

Abstract
Background
Increasing attention to liver-bone crosstalk has spurred interest in targeted interventions for various forms of osteoporosis. Liver injury induced by different liver diseases can cause an imbalance in bone metabolism, indicating a novel regulatory paradigm between the liver and bone. However, the role of the liver-bone axis in both primary and secondary osteoporosis remains inadequately elucidated. Therefore, exploring the exact regulatory mechanisms of the liver-bone axis may offer innovative clinical approaches for treating diseases associated with the liver and bone.
Methods
Here, we summarize the latest research on the liver-bone axis by searching the PubMed and Web of Science databases and discuss the possible mechanism of the liver-bone axis in different types of osteoporosis. The literature directly reporting the regulatory role of the liver-bone axis in different types of osteoporosis from the PubMed and Web of Science databases has been included in the discussion of this review (including but not limited to the definition of the liver-bone axis, clinical studies, and basic research). In addition, articles discussing changes in bone metabolism caused by different etiologies of liver injury have also been included in the discussion of this review (including but not limited to clinical studies and basic research).
Results
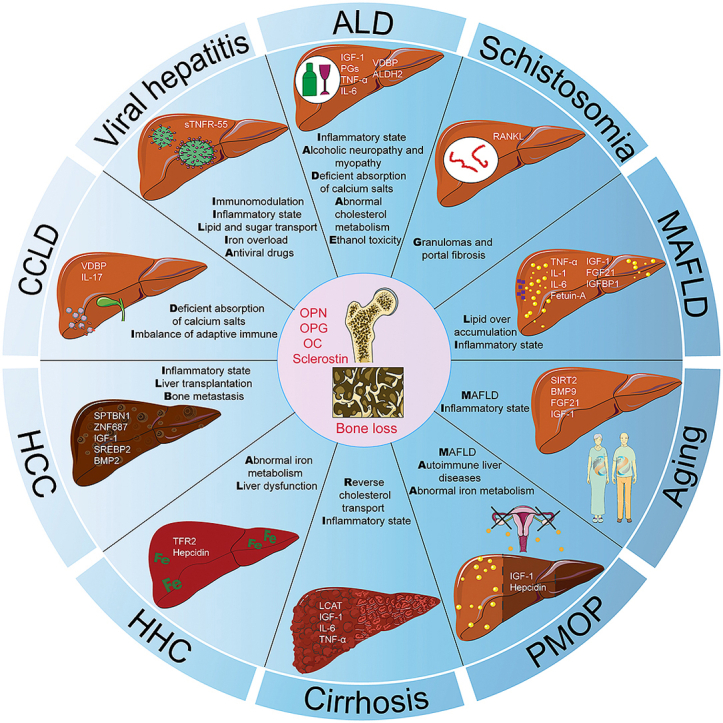
Several endocrine factors (IGF-1, FGF21, hepcidin, vitamin D, osteocalcin, OPN, LCAT, Fetuin-A, PGs, BMP2/9, IL-1/6/17, and TNF-α) and key genes (SIRT2, ABCB4, ALDH2, TFR2, SPTBN1, ZNF687 and SREBP2) might be involved in the regulation of the liver-bone axis. In addition to the classic metabolic pathways involved in inflammation and oxidative stress, iron metabolism, cholesterol metabolism, lipid metabolism and immunometabolism mediated by the liver-bone axis require more research to elucidate the regulatory mechanisms involved in osteoporosis.
Conclusion
During primary and secondary osteoporosis, the liver-bone axis is responsible for liver and bone homeostasis via several hepatokines and osteokines as well as biochemical signaling. Combining multiomics technology and data mining technology could further advance our understanding of the liver-bone axis, providing new clinical strategies for managing liver and bone-related diseases.
The translational potential of this article is as follows: Abnormal metabolism in the liver could seriously affect the metabolic imbalance of bone. This review summarizes the indispensable role of several endocrine factors and biochemical signaling pathways involved in the liver-bone axis and emphasizes the important role of liver metabolic homeostasis in the pathogenesis of osteoporosis, which provides novel potential directions for the prevention, diagnosis, and treatment of liver and bone-related diseases.
Keywords: Chronic liver diseases, Endocrine, Hepatokines, Liver-bone axis, Osteoporosis
Graphical abstract
1. Pathogenesis of osteoporosis
Osteoporosis is defined as a systemic skeletal disorder characterized by a decrease in bone mass and microarchitecture deterioration, with a consequent increase in bone fragility and susceptibility to fracture [1]. Traditionally, osteoporosis is divided into primary and secondary osteoporosis. Primary osteoporosis can be divided into postmenopausal osteoporosis, senile osteoporosis, and idiopathic osteoporosis [2]. Osteoporosis caused by endocrine disease, blood disease, malnutrition, drugs, or other diseases at various age levels in men and women was defined as secondary osteoporosis. With the increase in aging and the high incidence of chronic metabolic diseases, the incidence of osteoporosis has gradually increased [3], and the incidence of osteoporosis-related fractures has increased. Therefore, osteoporosis remains a growing problem worldwide.
Bone is a dynamic, mineralized tissue that performs vital functions in the body, including providing support, providing protection, storing calcium, housing bone marrow, and facilitating movement [4]. During human growth and development, bone needs continuous and dynamic remodeling to adapt to body changes [5]. Furthermore, homeostatic maintenance of dynamic alterations requires the concerted actions of bone-forming osteoblasts and bone-resorbing osteoclasts [6]. As shown in Fig. 1, when mechanical signals and/or microdamage were sensed by osteocytes, the bone turnover process, which includes the bone resorption phase, the reversal phase, the matrix deposition phase, and the mineralization phase, was maintained by osteoclasts derived from hematopoietic stem cells and osteoblasts derived from bone marrow-derived mesenchymal stem cells [7]. In the bone microenvironment, osteoblasts, osteoclasts, and mechanosensing osteocytes exhibited complex interactions. However, when the balance is disturbed, osteoporosis occurs due to a variety of pathological changes. A decreased balance during bone remodeling with aging and after menopause may result in either osteoporosis or osteopenia. In addition to the normal aging process and menopause, there are many other clinical, medical, behavioral, and nutritional risk factors involved in the etiology of bone loss.
Figure 1.

Five phases of the bone remodeling process. The bone surface is covered by lining cells; when mechanical signals and/or microdamage are sensed by osteocytes, bone resorption is initiated by osteoclasts. Osteoblasts derived from hematopoietic stem cells are responsible for reversal and matrix deposition followed by mineralization, after which osteoblasts eventually become bone lining cells or osteocytes.
2. Liver-bone axis
The liver is the largest internal organ in the human body and is responsible for the metabolism and storage of three principal nutrients: carbohydrates, fats, and proteins. In addition, the liver contributes to the breakdown and excretion of alcohol, medicinal agents, and toxic substances and the production and secretion of bile [8]. In addition, liver-derived proteins, known as hepatokines, can also regulate the metabolism of remote tissues [9]. Liver-centered organ crosstalk has received significant attention recently [[10], [11], [12], [13], [14]]. In addition, the liver is tightly regulated by bone [[15], [16], [17], [18]].
The physical distance separating the liver and bone prevents direct physical interaction between these two tissues, and proteins, enzymes, and cytokines secreted by the liver become important ways to influence bone metabolism (Table 1). Moreover, bone has recently been characterized as an endocrine organ that serves as a rheostat regulating glucose metabolism, and these factors derived from bones can regulate global energy homeostasis by altering insulin sensitivity, feeding behavior, and adipocyte commitment [19,20]. Osteokines derived from bone can also regulate liver metabolism via the endocrine system (Table 1). Therefore, crosstalk between the liver and bone has attracted increasing interest in recent years.
Table 1.
Hepatokines and osteokines involved in the liver-bone axis.
| Type and Head | Name | Secreted by | Effect on liver | Effect on bone |
|---|---|---|---|---|
| Hepatokines | Bone Morphogenetic Protein 9 (BMP 9) | Hepatic stellate cell | Inhibit hepatosteatosis | Increase bone formation and suppress bone resorption |
| Fibroblast growth factor 21 (FGF21) | Hepatocyte | Alleviate the development of metabolic dysfunction-associated steatohepatitis (MASH) | Maintain bone mass | |
| Insulin-like growth factor 1 (IGF-1) | Hepatocyte | Biomarker for metabolic dysfunction-associated fatty liver disease (MAFLD) Lipid regulation Downregulate liver fibrosis |
Promote the differentiation of osteoblasts | |
| Hepcidin | Hepatocyte | Supress iron overload and oxidative stress | Maintain bone mass | |
| Lecithin cholesterol acyltransferase (LCAT) | Hepatocyte | Reverse cholesterol transportation to liver and alleviate liver fibrosis | Promote the differentiation of osteoblasts and inhibited the differentiation of osteoclasts | |
| Fetuin A | Hepatocyte | Potential biomarker in the development of MAFLD | Modulation of bone mineralization | |
| Vitamin D | Hepatocyte | Inhibit insulin resistance, alleviate steatosis, necroinflammation and fibrosis in MAFLD | Stimulate the differentiation of osteoblasts and osteoclasts | |
| Prostaglandins (PGs) | Hepatocyte | Positively associate with and liver fat and insulin resistance | Regulatie the recruitment, differentiation, and activity of osteoblast and osteoclast | |
| Osteokines | Osteocalcin (OC) | Osteoblast | Supress accumulation of lipids in liver | Promote bone mineralization and increases bone strength |
| Sclerostin | Osteocyte | High sclerostin levels are related to fat deposition and associated with deranged liver function | Decrease bone formation | |
| Osteopontin (OPN) | Osteoblast &osteoclast |
Activate hepatic stellate cells which accelerating the development of MAFLD and even to hepatocellular carcinoma (HCC) | Activate osteoclastic bone resorption and reduce osteoblastic bone formation |
However, various liver injuries can impair the secretory and biosynthetic functions of the liver and further lead to abnormal bone metabolism. At the same time, abnormalities in the secretion of osteokines in a pathological state are vital for the regulation of the liver. This finding reminds us of a novel mode of the liver-bone axis under physiological and pathological conditions. Here, we summarize recent studies on the homeostatic regulation of the liver-bone axis in different types of osteoporosis, and understanding the regulatory mode of endocrine signaling between the liver and bone may improve the prevention, diagnosis, and treatment of liver-related bone metabolic diseases.
3. Regulation of the liver-bone axis in primary osteoporosis
3.1. Senile osteoporosis
Spontaneous bone loss is one of the main features of aging and can increase the risk of osteoporotic fracture and mortality [21]. With advancing age, bone resorption exceeds bone formation, leading to a gradual loss of bone mass and deterioration of bone microarchitecture. This imbalance is influenced by various factors, including hormonal changes, genetic predisposition, and lifestyle factors such as inadequate nutrition and physical inactivity [22]. Moreover, more attention should be given to the pathological changes in multiple organs, such as the liver, lungs, brain, and muscles, during aging, such as disorders of lipid metabolism and an inflammatory environment.
In old age, fat is redistributed outside the usual fat deposits, and lipids can accumulate in nonadipose tissues in the liver and are considered risk factors for aging-related diseases, including osteoporosis [23]. Abnormal fat accumulation in the liver ultimately leads to MAFLD [24,25]. However, the underlying mechanism of MAFLD and osteoporosis remains unclear. It has been reported that MAFLD and its severity are independently associated with an increased incidence of low bone mineral density (BMD) (in the lumbar spine and total hip) [26]. In addition, J. Jadzic [27,28] reported that MAFLD could be a contributing factor that negatively affects vertebral bone strength in postmenopausal women. However, a meta-analysis revealed no significant differences in the BMD (in the arms, ribs, lumbar spine, pelvis, or femur) between patients with MAFLD and those with non-MAFLD [29]. However, the underlying mechanism of MAFLD and osteoporosis remains unclear. In addition, the aging process is characterized by a chronic inflammatory state called “inflammaging” [23], which shares major molecular and cellular features with metabolism-induced inflammation called “metaflammation”. The excessive activation of osteoclasts by inflammation is also closely related to osteoporosis. In addition, chronic liver inflammation caused by aging may also participate in the occurrence of osteoporosis through the release of inflammatory factors.
Indeed, there has been little research on liver-bone crosstalk in aging-related osteoporosis. A previous study [30] showed that Sirtuin 2 (SIRT2) plays a role in liver-bone crosstalk. Aged SIRT2-KOhep mice had significantly greater bone mass than age-matched controls because of the inhibition of osteoclastogenesis. SIRT2 is a deacetylase that belongs to the class III family of histone deacetylases, which are located mainly in the cytoplasm and are abundantly expressed in the liver [31]. Recently, Lin [32] reported a new mechanism of liver-bone crosstalk regulated by hepatocyte SIRT2. They found that liver-specific SIRT2 deficiency inhibits osteoclastogenesis and alleviates bone loss in mouse models of senile osteoporosis through upregulating leucine-rich α-2-glycoprotein 1 (LRG1) levels in hepatocyte-derived small extracellular vehicles. SIRT2 has been shown to improve metabolic balance physiologically. It has been reported that SIRT2 can enhance the AKT signaling pathway and improve hepatic insulin sensitivity [33]. In addition to promoting glycolysis, SIRT2 can also promote gluconeogenesis [34]. SIRT2 can also promote fatty acid oxidation, thus relieving lipid metabolic stress in the liver, which may be useful for alleviating hepatic lipotoxicity [35]. Pathologically, SIRT2 can also alleviate inflammation, promote liver regeneration, maintain iron homeostasis, aggravate fibrogenesis and regulate oxidative stress in the liver [36]. Given the levels of oxidative stress and inflammation in the elderly liver, SIRT2 may also play an important role in alleviating oxidative stress and inflammation. Studies have reported that SIRT2 can effectively mitigate oxidative stress damage [37,38] and inflammation [[39], [40], [41]] within the liver. More intriguingly, SIRT2 is also capable of regulating iron metabolism homeostasis in the liver. It was reported that SIRT2 could inhibit iron export in primary hepatocytes and HepG2 cells [42], which helps to maintain iron homeostasis and improve cell viability. These findings suggested that SIRT2 may participate in the regulation of the liver-bone axis by maintaining liver homeostasis.
In addition, secreted hepatokines are mainly related to the maintenance of bone mass, suggesting an interesting relationship between the liver and bone. Xu [43] also demonstrated that BMP 9, which is produced and secreted by the liver, significantly increased bone mass and improved bone biomechanical properties in aged mice through the smad1-Stat1-P21 axis. Other hepatokines, such as FGF21, an endocrine hormone produced mainly by the liver, have been shown to regulate bone metabolism in animals through the FGF21-insulin-like growth factor binding protein 1 (IGFBP1) axis [44,45]. However, in a clinical study, FGF21 might be involved in the age-associated decrease in BMD (in the hip and spine), especially in the spine, through increased bone turnover, and IGFBP1 is unlikely to be the downstream effector of FGF21 [46]. The growth hormone (GH)-IGF-1 axis is also involved in bone remodeling and metabolism and has an essential role in the achievement and maintenance of bone mass throughout life. GH acts both directly on bones and indirectly through liver-derived IGF-1 or locally produced IGF-1 [47]. However, as individuals grow older, low levels of IGF-1 and GH can be detected in individuals aged ≥60 years [48].
These findings illustrated the interorgan action of the liver-bone axis in senile osteoporosis. However, fundamental laboratory research is still needed to prove the important regulatory role of the liver-bone axis in aging-related osteoporosis, thus providing a potential and promising therapeutic target.
