

Abstract
Background
Juvenile sudden cardiac death (SCD) remains unexplained in approximately 40% of cases, leading to a significant emotional burden for the victims’ families and society. Comprehensive investigations are essential to uncover its elusive causes and enable cascade family screening. This study aimed to enhance the identification of likely causative variants in juvenile SCD cases (age ≤ 50 years), particularly when autopsy findings are inconclusive.
Results
Autopsy revealed diagnostic structural abnormalities in 46%, non-diagnostic findings in 23%, and structurally normal hearts in 31% of cases. Whole-exome sequencing (WES), refined through a customized virtual gene panel was used to identify variants. These variants were then evaluated using a multidisciplinary approach and a structured variant prioritization scheme. Our extended approach identified likely causative variants in 69% of cases, outperforming the diagnostic yields of both the cardio panel and standard susceptibility gene analysis (50% and 16%, respectively). The extended cardio panel achieved an 80% diagnostic yield in cases with structurally normal hearts, demonstrating its efficacy in challenging scenarios. Notably, half of the positive cases harboured a single variant, while the remainder had two or more variants.
Conclusion
This study highlights the efficacy of a multidisciplinary approach employing WES and a tailored virtual gene panel to elucidate the aetiology of juvenile SCD. The findings support the expansion of genetic testing using tailored gene panels and prioritization schemes as part of routine autopsy evaluations to improve the identification of causative variants and potentially facilitate early diagnosis in first-degree relatives.
Supplementary Information
The online version contains supplementary material available at 10.1186/s40246-024-00657-x.
Keywords: Sudden cardiac death, Genetic, Molecular autopsy, NGS, Cardiomyopathy, Arrhythmia
Background
Sudden cardiac death (SCD) represents a dramatic event and a significant public health concern accounting for 15–20% of all deaths in the general population, with an estimated incidence of 1.3–2.8 per 100,000 person-years in individuals under the age of 50 [1, 2]. By definition, sudden natural death is presumed to be of cardiac cause when it occurs within 1 h from the onset of symptoms in witnessed cases, and within 24 h from the last time the subject was seen alive when it is unwitnessed [1, 2].
The aetiology of SCD is still largely unknown and varies with age. In individuals below the age of 50, SCD often stems from inherited disorders that induce structural and/or functional abnormalities triggering fatal arrhythmias [3, 4]. Such disorders encompass cardiomyopathies (CMP), heart structural abnormalities like hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), and arrhythmogenic cardiomyopathy (AC), and primary electrical disorders including Brugada syndrome (BrS), long QT syndrome (LQTS), short QT syndrome (SQTS), and catecholaminergic polymorphic ventricular tachyarrhythmias (CPVT) [4–6]. These conditions involve various genetic and clinical characteristics. HCM is characterized by unexplained left ventricular (LV) hypertrophy, myocyte disarray, and fibrosis. Hypertrophic cardiomyopathy (HCM) is typically associated with mutations in sarcomere and sarcomere-associated genes (e.g., Myosin Binding Protein C3 - MYBPC3 and Myosin Heavy Chain 7 - MYH7). Notably, non-sarcomere HCM and phenocopies are also reported, as well as digenic and polygenic risk factors [4, 5]. DCM features LV enlargement and fibrotic substitution, leading to systolic dysfunction and increased arrhythmic risk; common associated genes coding for cytoskeletal proteins among which Lamin A/C (LMNA) and Desmin (DES) are associated with a particularly arrhythmogenic phenotype [4, 5]. ACM is characterized by fibro-fatty replacement of the myocardium, usually due to genetic defects affecting cardiac desmosomes (e.g., Plakophilin-2 - PKP2 and Desmoplakin - DSP). Primary electrical diseases increase the risk of ventricular arrhythmias and SCD without apparent structural abnormalities. These include LQTS (often due to mutations in Potassium Voltage-Gated Channel Subfamily Q Member 1 - KCNQ1, Potassium Voltage-Gated Channel Subfamily H Member 2 - KCNH2, or Sodium Voltage-Gated Channel Alpha Subunit 5 - SCN5A), SQTS (associated with potassium channel gene mutations), BrS (often linked to SCN5A mutations), and CPVT (commonly caused by Ryanodine Receptor 2 - RYR2 and Calsequestrin 2 - CASQ2 mutations) [1, 3, 4]. Notably, all these cardiac disorders exhibit autosomal dominant pattern of inheritance with incomplete penetrance and variable expressivity making genetic assessment even more complex [1, 3–5, 7]. Other involved disorders are represented by myocarditis, congenital heart defects, including coronary artery anomalies and valve diseases, and storage cardiomyopathies. In the older individuals, coronary artery diseases and valve diseases represents the main cause of SCD [7, 8].
Autopsy plays a crucial role in identifying the cause of death, which is particularly relevant for risk prediction in family members. However, establishing a post mortem diagnosis remains challenging despite standardized autopsy guidelines [9, 10]. In particular, differentiating non-diagnostic findings from pathological abnormalities can be difficult, with up to 40% of SCD victims below the age of 50 remaining undiagnosed after a comprehensive autopsy [7, 8, 11]. Cases are classified as unexplained when autopsy reveals either non-diagnostic structural findings or no cardiac abnormalities, a scenario named sudden arrhythmic death (SAD) [1, 11, 12]. Notably, in 88% of autopsied SCD cases, the fatal event represents the first manifestation of an underlying, often asymptomatic and undiagnosed, life-threatening cardiac condition [3, 4]. Moreover, the concept of "concealed cardiomyopathy" has recently emerged, describing potentially fatal arrhythmias in inherited heart disorders that occur before visible structural changes. This highlights the complex interplay between genetic predisposition and phenotypic expression in SCD cases [11, 13, 14].
Consequently, a thorough investigation of young SCD victims is essential and post-mortem genetic testing may prove beneficial [12, 13, 15]. However, genetic testing has been limited for decades to four major susceptibility genes (KCNQ1, KCNH2, SCN5A, and RYR2), typically sequenced with the Sanger method [15, 16]. The advent of Next Generation Sequencing (NGS) has slightly enhanced diagnostic accuracy. The diagnostic yield increases by 25–40% with NGS, when a wider spectrum of genes linked to cardiomyopathies or channelopathies are included, analysing 40–200 genes depending on the method and panel used for sequencing. For instance, the TruSight Cardio Panel (Illumina, San Diego, California, U.S.) include 174 genes currently analysed for inherited heart disease (https://emea.illumina.com/products/by-type/clinical-research-products/trusight-cardio.html) [17, 18]. Current guidelines advocate for genetic testing in SCD cases with a probable genetic origin [1] but caution is advised against examining genes without a definitive link to the clinical phenotype [1]. This broader approach often results in the more frequent identification of variants of unknown significance (VUS), whit subsequent problems in interpreting the results [19–21]. American College of Medical Genetics (ACMG) guidelines for variant interpretation state that VUS are not clinically actionable but stress the importance of make an effort to reclassify VUS as either "pathogenic" or "benign [22]. In addition to globally standardized guidelines, diagnostic genetics laboratories often adopt supplementary practical guidelines, especially for managing VUS (https://www.acgs.uk.com/; https://sigu.net/). These laboratory-specific protocols guide practice and provide clarifications based on user experiences.
In light of these challenges and evolving practices, our study hypothesizes that employing whole exome sequencing (WES), refined through a bespoke virtual gene panel and a structured scheme for prioritizing variants (particularly VUS), coupled with a meticulous, case-specific variant evaluation using a multidisciplinary approach, could substantially increase the diagnostic performance of post-mortem genetic testing in SCD cases.
Methods
Study cohort
Our study is part of the "JUvenile Sudden cardiac deaTh: JUST know and treat" (JUST) project, which started in 2016 and involved both retrospective evaluation of old cases and prospective evaluations of new cases of juvenile SCD (age ≤ 50 years). In the former, young individuals were scrutinized from the Forensic Medicine Department of University Hospital of Pisa (1995–2016), while in the latter also cases from the Forensic Medicine Department of Lucca were included (2017–2023). In both cases, exclusion criteria were: non-cardiac death causes; ischemic heart and/or coronary disease; positive toxicology tests. According to forensic reports, all individuals were either completely asymptomatic or exhibited only nonspecific symptoms. Report of previously cardiological investigations with inconclusive results was reported for two subjects. None of the individuals had been diagnosed with a specific cardiac disorder prior to death (Supplemental Table 1). We secured consent for genetic testing and research use of data from the relatives of the deceased. This study was conducted in agreement with the Helsinki Declaration and received approval from the Ethic Committee of the Tuscany Region, Area Vasta Nord-Ovest (no. 14870).
Autopsy examinations
Autopsy examinations were conducted according to the latest guidelines [10]. The heart and lungs were removed "en bloc" and the pulmonary vessels were explored. Subsequently, an analysis of the right and left sections of the heart (atrial and ventricular) and the valve planes was carried out. Multiple transverse cuts at 3-mm intervals along the course of the main epicardial arteries, including branches, such as the diagonal and obtuse marginal, were performed to check patency. Finally, cuts were made along the short axis of the heart to obtain slices about 1 cm thick. Wall thickness was then verified: the endocardium was carefully inspected, and the thickness of the mid-cavity free wall of the left ventricle (LV), right ventricle (RV), and interventricular septum (IVS) (excluding trabeculae) was measured. At the end, we compared the measurements against tables of normal thickness by age, gender, and body weight [23]. The forensic experts (M.D.P., D.B.) retrospectively reviewed all the reports of macroscopic and histopathology examinations, and classified the cases into three categories: “diagnostic structural abnormalities”, if the macroscopic and/or histopathological alterations fell within the diagnostic criteria for a specific cardiomyopathy; “non-diagnostic autopsy findings”, if the macroscopic and/or histopathological alterations were subtler, such as non-diagnostic small areas of fibrosis, inflammation or fatty replacement; “structurally normal heart”, if no relevant cardiac alterations were found.
