

Abstract
Objective:
Multidisciplinary studies of posttraumatic stress disorder (PTSD) and major depressive disorder (MDD) implicate the dorsolateral prefrontal cortex (DLPFC) in disease risk and pathophysiology. Postmortem brain studies have relied on bulk-tissue RNA sequencing (RNA-seq), but single-cell RNA-seq is needed to dissect cell-type-specific mechanisms. The authors conducted the first single-nucleus RNA-seq postmortem brain study in PTSD to elucidate disease transcriptomic pathology with cell-type-specific resolution.
Method:
Profiling of 32 DLPFC samples from 11 individuals with PTSD, 10 with MDD, and 11 control subjects was conducted (~415K nuclei; >13K cells per sample). A replication sample included 15 DLPFC samples (~160K nuclei; >11K cells per sample).
Results:
Differential gene expression analyses identified significant single-nucleus RNA-seq differentially expressed genes (snDEGs) in excitatory (EX) and inhibitory (IN) neurons and astrocytes, but not in other cell types or bulk tissue. MDD samples had more false discovery rate–corrected significant snDEGs, and PTSD samples had a greater replication rate. In EX and IN neurons, biological pathways that were differentially enriched in PTSD compared with MDD included glucocorticoid signaling. Furthermore, glucocorticoid signaling in induced pluripotent stem cell (iPSC)–derived cortical neurons demonstrated greater relevance in PTSD and opposite direction of regulation compared with MDD, especially in EX neurons. Many snDEGs were from the 17q21.31 locus and are particularly interesting given causal roles in disease pathogenesis and DLPFC-based neuroimaging (PTSD: ARL17B, LINC02210-CRHR1, and LRRC37A2; MDD: LRRC37A and LRP4), while others were regulated by glucocorticoids in iPSC-derived neurons (PTSD: SLC16A6, TAF1C; MDD: CDH3).
Conclusions:
The study findings point to cell-type-specific mechanisms of brain stress response in PTSD and MDD, highlighting the importance of examining cell-type-specific gene expression and indicating promising novel biomarkers and therapeutic targets.
Posttraumatic stress disorder (PTSD) and major depressive disorder (MDD) are debilitating disorders associated with negative life outcomes. PTSD and MDD affect approximately 6.1% and 7.1% of the population, respectively (1, 2). Rates of PTSD, which develops after trauma exposure, are higher in combat-exposed veterans and at-risk civilians (36% and 46%, respectively) (3, 4). PTSD and MDD are highly comorbid, with ~50% of PTSD patients also meeting criteria for MDD (5). Functionally, these disorders converge in some pathophysiological processes, while other pathways are affected in opposing directions, for example, hypothalamic-pituitary-adrenal (HPA) axis and glucocorticoid (GC) activity (6–8).
The Psychiatric Genomics Consortium and the Million Veteran Program have conducted large-scale genome-wide association studies (GWASs) of PTSD and MDD. These GWASs have revealed significant heritability for PTSD and MDD, risk variants, and genetic correlation between them, as well as with other disorders, such as bipolar disorder and schizophrenia (9–12). They were also enriched in brain and neuronal gene sets, specifically from the dorsolateral prefrontal cortex (DLPFC) (10–12). The DLPFC, a region with reciprocal connections to the ventromedial PFC as well as the amygdala, moderates HPA axis function, is involved in emotional processing, memory, and executive control, and has various neuroanatomical and functional neuroimaging alterations in PTSD and MDD (13, 14).
Imputation of genetically regulated gene expression (GReX) has been applied to GWAS to conduct transcriptome-wide association studies (TWASs) for detecting gene-trait associations. TWASs in PTSD and MDD have predicted changes in gene expression in genes in the DLPFC and, more generally, in genes involved in stress response regulation. For instance, the Psychiatric Genomics Consortium PTSD TWAS predicted reduced expression of the stress-sensitive, and GC-regulated, minor splicing regulator SNRNP35 in the DLPFC (15). The Million Veteran Program PTSD TWAS (10) predicted differential brain expression of genes at the 17q21.31 locus (16), including CRHR1, encoding the CRHR1 receptor for corticotropin-releasing hormone, a major regulator of the stress response (17, 18), and other genes. Additionally, TWASs of MDD predicted alterations in DLPFC expression of CTC-467M3.3, FAM120A, and TMEM161B (11, 19). Emerging DLPFC bulk-tissue RNA sequencing (RNA-seq) studies have largely confirmed these gene signatures and have pointed to the involvement of interneuron and immune cell–associated pathways (20–24).
To make inferences of cell-type-specific effects, human postmortem bulk-tissue RNA-seq studies have largely relied on deconvolution and enrichment methods. Single-cell and single-nucleus RNA-seq methodologies allow direct analysis of the transcriptome at single-cell resolution and have revealed cell-type-specific expression signatures of disease. Studies in psychiatry have started to reveal such transcriptomic signatures associated with disease and pathology severity (25–28). For example, a recent study of male depressed suicide completers identified transcriptional dysregulation specific to clusters of oligodendrocyte precursor cells (OPCs) and deep-layer excitatory neurons (26). Here, we elucidate brain cell-type-specific mechanisms of stress response in PTSD and MDD. We report the first comprehensive single-nucleus transcriptomic characterization of postmortem human DLPFC from individuals with PTSD. We validate our pathway findings with an in vitro model and leverage neuropsychiatric and neuroimaging GWASs to assess the impact of our findings at the population level.
METHODS
In this study (Figure 1A), we generated a discovery data set of ~415K single-nucleus transcriptomes from 32 DLPFC brain samples from individuals with PTSD (N=11) and MDD (N=10) compared with neurotypical control subjects (N=11) from the VA’s National PTSD Brain Bank (NPBB) (29) to detect cell-type-specific expression alterations associated with PTSD and MDD. We also generated bulk-tissue RNA-seq data from the same samples to highlight the enhanced sensitivity of single-nucleus RNA-seq technologies in detecting disease-specific differentially expressed genes (DEGs) and dysregulated pathways. For replication, we generated a single-nucleus RNA-seq data set of 15 NPBB DLPFC samples (five per group), corresponding to ~160K single-nucleus transcriptomes. The newly identified cell-type-specific pathways of PTSD and MDD were evaluated in transcriptomic data from iPSC-derived neurons exposed to the potent glucocorticoid dexamethasone (DEX) in vitro. Large-scale DLPFC-based summary-data-based Mendelian randomization (SMR) and TWAS of PTSD (N~325K) and MDD (N~500K) were used to identify overlap of cell-type-specific DEGs with putative causal genes. Finally, the relevance of identified genes to brain structure and connectivity was evaluated with DLPFC-based TWASs of multimodal neuroimaging (N~33K). The study methods are described in detail in the online supplement.
FIGURE 1. Single-nucleus transcriptomics in PTSDa.

a Panel A shows the single-nucleus RNA sequencing (snRNA-seq) workflow. To create a discovery data set, postmortem brain samples were taken from dorsolateral prefrontal cortex (DLPFC) of 32 individuals: 11 with posttraumatic stress disorder (PTSD), 10 with major depressive disorder (MDD), and 11 neurotypical control subjects (CON). Nuclei were isolated and encapsulated with barcoded gel beads, using the 10x Genomics microfluidic chips and device. Encapsulation was followed by nucleus lysis, library preparation, and sequencing. The mapped reads corresponded to ~415K nuclei (>13K cells per sample). Nuclei passing quality-control (QC) metrics were clustered. Cell clusters were annotated for cell type and cell subtype identity. Differential gene expression analysis identified snRNA-seq differentially expressed genes (snDEGs) in the major cell types based on two outcomes, PTSD and MDD, and replication was assessed by generating an snRNA-seq data set from an independent set of 15 individuals (five subjects per group) containing ~160K nuclei (>10K cells per sample). Pathway analyses of the identified snDEGs were conducted, followed by validation of a disease-associated pathway in induced pluripotentstem cell–derived mature neurons exposed to glucocorticoids. The overlap of the identified snDEGs with the geneticrisk of PTSD and MDD was evaluated through large-scale DLPFC-based summary-data-based Mendelian randomization (SMR) and transcriptome-wide association study (TWAS), and the overlap with genetic underpinnings of brain structure and connectivity was evaluated through DLPFC-based TWAS of DLPFC-based neuroimaging. Integrating the results across all data modalities highlighted the most convergent loci and genes. Panel B is a t-distributed stochastic neighbor embedding (t-SNE) plot of the post-QC 362,996 cells from the discovery data set, annotated into eight major cell types. Cell type annotation relied on cell-type-specific canonical and previously reported markers in prior snRNA-seq studies. Cell type proportions are indicated as percentage in the full dataset. OPC=oligodendrocyte precursor cells. Panel C shows eightt-SNEplots of the post-QC 362,996 cells annotated by major cell-type-specific canonical markers. Above each plot is the marker used for annotation, and cells with expression are shown with their respective color. Greater color intensity denotes greater expression of the respective marker. Genes used for cell type annotation were PLP1 for oligodendrocytes, PDGFRA for OPCs, AQP4 for astrocytes, FLT1 for endothelial cells, DCN for pericytes, DOCK8 for microglia, SLC17A7 for excitatory (EX) neurons, and GAD1 for inhibitory (IN) neurons. Arrows indicate the smaller cell clusters. Panel D is a t-SNE plot of the post-QC 362,996 cells, annotated into 22 cellsubtypes. Cell subtype annotation relied on known cell-type-specific markers. Two cell clusters were assigned to astrocyte subtypes, eight to EX subtypes, and seven to IN subtypes. Panel E is a dendrogram depicting hierarchical clustering between the 22 cell clusters based on the overall gene expression, and panel Fisa dot plot depicting gene expression levels of cell-type-specific markers used to annotate the cell clusters into cell types and subtypes. The color of the lettering of the name of each cell cluster corresponds to the major cell type they belong to (colors are matched with panel D). At the bottom are the genes used for cell type annotation. The size of each dot represents the percentage of cells expressing the gene, where the lower threshold was set at 25%. The color intensity denotes the average scaled expression level. Panels Gand Hareviolinplotsofgeneexpressionofcell-type-specificmarkersusedtoannotate EX and IN cell subtypes, respectively. Markers used are labeled on the right of each plot. The eight EX clusters’ annotation was based on the expression of cortical layer markers. The IN clusters’ annotation was based on the expression of genes associated with developmental origin (e.g., LHX6, ADARB2), corticallayer, calciumbinding (e.g., PVALB), neuropeptide signaling(e.g., SST, TAC1, VIP), and other canonical and noncanonical marker expression. Panel I is abar plot of the proportions of each of the major cell types in the control (CON), MDD, and PTSD groups. Similar cell type composition is observed in all three groups. Proportions are expressed as ratios and add up to 1.
