(D) Locus plots for the eQTL analysis of the PRKX gene across cell types. R2, a measure of linkage disequilibrium (LD) to the rs6641874, is indicated by the color of the dots. Results of the colocalization analyses (PP.H4) with lymphocyte count GWASs in BBJ are indicated in the upper right of the plots.
(E) UMAPs represent the per-cell eQTL effect sizes of the variants in the single-cell-level eQTL analysis calculated (STAR Methods). Associations for PRKX genes rs6641874 (top) and rs6641601 (bottom) are indicated. The p values for the interaction between genotypes and batch-corrected PCs were 2.7 × 10−91 (top) and 2.4 × 10−51 (bottom).
(F) The effect sizes of the rs6641874 in the female-only (x axis) and male-only (y axis) lymphocyte count GWAS analyses in each cohort. The error bars indicate standard errors.
(G) The female/male effect size ratios of the rs6641874 in the lymphocyte count GWAS analyses in each cohort. The error bars indicate 95% CI.
Next, we evaluated the effects of escape on the genotype-phenotype association using the two independent biobank datasets. To focus on the association signals mediated by the expression of escapee genes, we evaluated the association between the eQTL variants and blood-related traits using the BioBank Japan (BBJ) dataset (N = 82,228–161,145; Tables S14 and S15).30,31 Nine associations satisfied the significance threshold, of which only an association between the eQTL variant for PRKX (escapee gene) and lymphocyte counts was replicated in the analysis of the UK Biobank (UKB) dataset (Figures 6C, S11A, and S11B; Table S15; http://www.nealelab.is/uk-biobank/). Pseudobulk and single-cell-level eQTL analyses revealed that two different eQTL signals existed in this region, namely a T/NK cell-specific one and a myeloid cell-specific one, and only the T/NK cell-specific eQTL signal colocalized with the GWAS signal (Figures 6D and 6E). Neither of the eQTL signals showed a difference in the effect sizes between sexes (Figure S11C). Interestingly, this locus was suggested to be associated with white blood cell counts via PRKX expression in a female-biased manner in a previous report on the UKB analysis.7 Given the results of the per-cell-type and single-cell-level eQTL analysis, this locus could affect the white blood cell counts via its effects on the lymphocytes. Then, we evaluated the effect sizes of the PRKX gene loci-lymphocyte counts association in each sex and found that effect sizes were significantly larger in females than in males (Figures 6F and 6G; Table S16). Although it was difficult to generalize the finding from a single locus, this result might be evidence for the effect of escape on the difference in the GWAS effect sizes between sexes.
Discussion
In this study, we quantitatively evaluated escape from XCI across multiple cell types with large-scale immune cell and multi-organ scRNA-seq datasets. The scLinaX method enabled us to directly evaluate escape across cell types, and both the DEG and scLinaX analyses revealed a stronger degree of escape in lymphocytes than in myeloid cells. We also implemented an extension of scLinaX for the multi-modal dataset, scLinaX-multi, and revealed a stronger degree of escape in lymphocytes at the chromatin-accessibility level. We also applied scLinaX to the multi-organ dataset, Tabula Sapiens, and found that lymphatic tissues and lymphocytes showed a stronger degree of escape in comparison to other tissues and cell types. Finally, we presented an example of how escape might have affected sex differences in genotype-phenotype association through the single-cell eQTL analysis and GWAS with two biobank datasets.
scLinaX is a method that enables direct observation of escape at the cell-cluster level, and its applicability to 10x data makes it highly versatile. Because 10x scRNA-seq data are sparser than plate-based scRNA-seq methods such as smart-seq, single-cell-level ASE profiles generated from 10x data are difficult to handle in the same way as plate-based scRNA-seq data. scLinaX resolves the technical difficulty associated with the sparsity of the data by generating pseudobulk ASE profiles for each SNP on the X chromosome and aggregating alleles on the same X chromosome based on the correlation of the pseudobulk ASE profiles of the SNPs. Since the raw output from scLinaX is single-cell-level data, it is possible to evaluate escape in any user-defined cluster, including cell types. This unique feature of scLinaX is useful for evaluating the heterogeneity of escape across various kinds of cells. Since scLinaX can quantify escape at individual levels, which cannot be achieved by DEG analysis, it can also be useful for evaluating the inter-individual differences of escape as long as the measurement errors due to the sparsity of scRNA-seq data are correctly considered.
scLinaX can map which X chromosome is inactivated for each cell based on the single-cell-level transcriptome data, and this information is also useful for evaluating escape at levels other than the transcriptome level, as demonstrated by the scLinaX-multi analysis with the 10x multiome dataset (RNA + ATAC). In addition to RNA + ATAC, single-cell joint measurements of RNA + other modalities, such as histone modifications,32 are currently being developed. Such technologies can enable us to directly observe escape at the level of the various X chromosome regulations, which will be useful to elucidate the biological mechanisms of escape.
Through a series of analyses, we identified a unique feature of the lymphocyte, a relatively strong degree of escape. In a previous analysis utilizing cell imaging, it was revealed that lymphocytes, especially naive ones, had an abnormally dispersed distribution of XIST RNA and reduced normal heterochromatin histone modifications.5,6 These results suggested that there may be a unique mode of the regulation of XCI in lymphocytes at the chromosome scale. In addition, a relatively strong degree of escape in lymphocytes may also be related to the sex differences in immune phenotype, which could be linked to the higher prevalence of autoimmune diseases in females33 and Klinefelter syndrome patients, where males have an extra X chromosome.34
How we should handle the allele dosage for males and females and whether allele dosage should be adjusted in the presence of escape is one of the technical difficulties associated with X chromosome analysis.8,9 Currently, many GWAS software, such as PLINK2,35 BOLT-LMM,36 and REGINIE,37 handle the dosage of alleles assuming the complete XCI as a default setting, while previous literature argued that, in the presence of escape, the effective dosage in the female should increase.8,9 In our comparisons of the eQTL effect sizes between sexes, we found no inter-sex differences in eQTL effects regardless of the quantified estimates of escape. Hence, it might be the case that the effective dosage between sexes could be explained by the sex term in a linear regression model, suggesting that it might not be necessary to alter the scale of the genotype term in the eQTL analysis of females (Figure S10D).
However, this holds true only for a limited trait, such as gene expression, and does not apply to more complex traits contributed by multiple genes. Indeed, in this study, the PRKX gene locus was associated with lymphocyte count likely via its eQTL effect in the lymphocytes, and the effect was larger in females than in males. This difference in the effect sizes between sexes might be linked to the increase in allele dosage and PRKX expression in females due to escape. Although the limited number of GWAS signals associated with the escapee gene and the complexity of the mode of genotype-phenotype associations made it difficult to generalize how escape affects the sex difference of the GWAS signal, it would be important to perform GWAS with care for the inter-sex heterogeneity (e.g., sex-stratified analysis8). Although the X chromosome has often been excluded from the largest-scale GWAS meta-analyses due to technical difficulties,38,39 there is a need to actively conduct GWAS of the X chromosome, share sumstats, and promote secondary use in order to overcome this technical difficulty.
In summary, we developed scLinaX, a method to directly evaluate escape at the cell-cluster level. We believe that scLinaX and the quantified catalog of escape identified the heterogeneity of escape across cell types and tissues and will contribute to expanding the current understanding of the XCI, escape, and sex differences in gene regulation.
Limitations of the study
Evaluation of the functional effects of the heterogeneity of escapes on cell phenotypes was out of the scope of this study because it is still technically difficult to manipulate escape from XCI.
Since scLinaX is derived from ASE analysis, it inherits the general limitations of ASE analysis, such as the requirement for transcribed SNPs and sufficient read coverage. Therefore, only samples with transcribed SNPs can be included in the scLinaX analysis, which might decrease the power of the case-control comparisons of escape from XCI (Figure S7). Also, it is still difficult to directly quantify escape for all the expressed genes, especially for rare cell populations with poor total read coverages and genes (Figure S6A; Table S6). We believe that future expansion of the scRNA-seq datasets or new technologies such as long-read scRNA-seq40 will be promising to address these limitations.
