

Abstract
Glucocorticoids are a major class of therapeutic anti-inflammatory and immunosuppressive drugs prescribed to patients with inflammatory diseases, to avoid transplant rejection, and as part of cancer chemotherapy. However, exposure to these drugs increases the risk of opportunistic infections such as with the fungus Aspergillus fumigatus, which causes mortality in >50% of infected patients. The mechanisms by which glucocorticoids increase susceptibility to A. fumigatus are poorly understood. Here, we used a zebrafish larva-Aspergillus infection model to identify innate immune mechanisms altered by glucocorticoid treatment. Infected larvae exposed to dexamethasone succumb to infection at a significantly higher rate than control larvae. However, both macrophages and neutrophils are still recruited to the site of infection and dexamethasone treatment does not significantly affect fungal spore killing. Instead, the primary effect of dexamethasone manifests later in infection with treated larvae exhibiting increased invasive hyphal growth. In line with this, dexamethasone predominantly inhibits neutrophil function, rather than macrophage function. Dexamethasone-induced mortality also depends on the glucocorticoid receptor. Dexamethasone partially suppresses NF-κB activation at the infection site by inducing the transcription of IκB via the glucocorticoid receptor. Independent CRISPR/Cas9 targeting of IKKγ to prevent NF-κB activation also increases invasive A. fumigatus growth and larval mortality. However, dexamethasone treatment of IKKγ crispant larvae further increases invasive hyphal growth and host mortality, suggesting that dexamethasone may suppress other pathways in addition to NF-κB to promote host susceptibility. Collectively, we find that dexamethasone acts through the glucocorticoid receptor to suppress NF-κB-mediated neutrophil control of A. fumigatus hyphae in zebrafish larvae.
Keywords: Animals-Fish, Cells-Neutrophils, Infections-Fungal, Molecules-Transcription Factors, Processes-Inflammation
Introduction
Glucocorticoids are potent immunosuppressive and anti-inflammatory drugs that are prescribed for a range of conditions, including chronic inflammation, lymphoid malignancies, autoimmune conditions, and to avoid rejection in bone marrow and solid organ transplant patients (1–3). However, prolonged use of glucocorticoids causes adverse effects such as metabolic disorders, hypertension, osteoporosis, and depression (4). The immunosuppressive effects of glucocorticoids also increase the risk of opportunistic infections (4). Invasive aspergillosis caused by Aspergillus fumigatus is the most common fungal infection associated with glucocorticoid therapy (5). While immunocompetent hosts effectively clear inhaled airborne A. fumigatus spores from the lungs and airways, in patients undergoing glucocorticoid therapy spores can germinate into invasive filamentous hyphae, destroying tissues and organs (6). Anti-fungal treatments are often ineffective, partially due to growing drug resistance among fungal pathogens, and as a result >50% of infected patients do not survive (7, 8). Glucocorticoids can inhibit multiple different molecular and cellular pathways, and it is not clear which of these effects is the main cause of susceptibility to invasive aspergillosis and other opportunistic infections. This knowledge is necessary to develop novel therapeutic approaches to treat patients with invasive aspergillosis who are undergoing glucocorticoid therapy or to develop safe glucocorticoid therapy with a lower risk of opportunistic infection.
Glucocorticoids exert their activity by binding to the glucocorticoid receptor (GR) which is a nuclear receptor (2). Upon binding to ligand in the cytosol, GR translocates to the nucleus and activates or represses gene transcription (2). GR can affect gene expression through three mechanisms: directly binding to glucocorticoid response elements (GRE) in the DNA sequence, trans-repression through binding to other transcription factors, or composite binding to both GREs and transcription factors at the same time (2, 3). It is thought that the immunosuppressive effects of glucocorticoids are mainly mediated by trans-repression of nuclear factor-κB (NF-κB) (1, 9–11). NF-κB is a family of transcription factors, including the canonical p65 and p50 subunits, that activate inflammatory responses by promoting transcription of various signaling molecules such as cytokines (12). Under resting conditions, p65/p50 heterodimers are bound by an inhibitor, IκB, and sequestered in the cytoplasm (13–17). Downstream of activation of pattern recognition receptors (PRRs), cytokine receptors, or T/B-cell receptors (18, 19), IκB is phosphorylated by the multi-subunit IκB kinase (IKK) complex, which is composed of IKKα, IKKβ, and a regulatory subunit IKKγ (NEMO) (17–20). This phosphorylation leads to degradation of IκB, releasing the NF-κB dimers which rapidly translocate to the nucleus to initiate target gene expression (12, 17–19). To inhibit this activation, GR can both directly bind and trans-repress NF-κB subunits and directly bind to the IκB gene promoter to induce transcription. The relative significance of each of these activities in NF-κB suppression by GR is debated (21–23). Additionally, if glucocorticoid-mediated suppression of NF-κB is the major mechanistic cause of susceptibility to invasive aspergillosis, rather than GR-mediated effects on other pathways, is not well understood. A. fumigatus can induce NF-κB activation in vitro in monocytes, in bronchial epithelial cells in mice, and at the site of infection in larval zebrafish (24–26). However, how NF-κB activation controls A. fumigatus and if glucocorticoids suppress these control mechanisms in vivo is not understood.
In humans and in mammalian models, alveolar macrophages are tissue-resident in the lungs and are likely the first immune cells to encounter inhaled A. fumigatus spores, and neutrophils can be quickly recruited from the blood (27). Macrophages and neutrophils thus act as the first line of defense against A. fumigatus infection and are able to kill this fungus even before the activation of specific adaptive responses. Glucocorticoids are known to be able to alter the activity of these phagocytes (5, 6), however, it is unclear which specific phagocyte mechanisms against A. fumigatus are suppressed by glucocorticoids to increase susceptibility to invasive aspergillosis in vivo. In this study, we investigate the effect of glucocorticoid treatment on macrophages and neutrophils in a zebrafish larva-Aspergillus infection model which allows us to non-invasively image this dynamic host-pathogen interaction inside of a live, intact host over multiple days. Using this model, we have previously shown that macrophages mainly respond to A. fumigatus spores and prevent germination, while neutrophils are only recruited after spore germination into hyphae (28–30). This is in line with previous findings that macrophages efficiently kill spores and neutrophils are efficient at killing hyphae in cell culture (31–33). The immune system of zebrafish is largely conserved with humans and during the larval stage primarily consists of macrophages and neutrophils, as the adaptive system is not functional until later in development (34). The zebrafish model has been instrumental in modeling a range of human infections to better understand host-pathogen mechanisms, such as mycobacterial granuloma formation (35).
We report that exposure to the glucocorticoid drug dexamethasone significantly increases the mortality of A. fumigatus-infected larvae, recapitulating the susceptibility of glucocorticoid-treated human patients. Through CRISPR/Cas9 targeting of GR, we demonstrate that dexamethasone activity is mediated via GR in larval zebrafish. To determine the host innate mechanisms that are inhibited by dexamethasone treatment we used daily, live imaging of infected larvae in combination with established innate immune cell-deficiency models. We report that the increased mortality of infected hosts is primarily due to a decrease in neutrophil-mediated control of invasive hyphal growth. Dexamethasone treatment induces IκB transcription and suppresses A. fumigatus-induced NF-κB activation. Although inhibition of other pathways may also promote susceptibility to A. fumigatus infection, CRISPR/Cas9 targeting of IKKγ phenocopies dexamethasone treatment, suggesting that the effects of glucocorticoids are largely due to the inhibition of NF-κB signaling and inhibition of neutrophil function.
Materials and Methods
Ethics statement
All experimental procedures of zebrafish embryos and larvae were performed, and adult zebrafish were maintained and handled, according to protocols approved by the Clemson University Institutional Animal Care and Use Committee (AUP2021–0109, AUP2022–0093, AUP2022–0111). Larvae were anesthetized using buffered tricaine prior to any experimental procedures. Larvae were euthanized at 4°C and adults were euthanized with buffered tricaine.
Zebrafish lines and maintenance
Adult zebrafish were maintained at 28°C at 14/10 hr light/dark cycles. All mutant and transgenic lines were maintained in the wild-type AB background, and are listed in Table I. Embryos were collected after natural spawning, and were maintained in E3 medium with methylene blue at 28°C. Embryos were manually dechorionated and anesthetized in 0.3 mg/mL buffered tricaine prior to any experimental procedures. Larvae used for imaging were treated with 200 μM N-phenylthiourea (PTU) starting from 24 hours post fertilization to inhibit pigment formation. All transgenic larvae were screened for fluorescent expression prior to infections. For most experiments in the irf8 mutant line, larvae from irf8+/− in-crosses also carrying transgenes expressing fluorescent markers in neutrophils (Tg(mpx:mCherry) or Tg(lyzC:BFP)) were screened for a high number of neutrophils to select for irf8−/− individuals prior to infections. To test NF-κB activation levels in irf8 mutant individuals, larvae from irf8+/− in-crosses also carrying the NF-κB RE:GFP transgene were used. After experiments were concluded, larvae were genotyped at the irf8 locus as previously described (36), using the primers F: 5’ CAGGAGAGTTCAGTAAATTGAGC 3’; R: 5’ CTTGTTTTCCCGCATGTTTCC 3’. During daily imaging experiments in the irf8 mutant line, larvae with uncontrolled fungal growth were euthanized to isolate genomic DNA prior to completion of the 5 day experiment.
Table I.
Zebrafish lines used in this study.
Generation of Tg(mfap4:BFP) fish line
First, a clean Tol2 vector containing just the mfap4 promoter was generated. Tol2-mpx:mCherry-2A-rac2 (40) (a gift from Anna Huttenlocher) was cut with NheI and SalI (Promega) to remove the mpx:mCherry-2A-rac2 insert. The mfap4 promoter was then amplified from p5E-mfap4 (Addgene #70052, a gift from David Tobin) for InFusion cloning (Takara Bio) into the digested Tol2 backbone (F: 5’ GAAGTAAAAGGCTAGCGCGTTTCTTGGTACAGCTG 3’; R: 5’ TTCTAGATCAGTCGACCACGATCTAAAGTCATGAAG 3’). To generate Tol2-mfap4:BFP, BFP was amplified from Tol2-lyz:BFP (26) (a gift from Anna Huttenlocher) for HiFi cloning (NEB) into the Tol2-mfap4 vector cut open with SalI (F: 5’ TGACTTTAGATCGTGGTCGACGGTACCTCGCCACCATGA 3’; R: 5’ CTATAGTTCTAGATCATCGACTCACTTGTGCCCCAGTTT 3’).
For integration of mfap4:BFP into the zebrafish genome, Tol2 transposase was in vitro transcribed from NotI-digested (NEB) pCS2-transposase (a gift from Anna Huttenlocher) using an mMESSAGE mMACHINE SP6 kit according to the manufacturer’s directions (Invitrogen). mRNA was purified with a MEGAclear kit (Invitrogen). 1–3 nl of an injection mix containing 20 ng/μl Tol2-mfap4:BFP plasmid and 10 ng/μl transposase mRNA was injected into the yolk of single cell embryos of the AB strain. Injected F0 embryos were grown to adulthood and a founder with integration of the DNA into the germline was determined by outcrossing single F0 adults and screening for BFP expression.
Aspergillus fumigatus strains and spore preparation for injections
The CEA10 strain (41) was used for most non-imaging experiments. CEA10-derived GFP-expressing TFYL49.1 (42) and RFP-expressing TDGC1.2 (43) (a gift from Nancy Keller) strains were used for imaging experiments. An AF293-derived mCherry-expressing TBK5.1 strain (26) was also used for survival experiments. Expression of a fluorescent protein does not affect pathogenesis in zebrafish larvae (26, 43). An AF293-derived mCherry-expressing TBK5.1 strain (26) was also used for survival experiments. The A. fumigatus strain used is mentioned in each figure legend. Spores were prepared as previously described (28). Briefly, 106 spores were spread on 10 cm plates with solid glucose minimal media (GMM) and were incubated at 37°C for 4 days. Spores were collected into sterile water with 0.01% Tween by scraping using a L spreader and were filtered through sterile miracloth (Sigma-Aldrich) into a 50 mL centrifuge tube. Spores were pelleted by centrifugation at 900 g for 10 mins, washed in sterile PBS, pelleted again, and finally resuspended in 5 mL of sterile PBS. This suspension was again filtered through miracloth into a new 50 mL tube. The spore concentration was determined using a hemocytometer and a suspension at 1.5 × 108/ mL was made in PBS and stored at 4°C for up to ~6 weeks.
In vitro fungal growth assay
10 cm solid GMM plates containing 20 mL GMM agar with 10 μM dexamethasone or 0.01% DMSO vehicle control were prepared and stored at 4°C for up to ~4 months. Spores of the GFP-expressing TFYL49.1 strain were prepared as described above and resuspended at 107/ mL and stored at 4°C for up to ~6 weeks. 2 μL was dispensed into the middle of the plate and plates were incubated at 37°C for 4 days. To quantify the growth in each condition, the diameter of the colony was measured daily. To quantify branching of the growing hyphae, at 2 days post culture, a piece of agar from the edge of each colony was cut out and placed on a glass slide and flattened using a coverslip. Hyphae in this piece were imaged using a Zeiss Cell Observer Spinning Disk confocal microscope with a Plan-Apochromat 20X (0.8 NA) objective using ZEN software. One slide per plate was used for imaging, and four microscopic fields were captured for each slide. Quantification of hyphal branching is described in the Image analysis section. This experiment was repeated twice with three plates/condition/experiment.
Live-dead spore labeling
Spores of the GFP-expressing TFYL49.1 strain were stained with AlexaFluor546 as described previously (39, 44, 45). Briefly, spores were incubated on a shaker with 0.5 mg/mL of biotin-XX, SSE (Molecular Probes) in 0.05M NaHCO3 at 4°C for 2 hrs. Spores were pelleted by centrifugation and washed twice with 100 mM Tris-HCl (pH 8.0) on a shaker at 4°C for 30 min to deactivate free-floating biotin. Spores were washed with 1X PBS twice. Spores were then resuspended in 1X PBS containing 20 μg/mL of streptavidin-AlexaFluor546 (Invitrogen) and incubated for 40 min at room temperature. Stained spores were then pelleted and resuspended in 1X PBS, spore concentration was enumerated, and a spore suspension was made at 1.5 × 108/ mL and stored at 4°C for up to ~4 weeks.
