

ABSTRACT
Elimination of the binding of immunoglobulin Fc to Fc gamma receptors is highly desirable for the avoidance of unwanted inflammatory responses to therapeutic antibodies and fusion proteins. Many different approaches have been used in the clinic, but they have not been systematically compared. We have now produced a matched set of anti-CD20 antibodies with different Fc subclasses and variants and compared their activity for binding to C1q, Fc-gamma receptors and in cell-based assays. Most of the variants still have significant levels of activity in one or more of these assays and many of them have impaired temperature stability compared with the corresponding wild-type antibody.
KEYWORDS: DSF, Fc region, Fc receptor, Fc silent, therapeutic antibody, antibody engineering, FcγRI, FcγRII, FcγRIII, C1q, CD16, CD32, CD64, antibody effector function
Introduction
Pharmacologic properties of immunoglobulins depend very much on their Fc region. Interaction with C1q initiates activation of complement. Binding to various Fc receptors on leucocytes induces antibody-dependent cell-mediated cytotoxicity (ADCC) or antibody-dependent cell-mediated phagocytosis (ADCP). Binding to the FcRn receptor is responsible for the comparatively long half-life of IgG. As previously reported, we cataloged the protein sequences of 819 antibodies and Fc fusion proteins which have been given international nonproprietary names (INNs).1 This dataset includes 756 human therapeutics which have an intact Fc region, of which about 45% have been designed to lack effector function, either by selection of a subclass (IgG2 or IgG4) believed to have reduced binding to C1q and Fcγ receptors or by introduction of specific mutations. The earliest of these variants included either mutation of N297, to eliminate glycosylation, or L234A/L235A (LALA).2,3 However, neither of these alterations completely eliminate binding to Fcγ receptors or the clinical sequelae which can be associated with unwanted inflammatory responses. Many other variants have since been developed,4–9 but few systematic comparisons of the activity of the different variants have been reported. Furthermore, the variety of methods used in different studies makes it difficult to assess which of them give the most effective silencing.
We have now constructed a matched set of IgG antibodies with identical Fab regions and with Fc regions representing each subclass and all of the variants in our INN database, as well as others described in the literature. We systematically compare their binding to human C1q and to Fcγ receptors from human, mouse and cynomolgus monkey and their activity in cell-based assays. We find that even a low level of binding activity can result in significant activity in the cell-based systems. Very few, if any, of the constructs used in the clinic to date are completely silenced. We also analyze the thermal stability of the different variants and find that many of them are impaired compared with wild-type antibodies.
Results
Catalog of immunoglobulin variants with reduced effector function
Our INN dataset (Supplementary Tables S1 and S2 of reference 1) was interrogated to identify all of the human antibodies and Fc fusion proteins with an intact Fc designed to have minimal or reduced effector function. There were 339 in total: 42 wild-type IgG2, 124 wild-type IgG4, 4 hybrid IgG2/IgG4 and 169 variant Fc regions. (For this analysis we ignored modifications designed to stabilize the antibody, extend half-life, or to create heterodimers or drug conjugates. Four proteins with chimeric Fc domains were excluded: efepoetin, eflenograstim, nemolizumab and satumomab). We also considered the list of engineered variants assembled by the ImMunoGeneTics (IMGT®) information system.10 Although this does not contain all of the variants found in the INN dataset, it includes some additional variants for which no therapeutic antibodies had been assigned an INN (as of April 2022). These variants, along with wild-type IgG1, IgG2, IgG3 and IgG4, negative controls of each subclass containing the 234S/235T/236 R (“STR”) mutations we previously identified11 and others that we obtained from the literature were included in the panel of 77 antibodies to be synthesized and tested, as listed in Table 1. We tried to find the earliest primary reference for each variant, but this was not always possible, as some were found only in the INN lists or secondary sources. This emphasized the problem that, while some variants are well characterized, many lack any detailed data on their reactivity with different receptors or other properties.
Table 1.
List of samples used in this study with the number of antibodies or fusion proteins that have been given an INN and the corresponding IMGT nomenclature (where available). So far as possible, citations are to the first description of the variant of which we are aware.
| Sample number | Isotype | Mutations | Number of INNs | IMGT Code | Citation | doi |
|---|---|---|---|---|---|---|
| mAb-024 | IgG1 | 402 | ||||
| mAb-075 | IgG1 | E233del L234del L235del | 0 | G1v65 | ||
| mAb-029 | IgG1 | E233P L234V L235A | 1 | Armour 1999 | 10 .1002/(SICI)1521-4141(199908)29:08 < 2613:AID-IMMU2613 > 3.0.CO;2-J | |
| mAb-030 | IgG1 | E233P L234V L235A G236del | 0 | G1v50 | Armour 1999 | 10 .1002/(SICI)1521-4141(199908)29:08 < 2613:AID-IMMU2613 > 3.0.CO;2-J |
| mAb-027 | IgG1 | E233P L234A L235A G236del P329A | 2 | |||
| mAb-028 | IgG1 | E233P L234V G236del S267K | 6 | |||
| mAb-033 | IgG1 | L234A L235A | 28 | G1v14 | Xu 2000 | 10 .1006/cimm.2000.1617 |
| mAb-034 | IgG1 | L234A L235A G237A | 7 | G1v14-1 | Bloom 2009 | Patent application WO2009143536 |
| mAb-066 | IgG1 | L234A L235A G237A P238S H268A A330S P331S | 0 | Tam 2017 | 10 .3390/antib6030012 | |
| mAb-036 | IgG1 | L234A L235A G237A N297A | 1 | |||
| mAb-035 | IgG1 | L234A L235A G237A K322A | 1 | Hernandez-Hoyos 2016 | 10 .1158/1535-7163.MCT-15-024 | |
| mAb-061 | IgG1 | L234A L235A D265A | 0 | |||
| mAb-056 | IgG1 | L234A L235A D265S | 1 | G1v14-67 | Edavettal 2022 | 10 .1016/j.medj.2022.09.007 |
| mAb-062 | IgG1 | L234A L235A K322A | 0 | Lin 2013 | 10 .4161/cbt.26106 | |
| mAb-068 | IgG1 | L234A L235A L328R | 0 | G1v14-48 | ||
| mAb-057 | IgG1 | L234A L235A P329A | 0 | G1v14-4 | ||
| mAb-037 | IgG1 | L234A L235A P329G | 10 | G1v14-49 | Schlothauer 2016 | 10 .1093/protein/gzw040 |
| mAb-058 | IgG1 | L234A L235A P329S | 0 | |||
| mAb-038 | IgG1 | L234A L235A P331S | 1 | G1v40 | Kurnellas 2023 | 10 .1186/s12967-023 -04,251-y |
| mAb-039 | IgG1 | L234A L235E | 1 | |||
| mAb-040 | IgG1 | L234A L235E G237A | 3 | G1v43 | Latour 2001 | 10 .4049/jimmunol.167.5.2547 |
| mAb-041 | IgG1 | L234A L235E G237A A330S P331S | 4 | Li 2018 | 10 .1186/s40425-018-0329-7 | |
| mAb-042 | IgG1 | L234A L235Q K322Q | 1 | Alvarado 2022 | 10 .1111/all.15262 | |
| mAb-031 | IgG1 | L234A G237A | 1 | Bloom 2009 | Patent application WO2009143526 | |
| mAb-032 | IgG1 | L234A G237A A330V | 1 | |||
| mAb-043 | IgG1 | L234F L235E D265A | 3 | Engleberts 2020 | 10 .1016/j.ebiom.2019.102625 | |
| mAb-044 | IgG1 | L234F L235E P331S | 7 | G1v39 | Oganesyan 2008 | 10 .1107/S0907444908007877 |
| mAb-064 | IgG1 | L234F L235Q K322Q | 0 | G1v53 | Borrok 2017 | 10 .1002/eji.1830230216 |
| mAb-067 | IgG1 | L234S L235T G236R | 0 | G1v59 | Wilkinson 2021 | 10 .1371/journal.pone.0260954 |
| mAb-045 | IgG1 | L235A G237A | 4 | Frewin 2002 | Patent application WO2002102853 | |
| mAb-069 | IgG1 | L235E | 0 | G1v23 | Alegre 1992 | 10 .4049/jimmunol.148.11.3461 |
| mAb-046 | IgG1 | L235G G236R | 1 | Geuijen 2018 | 10 .1016/j.ccell.2018.04.003 | |
| mAb-055 | IgG1 | L235R G236R S239K A327G A330S P331S | 1 | |||
| mAb-063 | IgG1 | G236R L328R | 0 | G1v52 | Horton 2008, Chu 2008 | 10 .1158/0008-5472.CAN-08-2268 |
| mAb-074 | IgG1 | P238S | 0 | G1v63 | ||
