

Abstract
Flavin cofactors offer a wide range of chemical mechanisms to support a great diversity in catalytic function. As a corollary, such diversity necessitates careful control within each flavoprotein to limit its function to an appropriate subset of possible reactions and substrates. This task falls to the protein environment surrounding the flavin in most enzymes. For iodotyrosine deiodinase that catalyzes a reductive dehalogenation of halotyrosines, substrates can dictate the chemistry available to the flavin. Their ability to stabilize the necessary one-electron reduced semiquinone form of flavin strictly depends on a direct coordination between the flavin and α-ammonium and carboxylate groups of its substrates. While perturbations to the carboxylate group do not significantly affect binding to the resting oxidized form of the deiodinase, dehalogenation (kcat/Km) is suppressed by over 2000-fold. Lack of the α-ammonium group abolishes detectable binding and dehalogenation. Substitution of the ammonium group with a hydroxyl group does not restore measurable binding but does support dehalogenation with an efficiency greater than those of the carboxylate derivatives. Consistent with these observations, the flavin semiquinone does not accumulate during redox titration in the presence of inert substrate analogues lacking either the α-ammonium or carboxylate groups. As a complement, a nitroreductase activity based on hydride transfer is revealed for the appropriate substrates with perturbations to their zwitterion.
Graphical Abstract

INTRODUCTION
Flavin-dependent catalysis is diverse and widespread and encompasses both oxidative and reductive transformations including, but certainly not limited to, hydrogenation, hydroxylation, halogenation, nitroreduction, and dehalogenation.1–3 The unusual breadth of flavin reactivity can be attributed to its ability to promote both single-electron and two-electron (hydride) transfers. The specific function of each enzyme is programmed by the environment surrounding the flavin to alternatively activate and suppress competing modes of reaction. Sterics, electrostatics, solvation, hydrogen bonding, and proton transfer in proximity to flavin are typically governed by the protein backbone and its side chains.4,5 To activate single-electron transfers, hydrogen bond donation to the flavin N5 appears most critical (Figure 1),6–8 and polar interactions to its N39,10 and carbonyl oxygens (O2, O4)11–13 typically contribute in subordinate roles.
Figure 1.

Polar contacts surround the flavin of IYD in the presence of a zwitterionic substrate or ligand.
The nature of the hydrogen bonding partners further dictates the type of chemistry catalyzed by flavoproteins. For example, the NfsA and NfsB subfamilies within the nitroreductase superfamily offer hydrogen bonding to the flavin N5 through their amide backbone to promote hydride transfer during reduction of nitroaromatics.14,15 BluB and IYD subfamilies within the same superfamily offer hydrogen bonding to the N5 position from side chains of Ser and Thr to induce single-electron chemistry necessary for oxygen activation and dehalogenation, respectively.16,17 Replacement of the Thr with Ala in iodotyrosine deiodinase (IYD) destabilizes the intermediate one-electron reduced flavin semiquinone (FMNsq) and relieves suppression of l-3-nitrotyrosine (O2N-Tyr) reduction presumably by hydride transfer (Scheme 1).18 Accordingly, the hydrogen bond donor to the flavin N5 was considered the key mechanistic determinant for IYD. At least for human IYD, this hydrogen bond is controlled by substrate binding and the subsequent closure of an active site lid (PDB 4TTB and 4TTC) (Figure 1).17 In a bacterial homologue, an equivalent lid closes around l-3-iodotyrosine (I-Tyr) to promote deiodination (PDB 5KO8, see the Supporting Information).19 2-Iodophenol also adopted an equivalent geometry of stacking over the flavin as observed by crystallography (PDB 5KRD).19 However, the lid remained too dynamic in the presence of 2-iodophenol since no electron density associated with this region was observed. In accordance with the trend of one- versus two-electron transfer for this system, 2-iodophenol was dehalogenated very inefficiently while 2-nitrophenol was easily reduced.20
Scheme 1.

Products of Single-Electron and Hydride Transfers Catalyzed by IYDa
a(A) Dehalogenation results from two successive one-electron transfers. (B) Nitroreduction results from hydride transfer and typically generates a hydroxylamine product after two turnovers. The oxidized, one-electron reduced, and two-electron reduced flavins are indicated by FMNox, FMNsq, and FMNhq, respectively.
Our initial emphasis on the hydrogen bonding to flavin N5 was supported from a preponderance of literature reports and a general emphasis on the role of proteins to control cofactor activity. Initial studies on IYD were consistent with this dogma. However, the relatively small decrease in dehalogenation efficiency (15-fold) was surprising when the N5 hydrogen bond was lost upon the Thr to Ala substitution.18 For context, dehalogenation was suppressed by ~16,000-fold when the native flavin was replaced with its 5-deaza derivative that is incapable of single-electron transfer.21 Similarly, the importance of the substrate-induced closure of the active site lid as first suggested by crystallography requires reevaluation based on recent NMR experiments that suggest both tyrosine and phenol derivatives induce similar changes to the active site lid and bind in similar environments.22 Furthermore, a new lid sequence was successfully engineered to close around 2-iodophenol, but only a modest gain in dehalogenation was achieved with this variant.23 Together, these data suggest that other features of IYD may be most important for stabilizing the FMNsq intermediate and promoting dehalogenation. The conserved Arg that interacts with the flavin N1 and C2 carbonyl (O2) had previously been deemed nonessential,24 and thus attention next turned to the contributions of the substrate as described below (Figure 1). A series of zwitterion derivatives of I-Tyr, L-3-fluorotyrosine (F-Tyr), and O2N-Tyr have been prepared and characterized with Haliscomenobacter hydrossis IYD to demonstrate the dominance of the substrate in controlling the chemistry of catalysis.
MATERIALS AND METHODS
General Materials.
3-Fluoro-l-tyrosine (F-Tyr) and (S)-3-(4-hydroxyphenyl)lactic acid (HPLA) were obtained from AstaTech, Inc. (Bristol, Pennsylvania). d-Tyrosine (d-Tyr), dl-tyrosine (dl-Tyr), di-tert-butyl dicarbonate (Boc2O), and xanthine were obtained from Alfa Aesar (Ward Hill, Massachusetts). Formic acid (88%), N-α-benzoyl-dl-arginine-4-nitroanalide hydrochloride (dl-BAPNA), and trifluoroacetic acid were obtained from Acros Organics (Morris Plains, New Jersey). l-Tyrosinamide, tyramine, trypsin from bovine pancreas, sodium hydrosulfite, 3-(4-hydroxyphenyl)propionic acid, 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (SelectFluor), flavin mononucleotide (FMN), N-iodosuccinimide, ortho-phenylenediamine, sodium pyrophosphate tetrabasic decahydrate, xanthine oxidase, methyl viologen, 3-nitro-l-tyrosine, peroxidase from horseradish, l-amino acid oxidase (LAAO) from Crotalus adamanteus, and d-amino acid oxidase (DAAO) from porcine kidney were obtained from Sigma-Aldrich (Madison, Wisconsin). Sodium acetate, ammonium hydroxide (28%), and sodium nitrite were obtained from Thermo Fisher Scientific (Waltham, Massachusetts). Nitric acid (68%, ACS grade) was obtained from VWR Scientific (Radnor, PA). SilicaFlash P60 silica was used for all column chromatography (SiliCycle Inc.).