3.2. Postmenopausal osteoporosis (PMOP)
PMOP, also known as osteoporosis after menopause, is a condition characterized by decreased bone density and an increased risk of fractures in women after they have gone through menopause [49]. During menopause, the levels of estrogen, a hormone that helps maintain bone density, decrease significantly, which disrupts the delicate balance between bone formation and resorption, resulting in a net loss of bone mass [50,51].
It is well known that estrogens influence the differentiation and lifespan of mature osteoclasts and osteoblasts, as well as the lifespan of osteocytes. Estrogens can directly regulate the activity of these three types of cells through estrogen receptors [52]. By activating the Src/Shc/ERK signaling pathway [53] and downregulating JNK, estrogen can inhibit the apoptosis of osteoblasts. Accumulating studies have shown that oxidative stress increases with age, which is related to the pathophysiology of PMOP [54,55]. In response to oxidative stress, nuclear factor erythroid 2-related factor 2 (Nrf2) is released from Kelch-like ECH-associated protein 1 (Keap1) and accumulates in the nucleus, followed by upregulation of Nrf2/ARE antioxidant pathways [56]. Renlei Yang [57] reported that 17β-estradiol plays an anti-osteoporosis role by promoting osteoblastic bone formation through the ESR1-Keap1-Nrf2 axis. In addition, estrogen inhibited proinflammatory cytokines, such as interleukin-1β (IL-1β), interleukin-6 (IL‐6), and TNF‐α, which promote osteoclast differentiation. In addition, estrogen can also increase TGF-β and osteoprotegerin (OPG) levels, which inhibit osteoclast differentiation [[58], [59], [60]].
Although estrogen directly regulates osteoblasts and osteoclasts, we cannot ignore the indirect regulation by which estrogen modulates the skeletal system by positively regulating liver metabolism. The liver is an estrogen-targeted organ [61], and estradiol has been shown to inhibit hepatic stellate cell proliferation and fibrogenesis in animal models [62]. Due to the lack of estrogen during menopause, the liver appears to be more vulnerable to developing liver diseases such as MAFLD and primary biliary cholangitis (PBC) (a condition of autoimmune liver diseases) in females [63]. Up to a point, heavier individuals tend to have a higher BMD, however, the correlation between the body mass index and bone mass appeared to be U-shaped [64]. Therefore, after taking the mechanical stimulation of body weight into account, the low bone mass in PMOP patients might be partially caused by metabolism abnormalities under MAFLD.
The impaired synthetic and secretory functions of the liver under these conditions could further lead to osteoporosis. It has been reported that the serum IGF-1 concentration is significantly decreased in PMOP patients [65]. In addition, the structure of IGF-1 is similar to that of insulin, which might indicate that the presence of qualitative bone abnormalities in postmenopausal women with T2DM may be associated with the presence of MAFLD after menopause [66].
In addition, osteoporosis is a common complication of autoimmune liver diseases. Umit Secil Demirdal [67] demonstrated the presence of autoantibodies associated with autoimmune liver diseases in postmenopausal women, which might indicate an association between autoimmune liver diseases and osteoporosis.
Moreover, altered iron metabolism is closely associated with osteoporosis initiation and progression [68]. The iron deposition of hepcidin, which is responsible for reducing intestinal iron absorption and inhibiting macrophage (macrophage/bone) iron deposition, is regulated by estrogen [69,70]. Yanli Hou [71] reported that estrogen could greatly contribute to iron homeostasis by regulating hepatic hepcidin expression directly through a functional ERE in the promoter region of the hepcidin gene. Thus, these studies indicated a regulatory relationship between the liver and bone postmenopause (Fig. 2).
Figure 2.

Potential liver-bone axis in patients with PMOP. Under normal physiological conditions, estrogens can directly and indirectly regulate the activity of osteoblasts and osteoclasts. However, CLD, such as MAFLD, autoimmune liver diseases and altered iron metabolism caused by estrogen deficiency, might contribute to abnormal bone metabolism during PMOP.
In conclusion, the relationship between bone metabolism and the liver is complex in patients with PMOP. However, the precise mechanisms involved in the regulation of the liver-bone axis in the context of PMOP have not been precisely established, and a vast amount of clinical and basic research is still needed, which might further help build a better understanding of the treatment for PMOP.
4. Regulation of the liver-bone axis in secondary osteoporosis
4.1. Cirrhosis-induced bone loss
Chronic liver disease frequently progresses to liver cirrhosis, following different processes that involve liver cell degeneration and extensive necrosis. Historically, the severity of liver cirrhosis is evaluated by the Child-Pugh classification and Model for End-Stage Liver Disease (MELD) score [72]. Patients with cirrhosis often develop osteoporosis, with an incidence of approximately 20%–50 %, which is defined as hepatic osteodystrophy (HOD) [73,74]. Nevertheless, R. Wakolbinger [75] found a significant loss of bone structural integrity at the early stage of cirrhosis which indicated a liver-bone axis in the development of cirrhosis. In our previous study [16], we identified a critical hepatokine, LCAT, which is involved in the regulation of the liver-bone axis in patients with cirrhosis. Briefly, the unusually high expression of PP2Acα in the liver of patients with cirrhosis leads to the downregulation of LCAT through the dephosphorylation of the transcription factor USF1. Surprisingly, exogenous supplementation with recombinant LCAT relieved liver fibrosis and increased the BMD (right femur of mice) in a mouse HOD model by promoting the reversal of cholesterol transport from the bone to the liver via the liver-bone axis.
In addition, 10 %–25 % of circulating IGF-1 is crucial for normal bone development, and decreased concentrations of IGF-1 are observed in individuals who develop liver cirrhosis compared with healthy individuals [[76], [77], [78]] and are negatively related to clinical liver impairment [79]. Shoshana Yakar [80] reported decreased circulating IGF-1 levels and attenuated bone growth in liver Igf-1 and acid labile subunit (ALS) double-knockout mice, which indicated the important role of the IGF-1/IGFBP3/ALS ternary complex in osteoporosis pathophysiology. Therefore, this finding implies that IGF-1 may also be involved in the regulation of the liver-bone axis in cirrhosis.
Cirrhosis typically follows chronic inflammation [81]. Inflammation has been identified as a potential risk factor for osteoporosis [82]. During cirrhosis, sinusoidal endothelial cells, Kupffer cells and hepatocytes release proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interleukin-1 beta (IL-1) [83], which cause an increase in overall inflammation levels in the body and might also result in decreased bone mass [84]. It has been reported that circulating IL-6 and TNF-α levels are significantly greater in patients with cirrhosis. However, another study showed that serum IL-1 and TNF-α levels were not different between patients with cirrhosis and controls, but the levels of interleukin-2 (IL-2) and IL-6 in the serum of patients with cirrhosis were significantly elevated. Therefore, more experiments are needed to demonstrate the specific mechanism by which inflammatory factors regulate cirrhosis-induced bone loss through the liver-bone axis.
Moreover, OC, an osteoblast-derived factor, tightly regulates multiple target organs, including the pancreas, liver, muscle, adipose tissue, testes, and central and peripheral nervous systems [85]. It has been reported [86] that the livers of Osc-/Osc- mice exhibit lipid accumulation and steatosis, and mice treated with OC exhibit no accumulation of lipids and exhibit normal liver morphology when fed a high-fat diet [87,88]. In patients with end-stage cirrhosis, plasma OC is decreased [[89], [90], [91]]. However, the mechanism by which OC participates in the progression of liver cirrhosis is still unclear.
Another endogenous Wnt antagonist secreted almost exclusively by osteocytes, sclerostin, decreases bone formation by repressing osteoblast differentiation and proliferation [[92], [93], [94]]. A study in human cadaveric donors with alcoholic liver cirrhosis (ALC) revealed that ALC induced an increase in osteocytic sclerostin expression, suggesting its role in mediating low bone formation among ALC individuals. Yumie Rhee [95] reported that the serum sclerostin level was significantly greater in patients with cirrhosis than in controls and that patients with Child‒Pugh class B or C cirrhosis had higher sclerostin levels than patients with class A cirrhosis or controls after subgroup analysis. Furthermore, the sclerostin level showed an inverse correlation with the serum albumin concentration, which is a marker of liver dysfunction. In addition, they also found that serum sclerostin levels seemed to be greater in patients with alcoholic cirrhosis than in patients with hepatitis B virus (HBV)-associated cirrhosis, which was consistent with a recent report by Gonza'lez-Reimers [96]. However, Robert Wakolbinger [97] reported a contrasting sclerostin level based on a pilot study of individuals with various forms of liver cirrhosis-induced bone loss (ALD, viral hepatitis, nonalcoholic fatty liver disease, hemochromatosis and autoimmune hepatitis). They found that patients with ALD had significantly lower sclerostin levels, probably because alcohol promotes osteocyte apoptosis [98]. Considering that osteoblast activity is reduced in hepatic cirrhosis [99], the low sclerostin levels observed in ALD patients could serve as an attempt to preserve osteogenesis. Therefore, to further investigate the specific mechanism of sclerostin in cirrhosis-induced bone loss, large-scale cohort studies and the consideration of the different etiologies of cirrhosis and ethnicities are essential.
In general, the abnormal secretion of hepatokines and osteokines indicated that the liver-bone axis is regulated by cirrhosis-induced bone loss. Certainly, there might be other relevant factors that contribute to the maintenance of bone tissues through the liver-bone axis. However, the specific mechanism is not yet clear. Therefore, screening potential regulatory factors with the help of comprehensive multiomics research may reveal new therapeutic targets for the treatment of HOD.
4.2. Chronic cholestatic liver disease (CCLD) induced bone loss
Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are important causes of chronic cholestasis and are the most common causes of cholestatic liver disease. It has been reported that the incidence of osteoporosis in patients with PBC and PSC ranges from 15 % to 40 % [[100], [101], [102]]. Chronic cholestasis results in reduced bile acid levels in the intestine and affects the absorption of vitamin D and vitamin K, which further leads to deficient absorption of calcium salts. Moreover, vitamin D and its metabolites must bind to vitamin D-binding protein (VDBP), which is expressed in the liver, to circulate in the blood. However, VDBP expression decreases with the progression of chronic liver disease [103] and might indirectly participate in the regulation of the liver-bone axis. In contrast to the occurrence of osteoporosis accompanied by low bone formation in PBC patients, patients with PSC often exhibit increased bone resorption.
T helper 17 (Th17) cells are adaptive immune cells that play myriad roles in the body and have been reported to mediate bone loss under different pathological conditions [104]. Using a classic PSC animal model, Tobias Schmidt [105] reported that Abcb4−/− mice exhibited an increased frequency of Th17 cells in the liver, which enhanced IL-17 production and thus led to increased bone resorption.
4.3. Hepatitis virus infection-induced bone loss
Approximately 5 % of the world's population (350–400 million people) and 2 % of the world's population (approximately 180 million people worldwide) are chronically infected with the hepatitis B virus (HBV) and hepatitis C virus (HCV), respectively. With long-term infections of HBV and HCV, bone loss is frequently reported in these individuals [[106], [107], [108], [109], [110]]. However, the mechanism of bone mass loss in patients with viral hepatitis is poorly understood.
It has been reported that the serum levels of soluble TNF receptor p55 (sTNFR-55) in patients with HBV- and HCV-related cirrhosis are inversely correlated with BMD (in the lumbar spine and femoral neck) and positively correlated with urinary deoxypyridinoline, a biochemical marker of bone resorption [111]. However, recent studies revealed that patients with noncirrhotic chronic hepatitis B infection also exhibited bone loss. Chien-Hua Chen [107] reported that patients with noncirrhotic CHB infection, especially males, had a significantly greater risk of osteoporosis. Yuan-Yuei Chen [112] also showed that in noncirrhotic CHB patients, the clearance rate of HBeAg in women was greater than that in men, and the detection rate of anti-HbeAg and HBsAg antibodies in premenopausal women was greater than that in postmenopausal women and men. In addition, a higher estradiol level in women increases the level of interferon-γ in lymphocytes and enhances the specific antigen response of monocytes in peripheral blood, which leads to greater inflammatory activity in male HBV patients than in female patients and contributes to a significant reduction in bone mineral density.
Currently, HBV-mediated immunomodulation and chronic inflammation are recognized as possible mechanisms for noncirrhotic CHB-induced osteoporosis. When HBV infection remains active for a long time, the receptor activator of nuclear factor-κB ligand (RANKL)-receptor activator of nuclear factor-κB (RANK)-OPG system is out of balance, thus facilitating the overactivation of osteoclasts [113]. Moreover, HBV can induce fatty changes in hepatocytes, regulate lipid and sugar transport by activating sterol regulatory element binding protein 1c and peroxisome proliferator-activated receptor γ, and ultimately lead to systemic insulin resistance and other metabolic disorders [114]. In addition, HBV- and HCV-mediated iron overload might contribute to bone loss after infection [115]. However, the precise regulatory mechanism involved remains unclear.
It is also worth mentioning that an increased risk of osteopenia and osteoporosis was associated with the use of antiviral drugs [116]. One study [117] raises the question of whether drugs used to treat the disease may also cause varying degrees of bone density loss in patients, but this remains to be studied.
4.4. MAFLD-induced bone loss
MAFLD, formerly known as nonalcoholic fatty liver disease, affects approximately a quarter of the world's adult population and poses a major health and economic burden to all societies [118,119]. Increasing experimental evidence supports a pathophysiological link between MAFLD and osteoporosis [120]. Interestingly, the two diseases share common risk factors, including but not limited to aging, a sedentary lifestyle, and sex hormone deficiencies, suggesting that they may be linked beyond simple coincidence. However, the precise mechanism underlying the association between MAFLD and decreased bone mass remains controversial.
A key feature of MAFLD is the low-grade inflammatory state, could not only developing metabolic dysfunction-associated steatohepatitis but also affecting extrahepatic organs [121]. During MAFLD, the production of proinflammatory cytokines such as TNF-α, IL-1, and IL-6, which are produced mainly by inflammatory cells infiltrating the liver, increases. As mentioned before, inflammation has been identified as a potential risk factor for osteoporosis through excessive activation of osteoclasts. Elevated TNF-α levels were observed in patients with MAFLD [122]. RANKL, which belongs to the TNF superfamily, binds to RANK on the surface of osteoclast precursors and promotes osteoclastogenesis and bone resorption [123]. In addition, as a soluble decoy receptor of RANKL, OPG can inhibit the RANKL-RANK interaction, thus preventing bone resorption. It has been reported that liver fibrosis in MAFLD patients is significantly correlated with low BMD (in the lumbar spine, femur neck, and total hip), suggesting an association between the aggravation of hepatic inflammation, fibrosis, and bone loss in MAFLD patients [124]. Under inflammatory conditions, activated T cells and B cells can also secrete other pro-osteoclastogenic molecules, such as RANKL and IL-17A, which leads to the induction of bone loss [125].
Fetuin-A, which is exclusively secreted by the liver, is reduced in MAFLD patients and might serve as a potential biomarker for the development of MAFLD [18]. Moreover, the serum fetuin-A level was also decreased in patients with osteoporosis, and fetuin-A-deficient Ahsg−/− mice exhibited impaired bone development. In addition, other hepatokines mentioned above, such as IGF-1, FGF21, and IGFBP1, might also be related to osteoporosis in MAFLD patients. Studies performed in patients with MAFLD have shown that the level of IGF-1 is lower in MAFLD patients than in healthy controls and is negatively associated with the histological severity of MAFLD [[126], [127], [128]]. IGF-1 also induces cellular senescence and inactivates hepatic stellate cells [129]. Notably, low levels of IGF-1 in MAFLD patients with osteoporosis directly impair osteoblast differentiation and growth. In contrast, both the serum FGF21 level and liver FGF21 mRNA level is increased in MAFLD patients as long as the level of IGFBP1 increases and serves as an independent predictor of hepatic steatosis [130,131]. Considering the regulatory effect of FGF21 on IGFBP1, the FGF21-IGFBP1 axis might also play an important role in MAFLD-induced osteoporosis.