While we have evaluated escape across blood cells with the current largest-scale datasets, some datasets (e.g., Tabula Sapiens and 10x multiome) have fewer samples compared to such PBMC datasets. This is because there are currently no available large-scale datasets for human multi-organ scRNA-seq data or 10x multiome, which is considered a limitation of current single-cell omics research. We believe that cooperative efforts on a community level, such as the Human Cell Atlas,41 are necessary to address this limitation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Human DNA extracted from blood | This study | N/A |
| Human peripheral blood mononuclear cells | This study | N/A |
| Deposited data | ||
| Genotype data of BioBank Japan | Nagai et al.42 | Japanese Genotype-phenotype Archive of Biobank Japan (JGA) with the accession ID JGAS000412, which is available through application at https://humandbs.biosciencedbc.jp/en/hum0311-latest |
| Genome-wide genotype imputation reference panel | Akiyama et al.43 | Japanese Genotype-phenotype Archive (JGA) with the accession ID JGAS000114, which is available through application at https://humandbs.biosciencedbc.jp/en/hum0014-latest |
| Whole-genome sequencing data of a general Japanese population | Okada et al.44 | Japanese Genotype-phenotype Archive (JGA) with the accession ID JGAD000220, which is available through application at https://humandbs.biosciencedbc.jp/en/hum0014-latest |
| Allele frequency reference panel of Tohoku Medical Megabank Project | Tadaka et al.45 | https://jmorp.megabank.tohoku.ac.jp/downloads |
| Japanese PBMC scRNA-seq dataset | Edahiro et al.22 | Japanese Genotype-phenotype Archive (JGA) with the accession ID JGAS000593/JGAD000722/JGAS000543/JGAD000662, which is available through application at https://humandbs.biosciencedbc.jp/en/hum0197-latest |
| Japanese SNP array data | Edahiro et al.22 | European Genome-Phenome Archive (EGA) with the accession ID EGAS00001006950, which is available through application at EGA |
| pbmc_multimodal.h5seurat | Hao et al.46 | https://satijalab.org/seurat/articles/multimodal_reference_mapping.html |
| PBMC scRNA-seq dataset for SLE patients | Perez et al.26 | GEO accession number GSE17418 |
| PBMC 10x multiome data | 10x Genomics | https://www.10xgenomics.com/resources/datasets/pbmc-from-a-healthy-donor-granulocytes-removed-through-cell-sorting-10-k-1-standard-2-0-0 |
| AIDA scRNA-seq/SNP array dataset | AIDA | https://data.humancellatlas.org/explore/projects/f0f89c14-7460-4bab-9d42-22228a91f185 |
| Tabula Sapiens | Tabula Sapiens Consortium20 | https://tabula-sapiens-portal.ds.czbiohub.org |
| UKB GWAS sumstats | Neale lab | Nealelab/UK_Biobank_GWAS: v2; Zenodo, https://doi.org/10.5281/zenodo.8011558 |
| Software and algorithms | ||
| Annovar | Wang et al.47 | https://annovar.openbioinformatics.org/en/latest/ |
| bcftools | Danecek et al.48 | https://samtools.github.io/bcftools/ |
| Cell Ranger | 10x Genomics | https://www.10xgenomics.com/jp/support/software/cell-ranger |
| cellsnp-lite | Huang et al.49 | https://github.com/single-cell-genetics/cellsnp-lite |
| Coloc | Giambartolomei et al.50 | https://chr1swallace.github.io/coloc/articles/a01_intro.html |
| DESeq2 | Love et al.51 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| DoubletFinder | McGinnis et al.52 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| DRAGEN software | Illumina | https://support.illumina.com/downloads.html |
| edgeR | Robinson et al.53 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| harmony | Korsunsky et al.54 | https://github.com/immunogenomics/harmony |
| harmonypy | Korsunsky et al.54 | https://github.com/slowkow/harmonypy |
| Michigan Imputation Server | Das et al.55 | https://imputationserver.sph.umich.edu |
| Minimac4 | Fuchsberger et al.56 | https://github.com/statgen/Minimac4 |
| pbmm2 | PacificBioScience | https://github.com/PacificBiosciences/pbmm2 |
| Picard | Broad Institute | https://github.com/broadinstitute/picard?tab=readme-ov-file |
| PLINK | Purcell et al.57 | https://www.cog-genomics.org/plink/1.9 |
| PLINK2 | Chang et al.35 | https://www.cog-genomics.org/plink/2.0 |
| Python | Python Software Foundation | https://www.python.org/downloads/release/python-376/ |
| R | The R Foundation for Statistical Computing | https://www.r-project.org |
| RCAv2 | Schmidt et al.58 | https://github.com/prabhakarlab/RCAv2 |
| Scds | Bais et al.59 | https://github.com/kostkalab/scds |
| scLinaX | This study | https://github.com/ytomofuji/scLinaX |
| Scrublet | Wolock et al.60 | https://github.com/swolock/scrublet |
| Seurat | Hao et al.46 | https://satijalab.org/seurat/ |
| SHAPEIT4 | Delaneau et al.61 | https://github.com/odelaneau/shapeit4 |
| Signac | Stuart et al.62 | https://stuartlab.org/signac/ |
| tensorQTL | Broad Institute | https://github.com/broadinstitute/tensorqtl |
| whatshap | Martin et al.63 | https://github.com/whatshap/whatshap |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yukinori Okada (yokada@sg.med.osaka-u.ac.jp).
Materials availability
The materials that support the findings of this study are available from the corresponding authors upon reasonable request. Please contact the lead contact, Yukinori Okada (yuki-okada@m.u-tokyo.ac.jp) for additional information.
Data and code availability
The AIDA Data Freeze v1 gene-cell matrix (1,058,909 cells from 503 Japan, Singaporean Chinese, Singaporean Malay, Singaporean Indian, and South Korea Asian donors and 5 distinct Lonza commercial controls), with BCR-seq and TCR-seq metadata, and donor age, sex, and self-reported ethnicity metadata, is available via the Chan Zuckerberg CELLxGENE data portal at https://cellxgene.cziscience.com/collections/ced320a1-29f3-47c1-a735-513c7084d508. The open-access AIDA datasets are available via the Human Cell Atlas Data Coordination Platform at https://data.humancellatlas.org/explore/projects/f0f89c14-7460-4bab-9d42-22228a91f185. Raw scRNA-seq sequencing data for the Japanese dataset are available at the Japanese Genotype-phenotype Archive (JGA) with accession codes JGAS000593/JGAD000722/JGAS000543/JGAD000662.22,23 All the raw sequencing data of Japanese scRNA-seq dataset can also be accessed through application at the NBDC with the accession code hum0197 (https://humandbs.biosciencedbc.jp/en/hum0197-latest). Genotype data for the Japanese dataset are available at European Genome-Phenome Archive (EGA) with the accession code EGAS00001006950 (https://ega-archive.org/studies/EGAS00001006950). scLinaX and scLinaX-multi is available as an R package from https://github.com/ytomofuji/scLinaX. Original version of scLinaX and scLinaX-multi used in this study are available from Zenodo (https://doi.org/10.5281/zenodo.11023040).
Experimental model and subject details
Subject participation
The Asian Immune Diversity Atlas dataset (v1) was composed of 503 donors of East Asian (Chinese, N = 75; Japanese, N = 149; Korean, N = 165), Southeast Asian (Malay, N = 54), and South Asian (Indian, N = 60) self-reported ethnicities from Japan, Singapore, and South Korea, and five commercially available European ancestry control samples (LONZA 4W-270). A detailed description of the dataset was included in the flagship manuscript of the Asian Immune Diversity Atlas Network.21
The PBMC scRNA-seq data of the Japanese was derived from the previously published study.22 Briefly, peripheral blood samples were obtained from patients with COVID-19 (N = 73) and healthy controls (N = 75) at Osaka University Hospital. Almost all cases were patients who were transferred from nearby general hospitals because of severe or potentially severe illness during treatment and already initiated with systemic corticosteroid therapy at other hospitals. We also used a male sample with a karyotype of XXY who was also in the remission phase of multiple sclerosis. The sample was collected at Osaka University Hospital in the same manner as the Japanese dataset.
Method details
Generation and pre-processing of the AIDA PBMC scRNA-seq data
The methods for generation and pre-processing of the AIDA PBMC scRNA-seq dataset (v1) are described in the flagship manuscript of the Asian Immune Diversity Atlas Network.21 Briefly, single-cell RNA-seq for PBMC was performed with 10x Genomics Chromium Controller and 10x Genomics Single Cell 5′ v2 chemistry. We used the DRAGEN Single-Cell RNA pipeline in the Illumina DRAGEN v3.8.4 software (version 07.021.602.3.8.4–20-g74395e76) for pre-processing and genetic demultiplexing. We performed quality control of our dataset in two stages.
We first performed library-level quality control. We started by filtering out cells for which fewer than 300 genes were detected. We then identified the top 2,000 highly variable features using the variance-stabilizing transformation option in Seurat,46 scaled the data using all genes, and then performed principal component analysis on these highly variable features. We performed nearest-neighbor analyses based on the resulting principal components, and ran Louvain clustering in Seurat at a resolution of 1.0. We annotated the resulting clusters based on a majority vote of the major cell type annotation labels assigned by RCAv2 software58 to cells within each cluster. We used the genetic doublet proportion for a library (proportions of mixed genetic identity + ambiguous identity droplets) to estimate the likely total doublet rate for that library.64 We used this estimate of total doublets in a library, as well as the RCAv2 reference projection-based annotation of clusters (for estimation of homotypic doublet proportion) as part of our input into DoubletFinder,52 which we used for identifying heterotypic doublets. We then removed cells that had more than 10 (HBA1 UMIs + HBB UMIs), since these cells could be red blood cells, or cells contaminated with red blood cell RNA transcripts.