General Methods.
NMR spectra were recorded on Bruker Avance 300 or 400 MHz spectrometers. Chemical shifts are reported in parts per million (ppm), and coupling constants (J) are reported in hertz (Hz). The residual 1H and 13C from solvent were used as references. 19F signals were referenced from an internal standard of trifluoroacetic acid. All UV–vis measurements were performed on an Agilent 8453 spectrophotometer. Fluorescence measurements were performed with a FluoroMax-4 spectrofluorometer (Horiba Scientific). Mass analysis was performed on a VG Analytical VG-70S magnetic sector mass spectrometer with electron impact (EI) ionization.
Expression and Mutation of Iodotyrosine Deiodinase.
IYD from Haliscomenobacter hydrossis was expressed with a C-terminal His6-tag, purified by Ni2+ affinity chromatography, and stored as described previously.19 The E91K/K116E variant was generated by two rounds of site-directed mutagenesis first using a forward primer 5′-GCT TTC AAA TTT CTC TTT TTC AGC AGC TTG TCG G-3′ and a reverse primer 3′-AAA GAG AAA TTT GAA AGC TAT AAT GGC CGC ATG TCC-5′ for the E91K mutation and subsequently using the forward primer 5′-AAA TGG TTC ATG CCA ATC GGT TCC AAA AGG-3′ and reverse primer 3′-TGG CAT GAA CCA TTT TTG GAA ATT GCT CCG TAT CTT ATT G-5′ for the K116E mutation. Mutations were confirmed by DNA sequencing, and the desired plasmids were transformed into Escherichia coli Rosetta 2(DE3) cells for expression and purification following standard protocols.19
Catalytic Deiodination.
Dehalogenation was initiated by adding sodium dithionite (5%) in sodium bicarbonate (5%, 100 μL) to a solution of IYD (final 100–250 nM) and the indicated concentration of substrate in 100 mM potassium phosphate pH 7.4 (900 μL). After 1–3 h (25 °C), the reaction was quenched by addition of 88% formic acid (50 μL) and products were quantified by their A280 during separation by reverse-phase HPLC using a Varian Microsorb MV 300–5 (C18 250 × 4.6 mm) column with a JASCO MW-1510 PDA.25 HPLC solvent A was 0.44% aqueous formic acid, and solvent B was 0.44% formic acid in acetonitrile. The solvent program began with 95% solvent A (0−10 min) and then increased linearly to 40% solvent A (10−25 min) and then to 5% solvent A (30−41 min) and finally 100% solvent A (41−55 min) at 1 mL/min. Quantification of the deiodinated product was performed as described previously by comparing its integrated signal (A280) with an internal standard (meta-cresol) that had been calibrated with each substrate.26
Affinity of Ligands to IYD Containing FMNox.
Ligands were titrated into a solution of IYD (3 μM, 2.5 mL) in 110 mM potassium phosphate pH 7.4 that was equilibrated for 20 min at 25 °C with gentle stirring. Fluorescence was measured 2 min after each addition of ligand to allow for equilibration. Fluorescence emission at 516 nm was recorded using an excitation of 450 nm, and Kd values were calculated by plotting relative fluorescence (ΔF/F0) with respect to total enzyme ([E]T) and total ligand ([L]) using eq 1 (Origin 6.0).19,27
| (1) |
Redox Titration of IYD in the Presence of Substrate Analogues.
Xanthine (900 μM), methyl viologen (2 μM), and substrate (concentration at 10× its Kd) in 110 mM potassium phosphate pH 7.4 were added to a sealed quartz cuvette at 25 °C, as previously described.17,28,29 The solution was bubbled with argon for 20 min to remove molecular oxygen, and then IYD (20 μM) was added. The solution was bubbled with argon for an additional 2 min followed by a flushing of the cuvette head space with argon for an additional 20 min. Reaction was initiated by xanthine oxidase (40 μg/mL), and spectra were recorded every 1 min for 3 h. Absorbance versus wavelength was exported with a background subtraction from absorbance from 800 to 900 nm and plotted with Origin 6.0. Titrations for measuring reduction potentials also included a redox dye (20 μM). Suitable dyes were selected based on their reduction potentials within approximately 30 mV of that measured for IYD;30 anthraquinone-2,6-disulfonate (AQDS, Em = −188 mV) and anthraquinone-2-sulfonate (AQS, Em = −225 mV) were used for IYD in the presence and absence of substrate analogues, respectively.31 Reduction of IYD and dyes was measured at wavelengths corresponding to the isosbestic point of the other chromophore. The known reduction potential of the dye along with the relative proportion of oxidized/reduced dye, and IYD was used to calculate the reduction potential of IYD using eq 2, as described previously.29,31
| (2) |
D = dye, n = number of electrons, F = Faraday constant, E = enzyme, Em = midpoint potential, R = ideal gas constant, and T = temperature. Reduction potentials are calculated from the average of two individual titrations.
Nitroreductase Activity of IYD.
A solution of potassium phosphate (110 mM, pH 7.4) was degassed in a quartz cuvette by bubbling with argon for 20 min at 25 °C, as previously described.18 IYD (15–18 μM) was added, and the solution (900 μL final) was bubbled with argon for an additional 2 min; then, the headspace of the cuvette was purged with argon for 20 min. Enzyme was reduced by titration with sodium dithionite (0.25%, 1 μL aliquots) to generate reduced IYD•FMNhq, as monitored by A450. Air-saturated buffer (20 μL) was added to ensure that a small fraction of the enzyme would reoxidize as expected when no excess dithionite was present. Aliquots of a substrate solution (4 μM) degassed with argon in a separate cuvette for 20 min were transferred to the enzyme-containing cuvette, and spectra were recorded every 30 s for 60 min until full reoxidation to FMNox was observed by A450. Spectra were corrected for a background signal recorded from 800 to 900 nm.
Limited Proteolysis of IYD in the Presence of Substrate Analogues.
IYD (5 μg) and its active site ligand (at concentrations of 10× its Kd) were incubated in 110 mM potassium phosphate pH 7.4 (25 μL final) at 25 °C for 5 min as previously described.23 Reaction was initiated by addition of trypsin (1:50 w:w), and aliquots (5 μL) were taken after the indicated times, quenched with 2× SDS loading dye (5 μL), and placed in a 90 °C heat block for 5 min. The aliquots were then analyzed with 15% SDS-PAGE (40 min, 200 V). Coomassie brilliant blue staining solution (100 mL) was mixed with the gel, microwaved for 45 s, and placed on a shaker for 15 min. The gel was destained after addition of an aqueous solution of 10% glacial acetic acid and 30% methanol (100 mL) followed by microwaving for 45 s and shaking for 12 h. The gel was scanned with an HP Scanjet G4050, and signals were quantified with ImageQuant Software. The signal corresponding to a control sample of IYD was normalized to 5 μg, providing the relative quantity of IYD at each time. The disappearance of the IYD was plotted vs time and fit to a first-order decay with Origin 6.0 to calculate the half-life. Each digestion was performed in duplicate.