Zebrafish hindbrain microinjections
A. fumigatus spores were injected into the hindbrain ventricle of 2 days post fertilization (dpf) larvae as described previously (46). The prepared 1.5 × 108/ mL spore suspension was mixed at 2:1 with filter-sterilized phenol red to achieve a final concentration of 1 × 108/ mL. Injection plates made of 2% agarose in E3 were coated with 2% filter-sterilized bovine serum albumin (BSA) to prevent larvae sticking to the agarose. Dechorionated and anesthetized 2 dpf larvae were transferred to and aligned on the injection plate. A microinjection setup supplied with pressure injector, micromanupulator, micropipet holder, footswitch, and back pressure unit (Applied Scientific Instrumentation) was used to inject 30–50 spores into individual larvae. Actual injection doses were monitored by CFU plating as described below and are reported in each Figure legend. PBS mixed with phenol red was used as a mock infection control. Injected larvae were then rinsed at least twice with E3 without methylene blue to remove tricaine and any free spores. For imaging experiments, larvae were returned to E3 containing 200 μM PTU. Larvae were transferred to 96-well plates for survival and CFU experiments, 48-well plates for daily imaging experiments, or 6-well plates for RNA extraction and single day imaging experiments.
Morpholino injections
A stock solution of pu.1 (spi1b) morpholino oligonucleotide (MO) (ZFIN MO1-spi1b: 5’ GATATACTGATACTCCATTGGTGGT 3’) (GeneTools) was made by resuspension in water to 1 mM and stored at 4°C (47, 48). For injections, the stock was diluted in water with 0.5X CutSmart Buffer (New England Biolabs) and 0.1% filter-sterilized phenol red. A standard control MO at matching concentrations was used as a control. 3 nL of the injection mix was injected into the yolk of 1–2 cell stage embryos. We used 2 different concentrations of pu.1 MO: low-dose 0.05 mM to prevent development of macrophages only (49) or high-dose 0.5 mM to prevent development of both macrophages and neutrophils (47). Efficacy of 0.5 mM pu.1 knockdown was determined by injection into embryos of macrophage- (Tg(mpeg1:H2B-GFP)) and neutrophil-labeled (Tg(mpx:mCherry)) zebrafish lines and larvae were screened for the loss of fluorescent signal using a fluorescent zoomscope (Zeiss SteREO Discovery.V12 PentaFluar with Achromat S 1.0x objective) prior to A. fumigatus infections. To identify a low concentration of pu.1 MO that only inhibits macrophage development but not neutrophil development, multiple different concentrations ranging from 0.05 mM to 0.5 mM were tested. Embryos of macrophage-labeled (Tg(mpeg1:H2B-GFP)) and neutrophil-labeled (Tg(mpx:mCherry)) zebrafish lines were injected with increasing concentrations of pu.1 MO and larvae at 2 dpf were screened for the loss of GFP signal but intact mCherry signal (Supplemental Fig. 3E) using a fluorescent zoomscope (Zeiss SteREO Discovery.V12 PentaFluar with Achromat S 1.0x objective). To determine for how long macrophage development is inhibited, 0.05 mM pu.1 MO- or control MO-injected larvae were imaged (as described in the Live imaging section) at 2, 3, 5, and 7 dpf. The images were used to enumerate the total number of macrophages in the whole body of each larva (as described in the Image analysis section; Supplemental Fig. 3H). At least 4 larvae were used for each condition, and the experiment was repeated twice. To further test if neutrophils are still active with low-dose pu.1 MO injections, we performed a tail wounding experiment as previously described (50). Briefly, the tails of Tg(mpx:mCherry) larvae injected with 0.05 mM pu.1 or control MO were transected using a no.10 Feather surgical blade (GF Health Products) and the larvae were confocal imaged (as described in the Live imaging section) at 2 hours post wounding. The number of neutrophils at the wounding site was enumerated (Supplemental Fig. 3F, 3G). 6 larvae were used for each condition, and the experiment was done once.
CRISPR gRNA design and injections
For each target gene, two guide RNAs (gRNA) were designed to bind to regions of the coding sequence that are required for the function of the protein and in which the translated amino acid sequence is conserved with the human protein. gRNA targets were identified with the CHOPCHOP web-based program (51–53). gRNA sequences are listed in Table II. To generate DNA templates for in vitro transcription of gRNAs, gene-specific oligo sequences were generated containing a T7 promoter (5’-TAATACGACTCACTATAG-3’), the target sequence, and an overlap region to pair with a constant oligo encoding the reverse complement of the Cas9 binding sequence (Integrated DNA Technologies)(constant oligo (5’-AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGC TATTTCTAGCTCTAAAAC −3’)). Gene-specific and constant oligos were annealed and T4 DNA polymerase (New England Biolabs) was added to generate the DNA template. Purified template was in vitro transcribed using T7 RNA polymerase (New England Biolabs), treated with DNase I (New England Biolabs), and purified using a Monarch RNA cleanup kit (New England Biolabs). Embryos were injected with both gRNAs targeting a single gene. An injection mix containing 75 ng/μL of each gRNA and 250 ng/μL Cas9 protein (PNA Bio) was used. Two control gRNAs targeting luciferase (luc) at matching concentrations were used as the control. 1–2 nL of injection mix was injected into the yolk of 1 cell stage embryos. Genomic DNA from individual larvae was extracted at 2 dpf in 50 mM NaOH at 95°C. The efficacy of gRNAs were tested by PCR using primers flanking the target sequences. We used two sets of PCR, one with primers flanking an individual target site (F1R1 or F2R2) and with primers flanking the two target sequences (F1R2) as shown in Supplemental Fig. 1C, 4F. For ikbkg1+2-injected larvae, a separate primer pair flanking the two target sites with matching Tm was designed (F1altR2alt). The primers used are listed in Table III. The PCR products were run on a 2.5% agarose gel to visualize alterations of genomic DNA as shown in Supplemental Fig. 1D, 4G, 4J.
Table II.
CRISPR gRNAs.
| gene | gRNA | gRNA sequence (5’ – 3’) |
|---|---|---|
| ikbkg | 1 | ATATTGCAGAGTGCAGCCAC |
| 2 | TCCCTGCTTGACTGACACTC | |
| 3 | GCAACTGCTCCAGTGCAGCG | |
| 4 | GGAACAGTGACAGCACACGT | |
| nr3c1 | 1 | ACCCAAAGTGAAGGGGACCA |
| 2 | ATTTGCGGAAACGACAGGCA | |
| luciferase | 1 | TTGGAAACGAACACCACGGT |
| 2 | ACAACTTTACCGACCGCGCC |
Table III.
Primers used to test successful alteration of DNA.
| gRNA | Primers used for PCR | Primer sequence (5’ – 3’) |
|---|---|---|
| ikbkg-1 | F1 | CAATGCGGTCTTTTGTTGTG |
| R1 | TTCTGAACCTGCGGTCTCTT | |
| Fs1 | AACGTATTACTCATTGCTAGTTAAAGC | |
| Rs1 | TCAACAAAAACACCCTGTTCA | |
| ikbkg-2 | F2 | AGATTTCGTGAGGCCAGAGA |
| R2 | TTGGTGCACTTCAGCTCTTG | |
| Fs2 | CATTTGATCAGTAGTGTATGTCTCACA | |
| Rs2 | AGTGTCTCCTTCAGCGCATT | |
| ikbkg-1 + ikbkg-2 | F1alt | GCACTGCAGTAGTCTTGGTGA |
| R2alt | AGTGTCTCCTTCAGCGCATT | |
| ikbkg-3 | F3 | CGGGCTTTAAGGAGGAAGAA |
| R3 | TGGTGTCAGTTTTCAGTGCAT | |
| ikbkg-4 | F4 | TGCTGTCTTTCTCGCATGTT |
| R4 | GACAGAGCTTCATGGTCAGC | |
| nr3c1-1 | F1 | CTCAAACTGCTTGGGAAGGA |
| R1 | GGGGTTGTTAAGGTCTGCAA | |
| Fs1 | ATGTCTGTGGCCCCTACATC | |
| Rs1 | GCATCGTCAGCAACGTAGAA | |
| nr3c1-2 | F2 | ACGATTGCATCATTGACAAAA |
| R2 | TGCAAGATTTCATGTTACCCTCT | |
| Fs2 | AGATCCATGCAGTGGGTTTC | |
| Rs2 | GGGGGAATTGGGATAAGTTAG |
The efficiency of gRNAs was also quantified using a web-based tool, Tracking of Indels by Decomposition (TIDE, http://tide.nki.nl) (54). A new set of primers was designed to amplify a larger >400 bp fragment around the gRNA target site (FsRs primers, Table III, Supplemental Figs. 1C and 4E). The target site was amplified from genomic DNA from larvae injected with single test gRNAs or the luciferase control gRNA. The PCR products were run on a 2.5% agarose gel to confirm amplification, purified using a NucleoSpin Gel and PCR Clean-up kit (Macherey-Nagel), and Sanger sequenced (Eton Bioscience). The resulting chromatogram sequence files (.ab1) were uploaded to the TIDE web-based tool (54) to quantify the percentage of indels introduced at the cut site (Supplemental Fig. 1E, 1F).
Clodronate liposome injections
Larvae expressing GFP in macrophages (Tg(mpeg1:H2B-GFP)) were manually dechorionated at 1 dpf. Clodronate or PBS liposomes (Liposoma) were stored at 4°C. Prior to injections, 10% of volume of filter-sterilized phenol red was added to the liposomes and 2 nL was i.v. injected into the caudal vein plexus of larvae. To confirm macrophage depletion, larvae were screened for the loss of GFP signal using a fluorescent zoomscope (Zeiss SteREO Discovery.V12 PentaFluar with Achromat S 1.0x objective) prior to A. fumigatus infection at 2 dpf.
Dexamethasone treatment
Infected larvae were exposed to dexamethasone (Sigma-Aldrich) at 10 μM, a concentration which was previously used in zebrafish larvae (26). A 1000X 10 mM stock was prepared in DMSO and 0.1% DMSO was used as the vehicle control. Directly after hindbrain injection, E3 was removed from dishes containing larvae and new E3 with pre-mixed dexamethasone or DMSO was added. Larvae were kept in the same solution for the entirety of the experiment. For daily imaging experiments, larvae were pipetted out of the drug treatment, imaged, and were transferred back into the same drug solution.
CFU counts
Single larvae were transferred to 1.7 mL microcentrifuge tubes into 90 μL of PBS containing 1 mg/mL ampicillin and 0.5 mg/mL of kanamycin. Larvae were euthanized at 4°C overnight and homogenized with a tissue lyser (Qiagen) at 1800 oscillations/min (30 Hz) for 6 mins. The suspension was then centrifuged at 17000 g for 30 seconds, resuspended by pipetting, and spread on a GMM plate. Plates were incubated at 37°C for 3 days and the number of colonies were counted. For survival experiments, 8 larvae for each condition were collected and euthanized immediately after injections, and were plated the next day to enumerate the actual injection dose. To monitor the fungal burden across multiple days, 8 larvae/condition/day were plated and CFU counts were normalized to the initial injection dose for each condition.
RNA extraction and RT-qPCR
To quantify cytokine gene expression, larvae were infected with TFYL49.1 spores and exposed to dexamethasone or DMSO. At 1 or 2 dpi, infected larvae were anesthetized and screened using a Zeiss Cell Observer Spinning Disk confocal microscope to split larvae into groups based on the presence of germinated spores. From this screening, the pooled no germination group contained 20 larvae and the pooled germination group contained 1–3 larvae per replicate. 500 ng of RNA was used for cDNA synthesis. To quantify nfkbiaa expression in wild-type larvae, larvae were injected with CEA10 spores or PBS and were exposed to dexamethasone or DMSO. 20 pooled larvae for each condition per replicate at 1 or 2 dpi were used for RNA extraction. 1000 ng of RNA was used for cDNA synthesis. To test nfkbiaa expression in glucocorticoid receptor targeted larvae, nr3c1 crispant or control larvae were exposed to dexamethasone or DMSO at 2 dpf. At 1 day post treatment (dpt), 20 pooled larvae from each condition per replicate were used for RNA extraction. 1000 ng of RNA was used for cDNA synthesis.
For RNA extraction, anesthetized larvae were transferred to a 1.7 mL microcentrifuge tube homogenized in 500 μL TRIzol (Invitrogen) on a disruptor genie for 10 minutes. RNA was extracted following the manufacturer’s instructions, using 4 μg of glycogen as a carrier. cDNA synthesis was done with iScript RT Supermix with oligo dT (Bio-Rad). cDNA was diluted 1:10 and 4 μL of diluted cDNA was used for qPCR in a 10 μL reaction, using SYBR Green Supermix (Bio-Rad) and primers listed in Table IV. Fold change was calculated with the ΔΔCq method, using rps11 as the housekeeping gene (55) and three independent replicates, each with two or three technical replicates were performed for each RT-qPCR.
Table IV.
Primers used for RT-qPCR and references.
| Name | Sequence (5’–3’) | Reference |
|---|---|---|
| qrps11_F | TAAGAAATGCCCCTTCACTG | (55) |
| qrps11_R | GTCTCTTCTCAAAACGGTTG | |
| qtnfa_F | AGGCAATTTCACTTCCAAGG | This study |
| qtnfa_R | CAAGCCACCTGAAGAAAAGG | |
| qil1b_F | GCCTGTGTGTTTGGGAATCT | (55, 56) |
| qil1b_R | TGATAAACCAACCGGGACA | |
| qirg1_F | ACTGCTGGCTTTCAATGTGG | This study |
| qirg1_R | AGACGCAGGAGTTTAGCTGT | |
| qarg1_F | GCCGATGTCTTACCTCATCC | This study |
| qarg1_R | CATCCTGAGCTGCTATGCAA | |
| qil10_F | AGCACTCCACAACCCCAAT | This study |
| qil10_R | TTCAAAGGGATTTTGGCAAG | |
| qtgfb1a_F | AACTACTGCATGGGGTCCTG | This study |
| qtgfb1a_R | ACCAGGGTTGTGGTGTTTGT | |
| qnfkbiaa_F | CATGGTGGAGAGTCTGGTCC | This study |
| qnfkbiaa_R | TGGATCTCGCTGTTGTGTCT |
Live imaging
Fluorescent positive larvae were screened on a fluorescent zoomscope (Zeiss SteREO Discovery.V12 PentaFluar with Achromat S 1.0x objective) prior to A. fumigatus infections. Larvae were imaged using a Zeiss Cell Observer Spinning Disk confocal microscope on a Axio Observer 7 microscope stand with a confocal scanhead (Yokogawa CSU-X) and a Photometrics Evolve 512 EMCCD camera. A Plan-Apochromat 20X (0.8 NA) or an EC Plan-Neofluar 40X (0.75 NA) objective and ZEN software were used to acquire Z-stack images of the hindbrain area with 2.5 or 5 μm distance between slices. For daily imaging experiments, larvae were pipetted out of 48-well plates one at a time, anesthetized in tricaine, and transferred to a zWEDGI device (57, 58). After imaging, larvae were rinsed in E3 with 200 μM PTU and transferred back into the original wells into the same drug solution. For single time point imaging, larvae were anesthetized in 6-well plates and were transferred to and imaged in a zWEDGI device (57, 58). For the experiment to test NF-κB activation levels in irf8 mutant larvae, larvae were mounted in 1% low-melting agarose (Sigma) on a 35 mm glass bottom dish (Greiner). To determine how long macrophage development is inhibited, 0.05 mM pu.1 MO- or control MO-injected larvae were transferred to and imaged in a zWEDGI device. 5–6 microscopic fields covering the whole body of each larva were acquired using the same Zeiss confocal microscope with a EC Plan-Neofluar 10X (0.3 NA) objective.
