

SUMMARY
Patients with small-cell lung cancer (SCLC) are in dire need of more effective therapeutic options. Frequent disruption of the G1 checkpoint in SCLC cells creates a dependency on the G2/M checkpoint to maintain genomic integrity. Indeed, in pre-clinical models, inhibiting the G2/M checkpoint kinase WEE1 shows promise in inhibiting SCLC growth. However, toxicity and acquired resistance limit the clinical effectiveness of this strategy. Here, using CRISPR-Cas9 knockout screens in vitro and in vivo, we identified multiple factors influencing the response of SCLC cells to the WEE1 kinase inhibitor AZD1775, including the GCN2 kinase and other members of its signaling pathway. Rapid activation of GCN2 upon AZD1775 treatment triggers a stress response in SCLC cells. Pharmacological or genetic activation of the GCN2 pathway enhances cancer cell killing by AZD1775. Thus, activation of the GCN2 pathway represents a promising strategy to increase the efficacy of WEE1 inhibitors in SCLC.
In brief
Drainas et al. identify GCN2 as a factor influencing small-cell lung cancer (SCLC) response to the WEE1 inhibitor AZD1775. AZD1775 directly activates the GCN2 stress response pathway, increasing its effectiveness in killing SCLC cells. Thus, activating the GCN2 pathway presents a novel opportunity to enhance WEE1 inhibitor efficacy in SCLC.
Graphical Abstract
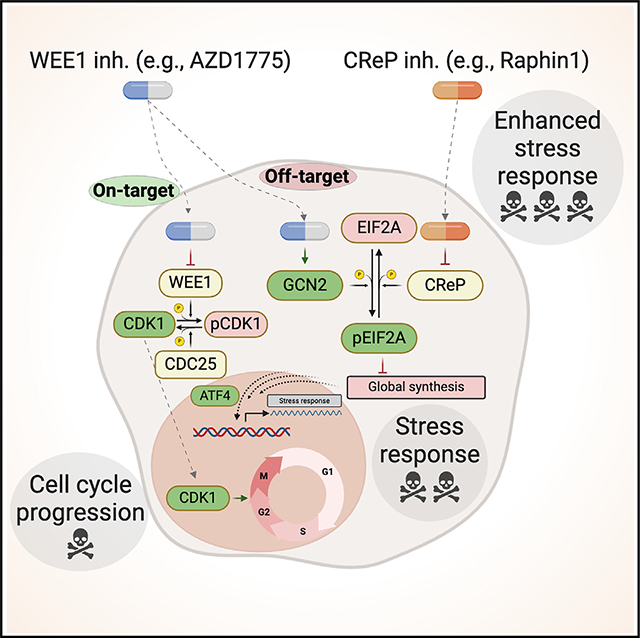
INTRODUCTION
Small-cell lung cancer (SCLC) is a high-grade neuroendocrine carcinoma that accounts for ~15% of all lung cancers. The 5-year survival rate for SCLC patients is only ~8%. For the past 40 years, SCLC has been managed with radiation and chemotherapy. However, the majority of patients suffer relapse within months of completing initial therapy.1,2 Even T cell-based immunotherapies benefit only a relatively small number of patients, highlighting a huge unmet need to identify novel effective therapeutic approaches, including approaches that could be used in combination with standard-of-care chemotherapy to prevent or delay resistance.1,3
Plasticity and multiple levels of heterogeneity likely contribute to therapy resistance in SCLC tumors. First, within the large group of p53 and RB (Retinoblastoma) mutant SCLC,4 additional events, such as amplification of MYC family genes or high expression of NFIB (coding for Nuclear Factor 1B), may define different genetic subgroups5–7 with different responses to therapeutic agents.6,8 Second, transcriptional networks driven by key transcription factors define subtypes of SCLC tumors, including the two major subtypes SCLC-A and SCLC-N, driven by ASCL1 (Achaete-scute Homolog 1) and NEUROD1 (Neuronal Differentiation 1) respectively6,9; these transcriptional programs can also influence the response of SCLC to therapy.10–12 Third, increased intratumoral heterogeneity has been linked to the ability of tumors to evade treatment.13–15 These observations indicate that new therapeutic strategies will need to be tested in several subtypes of SCLC to find the most appropriate context for effective antitumor responses; most likely, new combination therapies will also be required to effectively inhibit the growth of SCLC tumors.
Functional inactivation of p53 and RB in SCLC cells is thought to largely abrogate the G1/S checkpoint of the cell cycle.16 Consequently, the G2/M checkpoint becomes key to controlling genome integrity before cell division in these cells. Any additional impairment of the G2/M transition may thus lead to an unprepared entry into mitosis, accumulation of DNA damage, chromosomal instability, and death.17 These observations have led to the idea that targeting key G2/M checkpoints may help inhibit the growth of SCLC tumors. WEE1 is a tyrosine kinase that responds to stress and DNA damage downstream of CHK1 (checkpoint kinase 1) to disallow entry into mitosis by mainly phosphorylating Cyclin B1-CDK1 complexes.17–19 WEE1 is overexpressed and active in SCLC, and WEE1 inhibition leads to SCLC cell death in culture and in xenograft models.20–23 These pre-clinical experiments with specific WEE1 kinase inhibitors have led to clinical trials, but toxicities and the emergence of resistance are critical barriers limiting the use of such inhibitors in patients.24–27 Experiments in culture have identified some mechanisms of resistance,20,28 but our understanding of how SCLC cells respond to WEE1 inhibition and what drives resistance to WEE1 inhibition in SCLC remains incomplete.
Here, we conducted unbiased whole-genome knockout screens in human SCLC cell lines to identify genetic determinants of the response of SCLC cells to the WEE1 kinase inhibitor AZD1775. These experiments identified inactivation of the GCN2 kinase (General Control Non-derepressible 2, encoded by the EIF2AK4 gene) as a mechanism of protection against WEE1 kinase inhibition. Notably, pharmacological activation of the GCN2 signaling pathway results in a re-sensitization of cancer cells tolerant to AZD1775 treatment. Thus, the antitumor effects of WEE1 inhibitors may be potentiated by activation of the GCN2 pathway in SCLC.
RESULTS
Genetic determinants of the response to AZD1775
We first generated AZD1775-tolerant SCLC cell lines using increasing doses of this inhibitor. AZD1775-treated human NCI-H82 and NJH29 SCLC-N cells showed an increased tolerance to AZD1775 compared to their naïve counterparts, while NCI-H69 SCLC-A cells were intrinsically more resistant to AZD1775 (Figure S1A). AZD1775 treatment inhibited the phosphorylation of the WEE1 target CDK1 to the same extent in both naïve and tolerant cells (Figure S1B), indicating that the increased tolerance to AZD1775 was not due to a loss of WEE1-inhibitory activity.
We next performed genome-wide CRISPR-Cas9 screens on naïve and AZD1775-tolerant NCI-H82 cells (Figure 1A; Table S1). Among common hits between naïve and tolerant cells, knockout of EIF2AK4 was the most protective hit, and knockout of PKMYT1, which codes for a kinase that also phosphorylates CDK1 and can compensate for WEE1 inhibition,17,29 was the most sensitizing (Figures 1B and 1C). Gene ontology (GO) analysis (Figure 1D; Table S1) identified “cell cycle” as a top term in the sensitizing hits, while “TORC1 signaling” and “cellular response to amino acid starvation,” a pathway controlled by GCN2,30,31 were top terms in the protective hits. In the GCN2 pathway, inactivation of the genes coding for GCN2 and the direct GCN2 activator GCN1 had strong protective effects in the screens, while inactivation of the genes coding for two negative regulators of this pathway, CReP (constitutive reverter of EIF2A phosphorylation, also known as PPP1R15B) and PP1c (also known as PPP1CC) was sensitizing to AZD1775 treatment (Figure 1E). Notably, the GCN1 and GCN2 knockouts were enriched upon AZD1775 treatment but otherwise had no effect on the growth of NCI-H82 cells (Figure S1C). The CRISPR-Cas9 screens also identified genes coding for factors associated previously with resistance to WEE1 inhibitors, such as mTOR (mammalian Target Of Rapamycin),20,32 PARP-family proteins (Poly-ADP Ribose Polymerase),22,33 and CDK2 (Cyclin-Dependent Kinase 2)19,34 (Figure 1E; Table S1).
Figure 1. Genome-wide CRISPR-Cas9 screens identify the GCN2 pathway as a mediator of the response of SCLC cells to WEE1 kinase inhibition.