Potential Inhibition of Trypsin by Ligands of IYD.
A solution of trypsin (8 μM) and ligand (at a concentration of 10× its Kd for IYD) in 110 mM potassium phosphate pH 7.4 (65 μL) was incubated at 25 °C for 5 min, as previously described.23 Reaction was initiated by addition of DL-BAPNA (A315 = 0.45).32 Formation of 4-nitroaniline, the hydrolysis product of DL-BAPNA, was quantified by monitoring its absorbance at 405 nm (ε = 9920 M−1cm−1).32 Spectra were recorded every 15 s for 5 min. The rate of 4-nitroaniline formation over the first 2 min of reaction was plotted vs time, and linear regression was performed in Origin 6.0. These analyses were repeated independently twice.
Synthesis of Substrate Analogues.
Detailed syntheses of substrate analogues are described in the Supporting Information. Briefly, iodination of tyrosine analogues typically was performed with iodine in ammonium hydroxide, but (S)-3-(3-iodo-4-hydroxyphenyl)lactic acid (I-HPLA) was prepared with N-iodosuccinimide.33,34 Fluorination of N-Boc-protected tyrosine analogues utilized SelectFluor.35 Nitration of tyrosine analogues typically used nitric acid, but the lactic acid derivative, (S)-3-(4-hydroxy-3-nitrophenyl)lactic acid (O2N-HPLA), was generated by diazotization and hydrolysis of O2N-Tyr with sodium nitrite and sulfuric acid.36–38
RESULTS AND DISCUSSION
Effects of Zwitterion Modification on Substrate Binding and Turnover.
Most commonly, the role of polar interactions in controlling the chemistry of flavin is tested by site-directed mutagenesis since the overwhelming number of these interactions involves proteins. For IYD, significant contributions are proposed to involve the zwitterion of its substrates (Figure 1). Accordingly, a series of halotyrosine derivatives were prepared to systematically delete or modify the α-ammonium and carboxylate groups without altering the native protein (Figure 2). The most active substrates for human IYD are 3-iodo-, bromo-, and chlorotyrosines, and all generate tyrosine after dehalogenation.39–41 This profile is maintained in other IYDs from fruit flies and thermophilic bacteria.25,42 Because tyrosine binds relatively weakly to IYD relative to the halotyrosine substrates, the series of zwitterion derivatives of tyrosine (X = H, Figure 2) were iodinated (X = I) prior to studies with a bacterial IYD (Scheme S1).
Figure 2.

Structures of tyrosine derivatives used to probe flavin activation in IYD. X = I, F, NO2. X-HPPA, 3-(3-X-4-hydroxyphenyl)-propanoic acid; X-HPLA, (S) 3-(3-X-4-hydroxyphenyl)lactic acid.
Replacing the carboxylate of I-Tyr with a carboxamide group (I-tyrosinamide) removes the anionic charge but retains the potential for hydrogen bonding. This modification had little effect on affinity for IYD containing flavin in its oxidized and resting form (FMNox) (Table 1). Even the complete loss of the carboxylate group (I-tyramine) only diminished affinity by less than 18-fold. In contrast, loss of the α-ammonium group (3-(4-hydroxy-3-iodophenyl)propanoic acid, I-HPPA) decreased affinity by at least 700-fold and beyond a value that could be measured due to solubility limits of I-HPPA (Table 1). Substituting the α-ammonium group with an α-hydroxy group ((S)-3-(4-hydroxy-3-iodophenyl)lactic acid, I-HPLA) maintains hydrogen bonding, but the lack of charge was not sufficient to restore binding to IYD. Thus, coordination of the cationic ammonium group to the flavin O4 is critical for substrate coordination. This ammonium group also coordinates to a highly conserved carboxylate of an active site Glu, and conversion of this Glu to Gln in Drosophila IYD decreased the binding affinity of I-Tyr by 180-fold.25 Surprisingly, substitution of a conserved Lys that coordinates to the substrate carboxylate with Gln had a greater impact on I-Tyr binding, as evident from a 370-fold loss of affinity.25 This is contrary to the impact of losing the carboxylate from the substrate, and thus its zwitterion differentially interacts with the flavin and proximal amino acid side chains. Substrate recognition by wild-type IYD appears to demand the presence of an α-ammonium group on its substrates and is agnostic to the adjacent carboxylate, although both seem to interact with the flavin and the protein.
Table 1.
Binding and Deiodination of I-Tyr Zwitterionic Derivatives with IYDa
| compound | Kd (μM)b | kcat (s−1) × 10−2c | Km (μM)c | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| I-Tyrd | 5.7 ± 0.4 | 60 ± 0.5 | 49 ± 10 | (1.2 ± 0.2) × 104 |
| I-tyrosinamide | 7.0 ± 0.2 | 1.7 ± 0.2 | 2410 ± 410 | 5.7 ± 0.7 |
| I-tyramine | 99 ± 10 | 1.8 ± 0.3 | 1470 ± 450 | 13 ± 4.5 |
| I-HPPA | >4000 | <9.3 × 10−3e | ||
| I-HPLA | >3000 | 1.3 ± 0.1 | 270 ± 30 | 47 ± 5 |
| D-I-Tyr | 270 ± 15 | 2.7 ± 1.0 | 7900 ± 4000 | 3.4 ± 1 |
Despite the relatively mild loss of affinity for IYD caused by neutralization or deletion of the carboxylate group of I-Tyr, catalytic turnover was significantly impaired. For both I-tyrosinamide and I-tyramine, kcat values for dehalogenation decreased by more than 30-fold and their overall efficiency of turnover (kcat/Km) decreased by more than 900-fold (Table 1). Thus, the substrate carboxylate group that coordinates to the flavin N3 helps to stabilize intermediates formed during catalysis but offers little stabilization to the resting state. This is consistent with earlier investigations that demonstrated a lack of correlation between the binding of halophenol derivatives and their efficiency of dehalogenation by IYD.19 The α-ammonium group may also be necessary for stabilizing reaction intermediates, but the lack of binding by I-HPPA limited our ability to measure dehalogenation. The kcat value for catalysis can be no greater than 0.015% of that for I-Tyr, as set by our detection limit of the deiodinated product HPPA. While substitution of the α-ammonium group with an α-hydroxy group did not enhance binding to the resting enzyme, detectable catalytic activity was restored. I-HPLA was dehalogenated with a kcat that was similar to the carboxylate derivatives, and its kcat/Km surpassed those of the carboxylate derivatives by more than threefold. Coordination of the zwitterion to both the flavin O4 and N3 are consequently essential for efficient reductive dehalogenation. Note that the loss of catalysis experienced by the substrates lacking the full zwitterionic structure is significantly greater than the 15-fold loss in kcat/Km caused by either (i) a loss of hydrogen bonding to the flavin N5 resulting from a Thr to Ala substitution or (ii) a loss of coordination to the flavin O2 by a Lys to Gln substitution.18,24 The catalytic efficiency for substrates with perturbations to their zwitterions is also quite similar to that for 2-iodophenol (kcat/Km = 0.7 M−1 s−1),19 which is devoid of these groups entirely. Together, these comparisons suggest that the α-ammonium and carboxylate groups of the substrate are most critical for activating the flavin for sequential single-electron transfer and reductive dehalogenation. Thus, IYD appears distinct from the vast majority of flavoproteins that rely on protein, rather than substrate, interactions to modulate the catalytic activity of their bound flavin cofactor.