RESULTS
Profiling DLPFC Cell-Type-Specific Transcriptome in PTSD and MDD
The 32 samples were selected from the pool of NPBB brains, matched for age at death, sex, postmortem interval, and race (see Table S1 in the online supplement) and were profiled using droplet-based single-nucleus RNA-seq (~415K nuclei, ~12K nuclei per subject) at high sequencing depth (~46K reads per nucleus). Sequencing metrics did not differ between the groups (see Table S2A,B in the online supplement). Graph-based clustering identified 22 distinct cell clusters annotated into cell types and subtypes based on enrichment in well-established markers (see Table S3A–D in the online supplement). The 22 cell clusters were then annotated into eight major cell types (ordered by cell type proportion; see Figure 1B,C): excitatory (EX) and inhibitory (IN) neurons, oligodendrocytes, astrocytes, OPCs, microglia, endothelial cells, and pericytes. Across all samples, EX neurons were the most prevalent cell type (45.8% of cells) and pericytes the least (0.4%). The astrocyte clusters were further annotated into two astrocyte subtypes, EX clusters into eight EX subtypes, and IN clusters into seven IN subtypes (Figure 1D; see also Table S4 in the online supplement). Cell cluster annotation was corroborated by hierarchical clustering (Figure 1E), with cluster distances explained by cell type and lineage. Markers distinguishing cell types and EX and IN neurons are plotted in Figure 1F–H. Gene coexpression network analysis verified transcriptomic consistency between related cell types (see Supplementary Results in the online supplement). There were no statistically significant group differences in abundance of cell types and subtypes surviving multiple testing (Figure 1I; see also Table S5A,B in the online supplement).
Differential Gene Expression (DGE) Analysis in PTSD and MDD
We further conducted DGE analysis in the eight major cell types and bulk tissue (see Table S6A,B in the online supplement for PTSD and MDD). We identified 194 false discovery rate (FDR) corrected significant single-nucleus DEGs (snDEGs) in the discovery data set, termed D-snDEGs (Figure 2A, B): 38 in association with PTSD (89% in EX neurons) and 156 with MDD (87% in EX neurons). Sensitivity analysis confirmed that the results were not affected by RNA quality (see Supplementary Results in the online supplement).
FIGURE 2. Single-nucleus differentially expressed genes (snDEGs) in PTSDa.
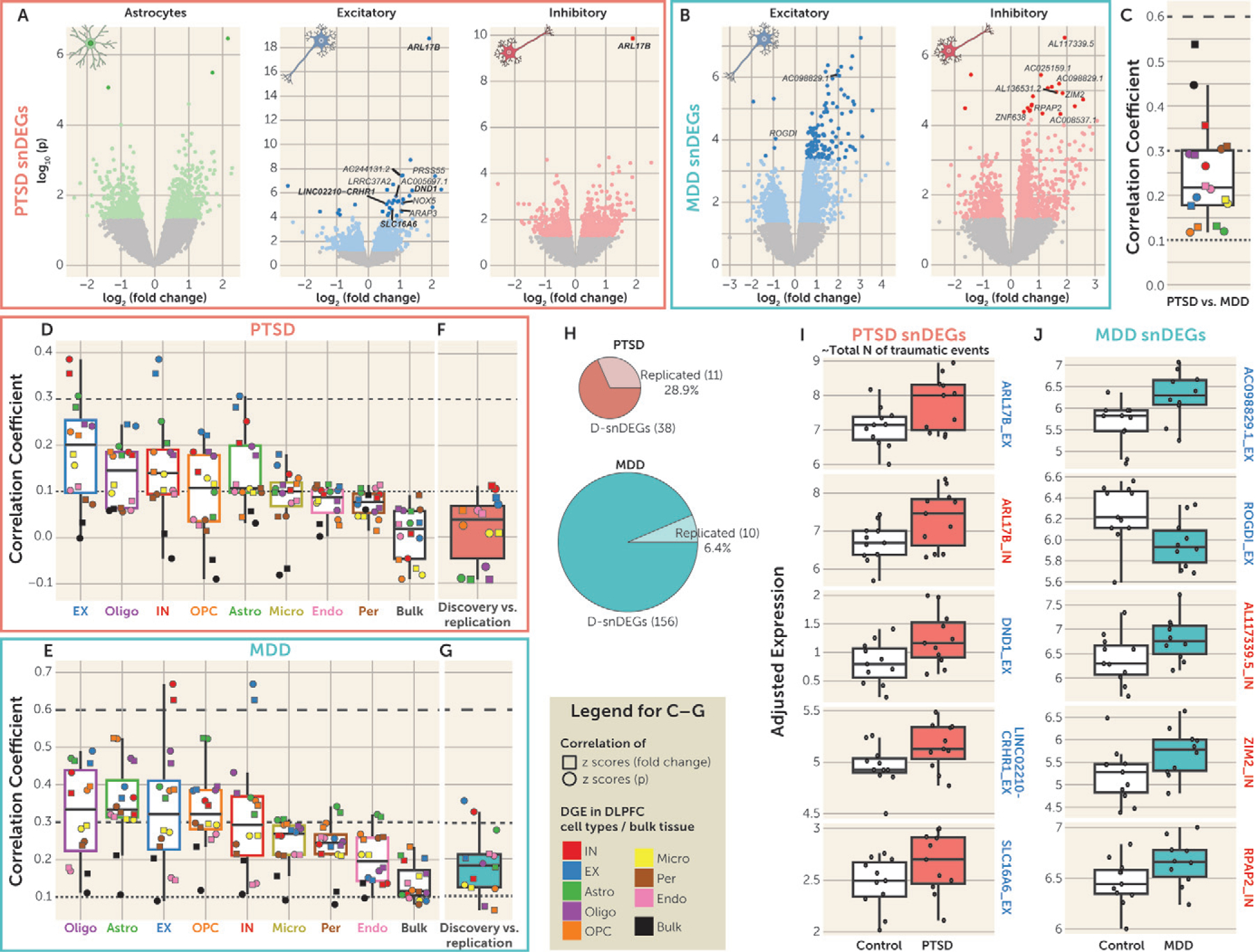
a Panel A shows volcano plots of the posttraumatic stress disorder (PTSD) differential gene expression (DGE) of the discovery sample in astrocytes and excitatory (EX) and inhibitory (IN) neurons. Colored dots denote nominally significant genes (p<0.05), and the darker colored dots denote genes with false discovery rate (FDR)–adjusted p<0.05 (termed discovery snDEGs: D-snDEGs). The genes that were found to be snDEGs in the replication data set are named on each plot. D-snDEGs that were also significantly associated with total number of traumatic events (FDR-adjusted p<0.05) are depicted as squares, and their names are in boldface if they were snDEGs in both discovery and replication. Panel B shows volcano plots of the major depressive disorder (MDD) differential gene expression of the discovery sample in EX and IN neurons. Colored dots denote nominally significant genes (p<0.05), and the darker colored dots denote genes with FDR-adjusted p<0.05 (D-snDEGs). The replicated D-snDEGs are named. Panel C shows the correlation of PTSD and MDD DGE in the discovery sample. Two types of correlation analyses were performed: Pearson correlation of standardized scores of fold changes (squares) and signed p values (circles). Data are represented as median ±1.5 interquartile range (IQR), and the individual points denote the correlation between PTSD and MDD for each cell type or bulk tissue. Dashed lines at coefficients 0.1, 0.3, and 0.6 annotate weak, moderate, and high strength of correlation, respectively. Panels D and E show the correlation of the DGE in association with PTSD and MDD, respectively, between cell types or bulk tissue in the discovery sample. Two types of correlation analyses were performed: Pearson correlation of standardized scores of fold changes (squares) and signed p values (circles). Cell type/bulk tissue box plots are ordered from highest (left) to lowest (right) median correlation. Data are represented as median ±1.5 IQR, and the individual points denote the correlation at each cell type or bulk tissue. Dashed lines at coefficients 0.1, 0.3, and 0.6 annotate weak, moderate, and high strength of correlation, respectively. Panels F and G show the correlation ofthe DGE in association with PTSD and MDD, respectively, between the discovery and replication samples. Two types of correlation analyses were performed: Pearson correlation of standardized scores of fold changes (squares) and signed p values (circles). Data are represented as median ±1.5 IQR, and the individual points denote the correlation at each cell type or bulk tissue. Dashed lines at coefficients 0.1, 0.3, and 0.6 annotate weak, moderate, and high strength of correlation, respectively. Panel H presents pie charts of the number of D-snDEGs and replicated D-snDEGs for PTSD and MDD. Panels I and J summarize the expression of the top five replicated D-snDEGs in association with PTSD/total number of traumatic events and MDD, respectively, in the discovery data set. The replicated D-snDEGs are ranked according to the p value in the discovery data set (lowest p values at the top). The red boxes correspond to the PTSD group (N=11), the green boxes to the MDD group (N=10), and the white boxes to the control group (N=11). The depicted genes are either EX (blue outline) or IN (red outline) neurons’ D-snDEGs. The y-axis depicts adjusted expression. Data are represented as median ±1.5 IQR, and the individual subject values are depicted as circles. For each gene, the t statistic and p value of the group differences (PTSD vs. control and MDD vs. control) were estimated by the limma-based DGE analysis and are included in Tables S6 and S7 in the online supplement.
Since trauma exposure events are significantly related to PTSD diagnosis, we evaluated the effects of number of traumatic events in the expression of the PTSD D-snDEGs. Indeed, in the entire brain cohort (N=380) from which the samples analyzed here were selected, there was a high correlation between total number of traumatic events and PTSD diagnosis (rho=0.87, p<1–25). Of the 38 D-snDEGs associated with PTSD, expression levels of 14 were also correlated with total number of traumatic events at an adjusted p threshold of 0.05 (see Table S7 in the online supplement).
Genome-Wide Comparison of DGE Signatures Between Cell Types and Disorders
Standardized scores of fold changes and signed p values from the DGE signatures of PTSD and MDD samples were correlated to characterize the relationships of the disorders within cell type and within bulk tissue (Figure 2C) and between cell types/bulk tissue within PTSD (Figure 2D) and within MDD (Figure 2E). We categorized the correlations into strong (r>0.6), moderate (0.3<r≤0.6), weak (0.1< r≤0.3), and no (r≤0.1) correlation. The between-disorder analysis revealed moderate correlations for bulk tissue, IN neurons, and pericytes, while correlations for all other cell types were weak, suggesting overall distinct cell-type-specific transcriptional signatures in PTSD compared with MDD (Figure 2C).
Between–cell type/bulk tissue correlations of DGE were lower in PTSD (including weak correlations for EX neurons, oligodendrocytes, IN neurons, OPCs, and astrocytes) (Figure 2D), compared with MDD (including moderate correlations for oligodendrocytes, astrocytes, EX neurons, and OPCs and weak correlations for IN neurons, microglia, pericytes, endothelial cells, and bulk tissue) (Figure 2E).