Image analysis
All image analysis was performed blinded and with Image J/Fiji (59). For any analysis where fluorescent intensity was quantified, images were not processed prior to analysis. To quantify the GFP signal from the NF-κB RE:GFP zebrafish line, the hindbrain area was manually identified using the polygon selection tool from the corresponding bright field image. The GFP signal was quantified within the identified area using maximum intensity projection of six z-slices containing A. fumigatus spores or hyphae. The displayed images show signal intensity with the 16 colors lookup table. To quantify phagocyte recruitment, images were processed with bilinear interpolation to increase pixel density two-fold prior to counting and the number of phagocytes and/or the phagocyte cluster area were quantified. Phagocyte numbers were manually counted across z-stacks using the Cell Counter plugin. Macrophage cluster area was measured in maximum intensity projections using the polygon selection tool. Displayed images were processed with bilinear interpolation to increase pixel density two-fold and maximum intensity projections of merged z-stacks were used. For live-dead staining, images were processed with bilinear interpolation to increase pixel density two-fold and live versus dead spores were counted using the Cell Counter plugin. The displayed images of live versus dead spores show a merged z-projection of 3 slices and were processed with gaussian blur (radius = 1) in Fiji to reduce noise. Fungal growth was manually categorized as germination or invasive hyphae. Any hyphal growth (branched or not) was considered an incidence of germination and the presence of branched hyphae was considered an incidence of invasive growth. 2D fungal area was measured by thresholding the fluorescent intensity from maximum intensity projections. To generate the heatmap of fungal growth, the severity was scored using pre-determined categories (44). The scoring was: 0 – no germination, 1 – at least one event of germination, 2 – at least one event of branched hyphae, but hyphae restricted to the infection site, 3 – at least one event of branched hyphae, but hyphae spreading in the hindbrain ventricle, 4 – hyphae invading into nearby tissue, and 5 – lethal. Representative images of each category are shown in Supplemental Fig. 2D. Maximum intensity projection of z-stacks was used for the displayed images. The same experiment and the same images were used to enumerate the number of phagocytes and fungal growth. For irf8−/− larvae imaging experiments, neutrophil cluster area was measured from maximum intensity projections using the polygon selection tool. Maximum intensity projections of z-stacks were used for the displayed images. To quantify the number of macrophages in the whole body of pu.1 MO-injected larvae, the 3D Objects Counter plugin was used, and the resulting Objects map was used to manually subtract any background signals the software counted by comparing with the original image. For the in vitro assay images, the fungal area was measured by quantifying GFP signal by thresholding the fluorescent intensity from the maximum intensity projection of all slices. The number of nodes/branching points were manually counted using the Cell Counter plugin.
Statistical analysis
For all experiments, unless stated otherwise, pooled data from at least three independent replicates were generated and the total Ns are given in each Figure. R version 4.1.0 was used for statistical analysis and GraphPad Prism version 10 (GraphPad Software) was used to generate graphs. Larval survival data and the cumulative percentage of larvae with fungal germination or invasive hyphae were analyzed by Cox proportional hazard regression to calculate P values and hazard ratios (HR). HR reports the likelihood of larvae succumbing to the infection in a particular condition as compared to the control. The statistical analysis considers variability within and between replicates to calculate the P values. Fluorescent intensity, phagocyte numbers and cluster area, fungal area, CFU counts, live-dead imaging analysis, and day of germination or invasive hyphae were analyzed with analysis of variance (ANOVA). For each condition, estimated marginal means (emmeans) and standard error (SEM) were calculated and pairwise comparisons were performed with Tukey’s adjustment. The statistical analysis considers variability within and between replicates to calculate the P values. The graphs for these analyses show values from individual larvae over time as individual lines or points in dot plots, and bars represent pooled emmeans ± SEM. The points in dot plots are color-coded by replicate. For RT-qPCR, the fold change was analyzed by t-test in Excel. For in vitro data, the radius of the colonies and the number of nodes normalized to the fungal area were compared by t-test in Excel. In the dot plot, each dot represents an individual plate, and the dots are color-coded by replicate. For macrophage counts in whole larvae, the total numbers were compared by t-test in Excel and the graph shows values from individual larvae over time as individual lines and bars represent means ± SEM.
Results
Dexamethasone exposure decreases survival of zebrafish larvae infected with A. fumigatus and suppresses NF-κB activation at the infection site
Glucocorticoid drugs such as dexamethasone increase host susceptibility to A. fumigatus infection, but the cellular mechanisms through which this occurs are largely unknown (6). To investigate the effects of glucocorticoids on innate immune responses, we used an established larval zebrafish host model (26, 28, 46). We injected A. fumigatus spores of the CEA10 strain into the hindbrain ventricle of 2 days post fertilization (dpf) wild-type larvae and immediately exposed larvae to 10 μM dexamethasone or a DMSO vehicle control. Dexamethasone-exposed larvae succumb to the infection at a significantly higher rate than control larvae, with a hazard ratio of 2.5, indicating that dexamethasone-exposed larvae are 2.5 times more likely to succumb to the infection as compared to control larvae (Fig. 1A), consistent with previous results (26). No significant survival defect due to dexamethasone treatment is observed in PBS-injected mock-infected larvae (Fig. 1A).
Figure 1. Dexamethasone suppresses NF-κB activation and increases susceptibility to Aspergillus fumigatus via the glucocorticoid receptor.
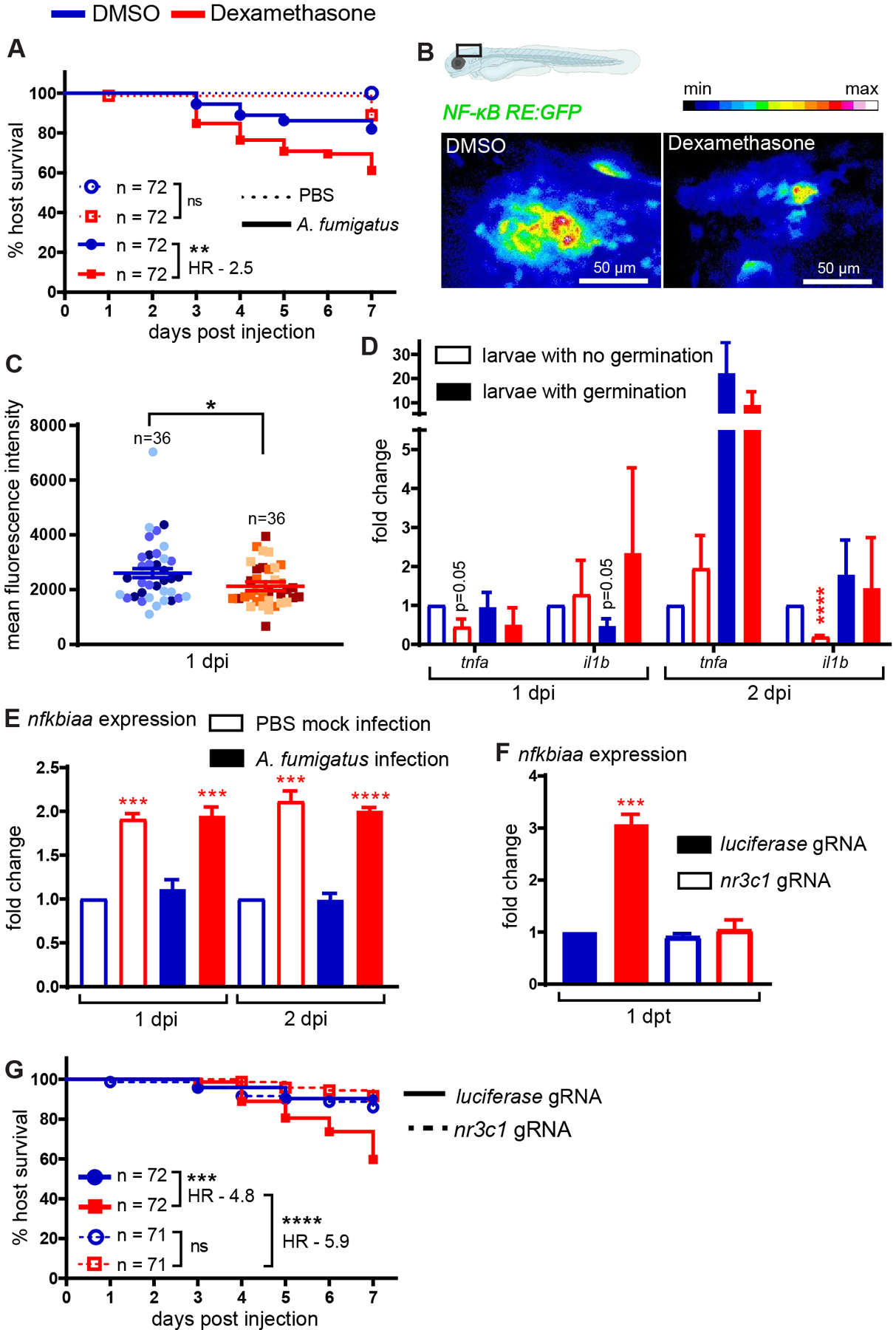
(A) Survival of wild-type larvae injected at 2 dpf with CEA10 A. fumigatus spores or PBS mock-infection in the presence of 10 μM dexamethasone or DMSO vehicle control. At least 24 larvae per condition, per replicate were used and the total larval N per condition is indicated. Cox proportional hazard regression analysis was used to calculate P values and hazard ratio (HR). Average injection CFUs: dexamethasone = 15, DMSO = 12. (B, C) Larvae of NF-κB reporter line (Tg(NF-κB RE:GFP)) were injected with CEA10 A. fumigatus spores and were exposed to 10 μM dexamethasone or DMSO. Larvae were imaged at 1 dpi. (B) Representative images showing relative GFP expression from z projection of 6 slices. Scale bar = 50 μm. (C) Quantification of fluorescent expression in the hindbrain ventricle at 1 dpi is shown with emmeans ± SEM from three independent replicates and the total larval N per condition is indicated. Each data point represents an individual larva, color-coded by replicate. P values were calculated by ANOVA. (D) Larvae were injected with GFP-expressing TFYL49.1 (CEA10) spores and exposed to 10 μM dexamethasone or DMSO. At 1 and 2 dpi, larvae were screened for germination and total RNA was extracted from each pooled group. RT-qPCR analysis of cytokine expression in pooled larvae is shown. Data are normalized to DMSO no germination control group. P values were calculated by Student’s t-test. Data are from three independent replicates. (E) NF-κB inhibitor nfkbiaa expression in larvae injected with CEA10 spores or PBS mock-infection and exposed to 10 μM dexamethasone or DMSO is shown. Total RNA was extracted at 1 and 2 dpi from 20 pooled larvae per condition per day. Data are normalized to DMSO PBS mock-infection at each day post injection. P values were calculated by Student’s t-test. Data are from three independent replicates. (F) Embryos at 1 cell stage were injected with gRNAs targeting glucocorticoid receptor gene nr3c1 or luciferase control together with Cas9 protein. At 2 dpf, larvae were treated with 10 μM dexamethasone or DMSO. Total RNA from 20 pooled larvae per condition was extracted at 1 day post treatment (dpt) and nfkbiaa expression was quantified using RT-qPCR. Data are normalized to luciferase gRNA + DMSO group. P values were calculated by Student’s t-test. Data are from three independent replicates. (G) Survival of nr3c1 mutant larvae injected with CEA10 spores and exposed to 10 μM dexamethasone or DMSO. Data are pooled from three independent replicates, at least 23 larvae per condition, per replicate and the total larval N per condition is indicated. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). Average injection CFUs: nr3c1 = 25 or luciferase = 26.
The A. fumigatus CEA10 strain induces NF-κB activation at the site of infection, and glucocorticoids can suppress NF-κB activity (1, 26). To test if dexamethasone suppresses NF-κB activation in this infection model, we used a previously published NF-κB reporter transgenic zebrafish line that express GFP under an NF-κB responsive promoter (Tg(NF-κB RE:GFP)) (37). We injected NF-κB RE:GFP larvae with A. fumigatus CEA10 spores, imaged the infection site 1 and 2 days post injection (dpi), and quantified GFP expression (Fig. 1B). Dexamethasone-exposed larvae have lower GFP expression than control larvae, although this difference is only statistically significant at 1 dpi (Fig. 1C and Supplemental Fig. 1A). We then tested if dexamethasone suppresses the expression of specific NF-κB target cytokine genes using RT-qPCR. Since fungal germination drives NF-κB activation we screened larvae by microscopy prior to RNA extraction and split larvae into two groups based on whether hyphae were present or not. At 1 dpi, the expression of NF-κB target genes tnfa and il1b was not yet induced by germination, although dexamethasone treatment significantly inhibited tnfa expression even in larvae without germination (Fig. 1D). At 2 dpi, germination increases tnfa expression ~20-fold in larvae exposed to DMSO, and this is only partially suppressed by dexamethasone treatment, potentially because dexamethasone treated larvae may experience more hyphal growth and therefore more immune activation overall (Fig. 1D). il1b is only induced ~2-fold by germination at 2 dpi but dexamethasone significantly inhibits il1b expression in larvae without germinated spores (Fig. 1D). Another marker of macrophage activation, irg1, is also induced by germination at 1 dpi and inhibited by dexamethasone treatment (Supplemental Fig. 1B). Additionally, the expression of anti-inflammatory genes il10 and tgfb are significantly inhibited by dexamethasone treatment at 1 dpi but increased at 2 dpi, while arg1 is not affected by germination or dexamethasone treatment (Supplemental Fig. 1B). Overall, these data demonstrate that dexamethasone can affect host gene expression of inflammatory markers, including NF-κB-regulated genes, although increased fungal germination and growth may override this suppression.