DNA extraction and whole exome sequencing
For retrospective cases, formalin-fixed, paraffin-embedded (FFPE) samples of the heart, kidney, or spleen were used. Conversely, for prospective cases, 5 to 10 mL of whole blood using a hypodermic syringe from either the femoral or inferior vena cava were collected, subsequently storing it in ethylenediaminetetraacetic acid (EDTA). The blood was refrigerated at 2 to 8 °C for analyses within 4 weeks, or frozen at − 20 °C or − 80 °C for later examinations.
DNA extraction from FFPE samples was performed using Promega's Maxwell 16 LEV DNA FFPE Purification Kit (Promega, Madison, Wisconsin U.S.). In prospective cases, DNA extraction from whole blood was done using the Maxwell® 16 LEV Blood DNA Kit (Promega, Madison, Wisconsin U.S.). Whole exome library preparation followed the manufacturer’s guidelines for the Illumina DNA Prep with Enrichment Kit (Illumina, San Diego, California, U.S.). Sequencing was conducted using Illumina’s NextSeq™ 500 instrument (Illumina, San Diego, California, U.S.).
Bioinformatic analysis and extended cardio panel
Primary exome data analysis was executed using the SeqMule pipeline [24]. FastQC and FastqScreen Quality Control (QC) systems identified and rectified errors, trimmed low-quality reads, and removed adaptors [25]. The reads were aligned to the reference human genome (GRCh37/hg19) using BWA-MEM software (https://github.com/MGPC-Nantes/MEM). Variant calling was conducted using the Genome Analysis Toolkit (GATK) [26], annotating only variants in genetic regions with a quality score ≥ 30 and a read depth ≥ 20 at the altered position [27]. VarAFT [28] and BaseSpace Variant Interpreter software (Illumina Inc., San Diego, CA, USA) were used to filter annotated variants, excluding those with a minor allele frequency (MAF) > 0.01 (1%) in GnomAD (http://gnomad.broad institute.org) and including only missense, nonsense, frameshift, and splice site variants. WES data were filtered to create a virtual panel specifically constructed from selected genes associated with cardiac diseases to expand the possible associations with SCD. Genes were first selected exploiting two databases:
Human Phenotype Ontology (HPO): this provides a standardized vocabulary of phenotypic abnormalities encountered in human disease. We searched for "Abnormal Myocardium Morphology" (HP:0001637), identifying 689 diseases and 565 associated genes, and "Abnormality of Cardiovascular System Electrophysiology" (HP:0030956), identifying 750 diseases and 524 associated genes.
Human Protein Atlas: this integrates various omics technologies to map all the human proteins in cells, tissues and organs. We searched for “heart-specific proteome” and selected the 419 genes with elevated expression in the heart compared to other tissue types. We have also checked genes used in previous NGS studies (13–36), such as those included in TruSight Cardio Panel (Illumina) (Supplemental Table 1). The final virtual panel used to filter the annotated variants thus included 1304 genes (Supplemental Table 2).
Variant interpretation
We prioritized filtered variants based on pathogenicity through VarSome (https://varsome.com) and Franklin by Genoox (https://franklin.genoox.com), which automatically classify variants according to the ACMG guidelines [22]. We also reconsidered the PM2 criterion, fulfilled for ACMG if the MAF was ≤ 0.01% (ultra-rare variants): after looking for a possible pathogenic variant among the rare variants (MAF < 1% during filtration), we evaluated the variants considering, together with the other ACMG criteria, the frequency of the different pathologies underlying SCD (e.g., HCM 1:625–1:344, MAF ⁓0.2%; DCM 1:250–400, MAF ⁓0.2 up to 1:2000, MAF ⁓0.005%; AC and channelopathies ⁓1/2000, MAF 0.005%). Splicing and frameshift variants’ functional impacts were further examined using Human Splicing Finder (https://hsf.genomnis.com) and Regulation Spotter (https://www.regulationspotter.org/) [29, 30]. We have sub-classified the VUS identified from VCF after excluding pathogenic (P) or likely pathogenic (LP). VUS that met specific criteria were classified as highly suspicious of pathogenicity and defined as VUS/LP:
Each ACMG criterion describing the variant falls within the pathogenic criteria set. This ensures that we do not include variants classified as VUS by ACMG solely due to conflicting evidence, even if some criteria suggest them as benign.
Franklin’s aggregated prediction, which combines results from various prediction tools, based on latest recommendations for PP3/BP4 rules [25], indicates a high likelihood of pathogenicity. If this criterion was not fully met, we considered whether the variants were in a recognised hot spot and/or in a critical functional domain (PM1).
The variant resides in a gene whose functional pathway aligns with the structural and/or functional cardiac alterations probably responsible for the SCD in the cases examined.
Using these prioritization schemes, variant reported as VUS based only on the PM2 criterion, when all other evidence suggests that it is benign, were considered VUS/likely to be benign and were not reported, in line with recent practical guidelines (https://www.acgs.uk.com/; https://sigu.net/). Variants reported underwent further scrutiny for clinically relevant information via ClinVar (https://www.ncbi.nlm.nih.gov/clinv), OMIM (https://www.omim.org/), ClinGen (https://clinicalgenome.org/), and PubMed (https://pubmed.ncbi.nlm.nih.gov), along with an evaluation of the variants’ presence in pertinent regions (e.g., protein functional domains, binding sites) via UniProt (https://www.uniprot.org/). Genotype associations were determined based on OMIM and ClinGen for genes already recognized in association with cardiomyopathy/channelopathies. For genes not previously linked, we considered whether the gene was reported in ClinGen as "challenged" or "limited" in association with the cardiac phenotype, previously published studies on cardiac involvement, the reported phenotype, and/or the pathway involved in cardiac function found on Human Phenotype Ontology and GeneCard (https://genecards.org/). The phenotype and/or pathway were retrieved from the Human Phenotype Ontology (HPO) and GeneCard (https://genecards.org/) databases. Globally, all information on identified variants and genes were interpreted by a multidisciplinary team, including geneticists, bioinformaticians, cardiologists and forensic medicine specialists.
We refer to the 4 main susceptibility genes as “Core genes”, the 174 genes from the TruSight Cardio Panel (Illumina, San Diego, California, USA) as “Cardio panel”, and the 1304 genes examined in this study as “Extended panel”.
Results
The final study cohort included 39 subjects (18 from the retrospective cohort and 21 from the prospective cohort; Table 1). Subjects were mainly male (n = 26, 67%), with an age of 33 ± 10 years. Autopsy results revealed a diagnostic structural abnormality in 18 cases (46%), which could be classified as HCM in 10 (56%), AC in 7 (39%), and DCM in 1 (6%). Additionally, 9 cases (23%) displayed non-diagnostic autopsy findings including fatty replacement (n = 4), mitral valve prolapse (n = 2), myocardial fibrosis (n = 2), and mild left ventricular dilation (n = 1). The remaining 12 cases (31%) displayed structurally normal hearts. Overall, 21 cases (54%) displayed non-diagnostic autopsy findings or structurally normal hearts.
Table 1.