In addition, OPN is a phosphorylated glycoprotein secreted mainly by osteoblasts and osteoclasts that was originally found in the bone marrow. OPN is a prerequisite for activating osteoclastic bone resorption and reducing osteoblastic bone formation in unloaded mice, and OPN-deficient mice are resistant to osteoporosis [132]. Patients with MAFLD were reported to have significantly greater serum OPN levels than patients in the control group [133]. OPN also negatively regulates the liver by activating hepatic stellate cells, which accelerates the development of MAFLD and even HCC [134]. Moreover, genetic depletion of OPN protected mice fed a high-fat diet from obesity-induced hepatic steatosis [135]. These findings also demonstrated that the liver-bone axis is regulated between MAFLD and osteoporosis.
Bone-derived OC [136] showed an unexpected regulatory effect on the liver. Clinical studies have shown that a reduction in the serum OC concentration is closely related to the occurrence and development of MAFLD [133,137]. Uncarboxylated OC can regulate insulin signaling, de novo lipogenesis, and endoplasmic reticulum stress through direct binding to its liver-specific receptor, G protein-coupled receptor family C Group 6 subtype A [138].
We likewise cannot ignore the important role of sclerostin in MAFLD-induced bone loss. It has been reported [99,139] that circulating sclerostin levels are significantly lower in MAFLD patients than in normal controls and are positively correlated with BMD (in the left femora of mice) and negatively correlated with many MAFLD-related biochemical parameters, including BMI, liver function tests, triglycerides, insulin and HOMA-IR. However, sclerostin levels are increased in patients with cirrhosis. This might be explained by “insulin-based”: increased insulin in MAFLD patients mimics the action of IGF1 through binding with IGF-1 receptors on osteocytes, further suppressing sclerostin production [140].
In conclusion, chronic inflammation, the fetuin-A level, the GH/IGF-1 axis and disturbances in the FGF21-IGFBP1 axis are the main pathophysiological factors linking MAFLD with abnormal bone metabolism. Some osteokines have also been shown to form a liver-bone axis in MAFLD. Furthermore, MAFLD is accompanied by increased inflammation [141], which impairs the synthetic and secretory functions of hepatokines in the liver. Therefore, although many issues related to the liver-bone axis in MAFLD patients with osteoporosis have not been elucidated to date, growing evidence suggests that screening and surveillance of abnormal bone metabolism and liver function in patients with MAFLD should be considered in future strategies and guidelines for identifying MAFLD-induced bone loss (Fig. 3).
Figure 3.

Potential role of the liver-bone axis in MAFLD-induced bone loss. Some molecules and proteins secreted by the liver, including IGF-1, FGF21−IGFBP1, TNF-α, OPN and fetuin A, contribute to bone loss in MAFLD. Besides, bone derived OC, OPN and sclerostin could also regulated the metabolism of liver during MAFLD.
4.5. Alcoholic liver disease (ALD)-induced bone loss
ALD is typically caused by long-term excessive alcohol consumption and has been described as an independent risk factor for osteoporosis [142]. Ethanol has a dose-dependent toxic effect on osteoblasts [143], and chronic alcohol consumption suppresses the osteoblastic differentiation of bone marrow cells and promotes adipogenesis, which further worsens bone metabolism [144]. ALD comprises a spectrum of liver injuries, including simple steatosis, acute alcoholic hepatitis, and cirrhosis, which might contribute to bone loss [145]. Therefore, this finding suggests a potential liver-bone axis during ALD. However, the regulation of the liver-bone axis under ALD-induced bone loss remains largely unknown.
As mentioned before, the GH/IGF-1 axis plays an important role in bone remodeling and metabolism. It was reported that IGF-1 was significantly decreased in alcohol abusers with liver cirrhosis [[146], [147], [148]]. Thus, excessive alcohol consumption could also lead to an imbalance between osteogenesis and adipogenesis through impairing the GH/IGF axis [149]. PGs, which are produced by hepatocytes, are cytokines that regulate the recruitment, differentiation, and activity of osteoblasts and osteoclasts [150]. Some data have shown that the synthesis of PGs in the liver is reduced due to strict restrictions on food intake in severe liver disease patients, and the downregulation of PGs could further contribute to bone loss [151,152]. Moreover, as a metabolite of arachidonic acid, PGs have been shown to promote the formation of new blood and lymphatic vessels, which are essential for supplying nutrients to bone [153]. Therefore, under pathological ALD conditions, the synthesis of PGs in the liver is blocked, which in turn leads to the activation of osteoclasts and bone loss. This might explain the mechanism of osteoporosis caused by ALD from a completely new perspective.
In addition, inflammatory factors are indispensable risk factors for osteoporosis. It has been reported that the concentrations of IL-1, IL-6, and TNF-α are increased in patients with ALD [154]. TNF-α and IL-6 levels caused the activation of osteoclastogenesis via the induction of RANKL. Therefore, the abnormal levels of inflammatory factors secreted by the liver caused by excessive alcohol intake might contribute partially to osteoporosis [155].
The imbalance of RANKL-OPG system might contribute to ALD-induced bone loss. RANKL levels are significantly increased in alcoholic cirrhotic patients [156]; however, García-Valdecasas-Campelo E [157] reported that RANKL levels are not altered in ALD patients. OPG acts as a decoy receptor for osteoclast activating factor and a receptor activator of RANKL and impairs osteoclast function [158]. Increased OPG levels were observed in alcohol-dependent subjects in several studies [157,159,160] and partly represent a compensatory mechanism for the negative balance of bone remodeling in patients with alcoholic cirrhosis.
As mentioned above, sclerostin decreases bone formation by repressing osteoblast differentiation and proliferation. It was reported that sclerostin levels tended (albeit not significantly) to be higher among individuals with cirrhosis and demonstrated a significant association with liver function impairment, with elevated levels observed among patients classified as Child C [161]. However, disparate results were observed in several studies. Wakolbinger [97] reported decreased sclerostin levels among 16 alcoholics, which might be explained by alcohol promoting osteocyte apoptosis. However, the exact role of sclerostin in ALD needs further investigation.
Interestingly, other factors, such as alcoholic neuropathy and myopathy, that contribute to bone loss in individuals with ALD indicate multiorgan crosstalk. Badrick reported [162] a 3 % increase in cortisol levels per unit of alcohol consumed, and the increase in cortisol observed in chronic alcoholics may indirectly cause osteopenia and aseptic necrosis [163]. Muscle mass and strength play crucial roles in determining bone mass, and muscle atrophy and bone loss are closely intertwined. In this context, muscle atrophy related to alcoholism serves as an additional contributing factor to decreased bone mass in alcoholic patients [164]. Vitamin D deficiency [165,166] and low testosterone levels are associated with muscle atrophy in alcoholics [167,168], which in turn leads to bone loss. In addition, vitamin D is closely related to Ca2+ levels and plays a vital role in bone development and calcium-dependent signaling [169]. Alcohol-induced liver injury can reduce the levels of VDBP and the hydroxylation of vitamin D by the liver [170,171]. In addition, deficient vitamin D levels are positively correlated with lower high-density lipoprotein levels, which might lead to abnormal cholesterol metabolism [172]. Abnormal upregulation of cholesterol leads to an increase in fat content in the bone marrow, which is also known to worsen osteoblast function and lead to osteopenia [173]. We hypothesize that the decrease in bone mass in ALD patients may be partially attributed to abnormalities in cholesterol metabolism caused by vitamin D deficiency.
The liver is the main organ of alcohol metabolism and the target organ of alcohol injury. Aldehyde dehydrogenase 2 (ALDH2), a key enzyme that detoxifies the ethanol metabolite acetaldehyde, is highly expressed in the liver, heart, and brain [[174], [175], [176]]. Compared with WT mice, Aldh2−/− mice had reduced cancellous bone volume and bone formation when exposed to ethanol, likely due to increased circulation levels of acetaldehyde. However, liver-specific Aldh2 knockout mice are needed to observe the regulation of the liver-bone axis under the influence of alcohol.
Furthermore, there is no doubt that ALD can be prevented by alcohol abstinence. It has been reported that decreased serum OC in alcoholics improves with abstinence after 6 months [177]. Another bone resorption marker, cross-lap, increased after 8 weeks of abstinence, indicating a high turnover rate [178]. In addition, the lower IGF-I bioavailability was also improved after abstention [147].
This evidence indicated the regulation of the liver-bone axis between ALD and osteopenia (Fig. 4). Therefore, elucidating the precise mechanism of ALD-induced bone loss will provide a more theoretical basis and potential treatments for the clinical treatment of osteoporosis caused by alcohol intake.
Figure 4.

Potential role of the liver-bone axis in ALD-induced bone loss. Alcohol-induced liver injury can reduce the levels of VDBP and the hydroxylation of vitamin D by the liver, which might further induce abnormal cholesterol metabolism and lead to bone loss. Other inflammatory factors, such as IL-1, IL-6 and TNF-α, as well as PGs, also contribute to bone loss. In addition, ALDH2 deficiency could increase the circulation levels of acetaldehyde and lead to bone loss.
4.6. Hereditary hemochromatosis (HHC)-induced bone loss
HHC is caused by mutations in the hereditary hemochromatosis protein, transferrin receptor 2 (TFR2), hemojuvelin, hepcidin, or ferroportin genes [179]. Most HHC subtypes result in low hepcidin levels and iron overload, and abnormal iron accumulation in various tissues can lead to fatal complications such as liver cirrhosis, heart failure, or diabetes [180]. Importantly, bone is highly susceptible to fluctuations in iron concentration [170,181]. Therefore, alterations in bone metabolism are certainly affected in HHC patients. Osteoporosis was detected in 29%–34 % and osteopenia in 74%–79 % of HHC patients, and the risk of vertebral fractures has been reported to reach 20 % [182]. Maintenance of systemic and cellular iron homeostasis predominantly regulated by the liver through the iron regulatory hormone hepcidin is vital for normal physiological functions in mammals [172]. To date, HHC-induced disease models have provided insights into the impact of HHC on bone metabolism, showing that, for example, hepcidin deficiency leads to bone loss via suppressed bone formation [183]. However, the molecular mechanism underlying hepcidin regulation by hemochromatosis proteins is incompletely understood. TFR2, which is predominantly expressed in the liver, serves as an important regulator of hepcidin [184]. It was reported that the hepatocyte-specific deletion of Tfr2 cells results in an iron overload phenotype [185]. However, the precise mechanism by which liver iron overload caused by Tfr2 deficiency leads to osteoporosis has not been thoroughly elucidated to date. In addition, based on a hindlimb unloading model, Zi Xu [186] observed an elevated hepcidin level in the liver and a reduced ferroportin-1 level in the duodenum, which led to abnormal iron metabolism, suggesting that unloading-induced bone loss was orchestrated by iron overload and coupled with the regulation of hepcidin by the liver.
Liver dysfunction is one of the leading pathologies in HHC. Therefore, the lower BMD may not be explained solely by iron toxicity. This has been confirmed by a cross-sectional study of 93 patients with HH, where bone fragility was observed in 25 % of patients with HH and was independent of the severity of iron overload but strongly associated with cirrhosis [187]. Moreover, patients homozygous for p.C282Y are at a high risk of developing general liver disease, with men additionally showing significantly increased rates of hepatocellular carcinoma [188].
An unappreciated potential mode of regulation of the liver-bone axis might contribute to bone loss in HHC. Therefore, elucidating the regulation of iron metabolism by liver damage is of great interest for targeted therapy for bone loss in HHC.
4.7. Schistosomiasis infection-induced bone loss
The neglected tropical disease schistosomiasis caused by blood flukes of the genus Schistosoma continues to scour humankind. Schistosomiasis affects approximately 200 million people, and approximately 700 million people are at risk in 74 countries [189,190]. Waterborne flatworm larvae penetrate the skin and move in the bloodstream through the heart and lungs to the liver [191]. Here, they mature and mate in the portal circulation before laying eggs that lodge in the liver. Parasite egg antigens can induce a severe host immune response resulting in granulomas and portal fibrosis in the liver, thus leading to schistosome-associated liver abnormalities.
Unexpectedly, schistosomiasis infection also leads to reduced bone mass. Indeed, the incidence of osteoporosis in schistosomiasis patients was considerably greater than that in healthy controls in nonendemic areas. Wei Li [192] demonstrated that the increase in RANKL produced by Tfh cells was correlated with host osteoclast-mediated bone loss in the context of schistosome infection. However, this study was unable to elucidate the entire mechanism of schistosomiasis infection-induced bone loss. As mentioned before, schistosomiasis infection invariably results in severe liver damage, such as liver fibrosis. This degree of liver injury results in the secretion of the corresponding liver-derived factors, thus further affecting bone homeostasis through the circulation. Therefore, we need to consider the effect on the skeletal system caused by liver damage. There are few clinical studies showing how long schistosome infection leads to bone loss. However, schistosome infection has been linked to growth inhibition in children [193], even those with low parasitic burdens. In addition, significant bone loss in mice during the chronic phase of infection has been reported to occur 11–13 weeks postinfection [192]. However, it should be noted that in areas where schistosomiasis is endemic, regular screening and treatment are necessary. Additionally, following the diagnosis of schistosome infection, immediate assessment of changes in bone metabolism is essential to prevent the occurrence of osteoporosis.
4.8. HCC induces bone loss
A subset of patients with MAFLD or hepatitis virus infection can develop progressive liver disease leading to cirrhosis and HCC [194]. Therefore, given the negative effects of MAFLD, hepatitis virus infection, and cirrhosis on bone metabolism, we cannot ignore the regulatory relationship between the liver and bone in the presence of HCC. According to clinical research in China, Fu [195] reported that the incidence of osteoporosis in patients with liver cancer significantly increased due to a decrease in serum calcium; metabolic disorders involving vitamin D, calcium, and phosphorus; and an increase in parathyroid hormone. There are few reports in the literature describing bone loss in HCC, let alone the precise mechanism involved.
The inflammatory environment is favorable for HCC development, and the oversecretion of inflammatory factors could further lead to bone loss. Stimulating the release of inflammatory cytokines is the key molecular mechanism by which spectrin, beta, and nonerythrocytic 1 (SPTBN1) induces the occurrence and disappearance of liver cancer [196]. SPTBN1 not only plays a powerful role in the occurrence and development of HCC but also affects bone health by promoting the proliferation and differentiation of osteoblasts and vascularization in bone through the TGF-β/Smad3 and STAT1/Cxcl9 pathways [197]. Targeting SPTBN1 may benefit fracture healing and drug discovery and might benefit patients with HCC and osteoporosis [198]. Similarly, comorbid genes might also indicate a liver-bone axis model. Sharon Russo [199] reported that mutations in the ZNF687 gene cause severe Paget's disease of bone, resulting in severe alterations in bone remodeling, and strikingly, in this mouse model, the mutation was also associated with high penetrance of HCC. This phenomenon suggests that liver-bone crosstalk may occur in HCC.
Secreted hepatokine levels are also decreased in HCC patients due to liver function damage. It has been reported that IGF-1 expression in the livers of HCC patients is significantly lower than that in the livers of non-HCC patients with cirrhosis and healthy controls. This might be one of the causes of HCC-induced osteoporosis. However, OPN, a protein secreted by osteoblasts, is highly expressed in HCC. Serum OPN could serve as a potential biomarker for HCC [200]. These results provide new ideas for the regulation of liver-bone crosstalk in HCC. However, more research is needed in the future to verify the regulatory mechanism of liver bone.
