

Abstract
Background
Common genetic variation at genes that are imprinted and exclusively maternally expressed could explain the apparent maternal-specific inheritance of low birthweight reported in large family pedigrees. We identified ten single nucleotide polymorphisms (SNPs) in H19, and we genotyped three of these SNPs in families from the contemporary ALSPAC UK birth cohort (1,696 children, 822 mothers and 661 fathers) in order to explore associations with size at birth and cord blood IGF-II levels.
Results
Both offspring's and mother's H19 2992C>T SNP genotypes showed associations with offspring birthweight (P = 0.03 to P = 0.003) and mother's genotype was also associated with cord blood IGF-II levels (P = 0.0003 to P = 0.0001). The offspring genotype association with birthweight was independent of mother's genotype (P = 0.01 to P = 0.007). However, mother's untransmitted H19 2992T allele was also associated with larger birthweight (P = 0.04) and higher cord blood IGF-II levels (P = 0.002), suggesting a direct effect of mother's genotype on placental IGF-II expression and fetal growth. The association between mother's untransmitted allele and cord blood IGF-II levels was more apparent in offspring of first pregnancies than subsequent pregnancies (P-interaction = 0.03). Study of the independent Cambridge birth cohort with available DNA in mothers (N = 646) provided additional support for mother's H19 2992 genotype associations with birthweight (P = 0.04) and with mother's glucose levels (P = 0.01) in first pregnancies.
Conclusion
The common H19 2992T allele, in the mother or offspring or both, may confer reduced fetal growth restraint, as indicated by associations with larger offspring birth size, higher cord blood IGF-II levels, and lower compensatory early postnatal catch-up weight gain, that are more evident among mother's smaller first-born infants.
Background
The maternal-uterine environment has a major influence on fetal growth and size at birth [1]. Early cross-breeding experiments in animals demonstrated that fetal growth could be restrained or enhanced from its genetic potential, according to the size of the mother [2,3]. In a normal human birth cohort study, we demonstrated that over 50% of all offspring may have experienced clinically significant restraint or enhancement of growth in utero, followed by compensatory catch-up or catch-down growth respectively during the first 2 years of postnatal life [4].
Birth order is an important common determinant of maternal-uterine restraint, as first-born infants are more likely to be restrained in utero and show compensatory postnatal catch-up growth, compared to subsequent-born infants [5]. Heritable or genetic factors may also contribute to degree of maternal restraint of fetal growth; from studies of birthweights in extended human families it has been suggested that low birthweight may be maternally transmitted [6]. We previously reported association between thinness at birth and the common 16189 variant in mitochondrial DNA, which is maternally transmitted [7]. Another possible mechanism to explain maternal inheritance of fetal growth restraint is common variation in genes that are imprinted and exclusively maternally expressed.
Many of the genes known to be imprinted and paternally or maternally expressed influence fetal growth [8]. An imprinted region on chromosome 11p15.5 in humans regulates the major fetal growth factor "insulin-like growth factor-II" (IGF-II). The IGF2 gene is imprinted and paternally expressed in both the mouse and human fetus [9,10]. The mechanisms of imprinting and expression of Igf2 have been thoroughly studied in the mouse. The gene is regulated by DNA elements close to Igf2, such as silencers [11] and activators [12], and particularly by enhancers located distal to the neighbouring maternally expressed gene H19 [13,14]. Access to these enhancers is restricted by an epigenetically controlled insulator upstream of H19 [15-18]. Finally, the maternally expressed H19 gene itself does not code for a protein, but the RNA has growth suppressing functions [19], potentially through inhibiting translation of IGF2 RNA [20].
We therefore aimed to demonstrate whether common genetic variation in human H19 may be associated with fetal growth restraint in a large representative birth cohort. We identified a common H19 2992 C>T SNP that was associated with offspring birth size, cord blood IGF-II levels, and infancy catch-up weight gain. These associations were more evident among first-born infants, and we propose that the common H19 2992T allele may confer reduced fetal growth restraint.
Results
Of the ten SNPs that we identified in the H19 region, we selected three SNPs (2992 C/T; 1737 A/G; and 3238 A/G) that marked different common H19 haplotypes for genotyping in ALSPAC offspring DNA samples for genetic association studies.
Associations with offspring H19 genotypes
Offspring H19 2992 SNP genotypes showed significant associations with size at birth (Table 1). However, neither H19 1737 nor 3231 genotypes were associated with birth size (data not shown).
Table 1.
Birth size and cord IGF-II levels by offspring H19 2992 genotype (ALSPAC cohort)
| CC | CT | TT | PA | PD | |
| (n = 1075) | (n = 518) | (n = 54) | |||
| Weight (gm) | 3468 ± 488 | 3522 ± 486 | 3524 ± 587 | 0.03 | 0.02 |
| Length (cm) | 50.7 ± 2.0 | 50.9 ± 2.0 | 50.9 ± 2.1 | 0.08 | 0.06 |
| Head circ. (cm) | 34.9 ± 1.2 | 34.9 ± 1.3 | 35.0 ± 1.6 | 0.3 | 0.4 |
| (n = 226) | (n = 110) | (n = 13) | |||
| Cord IGF-II (ng/dl) | 275 ± 78 | 277 ± 79 | 337 ± 91 | 0.1 | 0.3 |
Mean ± SD, adjusted for sex, gestation and parity.