Then, we performed cell type-specific quality control on our dataset. We removed doublets detected by the DRAGEN genetic demultiplexing workflow and/or DoubletFinder. We then combined single cells from multiple libraries across countries, performed reference projection of such combinations of cells to a reference panel of immune cell transcriptomes using the RCAv2 software,58 and performed nearest-neighbor analyses based on the principal components of the reference projection coefficients. We performed Louvain clustering and cluster annotation as done in the per-library quality control step. We performed cell type-specific quality control on all single cells across all libraries by applying number of detected genes (including <300 for platelets, <500 for myeloid cells, and <1,000 for other cell types) and percentage mitochondrial reads (>12.5% for plasma cells and platelets and >8% for other cell types) filters.
In this study, we removed samples with (i) mismatches between the scRNA-seq inferred sex and reported sex, (ii) < 500 cells per donor, (iii) European genetic ancestry, or (iv) missing/low-quality genotype data. We also removed platelets from the analysis. Finally, we used 896,511 cells from 489 individuals.
Generation and pre-processing of the PBMC scRNA-seq data of the Japanese healthy and COVID-19 subjects
Single-cell suspensions were processed through the 10x Genomics Chromium Controller following the protocol outlined in the Chromium Single Cell V(D)J Reagent Kits (v1.1 Chemistry) User Guide. Chromium Next GEM Single Cell 5′ Library & Gel Bead Kit v1.1 (PN-1000167), Chromium Next GEM Chip G Single Cell Kit (PN-1000127), and Single Index Kit T Set A (PN-1000213) were applied during the process. Samples were then sequenced on an Illumina NovaSeq 6000 in a paired-end mode.
Droplet libraries were processed using Cell Ranger 5.0.0 (10x Genomics). Filtered expression matrices generated using Cell Ranger count were used to perform the analysis. Cells that had fewer than the first percentile of UMIs or greater than the 99th percentile of UMIs in each sample were excluded. Cells with <200 genes expressed or >10% of reads from mitochondrial genes or hemoglobin genes were also excluded. Additionally, putative doublets were removed using Scrublet (v0.2.1)60 and scds (v1.10.0)59 for each sample.
The R package Seurat (v4.1.0)46 was used for data scaling, transformation, clustering, and dimensionality reduction. Data were scaled and transformed using the SCTransform() function, and linear regression was performed to remove unwanted variation due to cell quality (% mitochondrial reads). For integration, 3,000 shared highly variable genes (HVGs) were identified using SelectIntegrationFeatures() function. Principal component analysis (PCA) was run on gene expression, followed by batch correction using harmony (v0.1).54 UMAP dimension reduction was generated based on the first 30 harmony-adjusted principal components. A nearest-neighbor graph using the first 30 harmony-adjusted principal components was calculated using FindNeighbors() function, followed by clustering using FindClusters() function.
Cellular identity was determined by finding DEGs for each cluster using the FindMarkers() function with parameter ‘test.use = wilcox’, and comparing those markers to known cell type-specific genes. Two rounds of clustering were performed (1st, all cells; 2nd, separately for monocytes/DC, T/NK cells, and B cells) and cell type annotation was assigned at the three layers of the granularity based on the marker gene expression. In this study, we mainly used the coarsest annotation (L1) to maintain the number of cells per cluster. In this study, a male subject with COVID-19 was removed because of the aneuploidy of the X chromosome as done in the original study.
Generation and pre-processing of the AIDA genotype data
A genotyping of AIDA samples was performed using Infinium Global Screening Array (Illumina). SNPs on the nonPAR X chromosome were treated as diploid in males and heterozygous genotypes of such SNPs were converted into 'missing' with PLINK (v1.90b4.4).57 Then, we performed quality control of the genotype data with PLINK2 (v2.00a3 9 Apr 2020).35 We filtered out samples with a call rate of <0.98. Note that no samples deviated from the Asian sample clusters in a PCA analysis with the 1,000 Genomes (1KG) Project Phase3v5 samples (N = 2,504). We removed variants with a variant call rate of <0.99, deviation from Hardy–Weinberg equilibrium with p < 1.0 × 10−6 in each population, or significant allele frequency differences between sexes (p < 5.0 × 10−8). We also removed the variants whose MAF deviated from the reference panels (|MAF in the AIDA Japanese/Korean/Chinese - MAF in the 1KG EAS | > 0.15, |MAF in the AIDA Indian - MAF in the 1KG SAS | > 0.175, or |MAF in the AIDA Japanese - MAF in the 1KG Japanese | > 0.15). The genotype data after the QC was subjected to the genotype imputation in the Michigan Imputation Server.55 EAGLE (v2.4)65 was used for the haplotype phasing of genotype data and Minimac456 was used for genome-wide genotype imputation. We used the reference panels generated from 1KG Project Phase3v5 samples (N = 2,504) with high coverage (30×) sequencing. We set an imputation quality (R2) of 0.3 and 0.7, respectively for the scLinaX analysis and eQTL analysis. We used a relaxed threshold in the scLinaX analysis because the genotype could be also confirmed by the allele information of the scRNA-seq reads. In the eQTL analysis, we removed related samples with PI_HAT >0.17.
Generation and pre-processing of the Japanese genotype data
Imputed genotype data for the Japanese dataset was derived from the previously published study.22 A genotyping of COVID-19 and healthy samples was performed using Infinium Asian Screening Array (Illumina) through collaboration with Japan COVID-19 Task Force (https://www.covid19-taskforce.jp/en/home/). SNPs on the nonPAR X chromosome were treated as diploid in males and heterozygous genotypes of such SNPs were converted into 'missing'. We applied stringent quality control filters to the samples (sample call rate <0.98, related samples with PI_HAT >0.175 or outlier samples from East Asian clusters in PCA with HapMap project samples), and variants (variant call rate <0.99, deviation from Hardy–Weinberg equilibrium with p < 1.0 × 10−6, or minor allele count <5). We also excluded SNPs with >7.5% allele frequency difference with the representative reference datasets of Japanese ancestry, namely the used the population-specific imputation reference panel of Japanese (N = 1,037) combined with 1KG Project Phase3v5 samples (N = 2,504)43,44 and the allele frequency panel of Tohoku Medical Megabank Project.45 We used SHAPEIT4 software (v4.2.1)61 for the haplotype phasing of genotype data. After phasing, we used Minimac4 software for genome-wide genotype imputation. We used the aforementioned population-specific imputation reference panel of Japanese (N = 1,037) combined with 1KG Project Phase3v5 samples (N = 2,504). We set an imputation quality (R2) of 0.3 and 0.7, respectively for the scLinaX analysis and eQTL analysis. We used a relaxed threshold in the scLinaX analysis because the genotype can be also confirmed by the allele information of the scRNA-seq reads. Since scRNA-seq data was generated in the genome build of GRCh38, we performed a liftover with Picard software.
Pre-processing of the PBMC 10x multiome data
PBMC 10x multiome data was downloaded from the web repository of the 10x Genomics (https://www.10xgenomics.com/resources/datasets/pbmc-from-a-healthy-donor-granulocytes-removed-through-cell-sorting-10-k-1-standard-2-0-0). The count matrix for the RNA data and fragment data for the ATAC data were jointly processed with the Signac software (v1.9.0).62 First, cells satisfying all of the following criteria were kept for the analysis; ATAC tag count <100,000, ATAC tag count >25,000, RNA count <25,000, RNA count >1,000, nucleosome signal <2, TSS enrichment >1, percent mitochondrial genes ["ˆMT-"] < 25, percent hemoglobin genes ["ˆHB[ˆ(P)]"] < 0.1, and percent platelet genes (PECAM1 and PF4) < 0.25. Then, ATAC peaks were called with macs2 through the CallPeaks() function of the Signac and converted into a count matrix. Putative doublets were removed using DoubletFinder (v2.3.0) and scds (v1.14.0) based on the RNA information. RNA data were scaled and transformed using the SCTransform() function and subjected to a PCA analysis with the top 2,000 highly variable genes. ATAC data was subjected to normalization and dimension reduction based on the latent semantic indexing as implemented in the Signac. Cell type annotation was assigned to each cell by multimodal reference mapping with a Multimodal PBMC reference dataset (https://atlas.fredhutch.org/data/nygc/multimodal/pbmc_multimodal.h5seurat) using the FindTransferAnchors() and TransferData() functions. Cells predicted as platelets or erythrocytes were removed from the analysis. Finally, joint UMAP visualization from RNA (top 50 PCs) and ATAC (top 2–40 LSI components) data was generated by the FindMultimodalNeighbors() function followed by the RunUMAP() function. Peak information was visualized with the CoveragePlot() function in Signac.