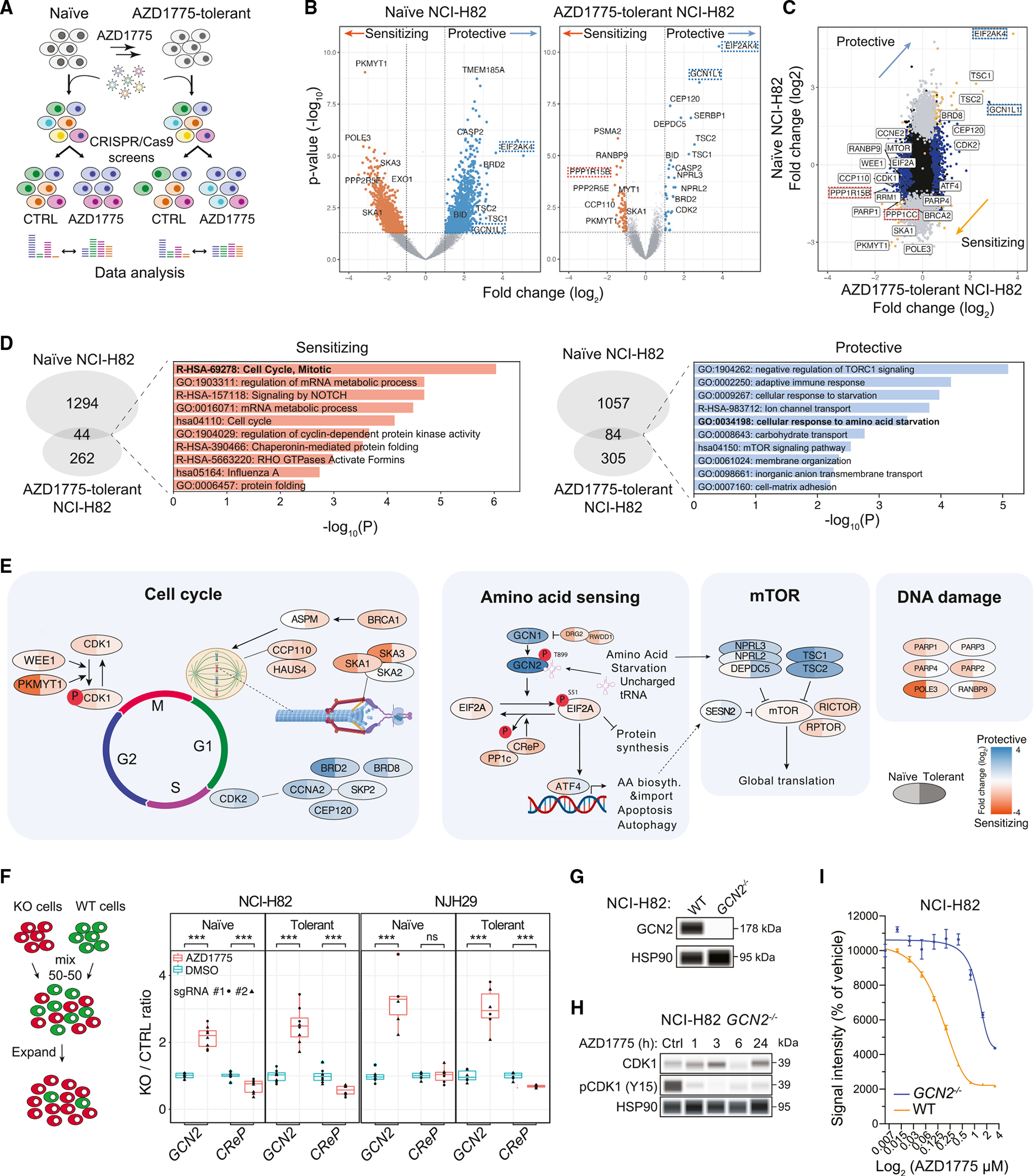
(A) CRISPR-Cas9 genome-wide screening strategy.
(B) Volcano plots from genome-wide CRISPR-Cas9 screens using naïve and AZD1775-tolerant NCI-H82 cells; AZD1775 (0.4 μM) vs. DMSO on day 21. Each screen was conducted in two replicates. Key genes in the GCN2 pathway are highlighted (EIF2AK4 = GCN2, GCN1L1 = GCN1, PPP1R15B = CreP). Fold change (log2) < −0.5 or > 0.5, ANOVA: p < 0.05 (MEMcrispR package).
(C) Fold changes of gene representations between naïve and AZD1775-tolerant NCI-H82 cells.
(D) GO enrichment terms for under- and over-represented gene hits.
(E) Representation of selected genes across the cell cycle, amino acid-sensing, mTOR, and DNA damage pathways from the CRISPR-Cas9 screens with NCI-H82 cells.
(F) Competition assay between knockout cells and control NCI-H82 cells. The ratio was calculated by the number of GFP+ and mCherry+ cells identified by flow cytometry. Two separate sgRNAs were used per gene (n = 4). t test: *p = 0.05–0.01, **p = 0.01–0.001, ***p < 0.001.
(G) GCN2 expression in NCI-H82 WT and NCI-H82 GCN2−/− cells measured by immunoassay. HSP90 is a loading control.
(H) Effect of CDK1 phosphorylation (at tyrosine 15) upon AZD1775 treatment in GCN2−/− cells (n = 1) measured by immunoassay. HSP90 is a loading control.
(I) AlamarBlue cell viability assays with wild-type and GCN2−/− NCI-H82 cells in a titration of AZD1775. n = 6 per condition, standard error of the mean (SEM). Treatment: 5 days.
We conducted similar genome-wide CRISPR-Cas9 screens in AZD1775-tolerant NJH29 cells and NCI-H69 cells (Figures S1D–S1H; Tables S2 and S3). PKMYT1 was a common hit among all cell lines. When we compared the two different SCLC-N cell lines, NCI-H82 and NJH29, the GCN2 knockout was the most protective hit (Figure S1E). However, GCN2 was not a hit in the intrinsically AZD1775-resistant NCI-H69 cell line.
Using individual sgRNAs (single guide RNAs), we confirmed that GCN2 loss protects NCI-H82 and NJH29 SCLC cells from AZD1775 treatment, whereas CReP loss is sensitizing (Figures 1F and S1I). GCN2 loss did not affect the ability of AZD1775 to inhibit WEE1 kinase activity on CDK1 in NCI-H82 cells (Figures 1G and 1H). A drug titration assay further confirmed the increased resistance to AZD1775 treatment in GCN2 knockout NCI-H82 cells compared to wild-type controls (Figure 1I).
In summary, genome-wide CRISPR-Cas9 screens identify a number of regulators of the response of SCLC cells to the AZD1775 WEE1 kinase inhibitor in culture, including the GCN2 nutrient-sensing pathway.
Rapid GCN2 activation upon WEE1 kinase inhibition
The data from the CRISPR-Cas9 screens raised the question of how the activity of the GCN2 pathway is affected by AZD1775 treatment. The GCN2 pathway can be normally activated by low levels of amino acids,35 which was observed in NCI-H82 cells upon arginine starvation (Figures 1E and S2A). Because the screens were analyzed after 4 weeks, we initially surmised that the GCN2 pathway would play an adaptive role in response to AZD1775 treatment. Unexpectedly, however, GCN2 was activated by just 1 h of AZD1775 treatment (Figure S2B). Inhibition of GCN2 kinase activity by GCN2iB36 blocked this activation of the GCN2 pathway by AZD1775, indicative of an activating effect by AZD1775 at the level of the kinase or above (Figure 2A). Treatment with PD407824, a structurally distinct WEE1 inhibitor (with some activity toward Checkpoint Kinase 1, CHK1),37 also rapidly activated GCN2 (Figure 2B), indicating that GCN2 activation was not due to specific features of AZD1775 molecules. GCN2 knockout in NCI-H69 cells did not increase the tolerance of these cells to AZD1775 treatment in the CRISPR-Cas9 screen (Table S3). Accordingly, GCN2 was not activated by AZD1775 or by amino acid starvation (Figures S2C and S2D), suggesting that this pathway is not active in this cell line (for reasons that remain unknown). However, AZD1775 treatment rapidly activated GCN2 in the SCLC-A cell line NCI-H2081 (Figure S2E), indicating that activation of GCN2 by AZD1775 treatment is not restricted to SCLC-N cells.
Figure 2. Direct activation of GCN2 by AZD1775 engages a stress response in SCLC cells.
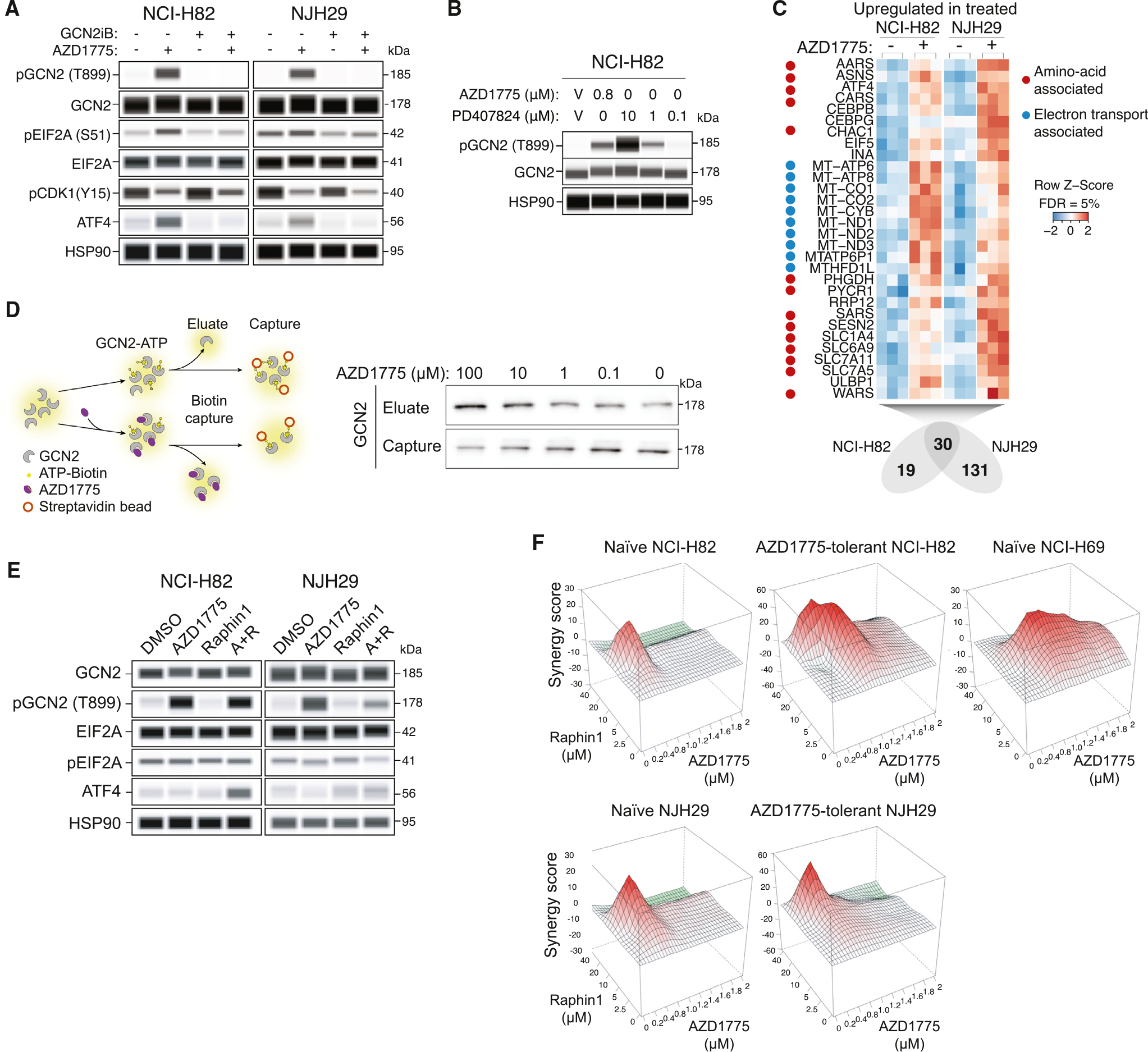
(A) Analysis of the GCN2 pathway by immunoassay after a 1-h treatment with AZD1775 (0.4 μM), the GCN2 inhibitor GCN2iB (1 μM), or both in NCI-H82 (n = 1) and NJH29 cells (n = 1). HSP90 is a loading control.