To determine the mechanism through which dexamethasone inhibits NF-κB activation, we tested the expression of the nfkbiaa gene which encodes IκBα, the inhibitor of NF-κB. Dexamethasone-treated larvae have higher expression of nfkbiaa, regardless of infection status (Fig. 1E). These results demonstrate that during A. fumigatus infection, one mechanism through which glucocorticoids inhibit NF-κB activation is by increasing transcription of this inhibitor.
Glucocorticoid receptor is required for dexamethasone-mediated immunosuppression
In mammals, glucocorticoids primarily mediate their effects through the glucocorticoid receptor (2). To determine if the increased susceptibility of dexamethasone-treated larvae to A. fumigatus CEA10 infection was due to signaling through this receptor, and not to off-target effects on either the host or pathogen, we used CRISPR/Cas9 to target the zebrafish glucocorticoid receptor gene nr3c1. We designed two gRNAs: one targeting exon 2 which encodes the N-terminal domain and the other targeting exon 4 which encodes part of the DNA binding domain (Supplemental Fig. 1C). We injected embryos at the 1 cell stage with both gRNAs targeting nr3c1 or control gRNAs targeting luciferase, together with Cas9 protein. PCR using primers flanking the target sites on genomic DNA isolated from 2 dpf larvae confirmed successful targeting of DNA (Supplemental Fig. 1D). TIDE analysis (54) estimated nr3c1 gRNAs 1 and 2 to be 62% and 84% efficient, respectively (Supplemental Figs. 1E, 1F).
In these same F0 injected crispants, we tested if dexamethasone can induce nfkbiaa expression as seen with wild-type larvae (Fig. 1E). Dexamethasone significantly induces nfkbiaa expression in control larvae but fails to induce any expression in nr3c1 crispant larvae (Fig. 1F), indicating that GR function is abolished in these crispant larvae and that IκB-mediated suppression of NF-κB activation by dexamethasone depends on the glucocorticoid receptor. Further, we tested the effects of nr3c1 mutation in survival of infected larvae. While dexamethasone-exposed control larvae succumb to A. fumigatus CEA10 infection, dexamethasone has no effect on survival of infected nr3c1 crispant larvae (Fig. 1G). Targeting of nr3c1 had no effect on the survival of PBS mock-infected larvae (Supplemental Fig. 1G). Additionally, direct exposure of A. fumigatus CEA10 spores to dexamethasone has no effect on spore germination or hyphal growth (Supplemental Fig. 2A–C). These data indicate that the immunosuppressive effects of dexamethasone in the context of A. fumigatus CEA10 infection depend solely on signaling through a functional glucocorticoid receptor to inhibit host immune mechanisms.
Dexamethasone partially suppresses macrophage recruitment, but not neutrophil recruitment
We next sought to understand how dexamethasone mediates phagocyte responses to A. fumigatus. As dexamethasone suppresses pro-inflammatory cytokine expression (Fig. 1D), we hypothesized that phagocyte recruitment would be inhibited by dexamethasone. We injected ~30 GFP-expressing CEA10 A. fumigatus spores into larvae expressing mCherry in macrophages (Tg(mpeg1:H2B-mCherry)) and BFP in neutrophils (Tg(lyz:BFP)) and treated larvae with dexamethasone or DMSO vehicle control. We enumerated macrophage and neutrophil recruitment to the infection site through daily, live confocal imaging of infected larvae. In line with previous findings (26, 44), macrophages arrive first and form clusters around spores starting at 1 dpi (Fig. 2A). A significantly higher number of macrophages arrive at 2 dpi in control larvae compared to dexamethasone-treated larvae, yet ~90 macrophages still arrive at the infection site in dexamethasone-treated larvae (Fig. 2B). Up to 3 dpi, macrophage cluster area is not significantly different between the two groups (Fig. 2C). Macrophage clusters resolve from 3–5 dpi in DMSO-exposed larvae (Fig. 2B, 2C). However, in dexamethasone-exposed larvae, more macrophages are recruited later in the infection with a significantly higher number of macrophages and larger cluster area at 5 dpi (Fig. 2B, 2C). Neutrophils respond starting at 2 dpi, primarily after spores start to germinate, and neutrophils are able to infiltrate into macrophage clusters (Fig. 2A). The number of recruited neutrophils is not significantly different between dexamethasone- and DMSO-treated larvae at 1, 2, or 3 dpi (Fig. 2D). At 5 dpi, similar to macrophages, a significantly higher number of neutrophils is present at the infection site in larvae exposed to dexamethasone compared to control larvae (Fig. 2D). This is likely due to increased fungal growth attracting more macrophages and neutrophils to the infection site, as described previously (44). Injected A. fumigatus spores can germinate, and after germination, the fungal filamentous growth can branch and form a network of hyphae, which we classified as invasive hyphae. To normalize for differences in this fungal growth, we analyzed the number of macrophages and neutrophils at specific stages of fungal germination and invasive hyphal growth. On the day that germination is first observed in larvae, the number of macrophages at the infection site is significantly lower in dexamethasone-exposed larvae compared to control larvae (Fig. 2E). However, macrophage numbers on the day before germination is observed and on the day that invasive hyphae is first observed are not significantly different between dexamethasone- and control-treated larvae (Fig. 2E), and neutrophil numbers are also similar between the two conditions throughout the infection (Fig. 2F). Overall, we find that many phagocytes are still recruited to the infection site in dexamethasone-treated larvae. Dexamethasone has no effect on neutrophil recruitment but can curb macrophage recruitment earlier in infection. However, immune activation from fungal germination can override dexamethasone-mediated suppression of macrophage recruitment at later stages of infection.
Figure 2. Dexamethasone moderately suppresses macrophage recruitment but not neutrophil recruitment.
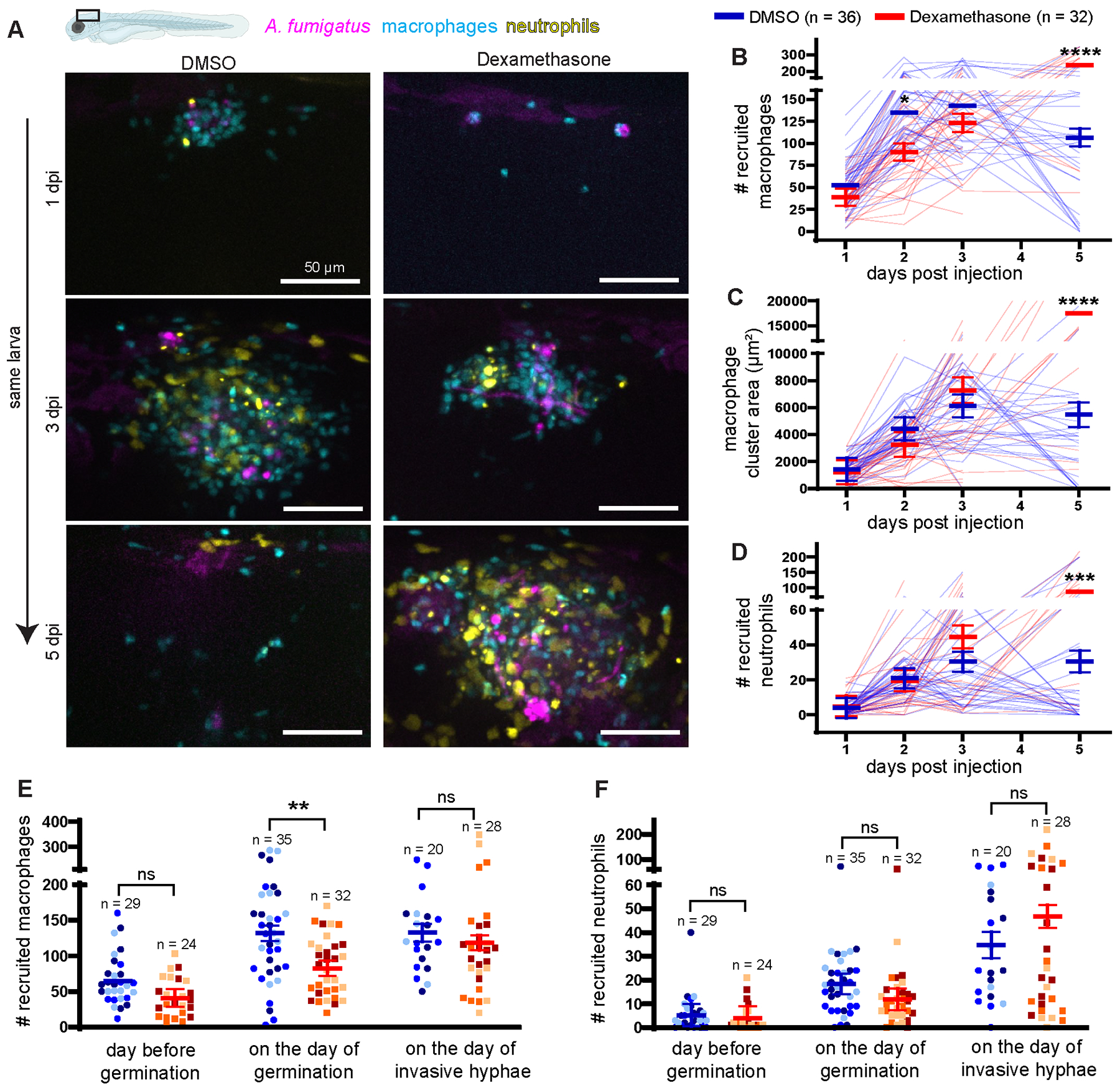
Larvae with labeled macrophages (Tg(mpeg1:H2B-mCherry)) and neutrophils (Tg(lyz:BFP)) were injected with GFP-expressing TFYL49.1 (CEA10) spores at 2 dpf, exposed to 10 μM dexamethasone or DMSO vehicle control and live imaged at 1, 2, 3, and 5 dpi. Data are pooled from three independent replicates, at least 10 larvae per condition, per replicate. (A) Representative images show phagocyte recruitment across multiple days in larvae exposed to dexamethasone or DMSO. Scale bar = 50 μm. (B) Number of macrophages recruited, (C) macrophage cluster area, and (D) number of neutrophils recruited were quantified from the images. (B–D) Bars represent emmeans ± SEM and P values were calculated by ANOVA. Each line represents an individual larva. (E, F) Number of recruited macrophages (E) and neutrophils (F) one day before germination occurred, on the day of germination, and on the day invasive hyphae occurred were plotted for larvae that experienced fungal growth. Bars represent emmeans ± SEM and P values were calculated by ANOVA. Each data point represents an individual larva, color-coded by replicate.
Spore killing is not significantly impacted by dexamethasone exposure
As phagocytes are able to migrate to the site of infection even in dexamethasone-treated larvae, we hypothesized that the functions of phagocyte-mediated control of A. fumigatus are impacted by dexamethasone. The first step in the phagocyte response is macrophage-mediated phagocytosis of spores and spore killing (27, 60). To test whether dexamethasone treatment decreases macrophage-mediated spore killing, we used an established live-dead staining method in which A. fumigatus CEA10 spores expressing GFP are coated with AlexaFluor546 via cell wall cross-linking (26, 44). We injected these spores into larvae expressing BFP in macrophages (Tg(mfap4:BFP)) and imaged the infection site at 2 dpi. We then quantified the percentage of live spores (AlexaFluor+ and GFP+) versus dead spores (AlexaFluor+ and GFP−) (Fig. 3A). Dexamethasone-treated larvae are slightly worse at killing injected CEA10 spores than control larvae, although this difference is not statistically significant (Fig. 3B). To further quantify spore burden over time, we homogenized and plated larvae to quantify CFUs from dexamethasone-treated or control larvae across 7 days of infection. In agreement with the live-dead staining results, we find no significant difference in CFU burden in dexamethasone-treated larvae compared to control larvae (Fig. 3C).
Figure 3. Dexamethasone does not affect spore killing.

(A, B) Macrophage-labeled larvae (Tg(mfap4:BFP)) were injected with GFP-expressing A. fumigatus TFYL49.1 (CEA10) spores coated with AlexaFluor546 at 2 dpf, exposed to 10 μM dexamethasone or DMSO vehicle control, and live imaged at 2 dpi. (A) Representative images of z projection of 3 slices showing live (filled arrow) and dead (open arrow) spores within a macrophage. Scale bar = 10 μm. (B) The percentage of live spores in the hindbrain per larvae is shown with bars representing emmeans ± SEM from three independent replicates, and the total larval N per condition is indicated. Each data point represents an individual larva, color-coded by replicate. P values were calculated by ANOVA. (C) Wild-type larvae were injected with CEA10 spores at 2 dpf, exposed to 10 μM dexamethasone or DMSO vehicle control, and fungal burden was quantified by homogenizing and plating individual larvae for CFUs at multiple days post injection. Eight larvae per condition, per dpi, per replicate were quantified, and the number of CFUs at each dpi is represented as a percentage of the initial spore burden. Bars represent emmeans ± SEM from three independent replicates, P values calculated by ANOVA. Average injection CFU: 32.
Invasive hyphal growth post-germination is increased in dexamethasone-treated larvae
As spore killing is only minorly inhibited by dexamethasone treatment, we hypothesized that in dexamethasone-exposed hosts phagocytes fail to control A. fumigatus germination and invasive hyphal growth. To quantify fungal growth over time, we went back to our daily imaging experiment (Fig. 2) and monitored spore germination and invasive hyphal growth from the GFP signal expressed by A. fumigatus. As these experiments were done with a fast-germinating CEA10-derived strain, spore germination occurs at high levels by 2 dpi and the rate of spore germination is not significantly different in dexamethasone-exposed larvae compared to control DMSO-exposed larvae (Fig. 4A, 4B). The cumulative percentage of larvae that experience invasive hyphal growth (presence of branched hyphae) is significantly higher with dexamethasone exposure compared to control conditions (Fig. 4B). We also quantified the fungal burden in larvae across the full 5 day experiment by measuring the GFP+ area, confirming that dexamethasone-treated larvae experience significantly more fungal growth compared to control larvae (Fig. 4C). Next, we rated the severity of fungal growth using a scoring system of 0 to 4, from no germination to invasive hyphal growth (Supplemental Fig. 2D). In larvae exposed to dexamethasone, invasive hyphal growth becomes severe quickly, within 2–3 days, and causes mortality, while many control larvae are able to delay this invasive growth (Fig. 4D). These data suggest that dexamethasone treatment decreases the ability of host immune cells to inhibit post-germination invasive hyphal growth of the CEA10 strain of A. fumigatus.