Characteristics of sudden cardiac death cases
| Sample ID | Gender | Age | Event at death | Symptoms at death | Weight (kg) × height (cm) | Heart weight (g) | Autoptic diagnosis | Sample type |
|---|---|---|---|---|---|---|---|---|
| SCD01 | M | 26 | Sport | Syncope | 85 × 175 | 700 | HCM | FFPET |
| SCD02 | M | 29 | Sleep | Dyspnoea | 71 × 174 | n/a | SAD | n/a |
| SCD03 | M | 22 | Sport | Syncope | 87 × 171 | 365 | HCM | FFPET |
| SCD04 | M | 36 | Light activity | n/a | n/a | n/a | SAD | n/a |
| SCD05 | M | 29 | Sleep | None | 70 × 177 | 340 | SAD | FFPET |
| SCD06 | M | 37 | Rest | None | 63 × 172 | 350 | SAD | FFPET |
| SCD07 | M | 20 | Sport | Syncope | n/a × 175 | 500 | AC (RV, LV) | FFPET |
| SCD08 | M | 25 | Sleep | None | 77 × n/a | 370 | SAD | FFPET |
| SCD09 | F | 25 | n/a | n/a | n/a | n/a |
Non-diagnostic autopsy findings (MVP) |
n/a |
| SCD10 | M | 21 | Sport | Syncope | 90 × 187 | 520 | DCM | FFPET |
| SCD11 | M | 37 | Sleep | None | n/a × 168 | 360 | AC (RV) | FFPET |
| SCD12 | M | 25 | Rest | Dyspnoea | n/a | 360 | AC (RV) | FFPET |
| SCD13 | F | 14 | Sleep | None | n/a | 260 | AC (RV, LV) | n/a |
| SCD14 | F | 21 | Light activity | none | n/a × 160 | 310 | HCM | FFPET |
| SCD15 | F | 22 | Sleep | n/a | n/a | 220 | HCM | n/a |
| SCD16 | F | 35 | Rest | n/a | 87 × 171 | 365 | HCM | n/a |
| SCD17 | F | 45 | Rest | Dyspnoea | n/a | 290 | AC (RV) | FFPET |
| SCD18 | M | 20 | Rest | None | n/a × 178 | 360 | HCM | FFPET |
| SCD19 | M | 40 | n/a | n/a | n/a × 180 | 530 |
Non-diagnostic autopsy findings (foci of fatty replacement of the myocardium, RV) |
Autoptic blood |
| SCD20 | F | 50 | Rest | n/a | n/a × 164 | 300 | AC (RV) | Autoptic blood |
| SCD21 | F | 29 | Light activity | Syncope | n/a | n/a | MVP (Myxomatous degeneration of the mitral valve leaflets) | Autoptic blood |
| SCD22 | M | 42 | Sleep | None | n/a | n/a | SAD | Autoptic blood |
| SCD23 | M | 34 | Light activity | Syncope | n/a | n/a |
Non-diagnostic autopsy findings (small foci of fibrosis) |
Autoptic blood |
| SCD24 | M | 31 | Rest | Fever | n/a × 188 | 415 |
Non-diagnostic autopsy findings (foci of fatty replacement of the myocardium, LV) |
Autoptic blood |
| SCD25 | F | 37 | Light activity | n/a | n/a | 530 | SAD | Autoptic blood |
| SCD26 | M | 23 | Rest | n/a | n/a × 190 | 565 |
Non-diagnostic autopsy findings (mild fatty replacement, RV, LV) |
Autoptic blood |
| SCD27 | M | 50 | Light activity | None | n/a | 630 | HCM | Autoptic blood |
| SCD28 | M | 29 | Sport | Syncope | n/a | 380 | SAD | Autoptic blood |
| SCD29 | M | 42 | Light activity | None | n/a | 540 | Non-diagnostic autopsy findings (slightly dilated chambers and mild fatty replacement of the myocardium) | Autoptic blood |
| SCD30 | M | 40 | Rest | Chest pain | n/a | 450 |
Non-diagnostic autopsy findings (small foci of fibrosis) |
Autoptic blood |
| SCD31 | F | 45 | Rest | Chest pain | n/a | 286 | SAD | Autoptic blood |
| SCD32 | M | 29 | Sport | n/a | n/a × 184 | 315 | AC (RV, LV) | Autoptic blood |
| SCD33 | M | 50 | Sport | n/a | n/a | 510 |
Non-diagnostic autopsy findings (mild fatty replacement, RV, LV) |
Autoptic blood |
| SCD34 | M | 45 | n/a | n/a | n/a | n/a | SAD | Autoptic blood |
| SCD35 | M | 39 | Light activity | Palpitations | 175 × n/a | 575 | HCM | Autoptic blood |
| SCD36 | F | 50 | Sport | n/a | 173 × 70 | 300 | HCM | Autoptic blood |
| SCD37 | M | 42 | Sleep | None | 185 × 90 | 610 | HCM | Autoptic blood |
| SCD38 | F | 50 | Rest | None | 160 × 80 | 400 | SAD | Autoptic blood |
| SCD39 | F | 45 | Rest | None | 160 × 60 | 200 | SAD | Autoptic blood |
AC arrhythmogenic cardiomyopathy, HCM hypertrophic cardiomyopathy, DCM dilated cardiomyopathy, SAD sudden arrhythmic death, MVP mitral valve prolapse, LV left ventricle, RV right ventricle, FFPET formalin-fixed and paraffin-embedded tissue, n/a not available
WES was performed in 32 cases (7 HCM, 6 AC, 1 DCM, 10 SAD, 8 with non-diagnostic autopsy findings), due to FFPE unavailability in 6 samples and bacterial DNA contamination in 1 FFPE sample, precluding the completion of the remaining retrospective cases. Likely causative variants were found in 22 cases, translating to a detection rate of 69%, outperforming both the Cardio panel and standard susceptibility gene analysis (50% and 16%, respectively, as shown in Fig. 1).
Fig. 1.
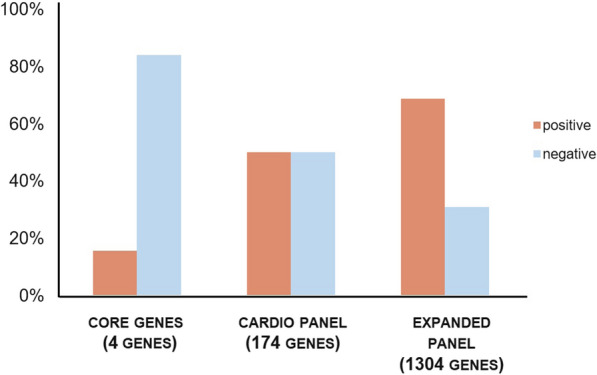
Added diagnostic value of the extended cardio panel. The variant detection rate (%) found using different virtual panels in all autopsy cases: core genes 16%, cardio panel 50%, extended cardio panel 69%. “Core genes” represent the 4 main susceptibility genes, “Cardio panel” includes the 174 genes from the TruSight Cardio Panel (Illumina, San Diego, California, USA) and the “extended panel” includes the 1304 genes selected in this study
Detailed variant descriptions are provided in Tables 2, 3 and 4. All identified variants were heterozygous. Half of positive cases (n = 11) harboured a single variant, while the remainder had ≥ 2 variants. P/LP and VUS/LP variants were distributed in the different autopsy groups as follows: SCD with diagnostic structural abnormalities, 8 out of 14 (57%; 6/7 in HCM, 2/6 in AC, 0/1 DCM); non-diagnostic autopsy findings, 6 out of 8 (75%); structurally normal heart, 8 out of 10 (80%; Fig. 2). Overall, 78% of cases with non-diagnostic autopsy findings or structurally normal heart had a positive genetic test with the extended cardio panel.
Table 2.
Variants assessed as P/LP or VUS/LP in SCD cases with diagnostic structural abnormalities
| Sample ID | Age | Genetic alteration (gene, nucleotide and aminoacidic change) | Variant type | ACMG | ACMG criteria | ClinVar | Variant literature (PMID) | Gene MIM number/cardiac association | ClinGen cardiac association | HP no. phenotype association/gene pathway from GeneCards | Protein |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HCM | |||||||||||
| SCD01 | 26 | n/a | / | / | / | / | / | / | / | / | / |
| SCD03 | 22 |
MYH7 (NM_000257.4): c.4258C > T; p.Arg1420Trp |
missense | LP/P |
PS4 PM1 PM2 PP3 PM5 |
P-HCM | (1–6) | 192600/HCM | HCM/definitive |
0001645-SCD/ Cytoskeleton remodeling: regulation of actin cytoskeleton |
Myosin Heavy Chain 7 |
| SCD14 | 21 | negative | / | / | / | / | / | / | / | / | / |
| SCD15 | 22 | n/a | / | / | / | / | / | / | / | / | / |
| SCD16 | 35 | n/a | / | / | / | / | / | / | / | / | / |
| SCD18 | 20 |
KCNH2 (NM_000238.4): c.1010C > G; p.Thr337Ser |
missense | VUS/LP |
PM2 PP3 PP2 |
u.s.-LQTS | n/a | 613695/LQTS |
LQTS/SQTS/definitive BrS/disputed |
0001645-SCD/ Cardiac conduction: phase3-rapid repolarisation |
Potassium Voltage-Gated Channel (Subfamily H Member 2) |
|
KCNE1 (NM_000219.6): c.95G > A; p.Arg32His |
missense | VUS |
PM2 PM5 |
u.s.-LQTS | (7,8) | 176261/LQTS | LQTS/limited |
0001645-SCD/ Cardiac conduction: phase2-plateau phase; phase3-rapid repolarisation |
Potassium Voltage-Gated Channel (Subfamily E Regulatory Subunit 1) | ||
| SCD27 | 50 |
FBN1 (NM_000138.4): c.979A > G; p.Arg327Gly |
missense | LP/P |
PP3 PP2 PS4 PM2 |
LP-Marfan | (9) | 134797/Marfan Syndrome | Marfan syndrome/ definitive |
0001640-Cardiomegaly; 0001635-Congestive heart failure/ ERK Signaling |
Fibrillin 1 |
| SCD35 | 39 |
SLC4A3 (NM_001326559.2): c.1318G > T; p.Asp440Tyr |
missense | VUS/LP |
PM2 PP3 |
n/a | n/a | 106195/SQT | SQTS/moderate |
0001645-SCD 0001962-Palpitations/ Transport of inorganic cations/anions and amino acids/oligopeptides |
Solute Carrier Family 4 Member 3 (Sodium-independent anion exchanger) |
|
PSEN2 (NM_000447.3): c.506A > G; p.His169Arg |
missense | VUS/LP |
PM2 PP3 PP2 |
u.s | n/a | 600759/DCM | DCM/limited |
0005110-Atrial fibrillation/ 0001279-Syncope ERK Signaling |
Presenilin 2 | ||
| SCD36 | 50 |
SCN10A (NM_001293307.2): c.916G > C; p.Asp306His |
missense | VUS/LP |
PP3 PM2 |
n/a | n/a | 604427/episodic pain/BrS/VT | BrS/disputed |
0001645-SCD/ Cardiac conduction: phase0-rapid depolarisation |
Sodium Voltage-Gated Channel (Alpha Subunit 10) |
|
KCNMA1 (NM_001161352.2): c.3457A > C; p.Lys1153Gln |
missense | VUS/LP |
PP3 PP2 PM2 |
u.s.-epilepsy | n/a | 600150/seizure | n/a |
0030680-Abnormality of cardiovascular system morphology/ Ca2 + activated K + channels |