Liver transplantation is the only effective way to treat a variety of end-stage liver diseases, such as HCC. The survival rate of transplant patients has increased exponentially, which has led to a greater understanding of long-term complications secondary to the underlying pathology or the various treatments that must be followed. Metabolic bone disease is a chronic complication of liver transplantation that reduces quality of life, and liver transplant recipients have a significantly increased risk of osteoporosis and fractures [201,202]. A clinical study revealed a decreased level of serum OPG and an increased level of serum Dickkopf-related protein 1 after living donor liver transplantation, which indicated enhanced osteoclast activity [203]. Moreover, recent research has shown that bone density can be used as an indicator to predict posttransplant survival in liver transplant recipients with HCC [204,205]. In addition, ischemia/reperfusion injury and the use of immunosuppressants [206,207] after liver transplantation, which can lead to bone loss, cannot be disregarded, and further research is needed to determine the underlying molecular mechanisms involved.
Bone is now the second major site and accounts for approximately 25 % of extrahepatic metastases from HCC [208]. The primary types of bone metastasis are osteolytic and osteoblastic according to the predominant activities of certain cell types, and an impaired balance between bone formation and resorption is frequently observed in both types of metastases. Yang Lu [209] reported that most (90.7 %) of the lesions were mixed osteolytic and osteoblastic based on a cohort of 43 consecutive patients who were diagnosed with bone metastasis from HCC. OB can be stimulated by metastatic tumor cell-derived factors, including FGFs, urokinase-type plasminogen activator, endothelin-1, IGF-1, bone morphogenic proteins, and vascular endothelial growth factor (VEGF) [[210], [211], [212], [213], [214]]. However, little is known about osteoblastic bone metastasis in HCC. Sterol regulatory element-binding protein 2 (SREBP2) is an important enzyme in cholesterol metabolism and is highly expressed in HCC [215]. It was rather surprising that activating Srebp2 promoted osteoclastogenic gene expression [216]. Moreover, BMP2 serves as a downstream effector of Srebp2, Bmp2 transcription is inhibited by SREBP2, and decreased BMP2 levels are negatively associated with osteoblast activity [217,218]. Therefore, we can reasonably consider that the abnormal cholesterol metabolism caused by the elevated SREBP2 level in HCC could disrupt the bone microenvironment, thus promoting HCC bone metastasis. In addition, osteoclast activation is a prerequisite for the occurrence of osteolytic bone metastasis. In addition to RANKL and OPG produced by osteoblasts, the activation of osteoclasts induced by inflammatory factors such as TNF-α and ILs is also a significant factor in osteolytic bone metastasis. In addition, Bixiang Zhang [219] reported that the long noncoding RNA H19 induced the epithelial to mesenchymal transition of HCC cells by sponging microRNA-200b-3p and aggravated osteolytic bone remodeling by reducing osteoprotegerin expression via the inactivation of p38 mitogen-activated protein kinase (MAPK) signaling. Soon after, they demonstrated that bone-metastasized HCC-derived EVs could localize to orthotopic HCC sites and promote HCC progression by transferring ALKBH5-targeting miR-3190-5p (miR-3190) [220]. Furthermore, Hao Zheng [221] also reported that increased osteoclast differentiation caused by a normal increase in the m6A modification of anillin actin-binding protein promoted HCC bone metastasis.
In conclusion, a complex regulatory network of the liver-bone axis is involved in HCC-related bone metabolism (including bone loss after liver transplantation, overuse of immunosuppressants, and HCC bone metastases). However, further research is needed to elucidate the detailed mechanisms of liver-bone crosstalk in HCC, thus providing a new therapeutic strategy for clinical treatment.
5. Diagnosis and treatment targeting the liver-bone axis
The diagnosis of osteoporosis is made when patients have fragility fracture or a T score ≤ −2.5 at the lumbar spine, femoral neck, total hip, or distal one-third of the radius on dual-energy X-ray absorptiometry (DXA) examination [222]. The lowest T score on an individual's DXA examination is used for the diagnosis of primary osteoporosis. However, it is not particularly precise to rely on DXA alone for diagnosis, as secondary osteoporosis frequently occurs. Thus, we should pay more attention to the assessment of liver function. A thorough history, physical examination, and laboratory evaluation should be performed for various extrahepatic and/or intrahepatic factors. Additional attention should be devoted to the factors that are abnormally expressed in the serum during various primary or secondary osteoporosis processes. By assessing the expression levels of these factors, such as IGF-1, FGF21 and LCAT, it is possible to predict the progression of osteoporosis and provide a potential additive therapeutic intervention.
Once osteoporosis has been confirmed, timely anti-osteoporosis treatment is crucial. Generally, osteoporosis therapy consists of pharmacologic and nonpharmacologic treatments [102]. For nonpharmacologic treatments, in addition to adequate calcium/vitamin D/protein intake, maintaining a healthy weight, remaining active with weight-bearing exercise, and avoiding excess alcohol and caffeine intake were also necessary. In addition, factors that might lead to liver damage should also be avoided. On the other hand, there are two groups of pharmacological osteoporosis treatments: antiresorptive and anabolic agents [223]. Bisphosphonates, denosumab, and raloxifene mainly target and block osteoclast activity to decrease bone resorption and bone loss. Moreover, anabolic agents such as teriparatide and abaloparatide can transiently stimulate the parathyroid hormone receptor to stimulate osteoblasts and bone formation [224]. In addition, treatments for liver injury under pathophysiological conditions cannot be ignored. As an association between the liver and bone is established, a medication that could target both diseases would be a great advancement. It was reported that treatment of hypogonadism and iron overload with testosterone replacement and phlebotomy was effective in improving bone mass in a small study of six hypogonadal men with hemochromatosis [225]. Vitamin E, a potent antioxidant, could resolve MAFLD at a dose of 800 IU/d [226] and effectively suppress bone resorption in postmenopausal women with osteopenia [227]. In addition, there are also some other emerging pharmacological options. Semaglutide, a glucagon-like peptide-1 receptor agonist (GLP-1RA), was shown to increase bone mass and reduce fracture risk in addition to resulting in increased rates of MASH resolution [228,229]. The FXR agonist obeticholic acid, which is a promising option for MAFLD management, might decrease osteoclastogenesis and prevent bone loss [230,231]. Therefore, targeting primary liver injury may provide novel insights into the treatment of osteoporosis in the clinic.
With in-depth research based on the liver-bone axis in different types of osteoporosis, more precise treatments targeting liver damage and osteoporosis should be developed. Currently, specific recombinant proteins, agonists, and inhibitors involved in the liver-bone axis might prove to be effective against osteoporosis. In addition, powerful multiomics approaches are creating new opportunities to annotate proteins involved in the regulation of the liver-bone axis, and the development of therapeutic targets based on these approaches will provide a solid theoretical basis for clinical practice.
6. Conclusion and perspectives
As a classical endocrine organ, the liver is involved in numerous pathophysiological regulatory processes. Liver-centered organ crosstalk has received significant attention, and concepts such as the “liver–gut axis”, “liver–brain axis”, “liver–lung axis”, and “liver–kidney axis” have been developed to illustrate the relationships between organs. In addition, the liver also showed tight regulation of the newly defined endocrine organ, bone, which could be defined as the “liver-bone axis”. Therefore, further elucidating the crosstalk between classical and newly defined endocrine organs could provide new ideas for basic and clinical research.
In this review, we discussed the potential regulation of the liver-bone axis under different physiopathological conditions both in primary osteoporosis and secondary osteoporosis (Fig. 5). Several endocrine factors (IGF-1, FGF21, hepcidin, vitamin D, OC, OPN, LCAT, fetuin-A, PGs, BMP2/9, IL-1/6/17, and TNF-α) and key genes (SIRT2, ALDH2, TFR2, SPTBN1, ZNF687 and SREBP2) might be involved in the regulation of the liver-bone axis. In addition to the classic metabolic pathways involved in inflammation and oxidative stress, iron metabolism, cholesterol metabolism, lipid metabolism and immunometabolism mediated by the liver-bone axis require more research to elucidate the regulatory mechanisms involved in osteoporosis. Multiomics technology and data mining technology could certainly advance our understanding of the liver-bone axis in the future, providing new clinical strategies for managing liver and bone-related diseases. Moreover, we cannot deny that multiple organ crosstalk networks are based on the liver-bone axis, and the regulatory functions of other organs, such as muscle, kidney, brain, and intestine, will also be evaluated in subsequent scientific research.
Figure 5.

Potential factors and mechanism involved in the liver-bone axis. Various endocrine factors (IGF-1, FGF21, hepcidin, vitamin D, OC, OPN, LCAT, fetuin-A, PGs, BMP2/9, IL-1/6/17, TNF-α) and key genes (SIRT2, ALDH2, TFR2, SPTBN1, ZNF687, SREBP2) might be involved in the regulation of liver-bone axis in different liver injuries induced bone loss. In addition to classical metabolic pathways such as inflammation and oxidative stress, iron metabolism, cholesterol metabolism, lipid metabolism and immune metabolism mediated by the liver-bone axis might also play some roles in regulating bone loss.
Conflicts of interest
A conflict of interest occurs when an individual's objectivity is potentially compromised by a desire for financial gain, prominence, professional advancement or a successful outcome. The Editors of the Journal of Orthopaedic Translation strive to ensure that what is published in the Journal is as balanced, objective and evidence-based as possible. Since it can be difficult to distinguish between an actual conflict of interest and a perceived conflict of interest, the Journal requires authors to disclose all and any potential conflicts of interest.
Funding statement
The research was supported by the Chinese National Science Foundation (32271187, 32071142, 32300646), Collaborative Innovation Center for Cancer Personalized Medicine - Clinical Research Fund of Hengrui Medicine (JZ21449020210617); The Basic Research Project of Changzhou Medical Center, Nanjing Medical University (CMCB202305);Natural Science Foundation of Xinjiang Uygur Autonomous Region (2023D01D05); Top innovative talent program of Nanjing Medical University (NJMUTY20230093); The Nature Science Foundation of Jiangsu Province (BK20230300);Medical research project of Jiangsu Provincial Health Commission (H2023103); Jiangsu Key Laboratory of Oral Diseases Research Open project fund (JSKLOD-KF-2303);Science and Technology Bureau self-funded project of Fuyang (FK202081049).
CRediT authorship contribution statement
Hongliang Gao: Conceptualization, Investigation, Writing – original draft, Visualization. Xing Peng: Conceptualization, Writing – original draft. Ning Li: Conceptualization, Writing – original draft. Liming Gou: Investigation, Writing – review & editing. Tao Xu: Investigation, Writing – review & editing. Yuqi Wang: Investigation, Writing – review & editing. Jian Qin: Investigation. Hui Liang: Investigation. Peiqi Ma: Investigation. Shu Li: Investigation. Jing Wu: Funding acquisition, Conceptualization, Supervision. Xihu Qin: Supervision. Bin Xue: Conceptualization, Funding acquisition, Supervision.
Declaration of competing interest
The authors declare no competing financial interest.
Acknowledgments
We greatly appreciate all the authors for their endeavors.
Footnotes
Review Article
Contributor Information
Shu Li, Email: wylishu@wnmc.edu.cn.
Jing Wu, Email: wujingshanx@163.com.
Xihu Qin, Email: qinxihu@126.com.
Bin Xue, Email: xuebin@njmu.edu.cn.