Figure 4. Dexamethasone suppresses immune control of A. fumigatus invasive hyphal growth.
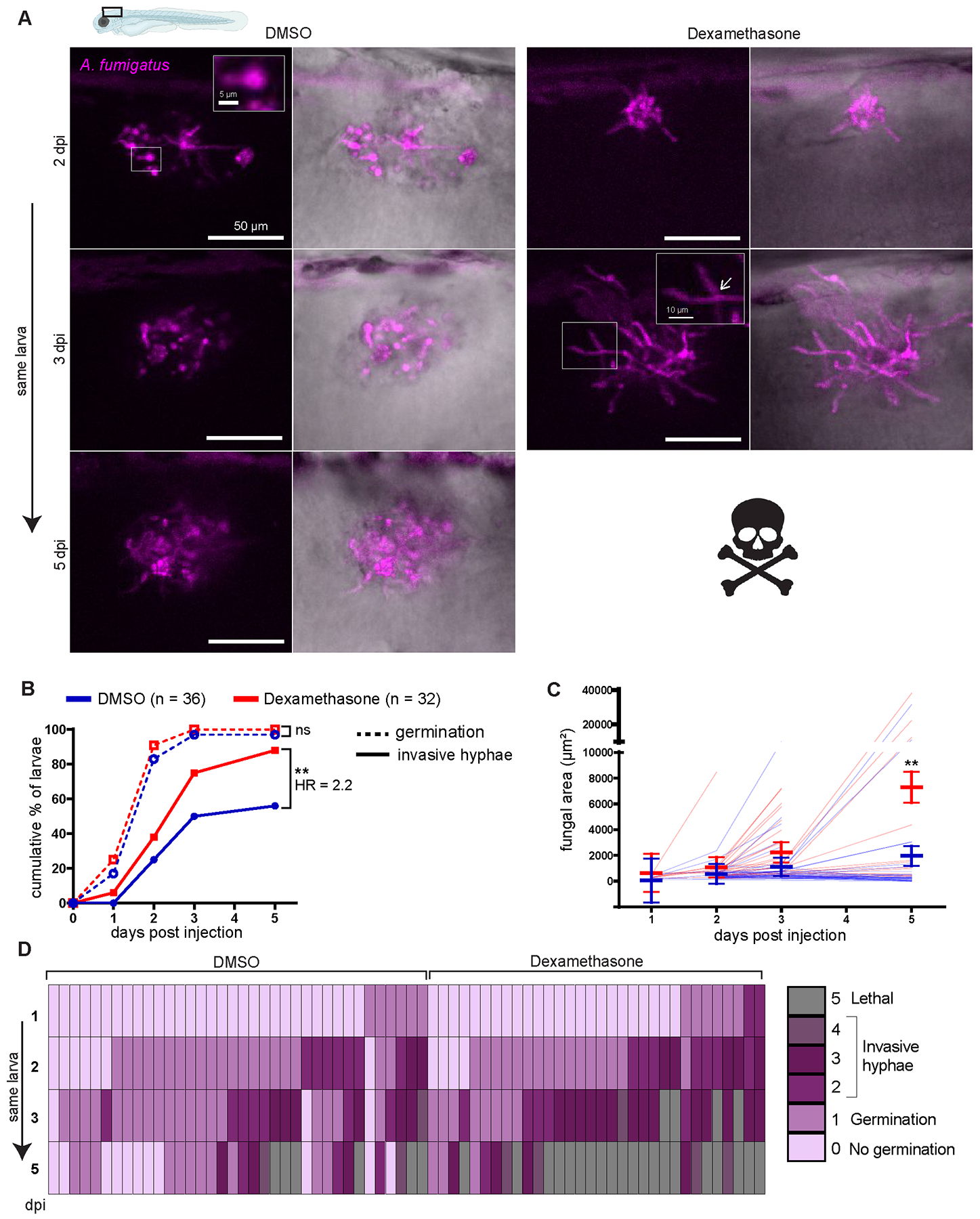
Wild-type larvae were injected with GFP-expressing TFYL49.1 (CEA10) spores at 2 dpf, exposed to 10 μM dexamethasone or DMSO vehicle control and imaged at 1, 2, 3, and 5 dpi. Data are pooled from three independent replicates, at least 10 larvae per condition, per replicate.
(A) Representative images show hyphal growth differences in larvae exposed to dexamethasone or DMSO. Insets show a germinated spore and branched invasive hyphae (open arrow). Scale bar = 50 μm (5 and 10 μm in insets). (B) Cumulative percentage of larvae with germination (dotted line) and invasive hyphae (solid line) through 5 dpi. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). (C) In larvae with fungal germination, fungal area was quantified from maximum intensity projection images. Each line represents an individual larva and bars represent emmeans ± SEM. (D) Severity of fungal growth was scored for all larvae and displayed as a heatmap. Representative images for each score can be found in Supplemental Fig. 2D.
Dexamethasone predominantly suppresses neutrophil function to cause host susceptibility to A. fumigatus CEA10 strain
To confirm that dexamethasone impacts the ability of host innate immune cells to control the invasive growth stages of A. fumigatus, we generated zebrafish larvae without macrophages and neutrophils, the primary innate immune cells active in zebrafish larvae(34). If dexamethasone increases host susceptibility by inhibiting the function of these cells, then in larvae that already lack these cells we expect that dexamethasone treatment would not significantly decrease host survival. However, in larvae with the pu.1 transcription factor knocked down, that do not develop phagocytes, >90% of larvae succumb to A. fumigatus CEA10 infection rapidly regardless of dexamethasone exposure, making it hard to determine any further effects of dexamethasone (Supplemental Fig. 3A), and we therefore decided to test the impact of dexamethasone on macrophage and neutrophil function separately.
While neutrophil function is thought to predominate during control of invasive growth post-germination, macrophages can also attack hyphae and may play a role in control of hyphal growth (26, 61, 62). We employed established models of neutrophil-defective or macrophage-deficient larvae. If dexamethasone predominantly suppresses neutrophil-mediated mechanisms, we expect dexamethasone to cause little additional survival defect in larvae that already do not have functional neutrophils. We infected neutrophil-defective (Tg(mpx:mCherry-2A-rac2D57N)) larvae, in which neutrophils are unable to migrate to the infection site (40), with A. fumigatus CEA10 spores, treated larvae with either dexamethasone or DMSO vehicle control, and monitored survival. Consistent with previous results (26), neutrophil-defective larvae succumb to the infection at a higher rate than control wild-type larvae (Fig. 5A). Dexamethasone further decreases survival of neutrophil-defective larvae (Fig. 5A). While wild-type larvae treated with dexamethasone are 4.3 times more likely succumb to the infection compared to vehicle control larvae, neutrophil-defective larvae are only 2.2 times more likely to succumb due to treatment. Dexamethasone treatment does not affect survival of PBS mock-infected larvae ((Supplemental Fig. 3B). These results suggest that dexamethasone can affect the function of other cell types remaining in these neutrophil-defective larvae but that neutrophils are also a target of dexamethasone.
Figure 5. Dexamethasone primarily suppresses neutrophil-mediated host protection.
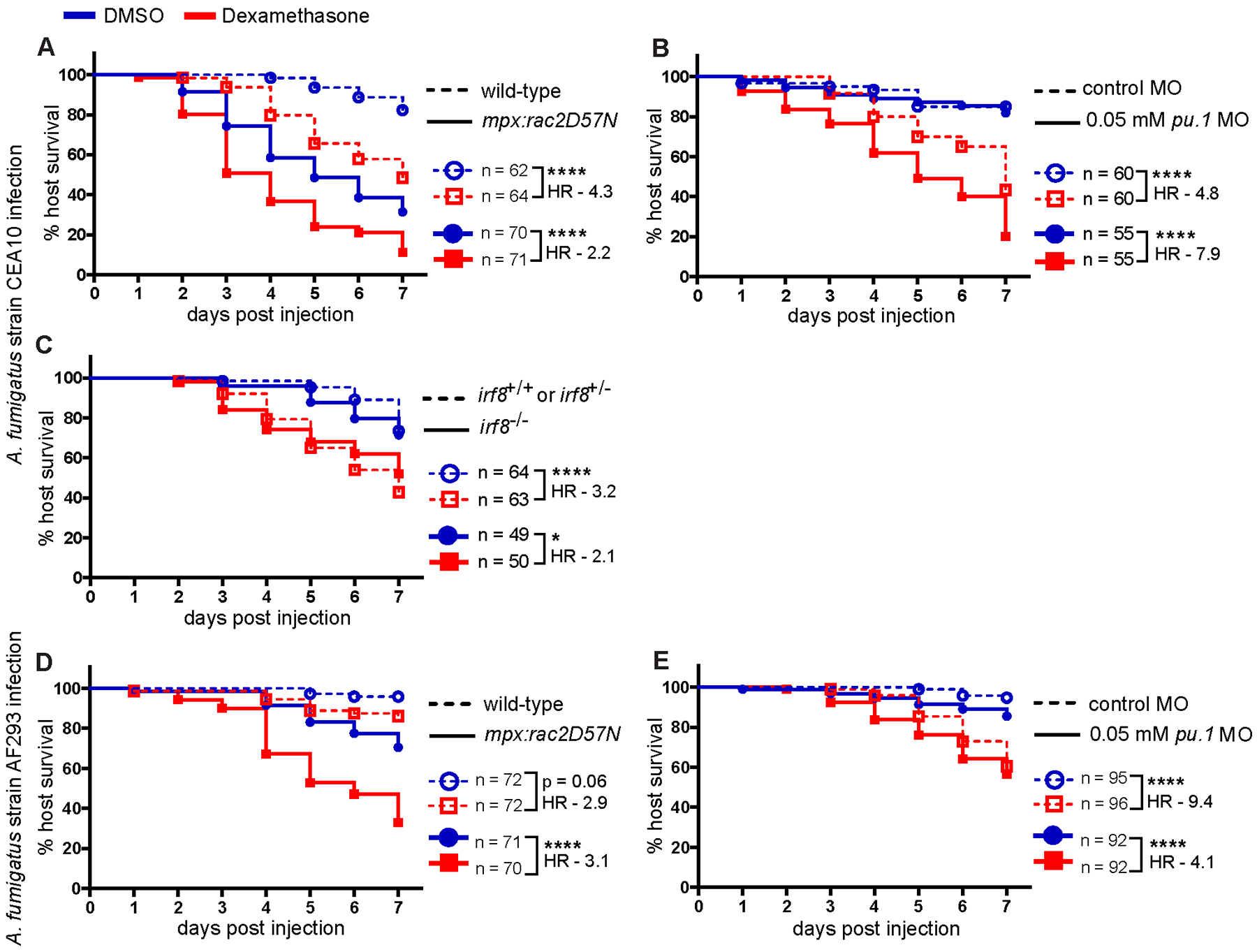
Survival of larvae injected at 2 dpf with CEA10 (A-C) or AF293 (D, E) A. fumigatus spores and exposed to 10 μM dexamethasone or DMSO vehicle control. Data are pooled from at least three independent replicates, at least 12 larvae per condition, per replicate and the total larval N per condition is indicated in each Figure. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). (A-C) Survival of larvae injected with CEA10 A. fumigatus spores. (A) Survival of neutrophil-defective larvae (mpx:rac2D57N) and wild-type larvae. Average injection CFUs: wild-type = 26, mpx:rac2D57N = 29. (B) Survival of macrophage-deficient or control larvae. Development of macrophages was inhibited by 0.05 mM pu.1 morpholino (MO). Control larvae received standard control MO. Average injection CFUs: control MO = 31, low-dose pu.1 MO = 25. (C) Survival of macrophage-deficient irf8−/− or control (irf8+/+ or irf8+/−) larvae. Average injection CFUs: irf8+/+ or irf8+/−= 71, irf8−/− = 50. (D, E) Survival of larvae injected with TBK5.1 (AF293) A. fumigatus spores. (D) Survival of neutrophil-defective larvae (mpx:rac2D57N) and wild-type larvae. Average injection CFUs: wild-type = 33, mpx:rac2D57N = 36. (E) Survival of pu.1 morphant macrophage-deficient or control larvae. Average injection CFUs: control MO = 39, low-dose pu.1 MO = 36.
Next, we performed these experiments in macrophage-deficient larvae. First, we used clodronate liposomes to deplete macrophages. Macrophage-depleted larvae succumb to CEA10 infection, and dexamethasone further increases mortality of these larvae (Supplemental Fig. 3C). However, dexamethasone causes significant mortality in macrophage-depleted PBS-injected mock-infected larvae (Supplemental Fig. 3D), making results from these experiments difficult to interpret. Instead, to prevent the development of macrophages, we used a low concentration of pu.1 morpholino that affects macrophage but not neutrophil development (49). We confirmed that at this concentration the development of macrophages is inhibited for at least 3 days post infection, but neutrophils remain intact and functional (Supplemental Fig. 3E–H). We have previously reported that in larvae without macrophages, CEA10 spores germinate rapidly and are cleared by neutrophils, leading to no significant decrease in host survival (26), and again we find that macrophage deficiency alone does not significantly affect the survival of larvae infected with the CEA10 strain of A. fumigatus (Fig. 5B). Additionally, dexamethasone does not affect the survival of PBS mock-infected low-dose pu.1 morphant larvae (Supplemental Fig. 3I). However, dexamethasone exposure increases the susceptibility of both control and macrophage-deficient A. fumigatus infected larvae (Fig. 5B). Dexamethasone-exposed macrophage-deficient larvae are 7.9 times more likely to succumb to the infection compared to vehicle exposure, while control larvae are 4.8 times more likely to succumb to the infection after dexamethasone treatment (Fig. 5B). Therefore, when macrophages are not present and neutrophils are the major immune cell present, dexamethasone has a greater impact on host survival. These results are in contrast to experiments in the neutrophil-defective line, where macrophages are the major immune cell present and dexamethasone has a lower impact on host survival. Overall, these data demonstrate that dexamethasone-mediated suppression of neutrophil function is a cause of host susceptibility to A. fumigatus CEA10 infection.