Potassium Calcium-Activated Channel (Subfamily M Alpha 1) | ||
| SCD37 | 50 |
BAG3 (NM_004281.3): c.67C > T; p.Arg203Trp |
missense | VUS/LP |
PM2 PP3 PM1 |
u.s-DCM/ CMP | n/a | 603883/DCM | DCM/definitive |
0030872-Abnormal cardiac ventricular function/ Cellular responses to stimuli and heat stress |
BAG Cochaperone 3 |
| AC | |||||||||||
| SCD07 | 20 | negative | / | / | / | / | / | / | / | / | / |
| SCD11 | 37 |
RYR2 (NM_001035.3): c.8779C > T; p.Gln2927Ter |
nonsense | LP/P |
PVS1 PM2 |
n/a | n/a | 180902/VA/CPVT |
CPVT/definitive HCM/limited |
0001645-SCD/ Cardiac conduction: ion homeostasis |
Ryanodine Receptor 2 (Calcium channel on sarcoplasmic reticulum) |
| SCD12 | 25 | negative | / | / | / | / | / | / | / | / | / |
| SCD13 | 14 | n/a | / | / | / | / | / | / | / | / | / |
| SCD17 | 45 | negative | / | / | / | / | / | / | / | / | / |
| SCD20 | 50 | negative | / | / | / | / | / | / | / | / | / |
| SCD32 | 29 | DSP (NM_004415.4): c.7000C > T; p.Arg2334Ter | Nonsense | LP/P |
PVS1 PP5 PM2 |
P-AC u.s.-CMP | n/a | 125647/AC |
AC/definitive HCM/disputed |
0001645-SCD/ Signaling by Rho GTPases;Apoptosis |
Desmoplakin |
| DCM | |||||||||||
| SCD10 | 21 | Negative | / | / | / | / | / | / | / | / | / |
AC arrhythmogenic cardiomyopathy, ACMG American College of Medical Genetics, BrS Brugada syndrome, CMP cardiomyopathy, CPVT catecholaminergic polymorphic ventricular tachyarrhythmias, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, LP likely pathogenic, LQTS long QT syndrome, n/a not available, P pathogenic, SCD sudden cardiac death, SQTS short QT syndrome, u.s. uncertain_significance on ClinVar, VUS/LP variant with unknown significance/likely pathogenic, PVS1 Very Strong—Null variants (nonsense, frameshift, canonical ± 1 or 2 splice sites, initiation codon, single exon or multiexon deletion) in a gene where loss of function is a known mechanism of disease, PM1 Mutational hot spot and/or critical and well-established functional domain, PM2 Absent from controls or at extremely low frequency in Exome Sequencing Project, 1000 Genomes or ExAC, PM5 Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before, PP5 Reputable source recently reports variant as pathogenic but the evidence is not available to the laboratory to perform an independent evaluation, PP2 Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease, PP3 Multiple lines of computational evidence support a deleterious effect on the gene or gene product
1.O’Hare BJ, Bos JM, Tester DJ, Ackerman MJ. Patients With Hypertrophic Cardiomyopathy Deemed Genotype Negative Based on Research Grade Genetic Analysis. Circ Genom Precis Med. 2020 Dec;13(6)
2.Patel AP, Dron JS, Wang M, Pirruccello JP, Ng K, Natarajan P, et al. Association of Pathogenic DNA Variants Predisposing to Cardiomyopathy With Cardiovascular Disease Outcomes and All-Cause Mortality. JAMA Cardiol. 2022 Jul 1;7(7):723
3.Stava TT, Leren TP, Bogsrud MP. Molecular genetics in 4408 cardiomyopathy probands and 3008 relatives in Norway: 17 years of genetic testing in a national laboratory. Eur J Prev Cardiol. 2022 Oct 18;29(13):1789–99
4.Park J, Packard EA, Levin MG, Judy RL, Damrauer SM, Day SM, et al. A genome-first approach to rare variants in hypertrophic cardiomyopathy genes MYBPC3 and MYH7 in a medical biobank. Hum Mol Genet. 2022 Mar 3;31(5):827–37
5.Walsh R, Mazzarotto F, Whiffin N, Buchan R, Midwinter W, Wilk A, et al. Quantitative approaches to variant classification increase the yield and precision of genetic testing in Mendelian diseases: the case of hypertrophic cardiomyopathy. Genome Med. 2019 Dec 29;11(1):5
6.Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy. Circulation. 2018 Oct 2;138(14):1387–98
7.Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound Mutations. Circulation. 2004 Apr 20;109(15):1834–41
8.Leinonen JT, Crotti L, Djupsjöbacka A, Castelletti S, Junna N, Ghidoni A, et al. The genetics underlying idiopathic ventricular fibrillation: A special role for catecholaminergic polymorphic ventricular tachycardia? Int J Cardiol. 2018 Jan;250:139–45
9.Mannucci L, Luciano S, Salehi LB, Gigante L, Conte C, Longo G, et al. Mutation analysis of the FBN1 gene in a cohort of patients with Marfan Syndrome: A 10-year single center experience. Clinica Chimica Acta. 2020 Feb;501:154–64
Table 3.
Variants assessed as P/LP or VUS/LP in SCD cases with non-diagnostic autopsy findings
| ID | Age | Genetic alteration (gene, nucleotide and aminoacidic change) |
Variant type | ACMG | ACMG criteria | ClinVar | Variant literature (PMID) | Gene MIM number/cardiac association | ClinGen cardiac association | HP no. phenotype association/gene pathway from GeneCards | Protein |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mild fatty replacement | |||||||||||
| SCD19 | 40 | CACNA1C (NM_000719.7): c.2558 T > C; p.Met853Thr | missense | VUS/LP |
PM2 PM1 PP3 PP2 |
u.s.-LQTS | n/a | 114205/BRs/LQTS |
LQTS/moderate BrS/SQTS/disputed |
0001645-SCD/ Cardiac conduction: phase 2-plateau phase |
Calcium Voltage-Gated Channel (Subunit Alpha1 C) |
| SCD24 | 31 | negative | / | / | / | / | / | / | / | / | / |
| SCD26 | 23 | ATP1A2 (NM_000702.4): c.1352A > C; p.Glu451Ala | missense | VUS/LP |
PP3 PP2 PM2 |
n/a | n/a | 182340/n/a | n/a |
0011675-Arrhythmia/ Cardiac conduction: ion homeostasis; ion transport by P-type ATPases |
ATPase Na+/K+ Transporting Subunit Alpha 2 |
| SCD33 | 50 | SNTA1 (NM_003098.3): c.583delC; p.Leu195Phefs*23 | frameshift | LP |
PVS1 PM2 |
n/a | n/a | 612955/LQTS | LQTS/disputed |
0001645-SCD/ NGF Pathway |
Syntrophin Alpha 1 |
| Mitral valve prolapse | |||||||||||
| SCD09 | 25 | n/a | / | / | / | / | / | / | / | / | / |
| SCD21 | 29 | TGFB2 (NM_001135599.3): c.1312 T > C; p.Ser410Pro | missense | LP |
PP3 PM2 |
n/a | n/a | 190220/Loeys-Dietz syndrome | Loeys-Dietz syndrome/definitive |
0001654-Abnormal heart valve morphology/ ERK Signaling |
Transforming growth factor beta 2 |
|
SCN5A (NM_001160161.2): c.1820G > A; p.Gly607Asp |
missense | VUS |
PM2 PP2 |
n/a | n/a | 600163/BrS/LQTS |
Brs/LQTS/DCM/ definitive AC/limited CPVT/SQTS/ disputed |
0001645-SCD/ Cardiac conduction: phase0-rapid depolarisation |
Sodium Voltage-Gated Channel (Alpha Subunit 5) | ||
| Small foci of fibrosis | |||||||||||
| SCD23 | 34 |
TTN (NM_001267550.2): c.69936C > G; p.Tyr23312Ter |
nonsense | P |
PVS1 PM2 |
n/a | n/a | 188840/DCM/HCM |
DCM/definitive HCM/AC/limited |
0001645-SCD/ Striated muscle contraction; cardiac conduction |
Titin |
| SCD30 | 40 | negative | / | / | / | / | / | / | / | / | / |
| Slightly dilated chambers | |||||||||||
| SCD29 | 42 |
LAMA4 (NM_002290.4): c.719-2A > G; |
splicing | LP |
PVS1 PM2 |
u.s-DCM | (1) | 600133/DCM | DCM/limited |
0001644-DCM; 0012664-Reduced left ventricular ejection fraction/ ERK Signaling |
Laminin (Subunit Alpha 4) |
|
CDH2 (NM_001792.4): c.1729G > C; p.Ala577Pro |
missense | VUS/LP |
PM2 PP3 |
u.s | n/a | 114020/AC | AC/limited |
0004756-Ventricular tachycardia; 0011675-Arrhythmia/ Cell junction organization; ERK Signaling |
Cadherin 2 | ||
AC arrhythmogenic cardiomyopathy, ACMG American College of Medical Genetics, BrS Brugada syndrome, CMP cardiomyopathy, CPVT catecholaminergic polymorphic ventricular tachyarrhythmias, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, LP likely pathogenic, LQTS long QT syndrome, n/a not available, P pathogenic, SCD sudden cardiac death, SQTS short QT syndrome, u.s. uncertain_significance on ClinVar, VUS/LP variant with unknown significance/likely pathogenic, PVS1 Very Strong—Null variants (nonsense, frameshift, canonical ± 1 or 2 splice sites, initiation codon, single exon or multiexon deletion) in a gene where loss of function is a known mechanism of disease, PM1 Mutational hot spot and/or critical and well-established functional domain, PM2 Absent from controls or at extremely low frequency in Exome Sequencing Project, 1000 Genomes or ExAC, PP3 Multiple lines of computational evidence support a deleterious effect on the gene or gene product, PP2 Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease
1.Verdonschot JAJ, Hazebroek MR, Krapels IPC, Henkens MTHM, Raafs A, Wang P, et al. Implications of Genetic Testing in Dilated Cardiomyopathy. Circ Genom Precis Med. 2020 Oct;13(5):476–87
Table 4.