Pre-processing of the scRNA-seq data for a sample with a karyotype of XXY
Library preparation, sequencing, and generation of the count matrix were performed as done for the Japanese dataset. Then a count matrix generated by Cell Ranger 6.0.0 was subjected to a QC with the Seurat R package (v4.3.0). First, cells satisfying all of the following criteria were kept for the analysis; RNA count <25,000, RNA count >1,000, RNA features >200, nucleosome percent mitochondrial genes ["ˆMT-"] < 12, percent hemoglobin genes ["ˆHB[ˆ(P)]"] < 0.1, and percent platelet genes (PECAM1 and PF4) < 0.25. Putative doublets were removed using DoubletFinder (2.3.0) and scds (v1.14.0) based on the RNA information. RNA data were scaled and transformed using the SCTransform() function and subjected to a PCA analysis with the top 2,000 highly variable genes. Cell type annotation was assigned to each cell by multimodal reference mapping with the Multimodal PBMC reference dataset using the FindTransferAnchors() and TransferData() functions. Cells predicted as platelets or erythrocytes were removed from the analysis.
Pseudobulk DEG analysis
First, pseudobulk raw UMI count data was generated by aggregating the raw UMI counts from all of the cells for each cell type. Samples with at least five cells were used for the analysis. Then, pseudobulk raw UMI count data was subjected to DESeq2 (v1.38.0)51 for the DEG analysis. The formulas for the DEG analysis were the following; gene expression ∼ sex + age + cell count + library (+ cell proportion of the CD4+ T, CD8+ T, gdT, MAIT, NK, B, Plasma B, Monocyte, cDC, and pDC in the cell proportion adjusted analysis; AIDA dataset), gene expression ∼ sex + disease (COVID-19 or healthy control) + age + cell count (Japanese dataset). DEGs were the genes satisfying FDR <0.05 calculated by the DESeq2. Throughout this paper, annotation from a previous study2 was used for the comparative analysis across the XCI statuses.
Single-cell level DEG analysis
We performed single-cell level regression analysis based on the linear mixed model by modifying the method implemented in a previous study.66 To represent the continuous state of each cell, we used batch-corrected PCs calculated by harmony (v0.1 for the Japanese dataset) or harmonypy (v 0.0.6 for the AIDA dataset) from the top 30 original PCs. The negative binomial model was fitted with the following formula using glmer.nb() function in the lme4 R library (1.1_31); gene expression (raw UMI count) ∼ sex + age + %mitochondrial gene + log10(total UMI count of the cell) + PC1-10 of the raw data + (1 | library) + (1 | individual) (for the evaluation of the main effect with the AIDA dataset), gene expression (raw UMI count) ∼ sex + age + %mitochondrial gene + log10(total UMI count of the cell) + PC1-10 of the raw data + batch corrected PC 1–10 + sex × batch corrected PC 1–10 + (1 | library) + (1 | individual) (for the evaluation of the interaction effect with the AIDA dataset), gene expression (raw UMI count) ∼ sex + age + disease + %mitochondrial gene + log10(total UMI count of the cell) + PC1-10 of the raw data + (1 | individual) (for the evaluation of the main effect with the Japanese dataset), gene expression (raw UMI count) ∼ sex + age + disease + %mitochondrial gene + log10(total UMI count of the cell) + PC1-10 of the raw data + batch corrected PC 1–10 + sex × batch corrected PC 1–10 + (1 | individual) (for the evaluation of the interaction effect with the Japanese dataset). In the evaluation for the main effect, the contribution of the sex to the model was evaluated by the likelihood ratio test. In the evaluation of the interaction effect, the contribution of the sex × batch corrected PC 1–10 to the model was evaluated by the likelihood ratio test. For the calculation of the single-cell level effect sizes of the sex, we summed up the effect sizes of the sex and sex × batch corrected PC 1–10 in the interaction effect analysis as done in the previous study.
Implementation of scLinaX and scLinaX-multi
Generation and QC of the single-cell level ASE profile
First, single-cell level ASE profiles were generated by cellsnp-lite software49 (v 1.2.3) for each sample. While cellsnp-lite takes genotype data as input, it can also call genotype data from scRNA-seq data. Therefore, we used imputed genotype data based on the SNP array when available, and used genotype data internally called from scRNA-seq data in other cases. Then, allele frequency and gene information were assigned to the SNPs included in the single-cell level ASE profiles by Annovar (Mon, 8 Jun 2020),47 and only the common SNPs (MAF >0.01 in the matched population of the 1KG dataset; AIDA dataset, EAS and SAS; Japanese dataset, EAS; Tabula Sapiens dataset, ALL; 10x multiome dataset, ALL; Asian sample in the SLE dataset, EAS; European sample in the SLE dataset, EUR; XXY sample, EAS) on the gene (intronic, UTR5, UTR3, exonic, ncRNA_exonic, ncRNA_intronic, and splicing) was retained for the analysis.
QC of the candidate reference genes used in scLinaX
In scLinaX, we used SNPs on the genes previously annotated as completely subjected to XCI (nonPAR inactive) as candidates for the reference SNPs.23 We also set QC criteria for these genes to exclude potentially escaping genes. First, SNPs on nonPAR inactive genes (candidate reference genes) expressed in more than 50 cells were extracted and designated as reference SNP candidates. For each SNP, pseudobulk ASE profiles across all the expressing SNPs were calculated separately for cells expressing the ref allele and alt allele, and these were added together after flipping the ref and alt allele counts for the cells expressing the alt allele. In other words, we made a completely skewed XCI in silico. For each sample-reference gene pair, the one with the highest number of cells was retained to remove the redundancy. For the pseudobulk ASE profiles, the SNPs with a total allele count of ≥10 were retained, and the minor allele count ratio was calculated as a ratio of the expression from Xi. The SNPs on the reference gene of each pseudobulk profile were excluded from the pseudobulk profiles to prevent the underestimation of the ratio of the expression from Xi. The following two metrics were then calculated for each candidate reference gene. (1) The average ratio of the expression from Xi for the gene when SNPs on the other candidate reference genes were used as references (2) The average of the ratio of the expression from Xi across the other candidate reference genes when the SNPs on the gene was used as reference. Note that when there were multiple SNPs on the same genes derived from the same sample and reference gene, only one with the highest total allele count was used for the calculation of the metrics. Since there could be a potential escape for genes with high metrics values, we used a threshold of 0.05, 0.075, and 0.1 respectively for the AIDA dataset, Japanese dataset, and SLE dataset, and filtered out the potential escapee genes from the candidate reference SNP list. For the Tabula Sapiens, 10x Multiome, and XXY karyotype data, we used the QC results from the AIDA dataset because there were a relatively small number of samples.
Grouping cells based on which X chromosome is inactivated
After defining the candidate reference gene set, we performed the scLinaX analysis. First, SNPs on the candidate reference genes expressed in more than 50 (PBMC scRNA-seq dataset), 30 (10x multiome dataset), or 100 (Tabula Sapiens dataset) cells were extracted for each sample. For each SNP, pseudobulk ASE profiles were calculated separately for cells expressing the ref alleles and alt alleles, and these were added together after flipping the ref and alt allele counts for the cells expressing alt alleles. Note that scLinaX had the option to remove known escapee genes from the pseudobulk ASE profiles (throughout this paper, this option was set as active). Then, pseudobulk ASE profiles generated from the same samples were subjected to the pairwise Spearman correlation calculation. We set a threshold for the P-values (<0.05 for all of the datasets) and correlation coefficients (absolute values >0.5 for the PBMC datasets and >0.3 for the Tabula Sapiens dataset) for defining the significant correlations. We generated a group of SNPs that had connected by at least one significant correlation. Then we defined a group of reference SNP alleles on the same X chromosome based on the significant correlations within the group. When assuming the XCI, a significant positive correlation meant that the reference alleles of the two reference SNPs were on the same X chromosomes and a significant negative correlation meant that the reference alleles of the two reference SNPs were on the different X chromosomes. If the contradiction happened during the processing of the correlation information within a group of SNPs (e.g., alternative alleles of the three reference SNPs are predicted to be on the different X chromosomes), such a group of SNPs was removed from the analysis. After defining the group of alleles on the same X chromosome, we divided the cells into three groups; (i) cells expressing only alleles of a group, (ii) cells expressing only alleles of another group, (iii) cells expressing no reference alleles or both groups of the reference alleles.