(B) Immunoassay for GCN2 phosphorylation after a 2-h treatment with the WEE1 inhibitors AZD1775 and PD407824 in NCI-H82 cells (n = 1). HSP90 is a loading control.
(C) Commonly upregulated genes in an RNA-seq analysis of NCI-H82 and NJH29 cells (n = 3 per condition) after a 24-h treatment with AZD1775 (0.4 μM). Heatmaps indicate gene counts in each replicate.
(D) Left: schematic of the biotin capture assay used to evaluate the competition between ATP and AZD1775 with recombinant GCN2. Right: immunoblot for GCN2 on the eluate and captured fractions with different concentrations of AZD1775 (independent replicate in Figure S2J).
(E) Analysis of the GCN2 pathway by immunoassay in NCI-H82 and NJH29 cells (n = 1 per cell line) upon treatment with AZD1775 (0.4 μM), Raphin1 (10 μM), or the combination of both molecules.
(F) Synergy plots from alamarBlue assays in human SCLC cell lines with various doses of AZD1775 (0–2 μM) and Raphin1 (0–40 μM) for 5 days (n = 1).
To determine the consequences of GCN2 pathway activation upon AZD1775 treatment, we performed RNA sequencing (RNA-seq) analysis of AZD1775-tolerant NCI-H82 and NJH29 cells treated with AZD1775 or vehicle control. We identified 30 and 26 commonly upregulated and downregulated genes, respectively (Figures 2C and S2F; Table S4). GO term analysis for the upregulated genes showed enrichment for oxidative phosphorylation as well as mTOR signaling and amino acid pathways, including tRNA synthetases and amino acid transporters, which have been identified as ATF4 (Activating Transcription Factor 4) transcriptional targets38,39 (Figures 2C and S2G; Table S4). Downregulated genes were enriched in hypoxia response and glycolysis terms (Table S4). Thus, in these cell lines, acute AZD1775 treatment caused a transcriptional response that resembles the stress response normally observed upon activation of the GCN2/EIF2A/ATF4 pathway.
We also examined the transcriptome of naïve and AZD1775-tolerant NCI-H82 and NJH29 cell lines (Figure 1). This analysis showed very few common genes that were differentially expressed between the two cell lines (Figure S2H; Table S5) and little to no overlap in GO terms for upregulated and downregulated genes (Table S5). Notably, however, transcriptional regulatory network analysis using transcriptional regulatory relationships unraveled by sentence-based text-mining40 identified the downstream effector of the GCN2 pathway ATF4 as a common candidate transcription factor (Figure S2I; Table S5). These data suggest that long-term adaptation to increasing doses of AZD1775 also involves GCN2 pathway activation, underscoring the significance of the activation of GCN2 by AZD1775.
These observations raised the question of the mechanisms underlying the acute activation of GCN2 by AZD1775. GCN2 has been shown previously to be activated by direct binding to ATP-competitive kinase inhibitors.41 Because both AZD1775 and PD407824 are also ATP-competitive kinase inhibitors,18 we tested whether AZD1775 and PD407824 directly bind to GCN2 using an ATP-GCN2 pull-down assay.41 Indeed, these ATP-competitive kinase inhibitors of WEE1 could compete with ATP for GCN2 binding (Figures 2D, S2J, and S2K). As shown previously for neratinib,41 binding of AZD1775 may lead to a conformational change, increasing the affinity to uncharged tRNAs, and activating GCN2. Together, these observations indicate that one link between WEE1 kinase inhibition and the GCN2 pathway is the direct activation of the GCN2 kinase by ATP-competitive kinase inhibitors.
GCN2 pathway activation sensitizes cells to AZD1775 treatment
EIF2A is phosphorylated by GCN2 but can be dephosphorylated by the catalytic subunit of protein phosphatase 1 (PP1c) in complex with either GADD34 (growth arrest and DNA damage 34, also known as PPP1R15A) or CreP42 (Figure 1E). The genes coding for PP1c and CReP were both sensitizing hits from the CRISPR-Cas9 knockout screens (Figure 1). Thus, we reasoned that prolonged EIF2A phosphorylation (and GCN2 pathway activation) upon PP1c inhibition may increase the sensitivity of SCLC cells to AZD1775 treatment.
Raphin1 is a small molecule that can inhibit the recruitment of PP1c to EIF2A; at lower doses, this inhibition is more specific to PP1c/CreP, while at doses higher than 10 μM, Raphin1 also inhibits PP1c/GADD34.43 NCI-H82 SCLC cells survived Raphin1 treatment up to ~10 μM in culture, suggesting that full inhibition of PP1c may be required to induce cell death in these cells (Figure S2L). Similarly, EIF2A phosphorylation only increased at doses higher than 10 μM in NCI-H82 and NJH29 cells (Figure S2M). When we combined Raphin1 and AZD1775 treatment, we observed increased ATF4 levels that were not observed with single treatment at the concentrations used (Figure 2E). Accordingly, AZD1775 and Raphin1 synergized across several concentrations in SCLC cell lines in culture, including AZD1775-tolerant cells (Figure 2F). In both NCI-H82 and NJH29 cells, knocking down WEE1 and Raphin1 treatment also had an additive effect on decreased cell viability across different concentrations (Figure S2N and S2O). We note that, compared to AZD1775, the antitumor effects of knocking down WEE1 are expected to be less pronounced, since AZD1775 enhances the stress response by activating the GCN2 pathway in addition to inhibiting WEE1 activity. Still, in this context, greater activation of the GCN2 pathway can again enhance the anti-cancer effects of AZD1775.
Activation of GCN2 enhances the anti-tumor effects of AZD1175 in vivo
The responses of cancer cells to perturbations may differ in culture and in vivo. We thus investigated the link between WEE1 kinase inhibition and GCN2 pathway activation in mice. First, we verified that AZD1775 treatment can inhibit the growth of NCI-H82 xenografts (Figures S3A and S3B). Next, we mixed GFP-expressing GCN2 knockout NCI-H82 cells with mCherry-expressing wild-type cells in subcutaneous tumors. In this assay, GCN2 knockout cells outcompeted wild-type cells upon treatment with AZD1775 (Figures 3A, S3C, and S3D).
Figure 3. Activation of the GCN2 pathway enhances the response to WEE1 kinase inhibition in vivo.
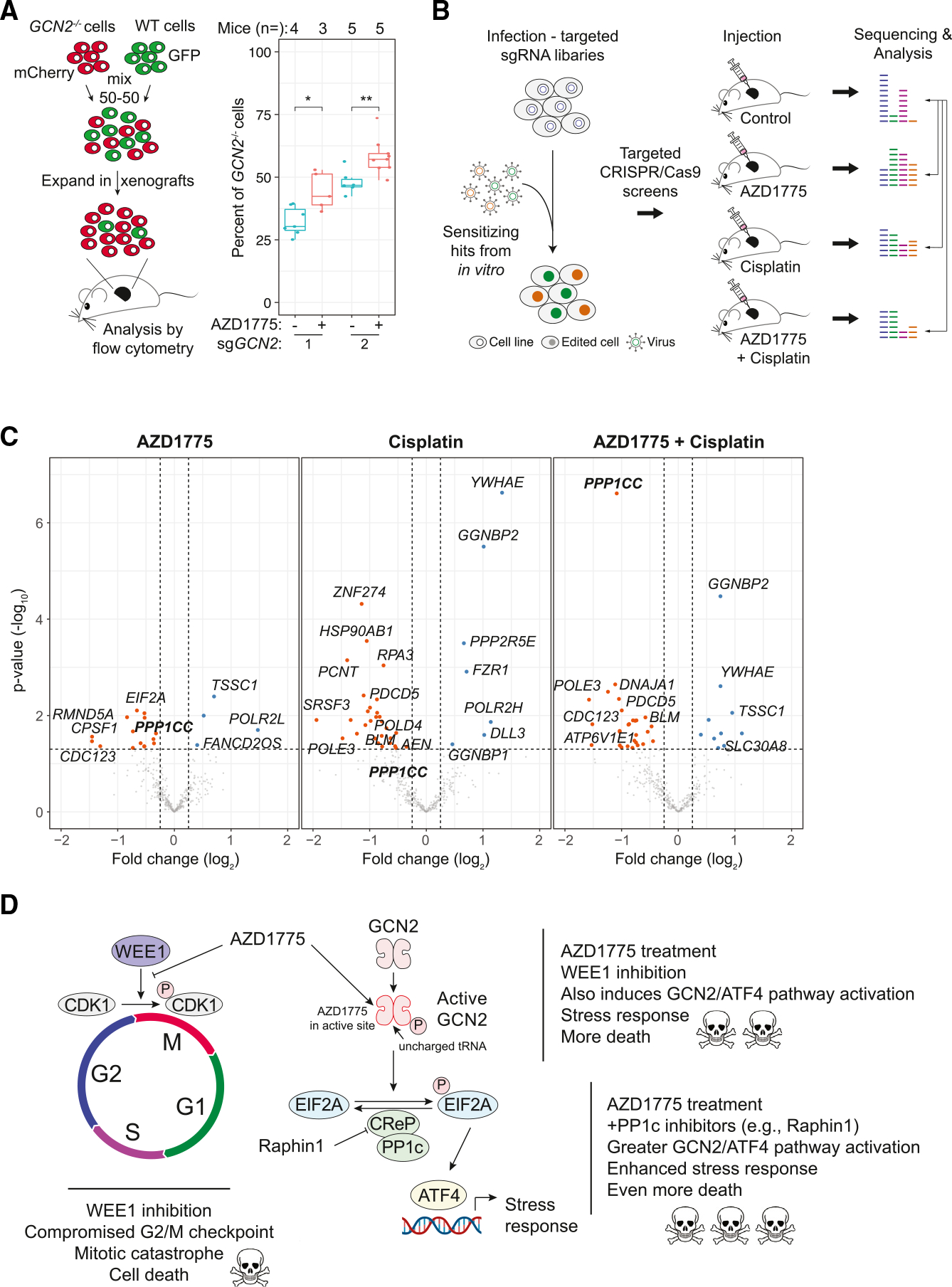
(A) Left: in vivo competition assay with GCN2−/− (mCherry+) and WT (GFP+) NCI-H82 AZD1775-naïve cells. Right: percent of GCN2−/− cells between the treated group and untreated group. t test, *p = 0.05–0.01, **p = 0.01–0.001, ***p < 0.001. Mouse numbers per condition are indicated in the plot (independent replicates in Figures S3C and S3D).