PA = P value for additive genetic model (per T allele)
PD = P value for dominant genetic model (CC versus T/*)
H19 2992, 1737 and 3231 genotypes were in significant linkage disequilibrium, and haplotype analysis revealed only 7 genotype combinations with frequency >1%. Only those two haplotypes that included a 2992T allele had birthweights that were above the average [see Additional file 1].
Associations with mother's H19 2992 genotype
The H19 2992 SNP was subsequently genotyped in DNA samples collected from mothers and fathers, for which the family relationships had been validated by genotyping. H19 2992 genotype in mothers, but not in fathers, showed associations with size at birth, and also with cord blood IGF-II levels (Table 2). Mother's H19 2992 genotype associations were independent of mother's pre-pregnancy weight and height (additive models: birthweight: P = 0.001; cord IGF-II levels: P = 0.0005). The association between mother's H19 2992 genotype and IGF-II levels appeared to vary with mother's parity (birth order), being evident only in first pregnancies (Figure 1), although formal test for interaction did not reach significance (P = 0.06). When both mother's and offspring's genotypes were included in a multivariate analysis, birthweight was significantly associated with both mother's H19 2992 genotype (P < 0.05) and also offspring's H19 2992 genotype (P = 0.01), suggesting that there were separate effects of mother's and offspring's genotype on birthweight.
Table 2.
Birth size by mothers or fathers H19 2992 genotype (ALSPAC cohort)
| Mothers genotype | CC | CT | TT | PA | PD |
| (n = 550) | (n = 242) | (n = 30) | |||
| Weight (gm) | 3457 ± 474 | 3543 ± 508 | 3595 ± 485 | 0.003 | 0.003 |
| Length (cm) | 50.7 ± 2.0 | 51.0 ± 1.9 | 51.1 ± 1.8 | 0.04 | 0.04 |
| Head circ. (cm) | 34.8 ± 1.2 | 35.1 ± 1.3 | 34.9 ± 1.4 | 0.07 | 0.03 |
| (n = 164) | (n = 61) | (n = 9) | |||
| Cord IGF-II (ng/dl) | 266 ± 72 | 312 ± 99 | 312 ± 92 | 0.0003 | 0.0001 |
| Fathers genotype | CC | CT | TT | PA | PD |
| (n = 442) | (n = 201) | (n = 18) | |||
| Weight (gm) | 3492 ± 491 | 3474 ± 472 | 3537 ± 519 | 0.9 | 0.7 |
| Length (cm) | 50.8 ± 1.9 | 50.8 ± 1.9 | 51.1 ± 1.7 | 0.8 | 0.9 |
| Head circ. (cm) | 34.9 ± 1.2 | 34.9 ± 1.2 | 35.0 ± 1.2 | 0.9 | 0.7 |
| (n = 76) | (n = 37) | (n = 3) | |||
| Cord IGF-II (ng/dl) | 270 ± 75 | 290 ± 93 | 245 ± 96 | 0.5 | 1.0 |
Mean ± SD, adjusted for sex, gestation and parity.
PA = P value for additive genetic model (per T allele)
PD = P value for dominant genetic model (CC versus T/*)
Figure 1.
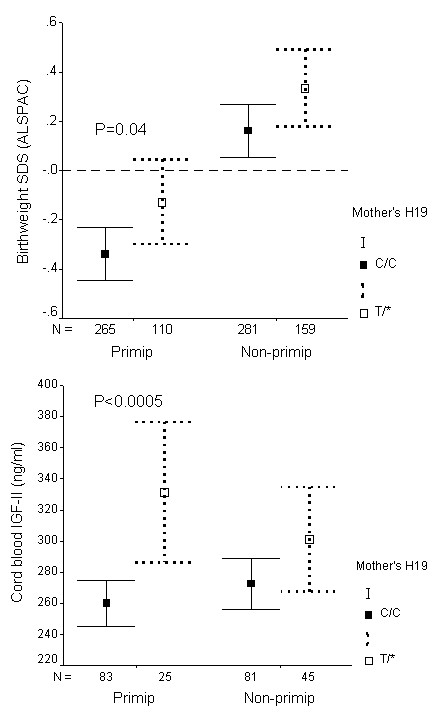
Birthweight SD score (A) and cord blood IGF-II levels at birth (B) in the ALSPAC cohort, by mother's H19 2992 genotype, and stratified by birth order ("Primip" = mother's first child; "Non-primip" = second or subsequent child). Mean ± 95% CI. First-born infants had lower birthweights than infants of subsequent pregnancies (Ref. 5). Associations with mother's genotype (CC vs. T* [CT or TT]) were only seen in first pregnancies.
Mother's untransmitted H19 2992 allele effects
Effects of mother's genotype, independent of transmission to the offspring, were examined by looking at association with mother's H19 2992 CC versus CT genotype within CC genotype offspring. Mother's untransmitted T versus C allele was associated with larger birthweight (P = 0.04) and higher cord blood IGF-II levels (P = 0.002; Table 3). As suggested by the overall associations with mother's genotype (in Figure 1), the effect of mother's untransmitted allele on IGF-II levels was clearer in first pregnancies (P = 0.0002) than in subsequent pregnancies (P = 0.36; P-interaction between mother's untransmitted allele and birth order = 0.03).