Calculation of the ratio of the expression from Xi
We calculated the pseudobulk ASE profiles across cell groups (i) and (ii) separately and combined them after flipping the ref and alt allele counts for the pseudobulk profiles from group (ii) cells. Then, we calculated the ratio of the expression from Xi as a ratio of the minor allele count under the assumption that the expression from Xi was lower than that from Xa.1 Only the positions with ≥10 total allele counts were considered. When multiple transcribed SNPs were detected for a gene in a sample, one with the deepest allele counts was selected to evaluate the ratio of the expression from Xi for the gene. When calculating the ratio of the expression from Xi per cell cluster, pseudobulk ASE profiles were generated from cells within the cell cluster while the definition of the Xi/Xa alleles was based on the pseudobulk ASE profiles from all cells.
Summarization of the scLinaX results for the AIDA and Japanese dataset
To obtain the ratio of the expression from Xi for each gene, we calculated the average across the samples that had the transcribed SNPs with ≥10 total allele counts on that gene. Only the genes for which ≥3 samples were used for calculating the average were considered.
Evaluation of the performance of scLinaX with the down-sampled Japanese dataset
To evaluate the performance of scLinaX with different cell numbers and UMI per cell, we performed scLinaX analysis with down-sampled Japanese dataset. We chose 22 samples which had ≥2,000 cells with at least 4,000 UMI counts. Bam files were down-sampled to the cell numbers of 100, 200, 300, 400, 500, 750, 1000, 1250, 1500, 1750, 2000, and UMI count per cell of 500, 1000, 1500, 2000, 2500, 3000, 3500, 4000. In the actual implementation, the number of extracted UMI for each cell were determined as original UMI count from the X chromosome × target UMI count/original all UMI count, which enabled us to perform analysis computationally efficiently with bam files only for X chromosome. Then, scLinaX was applied to the down-sampled data with the reference gene sets same to the original scLinaX analysis.
Implementation of scLinaX-multi and application to the PBMC 10x multiome data
scLinaX-multi is an extension of scLinaX to the multi-modal dataset. In this study, we estimated which X chromosome was inactivated from the RNA-level information and evaluated escape at the chromatin accessibility level by using the 10x multiome dataset. First, cells were grouped into the following three groups; (i) cells expressing only alleles of a group, (ii) cells expressing only alleles of another group, (iii) cells expressing no reference SNPs or both groups of the alleles, same as the scLinaX procedure. Then, single-cell level allele-specific chromatin accessibility profiles were generated by cellsnp-lite software. In this study, we used genotype data called from the single-cell ATAC data, while it can also take other types of genotype data. Allele frequency and gene information were assigned to the SNPs included in the single-cell level allele-specific chromatin accessibility profiles and only the common SNPs (MAF >0.01 in the 1KG ALL dataset) on the ATAC peaks were retained for the analysis. We calculated the pseudobulk allele-specific chromatin accessibility profiles across cell groups (i) and (ii) separately and combined them after flipping the ref and alt allele counts for the pseudobulk profiles from group (ii) cells. Finally, we calculated the ratio of the Xi-derived accessible chromatin as a ratio of the minor allele count. Only the positions with ≥10 total allele counts were considered. When calculating the ratio of the Xi-derived accessible chromatin per cell cluster, pseudobulk allele-specific chromatin accessibility profiles were generated from cells within the cell cluster while the definition of the Xi/Xa allele was based on the pseudobulk allele-specific chromatin accessibility profiles from all cells. When multiple transcribed SNPs were detected for a peak, one with the deepest allele counts was selected to evaluate the ratio of the Xi-derived accessible chromatin. Exceptionally, when visualizing escape at the chromatin accessibility level (Figure 4F), we retained both of the SNPs on the peaks at the TSS of the USP9X gene.
Summarization of the scLinaX results for the Tabula Sapiens dataset
We used the processed Tabula Sapiens dataset contributed by the Tabula Sapiens Consortium (https://tabula-sapiens-portal.ds.czbiohub.org).20 For the calculation of the ratio of the expression from Xi, we aggregated the allele counts from Xi and Xa across samples for summarization. The annotation of the organs and cell type was derived from the previous study, while the cell type of 'immune' was divided into the 'Lymphoid', 'Myeloid', and 'Other blood cell' considering the difference of escape across immune cells identified in this study. In the pairwise comparisons of escape across organs and cell types, genes detected in both organs/cell types 1 and 2 were extracted, and the ratio of the genes with a higher ratio of the expression from Xi in the organ/cell type 1 was used as an indicator of the difference of escape between the organs/cell types. In addition, comparisons of the ratio of the expression from Xi were performed at the individual level. We used only the TSP2 sample for the evaluation of the difference in escape across organs because major lymphoid tissues were derived solely from the TSP2.
Case-control comparisons of the ratio of the expression from Xi
For the generation of the scRNA-seq bam files of the SLE dataset,26 we downloaded the fastq files and processed them with Cell Ranger 6.1.2. For the case–control comparisons of escape from XCI with the COVID-19 and SLE datasets, we considered the transcribed SNPs with ≥5 total allele counts to increase the sample size. We evaluated the genes (i) considered in ≥5 case samples, (ii) considered in ≥5 control samples, and (iii) the ratio of the expression from Xi calculated from the aggregated allele count data across all samples was ≥0.1. We used a negative binomial model (glm.nb() function in the MASS R library [v7.3_58.1]) to evaluate the case–control differences of escape using the following formula; allele counts from Xi ∼ disease status + log(total allele count) (offset term).
scLinaX analysis with a male sample with a karyotype of XXY
As input genotype data for scLinaX, we used imputed genotype data of the X chromosome (non-PAR region) which were generated and processed in the same manner as the genotype data of the Japanese dataset. Since a single sample was available for this analysis, the ratio of the expression from Xi in the sample was presented as it was.
PacBio HiFi sequencing for phasing
To evaluate the accuracy of phase information inferred from scLinaX, PacBio HiFi long-read whole-genome-sequencing was performed for the four samples from the Japanese dataset at Takara Bio Corporation. DNA samples were sheared targeting the size of 20kb using Megaruptor 3 (Diagenode). SMRTbell libraries were prepared with the SMRTbell Express Template Prep Kit 2.0 according to the manufacturer’s protocols. Fragments were size-selected using SageELF (Sage Science). Libraries were sequenced on the Sequel II (Pacific Bioscience) system using the Sequel II Binding Kit 2.0 and Sequel II Sequencing Kit 2.0 (mean coverage = 16.0×). Based on the sequenced subreads, circular consensus sequence (CCS) reads were generated using SMRT Link (v9.0.0, Pacific Bioscience). CCS reads were aligned against GRCh38 reference genome using pbmm2 (v1.7.0) (https://github.com/PacificBiosciences/pbmm2). Then, generated bam files were utilized for physical phasing with whatshap63 (v1.4).
Pseudobulk eQTL analysis with the AIDA and Japanese dataset
Raw pseudobulk gene expression data was TMM-normalized and log2-transformed with the edgeR R library (v3.40.0).53 The genes with (i) raw UMI count ≥5 in more than 20% of the samples and (ii) count per million (CPM) ≥ 0.2 in more than 20% of the samples were filtered out as done in a previous study.67 Then cis-eQTL was identified by tensorQTL (v1.0.8)68 with the '--mode cis' option to obtain the list of the significant eQTL signals and with the '--mode cis_nomial' option to obtain the nominal P-values for all of the gene–cis-variant pairs. tensorQTL was applied for (i) all sample data, (ii) only female data, and (iii) only male data with the '--maf_threshold 0.05' option. Sex (only for all sample data analysis), age, cell count, library, genotype PCs 1–10, and gene expression PCs 1–10 were included as covariates for the AIDA dataset analysis. Sex (only for all sample data analysis), age, disease, cell count, genotype PCs 1–10, and gene expression PCs 1–10 were included as covariates for the Japanese dataset analysis. Genotype PCs were calculated from the SNP array data before imputation by using PLINK2. Gene expression PCs were calculated from the TMM-normalized gene expression data using the prcomp() function in the R. Genotypes of the variants on the X chromosome were coded as 0/1/2 in females and 0/2 in males. We defined eQTL signals satisfying p < 5 × 10−8 in the AIDA all sample analysis as significant eQTL signals.
Escape QTL analysis with the AIDA dataset
Escape QTL analysis was performed for the known escapee genes and the SEPTIN6 gene which were evaluated in ≥50 individuals. Then cis-escape QTL was identified by tensorQTL (v1.0.8)68 with the '--mode cis_nomial' and '--maf_threshold 0.05' options to obtain the nominal P-values for all of the gene–cis-variant pairs. Age, genotype PCs 1–10, SNPs represent the escapee genes, and total allele count of the SNPs were included as covariates. Genotype PCs were calculated from the SNP array data before imputation by using PLINK2 as described above. We defined the significance threshold as p < 5.1 × 10−7 (0.05/97,120).