| mAb-059 | IgG1 | D265A | 0 | G1v66 | Shields 2001 | 10 .1074/jbc.M009483200 |
| mAb-026 | IgG1 | D265A P329A | 1 | |||
| mAb-060 | IgG1 | D265S | 0 | G1v67 | ||
| mAb-072 | IgG1 | S267K | 0 | G1v51 | ||
| mAb-054 | IgG1 | V273E | 1 | Ye 2019 | 10 .1158/2326-6066.CIR-18-0805 | |
| mAb-047 | IgG1 | N297A | 12 | G1v29 | Bolt 1993 | 10 .1002/eji.1830230216 |
| mAb-048 | IgG1 | N297A K322A | 1 | Hoseini 2018 | 10 .1158/0008-5472.CAN-08-2268 | |
| mAb-049 | IgG1 | N297G | 16 | G1v30 | Leabman 2013 | 10 .4161/mabs.26436 |
| mAb-050 | IgG1 | N297H | 1 | Tao 1989 | 10 .4049/jimmunol.143.8.2595 | |
| mAb-051 | IgG1 | N297Q | 2 | G1v36 | Tao 1989 | 10 .4049/jimmunol.143.8.2595 |
| mAb-052 | IgG1 | N297S | 1 | He 2013 | 10 .4049/jimmunol.1300409 | |
| mAb-065 | IgG1 | S298N T299A Y300S | 0 | Zhou 2020 | 10 .1080/19420862.2020.1814583 | |
| mAb-053 | IgG1 | T299A | 1 | Sazinsky 2008 | 10 .1073/pnas.0809257105 | |
| mAb-070 | IgG1 | L328R | 0 | G1v48 | ||
| mAb-071 | IgG1 | P329G | 0 | G1v49 | Schlothauer 2016 | 10 .1093/protein/gzw040 |
| mAb-073 | IgG1 | A330S P331S | 0 | G1v60 | ||
| mAb-025 | IgG1 | A330V | 2 | Knopf 2007 | Patent application WO2007062188 | |
| mAb-001 | IgG2 | 42 | ||||
| mAb-008 | IgG2 | V234A G237A | 2 | Cole 1997 | 10 .4049/jimmunol.159.7.3613 | |
| mAb-009 | IgG2 | V234A G237A P238S H268A V309L A330S P331S | 1 | G2v3 | Vafa 2014 | 10 .1016/j.ymeth.2013.06.035 |
| mAb-011 | IgG2 | V234S A235T del236R | 0 | Wilkinson 2021 | 10 .1371/journal.pone.0260954 | |
| mAb-005 | IgG2 | D265A A330S P331S | 1 | Bloom 2009 | Patent application WO2009143526 | |
| mAb-076 | IgG2 | H268Q V309L A330S P331S | 0 | G2v2 | An 2009 | 10 .1111/all.15262 |
| mAb-006 | IgG2 | F296A N297Q | 0 | |||
| mAb-007 | IgG2 | K322A | 1 | |||
| mAb-004 | IgG2 | A330S P331S | 5 | Armour 1999 | 10 .1002/(SICI)1521-4141(199908)29:08 < 2613:AID-IMMU2613 > 3.0.CO;2-J | |
| mAb-010 | IgG2 | P331S | 2 | Yan 2011 | Patent application W2011147319 | |
| mAb-002 | IgG2/1 | F296A N297Q | 1 | Goodman 2009 | Patent application WO2009010290 | |
| mAb-003 | IgG2/4 | 4 | G2G4v1 | Mueller 1997 | 10 .1016/S0161-5890(97)00042-4 | |
| mAb-022 | IgG3 | 1 | ||||
| mAb-023 | IgG3 | L234S L235T G236R | 0 | Wilkinson 2021 | 10 .1371/journal.pone.0260954 | |
| mAb-012 | IgG4 | 29 | Bruggemann 1987 | 10 .1084/jem.166.5.1351 | ||
| mAb-013 | IgG4-P | 95 | Angal 1993 | 10 .1016/0161-5890(93)90432-B | ||
| mAb-018 | IgG4-P | E233P F234V L235A G236del | 4 | Armour 1999 | 10 .1002/(SICI)1521-4141(199908)29:08 < 2613:AID-IMMU2613 > 3.0.CO;2-J | |
| mAb-017 | IgG4-P | E233P F234V L235A D265A | 1 | Zhang 2018 | 10 .1007/s00262-018-2160-x | |
| mAb-014 | IgG4-P | F234A L235A | 17 | G4v4 | Alegre 1994 | 10 .1097/00007890 -199,457,110 -00,001 |
| mAb-020 | IgG4-P | F234A L235A G237A P238S | 0 | Tam 2017 | 10 .3390/antib6030012 | |
| mAb-021 | IgG4-P | F234S L235T G236R | 0 | Wilkinson 2021 | 10 .1371/journal.pone.0260954 | |
| mAb-015 | IgG4-P | L235A | 1 | Smith 2008 | Patent application WO2008129124 | |
| mAb-016 | IgG4-P | L235E | 8 | G4v3 | Alegre 1992 | 10 .4049/jimmunol.148.11.3461 |
| mAb-077 | IgG4-P | L235E P329G | 0 | G4v3-49 | Schlothauer 2016 | 10 .1093/protein/gzw040 |
| mAb-019 | IgG4-P | N297Q | 1 | G4v36 | Schlothauer 2016 | 10 .1093/protein/gzw040 |
Apart from wild-type IgG2 or IgG4, the variants most frequently appearing in the INN lists were IgG1 L234A/L235A (28), IgG4 F234A/L235A (17), IgG1 N297G (16), IgG1 N297A (12), IgG1 L234A/L235A/P329G (10), IgG4 L235E (8), IgG1 L234A/L235A/G237A (7) and IgG1 L234F/L235E/P331S (7). These 105 ‘most popular’ variants made up 62% of all variants.
Binding to Fc gamma receptors by surface plasmon resonance
Experiments were carried out using two independently prepared sets of CD20 antibodies, the first expressed in HEK cells and the second in Chinese hamster ovary (CHO) cells. Binding to a range of human, monkey and mouse Fc receptors was measured using a Biacore T200. Each sample was tested at least twice with the running order reversed for every second run in order to minimize any impact of drift in the system. Due to the large number of antibody-receptor combinations analyzed and the low affinity of many of the interactions, it was not practicable to measure individual binding affinities. Instead, the maximum binding at a high concentration (100 μg/mL) was measured relative to the binding of wild-type IgG1. A set of 67 samples (produced in HEK cells) was tested for binding to all of the human Fc receptors: FcγRI, FcγRIIa (131 H), FcγRIIa (131 R), FcγRIIb, FcγRIIIa (158F), FcγRIIIa (158 V), FcγRIIIb (NA1) and FcγRIIIb (NA2), the cynomolgus monkey receptors: FcγRI, FcγRIIa, FcγRIIb and FcγRIIIa and the mouse receptors: FcγRI and FcγRIV. A second set of 77 samples (produced in CHO cells), which included some additional controls and variants from the literature, was tested only on the FcγRI receptors from human, cynomolgus monkey and mouse. The results were in excellent agreement with the first set with correlation coefficients of 0.98 (Figure 1). Many of the variants showed higher relative binding to cynomolgus monkey FcγRI compared with human FcγRI, whereas they showed lower relative binding to mouse FcγRI.
Figure 1.
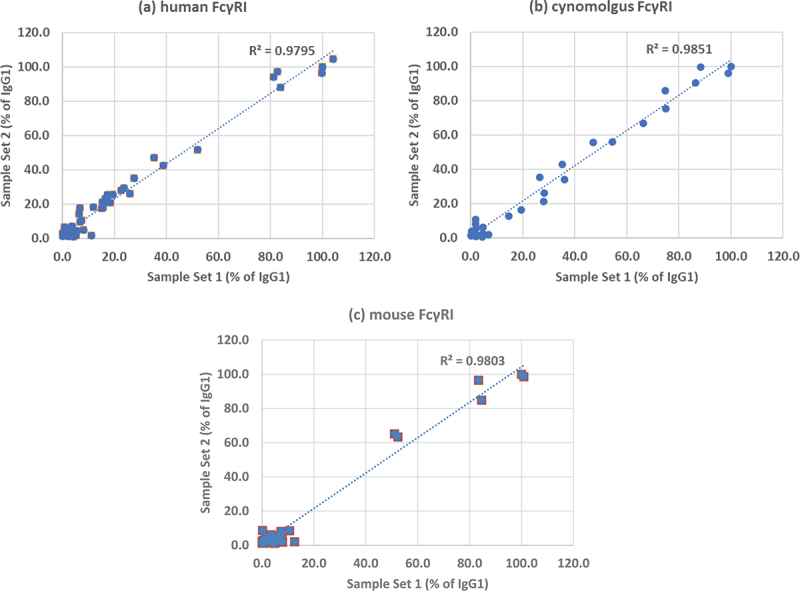
Comparison of two sets of CD20 antibodies binding to FcγRI from different species. (a) human, (b) cynomolgus monkey, (c) mouse. Binding was measured by surface plasmon resonance and expressed as a percentage of the binding of wild-type IgG1. Sample set 1 was prepared in HEK cells and sample set 2 in CHO cells.
The mean results for all experiments are summarized in Table 2. As expected, wild-type IgG1 and IgG3 showed substantial binding to all of the Fc gamma receptors. Wild-type IgG2 bound strongly to human FcγRIIa (131 H) and to cynomolgus monkey FcγRIIa and FcγRIIb, but only weakly to human FcγRIIa (131 R) and human FcγRIIb. Wild-type IgG4 bound strongly to FcγRI from all species, also to FcγRIIa and FcγRIIb. The S228P stabilizing mutation in IgG4 had negligible impact on FcγR binding. A hybrid IgG2/IgG4 antibody showed substantially reduced binding to human, but not to cynomolgus monkey receptors. The triple mutation 234S/235T/236 R (STR), which we previously described to completely abolish binding to human Fcγ receptors,11 gave no measurable binding to any of the receptors whether it was introduced into IgG1, IgG2, IgG3 or IgG4. Single point mutations at any of positions 235, 238, 265, 267, 273, 297, 299, 322, 328, 329, 330, 331 were insufficient to eliminate FcγR binding and the only examples where two mutations eliminated binding to all the receptors were F296A/N297Q (i.e., aglycosyl) in the context of either IgG2 or hybrid IgG2/1. Besides STR, the only IgG1 variant to completely eliminate FcγR binding was F233Δ/L234Δ/L235Δ. There were also two IgG2 and two IgG4 variants where we detected no binding in the surface plasmon resonance (SPR) experiments. However, based on the INN lists, none of these variants have been widely used in the clinic. In contrast, the ‘most popular’ variants which appeared in at least seven or more INNs and are based on mutations either in the region 233 to 237 (LALA or similar) or 297 (aglycosyl), all gave measurable levels of binding to two or more receptors.
Table 2.
Relative binding responses of CD20 antibodies binding to human, cynomolgus and mouse Fc receptors measured by surface plasmon resonance or by affinity HPLC. All results are normalized and expressed as a percentage of wild-type IgG1. Due to assay noise, some of the normalized responses of negative samples are less than 0%. The magnitude of the negative values gives an impression of the experimental error for each receptor. Cells are shaded to indicate the response range: green = negative (less than 2%), yellow = low (2% to 10%), orange = substantial (10% to 40%), red = high (more than 40%).