The correlation coefficients between EX and IN neurons were the highest for both analyses (moderate in PTSD and high in MDD). The median correlation of bulk-tissue DGE with DGE from the cell types was negligible in PTSD and weak in MDD, with all individual cell types showing either no correlation or weak correlation (Figure 2D,E). Rank-rank hypergeometric overlap (RRHO) provided alternative support and visualization of the same relationships (between PTSD and MDD, within PTSD, and within MDD).
Replication of Cell Cluster Annotation and Cell-Type-Specific Alterations
We profiled 15 additional DLPFC samples to capture ~160K DLPFC cells (~11K nuclei per subject; see Table S8A–C in the online supplement) that clustered into 17 cell clusters and seven major cell types (see Figure S4A,B in the online supplement). We confirmed correspondence of cell cluster annotations of the discovery data set with those of the replication data set (see Figure S4C in the online supplement), the prior autism spectrum disorder (ASD) study (see Figure S5 in the online supplement) (25), and the prior MDD study (see Figure S6 in the online supplement) (26) using the respective between-cluster DEGs (see Supplementary Results in the online supplement).
We generated DGE of PTSD and MDD in the replication data set (see Table S9A,B in the online supplement) to compare with the discovery DGE. We computed correlations of standardized scores of fold changes and signed p values (Figure 2F,G) as well as RRHO analyses (PTSD, MDD) to reveal a weak correlation in IN and EX neurons for PTSD and moderate correlations in IN and EX neurons and weak correlations in the other cell types for MDD.
Enrichment analysis of the cell-type-specific PTSD and MDD snDEGs (p<0.05) in the replication sample, based on a binomial distribution, revealed significant enrichments in astrocytes (PTSD: p=2.26e–6), EX neurons (PTSD: p=1.27e–32; MDD: p=3.84e–96), IN neurons (PTSD: p=9.15e–7; MDD: p=3.17e–90), and oligodendrocytes (PTSD: p=1.23e–2, MDD; p=7.24e–8). Notably, 11 of 38 (28.9%) D-snDEGs of PTSD, and 10 of 156 (6.4%) D-snDEGs of MDD were FDR significant in the replication data set, with the same direction of effect observed in the discovery data set (Figure 2H; see also Table S10A,B in the online supplement). In Figure 2I, the top replicated genes for PTSD that were also significantly correlated with number of traumatic events (see Table S10C in the online supplement) are plotted, while in Figure 2J, the top replicated MDD snDEGs (see also Table S10D in the online supplement) are plotted.
Differential Module Eigengene Expression Analysis in PTSD and MDD
We also conducted differential module eigengene expression analysis to identify networks associated with PTSD and/or MDD. No cell-type or bulk-tissue gene network was associated with PTSD (see Table S11A in the online supplement), while three EX networks and one IN network were FDR-significantly upregulated in association with MDD (see Table S11B in the online supplement). The EX “mediumorchid (ME22)” network is involved in regulation of innate immunity and includes eight D-snDEGs as hub genes (Figure 3A; see also Table S11C in the online supplement). Two gene networks were related to nucleic acid metabolism: the EX “antiquewhite2 (M10)” and IN “blue2 (M4)” with RPAP2, a replicated D-snDEG, as a hub gene (Figure 3A; see also Table S11C in the online supplement). Finally, the EX “blue (M6)” was involved in structure of intracellular organelles and included two D-snDEGs, MAPK14 and XRN1, as hub genes (Figure 3A; see also Table S11C in the online supplement).
FIGURE 3. Alterations in network, pathway, and upstream regulator activity in PTSD and MDDa.

a Panel A shows the module eigengene (ME) expression of coexpression networks associated with major depressive disorder (MDD). Expression of modules with false discovery rate (FDR)–adjusted p<0.05 is plotted. For each module, the t statistic and p value of the group differences (posttraumatic stress disorder [PTSD] vs. control and MDD vs. control) were estimated by the differential ME expression analysis and are included in Table S11B in the online supplement. Fold change values and p values are shown above each plot. Information about the number of coexpressed genes in each module and the cell type (excitatory [EX] or inhibitory [IN] neurons) is provided in the y-axis labels. Each module was subjected to Gene Ontology (GO) enrichment analysis, and the top three enrichment terms are shown together with the FDR-adjusted p value <0.05. The top 10 hub genes in each module are also displayed in the last column. The hub genes that are significant single-nucleus differentially expressed genes in the discovery data set (D-snDEGs) are in boldface, and replicated D-snDEGs are in boldface and underlined. In panel B, gene-set enrichment analysis identified 54 pathways with differential enrichments in PTSD compared with MDD in EX and/or IN neurons. Depicted pathways are from C2-curated gene sets from Molecular Signatures Database. Each cell represents the normalized enrichment score (NES) of the respective pathway, with red indicating positive enrichment and blue negative enrichment; the intensity of the color denotes the magnitude of the effect. An asterisk indicates that the enrichment of the respective gene set was nominally significant (p<0.05), and a hash sign indicates that the enrichment of the respective gene set was FDR significant (FDR-adjusted p<0.05). Biclustering was based on pathway NESs for PTSD and MDD in EX and IN neurons. The clustering of columns (MDDEX, MDDIN, PTSDIN, and PTSDEX) is depicted, and rows (pathways) are clustered in four blocks (separated by white space). In panel C, upstream regulator analysis highlights that networks of cytokines, hormones, and other regulatory molecules are differentially activated in MDD and PTSD. Upstream regulator analysis was performed using Ingenuity Pathway Analysis (IPA). The heat map shows the predicted activity of upstream regulators that were FDR significant in both cell types in at least one disorder. Each cell represents the activation z score of the respective upstream regulator, with red indicating positive z score (activated), blue negative z score (inhibited), and gray not predicted activity; the intensity of the color denotes the magnitude of the effect. An asterisk indicates that the upstream regulator was nominally significant (p<0.05), and a hash sign indicates that the upstream regulator was FDR significant (FDR-adjusted p<0.05). Biclustering was based on activation z scores for PTSD and MDD in EX and IN neurons. The columns (upstream regulators) are clustered within the functional class they belong to (cytokines, endogenous and exogenous chemicals, growth factors, transcription factors [TF], and kinases) and the rows are clustered by DGE signature (MDD EX, MDD IN, PTSD IN, and PTSD EX). CORT=corticosterone; DEX=dexamethasone; E2=estradiol; GH=growth hormone; LPS=liposaccharide; P4=progesterone; TF=transcription factors; TPA=12-O-tetrade-canoylphorbol-13-acetate. Panel D is a heat map with the gene overlap of target genes of upstream regulators with the respective pathways. The top five upstream regulators based on their overlap with pathways are depicted. Each cell represents the NES of the respective pathway, with the green color intensity denoting the level of significance of the overlap. DEX=dexamethasone; E2=estradiol; LPS=liposaccharide. Panel E shows mechanistic networks for dexamethasone effects in MDD IN neurons (upper) and PTSD EX neurons (lower). The mechanistic network of the upstream regulators and their relationship were predicted by IPA. The molecules (nodes) shown in blue are predicted to be inhibited, and those shown in orange are predicted to be activated; color intensity represents the level of inhibition or activation. Lines (edges) ending with an arrow indicate activation, and lines without an arrow indicate inhibition. Blue lines represent weakened relationship, orange lines represent strengthened relationship, yellow lines represent inconsistent effects relative to the state of downstream molecule, and gray lines represent interactions for which direction of change is not predicted. Continuous and dashed lines indicate direct and indirect effects, respectively. The shapes of the nodes reflect the functional class of molecule product: transcription regulator (ellipse), cytokine/growth factor (square), ligand-dependent nuclear receptor (rectangle), and complex/group/other (circle).
Cell Type Enrichment in PTSD and MDD GWAS
With the majority of D-snDEGs (98.4%) and disease-associated gene networks in EX and IN neurons, we sought to determine whether PTSD and MDD GWASs (Ns, ~325K and ~500K, respectively) would support the enrichment of disease-associated genes in DLPFC neuronal cell type markers. Using the cell type annotation in this study, we observed FDR-significant enrichments in EX and IN neurons and OPCs for both disorders, with EX and IN neurons being the most statistically significant for PTSD and MDD, respectively (see Figure S7A in the online supplement).
Cell-Type-Specific Pathways and Upstream Regulators
Since our D-snDEGs and GWAS cell type enrichments converged in the involvement of neuronal cell types in PTSD and MDD, we investigated enriched biological processes in these cell types (see Table S12A,B in the online supplement). More pathways from the Molecular Signatures Database were associated with MDD compared with PTSD (675 vs. 459, respectively), and 140 were associated with both disorders (see Figure S7B in the online supplement). To better differentiate PTSD and MDD, we calculated the difference of enrichment scores between the two disorders in 399 pathways with FDR-significant signals in both cell types (see Figure S7C in the online supplement). We observed the highest difference between PTSD and MDD in the “C2” pathway category, which included genetic/chemical perturbations and canonical pathways (see Figure S7D in the online supplement). These pathways are related to macrophage differentiation, metabolism and mitochondria biology, immune response, tumor differentiation, and multiple enzyme functions (Figure 3B).
We then identified upstream regulators associated with EX and IN DGE signatures of PTSD and MDD(see Table S13A,B in the online supplement). Among them, 24 regulators showed FDR significance in both cell types and predicted activation z score (Figure 3C). Tumor necrosis factor and brain-derived neurotrophic factor were predicted to have increased and decreased activity, respectively, in both traits and in both cell types (Figure 3C). Alterations in steroid hormone activity were also predicted, with increased progesterone activity in both cell types for both traits, differential estrogen activity based on both trait and cell type, and opposing GC activity in PTSD compared with MDD in both cell types (Figure 3C).
We ranked the significant upstream regulators based on the overlap of their target gene sets with the dysregulated pathways; the top five (LPS, MYC, TGFB1, E2, DEX) are depicted in Figure 3D. The differential involvement of GCs in PTSD and MDD was observed at the pathway level (i.e., “Rhein ALL Glucocorticoid Therapy DN”) and at the upstream regulator level (i.e., “DEX”). The predicted DEX-regulated transcriptional network in PTSD and in MDD can be found in Figure 3E. Our observations in the DLPFC neurons were concordant with previous studies relying on plasma/serum and peripheral blood mononuclear cells (PBMCs). These studies have associated heightened GC sensitivity of the HPA axis and low GC tone/signaling with PTSD, with the opposite effects reported in MDD (8, 30). Our observations here also prompted us to test further the GC signaling involvement and investigate whether it was specific to glucocorticoid receptor (GR) and mineralocorticoid receptor (MR). Using gene sets of in vivo GR- and MR-specific targets in the brain (31), we observed enrichment of direct targets of both receptors in the DGE signatures of PTSD and MDD in both EX and IN neurons, with opposing direction of enrichment in the two traits and stronger effects for GR target genes (see Figure S8A in the online supplement).