(B) Targeted in vivo CRISPR-Cas9 screening strategy. Cas9-expressing cells were infected with lentiviral particles containing the targeted library (focusing on sensitizing hits in culture) and subsequently injected into the flanks of mice. After tumors reached ~1 cm3 in size, the tumors were collected for analysis.
(C) Volcano plots from targeted in vivo CRISPR-Cas9 screens as in (B). Each screen was performed in replicates (n = 2). Mice (n = 12, 3 per group) were treated for 21 days. PPP1CC (PP1c) is bolded. Fold change (log2) < −1 or > 1, ANOVA: p < 0.05 (MEMcrispR package).
(D) Proposed model: inhibition of the WEE1 kinase by the ATP-competitive inhibitor AZD1775 not only inhibits WEE1 in SCLC cells but also activates GCN2 and the integrated stress response (ISR) pathway, which contributes to enhanced cell death. Further activation of the ISR pathway by inhibiting a phosphatase in the GCN2 pathway can lead to greater cancer cell death.
To gain a broader view of the response to AZD1775 in SCLC in vivo, we generated a CRISPR-Cas9 library targeting 364 genes from the top sensitizing hits in the genome-wide screens in culture (Figure 3B). Because previous studies with SCLC cell lines in culture showed that cisplatin treatment can significantly enhance cell death upon WEE1 kinase inhibition,28 we conducted three screens with NCI-H82 cells: AZD1775 treatment, cisplatin treatment, and combination treatment (Figure 3B). Many but not all gene knockouts were sensitizing in vivo, underscoring the importance of performing the screen in vivo (Figure 3C; Table S6). Notably, we observed decreased representation of sgRNAs targeting PPP1CC, the gene coding for PP1c, indicating that inhibition of the phosphatase counteracting the effects of GCN2 activation enhances tumor growth inhibition in vivo in the context of AZD1775 treatment (Figure 3C; Table S6). PPP1CC was the most significant sensitizing gene in the AZD1775+cisplatin combination treatment condition.
Altogether, these experiments identify the GCN2 pathway as a critical determinant of the response of SCLC cells to AZD1775 in culture and in mice. These experiments also suggest combination therapies between small-molecule inhibitors of the WEE1 kinase and activators of the GCN2/EIF2A/GCN4 pathway in SCLC (Figure 3D).
DISCUSSION
Here we investigated the genetic determinants of the response of SCLC cells to the WEE1 kinase inhibitor AZD1775. Our data identify a number of pathways genetically linked to WEE1 kinase inhibition. These pathways may serve as targets in the future to enhance the anti-tumor effects of WEE1 inhibition in SCLC.
Because SCLC cells have a deficient G1 checkpoint, treatment with inhibitors of other cell cycle checkpoints may result in cell death due to an accumulation of damage in these cells. A recent validation of this idea includes inhibition of Aurora kinase in SCLC tumors with high levels of MYC.6,44,45 An advantage of inhibiting G2/M checkpoint factors is also that the death induced by these inhibitors may be immunogenic.28,46–49 A major issue with this strategy, however, may be the toxicity associated with these G2/M checkpoint inhibitors as well as the rapid emergence of resistance.
Our CRISPR-Cas9 screens identified several mechanisms that could be engaged to enhance the effects of WEE1 kinase inhibition, either by preventing or delaying resistance or simply augmenting cell death. Notably, PKMYT1 has not been pursued as extensively as a target as other G2/M regulators, but, based on our data, PKMYT1 inhibitors could be tested in the future together with WEE1 inhibitors to acutely kill SCLC cells.50–52 Our screens also identified a number of additional targets, including CDK2, for which specific inhibitors are being developed;53 mTOR, whose activation was already known to be a possible mechanism of resistance20; and PARP family members, whose inhibition is effective in combination with WEE1 inhibitors in some pre-clinical cancer models.54,55 Phosphoproteomics analyses will certainly provide a powerful complementary strategy to identify key factors in the WEE1 network and new targets for combination therapy.56
Our screens identified GCN2 as a kinase whose loss increases the resistance of SCLC cells to AZD1775 treatment. GCN2 is a stress response protein that is activated in response to amino acid depletion.57 GCN2 phosphorylates EIF2A, which can subsequently lead to ATF4 expression and the initiation of the integrated stress response (ISR).42,58 Prolonged ISR can trigger cell death.59 Our data and previous work41 suggest that GCN2 is directly activated by AZD1775 by an off-target mechanism that is still related to the ATP competitor nature of the small molecule. This off-target effect may still be relevant to patients whose tumors are treated with AZD1775 or similar molecules, as these molecules may elevate the ISR and render cancer cells more prone to death. We note that GCN2 loss was protective in the two AZD1775-tolerant cell lines we examined, indicating that the ISR may still play a role in tumors that have become resistant to AZD1775. On the other hand, NCI-H69 cells seem to have inactivated this pathway, suggesting that any future plan to enhance cell death upon activation of the ISR may require prior validation that this pathway can be activated in tumors that are treated.
A recent study has identified several combination therapies with AZD1775.60 Based on our data, we sought to begin to exploit GCN2 activation as a therapeutic option using an inhibitor of the CReP phosphatase. Whether a similar strategy will inhibit tumor growth in pre-clinical models in vivo will require a number of experiments to determine optimal doses and regimens of Raphin1 or similar molecules, including in the background of standard-of-care treatment. Whether targeting CReP is the best way to enhance ISR activity in vivo also remains to be determined. In particular, ATF4 activation may induce cell death in some contexts but also protect from cell death in other contexts, which could lead to tumor promotion instead of tumor inhibition.61 Still, our work indicates that there may be ways to improve the efficacy of G2/M checkpoint inhibitors, such as WEE1 inhibitors, in the treatment of SCLC and other tumors.
Limitations of the study
The study is limited by the number of SCLC cell lines analyzed. Future work should include similar CRISPR-Cas9 screens across all SCLC subtypes to investigate the contributions of the GCN2 signaling pathway upon AZD1775 treatment in a broader context. It will also be important to investigate other cell lines intrinsically resistant to AZD1775 beyond NCI-H69 (including non-SCLC cancer cell lines) to gain a better understanding of mechanisms of resistance and how they may relate to the GCN2 pathway. There are also some additional technical limitations to our study as they pertain to CRISPR-Cas9 screening, including in vivo, where the analysis of a greater number of tumors would improve statistical analyses and may change the cutoff for top hits. Another limitation of the experiments performed in this study is the selection of time points, which may not fully capture the dynamics of GCN2 signaling and transcriptional output.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Julien Sage (julsage@stanford.edu).
Materials availability
No materials were newly generated for this manuscript.
Data and code availability
The RNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus64 and are accessible through GEO: GSE246354 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE246354). Additional raw data discussed in this publication have been deposited at Mendeley Data: https://doi.org/10.17632/f5jtt3s8hv.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Ethics statement
Mouse maintenance and experiments were conducted in accordance with practices prescribed by the NIH, the Institutional Animal Care and Use Committee (IACUC), and Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). The study protocol was approved by the Administrative Panel on Laboratory Animal Care (APLAC) at Stanford University (protocol APLAC-13565).
Mice and tumor initiation
Mus Musculus immunocompromised NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, NOD scid gamma) were used for transplantation studies with human cancer cells lines. 750,000 cells were expanded to be injected into mice to form subcutaneous flank tumors. Cells were first mixed with Matrigel in a 3:2 ratio (75 μL cells and 50 μL Matrigel) and then injected using Monoject 1mL insulin syringes (Covidien 1188128012). AZD1775 was diluted in methylcellulose 0.5%, the drug was sonicated and constantly inverted overnight. AZD1775 vials were used for 5 days. After 10 days, when tumors reached an average of 100 mm3, mice were randomly assigned to two experimental groups: AZD1775 or vehicle. Mice were then fed the drug orally with an oral gavage 5 consecutive days a week for 3 weeks. Tumors were measured by caliper.
Cell lines
NCI-H82, NCI-H69, and NCI-H2081 cells were purchased from ATCC (HTB-175 and HTB-119). NJH29 cells were developed at Stanford.65 The cell lines were grown in RPMI 1640 (Corning 15–040-CV) supplemented with 10% FBS (Thermo Fisher Scientific 10500064) or BGS (Thermo Fisher Scientific, SH3054103HI) and 1X Antibiotic-Anti-mycotic (Thermo Fisher Scientific 15240062) and L-glutamine (Corning 25–005-CI). 293T cells (ATCC CRL-3216) were grown in DMEM (Thermo Fisher Scientific 31966021) supplemented with 10% FBS and 1x Antibiotic-Antimycotic (Thermo Fisher Scientific, 15240062). All cell lines were maintained at appropriate densities and were incubated in a humidity-controlled environment (37°C, 5% CO2). All cell lines tested negative for mycoplasma contamination.