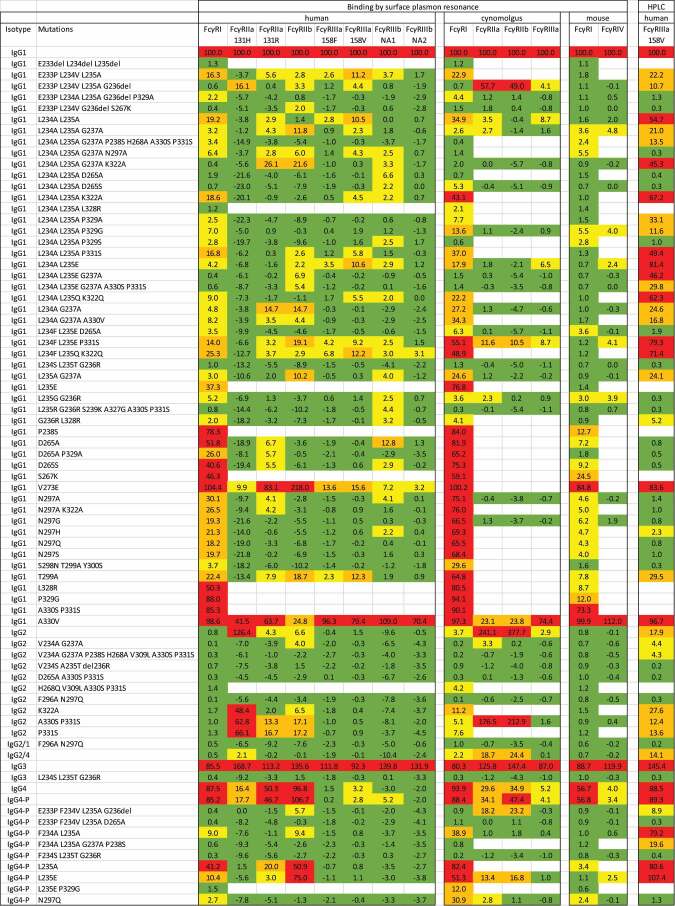 |
Binding to C1q by ELISA
The binding of the first set of 67 antibodies to human C1q was measured by ELISA. Microplates were coated with test samples (or buffer alone) and incubated with peroxidase-labeled C1q. Each sample was tested in triplicate in two separate experiments. The only samples to give a positive response were wild-type IgG3 (mean absorbance 2.970 and wild-type IgG1 (mean absorbance 0.579). The responses of all of the other samples were statistically indistinguishable from buffer alone (mean absorbance 0.020).
Binding to FcγRIIIa by affinity chromatography
An alternative method to measure binding to FcγRIIIa (158 V) was by affinity HPLC using a column containing the immobilized receptor.12 This method can discriminate the binding of different antibody glycoforms. Examples of typical elution profiles are shown in Figure 2. A trastuzumab IgG1 reference standard showed three principal peaks with elution times at 15.2, 16.7 and 18.5 min. These are believed to correspond to species with different levels of galactosylation, i.e., G0F, G1F, and G2F, respectively.12,13 The wild-type IgG1 CD20 antibody gave a similar profile with major peaks at 14.4, 16.2 and 17.9 min. Variants from the first set of 67 samples (produced in HEK cells) showed similar patterns except that the elution times were shortened to various extents and some were completely unretained and eluted in the void volume of the column at 2.0 min. To facilitate comparisons, a weighted mean elution time was calculated for each sample, being the mean of the elution times of each principal peak multiplied by the area of that peak. The results are summarized in Table 2.
Figure 2.
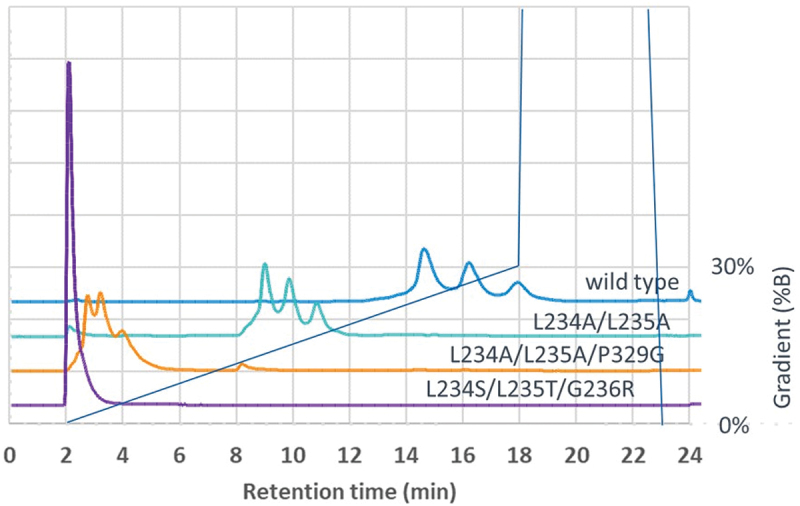
Affinity chromatography of CD20 antibodies on a column containing immobilized FcγRIIIa (158 V). Elution profiles of wild-type IgG1, variants L234A/L235A (LALA), L234A/L235A/P329G (LALAPG) and L234S/L235T/G236R (STR) are shown.
Wild-type IgG3 showed the highest affinity, giving a series of peaks between 20.4 and 22.0 min toward the end of the elution gradient. The STR variants, along with a number of others, showed no binding, being eluted at the void volume of the column. Many variants gave significant levels of binding by this method and among the ‘most popular’ (7 or more INNs), only the aglycosyl variants N297G and N297A showed no binding.
Affinity HPLC was capable of detecting positive binding responses for many variants where binding was undetectable by SPR. In particular, wild-type IgG4 and some of the IgG4 variants showed a high level of binding. This is believed to be caused by the fact that the commercial HPLC column is made using aglycosylated FcγRIIIa, which gives anomalous binding to IgG4.13 However, as shown in Figure 3, many of the IgG1 and IgG2 variants also showed significant binding by this method, whereas they had been weak or negative by SPR.
Figure 3.
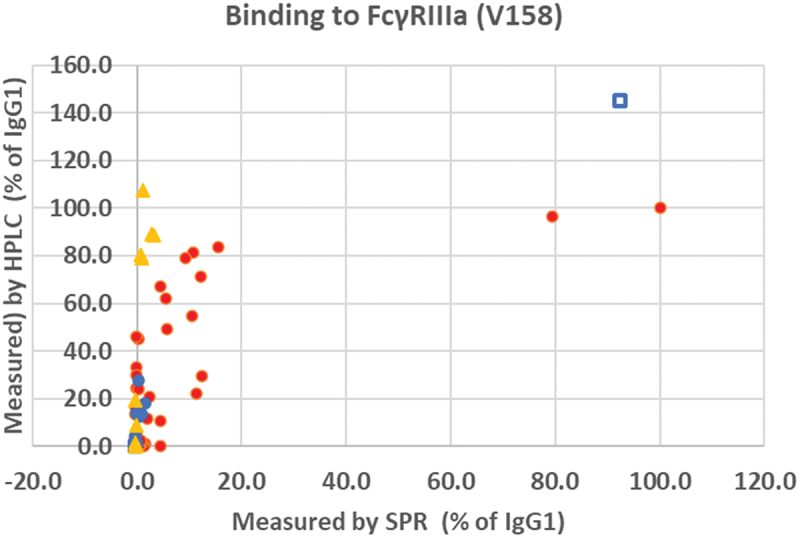
Comparison of the binding of CD20 antibodies to FcγRIIIa (158 V) measured by surface plasmon resonance compared with affinity HPLC. The SPR results are expressed as percentage of the binding of wild-type IgG1 and the HPLC results are expressed as a percentage of the retention time of wild-type IgG1. IgG1 samples filled red circles ●, IgG2 samples filled blue circles ●, IgG3 samples open blue squares □, IgG4 samples filled orange triangles ▲.
During the course of the affinity HPLC experiments, we noted that some samples displayed an unexpected set of minor peaks corresponding to either higher or lower affinities. These were not included in the calculations of mean elution volume, but on further investigation, we discovered that these samples had become cross-contaminated during the purification process due to re-use of Protein A affinity columns in a medium-throughput spin column process. (Despite following the manufacturer’s recommended cleaning procedure, this was insufficient to prevent low-level cross contamination, not detected by other methods.) The level of cross-contamination (<1%) did not influence the results of binding experiments, whether by affinity HPLC or SPR, but it could affect the more sensitive cell-based assays, where a small amount of contamination of a ‘negative’ sample with a ‘positive’ one, would give erroneous results. The second set of 77 samples, independently prepared from CHO cells, did not show this problem and were used for the subsequent experiments.
Fc effector cell bioassays
The ability of anti-CD20 antibodies to engage with cellular Fcγ receptors was measured using the Promega Fc effector cell bioassay reporter system. This indicates the potential for ADCP or ADCC. Target cells were Raji, a CD20+ human B lymphocyte cell line and effector cells were Jurkat, a human T cell line with a luciferase gene under the control of an NFAT promoter and transfected with the required human Fc receptor. Samples of variant antibodies were tested at a final concentration (10 µg/mL), previously established to give maximal response with wild-type IgG1.11 The second set of 77 antibodies prepared from CHO cells was tested with the complete panel of effector cells. The results are summarized in Table 3. Results from Biacore and cell-based assays for each receptor are compared in Figure 4.
Table 3.
Relative activity of CD20 antibodies in cell-based assays for ADCP, ADCC or CDC measured by luminescence using assay systems from Promega or Svar and thermal stability measured by DSF. The activity assays are normalized and expressed as a percentage of wild-type IgG1. Cells are shaded to indicate the response range: green = negative (less than 2%), yellow = low (2% to 10%), orange = substantial (10% to 40%), red = high (more than 40%). The onset of denaturation (Ton) and first melting point (Tm) are reported. Cells are shaded to indicate the temperature compared with wild-type IgG1 (Ton = 63.1°C, Tm = 73.2°C). Ton: green = greater than 61.9°C, yellow = 60°C to 61.9°C, orange = 56°C to 60°C, red = less than 56°C. Tm: green = greater than 72.6°C, yellow = 70°C to 72.6°C, orange = 66°C to70°C, red = less than 66°C.