Investigation of Neuron-Based GC Pathway Involvement in PTSD and MDD
To validate the GC pathway dysregulation observed in PTSD and MDD, we also created an appropriate in vitro system. PBMCs have been the most used cell type for researching PTSD in vitro. Yet, we found that PTSD and MDD GWAS genes are enriched in neuron-specific markers (Figure 4A; see also the curated marker list in Table S14 in the online supplement). Motivated by this and the neuron-based vulnerability highlighted by our D-snDEGs, we opted to use iPSC-derived neurons for modeling of the GC pathway in vitro.
FIGURE 4. In vitro validation of a single-nucleus RNA-seq–discovered pathway with iPSC-derived neuronsa.

a Panel A shows results of the enrichment of genome-wide association studies (GWASs) of posttraumatic stress disorder (PTSD) and major depressive disorder (MDD) summary statistics in cell-type-specific markers for induced pluripotent stem cells (iPSCs), peripheral blood mononuclear cells (PBMCs), and cortical neurons. Each point represents the negative logarithm of the MAGMA-based enrichment p value (y-axis) for each literature-reported marker gene set (x-axis) (see marker gene sets in Table S14 in the online supplement). The shapes denote the two different input GWASs’ summary statistics (square=PTSD; triangle=MDD), and error bars indicate standard error of the mean. Opaque dots denote cell-type-specific markers that were false discovery rate (FDR) significant. Panel B illustrates iPSCs being differentiated into cortical neurons. The top left panel is a bright-field image of an iPSC colony displaying nominal cellular morphology; the bottom left panel is an immunofluorescence image of iPSCs expressing the canonical nuclear pluripotency marker NANOG (green). The top right panel is a bright-field image of iPSC-derived cortical cultures after ~60 days of differentiation displaying expansive neurite arborization and connectivity; the bottom right panel is an immunofluorescence image of neurons expressing the mature neuron markers for cytoskeleton and nucleus, MAP2 (green), and NeuN (red). DAPI is used as a nuclear counterstain. Scale bars are 100 mm. Panel C shows gene expression in control iPSC-derived neurons of literature-reported cell-type-specific markers for iPSCs, PBMCs, and neurons (see Table S14 in the online supplement). Blue denotes the iPSCs’ marker set, orange the PBMCs’ marker set, and red the neurons’ marker set. Data are represented as box plots of normalized expression (log2 scale), with data points labeled with their corresponding marker gene. The between-gene-set differences in expression were estimated by two-tailed Wilcoxon signed-rank test. Panel D shows representative micrographs of glucocorticoid receptor (GR) in iPSC-derived neurons. Cells were stained with the neuronal nuclear marker NeuN (red), the GR protein (green), and the nuclear DNA marker DAPI (blue). The left panel is of iPSC-derived neurons in vehicle conditions, with minimal GR signal localized in the nucleus. The right panel is of iPSC-derived neurons exposed to 100 nM dexamethasone (DEX) for 1 hour, showing that GR signal has been translocated into the nuclei. White arrows indicate colocalization of GR with NeuN. Panel E shows nuclear GR levels, with quantification of the average ratio of GR signal to nucleus area for individual neuronal nuclei across control (0 nM) and 100 nM DEX conditions, with DEX-treated neurons having a higher ratio of GR signal in their nuclei (control group, N=122 nuclei; DEX group, N=157 nuclei). Data are represented as violin plots using a fold change (from the mean of the control group). The p value of the group difference was estimated by a two-tailed Wilcoxon signed-rank test. Panel F shows DEX-induced increased levels of phosphorylated GR at Ser211 (pGR-Ser211), indicative of an activated and nucleus-localized GR in iPSC-derived neurons (control group, 0 nM DEX; DEX group, 100 nM DEX). Data are represented as mean fold change (from the control group) ± standard error of the mean. The p value of the group difference was estimated by a two-tailed unpaired t test. Panel G shows DEX-induced increase in FKBP5 protein levels (in fold change), indicative of direct GR-binding to the FKBP5 gene leading to upregulation. FKBP5 protein was measured by capillary-based immunoblotting (control group, 0 nM DEX; DEX group, 100 nM DEX). Data are represented as mean fold change (from the control group) ± standard error of the mean. The p value of the group difference was estimated by a two-tailed unpaired t test. Panel H is a volcano plot illustrating the relationship of p value with fold changes of differential gene expression (DGE) results based on 100 nM versus 0 nM DEX in iPSC-derived neurons. Green denotes downregulation, and purple denotes upregulation. Dots with high-intensity color indicate differentially expressed genes with FDR-adjusted p<0.05. Low-intensity color denotes p<0.05. Gray dots indicate that the gene was not significant. The top genes with p<1.0–27 have been named. Panels I and J are rank-rank hypergeometric overlap (RRHO) plots between DEX-induced DGE in iPSC-derived neurons and PTSD and MDD neuronal DGE, respectively (excitatory neurons [EX] at left and inhibitory neurons[IN] at right). For this analysis, genes in each DGE list were ranked according to the product of the sign of log2(fold change) with −log10(p). The plotted RRHO heat map represents the extent of overlap with the colors based on −log10(p) of the hypergeometric test measuring the significance of overlap of gene lists. Warm colors in the bottom left and top right quadrants reflect overlap in genes with upregulation or downregulation, respectively, in both data sets. Warm colors in the top left and bottom right quadrants reflect overlaps in genes with opposite direction of effects in the two data sets. The coefficient (ρ) of Spearman correlations based on the ranking metrics are given as reference of effect size of the relationship. Panels Kand L present scatterplots of z statistics of the PTSD effect and MDD effect, respectively, in EX neurons and IN neurons with DEX effect in iPSC-derived neurons. Nominally significant genes in each pair of analyses were selected, and genes with z score (of signed p value) >3 in the PTSD and/or the MDD analysis, respectively, are named. Each red circle corresponds to a gene and denotes the PTSD effect or the MDD effect in EX neurons paired with the DEX effect in iPSC-derived neurons, and each blue triangle corresponds to a gene and denotes the PTSD effect or the MDD effect in IN neurons paired with the DEX effect in iPSC-derived neurons. For each panel, the regression line was plotted along with the confidence interval, and the Pearson correlation coefficient, the p value, and the size of the gene set are listed. Inserts show also the respective normalized enrichment score (NES) of PTSD and MDD DGE signatures for DEX-regulated genes (up or down) in iPSC-derived neurons’ DGE. An asterisk indicates significant enrichment.
iPSC-derived neurons expressed canonical neuron markers, MAP2 and NeuN (Figure 4B), high levels of cortical neuron markers (Figure 4C; see also Figure S8B,C in the online supplement), the GR gene (NR3C1), and, to a lesser extent, the MR gene (NR3C2; see Figure S8D in the online supplement). GR was expressed throughout their plasma membrane, cytoplasm, and nucleus in basal conditions at various levels (see Figure S8E in the online supplement). One hour of exposure to DEX (100 nM), a selective GR agonist, induced a 1.5-fold increase in average nuclear GR compared with vehicle-treated neurons (p=1.1e–14) (Figure 4D,E; see also Figure S8D and Table S15 in the online supplement), indicative of DEX-induced GR nuclear translocation. After 4 hours of DEX exposure, Western blotting showed a 2.5-fold increase in phosphorylated GR at Ser211 (Figure 4F), a strong indicator of GR nuclear translocation and activation (32), and increased FKBP5 protein (Figure 4G), indicative of fast induction of protein expression of a GC/GR target gene (8). RNA-seq identified 1,239 DEGs associated with DEX exposure (Figure 4H; see also Table S16A in the online supplement). Six DEX-induced DEGs were verified by qPCR (see Figure S9 in the online supplement), including the most upregulated DEG (SERPINE1), three canonical GC/GR targets (FKBP5, TSC22D3, SGK1), a PTSD D-snDEG (SLC16A6), and a PTSD/MDD TWAS gene (FURIN). The DEX-induced DEG signature included gene targets of GR binding (see Table S16B,C in the online supplement) and was associated with enrichment of GR and DEX as upstream regulators (see Table S16D in the online supplement).
We then tested whether GR-mediated DEX effects in iPSC-derived neuronal cultures are comparable to the PTSD and/or MDD DGE signatures in EX and IN neurons. We observed positive weak to moderate correlations of standardized fold changes and signed p values of in vitro DEX effects with PTSD effects in EX and IN neurons (EX neurons: r=0.09 and r=0.14, respectively; IN neurons: r=0.18 and r=0.25, respectively), and negative moderate correlations with MDD effects only in EX neurons (r=−0.14 and r=−0.18, respectively). When focusing on nominally significant genes across data sets, the correlation effect sizes became moderate to strong, and plotting revealed gene drivers of the correlations in PTSD and MDD (Figure 4K,L).
Next, gene-set enrichment analysis with the in vitro DEX-induced DEGs as a target gene set showed that in PTSD, genes downregulated in vitro by DEX were downregulated in EX and IN neurons (normalized enrichment scores, −1.76 and −1.99, respectively; see the enrichment plots in Figure S10A,B in the online supplement), and genes upregulated by DEX were upregulated only in IN neurons (normalized enrichment score=1.59; see Figure S10B in the online supplement). In MDD, only genes downregulated by DEX were upregulated in EX neurons (normalized enrichment score=1.49; see Figure S10C in the online supplement). The difference in directionality of enrichment of DEX-regulated genes in PTSD versus MDD in EX neurons was in line with the opposite direction in enrichments we predicted using gene-set enrichment analysis and upstream regulator analysis.
DLPFC-Based SMR and TWAS of PTSD and MDD
To integrate our PTSD and MDD snDEGs with PTSD and MDD GWAS, we conducted a combination of summary-data-based Mendelian randomization (SMR) and TWAS analyses. For SMR analysis, we used DLPFC bulk-tissue expression quantitative trait loci (eQTL) (33) and cell-type-specific eQTLs (matching the eight DLPFC cell types of the discovery data set [34]) to discover FDR-significant SMR genes with nonsignificant tests for heterogeneity in dependent instruments. At the bulk-tissue level, we identified 22 genes for PTSD and 42 for MDD, and at the cell-type level, we identified 36 genes for PTSD and 12 for MDD (see Figure S11 and Table S17 for PTSD and Table S18 for MDD in the online supplement). We next conducted TWASs of PTSD and MDD using GReX model weights, which we trained in a large DLPFC genotype expression panel, validated, and found superior to prior models (see Supplementary Results in the online supplement). Our TWAS revealed 72 FDR-significant associations for PTSD and 117 for MDD (see Figure S11 and Table S19A,B in the online supplement).