Single-cell level dynamic eQTL analysis
We performed a single-cell level dynamic eQTL analysis based on the linear mixed model by modifying the method implemented in the previous study66 to evaluate the heterogeneity of the effects of the eQTL variants (rs6641874 and rs6641601) on the PRKX gene expression. As done in the single-cell level DEG analysis, we used batch-corrected PCs calculated by harmonypy from the top 30 original PCs to represent the continuous state of each cell. The negative binomial model was fitted with the following formula using glmer.nb() function in the lme4 R library; gene expression (raw UMI count) ∼ genotype + sex + age + %mitochondrial gene + log10(total UMI count) + original PC1-10 of the scRNA-seq data + genotype PC 1–10 + batch corrected PC 1–10 of the scRNA-seq data + genotype × batch corrected PC 1–10 of the scRNA-seq data + (1 | library) + (1 | individual). Genotypes of the variants on the X chromosome were coded as 0/1/2 in females and 0/2 in males. In the evaluation of the interaction effect, the contribution of the genotype × batch corrected PC 1–10 to the model was evaluated by the likelihood ratio test. For the calculation of the single-cell level effect sizes of the eQTL effect, we summed up the effect sizes of the genotype and genotype × batch corrected PC 1–10 of the scRNA-seq data in the interaction effect analysis as done in the previous study.
GWAS for the blood-related traits with the BBJ cohort
BBJ is a prospective biobank that collaboratively recruited approximately 200,000 patients with ≥1 of 47 diseases and collected DNA, serum samples, and clinical information from 12 medical institutions in Japan between 2003 and 2007.42 The Japanese samples in BBJ were genotyped with the Illumina HumanOmniExpressExome BeadChip or a combination of the Illumina HumanOmniExpress and HumanExome BeadChips. Quality control of samples and genotypes was conducted as described elsewhere.43 We analyzed subjects of Japanese ancestry identified by a PCA analysis. Genotype data were imputed with the aforementioned 1KG Project phase3v5 genotype data and Japanese whole-genome sequencing data using Minimac3. As for the blood-related trait data (white blood cell number [WBC], lymphocyte number [LYM], monocyte number [Mono], eosinophils number [EOS], basophils number [BAS], neutrophils number [NEU], hemoglobin [Hb], hematocrit [Ht], mean corpuscular volume [MCV], red blood cell number [RBC], and platelet number [PLT]), we generally used the values measured at the participants’ first visit to the hospitals, and excluded values outside three times the interquartile range (IQR) of the upper or lower quartile across participants as previously described (Table S14).31 Then, blood-related trait data were subjected to the rank-based inverse normal transformation separately for males and females. We conducted X chromosome GWAS for each blood-related trait using REGENIE (v3.2.7).37 We included age, sex, and the top 20 principal components as covariates. Genotypes of the variants on the X chromosome were coded as 0/1/2 in females and 0/2 in males.
Comparisons of the GWAS effect sizes between sexes with the BBJ and UKB cohort
GWAS summary statistics for the UKB cohort were downloaded from the web repository (Nealelab/UK_Biobank_GWAS: v2; Zenodo, https://doi.org/10.5281/zenodo.8011558). Fixed-effect meta-analysis across sexes or cohorts was performed with the metafor R package (v4.2_0). The standard error of the ratio between the female effect sizes (βfemale) and male effect sizes (βmale) was calculated based on the law of error propagation as previously done.7
The significance of the difference between the female effect sizes (βfemale) and male effect sizes (βmale) was evaluated by calculating the following statistics which follow a χ2-distribution.
Evaluation of the colocalization between the GWAS and eQTL signals
To evaluate the colocalization between the lymphocyte count GWAS signals and PRKX gene eQTL signals, we used the coloc R package (v5.2.2).50 Since the reference human genome was different between the GWAS (GRCh37) and eQTL (GRCh38) analysis, we performed a liftover with the bcftools48 (v.1.16). Variants within 1,000,000 bp from rs6641874 were used as inputs and PP.H4 > 0.80 was considered as a colocalization of the signals.
Quantification and statistical analysis
Please refer to figure legends and method details for details of statistical analysis. Unless specified, statistical tests were conducted as two-sided. Number of the samples used in the analyses are described in Tables S1, S2, S7, and S14. Throughout this study, the boxplot indicates the median values (center lines) and IQRs (box edges), with the whiskers extending to the most extreme points within the range between (lower quantile − [1.5 × IQR]) and (upper quantile + [1.5 × IQR]).
Acknowledgments
We would like to thank all donors and participants in the studies constituting the Asian Immune Diversity Atlas. The Singapore donor samples were obtained through the Health for Life in Singapore (HELIOS) Study (Lee Kong Chian School of Medicine, Nanyang Technological University; National Healthcare Group, Singapore; Imperial College London). We would like to express our thanks to participants of the HELIOS study and the HELIOS operation team for recruitment, organization, and data/sample collection. This study (NTU IRB: 2016-11-030) is supported by Singapore Ministry of Health’s (MOH) National Medical Research Council (NMRC) under its OF-LCG funding scheme (MOH-000271-00) and intramural funding from Nanyang Technological University, Lee Kong Chian School of Medicine, and the National Healthcare Group. This project has been made possible in part by grant number CZF2019-002446 (to S.P., W.-Y.P., J.W.S., and John Chambers) from the Chan Zuckerberg Foundation, and grant numbers 2020-224570 (to S.P., W.-Y.P., Varodom Charoensawan, Ponpan Matangkasombut, and Partha P. Majumder) and 2021–240178 (to S.P., W.-Y.P., J.W.S., John Chambers, Varodom Charoensawan, Ponpan Matangkasombut, and Partha P. Majumder) from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation. This project was also supported by the Thailand Program Management Unit for National Competitiveness Enhancement (PMU-C) (C10F650132) (to Varodom Charoensawan, Ponpan Matangkasombut, Manop Pithukpakorn, and Bhoom Suktitipat) and Mahidol University’s Basic Research Fund: fiscal year 2021 (BRF1-017/2564) (to Varodom Charoensawan and Bhoom Suktitipat). We would like to thank Jennifer Zamanian, Jennifer Chien, and Jason Hilton from the Human Cell Atlas Lattice team (Stanford University) for their help with and work on data deposits and coordination for community access. B.L. is supported by the Ministry of Education, Singapore, under its Academic Research Fund Tier 1 (FY2023, 23-0434-A0001, and 22-5800-A0001) and Tier 2 (MOE-T2EP30123-0015), the Precision Medicine Translational Research Programme Core Funding (NUHSRO/2020/080/MSC/04/PM), NUS ODPRT Seed Funding, and NUS YLLSoM Seed Funding. We want to acknowledge the participants and investigators of BBJ and UKB study. We thank all the members of the Japan COVID-19 Task Force and the Asian Immune Diversity Atlas Network members for their support. We thank Prof. Keishi Fujio, Dr. Mineto Ota, Dr. Kazuyoshi Ishigaki, and Dr. Masahiro Nakano for the scientific discussion. Y.O. was supported by JSPS KAKENHI (22H00476), AMED (JP23km0405211/JP23km0405217/JP23ek0109594/JP23ek0410113/JP23kk0305022/JP223fa627002/JP223fa627010/JP233fa627011/JP23zf0127008/JP23tm0524002), JST Moonshot R&D (JPMJMS2021/JPMJMS2024), Takeda Science Foundation, Bioinformatics Initiative of Osaka University Graduate School of Medicine, Institute for Open and Transdisciplinary Research Initiatives, Center for Infectious Disease Education and Research, and Center for Advanced Modality and DDS, Osaka University.
Author contributions
Y.T. and Y.O. designed the study. Y.T., R.E., K.S., Y.S., K.H.K., Q.S.W., S.N., J.M., T.N., Q.X.X.L., E.V.B., R.S., K.Y.H., B.L., and C.-C.H. conducted the data analysis. Y.T. and Y.O. wrote the manuscript. R.E., Y.S., and L.M.T., conducted the experiments. Y.T., R.E., K.S., Y.S., S.N., Y.A., A.S., T.Y., K.O., H.N., H.T., H.L., and T.O. collected and managed the samples. B.L., K.M., K.F., H.M., W.-Y.P., K.Y., C.-C.H., J.W.S., S.P., A.K., and Y.O. supervised the study. All authors contributed to the article and approved the submitted version.
Declaration of interests
The authors declare no competing interests.
Published: July 30, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2024.100625.