References
- 1.Levin V.A., Jiang X., Kagan R. Estrogen therapy for osteoporosis in the modern era. Osteoporos Int. 2018;29(5):1049–1055. doi: 10.1007/s00198-018-4414-z. [DOI] [PubMed] [Google Scholar]
- 2.Sobh M.M., Abdalbary M., Elnagar S., Nagy E., Elshabrawy N., Abdelsalam M., et al. Secondary osteoporosis and metabolic bone diseases. J Clin Med. 2022;11(9) doi: 10.3390/jcm11092382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin X., Xiong D., Peng Y.Q., Sheng Z.F., Wu X.Y., Wu X.P., et al. Epidemiology and management of osteoporosis in the People's Republic of China: current perspectives. Clin Interv Aging. 2015;10:1017–1033. doi: 10.2147/CIA.S54613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng P., Chang I., Wang J., Badreldin A.A., Li X., Yu B., et al. Loss of KDM4B impairs osteogenic differentiation of OMSCs and promotes oral bone aging. Int J Oral Sci. 2022;14(1):24. doi: 10.1038/s41368-022-00175-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Florencio-Silva R., Sasso G.R., Sasso-Cerri E., Simões M.J., Cerri P.S., et al. Biology of bone tissue: structure, function, and factors that influence bone cells. BioMed Res Int. 2015;2015 doi: 10.1155/2015/421746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datta H.K., Ng W.F., Walker J.A., Tuck S.P., Varanasi S.S. The cell biology of bone metabolism. J Clin Pathol. 2008;61(5):577–587. doi: 10.1136/jcp.2007.048868. [DOI] [PubMed] [Google Scholar]
- 7.Kenkre J.S., Bassett J. The bone remodelling cycle. Ann Clin Biochem. 2018;55(3):308–327. doi: 10.1177/0004563218759371. [DOI] [PubMed] [Google Scholar]
- 8.Woo M., Noh J.S., Kim M.J., Song Y.O., Lee H. Magma seawater inhibits hepatic lipid accumulation through suppression of lipogenic enzymes regulated by SREBPs in thioacetamide-injected rats. Mar Drugs. 2019;17(6) doi: 10.3390/md17060317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z., Wen X., Li N., Zhong C., Chen L., Zhang F., et al. The roles of hepatokine and osteokine in liver-bone crosstalk: advance in basic and clinical aspects. Front Endocrinol. 2023;14 doi: 10.3389/fendo.2023.1149233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nomura K., Tatsumi S., Miyagawa A., Shiozaki Y., Sasaki S., Kaneko I., et al. Hepatectomy-related hypophosphatemia: a novel phosphaturic factor in the liver-kidney axis. J Am Soc Nephrol. 2014;25(4):761–772. doi: 10.1681/ASN.2013060569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ye D.W., Rong X.L., Xu A.M., Guo J. Liver-adipose tissue crosstalk: a key player in the pathogenesis of glucolipid metabolic disease. Chin J Integr Med. 2017;23(6):410–414. doi: 10.1007/s11655-017-2810-4. [DOI] [PubMed] [Google Scholar]
- 12.Hamoud A.R., Weaver L., Stec D.E., Hinds T.D., Jr. Bilirubin in the liver-gut signaling Axis. Trends Endocrinol Metabol. 2018;29(3):140–150. doi: 10.1016/j.tem.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arteel G.E. Liver-lung axes in alcohol-related liver disease. Clin Mol Hepatol. 2020;26(4):670–676. doi: 10.3350/cmh.2020.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsubara Y., Kiyohara H., Teratani T., Mikami Y., Kanai T. Organ and brain crosstalk: the liver-brain axis in gastrointestinal, liver, and pancreatic diseases. Neuropharmacology. 2022;205 doi: 10.1016/j.neuropharm.2021.108915. [DOI] [PubMed] [Google Scholar]
- 15.Nussler A.K., Wildemann B., Freude T., Litzka C., Soldo P., Friess H. Chronic CCl4 intoxication causes liver and bone damage similar to the human pathology of hepatic osteodystrophy: a mouse model to analyse the liver-bone axis. Arch Toxicol. 2014;88(4):997–1006. doi: 10.1007/s00204-013-1191-5. [DOI] [PubMed] [Google Scholar]
- 16.Lu K., Shi T.S., Shen S.Y., Shi Y., Gao H.L., Wu J. Defects in a liver-bone axis contribute to hepatic osteodystrophy disease progression. Cell Metabol. 2022;34(3):441–457 e7. doi: 10.1016/j.cmet.2022.02.006. [DOI] [PubMed] [Google Scholar]
- 17.Jin X., Li Y., Li J., Cheng L., Yao Y., Shen H., et al. Acute bone damage through liver-bone axis induced by thioacetamide in rats. BMC Pharmacol Toxicol. 2022;23(1):29. doi: 10.1186/s40360-022-00568-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J., Lei H., Wang T., Xiong X. Liver-bone crosstalk in non-alcoholic fatty liver disease: clinical implications and underlying pathophysiology. Front Endocrinol. 2023;14 doi: 10.3389/fendo.2023.1161402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee N.K., Sowa H., Hinoi E., Ferron M., Ahn J.D., Confavreux C., et al. Endocrine regulation of energy metabolism by the skeleton. Cell. 2007;130(3):456–469. doi: 10.1016/j.cell.2007.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang W., Wei T., Hu L., Chen M., Tong L., Zhou W., et al. An integrated multi-omics analysis reveals osteokines involved in global regulation. Cell Metabol. 2024;36(5):1144–1163 e7. doi: 10.1016/j.cmet.2024.03.006. [DOI] [PubMed] [Google Scholar]
- 21.Liu Z.Z., Hong C.G., Hu W.B., Chen M.L., Duan R., Li H.M., et al. Autophagy receptor OPTN (optineurin) regulates mesenchymal stem cell fate and bone-fat balance during aging by clearing FABP3. Autophagy. 2021;17(10):2766–2782. doi: 10.1080/15548627.2020.1839286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowden Davies K.A., Pickles S., Sprung V.S., Kemp G.J., Alam U., Moore D.R., et al. Reduced physical activity in young and older adults: metabolic and musculoskeletal implications. Ther Adv Endocrinol Metab. 2019;10 doi: 10.1177/2042018819888824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ponti F., Santoro A., Mercatelli D., Gasperini C., Conte M., Martucci M., et al. Aging and imaging assessment of body composition: from fat to facts. Front Endocrinol. 2019;10:861. doi: 10.3389/fendo.2019.00861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eslam M., Newsome P.N., Sarin S.K., Anstee Q.M., Targher G., Romero-Gomez M., et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73(1):202–209. doi: 10.1016/j.jhep.2020.03.039. [DOI] [PubMed] [Google Scholar]
- 25.Rinella M.E., Lazarus J.V., Ratziu V., Francque S.M., Sanyal A.J., Kanwal F., et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Ann Hepatol. 2024;29(1) doi: 10.1016/j.aohep.2023.101133. [DOI] [PubMed] [Google Scholar]
- 26.Shen Z., Cen L., Chen X., Pan J., Li Y., Chen W., et al. Increased risk of low bone mineral density in patients with non-alcoholic fatty liver disease: a cohort study. Eur J Endocrinol. 2020;182(2):157–164. doi: 10.1530/EJE-19-0699. [DOI] [PubMed] [Google Scholar]
- 27.Jadzic J., Milovanovic P., Tomanovic N., Zivkovic V., Djukic D., Nikolic S., et al. Micro-scale vertebral features in postmenopausal women with alcohol-associated and metabolic-associated fatty liver disease: ex vivo bone quality analyses. J Endocrinol Invest. 2024;47(1):131–140. doi: 10.1007/s40618-023-02130-3. [DOI] [PubMed] [Google Scholar]
- 28.Xie R., Liu M. Relationship between non-alcoholic fatty liver disease and degree of hepatic steatosis and bone mineral density. Front Endocrinol. 2022;13 doi: 10.3389/fendo.2022.857110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Upala S., Jaruvongvanich V., Wijarnpreecha K., Sanguankeo A. Nonalcoholic fatty liver disease and osteoporosis: a systematic review and meta-analysis. J Bone Miner Metabol. 2017;35(6):685–693. doi: 10.1007/s00774-016-0807-2. [DOI] [PubMed] [Google Scholar]
- 30.Jing Y., Jing Y., Zhou Y., Zhou F., Wang X., Tao B., et al. SIRT2 deficiency prevents age-related bone loss in rats by inhibiting osteoclastogenesis. Cell Mol Biol. 2019;65(7):66–71. [PubMed] [Google Scholar]
- 31.Wang Y., Yang J., Hong T., Chen X., Cui L. SIRT2: controversy and multiple roles in disease and physiology. Ageing Res Rev. 2019;55 doi: 10.1016/j.arr.2019.100961. [DOI] [PubMed] [Google Scholar]
- 32.Lin L., Guo Z., He E., Long X., Wang D., Zhang Y., et al. SIRT2 regulates extracellular vesicle-mediated liver-bone communication. Nat Metab. 2023;5(5):821–841. doi: 10.1038/s42255-023-00803-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lemos V., de Oliveira R.M., Naia L., Szegö É., Ramos E., Pinho S., et al. The NAD+-dependent deacetylase SIRT2 attenuates oxidative stress and mitochondrial dysfunction and improves insulin sensitivity in hepatocytes. Hum Mol Genet. 2017;26(21):4105–4117. doi: 10.1093/hmg/ddx298. [DOI] [PubMed] [Google Scholar]
- 34.Zhang M., Pan Y., Dorfman R.G., Yin Y., Zhou Q., Huang S., et al. Sirtinol promotes PEPCK1 degradation and inhibits gluconeogenesis by inhibiting deacetylase SIRT2. Sci Rep. 2017;7(1):7. doi: 10.1038/s41598-017-00035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnan J., Danzer C., Simka T., Ukropec J., Walter K.M., Kumpf S., et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012;26(3):259–270. doi: 10.1101/gad.180406.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S., Guo L. The role of Sirtuin 2 in liver - an extensive and complex biological process. Life Sci. 2024;339 doi: 10.1016/j.lfs.2024.122431. [DOI] [PubMed] [Google Scholar]
- 37.Wang Y.P., Zhou L.S., Zhao Y.Z., Wang S.W., Chen L.L., Liu L.X., et al. Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J. 2014;33(12):1304–1320. doi: 10.1002/embj.201387224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan Y., Zhang H., Zheng Y., Zhou J., Yuan J., Yu Y., et al. Resveratrol exerts antioxidant effects by activating SIRT2 to deacetylate Prx1. Biochemistry. 2017;56(48):6325–6328. doi: 10.1021/acs.biochem.7b00859. [DOI] [PubMed] [Google Scholar]
- 39.Rothgiesser K.M., Erener S., Waibel S., Lüscher B., Hottiger M.O. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci. 2010;123(Pt 24):4251–4258. doi: 10.1242/jcs.073783. [DOI] [PubMed] [Google Scholar]
- 40.Wang X., Buechler N.L., Long D.L., Furdui C.M., Yoza B.K., McCall C.E., et al. Cysteine thiol oxidation on SIRT2 regulates inflammation in obese mice with sepsis. Inflammation. 2019;42(1):156–169. doi: 10.1007/s10753-018-0881-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He M., Chiang H.H., Luo H., Zheng Z., Qiao Q., Wang L., et al. An acetylation switch of the NLRP3 inflammasome regulates aging-associated chronic inflammation and insulin resistance. Cell Metabol. 2020;31(3):580–591 e5. doi: 10.1016/j.cmet.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang X., Park S.H., Chang H.C., Shapiro J.S., Vassilopoulos A., Sawicki K.T., et al. Sirtuin 2 regulates cellular iron homeostasis via deacetylation of transcription factor NRF2. J Clin Invest. 2017;127(4):1505–1516. doi: 10.1172/JCI88574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu J.Z., Zhou Y.M., Zhang L.L., Chen X.J., Yang Y.Y., Zhang D., et al. BMP9 reduces age-related bone loss in mice by inhibiting osteoblast senescence through Smad1-Stat1-P21 axis. Cell Death Dis. 2022;8(1):254. doi: 10.1038/s41420-022-01048-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang X., Wei W., Krzeszinski J.Y., Wang Y., Wan Y. A liver-bone endocrine relay by IGFBP1 promotes osteoclastogenesis and mediates FGF21-induced bone resorption. Cell Metabol. 2015;22(5):811–824. doi: 10.1016/j.cmet.2015.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Talukdar S., Zhou Y., Li D., Rossulek M., Dong J., Somayaji V., et al. A long-acting FGF21 molecule, PF-05231023, decreases body weight and improves lipid profile in non-human primates and type 2 diabetic subjects. Cell Metabol. 2016;23(3):427–440. doi: 10.1016/j.cmet.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Lee S.Y., Fam K.D., Chia K.L., Yap M.M.C., Goh J., Yeo K.P., et al. Age-related bone loss is associated with FGF21 but not IGFBP1 in healthy adults. Exp Physiol. 2020;105(4):622–631. doi: 10.1113/EP088351. [DOI] [PubMed] [Google Scholar]
- 47.Perrini S., Laviola L., Carreira M.C., Cignarelli A., Natalicchio A., Giorgino F. The GH/IGF1 axis and signaling pathways in the muscle and bone: mechanisms underlying age-related skeletal muscle wasting and osteoporosis. J Endocrinol. 2010;205(3):201–210. doi: 10.1677/JOE-09-0431. [DOI] [PubMed] [Google Scholar]
- 48.Junnila R.K., List E.O., Berryman D.E., Murrey J.W., Kopchick J.J. The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol. 2013;9(6):366–376. doi: 10.1038/nrendo.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Munoz M., Robinson K., Shibli-Rahhal A. Bone health and osteoporosis prevention and treatment. Clin Obstet Gynecol. 2020;63(4):770–787. doi: 10.1097/GRF.0000000000000572. [DOI] [PubMed] [Google Scholar]
- 50.Frost H.M. On the estrogen-bone relationship and postmenopausal bone loss: a new model. J Bone Miner Res. 1999;14(9):1473–1477. doi: 10.1359/jbmr.1999.14.9.1473. [DOI] [PubMed] [Google Scholar]
- 51.Grumbach M.M. Estrogen, bone, growth and sex: a sea change in conventional wisdom. J Pediatr Endocrinol Metab. 2000;13(Suppl 6):1439–1455. doi: 10.1515/jpem-2000-s619. [DOI] [PubMed] [Google Scholar]
- 52.Imai Y., Youn M.Y., Kondoh S., Nakamura T., Kouzmenko A., Matsumoto T., et al. Estrogens maintain bone mass by regulating expression of genes controlling function and life span in mature osteoclasts. Ann N Y Acad Sci. 2009;1173(Suppl 1):E31–E39. doi: 10.1111/j.1749-6632.2009.04954.x. [DOI] [PubMed] [Google Scholar]
- 53.Kousteni S., Bellido T., Plotkin L.I., O'Brien C.A., Bodenner D.L., Han L., et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104(5):719–730. [PubMed] [Google Scholar]
- 54.Manolagas S.C. From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev. 2010;31(3):266–300. doi: 10.1210/er.2009-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hajam Y.A., Rani R., Ganie S.Y., Sheikh T.A., Javaid D., Qadri S.S., et al. Oxidative stress in human pathology and aging: molecular mechanisms and perspectives. Cells. 2022;11(3) doi: 10.3390/cells11030552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bellezza I., Giambanco I., Minelli A., Donato R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim Biophys Acta Mol Cell Res. 2018;1865(5):721–733. doi: 10.1016/j.bbamcr.2018.02.010. [DOI] [PubMed] [Google Scholar]
- 57.Yang R., Li J., Zhang J., Xue Q., Qin R., Wang R., et al. 17beta-estradiol plays the anti-osteoporosis role via a novel ESR1-Keap1-Nrf2 axis-mediated stress response activation and Tmem119 upregulation. Free Radic Biol Med. 2023;195:231–244. doi: 10.1016/j.freeradbiomed.2022.12.102. [DOI] [PubMed] [Google Scholar]
- 58.Vaananen H.K., Harkonen P.L. Estrogen and bone metabolism. Maturitas. 1996;23(Suppl):S65–S69. doi: 10.1016/0378-5122(96)01015-8. [DOI] [PubMed] [Google Scholar]
- 59.Cheng C.H., Chen L.R., Chen K.H. Osteoporosis due to hormone imbalance: an overview of the effects of estrogen deficiency and glucocorticoid overuse on bone turnover. Int J Mol Sci. 2022;23(3) doi: 10.3390/ijms23031376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hughes D.E., Dai A., Tiffee J.C., Li H.H., Mundy G.R., Boyce B.F. Estrogen promotes apoptosis of murine osteoclasts mediated by TGF-beta. Nat Med. 1996;2(10):1132–1136. doi: 10.1038/nm1096-1132. [DOI] [PubMed] [Google Scholar]
- 61.Tian W.H., Wang Z., Yue Y.X., Li H., Li Z.J., Han R.L., et al. miR-34a-5p increases hepatic triglycerides and total cholesterol levels by regulating ACSL1 protein expression in laying hens. Int J Mol Sci. 2019;20(18) doi: 10.3390/ijms20184420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yasuda M., Shimizu I., Shiba M., Ito S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology. 1999;29(3):719–727. doi: 10.1002/hep.510290307. [DOI] [PubMed] [Google Scholar]
- 63.Cooper K.M., Delk M., Devuni D., Sarkar M. Sex differences in chronic liver disease and benign liver lesions. JHEP Rep. 2023;5(11) doi: 10.1016/j.jhepr.2023.100870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rikkonen T., Sund R., Sirola J., Honkanen R., Poole K.E.S., Kröger H. Obesity is associated with early hip fracture risk in postmenopausal women: a 25-year follow-up. Osteoporos Int. 2021;32(4):769–777. doi: 10.1007/s00198-020-05665-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y., Huang X., Sun K., Li M., Wang X., Han T., et al. The potential role of serum IGF-1 and leptin as biomarkers: towards screening for and diagnosing postmenopausal osteoporosis. J Inflamm Res. 2022;15:533–543. doi: 10.2147/JIR.S344009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mantovani A., Sani E., Fassio A., Colecchia A., Viapiana O., Gatti D., et al. Association between non-alcoholic fatty liver disease and bone turnover biomarkers in post-menopausal women with type 2 diabetes. Diabetes Metab. 2019;45(4):347–355. doi: 10.1016/j.diabet.2018.10.001. [DOI] [PubMed] [Google Scholar]