DLPFC-Based TWAS of DLPFC-Based Neuroimaging
To examine the relevance of our PTSD and MDD snDEGs for brain structure and connectivity, we used the UK Biobank GWAS results of multimodal imaging-derived phenotypes (IDPs) from ~33K subjects (35) to conduct neuroimaging TWASs. IDPs belonged to 14 structural MRI and diffusion MRI categories. Since our single-nucleus RNA-seq study was in DLPFC, we compared DLPFC-based TWAS signals for 392 IDPs that were neuroanatomically DLPFC based against the respective signals of 903 non-DLPFC-based IDPs (see Figure S12A and Table S20 in the online supplement). For 59 genes, there was a higher percentage of Bonferroni-significant TWAS signals in DLPFC-based IDPs compared with non-DLPFC-based IDPs, especially in particular white matter tracts, with 19 genes localized at the 17q21.31 locus (see Figure S12B in the online supplement). LINC02210 showed the most extreme contrast, with significant gene-trait associations in 20.4% of DLPFC-based IDPs, compared with 7.1% of non-DLPFC-based IDPs (see Figure S12C in the online supplement).
Integration of Findings
We integrated gene-level findings across all data modalities (our snRNA-seq PTSD/MDD study, SMR/TWAS of PTSD/MDD, neuroimaging TWAS, in vitro DEX study, and the published bulk-tissue PTSD/MDD studies [22–24]) to reveal the most convergent genomic loci and genes (Figure 5A). Genes located in the 17q21.31 locus showed greater convergence mostly at the level of postmortem findings for PTSD and genetic-based findings. Genes in other loci showed convergence at the level of postmortem PTSD or MDD findings and DEX findings (Figure 5B).
FIGURE 5. Highlighting the most important genes upon integration of all the reported data modalities in this studya.
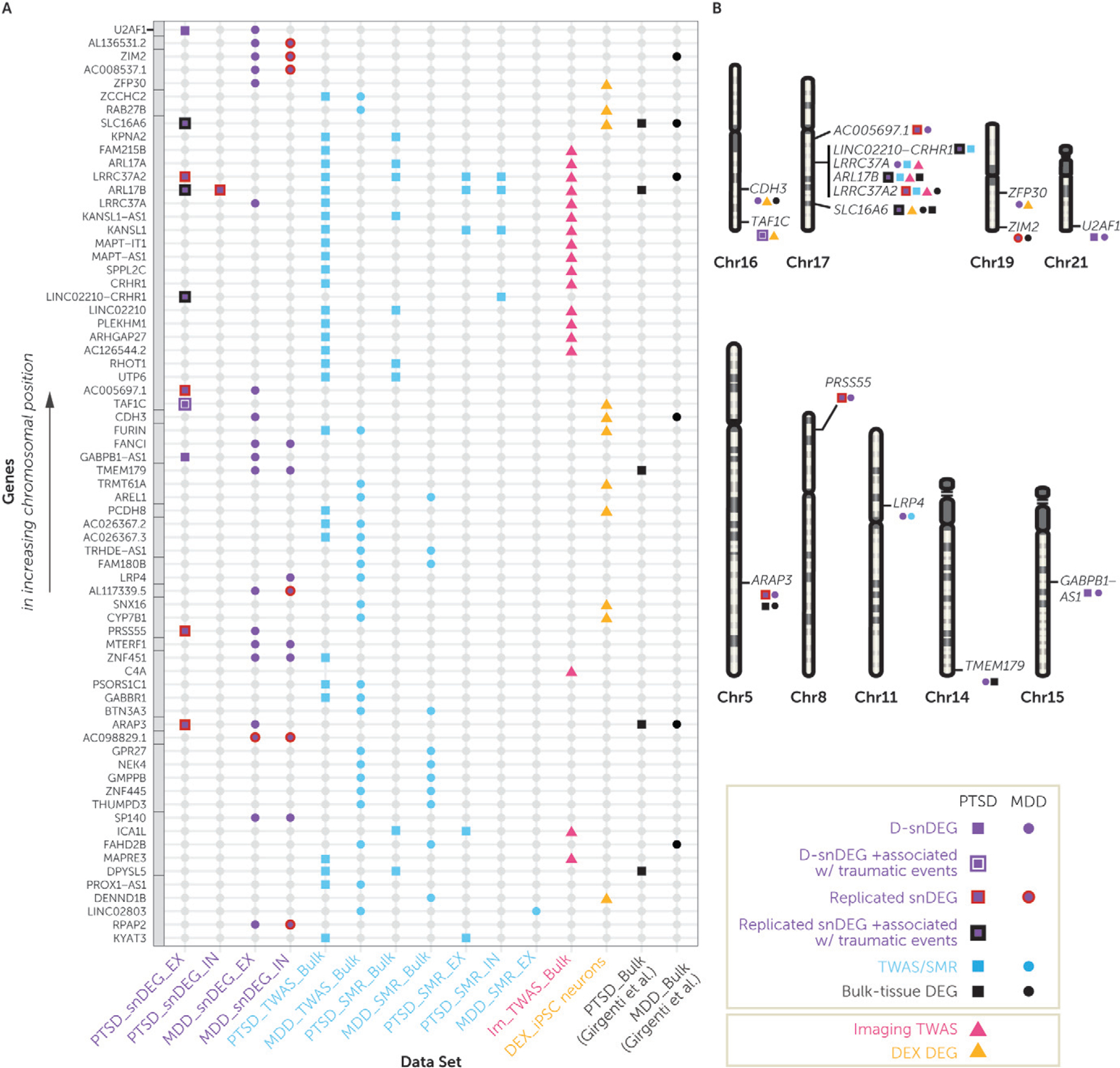
a Panel A is an integrative dot plot with all genes that were false discovery rate (FDR) significant in at least two analyses used in this study. The genes are listed from bottom to top in increasing chromosomal position order, with the gray boxes annotating genes that belong to the same chromosome. Purple denotes significant results from the dorsolateral prefrontal cortex (DLPFC) single-nucleus RNA-seq analysis; squares depict significant posttraumatic stress disorder (PTSD) single-nucleus differentially expressed genes in the discovery data set (D-snDEGs), and circles depict major depressive disorder (MDD) D-snDEGs. A purple line surrounding the square indicates that the PTSD D-snDEG was also associated with number of traumatic events, and a black line surrounding the square indicates that a replicated PTSD D-snDEG was also associated with number of traumatic events. Blue denotes significant results from DLPFC-based transcriptome-wide association study (TWAS) or summary-data-based Mendelian randomization (SMR) analyses; squares depict PTSD FDR-significant genes, and circles depict MDD FDR-significant genes. Pink triangles denote TWAS genes of DLPFC-based neuroimaging, and orange triangles denote FDR-significant genes based on dexamethasone (DEX) treatment in iPSC-derivedneurons. Black denotes significant results from the previous DLPFC bulk-tissue RNA-seq study (22), with squares and circles depicting PTSD and MDD FDR-significant differentially expressed genes, respectively. In panel B, D-snDEG genes that were significant in more than one data modality or in both disorders in any modality are depicted on their chromosomal location. The 17q21.31 locus had four of such genes, and there was no other locus with more than one such gene.
The genes at the 17q21.31 locus included ARL17B as a replicated, upregulated neuronal and nonneuronal D-snDEG in PTSD that was associated with number of traumatic events, and a bulk-tissue PTSD DEG. ARL17B was also a neuronal and nonneuronal PTSD SMR gene, a bulk-tissue PTSD TWAS gene, and a bulk-tissue TWAS gene of DLPFC-based neuroimaging. LINC02210-CRHR1 was a replicated, upregulated PTSD snDEG in EX neurons, was associated with number of traumatic events, and is a neuronal PTSD SMR gene. LINC02210 and CRHR1, which share coding and noncoding sequences with LINC02210-CRHR1, were bulk-tissue PTSD SMR/TWAS genes and bulk-tissue TWAS genes of DLPFC-based neuroimaging. LRRC37A2 was a replicated, upregulated PTSDD-snDEGin EXneurons, buta bulk-tissue DEGin MDD. It was also a neuronal/nonneuronal/bulk-tissue PTSD SMR gene, a bulk-tissue PTSD TWAS gene, and a bulk-tissue TWAS gene of DLPFC-based neuroimaging. Finally, LRRC37A was an MDD D-snDEG, a PTSD TWAS gene, and a bulk-tissue TWAS gene from DLPFC-based neuroimaging.
Outside of the genes identified at the 17q21.31 locus, LRP4 was a downregulated MDD gene and a bulk-tissue TWAS gene. SLC16A6 was a replicated, upregulated PTSD D-snDEG in EX neurons that was associated with number of traumatic events, was an upregulated PTSD and MDD bulk-tissue DEG, and was downregulated by DEX. TAF1C was an upregulated PTSD D-snDEG in EX neurons that was associated with number of traumatic events and was upregulated by DEX. MDD convergent findings included CDH3, which was an upregulated MDD D-snDEG in EX neurons, was an upregulated MDD bulk-tissue DEG, and was upregulated by DEX.
DISCUSSION
This is the first single-nucleus RNA-seq study that dissects cell-type-specific transcriptomic alterations in PTSD. We profiled ~415K single nuclei belonging to eight cell types from postmortem samples of individuals with PTSD, individuals with MDD, and control subjects. The disorders were similar in cell type proportions and differed in specific disease-associated transcriptomic alterations in neurons and astrocytes, observations replicated in an independent data set (N=15 per group, ~160K single nuclei). Among these different expression patterns in PTSD compared with MDD, we associated distinct biological pathways in neuronal cell types, including the GC pathway. We further compared the expression signatures of PTSD and MDD in EX and IN neurons with GR-dependent GC-induced DEGs of iPSC-derived neurons and demonstrated opposite directions of enrichment between the two disorders. To prioritize top genes for PTSD and MDD, we generated sets of putative causal genes based on SMR and/or TWAS analyses for PTSD and MDD, and TWAS analysis of DLPFC-based neuroimaging.
This study, along with a schizophrenia study in preprint (28), profiled significantly more cells (~500K) compared with earlier DLPFC single-nucleus RNA-seq studies in ASD (~100K [25]) and MDD (~80K [26]). Using similar technical and bioinformatic pipelines, we confirmed consistent cell subtype annotations. It is worth noting that our study was concordant with the astrocyte subtyping of the ASD study (25) and agreed on the development-based pseudo time distinctions between OPC and oligodendrocyte cell clusters of the MDD study (26).
While large-scale, deeply sequenced bulk-tissue RNA-seq study designs can provide substantial insight into the pathophysiology of diseases, our study design showed higher discoverability of DEGs in the snRNA-seq portion of the data, compared with the bulk-tissue RNA-seq, within a relatively limited sample size. This demonstrates the benefits of single-cell transcriptomic approaches in the field of complex psychiatric disorders, where it remains to be fully exploited. PTSD and MDD D-snDEGs were found almost entirely in EX and IN neurons compatible with the cell type enrichment of PTSD and MDD GWASs and bulk-tissue RNA-seq studies showing downregulation of interneuron markers in PTSD (22–24).