Variants assessed as P/LP or VUS/LP in SCD cases with structurally normal heart: SAD (sudden arrhythmic death)
| Sample ID | Age | Genetic alteration (gene, nucleotide and aminoacidic change) |
Protein impact | ACMG | ACMG criteria | ClinVar | Variant literature (PMID) | Gene MIM number/Cardiac association | ClinGen cardiac association | HP no. phenotype association/ gene pathway from GeneCards |
Protein |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SCD02 | 29 | n/a | / | / | / | / | / | / | / | / | / |
| SCD04 | 36 | n/a | / | / | / | / | / | / | / | / | / |
| SCD05 | 29 |
HCN1 (NM_021072.3): c.2560C > T; p.Arg854Ter |
nonsense | P/LP |
PVS1 PM2 |
n/a | n/a | 602780/seizure | seizure/definitive |
n/a Contributes to the native pacemaker currents in heart/ Potassium Channels |
Hyperpolarization Activated Cyclic Nucleotide Gated Potassium Channel 1 |
|
KCND3 (NM_004980.5): c.416G > A; p.Arg139His |
missense | VUS |
PM2 PP2 |
n/a | n/a | 616399/BrS | BrS/disputed |
0001695-Cardiac arrest/ Cardiac conduction: phase1-inactivation of fast Na + channels |
Potassium Voltage-Gated Channel (Subfamily D Member 3) | ||
| SCD06 | 37 |
MYBPC3 (NM_000256.3): c.1255C > T; p.Arg419Cys |
missense | VUS/LP |
PM2 PM1 PP3 PP2 |
us-HCM us-CMP |
(1,2) | 600958/HCM/DCM/ LVNC |
HCM/definitive DCM/AC/limited |
0001695-Cardiac arrest/ Striated muscle contraction pathway; Cardiac conduction |
Myosin Binding Protein C3 |
|
RYR2 (NM_001035.2): c.12168G > T;p.Lys4056Asn |
missense | VUS/LP |
PM2 PM1 PP2 |
us-CMP | n/a | 180902/VA/CPVT |
CPVT/definitive HCM/limited |
0001645-SCD/ Cardiac conduction: ion homeostasis |
Ryanodine Receptor 2 (Calcium channel on sarcoplasmic reticulum) |
||
| SCD08 | 25 |
CALM1 (NM_006888.4): c.10C > T; p.Gln4Ter |
nonsense | P/LP |
PVS1 PM2 |
n/a | n/a | 614916/CPVT/LQT |
LQTS/definitive CPVT/moderate |
0001645-SCD/ Cardiac conduction:ion homeostasis |
Calmodulin 1 (calcium signal transduction pathway) |
|
RYR2 (NM_001035.2): c.7034C > T; p.Ala2345Val |
missense | VUS/LP |
PM2 PM1 PP2 |
n/a | n/a | 180902/VA/CPVT |
CPVT/definitive HCM/limited |
0001645-SCD/ Cardiac conduction:Ion homeostasis |
Ryanodine Receptor 2 (Calcium channel on sarcoplasmic reticulum) |
||
| SCD22 | 42 | negative | / | / | / | / | / | / | / | / | / |
| SCD25 | 37 | negative | / | / | / | / | / | / | / | / | / |
| SCD28 | 29 |
RYR2 (NM_001035.2): c.4972 C > G; p.Leu1658Val |
missense | VUS/LP |
PM2 PP3 PP2 |
n/a | n/a | 180902/VA/CPVT |
CPVT/definitive HCM/limited |
0001645-SCD/ Cardiac conduction:Ion homeostasis |
Ryanodine Receptor 2 (Calcium channel on sarcoplasmic reticulum) |
|
CACNA2D1 (NM_000722.4): c.1393C > A;p.Pro465Thr |
missense | VUS/LP |
PP3 PP2 PM2 |
n/a | n/a | 114204/– | BrS/SQTS/disputed |
0001645-SCD/ CREB Pathway: intracellular calcium signaling |
Calcium Voltage-Gated Channel (Auxiliary Subunit Alpha2delta 1) | ||
|
LMNA (NM_170707.3): c.1122 C > G; p.His374Gln |
missense | VUS/LP |
PM2 PM1 PP2 |
n/a | n/a | 150330/DCM |
DCM/definitive AC/limited |
0001645-SCD/ Apoptosis and survival FAS signaling cascades |
Lamin A/C | ||
| SCD31 | 45 |
TTN (NM_001267550.2): c.97556_97557insA; p.Trp32520LeufsTer7 |
frameshift | P/LP |
PVS1 PM2 |
n/a | n/a | 188840/DCM | DCM/definitive |
0001645-SCD/ Striated muscle contraction;Cardiac conduction |
Titin |
| SCD34 | 45 |
KCNJ14 (NM_013348.4): c.599 T > C; p.Phe200Ser |
missense | VUS/LP |
PM2 PP3 |
n/a | n/a | 603953/– | n/a |
n/a Cardiac conduction: classical Kir channels; phase4-resting membrane potential |
Potassium Inwardly Rectifying Channel (Subfamily J Member 14) |
|
SCN5A (NM_001160161.2): c.1820G > A; p.Gly607Asp |
missense | VUS |
PM2 PP2 |
us-BrS | n/a | 600163/BrS/LQTS/AF/VF |
BrS/LQTS/DCM/ definitive AC/limited CPVT/SQTS/disputed |
0001645-SCD/ Cardiac conduction: phase0-rapid depolarisation |
Sodium Voltage-Gated Channel (Alpha Subunit 5) | ||
| SCD38 | 50 |
KCND3 (NM_004980.5): c.905G > A; p.Arg302His |
missense | P/LP |
PM1 PM5 PP3 PM2 |
n/a | n/a | 605411/BRS | BrS/disputed |
0001695-Cardiac arrest/ Cardiac conduction: phase1-inactivation of fast Na+ channels |
Potassium Voltage-Gated Channel (Subfamily D Member 3) |
|
MYH7 (NM_000257.4): c.5606A > G; p.Asp1869Gly |
missense | VUS/LP |
PP3 PM2 |
us | (3,4) | 192600/HCM | HCM/definitive |
0001645-SCD/ Cytoskeleton remodeling: regulation of actin cytoskeleton |
Myosin Heavy Chain 7 | ||
| SCD39 | 45 |
SCN9A (NM_002977.3): c.2633G > C; p.Gly878Ala |
missense | VUS/LP | PP3 PM2 | n/a | n/a | 603415/– | n/a |
0011675-Arrhythmia/ Cardiac conduction: phase0-rapid depolarisation |
Sodium Voltage-Gated Channel (Alpha Subunit 9) |
AC arrhythmogenic cardiomyopathy, ACMG American College of Medical Genetics, BrS Brugada syndrome, CMP cardiomyopathy, CPVT catecholaminergic polymorphic ventricular tachyarrhythmias, DCM dilated cardiomyopathy, HCM hypertrophic cardiomyopathy, LP likely pathogenic, LQTS long QT syndrome, n/a not available, P pathogenic, SCD sudden cardiac death, SQTS short QT syndrome, u.s. uncertain_significance on ClinVar, VUS/LP variant with unknown significance/likely pathogenic, PVS1 Very Strong—Null variants (nonsense, frameshift, canonical ± 1 or 2 splice sites, initiation codon, single exon or multiexon deletion) in a gene where loss of function is a known mechanism of disease, PM5 Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before, PM1 Mutational hot spot and/or critical and well-established functional domain, PM2 Absent from controls or at extremely low frequency in Exome Sequencing Project, 1000 Genomes or ExAC, PP3 Multiple lines of computational evidence support a deleterious effect on the gene or gene product, PP2 Missense variant in a gene that has a low rate of benign missense variation and where missense variants are a common mechanism of disease
1.van Lint FHM, Mook ORF, Alders M, Bikker H, Lekanne dit Deprez RH, Christiaans I. Large next-generation sequencing gene panels in genetic heart disease: yield of pathogenic variants and variants of unknown significance. Netherlands Heart Journal. 2019 Jun 7;27(6):304–9
2.Bourfiss M, van Vugt M, Alasiri AI, Ruijsink B, van Setten J, Schmidt AF, et al. Prevalence and Disease Expression of Pathogenic and Likely Pathogenic Variants Associated With Inherited Cardiomyopathies in the General Population. Circ Genom Precis Med. 2022 Dec;15(6)
3.Homburger JR, Green EM, Caleshu C, Sunitha MS, Taylor RE, Ruppel KM, et al. Multidimensional structure–function relationships in human β-cardiac myosin from population-scale genetic variation. Proceedings of the National Academy of Sciences. 2016 Jun 14;113(24):6701–6
4.Marstrand P, Han L, Day SM, Olivotto I, Ashley EA, Michels M, et al. Hypertrophic Cardiomyopathy With Left Ventricular Systolic Dysfunction. Circulation. 2020 Apr 28;141(17):1371–83
Fig. 2.