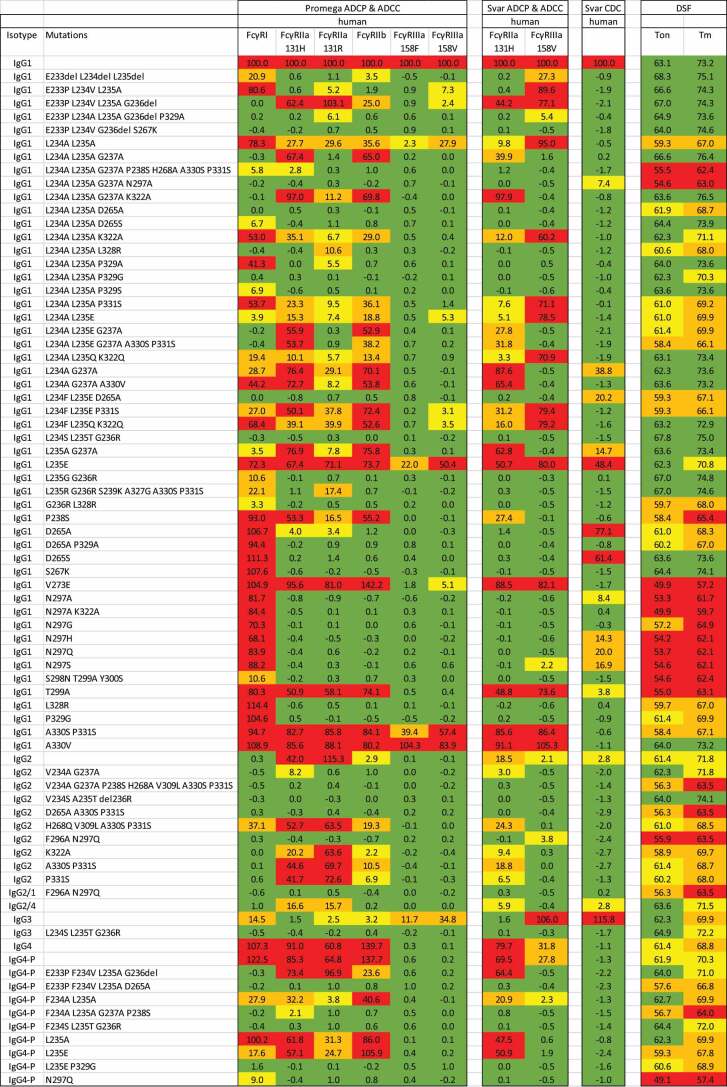 |
Figure 4.
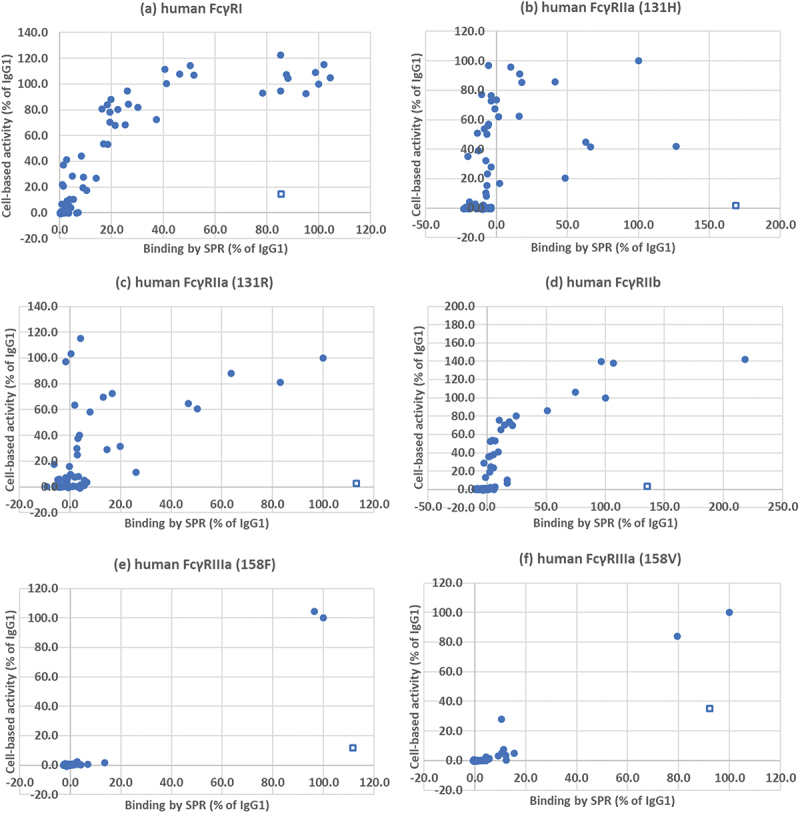
Comparison of the binding of CD20 antibodies to human fc receptors and their activity in a cell-based luminescence assay. (a) FcγRI, (b) FcγRIIa (131 H), (c) FcγRIIa (131 R), (d) FcγRIIb, (e) FcγRIIIa (158F), (f) FcγRIIIa (158 V). Binding and activity are expressed as a percentage of wild-type IgG1. Wild-type IgG3 is indicated with an open square □, other samples with a closed circle ●.
Wild-type IgG3, which had given binding levels as high or higher than IgG1, showed much reduced activity in all of the cell-based assays. In contrast, many of the variants which showed comparatively low levels of binding activity to FcγRI or FcγRII, showed higher levels of relative activity in the cell-based assays. There was virtually no binding to FcγRIIIa by Biacore nor ADCC activity in the Promega assays with any of the variants.
The samples were also analyzed using an alternative ADCP and ADCC cell-based luminescence assay system from Svar. Although the system is similar to the Promega effector cells, there are some differences. Raji target cells were engineered to specifically express human CD20 and the Jurkat-based ADCC effector cells were engineered to express firefly luciferase under the control of a promoter inspired by a target gene of FcγRIIIa-dependent signaling, consisting of binding sites for five transcription factors instead of NFAT only. By contrast, Svar’s ADCP effector cells use an NFAT-driven reporter gene promoter. Svar reporter cells also contain a second reporter gene (renilla luciferase) under control of a constitutive promoter. Use of two different luciferases allowed normalization of the results to the number of cells in each microwell. Two effector cells were available, transfected either with FcγRIIa (131 H) or FcγRIIIa (158 V). Samples were tested at a final concentration of 15 μg/mL. The results are summarized in Table 3 and compared with the results from the corresponding Promega assays in Figure 5. There was a good correlation between the results for ADCP with FcγRIIa, though the relative responses of variants in the Svar assay were slightly lower. However, the SVAR ADCC assay with FcγRIIIa gave much higher responses with many of the variants compared with the Promega assay, similar to the results from affinity HPLC.
Figure 5.
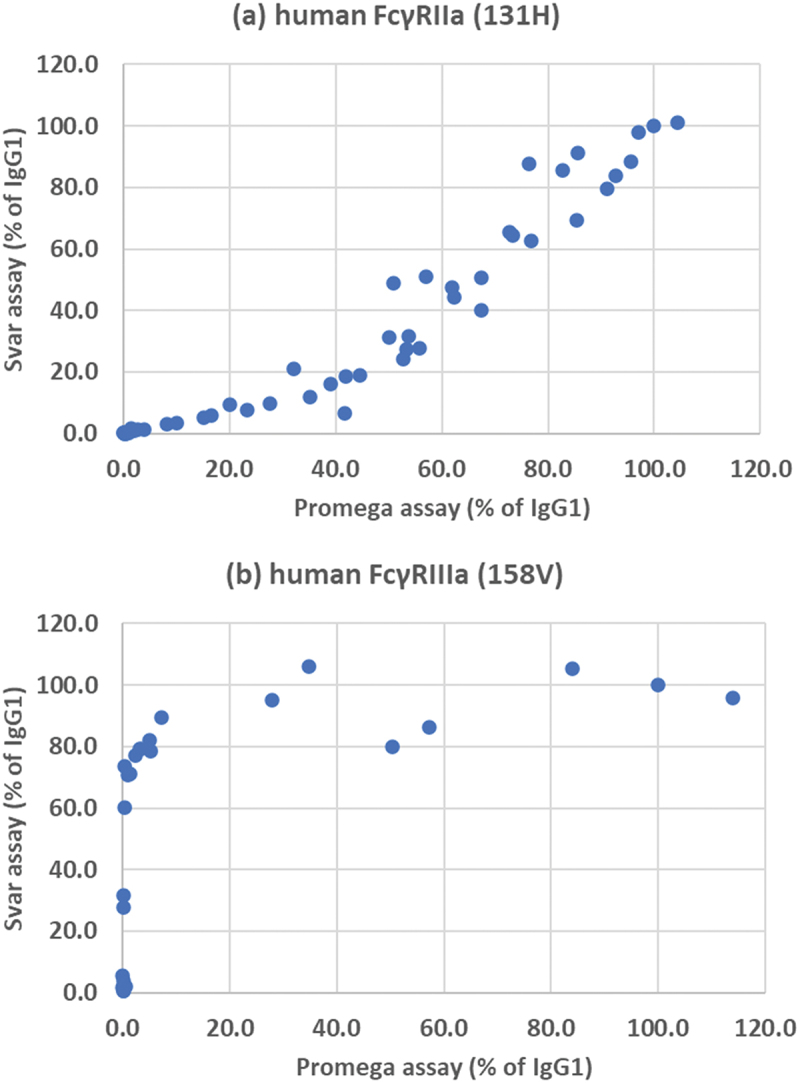
Comparison of Promega and Svar cell-based assay systems. (a) FcγRIIaγ (131 H), (b) FcγRIIIa (158 V). Activity is expressed as a percentage of wild-type IgG1.
Among the ‘most popular’ variants (7 or more INNs), only L234A/L235A/P329G (LALAPG) gave negative responses in all of the cell-based assays. Aglycosyl variants N297G and N297A gave high levels of ADCP activity with FcγRI, while the others gave substantial ADCP activity with two or more receptors. There were a few ‘less popular’ variants besides LALAPG which gave negative responses in all of the cell-based assays for ADCP and ADCC. These included: STR in the context of all four Ig subclasses, as well as 4 IgG1 variants, 2 IgG2 variants, 2 IgG4 variants and the IgG2/1 variant. Together, these represent a total of 11 mutations from the 72 different mutations which were tested.