To further study the effect of dexamethasone on neutrophil function, we used an irf8 mutant line, which lacks macrophages and has a compensating increase in neutrophil numbers (36). Irf8 drives macrophage development, therefore, when it is not present, myeloid-committed progenitors develop into neutrophils, increasing neutrophil numbers in irf8 mutant larvae (36). irf8−/− larvae are significantly more susceptible to infection with CEA10 when exposed to dexamethasone compared to vehicle control (Fig. 5C). However, the relative increase in susceptibility is similar to that of irf8+/− and irf8+/+ control larvae, suggesting that an increased number of neutrophils can partially compensate for the suppressive effects of dexamethasone on neutrophil function (Fig. 5C).
We wondered if this neutrophil-biased response is due to the fast-germination of the CEA10 strain, which leads to hyphae that are predominantly controlled by neutrophils (26). In contrast, against the slow-germinating AF293 strain, macrophages play a larger role (26). To test the relative effect of dexamethasone on these cell types in the context of AF293 infection, we injected neutrophil-defective and macrophage-deficient larvae with AF293 spores and then treated larvae with dexamethasone. Neutrophil-defective larvae are 3.1 times more likely to succumb to AF293 infection with dexamethasone treatment compared to vehicle treatment, while dexamethasone makes wild-type larvae 2.9 times more likely to succumb (Fig. 5D). This suggests that, in larvae with only macrophages at the infection site, dexamethasone increases host susceptibility to AF293 infection. In contrast, dexamethasone-exposed macrophage-deficient larvae are 4.1 times more likely to succumb to the infection compared to vehicle exposure, while control larvae are 9.4 times more likely to succumb to the infection after dexamethasone treatment (Fig. 5E), indicating that the effect of dexamethasone treatment on susceptibility to AF293 goes down when neutrophils are the major immune cell present. Overall, these results suggest that with a fast-germinating CEA10 strain, in which neutrophil function is required to control hyphal growth, dexamethasone-mediated suppression of neutrophil function is the major cause of host susceptibility. This is consistent with our finding that dexamethasone exposure leads to increased invasive hyphal growth of the CEA10 strain inside of larvae (Fig. 4), as neutrophils are the primary innate immune cell responsible for clearing A. fumigatus hyphae (26, 31). However, after infection with a slow-germinating AF293 strain, in which macrophage function is required to control spore germination, dexamethasone-mediated suppression of macrophage function is a major cause of host susceptibility.
Neutrophils fail to control invasive hyphal growth in dexamethasone-treated larvae.
We wanted to further investigate the effect of dexamethasone on the function of neutrophils. We therefore decided to further characterize the effects of dexamethasone exposure on CEA10-infected irf8−/− larvae which lack macrophages and have an excess of neutrophils. However, in addition to its requirement in macrophage development, Irf8 is also an interferon response factor that can regulate a range of inflammatory pathways including NF-κB activation (63). Therefore, we first checked whether irf8 mutation significantly affects NF-κB activation in our model. Contrary to in vitro findings (64), we find no difference in basal NF-κB activation levels across irf8 genotypes after control PBS injection (Supplemental Fig. 4A). NF-κB activation is slightly lower in A. fumigatus CEA10-injected irf8−/− larvae as compared to irf8+/+ and irf8+/− siblings, however, this could be due to either a lack of macrophages at the infection site or a direct role for Irf8 in NF-κB activation, and these differences are not statistically significant (Supplemental Fig. 4A). Levels of NF-κB activation are similar in all irf8 genotypes after dexamethasone treatment (Supplemental Fig. 4A). Overall, these data suggest that in our model, Irf8 plays only a minor role in NF-κB activation, and therefore irf8−/− larvae can be used as a model to investigate neutrophil-specific mechanisms against A. fumigatus infection.
We infected irf8−/− larvae with labeled neutrophils (Tg(lyz:BFP)) with GFP-expressing CEA10 spores of A. fumigatus, treated larvae with dexamethasone or vehicle control, and performed live imaging at 1, 2, 3, and 5 dpi (Fig. 6). As none of these larvae have macrophages which are the cell type primarily responsible for preventing spore germination (26), we observed similar levels of germination in larvae exposed to both dexamethasone and DMSO, with all larvae harboring germinated spores by 3 dpi (Fig. 6A–C). However, the cumulative percentage of larvae harboring invasive hyphae is significantly higher in dexamethasone-treated larvae compared to vehicle control-treated larvae (Fig. 6B), indicating a difference in control of fungal growth post-germination. There is no significant effect of dexamethasone treatment on the day that germination occurs, with the majority of larvae experiencing germination at 1 dpi regardless of treatment group (Fig. 6D). However, dexamethasone-exposed larvae develop initial invasive hyphae as early as ~2 dpi on average, while it takes until ~3 dpi for DMSO-exposed larvae to develop invasive hyphae (Fig. 6E). Once germination occurs, invasive hyphae appear on average 1 day later in dexamethasone-exposed larvae as compared to an average of 1.5 days later in DMSO-exposed larvae (Fig. 6F). Analysis of the severity of fungal growth or quantification of fungal area in all individual larvae across all days of the experiment demonstrates that dexamethasone-treated irf8 mutant larvae experience uncontrolled hyphal growth (Fig. 6C and Supplemental Fig. 4B). Although germination occurs in all larvae, neutrophils in DMSO-treated larvae are able to delay the development of severe invasive growth of hyphae, while dexamethasone-exposed larvae quickly develop severe invasive hyphae and succumb to the infection (Fig. 6C).
Figure 6. Dexamethasone suppresses neutrophil-mediated control of A. fumigatus invasive hyphal growth.
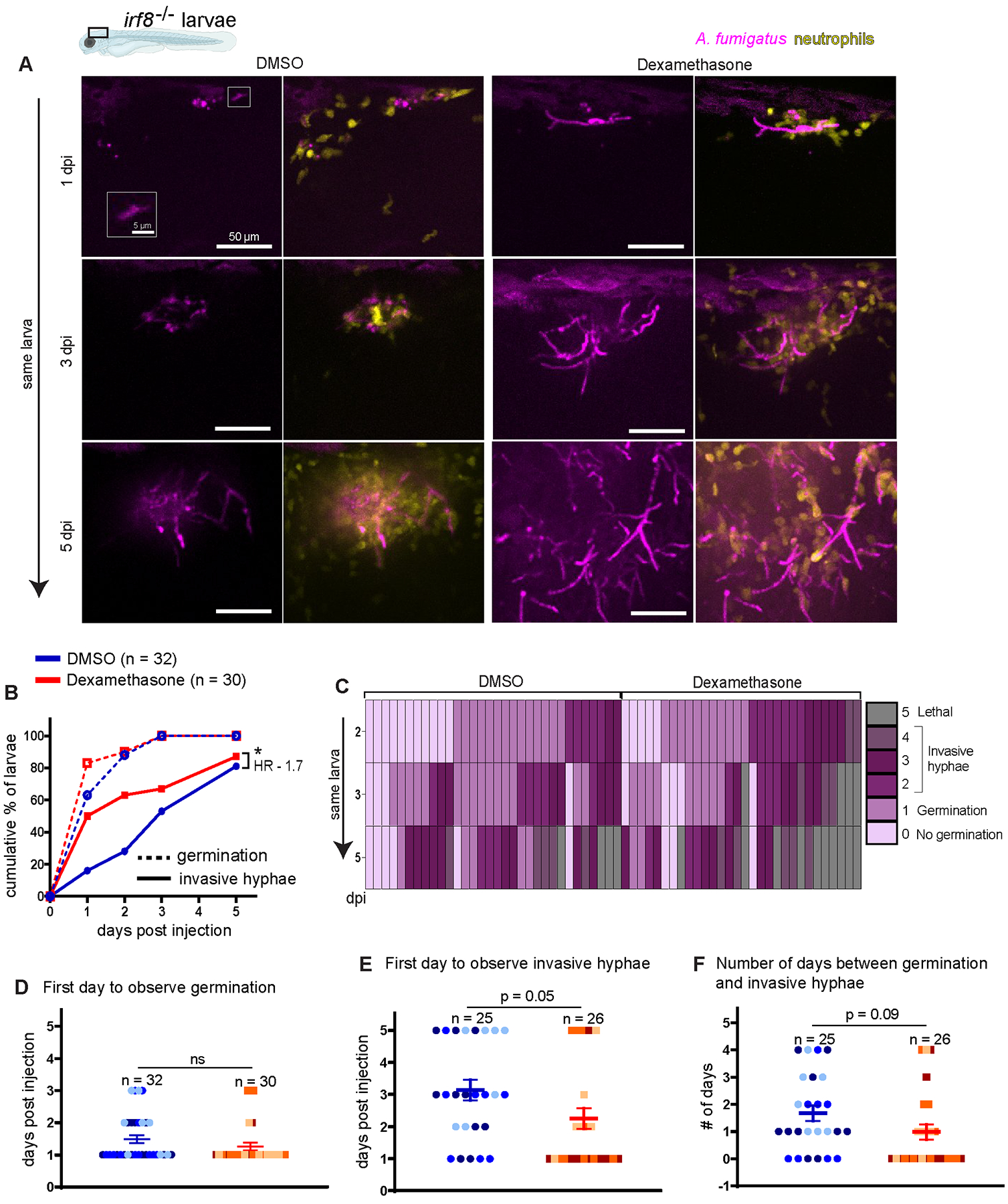
Macrophage-deficient irf8−/− larvae with labeled neutrophils (Tg(lyz:BFP)) were injected with GFP-expressing TFYL49.1 (CEA10) spores at 2 dpf, exposed to 10 μM dexamethasone or DMSO vehicle control and live imaged at 1, 2, 3, and 5 dpi. Data are pooled from three independent replicates, at least 10 larvae per condition, per replicate. (A) Representative images show hyphal growth differences and neutrophil recruitment in larvae exposed to dexamethasone or DMSO. Inset shows a germinated spore. Scale bar = 50 μm (inset 5 μm). (B) Cumulative percentage of larvae with germination (dotted line) and invasive hyphae (solid line) through 5 dpi. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). (C) Severity of fungal growth was scored for all larvae and displayed as a heatmap. Representative images for each score can be found in Supplemental Fig. 2D. (D-F) In larvae in which fungal growth occurred, the day on which germination (D) and invasive hyphae (E) was first observed and the number of days between germination and invasive hyphae (F) are plotted. Bars represent emmeans ± SEM and P values were calculated by ANOVA. Each data point represents an individual larva, color-coded by replicate.
In dexamethasone-treated irf8−/− larvae, we observed many neutrophils at the infection site, clustering around the fungus (Fig. 6A), suggesting that in these larvae dexamethasone inhibits neutrophil function rather than neutrophil recruitment, as we observed in wild-type larvae (Fig. 2). To confirm this, we quantified neutrophil cluster area both across all days of imaging and normalized to the germination status of larvae, finding no statistically significant differences between larvae treated with dexamethasone or vehicle control (Supplemental Fig. 4C, 4D). As expected, there is a positive correlation between fungal area and neutrophil cluster area in both vehicle- and dexamethasone-treated larvae (Supplemental Fig. 4E). However, when we plotted the neutrophil cluster area relative to the fungal area, the slope of this correlation is slightly lower in dexamethasone-treated larvae, suggesting that for a given fungal load these larvae may recruit fewer neutrophils (Supplemental Fig. 4E). Overall, a decrease in neutrophil recruitment may play a minor role in the lack of fungal control in irf8 mutant dexamethasone-treated larvae, but an inhibition of neutrophil function against hyphae by this drug is likely the major factor leading to host susceptibility to CEA10 infection.
Neutrophil-mediated control of invasive hyphal growth requires NF-κB
Our results thus far demonstrate that 1) dexamethasone treatment inhibits NF-κB at the site of A. fumigatus infection and 2) dexamethasone primarily impacts neutrophil function to cause host susceptibility to the A. fumigatus CEA10 strain. We therefore wanted to confirm that NF-κB signaling is required for neutrophil function against A. fumigatus growth, independent of dexamethasone treatment. We used CRISPR/Cas9 to target ikbkg (inhibitor of nuclear factor kappa B kinase regulatory subunit gamma), which encodes IKKγ (NEMO) and is required for canonical NF-κB activation (20). We designed two gRNAs targeting exons 2 and 3, both of which are part of the N-terminal IKKαβ binding domain (Supplemental Fig. 4F). We injected embryos at the 1 cell stage with both gRNAs targeting ikbkg or control gRNAs targeting luciferase, together with Cas9 protein. PCR using primers flanking the target sites on genomic DNA isolated from 2 dpf larvae confirmed successful targeting of DNA, with predicted efficiencies of 85.7% and 40.4% with gRNAs 1 and 2, respectively (Supplemental Fig. 4G–I). To test the survival of both control and macrophage-deficient larvae, we injected gRNAs and Cas9 into embryos from an irf8+/− in-cross and infected the resulting larvae with A. fumigatus CEA10. ikbkg crispant larvae in an irf8+/+ or irf8+/− background succumb to the infection at a significantly higher rate than larvae injected with control gRNAs (Fig. 7A). We observed similar results after targeting ikbkg with two independent gRNAs in wild-type larvae, demonstrating that this decreased survival is due to inhibition of IKKγ function, rather than any off-target effect (Supplemental Fig. 4F, 4J, 4K). In irf8−/− larvae, ikbkg targeting also has a large effect on survival, suggesting that when neutrophils are the primary immune cell type present, NF-κB activation plays a major role in promoting host survival to CEA10 infection (Fig. 7A).
Figure 7. Neutrophils fail to control invasive hyphal growth in irf8−/− IKKγ crispant larvae.