Table 3.
Birthweight and IGF-II levels by maternal H19 2992 genotype, within CC genotype offspring (i.e. effect of mother's untransmitted allele), (ALSPAC cohort)
| Mother's Genotype | ANOVA | ||
| CC | CT | ||
| Birthweight (gm) | 3431 ± 474 | 3533 ± 505 | P = 0.04 |
| (n = 407) | (n = 96) | ||
| Cord IGF-II (ng/dl) | 264 ± 72 | 320 ± 108 | P = 0.002 |
| (n = 124) | (n = 22) | ||
Mean ± SD, adjusted for sex, gestation and parity.
Those results indicate that the H19 2992 SNP associations may depend on mother's genotype. However, offspring H19 2992 SNP associations were also seen independent of mother's genotype; among CC genotype mothers, offspring CC vs. CT genotype was also associated with birthweight (P = 0.007, Table 4).
Table 4.
Birthweight and IGF-II levels by offspring H19 2992 genotype, within CC genotype mother's (i.e. effect of fetal genotype), (ALSPAC cohort)
| Offspring Genotype | ANOVA | ||
| CC | CT | ||
| Birthweight (gm) | 3425 ± 490 | 3535 ± 495 | P = 0.007 |
| (n = 408) | (n = 126) | ||
| Cord IGF-II (ng/dl) | 262 ± 80 | 266 ± 98 | P = 0.8 |
| (n = 125) | (n = 36) | ||
Mean ± SD, adjusted for sex, gestation and parity.
H19 2992 allele transmission and parent-of-origin effects
Among the validated parent-offspring trios, comparison of allele transmission from heterozygous parents to their offspring showed that T versus C allele transmission was not associated with birthweight (P = 0.22, number of observations = 204), nor with cord blood IGF-II levels (P = 0.52, number of observations = 38). Nor was there any difference between paternal and maternal T allele transmission (parent-of-origin effects) on birthweight (P = 0.92, number of observations = 87), or on IGF-II levels (P = 0.81, number of observations = 15). Data on mother's parents' genotypes were unavailable.
Genotype associations with postnatal weight gain
1,449 ALSPAC children were followed-up to age 3 years. Neither offspring nor parental H19 genotypes showed association with offspring body size at age 3 years (not shown). The apparent loss of T allele-related size advantage between birth and age 3 years was explained by slower rates of weight gain during this period (Table 5). As with birthweight and IGF-II levels, the H19 2992 genotype associations with postnatal weight gain were more apparent in offspring of first pregnancies (Figure 2).
Table 5.
Size at 3 years, and growth from birth, by offspring H19 2992 genotype, (ALSPAC cohort)
| Offspring genotype | CC | CT | TT | PA | PD |
| (n = 946) | (n = 462) | (n = 41) | |||
| Weight at 3 y (kg) | 15.0 ± 1.7 | 14.9 ± 1.7 | 14.8 ± 1.7 | 0.4 | 0.4 |
| Height at 3 y (cm) | 95.6 ± 3.6 | 95.3 ± 3.5 | 95.2 ± 3.7 | 0.2 | 0.2 |
| Change in Weight SDS 0–3 y | 0.01 ± 1.17 | -0.13 ± 1.07 | -0.10 ± 1.29 | 0.01 | 0.02 |
| Change in Height SDS 0–3 y | 0.06 ± 1.12 | -0.10 ± 1.02 | -0.17 ± 1.26 | 0.01 | 0.01 |
Mean ± SD, adjusted for sex and age
PA = P value for additive genetic model (per T allele)
PD = P value for dominant genetic model (CC versus T/*)
Figure 2.
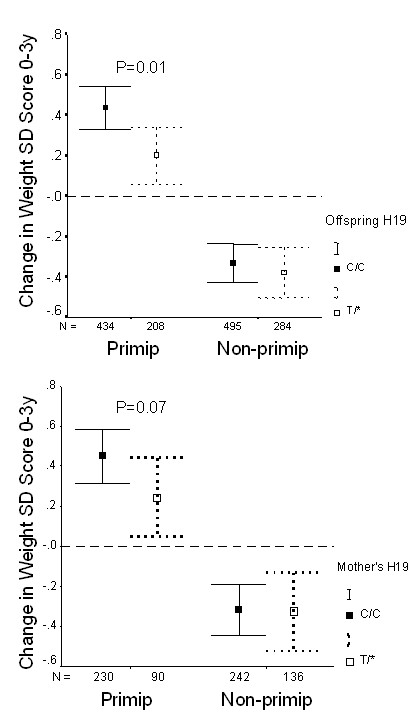
Postnatal weight gain (change in weight SD score 0–3 years) in the ALSPAC cohort, by offspring's (A) or mother's (B) H19 2992 genotype, and stratified by birth order ("Primip" = mother's first child; "Non-primip" = second or subsequent child). Mean ± 95% CI. The overall association between weight gain and offspring genotype (CC vs. T* [CT or TT], P = 0.01; Table 5) was only apparent in first-born offspring.