METHOD DETAILS
AlamarBlue assays
AlamarBlue Cell Viability Agent (Thermo Fisher Scientific DAL1100) assays were performed in 96-well plates. 20,000 cells were plated in each well and selected using a serial dilution of AZD1775 (Selleckchem S1525, 0–6.4μM) or Raphin1 (Selleckchem S0528, 0–256μM) in 200 μL of RPMI 1640 (Corning 15–040-CV) supplemented with 10% FBS (Thermo Fisher Scientific 10500064) and Antibiotic-Antimycotic for 5 days. After the selection period, 20 μL of 1:1 mixture of alamarBlue (0.5%) and PBS (Corning 21–040-CV) was added to each well and incubated for 6 h in a humidity-controlled environment (37°C, 5% CO2). Cell count was measured using a Synergy H1 Hybrid Reader (BioTekTM). Analysis was conducted with PRISM (https://www.graphpad.com/), R scripts, or with SynergyFinder (synergyfinder.fimm.fi).
Selection of cells with increased tolerance to AZD1775 treatment
Cell lines were selected in 0.4–1.2 μM AZD1775 depending on the cell line’s IC50 AZD1775 for a period of one week to four weeks to create tolerant cell lines. IC50 concentrations were determined using alamarBlue assays.
Genome-wide CRISPR-Cas9 screens
Cas9-expressing cell lines
Cells were cultured in 15-cm culture dishes in RPMI without antibiotics and infected with plentiCas9-Blast (Addgene 52962) for 24 h at 37°C, with the addition of 8 ng/mL polybrene (Sigma-Aldrich TR-1003). Cells were then selected for 1 week in 5 μg/mL blasticidin (Gibco A11139–03). Cells were tested for Cas9 functionality by infecting cells with a lentivirus expressing sgRNA molecules targeting GFP together with a plasmid expressing GFP (vector backbone.
Virus was produced by co-transfecting 293T cells with 100 mg of lentiviral vector library and 100 mg of 3G mix (at a 1:1:1 ratio of VSV-g (Addgene 1733), pMDLg/pRRE (Addgene 12251), and pRSV-Rev (Addgene 12253)) with Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific L3000015). Ten 15-cm plates per replicate were transfected for the genome-wide libraries and two 15-cm plates were transfected for the targeted libraries. After 24 h, the medium was changed with fresh DMEM. After an additional 48 h, viral supernatant was collected, centrifuged to remove cell debris, and diluted 4:1 with 5 M PEG (Sigma p5413) to precipitate the virus. After 24 h at 4°C, the precipitated virus was centrifuged at 3500rpm for 30 min at 4°C. The supernatant was discarded, and the viral pellet was resuspended in 1.5 mL of PBS generating a 20x concentrated viral solution. Aliquots were stored at −80°C.
Cell lines with functional Cas9 were cultured in 15-cm culture dishes in RPMI without antibiotics and infected with the CRISPR lentiviral libraries for 24 h at 37°C at a multiplicity of infection between 20 and 40%, with the addition of 8 ng/mL polybrene. The medium was replaced after 24 h with RPMI with antibiotics. Cells recovered for 24 h at 37°C, 5% CO2. The infected cells were selected with 0.5–2 μm/mL puromycin (Thermo Fisher Scientific #A1113803), depending on the cell line and selection was maintained for one week.
After selection, 50 × 106 cells were collected for the initial representation of the sgRNAs (noted as day 0). Cells were transferred to eight 5-layer flasks (Corning 353144) with medium supplemented with either DMSO (vehicle) or with 0.8 μg/mL AZD1775. Cells were incubated at 37°C, 5% CO2 and split every 2–3 days to maintain appropriate cell densities of approximately 200 × 106 cells per 250 mL. Two biological replicates were split into two technical replicates each. Selection was maintained for 28 days (~12 population doublings).
For the targeted screens, virus containing sgRNA against 364 genes and 10% safe sgRNAs were produced as for the genome-wide screens. Infected cells were selected with puromycin and then 2 × 106 of cells were mixed with Matrigel in a 3:2 ratio (75 μL cells and 50 μL Matrigel). The cells were injected into the flank of each mouse, with 7 mice per group. There were for 4 groups treated: treatment with AZD1775 (90 mg/kg, mice were then fed the drug orally with an oral gavage 5 consecutive days a week), treatment with cisplatin (5 mg/kg, once a week) treatment with a combination of AZD1775 and cisplatin, and a control group with vehicle administration.
PCR amplification of the genome-wide CRISPR screen libraries for sequencing and analysis
After selection, cells were frozen in pellets of 100 × 106 cells per condition. For the in vivo screens, tumors were blended and then processed for genomic DNA extraction. Genomic DNA was isolated from the initial and final time point collected cells using QIAamp DNA Blood Maxi Kit (Qiagen 51194). DNA was amplified for 18 cycles using plasmid-specific primers for the amplification of the sgRNA coding sequences (oMCB1562: 5′-AGGCTTGGATTTCTATAACTTCGTATAGCATACATTATAC-3′, oMCB1563: 5′-ACATGCATGGCGGTAATACGGTTATC-3′). An additional round of PCR amplification was run for 8 cycles to ensure the addition of Illumina adaptors and barcodes (oMCB1439: 5′-CAAGCAGAAGACGGCATACGAGATGCACAAAAGGAAACTCACCCT-3′, R: 5′-AATGATACGGCGACCACCGAGATCTACACGATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNCGACTCGGTGCCACTTTTTC-3′). Herculase II Fusion DNA Polymerase (Agilent 600677) was utilized for the PCR reactions. Samples were run on a 1% agarose gel and extracted and purified using QIAquick Gel Extraction Kit (Qiagen 28706X4). All samples were pooled and sequenced using Illumina NextSeq with a custom sequencing primer: 5′-GCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTAAACTTGCTATGCTGTTTCCAGCTTAGCTCTTAAAC-3’. The data were aligned against the sgRNAs of the designed library. Counts of each sgRNA were calculated and the representation of genes of each condition were analyzed using the MEMcrispR package.63
Methods for gene ontology
Gene ontology analysis was performed using Metascape.66 Genes were inputted to Metascape and custom analyses were conducted. Different gene ontology sets were used such as: Functional Set (GO Molecular Functions), Pathway (GO Biological Processes, Canonical Pathways, Reactome Gene sets, KEGG pathway), Structural component (GO Cellular Components), and TRRUST (transcriptional regulatory relationships unraveled by sentence-based text-mining).
RNA sequencing
Cells for RNA-seq were collected by centrifuging 106 cells and flash freezing the pellet. Samples were sent to Novogene for RNA isolation, library preparation, and sequencing. RNA counts were quantified using Salmon67 and differential RNA-seq analysis was conducted using DESeq2.68
Cell preparation for time-course treatment
Cells were incubated with AZD1775, Raphin1 (Tocris 1A/242832) or a combination of both for time periods of 1, 3, 6 or 24 h with a control of 1 h in DMSO (Sigma-Aldrich D8418–500mL). Cells were washed with PBS and the pellets stored at −80°C.
Protein extraction and immunoassays
Total protein was extracted from cells using RIPA buffer (Cell Signaling Technology 9806) and Benzonase endonuclease (Merck-Millipore 71206–3). Protein was quantified using Protein Assay Dye Reagent (Bio-Rad 5000006). Samples were quantified using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific 23227). Protein levels were measured using a Synergy H1 Hybrid Reader (BioTekTM). Protein extracts were analyzed using WES (Protein SimpleTM) or immunoblot following standard protocol (the R&D Systems Quality Control Western Blot Protocol). Semi-dry transfer system was used for the immunoblotting (iBlot 3 Western Blot Transfer System, Thermo Fisher Scientific).X Antibodies used were directed against GCN2 (Abcam ab157775), phospho-GCN2 (Abcam ab75836), EIF2a (Cell Signaling Technology 5324T), phospho-EIF2a (Cell Signaling 3398S), ATF4 (Cell Signaling Technology 11815S), WEE1 (Cell Signaling Technology 13084T), CDK1 (Cell Signaling Technology 28439S), phospho-CDK1 (Cell Signaling Technology 9111S). HSP90 as a loading control (Cell Signaling Technology 4877S).
GCN2 ATP-binding assay
This protocol was adapted from.41 In the bead-based binding assay, low protein binding Eppendorf tubes (VWR 525–0133) were used. Streptavidin beads (Thermo Fisher Scientific 20357) (15 μL per sample) were incubated with 100 μL of 1% (w/v) bovine serum albumin (BSA) (Millipore-Sigma A3059) diluted in PBS on a rotator for 30 min. The beads were centrifuged at 1000 rcf and the supernatant was aspirated without disturbing the pellet. The reaction mix for each sample consisted of: H2O 8.8 μL, 10X drug in kinase buffer (0.5 M Tris-HCl, 0.3 M MgCl2, 10 mM DTT) 2.5 μL, GCN2 (Sigma 14–934) (0.1 μg/μL) 2.5 μL, MgCl2 (Sigma M8266) (1M) 0.625 μL, RNA extracted from NCI-H82 cells 25 ng, AZD1775 (used at the final concentration of 100 μM, 10 μM, 1 μM) 2.5 μL, and ActivX Desthiobiotin-ATP Probe (Thermo Fisher Scientific 88311) (46.5 μM) 2.5 μL and add water to 25 μL. The mixture was incubated at room temperature for 20 min. The reaction mix was then added to the beads pellet, and were incubated on a rotator for 1 h at room temperature. The streptavidin beads were subsequently washed three times with ice-cold 1% (w/v) BSA PBS and eluted with 2x Laemmli sample buffer (Bio-Rad #1610737) for subsequent analysis by immunoblot.
Flow cytometry and cell sorting
Flow cytometry was conducted on a CytoFLEX (Beckman Coulter) and on a FACSAria (BD Biosciences). Analysis of the data was done using Cytobank and R software.
sgRNA cloning
The backbone vectors pMCB320 (expresses mCherry) or pMCB306 (expresses GFP) were digested with BstXI (NEB R0113S) and BlpI (NEB R0585S) overnight: The linearized plasmids were purified using the QIAquick Gel Extraction Kit (Qiagen 28704) before ligation with sgRNAs (ligation buffer NEB B0202S). The ligation products were transformed into competent bacteria (Endura, Lucigen 60240–1) before antibiotic selection on agar plates.