Binding and turnover of the unnatural enantiomer d-3-iodotyrosine (d-I-Tyr) was also examined to test the spatial hierarchy of interactions established by the substrate zwitterion. In this example, electrostatic repulsion could be expected for interactions between d-I-Tyr and the flavin O4 and N3 groups as well as the Glu and Lys side chains described above. Instead, binding of d-I-Tyr to native IYD was moderately weakened but not nearly to the extent of that measured for I-HPPA or I-HPLA (Table 1). Perhaps the active site has a flexibility to accommodate the reorientation of charged groups in the substrate, but only a low basal level of catalysis was observed similar to that of the natural enantiomer of I-tyrosinamide. The values measured for d-I-Tyr are expected to reflect properties of this enantiomer rather than those arising from a trace impurity of the natural l-enantiomer. d-I-Tyr was treated with l-amino acid oxidase to remove any l-enantiomer that originated in the commercial starting material d-tyrosine (see the Supporting Information). Complementary analysis with l- and d-amino acid oxidases also indicated that d-I-Tyr had an enantiomeric purity of 99 ± 1% (Figures S3).
To probe the plasticity of the IYD active site further, the Glu and Lys that provide complementary interactions to the α-ammonium and carboxylate groups of (l-)I-Tyr were switched by site-directed mutagenesis to their E91K and K116E counterparts. This variant containing the double substitution expressed a diminished ability to discriminate between the substrate enantiomers based on Kd values for (l-) and d-I-Tyr of 85 ± 14 and 200 ± 30 μM, respectively (Figure S4). No catalytic dehalogenation could be detected for either enantiomer with this IYD variant, demonstrating once again that binding affinity is not predictive of catalytic activity. The placement and nature of the polar substrate and protein side chains are hence immutable for effective turnover.
Closure of the Active Site Lid Induced by Ligand Binding.
A correlation between the catalytic efficiency of IYD and closure of its active site lid was previously suggested by the contrasting catalytic and crystallographic information on I-Tyr and 2-iodophenol.19 More recent data based on engineering of the active site lid sequence and measurement of the dynamics of lid closure by NMR began to question the significance of this correlation.22,23 The zwitterion derivatives described here offer an opportunity to clarify the role of lid closure for catalysis. Limited proteolysis has been a convenient tool for characterizing solvent accessibility of the lid.32,42,43 In the absence of ligand, an Arg and a Lys within the lid are most facile targets of trypsin.22 These same residues gain protection from trypsin as the lid condenses around its substrates and ligands. IYD in the absence of ligand was consumed by trypsin (1:50, w:w) with a half-life of 10 min (Table 2 and Figures S5 and S6). Resistance to proteolysis increased over twofold in the presence of a saturating concentration of I-Tyr that is consistent with lid closure.22 This effect was not due to general inhibition of trypsin, as determined by its turnover of N-benzoyl-dl-arginine- 4-nitroanilide hydrochloride (dl-BAPNA, Figure S7). A slightly greater level of protection was gained from the presence of either I-tyrosinamide or I-tyramine (Table 2). These observations may not be surprising based on their reasonable affinity for IYD. Thus, active site binding was sufficient to secure the lid but not guarantee dehalogenation activity.
Table 2.
Half-Lives of IYD Proteolysis by Trypsin in the Presence and Absence of Iodinated Ligandsa
| ligand | t1/2 (min)b | fold change in t1/2 |
|---|---|---|
| none | 10 ± 1 | |
| I-Tyr | 22 ± 2 | 2.2 |
| I-tyrosinamide | 27 ± 2 | 2.7 |
| I-tyramine | 27 ± 8 | 2.7 |
| d-I-Tyr | 6.2 ± 1.2 | 0.62 |
Values and error were obtained from fitting two independent measurements to a first-order decay in Origin 6.0.
See Figures S5 and S6.
d-I-Tyr is the only ligand to date that appears to promote trypsin-dependent hydrolysis of IYD relative to that in its absence. A similar activation by d-I-Tyr is not observed when dl-BAPNA is used as a substrate for trypsin and more likely d-I-Tyr is a mild inhibitor of trypsin (Table 2 and Figures S5–S7). Perhaps the aromatic ring of d-I-Tyr stacks over the flavin in analogy to (l-)I-Tyr and 2-iodophenol and orients the zwitterion in a manner that prevents lid closure and enhances its exposure relative to the dynamic range of conformations available in its absence.19 The E91K/K116E variant of IYD is somewhat more susceptible than the wild-type IYD to trypsin in the absence of ligands but gains over a twofold protection in the presence of either (l-)I-Tyr or d-I-Tyr (Table S1 and Figures S8 and S9). The protection gained in the presence of native (l-)I-Tyr suggests that active site binding and lid closure are maintained despite the lack of subsequent dehalogenation. The results with d-I-Tyr are more ambiguous since this enantiomer inhibits trypsin and may protect IYD through a nonspecific mechanism (Figure S7).
Substrate-Induced Stabilization of the FMNsq.
In-efficient dehalogenation of substrates lacking the ability to coordinate with the flavin O4 and N3 is best explained by their inability to stabilize the intermediate FMNsq that is critical for catalysis. While direct observation of FMNsq during turnover can be difficult, prior investigations have utilized the binding of F-Tyr to IYD. This is an inert substrate analogue known to bind to the active site in analogy to I-Tyr and stabilize FMNsq during reductive titration.17,20 Accordingly, L-tyrosinamide and L-tyrosamine were fluorinated to prepare F-Tyr derivatives containing perturbations to the carboxylate group (see the Supporting Information). Equivalent fluorination of HPPA and HPLA was not pursued since neither of their iodinated derivatives were observed to bind IYD (Table 1). As expected from prior investigations,17 the affinity of F-Tyr for IYD (Kd = 97 ± 3 μM) is more than 10-fold weaker than that of I-Tyr (Table 1 and Figure S10). However, the trend in affinity after modification of its carboxylate group is similar to that of I-Tyr. F-tyrosinamide (X = F, Figure 2) exhibited a similar affinity (Kd = 77 ± 3 μM), as F-Tyr and F-tyramine affinity decreased by approximately 1 order of magnitude (Kd = 730 ± 140 μM) (Figure S10). Both protected IYD against trypsin as well, although the concentration necessary for saturating the active site with F-tyramine (7 mM) also inhibited trypsin (Figures S11–S13).