Contributor Information
Yoshihiko Tomofuji, Email: ytomofuji@sg.med.osaka-u.ac.jp.
Yukinori Okada, Email: yuki-okada@m.u-tokyo.ac.jp.
Supplemental information
References
- 1.Balaton B.P., Brown C.J. Escape Artists of the X Chromosome. Trends Genet. 2016;32:348–359. doi: 10.1016/j.tig.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 2.Tukiainen T., Villani A.-C., Yen A., Rivas M.A., Marshall J.L., Satija R., Aguirre M., Gauthier L., Fleharty M., Kirby A., et al. Landscape of X chromosome inactivation across human tissues. Nature. 2017;550:244–248. doi: 10.1038/nature24265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunford A., Weinstock D.M., Savova V., Schumacher S.E., Cleary J.P., Yoda A., Sullivan T.J., Hess J.M., Gimelbrant A.A., Beroukhim R., et al. Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet. 2017;49:10–16. doi: 10.1038/ng.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Souyris M., Cenac C., Azar P., Daviaud D., Canivet A., Grunenwald S., Pienkowski C., Chaumeil J., Mejía J.E., Guéry J.-C. TLR7 escapes X chromosome inactivation in immune cells. Sci. Immunol. 2018;3 doi: 10.1126/sciimmunol.aap8855. [DOI] [PubMed] [Google Scholar]
- 5.Syrett C.M., Paneru B., Sandoval-Heglund D., Wang J., Banerjee S., Sindhava V., Behrens E.M., Atchison M., Anguera M.C. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight. 2019;4 doi: 10.1172/jci.insight.126751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J., Syrett C.M., Kramer M.C., Basu A., Atchison M.L., Anguera M.C. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc. Natl. Acad. Sci. USA. 2016;113:E2029–E2038. doi: 10.1073/pnas.1520113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sidorenko J., Kassam I., Kemper K.E., Zeng J., Lloyd-Jones L.R., Montgomery G.W., Gibson G., Metspalu A., Esko T., Yang J., et al. The effect of X-linked dosage compensation on complex trait variation. Nat. Commun. 2019;10:3009. doi: 10.1038/s41467-019-10598-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keur N., Ricaño-Ponce I., Kumar V., Matzaraki V. A systematic review of analytical methods used in genetic association analysis of the X-chromosome. Brief. Bioinform. 2022;23 doi: 10.1093/bib/bbac287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khramtsova E.A., Wilson M.A., Martin J., Winham S.J., He K.Y., Davis L.K., Stranger B.E. Quality control and analytic best practices for testing genetic models of sex differences in large populations. Cell. 2023;186:2044–2061. doi: 10.1016/j.cell.2023.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L., Wang Z., Lu T., Manolio T.A., Paterson A.D. eXclusionarY: 10 years later, where are the sex chromosomes in GWASs? Am. J. Hum. Genet. 2023;110:903–912. doi: 10.1016/j.ajhg.2023.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Migeon B.R., Moser H.W., Moser A.B., Axelman J., Sillence D., Norum R.A. Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc. Natl. Acad. Sci. USA. 1981;78:5066–5070. doi: 10.1073/pnas.78.8.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shapiro L.J., Mohandas T., Weiss R., Romeo G. Non-inactivation of an X-Chromosome Locus in Man. Science. 1979;204:1224–1226. doi: 10.1126/science.156396. [DOI] [PubMed] [Google Scholar]
- 13.Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 14.Carrel L., Cottle A.A., Goglin K.C., Willard H.F. A first-generation X-inactivation profile of the human X chromosome. Proc. Natl. Acad. Sci. USA. 1999;96:14440–14444. doi: 10.1073/pnas.96.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larson N.B., Fogarty Z.C., Larson M.C., Kalli K.R., Lawrenson K., Gayther S., Fridley B.L., Goode E.L., Winham S.J. An integrative approach to assess X-chromosome inactivation using allele-specific expression with applications to epithelial ovarian cancer. Genet. Epidemiol. 2017;41:898–914. doi: 10.1002/gepi.22091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cotton A.M., Ge B., Light N., Adoue V., Pastinen T., Brown C.J. Analysis of expressed SNPs identifies variable extents of expression from the human inactive X chromosome. Genome Biol. 2013;14 doi: 10.1186/gb-2013-14-11-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauteraud R., Stahl J.M., James J., Englebright M., Chen F., Zhan X., Carrel L., Liu D.J. Inferring genes that escape X-Chromosome inactivation reveals important contribution of variable escape genes to sex-biased diseases. Genome Res. 2021;31:1629–1637. doi: 10.1101/gr.275677.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wainer Katsir K., Linial M. Human genes escaping X-inactivation revealed by single cell expression data. BMC Genom. 2019;20:201. doi: 10.1186/s12864-019-5507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garieri M., Stamoulis G., Blanc X., Falconnet E., Ribaux P., Borel C., Santoni F., Antonarakis S.E. Extensive cellular heterogeneity of X inactivation revealed by single-cell allele-specific expression in human fibroblasts. Proc. Natl. Acad. Sci. USA. 2018;115:13015–13020. doi: 10.1073/pnas.1806811115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The T.S.C.∗, Jones R.C., Karkanias J., Krasnow M.A., Pisco A.O., Quake S.R., Salzman J., Yosef N., Bulthaup B., Brown P., et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. 2022;376 doi: 10.1126/science.abl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kock K.H., Tan L.M., Han K.Y., Ando Y., Jevapatarakul D., Chatterjee A., Lin Q., Buyamin E.V., Sonthalia R., Rajagopalan D., et al. Single-cell analysis of human diversity in circulating immune cells. bioRxiv. 2024 doi: 10.1101/2024.06.30.601119. [DOI] [Google Scholar]
- 22.Edahiro R., Shirai Y., Takeshima Y., Sakakibara S., Yamaguchi Y., Murakami T., Morita T., Kato Y., Liu Y.-C., Motooka D., et al. Single-cell analyses and host genetics highlight the role of innate immune cells in COVID-19 severity. Nat. Genet. 2023;55:753–767. doi: 10.1038/s41588-023-01375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Namkoong H., Edahiro R., Takano T., Nishihara H., Shirai Y., Sonehara K., Tanaka H., Azekawa S., Mikami Y., Lee H., et al. DOCK2 is involved in the host genetics and biology of severe COVID-19. Nature. 2022;609:754–760. doi: 10.1038/s41586-022-05163-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.San Roman A.K., Godfrey A.K., Skaletsky H., Bellott D.W., Groff A.F., Harris H.L., Blanton L.V., Hughes J.F., Brown L., Phou S., et al. The human inactive X chromosome modulates expression of the active X chromosome. Cell Genom. 2023;3 doi: 10.1016/j.xgen.2023.100259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hagen S.H., Henseling F., Hennesen J., Savel H., Delahaye S., Richert L., Ziegler S.M., Altfeld M. Heterogeneous Escape from X Chromosome Inactivation Results in Sex Differences in Type I IFN Responses at the Single Human pDC Level. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez R.K., Gordon M.G., Subramaniam M., Kim M.C., Hartoularos G.C., Targ S., Sun Y., Ogorodnikov A., Bueno R., Lu A., et al. Single-cell RNA-seq reveals cell type–specific molecular and genetic associations to lupus. Science. 2022;376 doi: 10.1126/science.abf1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Balaton B.P., Brown C.J. Contribution of genetic and epigenetic changes to escape from X-chromosome inactivation. Epigenet. Chromatin. 2021;14:30. doi: 10.1186/s13072-021-00404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu B., Qi Y., Li R., Shi Q., Satpathy A.T., Chang H.Y. B cell-specific XIST complex enforces X-inactivation and restrains atypical B cells. Cell. 2021;184:1790–1803.e17. doi: 10.1016/j.cell.2021.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abascal F., Acosta R., Addleman N.J., Adrian J., Afzal V., Ai R., Aken B., Akiyama J.A., Jammal O.A., Amrhein H., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanai M., Akiyama M., Takahashi A., Matoba N., Momozawa Y., Ikeda M., Iwata N., Ikegawa S., Hirata M., Matsuda K., et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018;50:390–400. doi: 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- 31.Sakaue S., Kanai M., Tanigawa Y., Karjalainen J., Kurki M., Koshiba S., Narita A., Konuma T., Yamamoto K., Akiyama M., et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021;53:1415–1424. doi: 10.1038/s41588-021-00931-x. [DOI] [PubMed] [Google Scholar]