Replication of mother's H19 2992 genotype associations in the Cambridge cohort
In the independent Cambridge birth cohort (n = 646 births analysed), we obtained additional support for the association between mother's H19 2992T allele and larger birthweight in offspring of first pregnancies (P = 0.04, Figure 3A). Mother's glucose levels one hour post oral glucose at 27 to 32 weeks gestation (mean ± SD: 5.5 ± 1.2 mmol/l) were positively related to offspring birthweight (r = 0.14, P = 0.01), and mother's H19 2992 T/* genotype was also associated with higher mother's glucose levels during first pregnancies (P = 0.01, Figure 3B).
Figure 3.
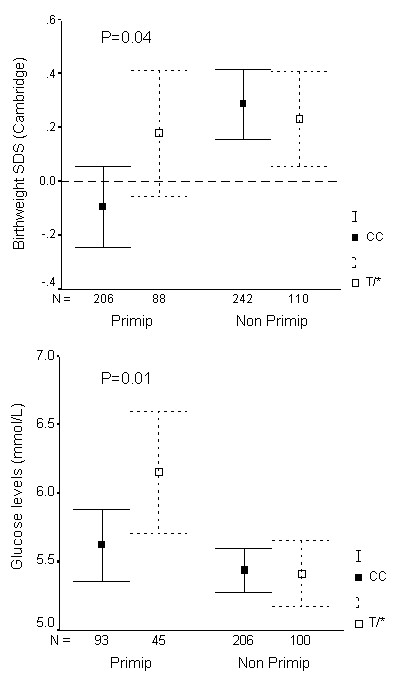
Birthweight SD score (A) and mother's glucose levels one hour post-oral glucose load at 27 to 32 weeks gestation (B) in the Cambridge cohort, by mother's H19 2992 genotype, and stratified by birth order ("Primip" = mother's first child; "Non-primip" = second or subsequent child). Mean ± 95% CI. Associations with mother's genotype (CC vs. T* [CT or TT]) were only seen in first pregnancies.
Discussion
In two separate contemporary birth cohorts we have identified associations between a common H19 2992 genotype and birthweight. This SNP is located in H19 exon 5 [see Additional file 2], however its effect on mRNA structure or function is unknown and H19 has no protein product. An insulator upstream of H19 [15-18] and enhancers downstream [13,14] regulate imprinting of IGF2, which encodes a major fetal growth factor [21]. In addition H19 RNA may suppress IGF2 expression in trans [20] and has tumour suppressor functions in cell transfection studies [19]. Consistent with the birthweight association, H19 2992 genotype was also associated with cord blood IGF-II protein levels at birth.
Birthweight associations were found with both mother's and offspring's H19 2992 genotypes, and it is not clear whether our findings represent direct effects of mother's genotype, offspring genotype, or both. As in other population studies, the low number of available DNA samples and genotypes from fathers contributed to relatively lower statistical power to formally detect effects of H19 2992 allele transmission. However, we did observe significant birthweight and cord blood IGF-II associations with mother's untransmitted allele, suggesting direct effects of mother's genotype. While in mice H19 and IGF2 are not expressed postnatally [22], in humans IGF2 expression continues into adulthood [23]. In most adult tissues IGF2 expression remains monoallelic as in the fetus, however biallelic expression is observed in adult human liver [23]. It is possible that mother's H19 2992 genotype may regulate IGF2 expression in maternally-derived placental tissues. Alternatively, H19 regulation of maternal IGF2 expression could influence mother's glucose metabolism [24] and thereby influence placental glucose transfer and fetal growth [25]. Offspring H19 2992 genotype was associated with birthweight, but not with IGF-II levels. It is possible that this discrepancy could relate to reduced power due to the smaller number of IGF-II samples available, or alternatively it could be possibly explained by a non-IGF-II mediated effect of H19 on birthweight. The association between birthweight and IGF-II levels in cord blood at birth is weak, and it is possible that the effects of IGF-II on fetal growth may be specific to certain tissues or developmental stages in fetal life [26].
The H19 2992 genotype associations with birthweight and IGF-II were independent of mother's body size and were more apparent in mother's first pregnancies. First-born offspring are more growth-restrained in utero than subsequent children, as evident by smaller birth size and compensatory rapid postnatal weight gain within the first 2 to 3 years [5]. The mechanism of growth restraint seen in first pregnancies is unknown, but could reflect a maternally inherited genetic trait with subsequent relaxation of restraint in later pregnancies [27]. We propose that the common H19 2992T allele, either in the mother or offspring, confers reduced fetal growth restraint particularly among first-born infants as indicated by larger birth size and less postnatal compensatory catch-up weight gain.
In subsequent pregnancies and in larger babies, a mendelian pattern of inheritance of birthweight is observed [27]. It was in these second and subsequent pregnancies that we saw association between size at birth and common allele class at the insulin gene minisatellite (INS VNTR) [28], which also regulates IGF2 expression [29]. Offspring INS VNTR class III alleles were associated with larger head size and higher IGF-II levels at birth, and this effect was masked in growth-restricted first pregnancies [28]. Thus, there may be important interaction between maternal parity and imprinted and non-imprinted genes involved in the regulation of IGF-II expression and fetal growth.