Complement-mediated cytotoxicity
We had already shown that nearly all of the variants failed to bind to human C1q in an ELISA assay. However, the avidity of binding C1q can be greatly increased by oligomerization of antibodies on the cell surface14 and so it was still possible that some variants could engage in complement-dependent cytotoxicity (CDC). Accordingly, we tested them using the Svar iLite assay system. This uses Ramos target cells which express CD20 and Svar luciferase (a modified form of Metridia longa luciferase). On lysis of the cells, this highly stabilized luciferase is released, accumulates in the cell medium over time and can be measured with a suitable substrate. The results are summarized in Table 3. As expected, the highest activity was given by wild-type IgG1 and IgG3 and none of the IgG2 or IgG4 variants showed any activity. However, several IgG1 variants showed significant activity, including most of the aglycosyl variants, also L234A/G237A, L234F/L235E/D265A and L235A/G237A. Three single-point variants, L235E, D265A and D265S gave high activity (these had not been included in the set which was previously tested for C1q binding). All of the other variants showed effective silencing of CDC activity. Results may depend on the source of complement. The reported data used fresh frozen human serum, but an aglycosyl variant did not show CDC activity when tested with freeze-dried complement from Sigma (data not shown).
Thermal stability by differential scanning fluorimetry
Antibodies were assessed using the SUPR-DSF system of Protein Stable. Intrinsic protein fluorescence was measured as an indicator of protein stability. Preliminary experiments with control samples established that a protein concentration of 1 mg/mL gave an adequate fluorescence signal which remained sufficient (more than 6-fold above background) as the temperature was ramped up to 97°C. The goal of these experiments was to compare the different antibody samples against each other. This is easier when the datasets share a common profile. Using the full spectrum range to quantify spectral shift, the melt curves showed different profiles and included both positive and negative gradient transitions. To simplify analysis, the wavelength range, was optimized to eliminate the negative transitions associated with long wavelengths. Comparing the first derivative melt curves, differences between samples were obvious. A selection is shown in Figure 6. Particularly notable is the first transition which is believed to be associated with the CH2 domain. The results are summarized in Table 3. Wild-type IgG1 gave a Ton of 63.1°C and Tm of 73.2°C. The mean fitting error of Ton was ± 0.4°C and of Tm ±0.2°C. The Ton of test samples was considered significantly lower if it was less than 61.9°C (i.e., lower by more than three times the fitting error). Similarly, the Tm was considered significantly lower if it was less than 72.6°C.
Figure 6.
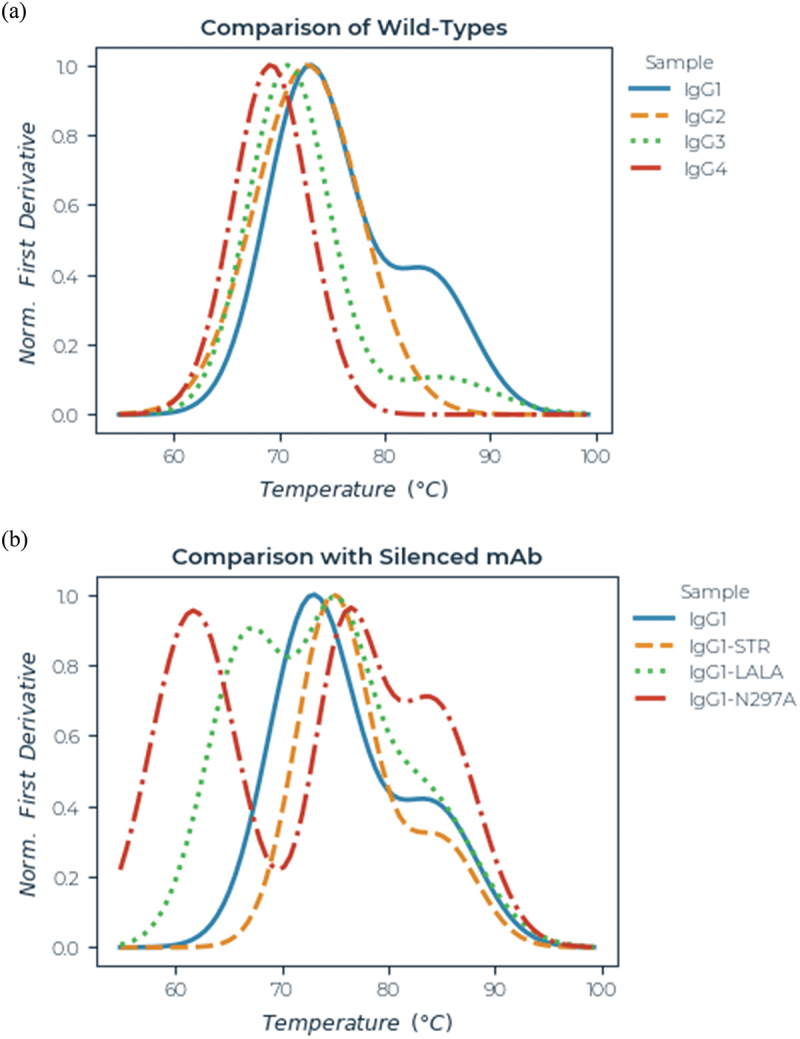
Thermal stability of CD20 antibodies measured by differential scanning fluorimetry. Representative melt curves (normalized first differential) are shown for: (a) wild-type IgG1, IgG2, IgG3 and IgG4-P and (b) wild-type IgG1, L234S/L235S/G236R (STR), L234A/L235A (LALA), and N297A (aglycosyl).
For most of the variants, two, and for some, three temperature transitions could be resolved, but for the purpose of comparison, only the onset of melting (Ton) and the first transition point (Tm) are reported. Wild-type IgG2, IgG3 and IgG4 all gave melting temperatures significantly lower than IgG1. Of the 51 IgG1 variants, 30 had significantly reduced Ton and/or Tm compared with wild-type. The most severely affected were the aglycosyl antibodies mutated at N297 or T299. Other mutations associated with reduced thermal stability included L234F, L235E, P238S, D265A, V273E, K322A, L328R and P329G. In contrast, the STR mutations 234S/235T/236 R gave about 2–4°C increase in both Ton and Tm relative to the corresponding wild-type control for all four subclasses.
Discussion
Since the first therapeutic monoclonal antibody, OKT3, entered the clinic in the early 1980s, side effects caused by unwanted inflammatory responses have been a complication of therapies which otherwise had great potential.15,16 Early research with recombinant antibodies suggested that the IgG4 isotype might be comparatively inert.17 As a consequence, IgG4 antibodies were selected for applications where inflammatory applications needed to be avoided.18,19 However, it was soon appreciated that the picture was not so simple due to the existence of multiple Fc gamma receptors, and different polymorphic forms, resulting in significant biologic activity for IgG4 in many situations.20,21 This became particularly evident with the disastrous results of administering the IgG4 anti-CD28 antibody TGN1412.22,23 Meanwhile, variant antibodies had been developed with Fc regions mutated to reduce binding to Fc gamma receptors and also to C1q.4–6 Among the first were N297A (aglycosyl)2 and L234A/L235A (LALA)24 and they remain in wide use to this day. However, they were incompletely silenced and therefore other variants have been developed which are described as “completely abolished”,25,26 “silent”27 or “no detectable binding”.28 The problem is that very rarely was one variant compared with another in the same assay system. From the plethora of information in the literature, it was impossible for investigators to select the most suitable variant for a particular application. And even had the methodology been comparable, differences in antibody variable regions, allotypic and other variations would still have made the results uncertain.
Therefore, we set out to systematically compare a set of antibodies with identical V regions and allotypes using straightforward experimental methods which can be replicated in most laboratories. We used V regions from the CD20 antibody rituximab because cell-based assays for anti-CD20 antibodies are readily available. Our analyses do not provide fine detail with regard to the exact affinity of each variant for each receptor because we were constrained to test the samples at a single concentration. Instead, we ranked the different variants in order of their residual activity in each assay system to identify the ones which were the most completely silenced. Because of the importance for animal models to mimic the human situation, we included binding studies with selected Fc receptors from cynomolgus monkeys and mice; at present the comparable cell-based assays are not commercially available.
Our panel of antibodies included the variants we previously identified from the WHO INN lists (up till April 2022)1 with the addition of those listed by the IMGT10 and some others. We attempted to identify a primary literature reference for each variant, but this was not always possible as some seemed to be mentioned only in reviews or in the INN lists. This emphasizes the difficulty of finding comprehensive data. A first set of 67 variants and controls prepared from HEK cells was used for initial experiments by SPR and affinity HPLC to measure binding to Fc receptors and by ELISA to measure binding to C1q. An enlarged set of 77 variants and controls prepared from CHO cells was used for cell-based assays, thermal stability studies and repeats of some SPR experiments.
The cornerstone of our studies was the cell-based assays for ADCP potential and ADCC potential using luminescence reporter assays. We compared two commercial assays systems, from Promega and Svar Life Sciences. (These assay systems do not measure phagocytosis or cytotoxicity directly, but provide sensitive measures of upstream events, namely the activation of gene transcription through the NFAT (nuclear factor of activated T cells) pathway in the effector cells which is triggered by cross-linking of Fc receptors.) The Promega systems covered the range of human receptors, including different alleles, whereas only two assays were available from Svar, for the 131 H allele of FcγRIIa (ADCP) and the 158 V allele of FcγRIIIa (ADCC). They showed comparable results to the Promega assays, though the Svar ADCP assay was a little less sensitive and the ADCC assay substantially more sensitive.
Wild-type IgG1 gave strong positive responses in every binding and cell-based assay and was used as the reference against which all other samples were compared. Wild-type IgG2 was virtually inactive in FcγRI assays, gave strong responses in FcγRIIa assays and was generally weak or negative in FcγRIIb and FcγRIII assays. It gave a very weak response in the CDC assay. Wild-type IgG3 was strongly positive in all the FcγR binding and CDC assays, but gave weaker responses in the other cell-based assays, except for the Svar ADCC assay which was strongly positive. Wild-type IgG4 was strongly positive in FcγRI and FcγRIIa assays. It was negative for CDC and generally gave weak responses in binding to FcγRIII or ADCC. It gave very strong binding to FcγRIIIa response by affinity chromatography, but this may not be representative of the normal situation since the recombinant receptor used in the commercial HPLC column is produced in E. coli and not glycosylated,12 which is thought to increase its affinity for IgG4.13 It is possible that some of the high binding results seen with variants of other subclasses might also be caused by the same anomalous binding of the aglycosyl receptor. Affinity chromatography is potentially a powerful and sensitive technique to characterize receptor binding because it can resolve different glycoforms and isoforms and detect very low affinity interactions. At present, only the one column is commercially available, but we are working to develop chromatographic methods for all of the Fc receptors expressed in mammalian cells.