Standard reductive titration of IYD with xanthine and xanthine oxidase under anaerobic conditions generated the fully reduced flavin (FMNhq) without a detectable FMNsq intermediate (Figure 3). In contrast, an equivalent titration in the presence of saturating F-Tyr generated a new chromophore before full reduction with a broad absorbance around 600 nm that is consistent with the neutral form of FMNsq and equivalent to that detected with IYD from mouse and human.17,44 This species increases during the initial phase of titration and then decreases to yield the spectrum of FMNhq (Figure 3). An equivalent formation of FMNsq is not detected during titrations in the presence of saturating concentrations of either F-tyrosinamide or F-tyramine despite their ability to bind to the IYD active site and likely induce closure of the active site lid. While the carboxylate charge was not critical for binding of I- or F-tyrosinamide, this charge still controls the accumulation of FMNsq. A similar lack of FMNsq accumulation was observed when a hydrogen bond to the flavin N5 was disrupted in human IYD,17,20 but surprisingly, this had much less impact on catalytic efficiency than the loss of the carboxylate group that interacts with the flavin N3. Coordination of F-tyrosinamide and F-tyramine also moderately destabilize the FMNhq form of IYD, as evident by their respective two-electron potentials of −236 and −225 mV that are lower than the −200 mV measured in the absence of ligands (Table S2 and Figure S14). This effect is surprising since a net positive electrostatic potential would usually be expected to relieve destabilization of the reduced FMNhq.45 In comparison to perturbations at the flavin N3, disruption of the hydrogen bond to flavin N5 in human IYD destabilized FMNhq to a lesser extent. In this case, the redox potential only decreased to −215 mV from the wild-type control of −200 mV.20 Thus, the substrate zwitterion again appears to dominate the properties of the flavin in IYD.
Figure 3.

Redox titration of IYD in the presence of F-Tyr and its zwitterionic derivatives. IYD containing FMNox (20 μM) in 110 mM potassium phosphate pH 7.4 at 25 °C was reduced in the alternative presence of (A) no substrate, (B) F-Tyr (1 mM), (C) F-tyrosinamide (1 mM), and (D) F-tyramine (7 mM) using the xanthine/xanthine oxidase system.20,28 Loss of absorbance at 450 nm corresponds to the reduction of FMNox, and an increase in absorbance at 600 nm corresponds to formation of FMNsq.
Hydrogen bonding to the flavin N3 is common in flavoproteins and their model systems, but rarely is this interaction considered the key to stabilizing FMNsq.4,10 Flavodoxin from Clostridium beijerinckii represented a possible exception by orienting a protein carboxylate side chain of Glu next to the N3 of its bound flavin in analogy to the substrate carboxylate for IYD. Initially, the Glu was considered critical for FMNsq formation based on its response to a Glu to Gln substitution. Further study reassigned the significance of the Glu from directly influencing N3 to indirectly positioning the FMN for maximizing the hydrogen bonding to its N5 position.9,46 As typical, interactions to N5 once again remained dominant. The carboxylate provided by substrates of IYD are not likely to function merely as an anchor for orienting substrate since the affinity of I-tyrosinamide is not significantly perturbed relative to that of I-Tyr (Table 1). Hydrogen bonding to the O4 of FMN offers an additional site for controlling its redox properties.13,47 Typically, interactions at this site are also established by the surrounding protein but for IYD these interactions derive from the α-ammonium group of substrate. Unfortunately, the significance of this group on the redox properties of FMN could not be characterized in IYD by the methods above since the ligands lacking the α-ammonium group had insufficient affinity for the active site. Hydrogen bonding to the O2 group of flavin represents yet another site for redox control.12 In IYD, an Arg coordinates to this region of FMN and remains constant in the presence and absence of ligand.17,19 This interaction represents the only polar interactions provided by the protein. All others are supplied by the substrate, and thus stabilization of the intermediate FMNsq and activation of catalysis depends on substrate structure.
Substrate-Induced Nitroreductase Activity of IYD.
If the substrate zwitterion is truly controlling the FMN chemistry to promote dehalogenation via a FMNsq intermediate, then derivatives of the zwitterion have the potential to change the chemoselectivity of catalysis. Previously, destabilization of FMNsq by loss of the hydrogen bond donor to the flavin N5 group suppressed dehalogenation and activated nitroreduction by IYD.18 Likewise, this same switch in catalytic specificity was observed in the absence of zwitterion interactions when studying turnover of 2-nitrophenol and 2-iodophenol.20 At the time, the assumed lack of active site lid closure was used to rationalize the complementary loss and gain in catalytic function. Perturbations of the zwitterion of O2N-Tyr offer an additional perspective for which binding and lid closure can be anticipated and nitroreductase activity may be inferred. To test these predictions, binding of the appropriate nitro compounds was characterized. IYD maintained a high affinity for both O2N-Tyr and O2N-tyrosinamide in analogy to I-Tyr and I-tyrosinamide (Tables 1 and 3 and Figure S15). In both examples, neutralization of the ligand carboxylate apparently had minimal impact on coordination to the N3 position of the resting-state FMNox. Coordination was more substantially perturbed by complete loss of the carboxylate group in O2N-tyramine. In this case, binding weakened by 70-fold relative to that of O2N-Tyr, well beyond the 17-fold loss for I-tyramine relative to that for I-Tyr. Unlike the iodinated derivatives, measurable binding was not lost upon substitution of the α-ammonium group with a hydroxy group. O2N-HPLA bound to IYD with even greater affinity than O2N-tyramine (Table 3 and Figure 2).
Table 3.
Binding of O2N-Tyr Zwitterionic Derivatives to IYDa
| A, B | Compound | kd(μM)b | |
|---|---|---|---|
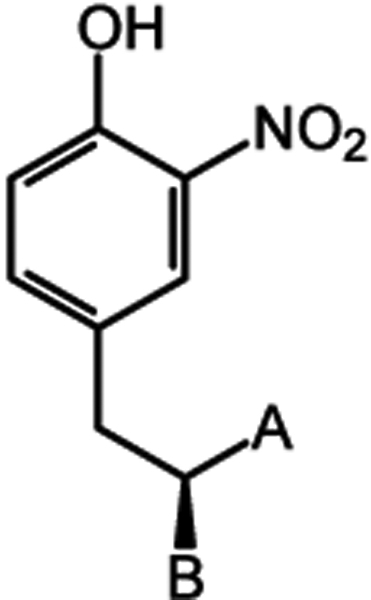
|
COO−, NH3+ | O2N-Tyr | 4.0 ± 0.9 |
| CONH2, NH3+ | O2N-tyrosinamide | 2.4 ± 0.1 | |
| H, NH3+ | O2N-tyraminc | 280 ± 10 | |
| COO−, OH | O2N-HPLA | 180 ± 4 |
Data represent the average of two independent measurements, and the error represents their range.