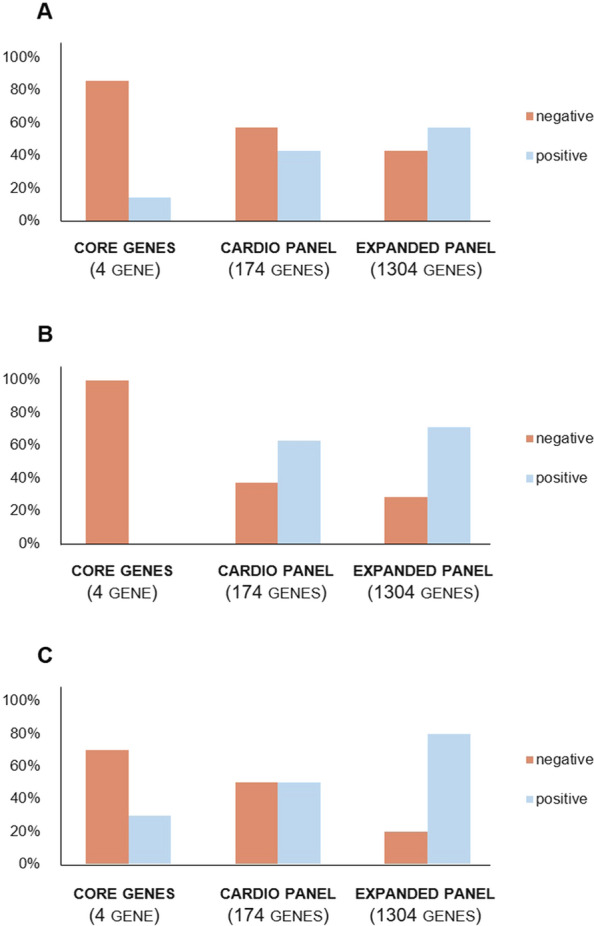
Variants diagnostic detection rate (%) in each autopsy group. The cohort of “SCD with diagnostic structural abnormalities” (A) (macroscopic and/or histopathological alterations fell within the diagnostic criteria for a specific cardiomyopathy) shows a detection rate of 14% (2/14) with the major susceptibility genes (core genes), 43% (6/14) with the cardio panel and 57% (8/14) with the extended cardio panel; the core genes have a detection rate of 0% in the group of “SCD with non-diagnostic autopsy findings” (B) (macroscopic and/or histopathological alterations were subtler) (0/8) and of 30% in the group of “SCD with structurally normal heart (SAD)” (3/10); the increase in the detection rate of likely causative variants ranges from 63% (5/8) with the cardio panel to 75% (6/8) with the extended panel for “SCD cases with inconclusive autopsy findings” (C) and from 60% (6/10) to 80% (8/10) for the SAD group
P/LP variants were found in 12 cases (38%): 7 in genes affecting myocardial structure/morphology and 4 in genes related to cardiac electrical function. VUS/LP variants were identified in 10 cases (31%): 6 in structural protein genes and 10 in ion channel-related genes. Fifty percent of sudden cardiac death (SCD) cases, whether they exhibit diagnostic structural abnormalities or have non-diagnostic autopsy findings, showed variants in cardiomyopathy genes. In contrast, 80% of cases with structurally normal hearts (SAD) presented variants in channelopathy genes (Fig. 3). Fifteen genes recognized for their association with cardiac disease were identified: 3 (RYR2, SCN5A, KCNH2) from the primary susceptibility panel and 12 (TTN, MYH7, MYBPC3, TGFB2, CACNA2D1, CALM1, LAMA4, DSP, CACNA1C, FBN1, SNTA1 BAG3) from the Cardio panel. Three cases with variants in Cardio panel genes had causative variants exclusively identified in the extended panel. Nine genes uniquely included in our extensive panel (HCN1, KCNJ14, SCN9A, SLC4A3, PSEN2, SCN10A, KCNMA1, ATP1A2, CDH2) were found to have a disease association labeled as “disputed” or “limited” (ClinGen) and/or cardiac-related pathways (GeneCards). No significant differences were found in the diagnostic yield across the different age groups (p = 1 for each age class; Fig. 4).
Fig. 3.

Cardiomyopathy versus channelopathy genes in different autopsy groups. Probable causative variants in genes associated with cardiomyopathy (orange) are observed more frequently in cases of SCD with diagnostic structural abnormalities (50%) (A) and with non-diagnostic autopsy findings (50%) (B), compared to those with structurally normal hearts (SAD) (25%) (C). Bars are marked with the same symbol when represent the same subject; genes marked with an asterisk present (P/LP) variant
Fig. 4.
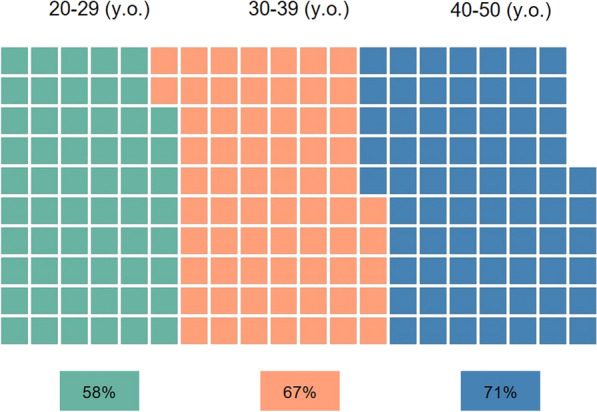
Diagnostic yield percentages across age groups. Waffle charts depicting diagnostic yield percentages for three age groups: 20–29 years (58%), 30–39 years (67%), and 40–50 years (71%). Each square represents 1% of the total sample. Despite apparent differences, statistical analysis revealed no significant variation in diagnostic yield across age groups (p = 1 for each age class)
Discussion
Juvenile SCD remains unexplained in approximately 40% of cases, despite forensic and molecular autopsy. This highlights the need for a more in-depth search for gene variants. Our study employed a multidisciplinary approach involving geneticists, cardiologists, bioinformaticians, and forensic medicine specialists. The expertise of this team was exploited in a stepwise manner: (1) review of macroscopic and microscopic findings from SCD cases, (2) comprehensive assessment of available guidelines and relevant literature to develop a robust variant prioritization scheme, (3) analysis of variants identified through WES. A multidisciplinary discussion was crucial to sub-classify VUS and evaluate prioritized variants in the context of individual case characteristics. This integrated approach enabled a thorough and nuanced interpretation of genetic findings in relation to the clinical and pathological features of each SCD case, identifying likely causative variants in 22 out of 32 cases (69%), compared to 16 cases (50%) when using the standard cardiac panel.
Our diagnostic yield is higher than previous studies. Among 26 other NGS studies that adhered to ACMG guidelines and with a sample size comparable to ours, reported diagnostic yields ranged from 6 to 44% [15, 17, 21, 31–33], as detailed in Supplemental Table 1. Several studies do not include VUS in the diagnostic yield reporting only their detection percentage [17, 21, 34–36]. Conversely, other studies have found that including VUSs in genetic evaluation increases the variant detection rates to approximately 50–80% [18, 31, 37]. However, these studies did not further categorize VUSs based on their probabilities of being benign or pathogenic, unlike our study. Some variants were classified as VUS based solely on the PM2 criterion, even when other criteria suggested benignity, or when the PP3 criterion was not fully satisfied. In our prioritization scheme, such VUS are considered 'cold,' which excludes variants that are more likely to be reclassified as benign over time. VUS are considered “not clinically actionable” and cannot be used directly for diagnostic purposes [22]. However, recent publications [38, 39] and practice guidelines (https://www.acgs.uk.com/; https://sigu.net/) advocate for a more nuanced approach to manage VUS, rather than simply categorizing them as uncertain. Multidisciplinary discussions to evaluate VUS in specific cases can help to determine which VUS should be reported to clinicians, particularly when there is high level of evidence supporting pathogenicity and potential for obtaining additional evidence [40]. Further studies are necessary to validate variants we have classified as VUS/LP and achieve final reclassification. However, our results underscore the importance of expanding genetic testing through tailored gene panels and specific prioritization, with variant assessment conducted in a multidisciplinary setting that considers the case context. Integrating this approach into laboratory practice facilitates comprehensive reporting to clinicians and enables further variant studies and patient follow-up. The data collected may contribute to future reclassification of the VUS/LP identified in this study. Furthermore, the stored raw exome data can be re-evaluated in future trio-family analyses.
All cases with positive genetic results exhibited heterozygous variants, and 27% had 2 or more variants, highlighting the complexity of the SCD phenotype. These observations support the theory that SCD might result from interactions among common variants with moderate impacts or clusters of rarer variants with more pronounced effects. Distinguishing pathogenic from benign variants is particularly challenging, given the potential for incomplete penetrance, variable expressivity, and phenotypic overlap in channelopathies and cardiomyopathies, which typically follow an autosomal dominant inheritance pattern. Combinations of known and unknown genetic and environmental factors may contribute to incomplete penetrance and variable expressivity [41–44].
In our study, 9 altered genes would not have been identified in the standard cardiac panel. Some of these genes— Solute Carrier Family 4 Member 3 - SLC4A3, Presenilin 2 - PSEN2, Sodium Voltage-Gated Channel Alpha Subunit 10 - SCN10A, and Cadherin 2 - CDH2—are currently under investigation, but are likely to be associated with SQTS, DCM, BrS and AC, respectively. The remaining genes are implicated in ion homeostasis and cardiac conditions. For instance, Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel 1 (HCN1), highly expressed in human sinoatrial node, could have a key role in pacemaker current [45–47]. ATPase Na+/K+ Transporting Subunit Alpha 2 (ATP1A2) had “cardiac conduction” among its related pathways, with a role in ion homeostasis and ion transport by P-type ATPases and recently, Staehr et al., suggesting its potential role in cardiac function and metabolism through the Src/Ras/Erk1/2 pathway [48]. Most variants in these genes are classified as VUS/LP, given the absence of definitive gene/disease associations. Current guidelines advocate genetic testing in SCD cases with a probable genetic basis, but also advice caution against analysing genes not definitively associated with the clinical phenotype [1]. This is a significant issue, particularly in cases with non-diagnostic autopsy findings or structurally normal hearts, when clinical and family histories are unavailable to guide post-mortem genetic testing. We propose a combined approach extending the search for genetic variants beyond specific panels, while limiting the likelihood of casual results from broad screenings.
The diagnostic yield found in SCD with diagnostic structural abnormalities (57%) aligns with results from earlier studies on inherited cardiomyopathies [49]. Ion channel-related genes were found in 4 cases revealed a potential overlapping phenotype. This result is consistent with previous studies that have detected variants in cardiac conduction-related genes in cases of SCD with autopsy diagnosis of cardiomyopathy [50, 51]. This could derive from the use of a broader gene panel including classical “channelopathies” genes to test subjects with structural alteration. On the other hand, structural and conduction alterations might coexist, and CPVT, LQTS, and SQTS can present as asymptomatic, but still lead to cardiac arrest as its first manifestation, as in our cohort.
Our results underscore the efficacy of a multidisciplinary approach to elucidate the aetiology of juvenile SCD, particularly in unexplained cases. We identified probable causative variants in 14 cases with either non-diagnostic autopsy findings or structurally normal hearts, achieving a yield of 78%. This approach reduced the proportion of unexplained cases from conventional autopsy to molecular autopsy. Variants in genes associated with channelopathies were found in 50% of cases, supporting previous findings [18, 44]. Notably, RYR2 VUS/LP variants were identified in 9% of SAD cases, aligning with previous reports that observed a prevalence of 5–10% [52, 53]. However, all three cases in our cohort also harboured additional probable causative variants, underscoring the importance of comprehensive genetic analysis. Conversely, variants in cardiomyopathy-associated genes were found in 28% of cases, supporting the hypothesis that variants in structural proteins can trigger functional abnormalities in cardiomyocytes before any macroscopic or histopathological changes are evident [11, 14, 44]. Sudden death in these individuals, who carry a likely causative variant in a cardiomyopathy-associated gene, could be attributed to a "concealed cardiomyopathy," where malignant arrhythmias occur in the absence of overt clinical disease. As instance, case [SCD29] had slightly dilated chambers found at autopsy and carried the splicing LP LAMA4:c.719-2A > G already found in a DCM patient [54], while case [SCD31] with structurally normal heart carried a frameshift LP variant in TTN. Therefore, these results support recent recommendations suggesting that genes linked to cardiomyopathy should be included in the molecular autopsy [1].