The IgG1 variants A330V and A330S/P331S were silenced in CDC, but gave high levels of activity in all of the Fc receptor assays. All other variants showed little or no activity with all forms of FcγRIII either by SPR or cell-based activity (ADCC), but three IgG4 variants, F234A/L234A, L235A and L235E, still showed substantial binding by affinity HPLC (as described above, this may not represent the normal situation). A much more mixed picture was seen in the FcγRI and FcγRII assays, where the majority of variants still showed some level of activity, often approaching that of the wild-type antibody. It was notable that a low level of residual binding in the SPR assay was often associated with a higher level of activity in the corresponding cell-based assay. It seems likely that even low-affinity interactions with the soluble receptor may lead to more substantial responses when both the antibody and receptor are immobilized on the surface of target and effector cells, respectively, thus increasing the functional affinity of the interactions. A few mutations discriminated between the different isoforms of FcγRII. For example, IgG1 L234A/L235A/G237A and L234A/L235E/G237A gave high levels of activity with FcγRIIa (131 H) and FcγRIIb, but none with FcγRIIa (131 R). However, there were no converse examples. Consistent with earlier reports, the 158 V allele of FcγRIIIa showed greater activity than the 158F allele.29,30
Binding to cynomolgus and mouse Fc receptors by SPR generally correlated with binding to the corresponding human receptors. Compared with human FcγRI, binding responses were higher on cynomolgus FcγRI and lower on mouse FcγRI. Human IgG2 and the IgG2 variant A330S/P331S gave high binding to human FcγRIIa (131 H), but low binding to human FcγRIIa (131 R) and FcγRIIb. They gave very high binding with both cynomolgus FcγRIIa and FcγRIIb. In most situations, the cynomolgus monkey appears to be a good model for human Fc receptors. (However, comparable cell-based assay methods are not yet available.) Our results are consistent with previous studies of the interaction of human subclasses and some Fc silenced variants, which found that human IgG generally bound more strongly to cynomolgus Fc receptors and less so to mouse Fc receptor, indicating the need for careful appraisal of the relevance of animal models when Fc silencing is not complete.31
It is important that Fc modifications to eliminate effector function do not have any other adverse effects, for example to increase the risk of unwanted immunogenicity, to reduce half-life, or to destabilize the antibody structure. Previously we showed that some of the ‘most popular’ mutations might theoretically increase the number of peptides available to be presented by MHC Class II antigens,11 but we are not aware of any reports of anti-drug antibodies which target particular Fc mutations. However, it is known that some silencing mutations could result in reduced half-life. In particular, lack of G236 in IgG2 and in G236Δ variants, as well as the mutations A330S/P332S, are associated with reduced binding to FcRn and reduced transplacental transport.32,33 Many silencing mutations have been made in IgG2 or IgG4 antibodies, but these subclasses are prone to disulfide interchange resulting in isoforms with differing activities and biophysical properties.34,35 In addition, IgG4 antibodies may be particularly prone to aggregation36 and in general aglycosylated antibodies are less stable. Substitution of D265 results in increased galactosylation and sialylation,37 which might reduce stability. Other studies have shown that mutations at D265, L328 and P331 are associated with reduced thermal stability as indicated by differential scanning fluorimetry (DSF) experiments.38 We found similar results and extended them to cover the full range of silenced variants, finding that the great majority show some degree of reduction in thermal stability compared with wild-type IgG1. We had insufficient material to carry out more detailed stability studies. None of the antibodies showed significant levels of aggregation by HPLC immediately after purification. However, others have shown that DSF correlates with stability-indicating measures such as differential scanning calorimetry and acid-induced aggregation, with IgG1 > IgG2 > IgG4, as we have seen.38
Somewhat to our surprise, the only variants to show a ‘green flag’ in every assay, were the four versions of STR, that is IgG1 and IgG3 L234S/L235T/G236R, IgG2 V234S/A235T/Δ236 R and IgG4 F234S/L235T/G236R. Other variants which combined nearly as low ADCP, ADCC and CDC activity with good thermal stability were E233P/L234A/L235A/G236Δ/P329A, E233P/L234V/G236Δ/S267K, L234A/L235A/P329S, L234A/L235A/D265S, and L235G/G236R
It is clear that Fc engineering to eliminate unwanted binding to Fc receptors and complement is necessary for many contemporary applications of therapeutic antibodies and fusion proteins, especially bispecific T-cell engagers, antibody-drug conjugates and whenever inflammatory responses must be avoided. Elimination of binding to Fc receptors may also be desirable for many research and diagnostic applications. Investigators have a wide choice of methodologies, but not all are equal. Many of the most popular variants were discovered before the full complexity of the Fc receptor system was appreciated and before the development of sensitive assay systems. With hindsight, they are not so effective as originally thought. Newer variants offer more comprehensive silencing, but the lack of comparative data and the thicket of patent applications associated with them may have been a deterrent to their optimal use. Here we have attempted to address the first of these problems. We have not been able to consider every relevant factor. Others have provided more detailed reviews of the literature and experimental methods to more comprehensively probe developability concerns.4–9,38,39 Nevertheless, we hope to have provided investigators with a database to enable them to identify a shortlist of silencing mutations suitable for their particular application.
Our studies here used a single anti-CD20 (rituximab) Fab region. We have previously reported similar results with a smaller set of variants that included CD3 and CD52 Fab regions.11 We expect that the results can be generalized to the great majority of Fab regions, though experience teaches us that antibodies can always be idiosyncratic. To take this work further, it would be helpful to analyze other parameters, such as different stability indicators or binding to FcRn. Researchers interested in such studies using this sample set are invited to contact us. An important question which we have not addressed here is “what degree of Fc silencing is really necessary in the clinic?”. The answer will depend very much on the particular context, the disease indication, antigenic target, antibody format and so on. As many more antibody variants pass through clinical trials, it may be possible to start to answer this question.40
Materials and methods
Nomenclature
The EU numbering system41 is used throughout this article. Amino acid alterations are described thus: XnnnY, where X is the single letter code for the residue in the native amino acid sequence, nnn is the EU index position and Y is the single letter code for the replacement amino acid residue. The symbol Δ refers to a residue which is deleted. Some variants are referred to in short form: LALA (L234A/L235A), LALAPG (L234A/L235A/P329G), aglycosyl (N297A, N297G, N297H, N297Q, or N297S), STR (234S/235T/236 R).
Antibody design, expression, and purification
The amino acid sequences of the heavy chains of the variant panel were based on the variable region sequence of rituximab linked to the desired constant region based on human IgG1, IgG2, IgG3 or IgG4. Heavy chain sequences were based on IGHG*01 (G1m(za)), IGHG2*01, IGHG3*01 (G3m(b*)) and IGHG4*01 (G4m(a)), corresponding to the allotypes most frequently used for therapeutic antibodies. Two versions of ‘wild-type’ IgG4 were made, one with the native S228 and one with the substitution S228P to prevent Fab arm interchange The S228P substitution was used in all of the variants. IgG3 heavy chains included a mutation R435H to facilitate purification using Protein A. A single light chain consisting of the variable region of rituximab with a kappa km1 constant region was used for all of the constructs. Variants of each IgG subclass with the mutations 234S/235T/236 R were included as negative controls. A first set of 67 samples was made by expression of the synthetic genes in HEK cells and purification by affinity chromatography on Protein A as previously described.11 A second independent set of 77 samples which included additional controls and variants described in the literature but not appearing in the INN lists was made by Sanyou Biopharmaceuticals Co. (Shanghai, China) according to their standard procedures using new preparations of synthetic genes expressed in CHO-K1 cells. The antibodies were > 95% pure by SDS gel electrophoresis (reducing and non-reducing) and contained > 90% monomer and < 4% aggregates by size-exclusion chromatography. The yield of antibody from a 30 mL culture varied between 0.86 mg and 11.17 mg (mean 4.0 mg), but there was no particular correlation with antibody subclass or mutations. 40 of the antibodies were produced again at 10 mL scale and the yield varied from 0.1 mg to 1.1 mg (mean 0.4 mg). There was no correlation between the yields of the same samples. The disproportionately lower yield at 10 mL scale is attributed to losses in purification and this probably accounts for most of the variability as well. All samples used for cell-based assays were checked to contain less than 1 EU/mL of endotoxin.
Binding to immobilized fc receptors by surface plasmon resonance
Binding analysis was carried out at 25°C using a Biacore T200 instrument with HBS-EP+ running buffer. The first set of 67 samples was analyzed according to the procedure previously described.11 Mouse monoclonal IgG1 anti-histidine antibody (Qiagen Cat. No. 34670) was immobilized to a CM5 chip by carbodiimide chemistry. A reference cell was activated and blocked with ethanolamine. Histidine-tagged recombinant Fc receptors were diluted to 5 µg/mL in HBS-EP+ and injected for 2 min at 10 µL/min. The following receptors were used.
| Receptor | Species | Supplier | Catalogue number |
| FcγRI (CD64) | human | Sino Biological | 10256-H08H |
| FcγRIIa (CD32a) 131 R allele | human | Sino Biological | 10374-H08H |
| FcγRIIa (CD32a) 131 H allele | human | Sino Biological | 10374-H08H1 |
| FcγRIIb (CD32b) | human | Sino Biological | 10259-H08H |
| FcγRIIIa (CD16a) 158F allele | human | Sino Biological | 10389-H08H |
| FcγRIIIa (CD16a) 158 V allele | human | Sino Biological | 10389-H08H1 |
| FcγRIIIb (CD16b) NA1 allele | human | Sino Biological | 11046-H08H1 |
| FcγRIIIb (CD16b) NA2 allele | human | Sino Biological | 11046-H08H |
| FcγRI (CD64) | cynomolgus | R&D Systems | 9239-FC |
| FcγRIIa (CD32a) | cynomolgus | Sino Biological | 90015-C08H |
| FcγRIIb (CD32b) | cynomolgus | Sino Biological | 90014-C08H |
| FcγRIIIa (CD16a) | cynomolgus | Sino Biological | 90013-C08H |
| FcγRI (CD64) | mouse | Sino Biological | 50086-M08H |
| FcγRIV (CD16-2) | mouse | Sino Biological | 50036-M08H |
Test samples were diluted with running buffer to 100 µg/mL and injected for 1 min at 30 µL/min. Between each cycle, the chip was regenerated by injection of 10 mM glycine pH 3.0 for 0.5 min at 30 µL/min and recharged with a fresh injection of Fc receptor. At the beginning and end of each experiment, blank cycles of buffer alone were run. The average SPR signal (relative to the reference cell) was measured approximately 5 seconds before the end of injection (response). The blank (buffer) response was subtracted to give a corrected response for each test sample. Due to random measurement errors, the corrected responses of some samples would be less than zero. The second set of 77 samples was analyzed by a similar method, except that anti-histidine antibody from Dianova (cat no DIA-910-200-ABF) was used as the capture reagent and the his-tagged receptors were injected for only 1 min instead of 2 min.