Altogether we identified ten SNPs in H19. Given that there is no confirmed function for the H19 transcript in vivo [30], it is difficult to assess the effect of the H19 2992 SNP. It is possible that this variant: (a) may alter the RNA itself, and might influence the putative growth suppressing function of the H19 RNA; (b) may influence levels of H19 transcripts, leading to an effect as above; (c) may be in linkage with an unidentified regulatory element in H19 acting on IGF2 in cis; (d) may be in linkage with variants in known cis regulatory elements for IGF2, such as the insulator or the enhancers. Further work, including systematic genotyping of other SNPs in the extended region and functional studies, will be required to identify the causal variant(s) and, hence, possible mechanisms by which they may influence birthweight and cord blood IGF-II levels. The identification of separate effects of H19 on fetal growth, by acting both within the mother and also within the fetus, could be confirmed by studying a larger population to provide greater power to detect an interaction between mother's and offspring's genotypes, and by exploration of grand-parental genotype and allele transmission to the mother. Intriguingly, while we confirmed that the offspring 2992 genotype (CC vs. CT) association with birthweight was independent of mother's genotype (within CC mothers; Table 4), that finding also raises the surprising suggestion of a paternal H19 allele transmission effect, however, we did not detect any formal association with paternal transmission. An alternative explanation would be that the H19 2992 is in LD with another paternally transmitted determinant of birthweight. Further studies with larger numbers of complete informative trios will be needed to clarify this issue.
Conclusion
In conclusion, the common H19 2992T allele, in the mother or offspring or both, may confer reduced fetal growth restraint, as indicated by associations with larger offspring birth size, higher cord blood IGF-II levels, and lower compensatory early postnatal catch-up weight gain. These observations may have implications for the early origins of adult disease hypothesis [31]. Following the original observations of association between small size at birth and increased later risks for cardiovascular disease and diabetes [32], recent studies have highlighted that these risks are strongest in subjects with the combination of fetal growth restraint followed by rapid postnatal catch-up growth [33]. Common factors that lead to fetal growth restraint in humans include mother's smoking and 1st pregnancies [5]. Common variants in exclusively maternally-expressed genes (such as H19) or maternally transmitted genes (such as in mitochondrial DNA) [7] could also contribute to fetal growth restraint and compensatory postnatal catch-up growth.
Methods
Subjects
The Avon Longitudinal Study of Parents and Children (ALSPAC) birth cohort numbers 14,185 children comprising over 80% of all births in three Bristol based health authorities between April 1991 and December 1992 [34]. Our studies are confined to a 10% sub-cohort ("Children in Focus") [4] and a second (control) cohort for which there were detailed data on size at birth and subsequent childhood growth. In these families, offspring DNA was extracted from cord blood samples collected at birth, mothers DNA from venous blood samples in pregnancy, and fathers DNA from mouthswab samples. H19 genotypes were analysed in available DNA samples from 1,696 children, 822 mothers and 661 fathers. Cord blood samples were collected at birth in 338 of these infants and assayed for IGF-II levels as previously reported [26]; these infants had larger birthweight (mean = 3546 g) than infants for whom cord blood was unavailable (3467 g), but were no different in H19 +2992 genotype (chi-square: P = 0.7)
The Cambridge (Wellbeing) birth cohort was studied to provide independent confirmation of mother's H19 genotype associations. This cohort was recruited from full-term, singleton deliveries at the Rosie Maternity Unit, Cambridge, 1999 to 2000. Routinely collected clinical data were available on offspring birthweight and mother's whole blood glucose levels measured at one hour following a standard dose oral glucose load at 27 to 32 weeks of gestation. Mother's were selected of full-term, singleton infants. Exclusion criteria were mother's hypertension and treatment for diabetes during pregnancy. We sought permission from the mother's General Practitioner to approach the mother to collect her DNA sample by postal mouthswab kits and questionnaire data on her other children. Mother's DNA was extracted and genotyped for the H19 2992 genotype only. Local ethical committee approval and signed informed consent from mothers were obtained.
SNP identification
In an independent cohort of 20 subjects, ten SNPs were identified in the H19 region using denaturing high performance liquid chromatography (Wave™) and were characterised by sequencing of PCR products (SNPs submitted to dbSNP). Confirmation and assessment of allele frequency was obtained by genotyping a second independent cohort of 100–200 subjects. We selected three H19 SNPs as these SNPs marked different haplotypes (2992 C/T [rs217727], 1737 A/G [rs2067051], and 3238 A/G [rs2839703]; the SNP numbers relate to their position in accession number M32053). We genotyped these three SNPs in offspring DNA samples from the ALSPAC cohort for genetic association studies. The H19 2992 SNP was subsequently genotyped in DNA samples from ALSPAC parents, and from mothers in the Cambridge cohort.
H19 2992 C/T genotyping
Genomic DNA (40 ng) was amplified in a reaction mixture containing 1x ammonium reaction buffer, 50 μM each dNTP, 500 μM magnesium, 225 ng each primer (forward: 5'-aaagacaccatcggaacagc-3', reverse: 5'-agcttccagact aggcgagg-3'), 10% (v/v) dimethylsulphoxide and 0.6 units DNA polymerase (Bioline, London, UK) in a 45 μL reaction volume. After an initial 5 min. incubation at 94°C, twenty amplification cycles of 94°C (65 sec.), 58°C (65 sec., dropping 0.5°C per cycle) and 72°C (140 sec.) were performed. This was followed by 14 cycles of 94°C (65 sec.), 48°C (65 sec.) and 72°C (140 sec.) and a final incubation of 72°C for 10 min. After overnight digestion with Fnu4HI (2 units per reaction; New England Biolabs, Hitchin, UK) at 37°C, PCR products were separated by agarose gel electrophoresis. This gave a 351 bp band for the 'T' allele and a 304 bp band for the 'C' allele. The observed genotype frequencies: CC: 65.3%, CT: 31.5%, TT: 3.3% (Table 1) were in Hardy Weinberg equilibrium (chi-square test: P = 0.99). The H19 2992 SNP was also genotyped in the parents DNA samples.