The co-segregation of the variant with the disease in family members is a powerful tool for identify the causative variant as well as for reclassification of VUSs. Functional validation studies of genetic variants represent another effective approach, though their expense and time-consuming nature limit their routine use, especially considering the vast number of variants uncovered by NGS. We did not perform either co-segregation or functional validation studies, and the small sample size and the limited information about previously clinical data and family history did not allow searching for associations between specific variants with clinical data. These limitations have led to a lack of definitive confirmation of the pathogenicity of the variants considered VUS/LP. However, we have tried to address this limitation through the specific VUS prioritization scheme and careful case-specific variant assessments by multidisciplinary team.
In conclusion, WES optimized with a customized virtual gene panel, a structured variant prioritization scheme and a multidisciplinary approach for case-specific variant evaluation can significantly improve the identification of likely causative variants in juvenile SCD cases, particularly when autopsy findings are inconclusive. This approach should be considered as a routine basis in this setting for achieving a thorough autopsy diagnosis and potentially facilitating early diagnosis in first-degree relatives.
Supplementary Information
Acknowledgements
We are grateful to Chiara Maria Mazzanti for her advices on experimantal design and to Sara Franceschi, Francesca Lessi and Michele Menicagli for their technical assistance. We would also thank all the medical legals that have performed autopsies during the retrospective and prospective period of the study.
Abbreviations
- AC
Arrhythmogenic cardiomyopathy
- ATP1A2
ATPase Na+/K+ Transporting Subunit Alpha 2
- ACMG
American College of Medical Genetics
- BrS
Brugada syndrome
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- CASQ2
Calsequestrin 2
- DES
Desmin
- DSP
Desmoplakin
- FFPE
Formalin-fixed: paraffin-embedded
- HCN1
Hyperpolarization-Activated Cyclic Nucleotide-Gated Channel 1
- IVS
Interventricular septum
- LMNA
Lamin A/C
- LQTS
Long QT syndrome
- LV
Left ventricle
- MAF
Minor allele frequency
- MYBPC3
Myosin Binding Protein C3
- MYH7
Myosin Heavy Chain 7
- PKP2
Plakophilin-2
- KCNQ1
Potassium Voltage-Gated Channel Subfamily Q Member 1
- KCNH2
Potassium Voltage-Gated Channel Subfamily H Member 2
- RYR2
Ryanodine Receptor 2
- RV
Right ventricle
- SAD
Sudden arrhythmic death
- SCD
Sudden cardiac death
- SCN5A
Sodium Voltage-Gated Channel Alpha Subunit 5
- SQTS
Short QT syndrome
- VUS
Variant of unknown significance
Author contributions
MM conceived the work, carried out the experimental analysis of the samples, performed the bioinformatic analysis and variant interpretation and drafted the manuscript. AG and AA conceived the work, interpreted the results, critically reviewed and edited the manuscript. ME and MDP conceived the work, supervised the study and critically revised the manuscript. PA performed the bioinformatics analysis. NB and SM critically reviewed the variant evaluation. MDP, DB and AS contributed to the autopsy diagnosis and critically reviewed the cases for classification and autopsy diagnosis. All authors contributed in a multidisciplinary team to the final variant evaluation on a case-by-case basis.
Funding
This study, part of the "JUvenile Sudden cardiac deaTh: JUST know and treat" (JUST) project, was funded without restrictions by Fondazione Pisa.
Availability of data and materials
All data described in this study are provided within the article and Supplemental Material. The raw DNA sequencing data for the cases are not available due to privacy and ethics restrictions.
Declarations
Ethical approval
We secured consent for genetic testing and research use of data from the relatives of the deceased. This study was conducted in agreement with the Helsinki Declaration and received approval from the Ethic Committee of the Tuscany Region, Area Vasta Nord-Ovest (no. 14870).
Consent to participate
We secured consent for genetic testing and research use of data from the relatives of the deceased.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Marco Di Paolo and Michele Emdin have contributed equally to this work.
References
- 1.Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA, et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) Endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2022;43(40):3997–4126. 10.1093/eurheartj/ehac262. [DOI] [PubMed] [Google Scholar]
- 2.Wong CX, Brown A, Lau DH, Chugh SS, Albert CM, Kalman JM, et al. Epidemiology of sudden cardiac death: global and regional perspectives. Heart Lung Circ. 2019;28(1):6–14. 10.1016/j.hlc.2018.08.026. [DOI] [PubMed] [Google Scholar]
- 3.Wilde AAM, Semsarian C, Márquez MF, Shamloo AS, Ackerman MJ, Ashley EA, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. EP Europace. 2022;24(8):1307–67. 10.1093/europace/euac030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scrocco C, Bezzina CR, Ackerman MJ, Behr ER. Genetics and genomics of arrhythmic risk: current and future strategies to prevent sudden cardiac death. Nat Rev Cardiol. 2021;18(11):774–84. 10.1038/s41569-021-00555-y. [DOI] [PubMed] [Google Scholar]
- 5.Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Barriales-Villa R, Basso C, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023;44(37):3503–626. 10.1093/eurheartj/ehad194. [DOI] [PubMed] [Google Scholar]
- 6.Wilde AAM, Semsarian C, Márquez MF, Shamloo AS, Ackerman MJ, Ashley EA, Sternick EB, Barajas-Martinez H, Behr ER, Bezzina CR, Breckpot J, Charron P, Chockalingam P, Crotti L, Gollob MH, Lubitz S, Makita N, Ohno S, Ortiz-Genga M, Sacilotto L, Schulze-Bahr E, Shimizu W, Sotoodehnia N, Tadros R, Ware JS, Winlaw DS, Kaufman ES; Document Reviewers; Aiba T, Bollmann A, Choi JI, Dalal A, Darrieux F, Giudicessi J, Guerchicoff M, Hong K, Krahn AD, MacIntyre C, Mackall JA, Mont L, Napolitano C, Ochoa JP, Peichl P, Pereira AC, Schwartz PJ, Skinner J, Stellbrink C, Tfelt-Hansen J, Deneke T; Developed in partnership with and endorsed by the European Heart Rhythm Association (EHRA), a branch of the European Society of Cardiology (ESC), the Heart Rhythm Society (HRS), the Asia Pacific Heart Rhythm Society (APHRS), and the Latin American Heart Rhythm Society (LAHRS). European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022 Sep 1;24(8):1307–1367. 10.1093/europace/euac030. Erratum in: Europace. 2022 Sep 1;24(8):1367. 10.1093/europace/euac106
- 7.Bagnall RD, Singer ES, Tfelt-Hansen J. Sudden cardiac death in the young. Heart Lung Circ. 2020;29(4):498–504. 10.1016/j.hlc.2019.11.007. [DOI] [PubMed] [Google Scholar]
- 8.Winkel BG, Holst AG, Theilade J, Kristensen IB, Thomsen JL, Ottesen GL, et al. Nationwide study of sudden cardiac death in persons aged 1–35 years. Eur Heart J. 2010;32(8):983–90. 10.1093/eurheartj/ehq428. [DOI] [PubMed] [Google Scholar]
- 9.Basso C, Burke M, Fornes P, Gallagher PJ, de Gouveia RH, Sheppard M, et al. Guidelines for autopsy investigation of sudden cardiac death. Virchows Arch. 2007;452(1):11–8. 10.1007/s00428-007-0505-5. [DOI] [PubMed] [Google Scholar]
- 10.Basso C, Aguilera B, Banner J, Cohle S, d’Amati G, de Gouveia RH, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch. 2017;471(6):691–705. 10.1007/s00428-017-2221-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yazdanfard PD, Christensen AH, Tfelt-Hansen J, Bundgaard H, Winkel BG. Non-diagnostic autopsy findings in sudden unexplained death victims. BMC Cardiovasc Disord. 2020;20(1):58. 10.1186/s12872-020-01361-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eckart RE, Shry EA, Burke AP, McNear JA, Appel DA, Castillo-Rojas LM, et al. Sudden death in young adults. J Am Coll Cardiol. 2011;58(12):1254–61. 10.1016/j.jacc.2011.01.049. [DOI] [PubMed] [Google Scholar]
- 13.Isbister JC, Semsarian C. The role of the molecular autopsy in sudden cardiac death in young individuals. Nat Rev Cardiol. 2024 Apr;21(4):215-216. 10.1038/s41569-024-00989-0. [DOI] [PubMed]
- 14.Isbister JC, Nowak N, Yeates L, Singer ES, Sy RW, Ingles J, et al. Concealed cardiomyopathy in autopsy-inconclusive cases of sudden cardiac death and implications for families. J Am Coll Cardiol. 2022;80(22):2057–68. 10.1016/j.jacc.2022.09.029. [DOI] [PubMed] [Google Scholar]
- 15.Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med. 2016;374(25):2441–52. 10.1056/NEJMoa1510687. [DOI] [PubMed] [Google Scholar]
- 16.Orland KM, Anderson KB. Molecular autopsy for sudden cardiac death: current state and considerations. Curr Genet Med Rep. 2019;7(3):145–52. 10.1007/s40142-019-00170-x. [Google Scholar]