Lentiviral production
HEK293T cells (ATCC, CRL-3216) were seeded in DMEM (Thermo Fisher Scientific, #11965092) with 10% fetal bovine serum (Thermo Fisher Scientific, # 16000044) at 106 cells per well of a 6-well plate. The cells were transfected with 0.5 μg of lentiviral vectors (key resources table) and 0.5 μg of 3rd generation lentiviral vectors (1:1:1 ratio of VSV-g, pMDLg/pRRE, and pRSV-Rev), 5 μL PEI (Polysciences, #23966–1) in 50 μL Opti-MEM (Thermo Fisher Scientific, # 31985088). The mixture added dropwise to cells after 10 min of incubation. 2 mL of fresh antibiotic-free media was added to the cells. 2 days later, lentiviruses were collected. Virus was concentrated by adding 1 volume of PEG (100g PEG (Sigma p5413), 6g NaCl, 250mL ddH20, pH to 7.2 and autoclave) to 4 volumes of virus containing media. The mixture was incubated overnight at 4°C and the next day the tubes were centrifuged at 3500rpm for 30 min at 4°C. The supernatant was removed, and PBS was added to generated 1:20 concentrated virus. Aliquots and stored at −80°C. Cells were then infected with 100 μL of concentrated virus per 4 × 106 cells for each lentiviral vector, with the addition of 8 μg/mL polybrene (Millipore-Sigma, #TR-1003-G). The next day, the media with the virus was removed, and fresh media was added containing penicillin/streptomycin (Thermo Fisher Scientific, #10378016). After 3 days, the multiplicity of infection was measured by FACS for GFP or mCherry. Then puromycin was added for approximately 1 week until over 95% of cells were positive expressing GFP or mCherry.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Wee1 (D10D2) mAb Rabbit | Cell Signaling Technology | CAT# 13084T; RRID: AB_2713924 |
| Anti-GCN2 (phospho T899) antibody | Abcam | CAT# ab75836; RRID: AB_1310260 |
| GCN2 antibody | Abcam | CAT# ab157775; RRID: |
| Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) XP® Rabbit | Cell Signaling Technology | CAT# 2211S; RRID: AB_331679 |
| HSP90 Rabbit mAb | Cell Signaling Technology | CAT# 4877S; RRID: AB_2233307 |
| ATF-4 (D4B8) Rabbit mAb | Cell Signaling Technology | CAT# 11815S; RRID: AB_2616025 |
| Phospho-eIF2α (Ser51) (D9G8) XP® Rabbit mAb | Cell Signaling Technology | CAT# 3398; RRID: AB_2096481 |
| eIF2α (D7D3) XP® Rabbit mAb | Cell Signaling Technology | CAT# 5324; RRID: AB_10692650 |
| CDK1 | Cell Signaling Technology | CAT# 28439S; RRID: AB_2798959 |
| phospho-CDK1 | Cell Signaling Technology | CAT# 9111S; RRID: AB_331460 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Endura™ Chemically Competent Cells | Lucigen | Cat# 60240-1 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Polyethylenimine (PEI) | Polysciences | Cat# 23966-1 |
| Polybrene | Millipore-Sigma | Cat# TR-1003-G |
| Opti-MEM | Thermo Fisher Scientific | Cat# 31985088 |
| RPMI 1640 | Corning | Cat# 15-040-CV |
| DMEM | Thermo Fisher Scientific | Cat# 11965092 |
| Bovine Growth Serum | Thermo Fisher Scientific | Cat# SH3054103HI |
| Penicillin-Streptomycin-Glutamine | Thermo Fisher Scientific | Cat# 10378016 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat#16000044 |
| Matrigel | BD biosciences | Cat# 356237 |
| Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) | Thermo Fisher Scientific | Cat# 77720 |
| PBS antibody stabilizer | Thermo Fisher Scientific | Cat# NC0414486 |
| Paraformaldehyde (PFA) | Thermo Fisher Scientific | Cat# 28906 |
| Saponin | Millipore-Sigma | Cat# S7900-100G |
| Bovine Serum Albumin (BSA) | Millipore-Sigma | Cat# A3059 |
| Sodium azide | Millipore-Sigma | Cat# 71289 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat# 201192B |
| L15 media | Millipore-Sigma | Cat# L1518 |
| Collagenase I | Millipore-Sigma | Cat# C0130 |
| Collagenase II | Millipore-Sigma | Cat# 6885 |
| Collagenase IV | Millipore-Sigma | Cat# 5138 |
| Elastase | CellSystems | Cat# LS002292 |
| DNAseI | Millipore-Sigma | Cat# 10104159 |
| RBC Lysis Buffer | Thermo Fisher Scientific | Cat# 00-4333-57 |
| AZD-1775 | SYNthesis | Cat# WZQ-0447-032 |
| Raphin1 | Tocris | Cat# 1A/242832 |
| Histogel | Thermo Fisher Scientific | Cat# NC9150318 |
| Triton X-100 | Millipore-Sigma | Cat# T8787-100ML |
| PD 407824 (Wee1 and Chk1 inhibitor) | Tocris | Cat# 2694 |
| DMSO | Sigma-Aldrich | Cat# D8418-500mL |
| RIPA buffer | Cell Signaling Technology | Cat# 9806 |
| Benzonase | Merck-Millipore | Cat# 71206-3 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23227 |
| Protein Assay Dye Reagent | Bio-Rad | Cat# 5000006 |
| Streptavidin beads | Thermo Fisher Scientific | Cat# 20357 |
| MgCl2 | Sigma | Cat# M8266 |
| ActivX™ Desthiobiotin-ATP Probe | Thermo Fisher Scientific | Cat# 88311 |
| 2x Laemmli sample buffer | Bio-Rad | Cat# 1610737 |
| PEG | Sigma | Cat# p5413 |
| QIAamp DNA Blood Maxi Kit | Qiagen | Cat# 51194 |
| BstXI | NEB | Cat# R0113S |
| BlpI | NEB | Cat# R0585S |
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28704 |
| T4 ligation buffer | NEB | Cat# B0202S |
| SOC media | Sigma | Cat# S1797 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Herculase II Fusion DNA Polymerase kit | Agilent | Cat# 600675 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq data | GEO | GSE246354 |
| Raw data | Mendeley Data | https://doi.org/10.17632/f5jtt3s8hv.1 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| 293T | ATCC® | HTB-3216™ |
| NCI-H82 | ATCC® | HTB-175™ |
| NJH29 | Sage Lab | N/A |
| NCI-H69 | ATCC® | HTB-119™ |
| NCI-H2081 | ATCC® | CRL-5920™ |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| NOD-scid IL2Rgammanull mice (NSG mice) | The Jackson Laboratory | #005557 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Genome-Wide CRISPR-KO library | Morgens et al.,62 | N/A |
| oMCB1562: 5′-AGGCTTGGATTTCTATAAC TTCGTATAGCATACATTATAC-3′ |
Morgens et al.,62 | N/A |
| oMCB1563: 5′-ACATGCATGGCGGTAAT ACGGTTATC-3′ |
Morgens et al.,62 | N/A |
| oMCB1439: 5′-CAAGCAGAAGACGGCAT ACGAGATGCACAAAAGGAAACTCACCCT-3′ |
Morgens et al.,62 | N/A |
| R: 5′-AATGATACGGCGACCACCGAGATC TACACGATCGGAAGAGCACACGTCTGAA CTCCAGTCACNNNNNNCGACTCGGTGC CACTTTTTC-3′ |
Morgens et al.,62 | N/A |
| Sequencing primer: 5′-GCCACTTTTTCAAG TTGATAACGGACTAGCCTTATTTAAACTTG CTATGCTGTTTCCAGCTTAGCTCTTAAAC-3′ |
Morgens et al.,62 | N/A |
| EIF2AK4_2 TTGGGAGGCCATTTACGGCG GTTTAAGAGC |
This paper | N/A |
| sgRNA cloning: EIF2AK4_2_R TTAGCTCTT AAACCGCCGTAAATGGCCTCCCAACAAG |
This paper | N/A |
| sgRNA cloning: EIF2AK4_4 TTGGCCGTTCT GTGTCCTCCTGTTTAAGAGC |
This paper | N/A |
| sgRNA cloning: EIF2AK4_4_R TTAGCTCTT AAACAGGAGGACACAGAACGGCCAACAAG |
This paper | N/A |
| sgRNA cloning: PPP1R15B_4 TTGGATCGC CGAGGGAAAAAGGGGTTTAAGAGC |
This paper | N/A |
| sgRNA cloning: PPP1R15B_4_R TTAGCTCT TAAACCCCTTTTTCCCTCGGCGATCCAACAAG |
This paper | N/A |
| sgRNA cloning: PPP1R15B_8 TTGGTTACTG AGTATTATATAAGGTTTAAGAGC |
This paper | N/A |
| sgRNA cloning: PPP1R15B_8_R TTAGCTCTT AAACCTTATATAATACTCAGTAACCAACAAG |
This paper | N/A |
| sgRNA safe: GCTAGCTTGTTACTGAGCAT | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pRSV-Rev | pRSV-Rev was a gift from Didier Trono | Addgene: #12253; RRID:Addgene_12253 |
| pMDLg/pRRE | pMDLg/pRRE was a gift from Didier Trono | Addgene: #12251; RRID:Addgene_12251 |
| pCI-VSVG | pCI-VSVG was a gift from Garry Nolan | Addgene: #1733; RRID:Addgene_1733 |
| pMCB306 | pMCB306 was a gift from Michael Bassik | Addgene: #89360; RRID:Addgene_89360 |
| pMCB320 | pMCB306 was a gift from Michael Bassik | Addgene: # 89359; RRID:Addgene_89359 |
|
| ||
| Software and algorithms | ||
|
| ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| R | R Core Team (2020) | https://www.R-project.org/. |
| CellEngine | CellCarta | https://cellcarta.com/ |
| Cytobank | Cytobank | https://cytobank.org/ |
| MEMcrispR | Drainas et al.63 | |
|
| ||
| Other | ||
|
| ||
| 0.22 μm filter | Millipore-Sigma | Cat# SLMP025SS |
| 70 μm strainer | Fisher Scientific | Cat# 08-771-2 |
| Superfrost Plus glass slides of 25 mm width and 75 mm length | Thermo Fisher Scientific | Cat#12-550-15 |
| Linear stainer | Leica | Cat# ST4020 |
| PT module | Thermo Fisher Scientific | Cat# A80400012 |
| CytoFLEX Flow Cytometer | Beckman Coulter | https://www.beckman.com/flow-cytometry/instruments/cytoflex |
| BD FACSAria™ III Cell Sorter | BD Biosciences | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-cell-sorters/facsaria-iii |
Competition assays
NCI-H82 and NJH29 cells expressed mCherry or GFP and co-expressed sgRNAs. Controls NCI-H82 and NJH29 cells expressing GFP and safe sgRNAs were mixed with GCN2−/− or PPP1R15B−/− cells 1:1. The cells were then cultured in vitro or injected subcutaneously in the flanks of NSG mice. Cells were collected when the tumors reached ~1 cm3. GFP- and mCherry-expressing cells were analyzed by flow cytometry.