Binding to immobilized fc receptors by affinity chromatography
Binding to FcγRIIIa was measured by affinity chromatography13 using a column of TSKgel FcγR-IIIA-NPR, 4.6 × 75 mm, 5 μm particle size (Tosoh cat. no. 0023513). This uses a non-glycosylated recombinant version of FcγRIIIa, 158 V allele. Unlike native, glycosylated FcγRIIIa, it shows significant binding to human IgG4. HPLC mobile phases consisted of 50 mM ammonium acetate pH 6.0 (A) and 50 mM ammonium acetate pH 4.0 (B). 25 μg of protein (1 mg/mL) was loaded and eluted at 300 μL/min as follows: 0% B for 2 min followed by a linear gradient from 0–30% B over 18 min, then a step to 100% B for 5 min, and finally to 0% B for 5 min to recondition the column. Wild-type trastuzumab (Herceptin) was used as a reference standard to define different integration areas and for quality control. Three major glycoforms could be distinguished, corresponding to low, mild and high affinity, with additional small pre-peaks and post-peaks. To assess the relative binding of test samples, a weighted mean elution time was calculated as the mean of the individual elution times multiplied by the corresponding fraction of the total peak area. The results were normalized to the unretained elution time (2 min = 0%) and the mean elution time of wild-type IgG1 (15.5 min = 100%).
Fc effector cell bioassays
Antibodies were assessed for their ability to engage in ADCP or ADCC using Promega Fc effector bioassay systems as previously described.11 The assay kit contained CD20+ Raji target cells (Promega cat. no. G7016) and engineered Jurkat effector cells which stably express the desired Fc receptor and an NFAT response element to drive expression of firefly luciferase. Experiments were carried out according to the manufacturer’s instructions using the following effector cells:
| Receptor | Species | Supplier | Catalogue number |
| FcγRI (CD64) | human | Promega | GA133A |
| FcγRIIa (CD32a) 131 H allele | human | Promega | G988A |
| FcγRIIa (CD32a) 131 R allele | human | Promega | CS1781B11 |
| FcγRIIb (CD32b) | human | Promega | CS1781E01 |
| FcγRIIIa (CD16a) 158F allele | human | Promega | G979A |
| FcγRIIIa (CD16a) 158 V allele | human | Promega | G701A |
Target cells, effector cells and sample dilutions were all prepared in RPMI1640 culture medium containing 4% low IgG bovine serum. Samples were diluted to 30 μg/mL in an off-line plate and 25 µL was transferred to a white flat-bottomed assay plate. 25 µL of target cell suspension was added and mixed on a plate shaker. 25 µL of effector cell suspension was added and mixed and the plate was incubated at 37°C in 5% CO2 for 6 h. 50 μL of luciferase assay substrate was added and luminescence was measured using a Glomax 96 luminometer (Promega). The results were normalized by dividing by the response of the wild-type IgG1 control.
Antibodies were also tested for ADCP and ADCC using the iLite reporter system of Svar Life Science (Malmo, Sweden). Experiments were carried out generally in accordance with the manufacturer’s instructions. Cells and sample dilutions were all prepared in RPMI1640 culture medium containing 4% low IgG bovine serum. Samples were diluted to 30 μg/mL in an off-line plate and 40 µL was transferred to a white flat-bottomed assay plate. iLite ADCP effector cells (cat. no. BM5004) or ADCC effector cells (cat. no. BM5001) and iLite CD20+ target cells (cat. no. BM5010) were thawed and mixed; 40 µL of cell suspension was added to each well and mixed very gently by tapping the plates. The plates were incubated at 37°C in 5% CO2 for 4 h. DualGlo substrate (Promega cat no E2920) was prepared according to the manufacturer’s instructions. 25 μL/well of firefly substrate was added and luminescence was measured after 10 min in the Glomax 96 luminometer. Then 25 μL/well of renilla substrate was added and luminescence was measured after 10 min. (Note, the substrate volumes were less than manufacturer’s recommendation, but still gave sufficient signals.) The results were normalized by dividing the firefly response by the renilla response then by dividing by the response of the IgG1 wild-type control.
Binding to C1q
Antibody binding to human C1q was measured by ELISA as previously described.11 Human C1q (Sigma Cat. No. 20476) was coupled to horseradish peroxidase (HRP) using Lightning-Link® HRP Conjugation Kit (AbCam Cat No ab102890). ELISA was carried out with shaking at ambient temperature. Samples were diluted to 10 µg/mL in phosphate-buffered saline (PBS). Negative controls consisted of PBS alone. Replicates of 100 µL each were added to 96-well microplates (Corning Cat. No. 9018) and incubated for 90 min. The samples were flicked out and 200 µL PBS containing 1% casein (block buffer) was added. The plates were incubated overnight at 4°C, then flicked out and 100 µL HRP-labeled C1q at 0.5 μg/mL in block buffer was added. The plates were incubated for 90 min then washed four times with PBS containing 0.05% (v/v) Tween 20 (wash buffer) and twice with water. 100 µL of 3,3’,5,5’-tetramethylbenzidine liquid substrate, supersensitive (TMB) (Sigma Cat. No. T4444) was added and the plates were incubated for 10 min. 50 µL of 1 M sulfuric acid was added to stop the reaction and the absorbance was read at 450 nm with 620 nm subtraction using a microplate spectrophotometer (Anthos Labtec HT). The whole experiment was carried out twice.
Complement-mediated cytotoxicity
Antibodies were tested for CDC using the iLite reporter system of Svar Life Science (Malmo, Sweden). Experiments were carried out in accordance with the manufacturer’s instructions. Low IgG FBS (Promega cat no G7110) was heat-inactivated by incubation at 56°C for 40 min. Dilution medium was prepared by adding the heat-inactivated FBS to RPMI culture medium (Promega cat no G708A) to give a final concentration of 9%. Samples were diluted to 100 μg/mL and 20 μL was added to white flat-bottomed microplates, followed by 20 μL of target cell suspension (iLite CD20+ Svar Luc cat. no. BM5028) and 10 μL of 25% human serum (Svar cat. no. 5980). The plates were incubated at 37°C and 5% CO2 for 4 h. 50 μL of Quanti-Luc 4 substrate (Invivogen cat. no. rep-qlc4lg1) was added and luminescence was read after 10 min. The responses were normalized with reference to the response of the wild-type IgG1 control.
Thermal stability by differential scanning fluorimetry
Antibodies were tested using the SUPR-DSF system (Protein Stable Ltd, Leatherhead, UK) in accordance with the manufacturer’s instructions. 10 μL of each test sample at 1 mg/mL in PBS was pipetted in triplicate into a black 384-well microplate (BioRad cat. no. HSP 3866). The plate was sealed using pressure-activated adhesive qPCR seals (Azenta Cat No 4ti-0560). The plate was placed in the SUPR-DSF and subjected to a thermal ramp from 20°C to 105°C at 1°C/min with a 25 ms integration time (these conditions had been optimized in a preliminary run with control samples). The fluorescence data were analyzed using the Protein Stable software, and corroborated using Python, to determine the first onset of melting (Ton) and the midpoint of each melting phase (Tm). The software default setting for the Barycentric Mean (BCM) is to use the wavelength range from 310 nm to 390 nm. However, this may not be ideal for all types of protein. To explore the parameter space, various ranges were tested and the range 310 nm to 350 nm was found to be optimal. Regardless of wavelength range, different samples exhibited different numbers of transitions. A Savitzky-Golay filter was used to determine the first derivative of the BCM melt curves. The derivative data was then modeled by a number of Gaussian curves by non-linear regression. This enabled the calculation of Ton values as the temperature where the normalized cumulative frequency of the first Gaussian curve reached 1%. The mean fitting error for Ton was 0.4°C and for Tm was 0.2°C.
Abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ADCP
antibody-dependent cell-mediated phagocytosis
- CDC
complement-dependent cytotoxicity
- CHO
Chinese hamster ovary
- DSF
differential scanning fluorimetry
- ELISA
enzyme-linked immunosorbent assay
- Fab
fragment antigen binding
- Fc
fragment crystallizable
- FcγR
Fc gamma receptor
- FcRn
neonatal Fc receptor
- HEK
human embryonic kidney
- HPLC
high pressure liquid chromatography
- NFAT
nuclear factor of activated T-cells
- HBS-EP+
hepes-buffered saline containing EDTA and P20 detergent
- IMGT
International ImMunoGeneTics information system
- INN
International nonproprietary name
- SPR
surface plasmon resonance
- WHO
World Health Organization
Supplementary Material
Acknowledgments
We acknowledge the support of Ullrich Mayer, Christophe Lallemand and Lone Frier Bovin at Svar Life Science who contributed reagents and advice to this study.