In line with the previous bulk-tissue RNA-seq studies (22–24), MDD had more D-snDEGs compared with PTSD. MDD had higher genome-wide correlations between the discovery and replication samples compared with PTSD, while there was a higher degree of replication of top snDEGs in PTSD compared with MDD. This was partially described in bulk-tissue RNA-seq studies, where low concordance in top DEGs was seen in MDD (20–22) and a higher level of concordance in top DEGs in PTSD (22, 23). Our study and the prior MDD single-nucleus RNA-seq study agreed on the reduced GR activity in MDD (26). We did not detect FDR-significant effects in OPCs as seen previously (26), but we showed a significant enrichment of MDD GWAS in OPC markers. It is worth noting that the prior study used a male cohort in which all MDD cases were suicide completers (only 20% of the subjects of the present study were suicide completers), conducted subclustering of OPC/oligodendrocyte cell clusters, and analyzed only highly expressed genes for differential expression between groups.
There were multiple dysregulated pathways in both EX and IN neurons across disorders. Importantly, there were pathways with a differential enrichment in PTSD compared with MDD. The involved pathways included immunologic, cancer-related, metabolic, and various ribosomal and enzymatic processes. Predicted upstream regulators tracked together with the cell-type-specific pathways. For instance, the GC treatment pathway differentiated the two traits, while GCs were also predicted to be upstream regulators. Moreover, neuronal GC-induced transcriptomic alterations were studied in iPSC-derived neuronal cultures, and these converged with gene set enrichment analyses and genome-wide correlations on DLPFC EX and IN neurons’ transcriptional signatures in PTSD and MDD in different directions. These transcriptional patterns showed GR activation in EX and IN neurons in PTSD and GRdeactivation in EXneurons in MDD, with the latter being generally weaker. These results are the first molecular demonstration of cell-type-specific directional differences in GC pathways between PTSD and MDD in a stress-related brain region. These differences are consistent with differences historically described on peripheral endocrine measures of the HPA axis and PBMC-based GC sensitivity assays (8, 30). Due to the maturity of our in vitro models, we observed FKBP5 and genome-wide transcriptomic responses to GC that overlapped with those described in iPSC-derived cerebral organoids (36) and were greater than those described in less mature iPSC-derived neural cells from embryoid body-based (37) and NGN2-based (38) neuralization protocols. Conceptually, our DEX-treated iPSC-derived neuron cultures represent an in vitro neuronal model optimized for studying a brain-based risk-associated pathway of PTSD. Future in vitro modeling studies in PTSD can examine GC responses in additional brain cell types and in novel cell lines that recapitulate PTSD’s genetic and epigenetic pathology, as well as further clarify whether they are mediated by GR and/or MR.
To determine the genes with a keyrole in disease states, we prioritized snDEGs that overlapped with bulk-tissue and/or cell-type-specific analyses of the largest GWASs of PTSD, MDD, and neuroimaging. To determine causation of genes for PTSD and/or MDD, we used SMR analysis on both bulk and cell-type-specific eQTL DLPFC databases (33, 34). To link the combination of genetic risk variants with changes in expression, we used TWAS. We trained a state-of-the-art DLPFC GReX model that was more accurate and predicted more genes than previous models generated by the GTEx project (39). We also revealed genes with preferential TWAS signals for DLPFC-based neuroimaging in UK Biobank (N~33K).
The observed overlap between snDEGs and genes discovered by the genetic analyses underscored the functional involvement of genes at the 17q21.31 locus, which are already implicated in neurodevelopmental (40) and neurodegenerative disorders (41). ARL17B was upregulated in PTSD across bulk-tissue, neuronal, and nonneuronal analyses and encodes for a GTP-binding protein involved in protein trafficking. ARL17B is upregulated in multiple cell types in carriers of a missense variant of SPPL2C associated with onset age in Alzheimer’s disease (42). LINC02210-CRHR1 was upregulated in EX neurons and is a fusion gene between LINC02210 and CRHR1. LINC02210 and CRHR1 were not differentially expressed in the postmortem data but were putative causal genes for PTSD and had effects on DLPFC-based neuroimaging. CRHR1 encodes the CRHR1 protein, a master regulator of the stress and fear response that has long been thought to be involved in PTSD and MDD. LINC02210 encodes a long noncoding RNA with a relatively unknown function, emphasizing the emerging role of noncoding RNAs in the pathophysiology of stress-related mental disorders (43). Long noncoding RNAs act as regulators of transcription, affecting many downstream target genes. Given associations of LINC02210 with DLPFC-based neuroimaging, future studies should expand the comparisons of coding versus noncoding genetic associations with neuroimaging. For instance, it would be very interesting to understand whether cell subtypes with dysregulation in noncoding genes are in a particular spatial brain compartment that would justify more influences in white matter specifically. LRRC37A2 was upregulated in PTSD EX neurons and encodes an immune-related leucine-rich repeat protein involved in the formation of protein-protein interactions. LRRC37A, a paralog of LRRC37A2, was an upregulated bulk-tissue TWAS gene for PTSD, while it was also downregulated in MDD EX neurons, indicating directional differences between PTSD and MDD in this genomic region.
Outside of the 17q21.31 locus, LRP4 was downregulated in MDD IN neurons and encodes for a member of the lipoprotein receptor-related protein family with known roles in neurodevelopment, synaptic remodeling, and Alzheimer’s disease (44). Furthermore, the upregulation in PTSD of the GC-regulated SLC16A6 in EX neurons might be a link to mitochondrial regulation of physiological and behavioral responses to psychological stress (45). SLC16A6 belongs to the monocarboxylate transporter (MCT) family that contributes to maintenance of intracellular pH; MCT7/SLC16A6 is expressed in the liver, brain, and endocrine tissues and is classified as a transporter for ketone bodies during mitochondrial ketogenesis. As such, it is expected to have a multitude of physiological roles (46). CDH3 was upregulated in MDD EX neurons and was induced by DEX in iPSC-derived neurons. CDH3 encodes a P-cadherin that has been associated with neuronal development and differentiation (47). The TAF1C gene was upregulated in PTSD IN neurons and by DEX in iPSC-derived neurons. This upregulation could result in proteomic alterations, since TAF1C encodes the largest subunit of the SL1 complex, which is required for RNA polymerase I to bind to promoters of ribosomal RNA genes (48).
A limitation of this single-nucleus RNA-seq study is the sample size of the discovery data set. We attempted to mitigate this limitation by profiling a high number of cells, using a replication data set, seeking biological validation of discovered pathways, convergence with putative causal genes, and assessing relevance to DLPFC-based neuroimaging. While these efforts contributed to the robustness of our findings, further validation is needed. Another limitation of postmortem analysis of PTSD is that while in-depth clinical characteristics of the PTSD collection were assessed systematically (49), assessments were based on interviews with next of kin and medical record review. We also acknowledge that the replication sample was also small and did not fully match our discovery sample. These demographic differences strengthened our observed levels of replication. Our data sets included only African and European ancestries, however, and generalizations to other populations warrant careful consideration. Finally, the evaluation of trauma exposure–based effects is a novel approach in postmortem research. Recent postmortem studies investigating trauma-based subgroup approaches across psychiatric disorders provide a level of precedence and confidence (50), although these approaches need to be further replicated in larger samples.
From a technical perspective, studying the gray versus white matter contributions to cell-type-specific transcriptomic disease signatures may be critical for revealing nonneuronal signals. To this end, single-nucleus RNA-seq may not adequately sample perivascular components, i.e., pericytes (e.g., detected in our discovery data set but not the replication data set). Future studies could use more refined techniques, such as vessel isolation and nucleus extraction for sequencing (VINE-seq), to enrich vascular and perivascular nuclei to profile endothelial cells and pericytes more comprehensively (51). Additionally, single-nucleus RNA-seq introduces some level of bias in the detection of certain gene types, as nonpolyadenylated transcripts cannot be detected (including ribosomal RNAs, small RNAs, histone mRNAs, and some long noncoding RNAs (52). As described by Thrupp et al. (53), a small but nonnegligible proportion of microglia genes (~1.1%) are not detected by single-nucleus RNA-seq, which can lead to underrepresentation of certain cellular states.
In summary, in this first single-cell transcriptomic investigation of PTSD, the comparison of PTSD with MDD results enabled us to identify shared and unique neuronal genes and targetable stress-related gene pathways, which we validated with in vitro modeling. Theoverlap of stress-related gene pathways with disease causal genes and specific brain connectivity measures provides an avenue for future translational investigation.
Supplementary Material
Acknowledgments
This work was supported by NIMH (grants P50-MH115874 to Drs. Carlezon and Ressler, R01-MH117291 to Dr. Kleinman, R01-MH117292 to Dr. Ressler, R01 R01-MH10659 to Drs. Nievergelt and Ressler, R21-MH102834 to Dr. Miller, and R21-MH121909 to Dr. Daskalakis). Dr. Chatzinakos was supported by the 2019 SPARED Center Seed Grant (through NIMH grant P50-MH115874). Dr. Pernia was supported by a Merrill Fellowship from McLean Hospital and an Administrative Diversity Supplement (through NIMH grant P50-MH115874). Dr. Morrison was supported by NIMH grant 5T32MH019836-18 while a postdoctoral fellow at Boston University and VA Boston Healthcare System. Dr. Logue was supported by VA grant I01BX003477. Dr. Wolf was supported by VA grant I01CX-001276-01, a Presidential Early Career Award for Scientists and Engineers (PECASE 2013A), and National Institute on Aging grant 1R21AG061367-01. Dr. Daskalakis was supported by 2015 and 2018 NARSAD Young Investigator grants from the Brain and Behavior Research Foundation, a Jonathan Edward Brooking mental health research fellowship from McLean Hospital, and a KL2 award from Harvard Catalyst/Harvard Clinical and Translational Science Center (NCATS KL2TR002542, UL1TR002541).