- 67.Demirdal U.S., Ciftci I.H., Kavuncu V. Markers of autoimmune liver diseases in postmenopausal women with osteoporosis. Clinics. 2010;65(10):971–974. doi: 10.1590/S1807-59322010001000008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Che J., Yang J., Zhao B., Zhang G., Wang L., Peng S., et al. The effect of abnormal iron metabolism on osteoporosis. Biol Trace Elem Res. 2020;195(2):353–365. doi: 10.1007/s12011-019-01867-4. [DOI] [PubMed] [Google Scholar]
- 69.Zhao G.Y., Di D.H., Wang B., Huang X., Xu Y.J. Effects of mouse hepcidin 1 treatment on osteoclast differentiation and intracellular iron concentration. Inflammation. 2015;38(2):718–727. doi: 10.1007/s10753-014-9982-2. [DOI] [PubMed] [Google Scholar]
- 70.Chen B., Li G.F., Shen Y., Huang X.I., Xu Y.J. Reducing iron accumulation: a potential approach for the prevention and treatment of postmenopausal osteoporosis. Exp Ther Med. 2015;10(1):7–11. doi: 10.3892/etm.2015.2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hou Y., Zhang S., Wang L., Li J., Qu G., He J., et al. Estrogen regulates iron homeostasis through governing hepatic hepcidin expression via an estrogen response element. Gene. 2012;511(2):398–403. doi: 10.1016/j.gene.2012.09.060. [DOI] [PubMed] [Google Scholar]
- 72.Kukla M., Waluga M., Żorniak M., Berdowska A., Wosiewicz P., Sawczyn T., et al. Serum omentin and vaspin levels in cirrhotic patients with and without portal vein thrombosis. World J Gastroenterol. 2017;23(14):2613–2624. doi: 10.3748/wjg.v23.i14.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goral V., Simsek M., Mete N. Hepatic osteodystrophy and liver cirrhosis. World J Gastroenterol. 2010;16(13):1639–1643. doi: 10.3748/wjg.v16.i13.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jeong H.M., Kim D.J. Bone diseases in patients with chronic liver disease. Int J Mol Sci. 2019;20(17) doi: 10.3390/ijms20174270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wakolbinger R., Muschitz C., Scheriau G., Bodlaj G., Kocijan R., Feichtinger X., et al. Bone microarchitecture and bone turnover in hepatic cirrhosis. Osteoporos Int. 2019;30(6):1195–1204. doi: 10.1007/s00198-019-04870-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Newman M.E., Shapira B., Lerer B. Regulation of 5-hydroxytryptamine1A receptor function in rat hippocampus by short- and long-term administration of 5-hydroxytryptamine1A agonist and antidepressants. J Pharmacol Exp Therapeut. 1992;260(1):16–20. [PubMed] [Google Scholar]
- 77.Vyzantiadis T., Theodoridou S., Giouleme O., Harsoulis P., Evgenidis N., Vyzantiadis A. Serum concentrations of insulin-like growth factor-I (IGF-I) in patients with liver cirrhosis. Hepato-Gastroenterology. 2003;50(51):814–816. [PubMed] [Google Scholar]
- 78.Ronsoni M.F., Lazzarotto C., Fayad L., Silva M.C., Nogueira C.L., Bazzo M.L., et al. IGF-I and IGFBP-3 serum levels in patients hospitalized for complications of liver cirrhosis. Ann Hepatol. 2013;12(3):456–463. [PubMed] [Google Scholar]
- 79.Colakoğlu O., Taşkiran B., Colakoğlu G., Kizildağ S., Ari Ozcan F., Unsal B. Serum insulin like growth factor-1 (IGF-1) and insulin like growth factor binding protein-3 (IGFBP-3) levels in liver cirrhosis. Turk J Gastroenterol. 2007;18(4):245–249. [PubMed] [Google Scholar]
- 80.Yakar S., Rosen C.J., Beamer W.G., Ackert-Bicknell C.L., Wu Y., Liu J.L., et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110(6):771–781. doi: 10.1172/JCI15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhou W.C., Zhang Q.B., Qiao L. Pathogenesis of liver cirrhosis. World J Gastroenterol. 2014;20(23):7312–7324. doi: 10.3748/wjg.v20.i23.7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim B.J., Lee S.H., Kwak M.K., Isales C.M., Koh J.M., Hamrick M.W. Inverse relationship between serum hsCRP concentration and hand grip strength in older adults: a nationwide population-based study. Aging (Albany NY) 2018;10(8):2051–2061. doi: 10.18632/aging.101529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fontes-Cal T.C.M., Mattos R.T., Medeiros N.I., Pinto B.F., Belchior-Bezerra M., Roque-Souza B., et al. Crosstalk between plasma cytokines, inflammation, and liver damage as a new strategy to monitoring NAFLD progression. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.708959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Adami G. Regulation of bone mass in inflammatory diseases. Best Pract Res Clin Endocrinol Metabol. 2022;36(2) doi: 10.1016/j.beem.2021.101611. [DOI] [PubMed] [Google Scholar]
- 85.Wang J.S., Mazur C.M., Wein M.N. Sclerostin and osteocalcin: candidate bone-produced hormones. Front Endocrinol. 2021;12 doi: 10.3389/fendo.2021.584147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferron M., Hinoi E., Karsenty G., Ducy P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc Natl Acad Sci U S A. 2008;105(13):5266–5270. doi: 10.1073/pnas.0711119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ferron M., McKee M.D., Levine R.L., Ducy P., Karsenty G. Intermittent injections of osteocalcin improve glucose metabolism and prevent type 2 diabetes in mice. Bone. 2012;50(2):568–575. doi: 10.1016/j.bone.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhou B., Li H., Xu L., Zang W., Wu S., Sun H. Osteocalcin reverses endoplasmic reticulum stress and improves impaired insulin sensitivity secondary to diet-induced obesity through nuclear factor-kappaB signaling pathway. Endocrinology. 2013;154(3):1055–1068. doi: 10.1210/en.2012-2144. [DOI] [PubMed] [Google Scholar]
- 89.Capra F., Casaril M., Gabrielli G.B., Stanzial A., Ferrari S., Gandini G., et al. Plasma osteocalcin levels in liver cirrhosis. Ital J Gastroenterol. 1991;23(3):124–127. [PubMed] [Google Scholar]
- 90.Brennan-Speranza T.C., Conigrave A.D. Osteocalcin: an osteoblast-derived polypeptide hormone that modulates whole body energy metabolism. Calcif Tissue Int. 2015;96(1):1–10. doi: 10.1007/s00223-014-9931-y. [DOI] [PubMed] [Google Scholar]
- 91.Mao H., Li L., Fan Q., Angelini A., Saha P.K., Coarfa C., et al. Endothelium-specific depletion of LRP1 improves glucose homeostasis through inducing osteocalcin. Nat Commun. 2021;12(1):5296. doi: 10.1038/s41467-021-25673-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Poole K.E., van Bezooijen R.L., Loveridge N., Hamersma H., Papapoulos S.E., Löwik C.W., et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. Faseb J. 2005;19(13):1842–1844. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 93.Baron R., Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148(6):2635–2643. doi: 10.1210/en.2007-0270. [DOI] [PubMed] [Google Scholar]
- 94.Jadzic J., Milovanovic P.D., Cvetkovic D., Zivkovic V., Nikolic S., Tomanovic N., et al. The altered osteocytic expression of connexin 43 and sclerostin in human cadaveric donors with alcoholic liver cirrhosis: potential treatment targets. J Anat. 2022;240(6):1162–1173. doi: 10.1111/joa.13621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rhee Y., Kim W.J., Han K.J., Lim S.K., Kim S.H. Effect of liver dysfunction on circulating sclerostin. J Bone Miner Metabol. 2014;32(5):545–549. doi: 10.1007/s00774-013-0524-z. [DOI] [PubMed] [Google Scholar]
- 96.González-Reimers E., Martín-González C., de la Vega-Prieto M.J., Pelazas-González R., Fernández-Rodríguez C., López-Prieto J., et al. Serum sclerostin in alcoholics: a pilot study. Alcohol Alcohol. 2013;48(3):278–282. doi: 10.1093/alcalc/ags136. [DOI] [PubMed] [Google Scholar]
- 97.Wakolbinger R., Muschitz C., Wallwitz J., Bodlaj G., Feichtinger X., Schanda J.E., et al. Serum levels of sclerostin reflect altered bone microarchitecture in patients with hepatic cirrhosis. Wien Klin Wochenschr. 2020;132(1–2):19–26. doi: 10.1007/s00508-019-01595-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maurel D.B., Boisseau N., Benhamou C.L., Jaffre C. Alcohol and bone: review of dose effects and mechanisms. Osteoporos Int. 2012;23(1):1–16. doi: 10.1007/s00198-011-1787-7. [DOI] [PubMed] [Google Scholar]
- 99.Pietschmann P., Resch H., Müller C., Woloszczuk W., Willvonseder R. Decreased serum osteocalcin levels in patients with liver cirrhosis. Bone Miner. 1990;8(2):103–108. doi: 10.1016/0169-6009(90)90113-t. [DOI] [PubMed] [Google Scholar]
- 100.Angulo P., Grandison G.A., Fong D.G., Keach J.C., Lindor K.D., Bjornsson E., et al. Bone disease in patients with primary sclerosing cholangitis. Gastroenterology. 2011;140(1):180–188. doi: 10.1053/j.gastro.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Keller S., Ittrich H., Schramm C., Lohse A.W., Amling M., Adam G., et al. Diffusion-weighted MRI for detection of hepatic osteodystrophy in primary sclerosing cholangitis: a comparison study with dual-energy X-ray absorptiometry. Jpn J Radiol. 2016;34(10):677–683. doi: 10.1007/s11604-016-0573-z. [DOI] [PubMed] [Google Scholar]
- 102.Akkawi I., Zmerly H. Osteoporosis: current concepts. Joints. 2018;6(2):122–127. doi: 10.1055/s-0038-1660790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pop T.L., Sîrbe C., Benţa G., Mititelu A., Grama A. The role of vitamin D and vitamin D binding protein in chronic liver diseases. Int J Mol Sci. 2022;23(18) doi: 10.3390/ijms231810705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zi C., Wang D., Gao Y., He L. The role of Th17 cells in endocrine organs: involvement of the gut, adipose tissue, liver and bone. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.1104943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schmidt T., Schwinge D., Rolvien T., Jeschke A., Schmidt C., Neven M., et al. Th17 cell frequency is associated with low bone mass in primary sclerosing cholangitis. J Hepatol. 2019;70(5):941–953. doi: 10.1016/j.jhep.2018.12.035. [DOI] [PubMed] [Google Scholar]
- 106.Gallego-Rojo F.J., Gonzalez-Calvin J.L., Muñoz-Torres M., Mundi J.L., Fernandez-Perez R., et al. Bone mineral density, serum insulin-like growth factor I, and bone turnover markers in viral cirrhosis. Hepatology. 1998;28(3):695–699. doi: 10.1002/hep.510280315. [DOI] [PubMed] [Google Scholar]
- 107.Chen C.H., Lin C.L., Kao C.H. Association between chronic hepatitis B virus infection and risk of osteoporosis: a nationwide population-based study. Medicine (Baltim) 2015;94(50):e2276. doi: 10.1097/MD.0000000000002276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wijarnpreecha K., Thongprayoon C., Panjawatanan P., Phatharacharukul P., Ungprasert P. Hepatitis C virus infection and risk of osteoporosis: a meta-analysis. Saudi J Gastroenterol. 2017;23(4):216–221. doi: 10.4103/sjg.SJG_452_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xie X., Huang R., Li X., Li N., Zhang H., Xu S., et al. Association between hepatitis B virus infection and risk of osteoporosis: a systematic review and meta-analysis: a protocol for systematic review. Medicine (Baltim) 2020;99(16) doi: 10.1097/MD.0000000000019719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tao J., Yan Z., Huang W., Feng T. Seropositive for hepatitis B and C viruses is associated with the risk of decreased bone mineral density in adults: an analysis of studies from the NHANES database. Front Med. 2023;10 doi: 10.3389/fmed.2023.1120083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gonzalez-Calvin J.L., Gallego-Rojo F., Fernandez-Perez R., Casado-Caballero F., Ruiz-Escolano E., Olivares E.G. Osteoporosis, mineral metabolism, and serum soluble tumor necrosis factor receptor p55 in viral cirrhosis. J Clin Endocrinol Metab. 2004;89(9):4325–4330. doi: 10.1210/jc.2004-0077. [DOI] [PubMed] [Google Scholar]
- 112.Chen Y.Y., Fang W.H., Wang C.C., Kao T.W., Chang Y.W., Yang H.F., et al. Crosssectional assessment of bone mass density in adults with hepatitis B virus and hepatitis C virus infection. Sci Rep. 2019;9(1):5069. doi: 10.1038/s41598-019-41674-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Byrne D.D., Newcomb C.W., Carbonari D.M., Nezamzadeh M.S., Leidl K.B., Herlim M., et al. Risk of hip fracture associated with untreated and treated chronic hepatitis B virus infection. J Hepatol. 2014;61(2):210–218. doi: 10.1016/j.jhep.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kim K.H., Shin H.J., Kim K., Choi H.M., Rhee S.H., Moon H.B., et al. Hepatitis B virus X protein induces hepatic steatosis via transcriptional activation of SREBP1 and PPARgamma. Gastroenterology. 2007;132(5):1955–1967. doi: 10.1053/j.gastro.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 115.Wang X.H., Cheng P.P., Jiang F., Jiao X.Y. The effect of hepatitis B virus infection on hepcidin expression in hepatitis B patients. Ann Clin Lab Sci. 2013;43(2):126–134. [PubMed] [Google Scholar]
- 116.Dalla Grana E., Rigo F., Lanzafame M., Lattuada E., Suardi S., Mottes M., et al. Relationship between vertebral fractures, bone mineral density, and osteometabolic profile in HIV and hepatitis B and C-infected patients treated with ART. Front Endocrinol. 2019;10:302. doi: 10.3389/fendo.2019.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Terrault N.A., Lok A.S.F., McMahon B.J., Chang K.M., Hwang J.P., Jonas M.M., et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67(4):1560–1599. doi: 10.1002/hep.29800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Eslam M., Sanyal A.J., George J. MAFLD: a consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. 2020;158(7):1999–2014 e1. doi: 10.1053/j.gastro.2019.11.312. [DOI] [PubMed] [Google Scholar]
- 119.Younossi Z., Anstee Q.M., Marietti M., Hardy T., Henry L., Eslam M., et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. doi: 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 120.Musio A., Perazza F., Leoni L., Stefanini B., Dajti E., Menozzi R., et al. Osteosarcopenia in NAFLD/MAFLD: an underappreciated clinical problem in chronic liver disease. Int J Mol Sci. 2023;24(8) doi: 10.3390/ijms24087517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu X.L., Ming Y.N., Zhang J.Y., Chen X.Y., Zeng M.D., Mao Y.M. Gene-metabolite network analysis in different nonalcoholic fatty liver disease phenotypes. Exp Mol Med. 2017;49(1):e283. doi: 10.1038/emm.2016.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tilg H., Moschen A.R., Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol. 2017;14(1):32–42. doi: 10.1038/nrgastro.2016.147. [DOI] [PubMed] [Google Scholar]
- 123.Lee H.L., Yi T., Woo K.M., Ryoo H.M., Kim G.S., Baek J.H. Msx2 mediates the inhibitory action of TNF-alpha on osteoblast differentiation. Exp Mol Med. 2010;42(6):437–445. doi: 10.3858/emm.2010.42.6.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim G., Kim K.J., Rhee Y., Lim S.K. Significant liver fibrosis assessed using liver transient elastography is independently associated with low bone mineral density in patients with non-alcoholic fatty liver disease. PLoS One. 2017;12(7):e0182202. doi: 10.1371/journal.pone.0182202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Filip R., Radzki R.P., Bienko M. Novel insights into the relationship between nonalcoholic fatty liver disease and osteoporosis. Clin Interv Aging. 2018;13:1879–1891. doi: 10.2147/CIA.S170533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liang S., Cheng X., Hu Y., Song R., Li G. Insulin-like growth factor 1 and metabolic parameters are associated with nonalcoholic fatty liver disease in obese children and adolescents. Acta Paediatr. 2017;106(2):298–303. doi: 10.1111/apa.13685. [DOI] [PubMed] [Google Scholar]
- 127.Yao Y., Miao X., Zhu D., Li D., Zhang Y., Song C., et al. Insulin-like growth factor-1 and non-alcoholic fatty liver disease: a systemic review and meta-analysis. Endocrine. 2019;65(2):227–237. doi: 10.1007/s12020-019-01982-1. [DOI] [PubMed] [Google Scholar]
- 128.Takahashi Y. The role of growth hormone and insulin-like growth factor-I in the liver. Int J Mol Sci. 2017;18(7) doi: 10.3390/ijms18071447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nishizawa H., Iguchi G., Fukuoka H., Takahashi M., Suda K., Bando H., et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci Rep. 2016;6 doi: 10.1038/srep34605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Dushay J., Chui P.C., Gopalakrishnan G.S., Varela-Rey M., Crawley M., Fisher F.M., et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. 2010;139(2):456–463. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Morris-Stiff G., Feldstein A.E. Fibroblast growth factor 21 as a biomarker for NAFLD: integrating pathobiology into clinical practice. J Hepatol. 2010;53(5):795–796. doi: 10.1016/j.jhep.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 132.Ishijima M., Rittling S.R., Yamashita T., Tsuji K., Kurosawa H., Nifuji A., et al. Enhancement of osteoclastic bone resorption and suppression of osteoblastic bone formation in response to reduced mechanical stress do not occur in the absence of osteopontin. J Exp Med. 2001;193(3):399–404. doi: 10.1084/jem.193.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Amin S.E.l., Amrousy D., Elrifaey S., Gamal R., Hodeib H. Serum osteocalcin levels in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr. 2018;66(1):117–121. doi: 10.1097/MPG.0000000000001768. [DOI] [PubMed] [Google Scholar]