- 17.Girolami F, Spinelli V, Maurizi N, Focardi M, Nesi G, Maio V, et al. Genetic characterization of juvenile sudden cardiac arrest and death in Tuscany: the ToRSADE registry. Front Cardiovasc Med. 2022;14:9. 10.3389/fcvm.2022.1080608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheiper-Welling S, Tabunscik M, Gross TE, Jenewein T, Beckmann BM, Niess C, et al. Variant interpretation in molecular autopsy: a useful dilemma. Int J Legal Med. 2022;136(2):475–82. 10.1007/s00414-021-02764-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campuzano O, Beltramo P, Fernandez A, Iglesias A, García L, Allegue C, et al. Molecular autopsy in a cohort of infants died suddenly at rest. Forensic Sci Int Genet. 2018;37:54–63. 10.1016/j.fsigen.2018.07.023. [DOI] [PubMed] [Google Scholar]
- 20.Lahrouchi N, Behr ER, Bezzina CR. Next-generation sequencing in post-mortem genetic testing of young sudden cardiac death cases. Front Cardiovasc Med. 2016;3(May):1–8. 10.3389/fcvm.2016.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ripoll-Vera T, Pérez Luengo C, Borondo Alcázar JC, García Ruiz AB, Sánchez Del Valle N, Barceló Martín B, et al. Sudden cardiac death in persons aged 50 years or younger: diagnostic yield of a regional molecular autopsy program using massive sequencing. Revista Española de Cardiología (English Edition). 2021;74(5):402–13. 10.1016/j.rec.2020.03.030. [DOI] [PubMed] [Google Scholar]
- 22.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaetano Thiene DCCB. Sudden cardiac death in the young and athletes: text atlas of pathology and clinical correlates. Berlin: Springer; 2016. 10.4081/jbr.2024.12184. [Google Scholar]
- 24.Guo Y, Ding X, Shen Y, Lyon GJ, Wang K. SeqMule: automated pipeline for analysis of human exome/genome sequencing data. Sci Rep. 2015;5:14283. 10.1038/srep14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, pp 1–3, 2013. 10.48550/arXiv.1303.3997
- 26.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koboldt DC. Best practices for variant calling in clinical sequencing. Genome Med. 2020;12(1):91. 10.1186/s13073-020-00791-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desvignes JP, Bartoli M, Delague V, Krahn M, Miltgen M, Béroud C, et al. VarAFT: a variant annotation and filtration system for human next generation sequencing data. Nucleic Acids Res. 2018;46(W1):W545. 10.1093/nar/gky471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67–e67. 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwarz JM, Hombach D, Köhler S, Cooper DN, Schuelke M, Seelow D. RegulationSpotter: annotation and interpretation of extratranscriptic DNA variants. Nucleic Acids Res. 2019;47(W1):W106–13. 10.1093/nar/gkz327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fadoni J, Santos A, Cainé L. Post-mortem genetic investigation in sudden cardiac death victims: complete exon sequencing of forty genes using next-generation sequencing. Int J Legal Med. 2022;136(2):483–91. 10.1007/s00414-021-02765-y [DOI] [PubMed] [Google Scholar]
- 32.Santori M, Blanco-Verea A, Gil R, Cortis J, Becker K, Schneider PM, et al. Sudden cardiac death in persons aged 50 years or younger: diagnostic yield of a regional molecular autopsy program using massive sequencing. Forensic Sci Int. 2016;18(4):888–96. 10.1016/j.forsciint.2014.01.014. [Google Scholar]
- 33.Rueda M, Wagner JL, Phillips TC, Topol SE, Muse ED, Lucas JR, et al. Molecular autopsy for sudden death in the young: is data aggregation the key? Front Cardiovasc Med. 2017;4:72. 10.3389/fcvm.2017.00072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christiansen SL, Hertz CL, Ferrero-Miliani L, Dahl M, Weeke PE, Lucamp L, et al. Genetic investigation of 100 heart genes in sudden unexplained death victims in a forensic setting. Eur J Hum Genet. 2016;24(12):1797–802. 10.1038/ejhg.2016.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hellenthal N, Gaertner-Rommel A, Klauke B, Paluszkiewicz L, Stuhr M, Kerner T, et al. Molecular autopsy of sudden unexplained deaths reveals genetic predispositions for cardiac diseases among young forensic cases. Europace. 2017;19(11):1881–90. 10.1093/europace/euw247. [DOI] [PubMed] [Google Scholar]
- 36.Nunn LM, Lopes LR, Syrris P, Murphy C, Plagnol V, Firman E, et al. Diagnostic yield of molecular autopsy in patients with sudden arrhythmic death syndrome using targeted exome sequencing. Europace. 2016;18(6):888–96. 10.1093/europace/euv285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neves R, Tester DJ, Simpson MA, Behr ER, Ackerman MJ, Giudicessi JR. Exome sequencing highlights a potential role for concealed cardiomyopathies in youthful sudden cardiac death. Circ Genom Precis Med. 2022;15(1):e003497. 10.1161/CIRCGEN.121.003497. [DOI] [PubMed] [Google Scholar]
- 38.Morales A, Hershberger RE. Variants of Uncertain Significance: Should We Revisit How They Are Evaluated and Disclosed? Circ Genom Precis Med. 2018 Jun;11(6):e002169. 10.1161/CIRCGEN.118.002169 [DOI] [PMC free article] [PubMed]
- 39.Garrett A, Durkie M, Callaway A, Burghel GJ, Robinson R, Drummond J, et al. Combining evidence for and against pathogenicity for variants in cancer susceptibility genes: CanVIG-UK consensus recommendations. J Med Genet. 2021;58(5):297–304. 10.1136/jmedgenet-2020-107248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen E, Facio FM, Aradhya KW, Rojahn S, Hatchell KE, Aguilar S, et al. Rates and classification of variants of uncertain significance in hereditary disease genetic testing. JAMA Netw Open. 2023;6(10):e2339571. 10.1001/jamanetworkopen.2023.39571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz PJ, Crotti L, George AL. Modifier genes for sudden cardiac death. Eur Heart J. 2018;39(44):3925. 10.1093/eurheartj/ehy502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz PJ, Ackerman MJ, Antzelevitch C, Bezzina CR, Borggrefe M, Cuneo BF, et al. Inherited cardiac arrhythmias. Nat Rev Dis Primers. 2020;6(1):58. 10.1038/s41572-020-0188-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bezzina CR, Lahrouchi N, Priori SG. Genetics of sudden cardiac death. Circ Res. 2015;116(12):1919–36. 10.1161/CIRCRESAHA.116.304030 [DOI] [PubMed] [Google Scholar]
- 44.Webster G, Puckelwartz MJ, Pesce LL, Dellefave-Castillo LM, Vanoye CG, Potet F, et al. Genomic autopsy of sudden deaths in young individuals. JAMA Cardiol. 2021;6(11):1247. 10.1001/jamacardio.2021.2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fenske S, Krause SC, Hassan SIH, Becirovic E, Auer F, Bernard R, et al. Sick sinus syndrome in HCN1-deficient mice. Circulation. 2013;128(24):2585–94. 10.1161/CIRCULATIONAHA.113.003712 [DOI] [PubMed] [Google Scholar]
- 46.Li N, Csepe TA, Hansen BJ, Dobrzynski H, Higgins RSD, Kilic A, et al. Molecular mapping of sinoatrial node HCN channel expression in the human heart. Circ Arrhythm Electrophysiol. 2015;8(5):1219–27. 10.1161/CIRCEP.115.003070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivolta I, Binda A, Masi A, DiFrancesco JC. Cardiac and neuronal HCN channelopathies. Pflugers Arch. 2020;472(7):931–51. 10.1007/s00424-020-02384-3. [DOI] [PubMed] [Google Scholar]
- 48.Staehr C, Rohde PD, Krarup NT, Ringgaard S, Laustsen C, Johnsen J, et al. Migraine-associated mutation in the Na, K-ATPase leads to disturbances in cardiac metabolism and reduced cardiac function. J Am Heart Assoc. 2022;11(7):e021814. 10.1161/JAHA.121.02181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mazzarotto F, Girolami F, Boschi B, Barlocco F, Tomberli A, Baldini K, et al. Defining the diagnostic effectiveness of genes for inclusion in panels: the experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet Med. 2019;21(2):284–92. 10.1038/s41436018-0046-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanchez O, Campuzano O, Fernández-Falgueras A, Sarquella-Brugada G, Cesar S, Mademont I, et al. Natural and undetermined sudden death: value of post-mortem genetic investigation. PLoS ONE. 2016;11(12):e0167358–e0167358. 10.1371/journal.pone.0167358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hertz CL, Ferrero-Miliani L, Frank-Hansen R, Morling N, Bundgaard H. A comparison of genetic findings in sudden cardiac death victims and cardiac patients: the importance of phenotypic classification. Europace. 2015;17(3):350–7. 10.1093/europace/euu210 [DOI] [PubMed] [Google Scholar]
- 52.Neubauer J, Lecca MR, Russo G, Bartsch C, Medeiros-Domingo A, Berger W, et al. Exome analysis in 34 sudden unexplained death (SUD) victims mainly identified variants in channelopathy-associated genes. Int J Legal Med. 2018;132(4):1057–65. 10.1007/s00414-018-1775-y. [DOI] [PubMed] [Google Scholar]
- 53.Bagnall RD, Crompton DE, Petrovski S, Lam L, Cutmore C, Garry SI, et al. Exome-based analysis of cardiac arrhythmia, respiratory control, and epilepsy genes in sudden unexpected death in epilepsy. Ann Neurol. 2016;79(4):522–34. 10.1002/ana.24596. [DOI] [PubMed] [Google Scholar]
- 54.Verdonschot JAJ, Hazebroek MR, Krapels IPC, Henkens MTHM, Raafs A, Wang P, et al. Implications of genetic testing in dilated cardiomyopathy. Circ Genom Precis Med. 2020;13(5):476–87. 10.1161/CIRCGEN.120.003031 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data described in this study are provided within the article and Supplemental Material. The raw DNA sequencing data for the cases are not available due to privacy and ethics restrictions.