See Figure S15.
Saturating concentrations of O2N-tyrosinamide, O2N-tyramine, and O2N-HPLA all protected IYD from proteolysis by trypsin (Table 4 and Figures S16 and S17). For each, the half-life of IYD was extended by at least twofold over that in the absence of an active site ligand (Table 2). Thus, these ligands appear to secure the lid sequence surrounding the active site similarly to the analogous fluoro and iodo derivatives described above. At least for O2N-tyrosinamide, protection of IYD was not the consequence of a general inhibition of trypsin as suggested by activity assays with DL-BAPNA as a substrate (Figure S18). Unfortunately, high concentrations required for saturation of the other ligands generated a very high background at A405 that is used to detect the product of dl-BAPNA hydrolysis. For O2N-HPLA, a concentration of only twice its Kd value could be examined with trypsin and these conditions resulted in no more than 25% inhibition of catalysis (Figure S18). With a confirmation that the nitro derivatives bound to IYD and secured closure of the lid, the nature of turnover could be examined.
Table 4.
Half-Lives of IYD Proteolysis by Trypsin Incubated with O2N Ligandsa
| ligand | t1/2 (min)b | fold increase in t1/2 |
|---|---|---|
| O2N-tyrosinamide | 30 ± 2 | 3.0 |
| O2N-tyramine | 27 ± 9 | 2.7 |
| O2N-HPLA | 21 ± 4 | 2.1 |
Values and error were obtained from fitting two independent measurements at each time point to a first-order decay in Origin 6.0.
See Figures S16 and S17.
Nitroreductase activity was detected by the ability of substrates to oxidize the reduced form of IYD, as previously described.18,21,48 IYD was first reduced by minimal dithionite, and then spectral changes were monitored after sequential addition of O2N-Tyr or its derivatives. Equimolar concentrations of O2N-Tyr were not capable of oxidizing IYD despite the potential for O2N-Tyr to consume two hydride equivalents while generating its hydroxylamine product (Figure 4). These conditions oxidized FMNhq by no more than approximately 30%, as evident from the gain in A450, and simultaneously generated only an intermediate level of FMNsq, as evident from the gain in A600 that was also observed in earlier studies.18 In contrast, equimolar concentrations of O2N-tyrosinamide, O2N-tyramine, and O2N-HPLA were each capable of oxidizing FMNhq to regenerate the initial FMNox spectrum of IYD with a λmax of ~450 nm, and this was accomplished with the latter two substrates at concentrations well below their Kd values (Table 3). The reciprocity between dehalogenation and nitroreductase can now be extended to substrates derivatized at their zwitterion. Loss of the carboxylate group by its conversion to a carboxamide analogue or its complete deletion destabilizes single-electron processes while concurrently stimulating a hydride transfer required for nitroreduction. Similarly, replacement of the α-ammonium group with a hydroxyl group activates hydride transfer and suppresses single-electron transfer (Figure 4 and Table 1). The polar interactions between the zwitterion and the N3 and O4 groups of flavin are thus key for supporting efficient dehalogenation by IYD. The absence of either the substrate α-ammonium or carboxylate group impacts catalysis to a far greater extent than loss of a hydrogen bond to its N5 position.
Figure 4.

Oxidation of FMNhq with O2N-Tyr and its zwitterionic derivatives. (A) IYD (oxidized, 18 μM, black trace) in 110 mM potassium phosphate pH 7.4 was reduced with a minimal amount of dithionite (red trace) under anaerobic conditions. This was confirmed to lack excess dithionite by adding 20 μL of air-saturated buffer and observing partial oxidation of IYD. Aliquots of 6 μM O2N-Tyr (green, blue, and cyan traces) were added sequentially at 30 min intervals for a final concentration of 18 μM. Independent samples of IYD (black traces, 15 μM) were reduced under equivalent conditions, and two aliquots each of 5 μM of (B) O2N-tyrosinamide, (C) O2N-tyramine, and (D) O2N-HPLA were alternatively added for final concentrations of 10 μM (green and blue traces, respectively).
The significance of polar interactions to the N3 and O4 groups of flavin serves IYD well. Catalysis is controlled by direct coordination between the substrate zwitterion and flavin to limit dehalogenation to halotyrosines at the amino acid level and avoid processing these residues embedded in peptides or proteins. At least for vertebrates, this mechanism of activation safeguards the iodotyrosyl residues that accumulate on the surface of thyroglobulin as intermediates during formation of thyroid hormone. The advantage for this same specificity in bacteria IYD as reported here will not be obvious until the physiological role of this enzyme can be identified. Whether a comparable substrate of bromotyrosine discovered in fruit flies is generated as an amino acid or derives from bromotyrosine containing proteins or peptides remains to be determined as well.49
When surveying conserved residues in the nitroreductase superfamily, structure−function relationships are most clearly established by residues adjacent to the flavin N5 and much less at the O2, N3, and O4.14 Consequently, the significance of the substrate zwitterion was unanticipated but now explains a variety of prior results including the relatively small loss in dehalogenase activity when hydrogen bonding to flavin N5 was removed.18 This discovery also explains why only a modest increase in dehalogenation of 2-iodophenol was achieved after redesigning the active site lid for stabilizing the association of this substrate with IYD.23 Future efforts for such engineering will require the additional positioning of side chains to establish polar interactions with the N3 and O4 of flavin. The mere proximity created by stacking of a phenolic derivative over FMNhq is not sufficient for single-electron transfer and dehalogenation. Stabilization of the FMNsq intermediate is also necessary whether this is supported by substrate or protein. In contrast, stacking and orientation may be sufficient to promote hydride transfer for nitroreduction by IYD.