- 134.Du M., Wang X., Yuan L., Liu B., Mao X., Huang D., et al. Targeting NFATc4 attenuates non-alcoholic steatohepatitis in mice. J Hepatol. 2020;73(6):1333–1346. doi: 10.1016/j.jhep.2020.07.030. [DOI] [PubMed] [Google Scholar]
- 135.Kiefer F.W., Neschen S., Pfau B., Legerer B., Neuhofer A., Kahle M., et al. Osteopontin deficiency protects against obesity-induced hepatic steatosis and attenuates glucose production in mice. Diabetologia. 2011;54(8):2132–2142. doi: 10.1007/s00125-011-2170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Neve A., Corrado A., Cantatore F.P. Osteocalcin: skeletal and extra-skeletal effects. J Cell Physiol. 2013;228(6):1149–1153. doi: 10.1002/jcp.24278. [DOI] [PubMed] [Google Scholar]
- 137.Yilmaz Y., Kurt R., Eren F., Imeryuz N. Serum osteocalcin levels in patients with nonalcoholic fatty liver disease: association with ballooning degeneration. Scand J Clin Lab Invest. 2011;71(8):631–636. doi: 10.3109/00365513.2011.604427. [DOI] [PubMed] [Google Scholar]
- 138.Pi M., Nishimoto S.K., Darryl Quarles L. Explaining divergent observations regarding osteocalcin/GPRC6A endocrine signaling. Endocrinology. 2021;162(4) doi: 10.1210/endocr/bqab011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Polyzos S.A., Anastasilakis A.D., Kountouras J., Makras P., Papatheodorou A., Kokkoris P., et al. Circulating sclerostin and Dickkopf-1 levels in patients with nonalcoholic fatty liver disease. J Bone Miner Metabol. 2016;34(4):447–456. doi: 10.1007/s00774-015-0687-x. [DOI] [PubMed] [Google Scholar]
- 140.Lau K.H., Baylink D.J., Zhou X.D., Rodriguez D., Bonewald L.F., Li Z., et al. Osteocyte-derived insulin-like growth factor I is essential for determining bone mechanosensitivity. Am J Physiol Endocrinol Metab. 2013;305(2):E271–E281. doi: 10.1152/ajpendo.00092.2013. [DOI] [PubMed] [Google Scholar]
- 141.Heida A., Gruben N., Catrysse L., Koehorst M., Koster M., Kloosterhuis N.J., et al. The hepatocyte IKK:NF-kappaB axis promotes liver steatosis by stimulating de novo lipogenesis and cholesterol synthesis. Mol Metabol. 2021;54 doi: 10.1016/j.molmet.2021.101349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kizilgul M., Ozcelik O., Delibasi T. Bone health and vitamin D status in alcoholic liver disease. Indian J Gastroenterol. 2016;35(4):253–259. doi: 10.1007/s12664-016-0652-1. [DOI] [PubMed] [Google Scholar]
- 143.Liu Y., Kou X., Chen C., Yu W., Su Y., Kim Y., et al. Chronic high dose alcohol induces osteopenia via activation of mTOR signaling in bone marrow mesenchymal stem cells. Stem Cell. 2016;34(8):2157–2168. doi: 10.1002/stem.2392. [DOI] [PubMed] [Google Scholar]
- 144.Chen X., Li M., Yan J., Liu T., Pan G., Yang H., et al. Alcohol induces cellular senescence and impairs osteogenic potential in bone marrow-derived mesenchymal stem cells. Alcohol Alcohol. 2017;52(3):289–297. doi: 10.1093/alcalc/agx006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chacko K.R., Reinus J. Spectrum of alcoholic liver disease. Clin Liver Dis. 2016;20(3):419–427. doi: 10.1016/j.cld.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 146.Santolaria F., González-González G., González-Reimers E., Martínez-Riera A., Milena A., Rodgíguez-Moreno F., et al. Effects of alcohol and liver cirrhosis on the GH-IGF-I axis. Alcohol Alcohol. 1995;30(6):703–708. [PubMed] [Google Scholar]
- 147.Rojdmark S., Brismar K. Decreased IGF-I bioavailability after ethanol abuse in alcoholics: partial restitution after short-term abstinence. J Endocrinol Invest. 2001;24(7):476–482. doi: 10.1007/BF03343879. [DOI] [PubMed] [Google Scholar]
- 148.Nedić O., Nikolić J.A., Hajduković-Dragojlović L., Todorović V., Masnikosa R. Alterations of IGF-binding proteins in patients with alcoholic liver cirrhosis. Alcohol. 2000;21(3):223–229. doi: 10.1016/s0741-8329(00)00090-2. [DOI] [PubMed] [Google Scholar]
- 149.Kasukawa Y., Miyakoshi N., Mohan S. The anabolic effects of GH/IGF system on bone. Curr Pharmaceut Des. 2004;10(21):2577–2592. doi: 10.2174/1381612043383764. [DOI] [PubMed] [Google Scholar]
- 150.Seddiqi H., Klein-Nulend J., Jin J. Osteocyte mechanotransduction in orthodontic tooth movement. Curr Osteoporos Rep. 2023;21(6):731–742. doi: 10.1007/s11914-023-00826-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Xu D., Cai J., Wan Z.K., Gao H., Sun Y. Pathophysiological role of prostaglandin E synthases in liver diseases. Prostag Other Lipid Mediat. 2021;154 doi: 10.1016/j.prostaglandins.2021.106552. [DOI] [PubMed] [Google Scholar]
- 152.Pan X.Y., Wang L., You H.M., Cheng M., Yang Y., Huang C., et al. Alternative activation of macrophages by prostacyclin synthase ameliorates alcohol induced liver injury. Lab Invest. 2021;101(9):1210–1224. doi: 10.1038/s41374-021-00531-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Furue A., Hattori K., Hosono K., Tanabe M., Sato E., Honda M., et al. Inhibition of TP signaling promotes endometriosis growth and neovascularization. Mol Med Rep. 2023;28(4) doi: 10.3892/mmr.2023.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Blaschke M., Koepp R., Cortis J., Komrakova M., Schieker M., Hempel U., et al. IL-6, IL-1beta, and TNF-alpha only in combination influence the osteoporotic phenotype in Crohn's patients via bone formation and bone resorption. Adv Clin Exp Med. 2018;27(1):45–56. doi: 10.17219/acem/67561. [DOI] [PubMed] [Google Scholar]
- 155.Wagner M.C., Rocha J.M., Gaio E.J., Cavagni J., Carrard V.C., Rösing C.K. Effect of 15% alcohol dependence on alveolar bone loss and TNF-alpha secretion in Wistar rats. Braz Dent J. 2016;27(2):135–140. doi: 10.1590/0103-6440201600545. [DOI] [PubMed] [Google Scholar]
- 156.Fábrega E., Orive A., García-Suarez C., García-Unzueta M., Antonio Amado J., Pons-Romero F. Osteoprotegerin and RANKL in alcoholic liver cirrhosis. Liver Int. 2005;25(2):305–310. doi: 10.1111/j.1478-3231.2005.01073.x. [DOI] [PubMed] [Google Scholar]
- 157.García-Valdecasas-Campelo E., González-Reimers E., Santolaria-Fernández F., De la Vega-Prieto M.J., Milena-Abril A., Sánchez-Pérez M.J., et al. Serum osteoprotegerin and RANKL levels in chronic alcoholic liver disease. Alcohol Alcohol. 2006;41(3):261–266. doi: 10.1093/alcalc/agl004. [DOI] [PubMed] [Google Scholar]
- 158.Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142(12):5050–5055. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- 159.Santori C., Ceccanti M., Diacinti D., Attilia M.L., Toppo L., D'Erasmo E., et al. Skeletal turnover, bone mineral density, and fractures in male chronic abusers of alcohol. J Endocrinol Invest. 2008;31(4):321–326. doi: 10.1007/BF03346365. [DOI] [PubMed] [Google Scholar]
- 160.Malik P., von Gleissenthall G., Gasser R.W., Moncayo R., Giesinger J.M., Mechtcheriakov S. Osteoprotegerin levels decrease in abstinent alcohol-dependent patients. Alcohol Clin Exp Res. 2016;40(6):1235–1240. doi: 10.1111/acer.13063. [DOI] [PubMed] [Google Scholar]
- 161.Martín González C., Fernández Rodríguez C.M., Abreu González P., García Rodríguez A., Alvisa Negrín J.C., Cabañas Perales E., et al. Sclerostin in excessive drinkers: relationships with liver function and body composition. Nutrients. 2022;14(13) doi: 10.3390/nu14132574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Badrick E., Bobak M., Britton A., Kirschbaum C., Marmot M., Kumari M. The relationship between alcohol consumption and cortisol secretion in an aging cohort. J Clin Endocrinol Metab. 2008;93(3):750–757. doi: 10.1210/jc.2007-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Rico H. Alcohol and bone disease. Alcohol Alcohol. 1990;25(4):345–352. [PubMed] [Google Scholar]
- 164.González-Reimers E., Quintero-Platt G., Rodríguez-Rodríguez E., Martínez-Riera A., Alvisa-Negrín J., Santolaria-Fernández F. Bone changes in alcoholic liver disease. World J Hepatol. 2015;7(9):1258–1264. doi: 10.4254/wjh.v7.i9.1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Miroliaee A., Nasiri-Toosi M., Khalilzadeh O., Esteghamati A., Abdollahi A., Mazloumi M., et al. Disturbances of parathyroid hormone-vitamin D axis in non-cholestatic chronic liver disease: a cross-sectional study. Hepatol Int. 2010;4(3):634–640. doi: 10.1007/s12072-010-9194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Girgis C.M., Clifton-Bligh R.J., Hamrick M.W., Holick M.F., Gunton J.E. The roles of vitamin D in skeletal muscle: form, function, and metabolism. Endocr Rev. 2013;34(1):33–83. doi: 10.1210/er.2012-1012. [DOI] [PubMed] [Google Scholar]
- 167.Horstman A.M., Dillon E.L., Urban R.J., Sheffield-Moore M. The role of androgens and estrogens on healthy aging and longevity. J Gerontol A Biol Sci Med Sci. 2012;67(11):1140–1152. doi: 10.1093/gerona/gls068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Santi D., Cignarelli A., Baldi M., Sansone A., Spaggiari G., Simoni M., et al. The chronic alcohol consumption influences the gonadal axis in men: results from a meta-analysis. Andrology. 2024;12(4):768–780. doi: 10.1111/andr.13526. [DOI] [PubMed] [Google Scholar]
- 169.Gusso D., Prauchner G.R.K., Rieder A.S., Wyse A.T.S., et al. Biological pathways associated with vitamins in autism spectrum disorder. Neurotox Res. 2023;41(6):730–740. doi: 10.1007/s12640-023-00674-z. [DOI] [PubMed] [Google Scholar]
- 170.Yamasaki K., Hagiwara H. Excess iron inhibits osteoblast metabolism. Toxicol Lett. 2009;191(2–3):211–215. doi: 10.1016/j.toxlet.2009.08.023. [DOI] [PubMed] [Google Scholar]
- 171.Khazai N., Judd S.E., Tangpricha V. Calcium and vitamin D: skeletal and extraskeletal health. Curr Rheumatol Rep. 2008;10(2):110–117. doi: 10.1007/s11926-008-0020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Li H., Jiang S., Yang C., Yang S., He B., Ma W., et al. Long-Term dexamethasone exposure down-regulates hepatic TFR1 and reduces liver iron concentration in rats. Nutrients. 2017;9(6) doi: 10.3390/nu9060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Papachristou N.I., Blair H.C., Kypreos K.E., Papachristou D.J. High-density lipoprotein (HDL) metabolism and bone mass. J Endocrinol. 2017;233(2):R95–R107. doi: 10.1530/JOE-16-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Yoshida A., Wang G., Dave V. Determination of genotypes of human aldehyde dehydrogenase ALDH2 locus. Am J Hum Genet. 1983;35(6):1107–1116. [PMC free article] [PubMed] [Google Scholar]
- 175.Seo W., Gao Y., He Y., Sun J., Xu H., Feng D., et al. ALDH2 deficiency promotes alcohol-associated liver cancer by activating oncogenic pathways via oxidized DNA-enriched extracellular vesicles. J Hepatol. 2019;71(5):1000–1011. doi: 10.1016/j.jhep.2019.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Xia P., Liu D.H., Wang D., Wen G.M., Zhao Z.Y. SLC3A2, as an indirect target gene of ALDH2, exacerbates alcohol-associated liver cancer via the sphingolipid biosynthesis pathway. Free Radic Biol Med. 2023;206:125–133. doi: 10.1016/j.freeradbiomed.2023.07.002. [DOI] [PubMed] [Google Scholar]
- 177.Alvisa-Negrín J., González-Reimers E., Santolaria-Fernández F., García-Valdecasas-Campelo E., Valls M.R., Pelazas-González R., et al. Osteopenia in alcoholics: effect of alcohol abstinence. Alcohol Alcohol. 2009;44(5):468–475. doi: 10.1093/alcalc/agp038. [DOI] [PubMed] [Google Scholar]
- 178.Malik P., Gasser R.W., Moncayo R., Kemmler G., Wolfgang Fleischhacker W. Markers of bone resorption and formation during abstinence in male alcoholic patients. Alcohol Clin Exp Res. 2012;36(12):2059–2064. doi: 10.1111/j.1530-0277.2012.01834.x. [DOI] [PubMed] [Google Scholar]
- 179.Gao Y., Zhan P., Hagen F., Menken S.B.J., Sun J., Rezaei-Matehkolaei A., et al. Molecular epidemiology and in vitro antifungal susceptibility of Trichophyton schoenleinii, agent of tinea capitis favosa. Mycoses. 2019;62(5):466–474. doi: 10.1111/myc.12889. [DOI] [PubMed] [Google Scholar]
- 180.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139(2):393–408 e1. doi: 10.1053/j.gastro.2010.06.013. 408 e2. [DOI] [PubMed] [Google Scholar]
- 181.Guggenbuhl P., Fergelot P., Doyard M., Libouban H., Roth M.P., Gallois Y., et al. Bone status in a mouse model of genetic hemochromatosis. Osteoporos Int. 2011;22(8):2313–2319. doi: 10.1007/s00198-010-1456-2. [DOI] [PubMed] [Google Scholar]
- 182.Valenti L., Varenna M., Fracanzani A.L., Rossi V., Fargion S., Sinigaglia L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int. 2009;20(4):549–555. doi: 10.1007/s00198-008-0701-4. [DOI] [PubMed] [Google Scholar]
- 183.Shen G.S., Yang Q., Jian J.L., Zhao G.Y., Liu L.L., Wang X., et al. Hepcidin1 knockout mice display defects in bone microarchitecture and changes of bone formation markers. Calcif Tissue Int. 2014;94(6):632–639. doi: 10.1007/s00223-014-9845-8. [DOI] [PubMed] [Google Scholar]
- 184.Bao W.D., Fan Y., Deng Y.Z., Long L.Y., Wang J.J., Guan D.X., et al. Iron overload in hereditary tyrosinemia type 1 induces liver injury through the Sp1/Tfr2/hepcidin axis. J Hepatol. 2016;65(1):137–145. doi: 10.1016/j.jhep.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 185.Rishi G., Secondes E.S., Wallace D.F., Subramaniam V.N. Normal systemic iron homeostasis in mice with macrophage-specific deletion of transferrin receptor 2. Am J Physiol Gastrointest Liver Physiol. 2016;310(3):G171–G180. doi: 10.1152/ajpgi.00291.2015. [DOI] [PubMed] [Google Scholar]
- 186.Xu Z., Sun W., Li Y., Ling S., Zhao C., Zhong G., et al. The regulation of iron metabolism by hepcidin contributes to unloading-induced bone loss. Bone. 2017;94:152–161. doi: 10.1016/j.bone.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 187.Nguyen C.D., Morel V., Pierache A., Lion G., Cortet B., Flipo R.M., et al. Bone and joint complications in patients with hereditary hemochromatosis: a cross-sectional study of 93 patients. Ther Adv Musculoskelet Dis. 2020;12 doi: 10.1177/1759720X20939405. 1759720X20939405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Grosse S.D., Gurrin L.C., Bertalli N.A., Allen K.J. Clinical penetrance in hereditary hemochromatosis: estimates of the cumulative incidence of severe liver disease among HFE C282Y homozygotes. Genet Med. 2018;20(4):383–389. doi: 10.1038/gim.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gryseels B., Polman K., Clerinx J., Kestens L. Human schistosomiasis. Lancet. 2006;368(9541):1106–1118. doi: 10.1016/S0140-6736(06)69440-3. [DOI] [PubMed] [Google Scholar]
- 190.McManus D.P., Dunne D.W., Sacko M., Utzinger J., Vennervald B.J., Zhou X.N. Schistosomiasis. Nat Rev Dis Primers. 2018;4(1):13. doi: 10.1038/s41572-018-0013-8. [DOI] [PubMed] [Google Scholar]
- 191.Barnett R. Schistosomiasis. Lancet. 2018;392(10163):2431. doi: 10.1016/S0140-6736(18)33008-3. [DOI] [PubMed] [Google Scholar]
- 192.Li W., Wei C., Xu L., Yu B., Chen Y., Lu D., et al. Schistosome infection promotes osteoclast-mediated bone loss. PLoS Pathog. 2021;17(3):e1009462. doi: 10.1371/journal.ppat.1009462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhou H., Ohtsuka R., He Y., Yuan L., Yamauchi T., Sleigh A.C. Impact of parasitic infections and dietary intake on child growth in the schistosomiasis-endemic Dongting Lake Region, China. Am J Trop Med Hyg. 2005;72(5):534–539. [PubMed] [Google Scholar]
- 194.Wei L., Lee D., Law C.T., Zhang M.S., Shen J., Chin D.W., et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun. 2019;10(1):4681. doi: 10.1038/s41467-019-12606-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.付士武 肝硬化. 肝癌患者骨代谢生化指标与骨质疏松的临床研究. 中国骨质疏松杂志. 2013;19(11):1177–1179. [Google Scholar]
- 196.Lin L., Chen S., Wang H., Gao B., Kallakury B., Bhuvaneshwar K., et al. SPTBN1 inhibits inflammatory responses and hepatocarcinogenesis via the stabilization of SOCS1 and downregulation of p65 in hepatocellular carcinoma. Theranostics. 2021;11(9):4232–4250. doi: 10.7150/thno.49819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Xu X., Yang J., Ye Y., Chen G., Zhang Y., Wu H., et al. SPTBN1 prevents primary osteoporosis by modulating osteoblasts proliferation and differentiation and blood vessels formation in bone. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.653724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Jung, J. and Q. Wu, Revealing the organ-specific expression of SPTBN1 using single-cell RNA sequencing analysis. bioRxiv, 2023(Preprint).doi:10.1101/2023.06.01.543198.