The authors acknowledge Kiki Galani, Bob Handsaker, Li-Lun Ho, Maria Kousi, Shahin Mohammadi, and Shreejoy Tripathy for suggestions in the choice of methods, technical assistance, and/or presentation of data. The authors acknowledge three working groups: The members of the Traumatic Stress Brain Research Group are Matthew Friedman, Neil Kowall, Christopher Brady, Ann McKee, Thor Stein, Bertrand Huber, Paul Holtzheimer, Victor Alvarez, David Benedek, Robert Ursano, Douglas Williamson, Dianne Cruz, Keith Young, John Krystal, Deborah Mash, Melanie Hardegree, William Scott, David Davis, Matthew Girgenti, Gayle Serlin, Brian Marx, Terence Keane, Mark Logue, Erika Wolf, and Mark Miller. The members of the PTSD BrainOmics Project of the PsychENCODE Consortium are Dhivya Arasappan, Sabina Berretta, Rahul Bharadwaj, Frances Champagne, Leonardo Collado-Torres, Christos Chatzinakos, Nikolaos Daskalakis, Chris DiPietro, Duc Duong, Amy Deep-Soboslay, Nick Eagles, Louise Huuki, Thomas Hyde, Artemis Iatrou, Aarti Jajoo, Joel Kleinman, Charles Nemeroff, Geo Pertea, Deanna Ross, Nicholas Seyfried, Joo Heon Shin, Kerry Ressler, Clara Snijders, Ran Tao, Daniel Weinberger, Stefan Wuchty, Dennis Wylie. The members of the PTSD Working Group of the Psychiatric Genomics Consortium are Adam X. Maihofer, Karmel W. Choi, Jonathan R.I. Coleman, Nikolaos P. Daskalakis, Christy A. Denckla, Elizabeth Ketema, Rajendra A. Morey, Renato Polimanti, Andrew Ratanatharathorn, Katy Torres, Aliza P. Wingo, Clement C. Zai, Allison E. Aiello, Lynn M. Almli, Ananda B. Amstadter, Soren B. Andersen, Ole A. Andreassen, Paul A. Arbisi, Allison E. Ashley-Koch, S. Bryn Austin, Esmina Avdibegovic, Anders D. Borglum, Dragan Babic, Marie Bækvad-Hansen, Dewleen G. Baker, Jean C. Beckham, Laura J. Bierut, Jonathan I. Bisson, Marco P. Boks, Elizabeth A. Bolger, Bekh Bradley, Meghan Brashear, Gerome Breen, Richard A. Bryant, Angela C. Bustamante, Jonas Bybjerg-Grauholm, Joseph R. Calabrese, J.M. Caldas-de-Almeida, Chia-Yen Chen, Anders M. Dale, Shareefa Dalvie, Jürgen Deckert, Douglas L. Delahanty, Michelle F. Dennis, Seth G. Disner, Katharina Domschke, Laramie E. Duncan, Alma Dzubur Kulenovic, Christopher R. Erbes, Alexandra Evans, Lindsay A. Farrer, Norah C. Feeny, Janine D. Flory, David Forbes, Carol E. Franz, Sandro Galea, Melanie E. Garrett, Aarti Gautam, Bizu Gelaye, Joel Gelernter, Elbert Geuze, Charles F. Gillespie, Aferdita Goci, Scott D. Gordon, Guia Guffanti, Rasha Hammamieh, Michael A. Hauser, Andrew C. Heath, Sian M.J. Hemmings, David Michael Hougaard, Miro Jakovljevic, Marti Jett, Eric Otto Johnson, Ian Jones, Tanja Jovanovic, Xue-Jun Qin, KarenInge Karstoft, Milissa L. Kaufman, Ronald C. Kessler, Alaptagin Khan, Nathan A. Kimbrel, Anthony P. King, Nastassja Koen, Henry R. Kranzler, William S. Kremen, Bruce R. Lawford, Lauren A.M. Lebois, Catrin Lewis, Israel Liberzon, Sarah D. Linnstaedt, Mark W. Logue, Adriana Lori, Bozo Lugonja, Jurjen J. Luykx, Michael J. Lyons, Jessica L. Maples-Keller, Charles Marmar, Nicholas G. Martin, D. Maurer, Matig R. Mavissakalian, Alexander McFarlane, Regina E. McGlinchey, Katie A. McLaughlin, Samuel A. McLean, Divya Mehta, Rebecca Mellor, Vasiliki Michopoulos, William Milberg, Mark W. Miller, Charles Phillip Morris, Ole Mors, P.B. Mortensen, Elliot C. Nelson, Merete Nordentoft, Sonya B. Norman, Meaghan O’Donnell, Holly K. Orcutt, Matthew S. Panizzon, Edward S. Peters, Alan L. Peterson, Matthew Peverill, Robert H. Pietrzak, Melissa A. Polusny, John P. Rice, Victoria B. Risbrough, Andrea L. Roberts, Alex O. Rothbaum, Barbara O. Rothbaum, P. Roy-Byrne, Kenneth J. Ruggiero, Ariane Rung, Bart P.F. Rutten, Nancy L. Saccone, Sixto E. Sanchez, Dick Schijven, S. Seedat, Antonia V. Seligowski, Julia S. Seng, Christina M. Sheerin, Derrick Silove, Alicia K. Smith, Jordan W. Smoller, Scott R. Sponheim, Dan J. Stein, Jennifer S. Stevens, Martin H. Teicher, Wesley K. Thompson, Edward Trapido, Monica Uddin, Robert J. Ursano, Leigh Luella van den Heuvel, Miranda Van Hooff, Eric Vermetten, Christiaan Vinkers, Joanne Voisey, Yunpeng Wang, Zhewu Wang, Thomas Werge, Michelle A. Williams, Douglas E. Williamson, Sherry Winternitz, Christiane Wolf, Erika J. Wolf, Rachel Yehuda, Keith A. Young, Ross McD. Young, Hongyu Zhao, Lori A. Zoellner, Magali Haas, Heather Lasseter, Allison C. Provost, Rany M. Salem, Jonathan Sebat, Richard A. Shaffer, Tianying Wu, Stephan Ripke, Mark J. Daly, Kerry J. Ressler, Karestan C. Koenen, Murray B. Stein, and Caroline M. Nievergelt.
Footnotes
The contents of this article do not represent the views of the U.S. Department of Veterans Affairs, NIH, or the U.S. government.
Dr. Morrison is currently an employee of Neumora Therapeutics. Dr. McCullough is currently an employee of Jazz Pharmaceuticals. Dr. Carlezon has served as a consultant for PSY Therapeutics. Dr. Krystal has served as a consultant for Aptinyx, Biogen Idec MA, Bionomics (Australia), Boehringer Ingelheim International, Epiodyne, EpiVario, Janssen Research and Development, Jazz Pharmaceuticals, Otsuka America Pharmaceutical, Spring Care, and Sunovion Pharmaceuticals; he has served on scientific advisory boards for Biohaven Pharmaceuticals, BioXcel Therapeutics (clinical advisory board), Cerevel Therapeutics, Delix Therapeutics, Eisai, EpiVario, Jazz Pharmaceuticals, Neumora Therapeutics, Neurocrine Biosciences, Novartis Pharmaceuticals, Psycho-Genics, Takeda Pharmaceuticals, Tempero Bio, and Terran Biosciences; he has been involved in studies that have received medications from AstraZeneca, Cerevel, and Novartis; he is cofounder of Freedom Biosciences; he holds stock in Biohaven Pharmaceuticals, Freedom Biosciences, and Spring Health and stock options in Biohaven Pharmaceuticals Medical Sciences, Cartego Therapeutics, Damona Pharmaceuticals, Delix Therapeutics, EpiVario, Neumora Therapeutics, Rest Therapeutics, Tempero Bio, Terran Biosciences, and Tetricus; he serves as Editor on the editorial board of Biological Psychiatry; and he is named on patents and patent applications related to psychiatric disorders and treatments. Dr. Ressler has served as a consultant for Acer, Alkermes, Bionomics, BioXcel, and Jazz Pharmaceuticals and on scientific advisory boards for Janssen, Boehringer Ingelheim, the Brain Research Foundation, Resilience Therapeutics, Sage, Senseye, and Verily, and he has received sponsored research support from Alto Neuroscience, BrainsWay, and Takeda. Dr. Daskalakis has served as a consultant for BioVie Pharma and Sunovion Pharmaceuticals and has served on scientific advisory boards for Circular Genomics and Sentio Solutions. The other authors report no financial relationships with commercial interests.
Contributor Information
Chris Chatzinakos, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.
Cameron D. Pernia, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.
Filomene G. Morrison, National Center for PTSD, VA Boston Healthcare System, Boston; Department of Psychiatry, Boston University School of Medicine, Boston
Artemis Iatrou, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.
Kenneth M. McCullough, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.
Heike Schuler, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.
Clara Snijders, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.; Department of Psychiatry and Neuropsychology, School for Mental Health and Neuroscience, Maastricht University, Maastricht, the Netherlands
Thomas Bajaj, RG Neurohomeostasis, Department of Psychiatry and Psychotherapy, Medical Faculty, University of Bonn, Bonn, Germany
Christopher P. DiPietro, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.
Marina Soliva Estruch, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.; Department of Psychiatry and Neuropsychology, School for Mental Health and Neuroscience, Maastricht University, Maastricht, the Netherlands
Nils C. Gassen, RG Neurohomeostasis, Department of Psychiatry and Psychotherapy, Medical Faculty, University of Bonn, Bonn, Germany
Constantin Anastasopoulos, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.; Department of Radiology, University Hospital Basel, University of Basel, Basel, Switzerland
Rahul A. Bharadwaj, Lieber Institute for Brain Development, Johns Hopkins Medical Campus, Baltimore
Benjamin C. Bowlby, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.
Jakob Hartmann, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.
Adam X. Maihofer, Department of Psychiatry, University of California San Diego, La Jolla; Center for Excellence in Stress and Mental Health, Veterans Affairs San Diego Healthcare System, San Diego; Research Service, Veterans Affairs San Diego Healthcare System, San Diego
Caroline M. Nievergelt, Department of Psychiatry, University of California San Diego, La Jolla; Center for Excellence in Stress and Mental Health, Veterans Affairs San Diego Healthcare System, San Diego; Research Service, Veterans Affairs San Diego Healthcare System, San Diego
Nicholas M. Ressler, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.
Erika J. Wolf, National Center for PTSD, VA Boston Healthcare System, Boston; Department of Psychiatry, Boston University School of Medicine, Boston
William A. Carlezon, Jr., Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.
John H. Krystal, Department of Psychiatry, Yale University School of Medicine, New Haven, Conn.; Psychiatry Service, VA Connecticut Healthcare System, West Haven; National Center for PTSD, Clinical Neurosciences Division, U.S. Department of Veterans Affairs, West Haven, Conn.
Joel E. Kleinman, Lieber Institute for Brain Development, Johns Hopkins Medical Campus, Baltimore; Department of Psychiatry and Behavioral Sciences, Johns Hopkins School of Medicine, Baltimore
Matthew J. Girgenti, Department of Psychiatry, Yale University School of Medicine, New Haven, Conn.; Psychiatry Service, VA Connecticut Healthcare System, West Haven; National Center for PTSD, Clinical Neurosciences Division, U.S. Department of Veterans Affairs, West Haven, Conn.