H19 1737 A/G genotyping
The reaction mix for the H19 1737 genotyping was the same as for the 2992 SNP, with the exception of the oligonucleotide primer sequences (forward: 5'-aaggtgacatcttctcgggg-3'; reverse: 5'-tgagagctcattcactccgc-3'). The amplification conditions were the same as those for the 2992 SNP except that the annealing temperatures were 4°C higher throughout. Overnight digestion with Tsp45I (2 units per reaction; New England Biolabs) produced 474 and 131 bp bands for the 'A' allele and 329, 145 and 131 bp fragments for the 'G' allele. The observed genotype frequencies (AA: 23.1%, AG: 52.6%, GG: 24.4%) were in Hardy Weinberg equilibrium (chi-square test: P = 0.5).
H19 3238 A/G genotyping
The reaction mix for the H19 3238 genotyping was the same as for the 2992 SNP, with the exception of the oligonucleotide primer sequences (forward: 5'-aaagacaccatcggaacagc-3'; reverse: 5'-agcttccagactaggcgagg-3'). Amplification conditions were the same as those for the 2992 SNP except that the annealing temperatures were 4°C higher throughout. Overnight digestion with DdeI (2 units per reaction; New England Biolabs) produced 137 and 44 bp bands and multiple small bands (116, 76, 60 and 26 bp) for the 'A' allele and a 181 bp band along with the same multiple small bands for the 'G' allele. The observed genotype frequencies (AA: 38.0%, AG: 48.5%, GG: 13.5%) were in Hardy Weinberg equilibrium (chi-square test: P = 0.8).
Microsatellite validation of parental DNA samples
Parental DNA samples were validated using amplification of markers D16S539, D7S820, D13S317 and D5S818 (Geneprint Multiplex-GammaSTR kit, Promega, Southampton, UK) on an ABI 3700 instrument as previously described [28]. We had DNA samples from 597 complete and validated parent-offspring trios.
Calculations and statistics
Postnatal weight and length gains were calculated by transforming weight and length data into SD scores allowing for variation relating to (gestational) age and gender, using the formula [SD score = individual value – group mean / group SD] [35], and then calculating changes in weight and length SD score between birth to 3 years.
Associations between offspring or mother's genotype and size at birth were determined by ANOVA. Multivariate analysis (general linear models) was used to identify independent effects of mother's and offspring genotypes. Associations with H19 2992 allele transmission were assessed by TDT using both quantitative and qualitative methods. Parent-of-origin effects were sought using quantitative and qualitative methods, allowing for possible confounding by the effects of non-transmission of mother's alleles [36,37].
Abbreviations
ALSPAC – Avon Longitudinal Study of Parents and Children
IGF – insulin-like growth factor
Primip – primiparous
SNP – single nucleotide polymorphism
Authors' contributions
CP performed genotyping and drafted the manuscript. KO contributed to the study design, and co-ordination, performed statistical analyses and drafted the manuscript. BB performed SNP identification, and contributed to the study design. DW performed Cambridge cohort recruitment, DNA preparation and genotyping. HC performed statistical analyses. SR supervised preparation of ALSPAC DNA samples and validation of genotyping results. MR participated in the design and co-ordination of the ALSPAC study, and supervised DNA management. WR participated in the conception and design of the study. JT supervised SNP identification, genotyping and data analyses. DD was responsible for the study conception, and contributed to the study coordination, analyses and drafting the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Birthweight SD score (adjusted for sex and gestational age) by combination of H19 2992/1737/3238 genotypes (for each SNP, 1 = more common homozygote, 2 = heterozygote, 3 = less common homozygote).
Schematic map of H19 indicating exons, SNPs published on dbSNP and also published SNPs identified by the Juvenile Diabetes Research Foundation/Wellcome Trust Diabetes and Inflammation Laboratory (DIL) T1Dbase http://www.t1dbase.org/cgi-bin/welcome.cgi. H19 2992 is indicated by the labels rs217727 and DIL1049.
{kind=link}
Acknowledgments
Acknowledgements
We are extremely grateful to all the children and parents who took part in the ALSPAC and Cambridge cohorts and to the study teams, nurses and midwives who supported these studies. The whole ALSPAC study team comprises interviewers, computer technicians, laboratory technicians, clerical workers, research scientists, volunteers, managers and also the staff of the ALSPAC research clinics. We also thank Miguel Constancia for helpful comments.
ALSPAC is supported by the Medical Research Council (MRC), the Wellcome Trust, the Department of Health, the Department of the Environment and many others. The Cambridge cohort is supported by Wellbeing, UK. KKO was supported by a MRC Clinical Training Fellowship. HJC, JAT and DBD are supported by the Wellcome Trust and the Juvenile Diabetes Research Foundation. WR is supported by the Biotechnology and Biological Sciences Research Council and the European Union.