Software
We used R (https://www.R-project.org), Cytoscape (https://cytoscape.org), GraphPad Prism 9 (https://www.graphstats.net/), Python (https://www.python.org).
QUANTIFICATION AND STATISTICAL ANALYSIS
CRISPR-Cas9 screens were analyzed with MEMcrispR.63 This software utilizes mixed-effects models to assess sgRNA count data, accounting for both fixed (e.g., treatment) and random effects (e.g., batch) through model fitting. MEMcrispR performs an ANOVA likelihood ratio test to identify statistically significant gene knockouts (~10 sgRNAs targeting each gene). The test compares the alternative model (M1) that contains treatment terms to a null model (M0) lacking the treatment effect. Significant p-values from these tests indicate treatment-dependent gene knockouts. Student’s t-tests were applied to data in Figures 1F, 3A, S3A, and S3D.
Supplementary Material
Highlights.
CRISPR-Cas9 screens identify genetic determinants of response to the WEE1 inhibitor AZD1775
GCN2 inactivation leads to resistance to AZD1775 treatment in vitro and in vivo
GCN2 gets rapidly activated upon WEE1 inhibition by AZD1775
Activation of the GCN2 pathway sensitizes cells to AZD1775 treatment
ACKNOWLEDGMENTS
We thank all the members of the Bassik and Sage labs for their help. We thank Alyssa Ray for administrative support, Pauline Chu from the Stanford Histology Service Center, Catherine Carswell-Crumpton and Cheng Pan from the FACS core in the Institute for Stem Cell Biology and Regenerative Medicine, Stanford’s animal mouse facility staff, Thuyen Nguyen for help with mice and for technical contributions, and Debadrita Bhattacharya for computational support. The research reported in this publication was supported by the Tobacco-Related Disease Research Program (T30FT0824 to A.P.D.) and the NIH (CA213273 to J.S.). J.S. is the Elaine and John Chambers Professor in Pediatric Cancer.
Footnotes
DECLARATION OF INTERESTS
J.S. has equity in and is an advisor for DISCO Pharmaceuticals.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114606.
REFERENCES
- 1.Rudin CM, Brambilla E, Faivre-Finn C, and Sage J (2021). Small-cell lung cancer. Nat. Rev. Dis. Primers 7, 3. 10.1038/s41572-020-00235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Megyesfalvi Z, Gay CM, Popper H, Pirker R, Ostoros G, Heeke S, Lang C, Hoetzenecker K, Schwendenwein A, Boettiger K, et al. (2023). Clinical insights into small cell lung cancer: Tumor heterogeneity, diagnosis, therapy, and future directions. CA. Cancer J. Clin. 73, 620–652. 10.3322/caac.21785. [DOI] [PubMed] [Google Scholar]
- 3.Moliner L, Zhang B, Lamberti G, Ardizzoni A, Byers LA, and Califano R (2023). Novel therapeutic strategies for recurrent SCLC. Crit. Rev. Oncol. Hematol. 186, 104017. 10.1016/j.critrevonc.2023.104017. [DOI] [PubMed] [Google Scholar]
- 4.George J, Lim JS, Jang SJ, Cun Y, Ozretić L, Kong G, Leenders F, Lu X, Fernández-Cuesta L, Bosco G, et al. (2015). Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53. 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D, et al. (2019). Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat. Rev. Cancer 19, 289–297. 10.1038/s41568-019-0133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, et al. (2017). MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to Aurora kinase inhibition. Cancer Cell 31, 270–285. 10.1016/j.ccell.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang D, Denny SK, Greenside PG, Chaikovsky AC, Brady JJ, Ouadah Y, Granja JM, Jahchan NS, Lim JS, Kwok S, et al. (2018). Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov. 8, 1316–1331. 10.1158/2159-8290.CD-17-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Böttger F, Semenova EA, Song JY, Ferone G, van der Vliet J, Cozijnsen M, Bhaskaran R, Bombardelli L, Piersma SR, Pham TV, et al. (2019). Tumor Heterogeneity Underlies Differential Cisplatin Sensitivity in Mouse Models of Small-Cell Lung Cancer. Cell Rep. 27, 3345–3358.e3344. 10.1016/j.celrep.2019.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borromeo MD, Savage TK, Kollipara RK, He M, Augustyn A, Osborne JK, Girard L, Minna JD, Gazdar AF, Cobb MH, and Johnson JE (2016). ASCL1 and NEUROD1 reveal heterogeneity in pulmonary neuroendocrine tumors and regulate distinct genetic programs. Cell Rep. 16, 1259–1272. 10.1016/j.celrep.2016.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E, Nguyen T, et al. (2017). Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell 31, 286–299. 10.1016/j.ccell.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mahadevan NR, Knelson EH, Wolff JO, Vajdi A, Saigí M, Campisi M, Hong D, Thai TC, Piel B, Han S, et al. (2021). Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov. 11, 1952–1969. 10.1158/2159-8290.CD-20-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gay CM, Stewart CA, Park EM, Diao L, Groves SM, Heeke S, Nabet BY, Fujimoto J, Solis LM, Lu W, et al. (2021). Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 39, 346–360.e7. 10.1016/j.ccell.2020.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart CA, Gay CM, Xi Y, Sivajothi S, Sivakamasundari V, Fujimoto J, Bolisetty M, Hartsfield PM, Balasubramaniyan V, Chalishazar MD, et al. (2020). Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 1, 423–436. 10.1038/s43018-019-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pal Choudhuri S, Girard L, Lim JYS, Wise JF, Freitas B, Yang D, Wong E, Hamilton S, Chien VD, Kim YJ, et al. (2024). Acquired Cross-resistance in Small Cell Lung Cancer due to Extrachromosomal DNA Amplification of MYC paralogs. Cancer Discov. 14, 804–827. 10.1158/2159-8290.CD-23-0656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lissa D, Takahashi N, Desai P, Manukyan I, Schultz CW, Rajapakse V, Velez MJ, Mulford D, Roper N, Nichols S, et al. (2022). Heterogeneity of neuroendocrine transcriptional states in metastatic small cell lung cancers and patient-derived models. Nat. Commun. 13, 2023. 10.1038/s41467-022-29517-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sherr CJ, and McCormick F (2002). The RB and p53 pathways in cancer. Cancer Cell 2, 103–112. [DOI] [PubMed] [Google Scholar]
- 17.Schmidt M, Rohe A, Platzer C, Najjar A, Erdmann F, and Sippl W (2017). Regulation of G2/M transition by inhibition of WEE1 and PKMYT1 kinases. Molecules 22, 2045. 10.3390/molecules22122045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Do K, Doroshow JH, and Kummar S (2013). Wee1 kinase as a target for cancer therapy. Cell Cycle 12, 3159–3164. 10.4161/cc.26062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moiseeva TN, Qian C, Sugitani N, Osmanbeyoglu HU, and Bakkenist CJ (2019). WEE1 kinase inhibitor AZD1775 induces CDK1 kinase-dependent origin firing in unperturbed G1- and S-phase cells. Proc. Natl. Acad. Sci. USA 116, 23891–23893. 10.1073/pnas.1915108116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sen T, Tong P, Diao L, Li L, Fan Y, Hoff J, Heymach JV, Wang J, and Byers LA (2017). Targeting AXL and mTOR pathway overcomes primary and acquired resistance to WEE1 inhibition in small-cell lung cancer. Clin. Cancer Res. 23, 6239–6253. 10.1158/1078-0432.CCR-17-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byers LA, Wang J, Nilsson MB, Fujimoto J, Saintigny P, Yordy J, Giri U, Peyton M, Fan YH, Diao L, et al. (2012). Proteomic profiling identifies dysregulated pathways in small cell lung cancer and novel therapeutic targets including PARP1. Cancer Discov. 2, 798–811. 10.1158/2159-8290.CD-12-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lallo A, Frese KK, Morrow CJ, Sloane R, Gulati S, Schenk MW, Trapani F, Simms N, Galvin M, Brown S, et al. (2018). The combination of the PARP inhibitor olaparib and the WEE1 inhibitor AZD1775 as a new therapeutic option for small cell lung cancer. Clin. Cancer Res. 24, 5153–5164. 10.1158/1078-0432.ccr-17-2805. [DOI] [PubMed] [Google Scholar]
- 23.Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, Olawaiye AB, Ellard SL, Weberpals JI, Wahner Hendrickson AE, et al. (2021). Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 397, 281–292. 10.1016/S0140-6736(20)32554-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vermeulen K, Van Bockstaele DR, and Berneman ZN (2003). The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Prolif. 36, 131–149. 10.1046/j.1365-2184.2003.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pines J (2006). Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 16, 55–63. 10.1016/j.tcb.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 26.Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115. 10.1038/nrc.2016.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bauer TM, Moore KN, Rader JS, Simpkins F, Mita AC, Beck JT, Hart L, Chu Q, Oza A, Tinker AV, et al. (2023). A Phase Ib Study Assessing the Safety, Tolerability, and Efficacy of the First-in-Class Wee1 Inhibitor Adavosertib (AZD1775) as Monotherapy in Patients with Advanced Solid Tumors. Target. Oncol. 18, 517–530. 10.1007/s11523-023-00965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taniguchi H, Caeser R, Chavan SS, Zhan YA, Chow A, Manoj P, Uddin F, Kitai H, Qu R, Hayatt O, et al. (2022). WEE1 inhibition enhances the antitumor immune response to PD-L1 blockade by the concomitant activation of STING and STAT1 pathways in SCLC. Cell Rep. 39, 110814. 10.1016/j.celrep.2022.110814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Toledo CM, Ding Y, Hoellerbauer P, Davis RJ, Basom R, Girard EJ, Lee E, Corrin P, Hart T, Bolouri H, et al. (2015). Genome-wide CRISPR-Cas9 Screens Reveal Loss of Redundancy between PKMYT1 and WEE1 in Glioblastoma Stem-like Cells. Cell Rep. 13, 2425–2439. 10.1016/j.celrep.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masson GR (2019). Towards a model of GCN2 activation. Biochem. Soc. Trans. 