- 32.Rang F.J., de Luca K.L., de Vries S.S., Valdes-Quezada C., Boele E., Nguyen P.D., Guerreiro I., Sato Y., Kimura H., Bakkers J., Kind J. Single-cell profiling of transcriptome and histone modifications with EpiDamID. Mol. Cell. 2022;82:1956–1970.e14. doi: 10.1016/j.molcel.2022.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gleicher N., Barad D.H. Gender as risk factor for autoimmune diseases. J. Autoimmun. 2007;28:1–6. doi: 10.1016/j.jaut.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 34.Scofield R.H., Bruner G.R., Namjou B., Kimberly R.P., Ramsey-Goldman R., Petri M., Reveille J.D., Alarcón G.S., Vilá L.M., Reid J., et al. Klinefelter’s syndrome (47,XXY) in male systemic lupus erythematosus patients: Support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum. 2008;58:2511–2517. doi: 10.1002/art.23701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4 doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loh P.-R., Tucker G., Bulik-Sullivan B.K., Vilhjálmsson B.J., Finucane H.K., Salem R.M., Chasman D.I., Ridker P.M., Neale B.M., Berger B., et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 2015;47:284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mbatchou J., Barnard L., Backman J., Marcketta A., Kosmicki J.A., Ziyatdinov A., Benner C., O’Dushlaine C., Barber M., Boutkov B., et al. Computationally efficient whole-genome regression for quantitative and binary traits. Nat. Genet. 2021;53:1097–1103. doi: 10.1038/s41588-021-00870-7. [DOI] [PubMed] [Google Scholar]
- 38.Harroud A., Stridh P., McCauley J.L., Saarela J., van den Bosch A.M.R., Engelenburg H.J., Beecham A.H., Alfredsson L., Alikhani K., Amezcua L., et al. Locus for severity implicates CNS resilience in progression of multiple sclerosis. Nature. 2023;619:323–331. doi: 10.1038/s41586-023-06250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mahajan A., Spracklen C.N., Zhang W., Ng M.C.Y., Petty L.E., Kitajima H., Yu G.Z., Rüeger S., Speidel L., Kim Y.J., et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 2022;54:560–572. doi: 10.1038/s41588-022-01058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al’Khafaji A.M., Smith J.T., Garimella K.V., Babadi M., Popic V., Sade-Feldman M., Gatzen M., Sarkizova S., Schwartz M.A., Blaum E.M., et al. High-throughput RNA isoform sequencing using programmed cDNA concatenation. Nat. Biotechnol. 2024;42:582–586. doi: 10.1038/s41587-023-01815-7. [DOI] [PubMed] [Google Scholar]
- 41.Rozenblatt-Rosen O., Stubbington M.J.T., Regev A., Teichmann S.A. The Human Cell Atlas: from vision to reality. Nature. 2017;550:451–453. doi: 10.1038/550451a. [DOI] [PubMed] [Google Scholar]
- 42.Nagai A., Hirata M., Kamatani Y., Muto K., Matsuda K., Kiyohara Y., Ninomiya T., Tamakoshi A., Yamagata Z., Mushiroda T., et al. Overview of the BioBank Japan Project: Study design and profile. J. Epidemiol. 2017;27:S2–S8. doi: 10.1016/j.je.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Akiyama M., Ishigaki K., Sakaue S., Momozawa Y., Horikoshi M., Hirata M., Matsuda K., Ikegawa S., Takahashi A., Kanai M., et al. Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat. Commun. 2019;10:4393. doi: 10.1038/s41467-019-12276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okada Y., Momozawa Y., Sakaue S., Kanai M., Ishigaki K., Akiyama M., Kishikawa T., Arai Y., Sasaki T., Kosaki K., et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 2018;9:1631. doi: 10.1038/s41467-018-03274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tadaka S., Katsuoka F., Ueki M., Kojima K., Makino S., Saito S., Otsuki A., Gocho C., Sakurai-Yageta M., Danjoh I., et al. 3.5KJPNv2: an allele frequency panel of 3552 Japanese individuals including the X chromosome. Hum. Genome Var. 2019;6:28. doi: 10.1038/s41439-019-0059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang X., Huang Y. Cellsnp-lite: an efficient tool for genotyping single cells. Bioinformatics. 2021;37:4569–4571. doi: 10.1093/bioinformatics/btab358. [DOI] [PubMed] [Google Scholar]
- 50.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian Test for Colocalisation between Pairs of Genetic Association Studies Using Summary Statistics. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh P., Raychaudhuri S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 2019;16:1289–1296. doi: 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das S., Forer L., Schönherr S., Sidore C., Locke A.E., Kwong A., Vrieze S.I., Chew E.Y., Levy S., McGue M., et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fuchsberger C., Abecasis G.R., Hinds D.A. minimac2: faster genotype imputation. Bioinformatics. 2015;31:782–784. doi: 10.1093/bioinformatics/btu704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmidt F., Ranjan B., Lin Q.X.X., Krishnan V., Joanito I., Honardoost M.A., Nawaz Z., Venkatesh P.N., Tan J., Rayan N.A., et al. RCA2: a scalable supervised clustering algorithm that reduces batch effects in scRNA-seq data. Nucleic Acids Res. 2021;49:8505–8519. doi: 10.1093/nar/gkab632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bais A.S., Kostka D. scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics. 2020;36:1150–1158. doi: 10.1093/bioinformatics/btz698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wolock S.L., Lopez R., Klein A.M. Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 2019;8:281–291.e9. doi: 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delaneau O., Zagury J.-F., Robinson M.R., Marchini J.L., Dermitzakis E.T. Accurate, scalable and integrative haplotype estimation. Nat. Commun. 2019;10:5436. doi: 10.1038/s41467-019-13225-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stuart T., Srivastava A., Madad S., Lareau C.A., Satija R. Single-cell chromatin state analysis with Signac. Nat. Methods. 2021;18:1333–1341. doi: 10.1038/s41592-021-01282-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin M., Patterson M., Garg S., Fischer S.O., Pisanti N., Klau G.W., Schöenhuth A., Marschall T. WhatsHap: fast and accurate read-based phasing. bioRxiv. 2016 doi: 10.1101/085050. [DOI] [Google Scholar]
- 64.Kang H.M., Subramaniam M., Targ S., Nguyen M., Maliskova L., McCarthy E., Wan E., Wong S., Byrnes L., Lanata C.M., et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 2018;36:89–94. doi: 10.1038/nbt.4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Loh P.-R., Danecek P., Palamara P.F., Fuchsberger C., A Reshef Y., K Finucane H., Schoenherr S., Forer L., McCarthy S., Abecasis G.R., et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nathan A., Asgari S., Ishigaki K., Valencia C., Amariuta T., Luo Y., Beynor J.I., Baglaenko Y., Suliman S., Price A.L., et al. Single-cell eQTL models reveal dynamic T cell state dependence of disease loci. Nature. 2022;606:120–128. doi: 10.1038/s41586-022-04713-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ota M., Nagafuchi Y., Hatano H., Ishigaki K., Terao C., Takeshima Y., Yanaoka H., Kobayashi S., Okubo M., Shirai H., et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell. 2021;184:3006–3021.e17. doi: 10.1016/j.cell.2021.03.056. [DOI] [PubMed] [Google Scholar]
- 68.Taylor-Weiner A., Aguet F., Haradhvala N.J., Gosai S., Anand S., Kim J., Ardlie K., Van Allen E.M., Getz G. Scaling computational genomics to millions of individuals with GPUs. Genome Biol. 2019;20:228. doi: 10.1186/s13059-019-1836-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The AIDA Data Freeze v1 gene-cell matrix (1,058,909 cells from 503 Japan, Singaporean Chinese, Singaporean Malay, Singaporean Indian, and South Korea Asian donors and 5 distinct Lonza commercial controls), with BCR-seq and TCR-seq metadata, and donor age, sex, and self-reported ethnicity metadata, is available via the Chan Zuckerberg CELLxGENE data portal at https://cellxgene.cziscience.com/collections/ced320a1-29f3-47c1-a735-513c7084d508. The open-access AIDA datasets are available via the Human Cell Atlas Data Coordination Platform at https://data.humancellatlas.org/explore/projects/f0f89c14-7460-4bab-9d42-22228a91f185. Raw scRNA-seq sequencing data for the Japanese dataset are available at the Japanese Genotype-phenotype Archive (JGA) with accession codes JGAS000593/JGAD000722/JGAS000543/JGAD000662.22,23 All the raw sequencing data of Japanese scRNA-seq dataset can also be accessed through application at the NBDC with the accession code hum0197 (https://humandbs.biosciencedbc.jp/en/hum0197-latest). Genotype data for the Japanese dataset are available at European Genome-Phenome Archive (EGA) with the accession code EGAS00001006950 (https://ega-archive.org/studies/EGAS00001006950). scLinaX and scLinaX-multi is available as an R package from https://github.com/ytomofuji/scLinaX. Original version of scLinaX and scLinaX-multi used in this study are available from Zenodo (https://doi.org/10.5281/zenodo.11023040).