Contributor Information
Clive J Petry, Email: cjp1002@mole.bio.cam.ac.uk.
Ken K Ong, Email: ko224@mrc-epid.cam.ac.uk.
Bryan J Barratt, Email: Bryan.Barratt@astrazeneca.com.
Diane Wingate, Email: dw251@cam.ac.uk.
Heather J Cordell, Email: Heather.Cordell@cimr.cam.ac.uk.
Susan M Ring, Email: S.M.Ring@bristol.ac.uk.
Marcus E Pembrey, Email: M.Pembrey@bristol.ac.uk.
The ALSPAC Study Team, Email: B.J.Stowe@bris.ac.uk.
Wolf Reik, Email: Wolf.Reik@bbsrc.ac.uk.
John A Todd, Email: John.Todd@cimr.cam.ac.uk.
David B Dunger, Email: dbd25@cam.ac.uk.
References
- Ounsted M, Ounsted C. Maternal regulation of intrauterine growth. Nature. 1966;111:995–997. doi: 10.1038/212995a0. [DOI] [PubMed] [Google Scholar]
- Walton A, Hammond J. The maternal effects on growth and conformation in Shire horse-Shetland pony crosses. Proceedings of the Royal Society of London. 1938;125:311–335. [Google Scholar]
- Joubert DM, Hammond J. A cross breeding experiment with cattle, with special reference to the maternal effect in South Devon-Dexter crosses. Journal of Agricultural Science. 1958;51:325–341. [Google Scholar]
- Ong KK, Ahmed ML, Emmett PM, Preece MA, Dunger DB, the-ALSPAC-Study-Team Association between postnatal catch-up growth and obesity in childhood: prospective cohort study. BMJ. 2000;320:967–971. doi: 10.1136/bmj.320.7240.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong KK, Preece MA, Emmett PM, Ahmed ML, Dunger DB. Size at birth and early childhood growth in relation to maternal smoking, parity and infant breast-feeding: longitudinal birth cohort study and analysis. Pediatric Research. 2002;52:863–867. doi: 10.1203/01.PDR.0000036602.81878.6D. [DOI] [PubMed] [Google Scholar]
- Ounsted M, Scott A, Ounsted C. Transmission through the female line of a mechanism constraining human fetal growth. Annals of Human Biology. 1986;13:143–151. doi: 10.1080/03014468600008281. [DOI] [PubMed] [Google Scholar]
- Casteels K, Ong KK, Phillips DI, Bednarz A, Bendall H, Woods KA, Sherriff A, ALSPAC_Study_Team. Golding J, Pembrey ME, Poulton J, Dunger DB. Mitochondrial 16189 variant, thinness at birth and type 2 diabetes. Lancet. 1999;353:1499–1500. doi: 10.1016/S0140-6736(98)05817-6. [DOI] [PubMed] [Google Scholar]
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nature Reviews Genetics. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- Giannoukakis N, Deal C, Paquette J, Goodyer CG, Polychronakos C. Parental genomic imprinting of the human IGF2 gene. Nature Genetics. 1993;4:98–101. doi: 10.1038/ng0593-98. [DOI] [PubMed] [Google Scholar]
- Ohlsson R, Nystrom A, Pfeifer-Ohlsson S, Tohonen V, Hedborg F, Schofield P, Flam F, Ekstrom TJ. IGF2 is parentally imprinted during human embryogenesis and in the Beckwith-Wiedemann syndrome. Nature Genetics. 1993;4:94–97. doi: 10.1038/ng0593-94. [DOI] [PubMed] [Google Scholar]
- Constancia M, Dean W, Lopes S, Moore T, Kelsey G, Reik W. Deletion of a silencer element in Igf2 results in loss of imprinting independent of H19. Nature Genetics. 2000;26:203–206. doi: 10.1038/79930. [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Bowden L, Constancia M, Dean W, Kelsey G, Reik W. An intragenic methylated region in the imprinted Igf2 gene augments transcription. EMBO Reports. 2001;2:1101–1106. doi: 10.1093/embo-reports/kve248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton PA, Saam JR, Ingram RS, Stewart CL, Tilghman SM. An enhancer deletion affects both H19 and Igf2 expression. Genes & Development. 1995;9:2079–2089. doi: 10.1101/gad.9.17.2079. [DOI] [PubMed] [Google Scholar]
- Davies K, Bowden L, Smith P, Dean W, Hill D, Furuumi H, Sasaki H, Cattanach B, Reik W. Disruption of mesodermal enhancers for Igf2 in the minute mutant. Development. 2002;129:1657–1668. doi: 10.1242/dev.129.7.1657. [DOI] [PubMed] [Google Scholar]
- Murrell A, Heeson S, Reik W. Interaction between differentially methylated regions partitions the imprinted genes Igf2 and H19 into parent-specific chromatin loops. Nature Genetics. 2004;36:889–893. doi: 10.1038/ng1402. [DOI] [PubMed] [Google Scholar]
- Thorvaldsen JL, Duran KL, Bartolomei MS. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes & Development. 1998;12:3693–3702. doi: 10.1101/gad.12.23.3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–489. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Hao Y, Crenshaw T, Moulton T, Newcomb E, Tycko B. Tumour-suppressor activity of H19 RNA. Nature. 1993;365:764–767. doi: 10.1038/365764a0. [DOI] [PubMed] [Google Scholar]