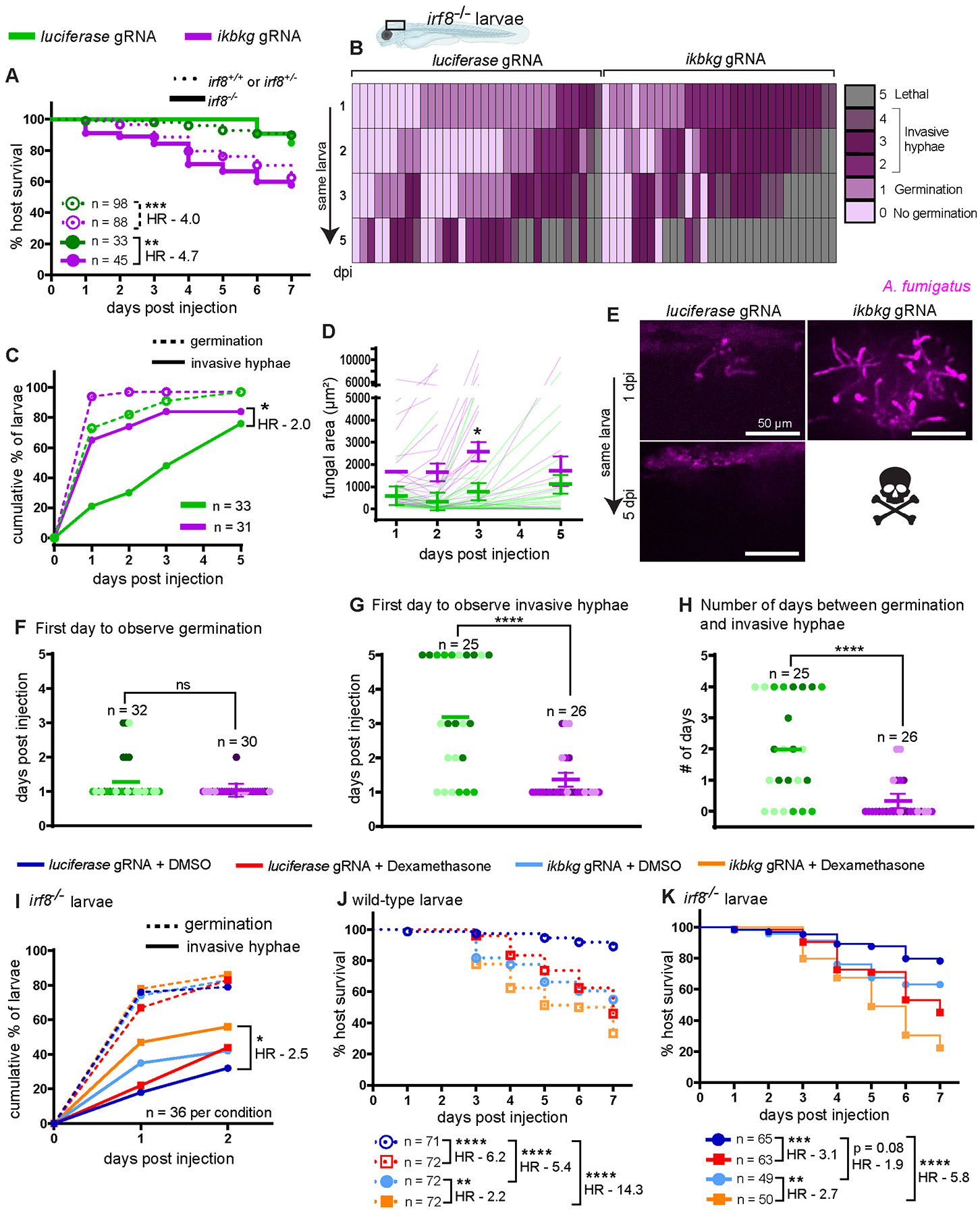
Macrophage-sufficient (irf8+/+ or irf8+/−) and macrophage-deficient (irf8−/−) embryos at 1 cell stage were injected with Cas9 protein and 2 gRNAs targeting the IKKγ gene ikbkg or control gRNAs targeting luciferase. (A) Survival of larvae after injection with CEA10 spores at 2 dpf. Data are pooled from three independent replicates and the total larval N per condition is indicated. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). Average injection CFUs: control + luciferase gRNA = 32, control + ikbkg gRNA = 30, irf8−/− + luciferase gRNA = 31, irf8−/− + ikbkg gRNA = 29. (B–H) irf8−/− embryos with labeled neutrophils (Tg(lyz:BFP)), injected with ikbkg or control gRNAs, were injected at 2 dpf with GFP-expressing TFYL49.1 (CEA10) spores and live imaged at 1, 2, 3, and 5 dpi. Data are pooled from three independent replicates, at least 10 larvae per condition, per replicate. (B) Representative images show hyphal growth differences in larvae injected with ikbkg or control gRNAs. Scale bar = 50 μm. (C) Cumulative percentage of larvae with germination (dotted line) and invasive hyphae (solid line) through 5 dpi. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). (D) Fungal area was quantified from maximum intensity projection images. Bars represent emmeans ± SEM and P values were calculated by ANOVA. Each line represents an individual larva. (E) Severity of fungal growth was scored for all larvae and displayed as a heatmap. Representative images for each score can be found in Supplemental Fig. 2D. (F–H) In larvae in which fungal growth occurred, the day on which germination (F) and invasive hyphae (G) was first observed and the number of days between germination and invasive hyphae (H) are plotted. Bars represent emmeans ± SEM and P values calculated by ANOVA. Each data point represents an individual larva, color-coded by replicate. (I) irf8−/− larvae injected with ikbkg or control gRNAs were injected at 2 dpf with GFP-expressing TFYL49.1 (CEA10) spores, treated with 10 μM dexamethasone or DMSO, and live imaged at 1 and 2 dpi. The cumulative percentage of larvae with germination (dotted line) and invasive hyphae (solid line) through 2 dpi is shown. Data are pooled from three independent replicates, 12 larvae per condition, per replicate. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). (J, K) Survival of wild-type (J) or irf8−/− (K) larvae injected with ikbkg or control gRNAs, injected with TFYL49.1 (CEA10) spores and treated with 10 μM dexamethasone or DMSO. Data are pooled from three independent replicates and the total larval N per condition is indicated. Cox proportional hazard regression analysis was used to calculate P values and hazard ratios (HR). Average injection CFUs: wild-type + luciferase gRNA = 28, wild-type + ikbkg gRNA = 20, irf8−/− + luciferase gRNA = 27, irf8−/− + ikbkg gRNA = 21.
To determine the requirement for NF-κB signaling in neutrophil-mediated control of invasive hyphae, we focused again on irf8−/− larvae and performed daily live imaging of ikbkg crispant larvae or control luciferase-targeted larvae after infection with a GFP-expressing CEA10 strain of A. fumigatus (Fig. 7B–H). As seen previously, almost all irf8−/− larvae experience germination, regardless of gRNA injection (Fig. 7B). However, the percentage of larvae with invasive hyphae is significantly higher in ikbkg crispant larvae compared to luciferase-targeted controls (Fig. 7C). Additionally, ikbkg crispant larvae have higher fungal burdens as measured by fungal area, with statistically significantly higher burden at 3 dpi compared to control larvae (Fig. 7D). Analysis of the severity of fungal growth indicates that control larvae are able to delay the development of invasive hyphal growth while ikbkg crispant larvae are not (Fig. 7B, 7E). While germination appears at 1 dpi on average in both ikbkg crispant and control larvae (Fig. 7F), invasive hyphae also appear as early as 1 dpi in ikbkg crispant larvae (Fig. 7G). In control larvae, A. fumigatus takes ~2 days to develop invasive hyphae after germination while this occurs on the same day in ikbkg crispant larvae (Fig. 7H). Together, these data demonstrate that in irf8 mutant larvae in which neutrophils are solely responsible for fungal control, genetic NF-κB pathway inhibition through ikbkg (IKKγ) targeting with CRISPR/Cas9 phenocopies dexamethasone treatment. The effect of ikbkg targeting on neutrophil recruitment to the infection site is also similar to effect of dexamethasone (Supplemental Fig. 4L–N). These data are consistent with the conclusion that the major signaling pathway inhibited by dexamethasone to inhibit neutrophil-mediated control of invasive hyphal growth is NF-κB.
To test if dexamethasone treatment has other NF-κB independent effects, we exposed infected irf8−/−, ikbkg crispant or control luciferase-targeted larvae to dexamethasone or vehicle control. We monitored GFP-expressing CEA10 fungal germination and hyphal growth by live imaging at 1 and 2 dpi. As expected, A. fumigatus CEA10 germinates readily in larvae in all four conditions (Fig. 7I). However, we did observe minor differences in the percentage of larvae harboring invasive hyphal growth. Control luciferase gRNA + DMSO larvae have the lowest rate of hyphal growth, while either dexamethasone treatment or ikbkg targeting alone increases this rate (Fig. 7I). The combination of both ikbkg gRNA and dexamethasone exposure increases the percentage of larvae with invasive hyphae further (Fig. 7I). We also observed similar results monitoring the survival of both wild-type and irf8−/− ikbkg crispants after dexamethasone or DMSO exposure (Fig. 7J, 7K). Both wild-type and irf8−/− larvae succumb to CEA10 infection at a much higher rate with the combination of both ikbkg gRNA and dexamethasone exposure than with each individual condition (Fig. 7J, 7K). These data suggest that dexamethasone inhibits other pathways besides NF-κB that have minor roles in promoting neutrophil control of invasive hyphal growth and host survival. Overall, however, our data demonstrate that the primary effect of dexamethasone in causing host susceptibility to A. fumigatus CEA10 infection is through inhibition of NF-κB-activated neutrophil functions against invasive fungal growth.
Discussion
Prolonged glucocorticoid therapy is one of the major risk factors for invasive aspergillosis (7, 65). The anti-inflammatory and immunosuppressive effects of glucocorticoids are largely caused by inhibition of NF-κB transcription factor activation and signaling (1). A. fumigatus infection activates NF-κB in vitro and in vivo (26, 66), but it has been unclear if NF-κB suppression is the main mechanism of glucocorticoid-mediated susceptibility to invasive aspergillosis in these patients. Here, we used an A. fumigatus-zebrafish larvae infection system to demonstrate that glucocorticoids induce susceptibility in infected larvae through glucocorticoid receptor-mediated suppression of NF-κB. To elucidate this pathway, we used CRISPR/Cas9 to target the glucocorticoid receptor and the NF-κB pathway activator IKKγ. Glucocorticoid receptor and IKKγ are essential genes during development and cannot be easily targeted for infection studies in mice as mice lacking these genes are not viable (67, 68). By using F0 mosaic crispant zebrafish larvae, we were able to inhibit the function of these genes in viable, morphologically normal, developing organisms. We find similar disease phenotypes in larvae exposed to dexamethasone and in IKKγ crispant larvae, demonstrating that inhibition of NF-κB is the major mechanism responsible for glucocorticoid-mediated susceptibility to A. fumigatus infection. However, IKKγ crispant larvae have a more severe disease phenotype when combined with dexamethasone, compared to either IKKγ mutation or dexamethasone alone, suggesting that dexamethasone also suppresses other pathways that contribute to control of fungal growth. One possible pathway that remains to be tested is activator protein-1 (AP-1) activation, which can be suppressed by glucocorticoids (69), and can be activated by A. fumigatus hyphae stimulation of mouse dendritic cells and alveolar macrophages (70, 71).
The key cellular immune mechanisms against A. fumigatus that are inhibited by glucocorticoid therapy have also been unclear. Glucocorticoids can affect all immune cell types to alter an inflammatory response (6). Glucocorticoids potently suppress T-cell mediated responses, which are an integral component of anti-fungal immune mechanisms (6, 72). However, larval zebrafish at the stage that was used for our study have not yet developed an adaptive system, and we have focused on the effects of dexamethasone on macrophages and neutrophils, cells that play key roles in host defense against different stages of fungal growth (26, 32, 33, 73). Our results demonstrate that dexamethasone can still worsen A. fumigatus pathogenesis even before the involvement of the adaptive system (34). To address whether dexamethasone primarily inhibits the function of macrophages or neutrophils or both, we used zebrafish larvae deficient for specific phagocyte populations. We find that dexamethasone does increase mortality in neutrophil-defective larvae, in which macrophages are the primary immune cells present, indicating that glucocorticoids can suppress macrophage-specific mechanisms. In monocytes and macrophages, glucocorticoids can suppress monocyte-to-macrophage maturation, chemotaxis to the infection site, phagocytosis, phagolysosomal fusion, oxidative killing, and pro-inflammatory cytokine secretion (6, 74). In particular, in response to Aspergillus infections, glucocorticoids curb reactive oxygen species (ROS) production and autophagy-related protein recruitment to phagosomes downstream of ROS in vitro (32, 75). Glucocorticoids can also alter macrophage M1 to M2 polarization, which can inhibit the ability of macrophages to prevent spore germination (76, 77). In our study, we do find that dexamethasone treatment moderately inhibits macrophage recruitment to the infection site, but only at specific stages of infection, and even then, many macrophages are still present. We find no effect of dexamethasone on the ability of macrophages to kill or prevent germination of CEA10 spores in vivo.
In macrophage-deficient larvae, in which neutrophils are the primary immune cell present, dexamethasone increases susceptibility to the CEA10 A. fumigatus strain even more than in wild-type larvae, demonstrating that the inhibition of neutrophil function is a major target of dexamethasone-mediated susceptibility to fast-germinating A. fumigatus spores. In line with this conclusion, we report that dexamethasone curbs immune control of invasive hyphal growth, which is predominantly mediated by neutrophils. However, dexamethasone treatment does not prevent neutrophil migration to the infection site. We find large numbers of neutrophils in response to invasive hyphae in both wild-type and macrophage-deficient larvae in the presence of dexamethasone. Similarly, in rabbits treated with glucocorticoids, pulmonary lesions of invasive aspergillosis comprise neutrophil and monocyte infiltration and tissue necrosis (78). Therefore, we conclude that dexamethasone inhibits the function of these cells against fungal infection. Neutrophils are efficient killers of hyphae and use multiple extracellular killing mechanisms such as degranulation, neutrophil extracellular traps (NETs), and the production of ROS (79). Which of these mechanisms are most important against A. fumigatus and which are inhibited by dexamethasone treatment remains unclear. Neutrophils can undergo NETosis in response to A. fumigatus hyphae to control further growth (80–82), but the role of neutrophil-mediated ROS in A. fumigatus hyphal control is inconclusive (83, 84).
While our results demonstrate that dexamethasone inhibits NF-κB activation and this NF-κB activation promotes neutrophil functions against A. fumigatus hyphae, we cannot yet conclude whether this NF-κB activation is cell-intrinsic to neutrophils or non-cell-autonomous. It should also be noted that the CRISPR-mediated gene editing is mosaic in F0 crispant larvae and that not all cells or tissues will have the same editing efficiency. We hypothesize that glucocorticoids inhibit activation of NF-κB in neutrophils, but cannot rule out the possibility that glucocorticoids inhibit activation of epithelial cells or macrophages, decreasing signals from these cells that activate neutrophils. Germination of spores exposes immunogenic ligands or pathogen-associated molecular patterns (PAMPs) such as β-glucans, which are otherwise masked by melanin and rodlet proteins in A. fumigatus spores, and this PAMP exposure induces a robust pro-inflammatory response that is likely mediated by all of these cells (24, 85, 86). In our experiments, spore germination is the primary factor driving phagocyte recruitment, activation of an NF-κB reporter line, and induction of inflammatory cytokine expression, regardless of dexamethasone treatment. Our results demonstrate that excessive hyphal growth can override the suppression of NF-κB by dexamethasone.