Disclosure statement
This research was sponsored by mAbsolve Limited. mAbsolve has a proprietary interest in STR mutations and licenses them to the biotech and pharmaceutical industry. Geoff Hale and Ian Wilkinson have financial interests in mAbsolve.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/19420862.2024.2402701
References
- 1.Wilkinson I, Hale G.. Systematic analysis of the varied designs of 819 therapeutic antibodies and Fc fusion proteins assigned international non-proprietary names. mAbs. 2022;14(1):2123299. doi: 10.1080/19420862.2022.2123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolt S, Routledge E, Lloyd I, Chatenoud L, Pope H, Gorman SD, Clark M, Waldmann H. The generation of a humanised, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol. 1993;23(2):403–15. doi: 10.1002/eji.1830230216. [DOI] [PubMed] [Google Scholar]
- 3.Lund J, Winter G, Jones PT, Pound JD, Tanaka T, Walker MR, Artymiuk PJ, Arata Y, Burton DR, Jefferis R. et al. Human fc gamma RI and Fc gamma RII interact with distinct but overlapping sites on human IgG. J Immunol. 1991;147(8):2657–2662. doi: 10.4049/jimmunol.147.8.2657. [DOI] [PubMed] [Google Scholar]
- 4.Strohl WR. Current progress in innovative engineered antibodies. Protein Cell. 2018;9(1):86–120. doi: 10.1007/s13238-017-0457-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang XM, Brezski M, Brezski RJ. IgG fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi: 10.1007/s13238-017-0473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abdeldaim DT, Schindowski K. Fc-engineered Therapeutic antibodies: recent advances and future directions. Pharmaceutics. 2023;15(10):2402. doi: 10.3390/pharmaceutics15102402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Damelang T, Brinkhaus M, van Oschl Tlj, Schuurman J, Labrijn AF, Rispens T, Vidarsson G, van Osch TLJ. Impact of structural modifications of IgG antibodies on effector functions. Front Immunol. 2023;14:1304365. doi: 10.3389/fimmu.2023.1304365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delidakis G, Kim JE, George K, Georgiou G. Improving antibody therapeutics by manipulating the fc domain: immunological and structural considerations. Annu Rev Biomed Eng. 24:249–274. doi: 10.1146/annurev-bioeng-082721-024500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefranc M-O, Lefranc G. IMGT® nomenclature of engineered IGHG variants involved in antibody effector properties and formats. Antibodies. 2022;11(4):65. doi: 10.3390/antib11040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson I, Anderson S, Fry J, Julien LA, Neville D, Qureshi O, Watts G, Hale G, Karagiannis SN. Fc-engineered antibodies with immune effector functions completely abolished. PLOS ONE. 2021;16(12):e0260954. doi: 10.1371/journal.pone.0260954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiyoshi M, Caaveiro JMM, Tada M, Tamura H, Tanaka T, Terao Y, Morante K, Harazono A, Hashii N, Shibata H. et al. Assessing the heterogeneity of the Fc-glycan of a therapeutic antibody using an engineered FcγReceptor IIIa-immobilized column. Sci Rep. 2018;8(1):3955. doi: 10.1038/s41598-018-22199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodall DW, Dillon TM, Kalenian K, Padaki R, Kuhns S, Semin DJ, Bondarenko, Bondarenko PV. Non-targeted characterization of attributes affecting antibody-FcγRIIIa V158 (CD16a) binding via online affinity chromatography-mass spectrometry. mAbs. 2022;14(1):e2004982. doi: 10.1080/19420862.2021.2004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frischauf N, Strasser J, Borg EGF, Labrijn AF, Beurskens FJ, Preiner J. Complement activation by IgG subclasses is governed by their ability to oligomerize upon antigen binding. bioRxiv Prepr. 2024; doi: 10.1101/2024.03.26.586731. [DOI] [Google Scholar]
- 15.Gaston RS, Deierhoi MH, Patterson T, Prasthofer E, Julian BA, Barber WH, Laskow DA, Diethelm AG, Curtis JJ. OKT3 first-dose reaction: association with T cell subsets and cytokine release. Kidney Int. 1991;39(1):141–148. doi: 10.1038/ki.1991.18. [DOI] [PubMed] [Google Scholar]
- 16.Chatenoud L, Ferran C, Bach JF. The anti-CD3-induced syndrome: a consequence of massive in vivo cell activation. In: Fleischer B, and Sjögren H. editors. Superantigens. Current topics in microbiology and immunology. Vol. 174. Berlin, Heidelberg: Springer-Verlag; 1991. p. 121–134. doi: 10.1007/978-3-642-50998-8_9. [DOI] [PubMed] [Google Scholar]
- 17.Bruggemann M, Williams GT, Bindon CI, Clark MR, Walker MR, Jefferis R, Waldmann H, Neuberger MS. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med. 1987;166(5):1351–1361. doi: 10.1084/jem.166.5.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curtin F, Perron H, Kromminga A, Porchet H, Lang AB. Preclinical and early clinical development of GNbAC1, a humanized IgG4 monoclonal antibody targeting endogenous retroviral MRRV-Env protein. mAbs. 2015;7(1):265–275. doi: 10.4161/19420862.2014.985021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salfeld J. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25(12):1369–1372. doi: 10.1038/nbt1207-1369. [DOI] [PubMed] [Google Scholar]
- 20.Greenwood J, Clark M, Waldmann H. Structural motifs involved in human IgG antibody effector functions. Eur J Immunol. 1993;23(5):1098–1104. doi: 10.1002/eji.1830230518. [DOI] [PubMed] [Google Scholar]
- 21.Isaacs JD, Wing MG, Greenwood JD, Hazleman BL, Hale G, Waldmann H. A therapeutic human IgG4 monoclonal antibody that depletes target cells in humans. Clin Exp Immunol. 1996;106(3):427–433. doi: 10.1046/j.1365-2249.1996.d01-876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N. Cytokine storm in a phase I trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355(10):1018–1028. doi: 10.1056/NEJMoa06384. [DOI] [PubMed] [Google Scholar]
- 23.Bartholomaeus P, Semmler LY, Bukur T, Boisguerin V, Romer PS, Tabares P, Chuvpilo S, Tyrsin DY, Matskevich A, Hengel H. et al. Cell contact–dependent priming and fc interaction with CD32+ immune cells contribute to the TGN1412-triggered cytokine response. J Immunol. 2014;192(5):2091–2098. doi: 10.4049/jimmunol.1302461. [DOI] [PubMed] [Google Scholar]
- 24.Xu D, Alegre ML, Varga SS, Rothermel AL, Collins AM, Pulito VL, Hanna LS, Bluestone Ja DKPP, Parren PWHI, Bluestone JA. et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. 2000;200(1):16–26. doi: 10.1006/cimm.2000.1617. [DOI] [PubMed] [Google Scholar]
- 25.Schlothauer T, Herter S, Koller CF, Grau-Richards S, Steinhart V, Spick C, Kubbies M, Klein C, Umaña P, Mössner E. et al. Novel human IgG1 and IgG4 fc-engineered antibodies with completely abolished immune effector functions. Protein Eng Des Selection. 2016;29(10):457–466. doi: 10.1093/protein/gzw040. [DOI] [PubMed] [Google Scholar]
- 26.Hezareh M, Hessell AJ, Jensen RC, van de Winkel Jgj, Parren P, Pwhi JGJ. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J Virol. 2001;75(24):12161–12168. doi: 10.1128/JVI.75.24.12161-12168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tam SH, McCarthy SG, Armstrong A, Somani S, Wu S-J, Liu X, Gervais A, Ernst R, Saro D, Decker R. et al. Functional biophysical and structural characterization of human IgG1 and IgG4 fc variants with ablated immune functionality. Antibodies. 2017;6(3):12. doi: 10.3390/antib6030012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, Scallon B, Teplyakov A, Malia TJ, Strohl WR. An engineered fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65(1):114–126. doi: 10.1016/j.ymeth.2013.06.035. [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Edberg JC, Redacha PB, Bansal V, Guyra PM, Coleman K, Salmon JE, Kimberly RP. A novel polymorphism of FcγRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100:1059–1070. doi: 10.1172/JCI119616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koene HR, Kleijer M, Algra J, Roos D, von Dem Borne Aeg, de Haas M. FcγRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell FcγRIIIa, independently of the FcγRIIIa-48L/R/H phenotype. Blood. 1997;90(3):1109–1114. doi: 10.1182/blood.V90.3.1109. [DOI] [PubMed] [Google Scholar]
- 31.Derebe MG, Nanjunda RK, Gilliland GL, Lacy ER, Chiu ML. Human IgG subclass cross-species reactivity to mouse and cynomolgus monkey Fcγ receptors. Immunol Lett. 2018;197:1–8. doi: 10.1016/j.imlet.2018.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Stapleton NM, Brinkhaus M, Armour KL, Bentiage AEH, de Taeye SW, Temming AR, Mok JY, Brasser G, Maas M, van Esch WJW. et al. Reduced FcRn-mediated transcytosis of IgG2 due to a missing glycine in its lower hinge. Nat Sci Rep. 2019;9(1):7363. doi: 10.1038/s41598-019-40731-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stapleton NM, Armstrong-Fisher SS, Andersen JT, van der Schoot CE, Porter C, Page KR, Falconer D, de Haas M, Williamson LM, Clark MR. et al. Human IgG lacking effector functions demonstrate lower FcRn-binding and reduced transplacental transport. Mol Immunol. 2018;95:1–9. doi: 10.1016/j.molimm.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 34.Dillon TM, Ricci MS, Vesina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S. et al. Structural and functional characterization of disulphide isoforms of the human IgG2 subclass. J Biol Chem. 2008;283(23):16205–16215. doi: 10.1074/jbc.M709988200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu H, May K. Disulfide bond structures of IgG molecules. In: mAbs. 2012;4:17–23. doi: 10.4161/mabs.4.1.18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Namisaki H, Salto S, Hiraishi K, Haba T, Tanaka Y, Yoshida H, Iida S, Takahashi H. R409K mutation prevents acid-induced aggregation of human IgG4. PLOS ONE. 2020;15(3):e0229027. doi: 10.1371/journal.pone.0229027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lund J, Takahashi N, Pound JD, Goodall M, Jeffris R. Multiple interactions of IgG with its core oligosaccharide can modulate recognition by complement and human Fc gamma receptor I and influence the synthesis of its oligosaccharide chains. J Immunol. 1996;157(11):4963–4969. doi: 10.4049/jimmunol.157.11.4963. [DOI] [PubMed] [Google Scholar]
- 38.Pejchal R, Cooper AB, Brown ME, Vasquez M, Krauland EM. Profiling the biophysical developability properties of common IgG1 fc effector silencing variants. Antibodies. 2023;12(3):54. doi: 10.3390/antibod12030054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito T, Tsumoto K. Effects of subclass change on the structural stability of chimeric, humanized and human antibodies under thermal stress. Protein Sci. 2013;22(11):1542–1551. doi: 10.1002/pro.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hale G. Living in LALA land? Forty years of attenuating fc effector functions. Immunol Rev. 2024;0:1–16. doi: 10.1111/imr.13379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest, 5th.; Bethesda (MD): National Institutes of Health; 1991. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.