- Li YM, Franklin G, Cui HM, Svensson K, He XB, Adam G, Ohlsson R, Pfeifer S. The H19 transcript is associated with polysomes and may regulate IGF2 expression in trans. Journal of Biological Chemistry. 1998;273:28247–28252. doi: 10.1074/jbc.273.43.28247. [DOI] [PubMed] [Google Scholar]
- Leighton PA, Ingram RS, Eggenschwiler J, Efstratiadis A, Tilghman SM. Disruption of imprinting caused by deletion of the H19 gene region in mice. Nature. 1995;375:34–39. doi: 10.1038/375034a0. [DOI] [PubMed] [Google Scholar]
- Stylianopoulou F, Efstratiadis A, Herbert J, Pintar J. Pattern of the insulin-like growth factor II gene expression during rat embryogenesis. Development. 1988;103:497–506. doi: 10.1242/dev.103.3.497. [DOI] [PubMed] [Google Scholar]
- Wu HK, Squire JA, Song Q, Weksberg R. Promoter-dependent tissue-specific expressive nature of imprinting gene, insulin-like growth factor II, in human tissues. Biochemical and Biophysical Research Communications. 1997;233:221–226. doi: 10.1006/bbrc.1997.6431. [DOI] [PubMed] [Google Scholar]
- Zapf J. Role of insulin-like growth factor II and IGF binding proteins in extrapancreatic tumor hypoglycemia. Hormone Research. 1994;42:20–26. doi: 10.1159/000184139. [DOI] [PubMed] [Google Scholar]
- Catalano PM, Thomas AJ, Huston LP, Fung CM. Effect of maternal metabolism on fetal growth and body composition. Diabetes Care. 1998;21 Suppl 2:B85–90. [PubMed] [Google Scholar]
- Ong KK, Kratzsch J, Kiess W, the-ALSPAC-Study-Team. Costello M, Scott CD, Dunger DB. Size at birth and cord blood levels of insulin, insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-1 (IGFBP-1), IGFBP-3, and the soluble IGF-II/mannose-6-phosphate receptor in term human infants. Journal of Clinical Endocrinology & Metabolism. 2000;85:4266–4269. doi: 10.1210/jc.85.11.4266. [DOI] [PubMed] [Google Scholar]
- Ounsted M, Scott A, Moar VA. Constrained and unconstrained fetal growth: associations with some biological and pathological factors. Annals of Human Biology. 1988;15:119–129. doi: 10.1080/03014468800009541. [DOI] [PubMed] [Google Scholar]
- Ong KK, Petry CJ, Barratt BJ, Ring S, Cordell HJ, Wingate DL, Pembrey ME, Todd JA, Dunger DB. Maternal-fetal interactions and birth order influence insulin variable number of tandem repeats allele class associations with head size at birth and childhood weight gain. Diabetes. 2004;53:1128–1133. doi: 10.2337/diabetes.53.4.1128. [DOI] [PubMed] [Google Scholar]
- Paquette J, Giannoukakis N, Polychronakos C, Vafiadis P, Deal C. The INS 5' variable number of tandem repeats is associated with IGF2 expression in humans. Journal of Biological Chemistry. 1998;273:14158–14164. doi: 10.1074/jbc.273.23.14158. [DOI] [PubMed] [Google Scholar]
- Jones BK, Levorse JM, Tilghman SM. Igf2 imprinting does not require its own DNA methylation or H19 RNA. Genes & Development. 1998;12:2200–2207. doi: 10.1101/gad.12.14.2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker DJ. Fetal growth and adult disease. B J Obstet Gynaecol. 1992;99:275–276. doi: 10.1111/j.1471-0528.1992.tb13719.x. [DOI] [PubMed] [Google Scholar]
- Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–1022. doi: 10.1136/bmj.303.6809.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ. Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ. 1999;318:427–431. doi: 10.1136/bmj.318.7181.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding J, Pembrey ME, Jones R. ALSPAC--the Avon Longitudinal Study of Parents and Children. I. Study methodology. Paediatric and Perinatal Epidemiology. 2001;15:74–87. doi: 10.1046/j.1365-3016.2001.00325.x. [DOI] [PubMed] [Google Scholar]
- Cole TJ. Do growth chart centiles need a face lift? BMJ. 1994;308:641–642. doi: 10.1136/bmj.308.6929.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker JC, Gharani N, Hindmarsh P, McCarthy MI. Estimation and testing of parent-of-origin effects for quantitative traits. American Journal of Human Genetics. 2003;72:1035–1039. doi: 10.1086/374382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauderman WJ. Candidate gene association analysis for a quantitative trait, using parent-offspring trios. Genetic Epidemiology. 2003;25:327–338. doi: 10.1002/gepi.10262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Birthweight SD score (adjusted for sex and gestational age) by combination of H19 2992/1737/3238 genotypes (for each SNP, 1 = more common homozygote, 2 = heterozygote, 3 = less common homozygote).
Schematic map of H19 indicating exons, SNPs published on dbSNP and also published SNPs identified by the Juvenile Diabetes Research Foundation/Wellcome Trust Diabetes and Inflammation Laboratory (DIL) T1Dbase http://www.t1dbase.org/cgi-bin/welcome.cgi. H19 2992 is indicated by the labels rs217727 and DIL1049.