- 199.Russo S., Scotto di Carlo F., Maurizi A., Fortunato G., Teti A., Licastro D., et al. A mutation in the ZNF687 gene that is responsible for the severe form of Paget's disease of bone causes severely altered bone remodeling and promotes hepatocellular carcinoma onset in a knock-in mouse model. Bone Res. 2023;11(1):16. doi: 10.1038/s41413-023-00250-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Abdel-Hafiz S.M., Hamdy H.E., Khorshed F.M., Aboushousha T.S., Safwat G., Saber M.A., et al. Evaluation of osteopontin as a biomarker in hepatocellular carcinomas in Egyptian patients with chronic HCV cirrhosis. Asian Pac J Cancer Prev APJCP. 2018;19(4):1021–1027. doi: 10.22034/APJCP.2018.19.4.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rodríguez-Aguilar E.F., Pérez-Escobar J., Sánchez Herrera D., García-Alanis M., Toapanta-Yanchapaxi L., Gonzalez-Flores E., et al. Bone disease and liver transplantation: a review. Transplant Proc. 2021;53(7):2346–2353. doi: 10.1016/j.transproceed.2021.07.049. [DOI] [PubMed] [Google Scholar]
- 202.Chang J.W., Yang H.H., Lin N.C., Kuo F.C., Lin T.C., Tsai H.L. Risk factors for fractures following liver transplantation: a population-based cohort study. Ann Med. 2023;55(1) doi: 10.1080/07853890.2023.2230871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kuo S.J., Chen C.L., Chen S.H., Ko J.Y. Changes in serum bone metabolism markers after living donor liver transplantation (LDLT) and their association with fracture occurrences. Life. 2023;13(7) doi: 10.3390/life13071438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sharma P., Parikh N.D., Yu J., Barman P., Derstine B.A., Sonnenday C.J., et al. Bone mineral density predicts posttransplant survival among hepatocellular carcinoma liver transplant recipients. Liver Transplant. 2016;22(8):1092–1098. doi: 10.1002/lt.24458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Toshima T., Yoshizumi T., Kosai-Fujimoto Y., Inokuchi S., Yoshiya S., Takeishi K., et al. Prognostic impact of osteopenia in patients who underwent living donor liver transplantation for hepatocellular carcinoma. World J Surg. 2020;44(1):258–267. doi: 10.1007/s00268-019-05206-5. [DOI] [PubMed] [Google Scholar]
- 206.Nachef C., Bousson V., Belmatoug N., Cohen-Solal M., Vilgrain V., Roux O., et al. Osteoporosis and fragility fractures in patients with cirrhosis evaluated for liver transplantation: identification of high-risk patients based on computed tomography at evaluation. Am J Gastroenterol. 2023;119(2):367–370. doi: 10.14309/ajg.0000000000002507. [DOI] [PubMed] [Google Scholar]
- 207.Liu G., Lipari P., Mollin A., Jung S., Teplova I., Li W., et al. Comparison of pharmaceutical properties and biological activity of prednisolone, deflazacort, and vamorolone in DMD disease models. Hum Mol Genet. 2023;33(3):211–223. doi: 10.1093/hmg/ddad173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Uchino K., Tateishi R., Shiina S., Kanda M., Masuzaki R., Kondo Y., et al. Hepatocellular carcinoma with extrahepatic metastasis: clinical features and prognostic factors. Cancer. 2011;117(19):4475–4483. doi: 10.1002/cncr.25960. [DOI] [PubMed] [Google Scholar]
- 209.Lu Y., Hu J.G., Lin X.J., Li X.G. Bone metastases from hepatocellular carcinoma: clinical features and prognostic factors. Hepatobiliary Pancreat Dis Int. 2017;16(5):499–505. doi: 10.1016/S1499-3872(16)60173-X. [DOI] [PubMed] [Google Scholar]
- 210.Bhattacharyya R.S., Stern P.H. IGF-I and MAP kinase involvement in the stimulatory effects of LNCaP prostate cancer cell conditioned media on cell proliferation and protein synthesis in MC3T3-E1 osteoblastic cells. J Cell Biochem. 2003;90(5):925–937. doi: 10.1002/jcb.10726. [DOI] [PubMed] [Google Scholar]
- 211.Feeley B.T., Gamradt S.C., Hsu W.K., Liu N., Krenek L., Robbins P., et al. Influence of BMPs on the formation of osteoblastic lesions in metastatic prostate cancer. J Bone Miner Res. 2005;20(12):2189–2199. doi: 10.1359/JBMR.050802. [DOI] [PubMed] [Google Scholar]
- 212.Kitagawa Y., Dai J., Zhang J., Keller J.M., Nor J., Yao Z., et al. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005;65(23):10921–10929. doi: 10.1158/0008-5472.CAN-05-1809. [DOI] [PubMed] [Google Scholar]
- 213.Guise T.A., Yin J.J., Mohammad K.S. Role of endothelin-1 in osteoblastic bone metastases. Cancer. 2003;97(3 Suppl):779–784. doi: 10.1002/cncr.11129. [DOI] [PubMed] [Google Scholar]
- 214.Smollich M., Wulfing P. The endothelin axis: a novel target for pharmacotherapy of female malignancies. Curr Vasc Pharmacol. 2007;5(3):239–248. doi: 10.2174/157016107781024082. [DOI] [PubMed] [Google Scholar]
- 215.Zhang F., Gao J., Liu X., Sun Y., Liu L., Hu B., et al. LATS-regulated nuclear-cytoplasmic translocation of SREBP2 inhibits hepatocellular carcinoma cell migration and invasion via epithelial-mesenchymal transition. Mol Carcinog. 2023;62(7):963–974. doi: 10.1002/mc.23538. [DOI] [PubMed] [Google Scholar]
- 216.Cheng H.M., Xing M., Zhou Y.P., Zhang W., Liu Z., Li L., et al. HSP90beta promotes osteoclastogenesis by dual-activation of cholesterol synthesis and NF-kappaB signaling. Cell Death Differ. 2023;30(3):673–686. doi: 10.1038/s41418-022-01071-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mai S., Zhu X., Wan E.Y.C., Wu S., Yonathan J.N., Wang J. Postnatal eye size in mice is controlled by SREBP2-mediated transcriptional repression of Lrp2 and Bmp2. Development. 2022;149(14) doi: 10.1242/dev.200633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Yang Z., Li X., Gan X., Wei M., Wang C., Yang G., et al. Hydrogel armed with Bmp2 mRNA-enriched exosomes enhances bone regeneration. J Nanobiotechnol. 2023;21(1):119. doi: 10.1186/s12951-023-01871-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Huang Z., Chu L., Liang J., Tan X., Wang Y., Wen J., et al. H19 promotes HCC bone metastasis through reducing osteoprotegerin expression in a protein phosphatase 1 catalytic subunit alpha/p38 mitogen-activated protein kinase-dependent manner and sponging microRNA 200b-3p. Hepatology. 2021;74(1):214–232. doi: 10.1002/hep.31673. [DOI] [PubMed] [Google Scholar]
- 220.Han S., Xue L., Wei Y., Yong T., Jia W., Qi Y., et al. Bone lesion-derived extracellular vesicles fuel prometastatic cascades in hepatocellular carcinoma by transferring ALKBH5-targeting miR-3190-5p. Adv Sci. 2023;10(17):e2207080. doi: 10.1002/advs.202207080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zheng H., Cheng Z.J., Liang B., Wang Z.G., Tao Y.P., Huang S.Y., et al. N(6)-Methyladenosine modification of ANLN enhances hepatocellular carcinoma bone metastasis. Int J Biol Sci. 2023;19(4):1009–1023. doi: 10.7150/ijbs.73570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Głuszko P., Sewerynek E., Misiorowski W., Konstantynowicz J., Marcinowska-Suchowierska E., Blicharski T., et al. Guidelines for the diagnosis and management of osteoporosis in Poland. Update 2022. Endokrynol Pol. 2023;74(1):5–15. doi: 10.5603/EP.a2023.0012. [DOI] [PubMed] [Google Scholar]
- 223.Yang S.J., Chang C.H., Young T.H., Wang C.H., Tseng T.H., Wang M.L. Human serum albumin-based nanoparticles alter raloxifene administration and improve bioavailability. Drug Deliv. 2022;29(1):2685–2693. doi: 10.1080/10717544.2022.2111479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.LeBoff M.S., Greenspan S.L., Insogna K.L., Lewiecki E.M., Saag K.G., Singer A.J., et al. The clinician's guide to prevention and treatment of osteoporosis. Osteoporos Int. 2022;33(10):2049–2102. doi: 10.1007/s00198-021-05900-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Diamond T., Stiel D., Posen S. Effects of testosterone and venesection on spinal and peripheral bone mineral in six hypogonadal men with hemochromatosis. J Bone Miner Res. 1991;6(1):39–43. doi: 10.1002/jbmr.5650060108. [DOI] [PubMed] [Google Scholar]
- 226.LeBoff M.S., Greenspan S.L., Insogna K.L., Lewiecki E.M., Saag K.G., Singer A.J., et al. Current and emerging pharmacological options for the treatment of nonalcoholic steatohepatitis. Metabolism. 2020;111S doi: 10.1016/j.metabol.2020.154203. [DOI] [PubMed] [Google Scholar]
- 227.Shen C.L., Yang S., Tomison M.D., Romero A.W., Felton C.K., Mo H. Tocotrienol supplementation suppressed bone resorption and oxidative stress in postmenopausal osteopenic women: a 12-week randomized double-blinded placebo-controlled trial. Osteoporos Int. 2018;29(4):881–891. doi: 10.1007/s00198-017-4356-x. [DOI] [PubMed] [Google Scholar]
- 228.Cai T.T., Li H.Q., Jiang L.L., Wang H.Y., Luo M.H., Su X.F., et al. Effects of GLP-1 receptor agonists on bone mineral density in patients with type 2 diabetes mellitus: a 52-week clinical study. BioMed Res Int. 2021;2021 doi: 10.1155/2021/3361309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kong Q.X., Ruan Q., Fan C., Liu B.L., Reng L.P., Xu W. Evaluation of the risk of fracture in type 2 diabetes mellitus patients with incretins: an updated meta-analysis. Endokrynol Pol. 2021;72(4):319–328. doi: 10.5603/EP.a2021.0031. [DOI] [PubMed] [Google Scholar]
- 230.Venetsanaki V., Karabouta Z., Polyzos S.A. Farnesoid X nuclear receptor agonists for the treatment of nonalcoholic steatohepatitis. Eur J Pharmacol. 2019;863 doi: 10.1016/j.ejphar.2019.172661. [DOI] [PubMed] [Google Scholar]
- 231.Li Z., Huang J., Wang F., Li W., Wu X., Zhao C., et al. Dual targeting of bile acid receptor-1 (TGR5) and farnesoid X receptor (FXR) prevents estrogen-dependent bone loss in mice. J Bone Miner Res. 2019;34(4):765–776. doi: 10.1002/jbmr.3652. [DOI] [PubMed] [Google Scholar]