CONCLUSIONS
The broad range of reactions promoted by flavin cofactors provide biology with great opportunities but also significant risks. The chemistry of this cofactor requires exquisite control to ensure a high specificity for the desired catalysis and protection against undesirable and often detrimental processes. Typically, such control is the responsibility of the surrounding protein that variably employs sterics, solvation, electrostatics, hydrogen bonding, acid–base chemistry, and proximity to manipulate the many modes of flavin reactivity. IYD offers one of the few examples of a flavoprotein that establishes direct coordination between polar functional groups of the substrate and flavin. Initially, these features were thought to help template active site organization and lid closure to align a Thr side chain for hydrogen bonding to the all-important flavin N5 position.17 This was further supported by crystallographic and kinetic studies connecting lid closure with efficient dehalogenation and lid disorder with a significant decrease in dehalogenation and increase in nitroreduction.19,20 However, solution studies by NMR revealed significant closure of the lid even with phenolic ligands and thus an alternative basis of FMN activation was necessary.22 The zwitterion ion derivatives of I-Tyr, F-Tyr, and O2N-Tyr have all now generated data consistent with a control of catalytic specificity that is dominated by interactions between the substrate zwitterion and the N3 and O4 groups of flavin. Although active site binding tolerates modifications of the substrate’s carboxylate group, efficient dehalogenation requires the presence of both the α-ammoniun and carboxylate groups. Together, these stabilize the FMNsq required for a mechanism of dehalogenation involving single-electron transfer. Such interactions are not required for a basal nitroreductase activity that is apparent only when formation of FMNsq is suppressed. The nature of the substrate consequently dictates the chemistry of catalysis supported by IYD in a manner that is highly distinct from most other flavoproteins.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by the National Institute of General Medicine (GM130937 to S.E.R.), and the authors would like to thank Jimin Hu and Fazel Fakhari for their preliminary investigations.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.4c00357.
Experimental methods, synthetic protocols for substrate analogues, binding, catalysis, limited proteolysis, and redox properties of IYD with 3-substituted tyrosine derivatives (PDF)
Accession Codes
IYD from Haliscomenobacter hydrossis, Uniprot F4KU78; IYD from Homo sapiens, Uniprot Q6PHW0; IYD from Drosophila melanogaster, Uniprot E1JIB2; Flavodoxin from Clostridium beijerinckii, Uniprot P00322.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biochem.4c00357
The authors declare no competing financial interest.
Contributor Information
Daniel Lemen, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States;.
Steven E. Rokita, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States;
REFERENCES
- (1).Fagan RL; Palfey BA Flavin-Dependent Enzymes. In Ch. 3 in Comprehensive Natural Products II; Begley TP Ed.; Vol. 7; Elsevier: 2010; pp 37–114. [Google Scholar]
- (2).Walsh CT; Wencewicz TA Flavoenzymes: Versatile catalysts in biosynthetic pathways. Nat. Prod. Rep 2013, 30, 175–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Romero E; Gómez Castellanos JR; Gadda G; Fraaije MW; Mattevi A Same substrate, many reactions: oxygen activation in flavoenzymes. Chem. Rev 2018, 118, 1742–1769. [DOI] [PubMed] [Google Scholar]
- (4).Fox KM; Karplus PA The flavin environment in old yellow enzyme. J. Biol. Chem 1999, 274, 9357–9362. [DOI] [PubMed] [Google Scholar]
- (5).Lyubimov AY; Lario PI; Moustafa I; Vrielink A Atomic resolution crystallography reveals how changes in pH shape the protein microenvironment. Nat. Chem. Biol 2006, 2, 259–264. [DOI] [PubMed] [Google Scholar]
- (6).Smith WW; Burnett RM; Darling GD; Ludwig ML Structure of the semiquinone form of flavodoxin from Clostridium MP. J. Mol. Biol 1977, 117, 195–225. [DOI] [PubMed] [Google Scholar]
- (7).Fraaije MW; Mattevi A Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem. Sci 2000, 25, 126–132. [DOI] [PubMed] [Google Scholar]
- (8).Yang K-Y; Swenson RP Modulation of the redox properties of the flavin cofactor through hydrogen-bonding interactions with the N(5) atom: role of αser254 in the electron-transfer flavoprotein from the methylotrophic bactgerium W3A1. Biochemistry 2007, 46, 2289–2297. [DOI] [PubMed] [Google Scholar]
- (9).Bradley LH; Swenson RP Role of hydrogen bonding Interactions to N(3)H of the flavin mononucleotide cofactor in the modulation of the redox potentials of the Clostridium beijerinckii flavodoxin. Biochemistry 2001, 40, 8686–8695. [DOI] [PubMed] [Google Scholar]
- (10).Cuello AO; McIntosh CM; Rotello VM Model systesm for flavoenzyme activity. The role of N93)-H hydrogen bonding in flavin redox processes. J. Am. Chem. Soc 2000, 122, 3517–3521. [Google Scholar]
- (11).González-Viegas M; Kar RK; Miller A-F; Mroginski M-A Noncovalent interactions that tune the reactivities of the flavins in bifurcating electron transferring flavoprotein. J. Biol. Chem 2023, 299, No. 104762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Das D; Miller A-F A single hydrogen bond that tunes flavin redox reactivity and activates it for modification. Chem. Sci 2024, 15, 7610–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Xu D; Kohli RM; Massey V The role of threonine 37 in flavin reactivity of the old yellow enzyme. Proc. Nat. Acad. Sci. (USA) 1999, 96, 3556–3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Akiva E; Copp JN; Tokuriki N; Babbit PC Evolutionary and molecular foundations of multiple contemporary functions of the nitroreductase superfamily. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E9549–E9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Koder RL; Haynes CA; Rodgers ME; Rodgers DW; Miller A-F Flavin thermodynamics explain the oxygen insensitivity of enteric nitroreductase. Biochemistry 2002, 41, 14197–14205. [DOI] [PubMed] [Google Scholar]
- (16).Taga ME; Larsen NA; Howard-Jones AR; Walsh CT; Walker GC BluB cannibalizes flavin to form the lower ligand of vitamin B12. Nature 2007, 446, 449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hu J; Chuenchor W; Rokita SE A switch between one- and two-electron chemistry of the human flavoprotein iodotyrosine deiodinase is controlled by substrate. J. Biol. Chem 2015, 290, 590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mukherjee A; Rokita SE Single amino acid switch between a flavin-dependent dehalogenase and nitroreductase. J. Am. Chem. Soc 2015, 137, 15342–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ingavat N; Kavran JM; Sun Z; Rokita SE Active site binding is not sufficient for reductive deiodination by iodotyrosine deiodinase. Biochemistry 2017, 56, 1130–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hu J; Su Q; Schlessman JL; Rokita SE Redox control of iodotyrosine deiodinase. Protein Sci. 2019, 28, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Su Q; Boucher PA; Rokita SE Conversion of a dehalogenase to a nitroreductase by swapping its flavin cofactor with a 5-deazaflavin analog. Angew. Chem., Int. Ed 2017, 56, 10862–10866. [DOI] [PubMed] [Google Scholar]