Bertrand R. Huber, Department of Neurology, Boston University School of Medicine, Boston; Pathology and Laboratory Medicine, VA Boston Healthcare System, Boston
Manolis Kellis, Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, and Broad Institute of MIT and Harvard, Cambridge, Mass.
Mark W. Logue, National Center for PTSD, VA Boston Healthcare System, Boston; Department of Psychiatry, Boston University School of Medicine, Boston; Department of Biomedical Genetics, Boston University School of Medicine, Boston; Department of Biostatistics, Boston University School of Public Health, Boston
Mark W. Miller, National Center for PTSD, VA Boston Healthcare System, Boston; Department of Psychiatry, Boston University School of Medicine, Boston
Kerry J. Ressler, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.
Nikolaos P. Daskalakis, Department of Psychiatry, McLean Hospital, Harvard Medical School, Belmont, Mass.; Stanley Center for Psychiatric Research, Broad Institute of MIT and Harvard, Cambridge, Mass.
REFERENCES
- 1.Substance Abuse and Mental Health Services Administration: Results From the 2010 National Survey on Drug Use and Health: Summary of National Findings. Rockville, Md, Substance Abuse and Mental Health Services Administration, September 2011. https://www.samhsa.gov/data/sites/default/files/NSDUHNationalFindingsResults2010-web/2k10ResultsRev/NSDUHresultsRev2010.pdf [Google Scholar]
- 2.Goldstein RB, Smith SM, Chou SP, et al. : The epidemiology of DSM-5 posttraumatic stress disorder in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions–III. Soc Psychiatry Psychiatr Epidemiol 2016; 51:1137–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wisco BE, Marx BP, Wolf EJ, et al. : Posttraumatic stress disorder in the US veteran population: results from the National Health and Resilience in Veterans Study. J Clin Psychiatry 2014; 75:1338–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gillespie CF, Bradley B, Mercer K, et al. : Trauma exposure and stress-related disorders in inner city primary care patients. Gen Hosp Psychiatry 2009; 31:505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flory JD, Yehuda R: Comorbidity between post-traumatic stress disorder and major depressive disorder: alternative explanations and treatment considerations. Dialogues Clin Neurosci 2015; 17:141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stupin KN, Zenko MY, Rybnikova EA: Comparative analysis of pathobiochemical changes in major depression and post-traumatic stress disorder. Biochemistry (Mosc) 2021; 86:729–736 [DOI] [PubMed] [Google Scholar]
- 7.Murrough JW, Russo SJ: Theneurobiologyof resilience: complexity and hope. Biol Psychiatry 2019; 86:406–409 [DOI] [PubMed] [Google Scholar]
- 8.Binder EB: The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009; 34:S186–S195 [DOI] [PubMed] [Google Scholar]
- 9.Maihofer AX, Choi KW, Coleman JRI, et al. : Enhancing discovery of genetic variants for posttraumatic stress disorder through integration of quantitative phenotypes and trauma exposure information. Biol Psychiatry 2022; 91:626–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stein MB, Levey DF, Cheng Z, et al. : Genome-wide association analyses of post-traumatic stress disorder and its symptom subdomains in the Million Veteran Program. Nat Genet 2021; 53: 174–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levey DF, Stein MB, Wendt FR, et al. : Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in .1.2 million individuals highlight new therapeutic directions. Nat Neurosci 2021; 24:954–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howard DM, Adams MJ, Clarke TK, et al. : Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci 2019; 22:343–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hartley CA, Phelps EA: Changing fear: the neurocircuitry of emotion regulation. Neuropsychopharmacology 2010; 35:136–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northoff G, Sibille E: Why are cortical GABA neurons relevant to internal focus in depression? A cross-level model linking cellular, biochemical, and neural network findings. Mol Psychiatry 2014; 19:966–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huckins LM, Chatzinakos C, Breen MS, et al. : Analysis of genetically regulated gene expression identifies a prefrontal PTSD gene, SNRNP35, specific to military cohorts. Cell Rep 2020; 31:107716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boettger LM, Handsaker RE, Zody MC, et al. : Structural haplotypes and recent evolution of the human 17q21.31 region. Nat Genet 2012; 44:881–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joels M, Baram TZ: The neuro-symphony of stress. Nat Rev Neurosci 2009; 10:459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herman JP, McKlveen JM, Ghosal S, et al. : Regulation of the hypothalamic-pituitary-adrenocortical stress response. Compr Physiol 2016; 6:603–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerring ZF, Gamazon ER, Derks EM, et al. : A gene co-expression network-based analysis of multiple brain tissues reveals novel genes and molecular pathways underlying major depression. PLoS Genet 2019; 15:e1008245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labonte B, Engmann O, Purushothaman I, et al. : Sex-specific transcriptional signatures in human depression. Nat Med 2017; 23: 1102–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gandal MJ, Haney JR, Parikshak NN, et al. : Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018; 359:693–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girgenti MJ, Wang J, Ji D, et al. : Transcriptomic organization of the human brain in post-traumatic stress disorder. Nat Neurosci 2021; 24:24–33 [DOI] [PubMed] [Google Scholar]
- 23.Logue MW, Zhou Z, Morrison FG, et al. : Gene expression in the dorsolateral and ventromedial prefrontal cortices implicates immune-related gene networks in PTSD. Neurobiol Stress 2021; 15: 100398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jaffe AE, Tao R, Page SC, et al. : Decoding shared versus divergent transcriptomic signatures across cortico-amygdala circuitry in PTSD and depressive disorders. Am J Psychiatry 2022; 179:673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Velmeshev D, Schirmer L, Jung D, et al. : Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019; 364:685–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagy C, Maitra M, Tanti A, et al. : Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat Neurosci 2020; 23:771–781 [DOI] [PubMed] [Google Scholar]
- 27.Brenner E, Tiwari GR, Kapoor M, et al. : Single cell transcriptome profiling of the human alcohol-dependent brain. Hum Mol Genet 2020; 29:1144–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruzicka WB, Mohammadi S, Fullard JF, et al. : Single-cell multi-cohort dissection of the schizophrenia transcriptome. medRxiv, September 2, 2022. https://www.medrxiv.org/content/10.1101/2022.08.31.22279406v1 [DOI] [PubMed]
- 29.Friedman MJ, Huber BR, Brady CB, et al. : VA’s National PTSD Brain Bank: a national resource for research. Curr Psychiatry Rep 2017; 19:73. [DOI] [PubMed] [Google Scholar]
- 30.Daskalakis NP, Lehrner A, Yehuda R: Endocrine aspects of post-traumatic stress disorder and implications for diagnosis and treatment. Endocrinol Metab Clin North Am 2013; 42: 503–513 [DOI] [PubMed] [Google Scholar]
- 31.Viho EMG, Buurstede JC, Berkhout JB, et al. : Cell type specificity of glucocorticoid signaling in the adult mouse hippocampus. J Neuroendocrinol 2022; 34:e13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Z, Frederick J, Garabedian MJ: Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J Biol Chem 2002; 277:26573–26580 [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Liu S, Warrell J, et al. : Comprehensive functional genomic resource and integrative model for the human brain. Science 2018; 362:eaat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bryois J, Calini D, Macnair W, et al. : Cell-type-specific cis-eQTLs in eight human brain cell types identify novel risk genes for psychiatric and neurological disorders. Nat Neurosci 2022; 25:1104–1112 [DOI] [PubMed] [Google Scholar]
- 35.Smith SM, Douaud G, Chen W, et al. : An expanded set of genome-wide association studies of brain imaging phenotypes in UK Biobank. Nat Neurosci 2021; 24:737–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cruceanu C, Dony L, Krontira AC, et al. : Cell-type-specific impact of glucocorticoid receptor activation on the developing brain: a cerebral organoid study. Am J Psychiatry 2022; 179:375–387 [DOI] [PubMed] [Google Scholar]
- 37.Lieberman R, Kranzler HR, Levine ES, et al. : Examining FKBP5 mRNA expression in human iPSC-derived neural cells. Psychiatry Res 2017; 247:172–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seah C, Breen MS, Rusielewicz T, et al. : Modeling gene 3 environment interactions in PTSD using human neurons reveals diagnosis-specific glucocorticoid-induced gene expression. Nat Neurosci 2022; 25: 1434–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barbeira AN, Bonazzola R, Gamazon ER, et al. : Exploiting the GTExresources to decipher the mechanisms at GWASloci. Genome Biol 2021; 22:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gandal MJ, Zhang P, Hadjimichael E, et al. : Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 2018; 362:eaat8127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JA, Chen Z, Won H, et al. : Jointgenome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol Neurodegener 2018; 13:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He L, Loika Y, Park Y, et al. : Exome-wide age-of-onset analysis reveals exonic variants in ERN1 and SPPL2C associated with Alzheimer’s disease. Transl Psychiatry 2021; 11:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daskalakis NP, Provost AC, Hunter RG, et al. : Noncoding RNAs: stress, glucocorticoids, and posttraumatic stress disorder. Biol Psychiatry 2018; 83:849–865 [DOI] [PubMed] [Google Scholar]
- 44.DePew AT, Mosca TJ: Conservation and innovation: versatile roles for LRP4 in nervous system development. J Dev Biol 2021; 9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Picard M, McEwen BS, Epel ES, et al. : An energetic view of stress: focus on mitochondria. Front Neuroendocrinol 2018; 49:72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Higuchi K, Sugiyama K, Tomabechi R, et al. : Mammalian monocarboxylate transporter 7 (MCT7/Slc16a6) is a novel facilitative taurine transporter. J Biol Chem 2022; 298:101800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Punovuori K, Malaguti M, Lowell S: Cadherins in early neural development. Cell Mol Life Sci 2021; 78:4435–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Di Pietro C, Rapisarda A, Amico V, et al. : Genomic localization of the human genes TAF1A, TAF1B and TAF1C, encoding TAF(I)48, TAF(I)63 and TAF(I)110 subunits of class I general transcription initiation factor SL1. Cytogenet Cell Genet 2000; 89:133–136 [DOI] [PubMed] [Google Scholar]
- 49.Mighdoll MI, Deep-Soboslay A, Bharadwaj RA, et al. : Implementation and clinical characteristics of a posttraumatic stress disorder brain collection. J Neurosci Res 2018; 96:16–20 [DOI] [PubMed] [Google Scholar]
- 50.Kaul D, Smith CC, Stevens J, et al. : Severe childhood and adulthood stress associates with neocortical layer-specific reductions of mature spines in psychiatric disorders. Neurobiol Stress 2020; 13:100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang AC, Vest RT, Kern F, et al. : A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022; 603:885–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang L, Duff MO, Graveley BR, et al. : Genomewide characterization of non-polyadenylated RNAs. Genome Biol 2011; 12:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thrupp N, Sala Frigerio C, Wolfs L, et al. : Single-nucleus RNA-seq is not suitable for detection of microglial activation genes in humans. Cell Rep 2020; 32:108189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.