47, 1481–1488. 10.1042/BST20190331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang P, McGrath BC, Reinert J, Olsen DS, Lei L, Gill S, Wek SA, Vattem KM, Wek RC, Kimball SR, et al. (2002). The GCN2 eIF2alpha kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell Biol. 22, 6681–6688. 10.1128/MCB.22.19.6681-6688.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang B, Kannan A, Guo T, and Gao L (2018). Simultaneously targeting DNA damage repair pathway and mTORC1/2 results in small cell lung cancer growth arrest via ER stress-induced apoptosis. Int. J. Biol. Sci. 14, 1221–1231. 10.7150/ijbs.25488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palve V, Knezevic CE, Bejan DS, Luo Y, Li X, Novakova S, Welsh EA, Fang B, Kinose F, Haura EB, et al. (2022). The non-canonical target PARP16 contributes to polypharmacology of the PARP inhibitor talazoparib and its synergy with WEE1 inhibitors. Cell Chem. Biol. 29, 202–214.e7. 10.1016/j.chembiol.2021.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P, Foijer F, and van Vugt MATM (2015). A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc. Natl. Acad. Sci. USA 112, 15160–15165. 10.1073/pnas.1505283112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Missiaen R, Anderson NM, Kim LC, Nance B, Burrows M, Skuli N, Carens M, Riscal R, Steensels A, Li F, and Simon MC (2022). GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. 34, 1151–1167.e7. 10.1016/j.cmet.2022.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, Tomita D, Fujimoto J, Kurasawa O, Takahara C, et al. (2018). Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc. Natl. Acad. Sci. USA 115, E7776–E7785. 10.1073/pnas.1805523115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronco C, Martin AR, Demange L, and Benhida R (2017). ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. Medchemcomm 8, 295–319. 10.1039/c6md00439c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wortel IMN, van der Meer LT, Kilberg MS, and van Leeuwen FN (2017). Surviving Stress: Modulation of ATF4-Mediated Stress Responses in Normal and Malignant Cells. Trends Endocrinol. Metab. 28, 794–806. 10.1016/j.tem.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neill G, and Masson GR (2023). A stay of execution: ATF4 regulation and potential outcomes for the integrated stress response. Front. Mol. Neurosci. 16, 1112253. 10.3389/fnmol.2023.1112253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, et al. (2018). TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386. 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang CP, Clark O, Ferrarone JR, Campos C, Lalani AS, Chodera JD, Intlekofer AM, Elemento O, and Mellinghoff IK (2022). GCN2 kinase activation by ATP-competitive kinase inhibitors. Nat. Chem. Biol. 18, 207–215. 10.1038/s41589-021-00947-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costa-Mattioli M, and Walter P (2020). The integrated stress response: From mechanism to disease. Science 368, eaat5314. 10.1126/science.aat5314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krzyzosiak A, Sigurdardottir A, Luh L, Carrara M, Das I, Schneider K, and Bertolotti A (2018). Target-Based Discovery of an Inhibitor of the Regulatory Phosphatase PPP1R15B. Cell 174, 1216–1228.e19. 10.1016/j.cell.2018.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Johnson ML, Wang JS, Falchook G, Greenlees C, Jones S, Strickland D, Fabbri G, Kennedy C, Elizabeth Pease J, Sainsbury L, et al. (2023). Safety, tolerability, and pharmacokinetics of Aurora kinase B inhibitor AZD2811: a phase 1 dose-finding study in patients with advanced solid tumours. Br. J. Cancer 128, 1906–1915. 10.1038/s41416-023-02185-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stefani A, Piro G, Schietroma F, Strusi A, Vita E, Fiorani S, Barone D, Monaca F, Sparagna I, Valente G, et al. (2022). Unweaving the mitotic spindle: A focus on Aurora kinase inhibitors in lung cancer. Front. Oncol. 12, 1026020. 10.3389/fonc.2022.1026020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sen T, Della Corte CM, Milutinovic S, Cardnell RJ, Diao L, Ramkumar K, Gay CM, Stewart CA, Fan Y, Shen L, et al. (2019). Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 14, 2152–2163. 10.1016/j.jtho.2019.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, Cristea S, Nguyen T, Diao L, Li L, et al. (2019). Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 9, 646–661. 10.1158/2159-8290.CD-18-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X, Li Y, Zhao Z, Miao N, Liu G, Deng L, Wei S, and Hou J (2023). Immunogenicity of small-cell lung cancer associates with STING pathway activation and is enhanced by ATR and TOP1 inhibition. Cancer Med. 12, 4864–4881. 10.1002/cam4.5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schultz CW, Zhang Y, Elmeskini R, Zimmermann A, Fu H, Murai Y, Wangsa D, Kumar S, Takahashi N, Atkinson D, et al. (2023). ATR inhibition augments the efficacy of lurbinectedin in small-cell lung cancer. EMBO Mol. Med. 15, e17313. 10.15252/emmm.202217313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benada J, Bulanova D, Azzoni V, Petrosius V, Ghazanfar S, Wennerberg K, and Sørensen CS (2023). Synthetic lethal interaction between WEE1 and PKMYT1 is a target for multiple low-dose treatment of high-grade serous ovarian carcinoma. NAR Cancer 5, zcad029. 10.1093/narcan/zcad029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qi X, Li G, Liu J, Mou L, Zhang Y, Guo S, Chen X, and Li W (2024). Structural and energetic insights into the selective inhibition of PKMYT1 against WEE1. J. Biomol. Struct. Dyn. 42, 3010–3018. 10.1080/07391102.2023.2225106. [DOI] [PubMed] [Google Scholar]
- 52.Szychowski J, Papp R, Dietrich E, Liu B, Vallée F, Leclaire ME, Fourtounis J, Martino G, Perryman AL, Pau V, et al. (2022). Discovery of an Orally Bioavailable and Selective PKMYT1 Inhibitor, RP-6306. J. Med. Chem. 65, 10251–10284. 10.1021/acs.jmed-chem.2c00552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faber EB, Sun L, Tang J, Roberts E, Ganeshkumar S, Wang N, Rasmussen D, Majumdar A, Hirsch LE, John K, et al. (2023). Development of allosteric and selective CDK2 inhibitors for contraception with negative cooperativity to cyclin binding. Nat. Commun. 14, 3213. 10.1038/s41467-023-38732-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Teo ZL, O’Connor MJ, Versaci S, Clarke KA, Brown ER, Percy LW, Kuykhoven K, Mintoff CP, Savas P, Virassamy B, et al. (2023). Combined PARP and WEE1 inhibition triggers anti-tumor immune response in BRCA1/2 wildtype triple-negative breast cancer. NPJ Breast Cancer 9, 68. 10.1038/s41523-023-00568-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, et al. (2019). Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 35, 851–867.e7. 10.1016/j.ccell.2019.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrosius V, Benada J, Nielsen O, Schoof EM, and Sørensen CS (2023). Temporal phosphoproteomics reveals WEE1-dependent control of 53BP1 pathway. iScience 26, 105806. 10.1016/j.isci.2022.105806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dong J, Qiu H, Garcia-Barrio M, Anderson J, and Hinnebusch AG (2000). Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279. 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 58.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, and Gorman AM (2016). The integrated stress response. EMBO Rep. 17, 1374–1395. 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tian X, Zhang S, Zhou L, Seyhan AA, Hernandez Borrero L, Zhang Y, and El-Deiry WS (2021). Targeting the Integrated Stress Response in Cancer Therapy. Front. Pharmacol. 12, 747837. 10.3389/fphar.2021.747837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bashi AC, Coker EA, Bulusu KC, Jaaks P, Crafter C, Lightfoot H, Milo M, McCarten K, Jenkins DF, van der Meer D, et al. (2024). Large-scale Pan-cancer Cell Line Screening Identifies Actionable and Effective Drug Combinations. Cancer Discov. 14, 846–865. 10.1158/2159-8290.CD-23-0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen C, Zhang Z, Liu C, Wang B, Liu P, Fang S, Yang F, You Y, and Li X (2022). ATF4-dependent fructolysis fuels growth of glioblastoma multiforme. Nat. Commun. 13, 6108. 10.1038/s41467-022-33859-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morgens DW, Wainberg M, Boyle EA, Ursu O, Araya CL, Tsui CK, Haney MS, Hess GT, Han K, Jeng EE, et al. (2017). Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 8, 15178. 10.1038/ncomms15178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Drainas AP, Lambuta RA, Ivanova I, Serçin Ö, Sarropoulos I, Smith ML, Efthymiopoulos T, Raeder B, Stütz AM, Waszak SM, et al. (2020). Genome-wide screens implicate loss of Cullin ring ligase 3 in persistent proliferation and genome instability in TP53-deficient cells. Cell Rep. 31, 107465. 10.1016/j.celrep.2020.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barrett T, and Edgar R (2006). Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods Enzymol. 411, 352–369. 10.1016/S0076-6879(06)11019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jahchan NS, Lim JS, Bola B, Morris K, Seitz G, Tran KQ, Xu L, Trapani F, Morrow CJ, Cristea S, et al. (2016). Identification and Targeting of Long-Term Tumor-Propagating Cells in Small Cell Lung Cancer. Cell Rep. 16, 644–656. 10.1016/j.celrep.2016.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus64 and are accessible through GEO: GSE246354 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE246354). Additional raw data discussed in this publication have been deposited at Mendeley Data: https://doi.org/10.17632/f5jtt3s8hv.1.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.