- (22).Greenberg HC; Majumdar A; Cheema EK; Kozyryev A; Rokita SE 19F NMR reveals the dynamics of substrate binding and lid closure for iodotyrosine deiodinase as a complement to steady-state kinetics and crystallography. Biochemistry 2024, 63. asap. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sun Z; Rokita SE Towards a halophenol dehalogenase from iodotyrosine deiodinase via computational design. ACS Catal. 2018, 8, 11783–11793. [Google Scholar]
- (24).Musila JM; Rokita SE Sequence conservation does not always signify a functional imperative as observed in the nitroreductase superfamily. Biochemistry 2022, 61, 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Phatarphekar A; Rokita SE Functional analysis of iodotyrosine deiodinase from Drosophila melanogaster. Protein Sci. 2016, 25, 2187–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kozyryev A; Lemen D; Dunn J; Rokita SE Substrate electronics dominate the rate of reductive dehalogenation promoted by the flavin-dependent iodotyrosine deiodinase. Biochemistry 2023, 62, 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Warner JR; Copley SD Pre-steady-state kinetic studies of the reductive dehalogenation catalyzed by tetrachlorohydroquinone dehalogenase. Biochemistry 2007, 46, 13211–13222. [DOI] [PubMed] [Google Scholar]
- (28).Massey V A simple method for the determination of redox potentials. In Flavins Flavoproteins, Proceedings of the 10th International Symposium, Como, Italy, July 15–20, 1990; Curti B; Ronchi S; Zanetti G, Eds.; Gruyter & Co.: 1991; pp 59–66. [Google Scholar]
- (29).van den Heuvel RHH; Fraaije MW; Van Berkel WJH Redox Properties of Vanillyl-alcohol Oxidase. Methods Enzymol. 2002, 353, 177–186. [DOI] [PubMed] [Google Scholar]
- (30).Christgen SL; Becker SM; Becker DF Methods for Determining the Reduction Potentials of Flavin Enzymes. Methods in Enzymol. 2019, 620, 1–25. [DOI] [PubMed] [Google Scholar]
- (31).Clark WM Oxidation-Reduction Potentials of Organic Systems; Williams & Wilkins: 1960. [Google Scholar]
- (32).Yamaguchi H; Miyazaki M; Maeda H Limited proteolysis in proteomics using protease-immobilized microreactors. Methods Mol. Biol 2012, 815, 187–198. [DOI] [PubMed] [Google Scholar]
- (33).Cochrane JR; White JM; Wille U; Hutton CA Total synthesis of mycocyclosin. Org. Lett 2012, 14, 2402–2405. [DOI] [PubMed] [Google Scholar]
- (34).Bergström M; Suresh G; Naidu VR; Unelius CR N-Iodosuccinimide (NIS) in Direct Aromatic Iodination. Eur. J. Org. Chem 2017, 2017, 3234–3239. [Google Scholar]
- (35).Flanagan JH; Owens CV; Romero SE; Waddel E; Kahn SH; Hammer RP; Soper SA Near-infrared heavy-atom-modified fluorescent dyes for base-calling in DNA-sequencing applications using temporal discrimination. Anal. Chem 1998, 70, 2676–2684. [DOI] [PubMed] [Google Scholar]
- (36).ndel W-H; Kramer W Untersuchungen an quartären pyridinium-salzen, VIII. cyclotetrakondensate mit reaktionsfähigen gruppen aus quartären nicotinamid-salzen. Chem. Ber 1978, 111, 2594–2604. [Google Scholar]
- (37).Washburn NW; Harper TW; Wu G; Godfrey JD; McCann P; Girotra R; Shao C; Zhang H; Gavai A; Mikkilineni A; Dejneka T; Ahmed S; Caringal Y; Hangeland J; Zhang M; Cheng PTW; Russell AD; Skwish S; Slusarchyk DA; Allen GT; Frohlich BH; Abboa-Offei BE; Cap M; Waldron TL; George RJ; Tesfamariam B; Dickinson KE; Seymour AA; Sher PM Arylpropanolamines: selective B3 agonists arising from strategies to mitigate phase I metabolic transformations. Bioorg. Med. Chem. Lett 2007, 17, 4290–4296. [DOI] [PubMed] [Google Scholar]
- (38).Podunavac M; Lacharity JJ; Jones KE; Zakarian A Stereodivergence in the Ireland–Claisen rearrangement of α-alkoxy esters. Org. Lett 2018, 20, 4867–4870. [DOI] [PubMed] [Google Scholar]
- (39).González-Guerrero C; Borsò M; Alkihani P; Alcaina Y; Salas-Lucia F; Liao X-H; García-Giménez J; Bertolini A; Martin D; Moratilla A; Mora R; Bunño-Soto A; Mani AR; Bernal J; Saba A; De Miguel MP; Refetoff S; Zuchi R; Moreno JC Iodotyrosines are biomarkers for preclinical stages of iodine-deficient hypothyroidism in Dehal1-knockout mice. Thyroid 2023, 33, 752–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mani AR; Ippolito S; Moreno JC; Visser TJ; Moore KP The metabolism and dechlorination of chlorotyrosine in vivo. J. Biol. Chem 2007, 282, 29114–29121. [DOI] [PubMed] [Google Scholar]
- (41).Mani AR; Moreno JC; Visser TJ; Moore KP The metabolism and debromination of bromotyrosine in vivo. Free Radical Biol. Med 2016, 90, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sun Z; Xu B; Spisak S; Kavran JM; Rokita SE The minimal structure for iodotyrosine deiodinase function is defined by an outlier protein from thermophilic bacteria Thermotoga neapolitana. J. Biol. Chem 2021, 297, No. 101385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Fontana A; De Laureto PP; Spolaore B; Frare E; Picotti P; Zambonin M Probing protein structure by limited proteolysis. Acta Biochim. Pol 2004, 51, 299–321. [PubMed] [Google Scholar]
- (44).McTamney PM; Rokita SE A mammalian reductive deiodinase has broad power to dehalogenate chlorinated and brominated substrates. J. Am. Chem. Soc 2009, 131, 14212–14213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhou Z; Swenson RP Electrostatic effects of surface acidic amino acids residues on the oxidation-reduction potentials of the flavodoxin from Desulfovibrio vulgaris (Hildenborough). Biochemistry 1995, 34, 3183–3192. [DOI] [PubMed] [Google Scholar]
- (46).Bradley LH; Swenson RP Role of glutamate-59 hydrogen bonded to N(3)H of the flavin mononucleotide cofactor in the modulation of the redox potentials of the Clostridium beijerinckii flavodoxin. Glutamate-59 is not responsible for the pH dependency but contributes to the stabilization of the flavin semiquinone. Biochemistry 1999, 38, 12377–12386. [DOI] [PubMed] [Google Scholar]
- (47).Tegoni M; Gervais M; Desbois A Resonance Raman study on the oxidized and anionic semiquinone forms of flavocytochrome b2 and L-lactate monooxygenase. Influence of the structure and environment of the isoalloxazine ring on the flavin function. Biochemistry 1997, 36, 8932–8946. [DOI] [PubMed] [Google Scholar]
- (48).Kozyryev A; Boucher PA; Quinñones-Jurgensen CM; Rokita SE The 2’-hydroxy group of flavin mononucleotide influences the catalytic function and promiscuity of the flavoprotein iodotyrosine dehalogenase. RSC Chem. Biol 2023, 4, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Su Q; Xu B; Chen X; Rokita SE Misregulation of endogenous bromotyrosine compromises fertility in male Drosophila. Proc. Natl. Acad. Sci. U. S. A 2024, 121, No. e2322501121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.