Innate immune control of A. fumigatus varies with strain differences (26, 87). In our experiments, we primarily used spores derived from the CEA10 strain that was isolated from a patient (41). CEA10-derived strains have relatively faster germination both in vitro and in vivo than spores of the other commonly studied AF293 strain (26, 87, 88). As CEA10 is faster-germinating, resistance to infection with this strain in wild-type animals is more dependent on neutrophils and CEA10 is susceptible to neutrophil-mediated killing in vitro and in vivo (26, 33, 84). In line with this, macrophage-deficient and wild-type hosts are equally resistant to CEA10-derived infections. Therefore, it is not surprising that the primary activity of dexamethasone in causing susceptibility to CEA10 infection is to inhibit neutrophil functions against invasive hyphal growth. AF293 spores can reside within macrophage clusters, which inhibit their germination, and therefore evade neutrophil-mediated killing and persist in the host for a long time (26). Consequently, in our experiments with AF293 infection, dexamethasone treatment has a larger effect when macrophages are the primary immune cells present. Therefore, the relative importance of the inhibition of neutrophil and macrophage function by dexamethasone likely depends on the specific fungal strain and how readily it germinates.
Glucocorticoid therapy is widely used in patients to decrease morbidity and mortality due to a variety of causes including transplant rejection and autoimmune disorders. However, a by-product of this therapy is increased susceptibility to infectious organisms, including fungi like Aspergillus spp. In order to better treat these opportunistic infections, it is important to fully understand the molecular and cellular mechanisms that are inhibited by glucocorticoid therapy that are the proximate cause of susceptibility to infection. Here, using a larval zebrafish host, we find that a major innate immune cell type inhibited by this therapy is neutrophils and that this inhibition allows for uncontrolled invasive hyphal growth in infected animals. However, the specific functions of neutrophils that are inhibited remain to be uncovered. In this model, NF-κB is the major molecular pathway inhibited by glucocorticoid treatment. However, the role of NF-κB in human infection is unclear, as the risk of invasive aspergillosis did not depend on single nucleotide polymorphisms (SNPs) of NF-κB and NF-κB pathway components in a cohort of hematopoietic stem-cell transplant patients (89), and the effect of glucocorticoids on other pathways should also be an area of future study.
Supplementary Material
Key Points:
-GCs inhibit the function, but not recruitment, of innate cells, primarily neutrophils
-GCs inhibit neutrophil activity against hyphae without affecting spore killing
-Inhibition of NF-κB through the GC receptor largely accounts for the effects of GCs
Acknowledgements
We thank Celia Shiau for providing the irf8 mutant zebrafish line. We thank John Rawls for sharing the NF-κB reporter line. We thank Anna Huttenlocher for sharing all other transgenic lines. We thank Nancy Keller for sharing the A. fumigatus TDGC1.2 strain. We thank Sourabh Dhingra for helping to design the A. fumigatus in vitro assay. We thank all the members of the Rosowski Lab for helpful discussions and for assistance with zebrafish care.
This research was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number K22AI134677 to E.E.R. and by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM147464 to E.E.R. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. M.H.A was supported by the Clemson University Eukaryotic Pathogens Innovation Center Seifert Scholars Summer Research Program. The funders had no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, and in the preparation, review, or approval of the manuscript.
References
- 1.Cain DW and Cidlowski JA. 2017. Immune regulation by glucocorticoids. Nature Reviews Immunology. 17: 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhen T and Cidlowski JA. 2005. Antiinflammatory action of glucocorticoids -- New mechanisms for old drugs. The New England Journal of Medicine. 353: 1711–1723. [DOI] [PubMed] [Google Scholar]
- 3.Oakley RH and Cidlowski JA. 2013. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. Journal of Allergy and Clinical Immunology. 132: 1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yasir M, Amandeep G, Pankaj B and Sidharth S. 2022. Corticosteroid adverse effects. In StatPearls. Treasure Island, FL, USA, [Google Scholar]
- 5.Latgé J 1999. Aspergillus fumigatus and Aspergillosis. Clinical Microbiology Reviews. 12: 310–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lionakis MS and Kontoyiannis DP. 2003. Glucocorticoids and invasive fungal infections. The Lancet. 362: 1828–1838. [DOI] [PubMed] [Google Scholar]
- 7.Baddley JW 2011. Clinical risk factors for invasive aspergillosis. Medical Mycology. 49: S7–S12. [DOI] [PubMed] [Google Scholar]
- 8.Lin S, Schranz J, and Teutsch SM. 2001. Aspergillosis case-fatality rate: Systematic review of the literature. Clinical Infectious Diseases. 32: 358–366. [DOI] [PubMed] [Google Scholar]
- 9.Nissen RM and Yamamoto KR. 2000. The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes & Development. 14: 2314–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray A and Prefontaine KE. 1994. Physical association and functional antagonism between the p65 subunit of transcription factor NF-cB and the glucocorticoid receptor. Proceedings of the National Academy of Sciences. 91: 752–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caldenhoven E 1995. Negative cross-talk between RelA and the glucocorticoid receptor: a possible mechanism for the antiinflammatory action of glucocorticoids. Molecular Endocrinology. 9: 401–412. [DOI] [PubMed] [Google Scholar]
- 12.Liu TL, Zhang D. Joo, and Sun S. 2017. NF-κB signaling in inflammation. Signal Transduction Targeted Therapy. 2:17203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baeuerle PA and Baltimore D. 1988. IkappaB: a specific inhibitor of the NF-kappaB transcription factor. Science. 242: 540–546. [DOI] [PubMed] [Google Scholar]
- 14.Henkel T, Zabel U, van Zee K, Müller JM, Fanning E, and Baeuerle PA. 1992. Intramolecular masking of the nuclear location signal and dimerization domain in the precursor for the p50 NF-κB subunit. Cell. 68: 1121–1133. [DOI] [PubMed] [Google Scholar]
- 15.Beg AA, Ruben SM, Scheinman RI, Haskill S, Rosen CA, and Baldwin AS. 1992. I kappa B interacts with the nuclear localization sequences of the subunits of NF-kappa B: a mechanism for cytoplasmic retention. Genes & Development. 6: 1899–1913. [DOI] [PubMed] [Google Scholar]
- 16.Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, and Dargemont C. 1997. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. Journal of Cell Science. 110: 369–378. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh S, May MJ, and Kopp EB. 1998. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annual Review of Immunology. 16: 225–260. [DOI] [PubMed] [Google Scholar]
- 18.Vallabhapurapu S and Karin M. 2009. Regulation and Function of NF-κB transcription factors in the immune system. Annual Review of Immunology. 27: 693–733. [DOI] [PubMed] [Google Scholar]
- 19.Karin M and Ben-Neriah Y. 2000. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annual Review of Immunology. 18: 621–663. [DOI] [PubMed] [Google Scholar]
- 20.Israel A 2010. The IKK complex, a central regulator of NF-B activation. Cold Spring Harbor Perspectives in Biology. 2: a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auphan N, DiDonato JA, Rosette C, Helmberg A, and Karin M. 1995. Immunosuppression by glucocorticoids: inhibition of NF-kappaB activity through induction of IkappaB synthesis. Science. 270: 286–290. [DOI] [PubMed] [Google Scholar]
- 22.Scheinman RI, Cogswell PC, Lofquist AK, and Baldwin AS. 1995. Role of transcriptional activation of IκBα in mediation of immunosuppression by glucocorticoids. Science. 270: 283–286. [DOI] [PubMed] [Google Scholar]
- 23.Heck S, Bender K, Kullmann M, Göttlicher M, Herrlich P, and Cato ACB. 1997. IκBα-independent downregulation of NF-κB activity by glucocorticoid receptor. The EMBO Journal. 16: 4698–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inoue K, Koike E, Yanagisawa R, Adachi Y, Ishibashi K, Ohno N, and Takano H. 2009. Pulmonary exposure to soluble cell wall β(l, 3)-glucan of Aspergillus induces proinflammatory response in mice. International Journal of Immunopathology and Pharmacology. 22: 287–297. [DOI] [PubMed] [Google Scholar]
- 25.Sun H, Xu X, Tian X, Shao H, Wu X, Wang Q, Su X, and Shi Y. 2014. Activation of NF-κB and respiratory burst following Aspergillus fumigatus stimulation of macrophages. Immunobiology. 219: 25–36. [DOI] [PubMed] [Google Scholar]
- 26.Rosowski EE, Raffa N, Knox BP, Golenberg N, Keller NP, and Huttenlocher A. 2018. Macrophages inhibit Aspergillus fumigatus germination and neutrophil-mediated fungal killing. PLoS Pathogens. 14:e1007229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dagenais TRT and Keller NP. 2009. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clinical Microbiology Reviews. 22: 447–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Knox BP, Deng Q, Rood M, Eickhoff JC, Keller NP, and Huttenlocher A. 2014. Distinct innate immune phagocyte responses to Aspergillus fumigatus conidia and hyphae in zebrafish larvae. Eukaryotic Cell. 13: 1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosowski EE 2020. Determining macrophage versus neutrophil contributions to innate immunity using larval zebrafish. Disease Models and Mechanisms 13: dmm041889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knox BP, Huttenlocher A, and Keller NP. 2017. Real-time visualization of immune cell clearance of Aspergillus fumigatus spores and hyphae. Fungal Genetics and Biology. 105: 52–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gazendam RP, van Hamme JL, Tool ATJ, Hoogenboezem M, van den Berg JM, Prins JM, Vitkov L, van de Veerdonk FL, van den Berg TK, Roos D, and Kuijpers TW. 2016. Human neutrophils use different mechanisms to kill Aspergillus fumigatus conidia and hyphae: Evidence from phagocyte defects. The Journal of Immunology. 196: 1272–1283. [DOI] [PubMed] [Google Scholar]
- 32.Philippe B, Ibrahim-Granet O, Prevost MC, Gougerpot-Pocidal MA, Sanchez Perez M, Van Der Meeren A, and Latge JP. 2003. Killing of Aspergillus fumigatus by alveolar macrophages is mediated by reactive oxidant intermediates. Infection and Immunity. 71: 3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaffner A, Douglas H, and Braude A. 1982. Selective protection against conidia by mononuclear and against mycelia by polymorphonuclear phagocytes in resistance to Aspergillus. Observations on these two lines of defense in vivo and in vitro with human and mouse phagocytes. The Journal of Clinical Investigation. 69: 617–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Traver D, Herbomel P, Patton EE, Murphey RD, Yoder JA, Litman GW, Catic A, Amemiya CT, Zon LI, and Trede NS. 2003. The zebrafish as a model organism to study development of the immune system. Advances in Immunology. 81: 253–330. [PubMed] [Google Scholar]
- 35.Gomes MC and Mostowy S. 2020. The case for modeling human infection in zebrafish. Trends in Microbiology. 28: 10–18. [DOI] [PubMed] [Google Scholar]
- 36.Shiau CE, Kaufman Z, Meireles AM, and Talbot WS. 2015. Differential requirement for irf8 in formation of embryonic and adult macrophages in zebrafish. PLoS ONE. 10: e0117513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanther M, Sun X, Mühlbauer M, Mackey LC, Flynn EJ, Bagnat M, Jobin C, and Rawls JF. 2011. Microbial colonization induces dynamic temporal and spatial patterns of NF-κB activation in the zebrafish digestive tract. Gastroenterology. 141: 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miskolci V, Squirrell J, Rindy J, Vincent W, Sauer JD, Gibson A, Eliceiri KW, and Huttenlocher A. 2019. Distinct inflammatory and wound healing responses to complex caudal fin injuries of larval zebrafish. eLife 8:e45976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vincent WJB, Freisinger CM, Lam P, Huttenlocher A, and Sauer J. 2015. Macrophages mediate flagellin induced inflammasome activation and host defense in zebrafish. Cellular Microbiology. 18: 591–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng Q, Yoo S, Cavnar P, Green J, and Huttenlocher A. 2011. Dual roles for Rac2 in neutrophil motility and active retention in zebrafish hematopoietic tissue. Developmental Cell. 21: 735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monod M, Paris S, Sarfati J, Jaton-Ogay K, Ave P, and Latgé J. 1993. Virulence of alkaline protease-deficient mutants of Aspergillus fumigatus. FEMS Microbiol. Lett 106: 39–46. [DOI] [PubMed] [Google Scholar]
- 42.Lim FY, Ames B, Walsh CT, and Keller NP. 2014. Co‐ordination between BrlA regulation and secretion of the oxidoreductase FmqD directs selective accumulation of fumiquinazoline C to conidial tissues in Aspergillus fumigatus. Cellular Microbiology. 16: 1267–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoen TJ, Calise DG, Bok JW, Giese MA, Nwagwu CD, Zarnowski R, Andes D, Huttenlocher A, and Keller NP. 2023. Aspergillus fumigatus transcription factor ZfpA regulates hyphal development and alters susceptibility to antifungals and neutrophil killing during infection. PLoS Pathogens. 19: e1011152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thrikawala S, Niu M, Keller NP, and Rosowski EE. 2022. Cyclooxygenase production of PGE2 promotes phagocyte control of A. fumigatus hyphal growth in larval zebrafish. PLoS Pathogens. 18: e1010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jhingran A, Mar KB, Kumasaka DK, Knoblaugh SE, Ngo LY, Segal BH, Iwakura Y, Lowell CA, Hamerman JA, Lin X, and Hohl TM. 2012. Tracing conidial fate and measuring host cell antifungal activity using a reporter of microbial viability in the lung. Cell Reports. 2: 1762–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thrikawala S and Rosowski EE. 2020. Infection of zebrafish larvae with Aspergillus spores for analysis of host-pathogen interactions. Journal of Visualized Experiments. 16: 10.3791/61165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhodes J, Hagen A, Hsu K, Deng M, Liu TX, Look AT, and Kanki JP. 2005. Interplay of Pu.1 and Gata1 determines myelo-erythroid progenitor cell fate in zebrafish. Developmental Cell. 8: 97–108. [DOI] [PubMed] [Google Scholar]
- 48.Su F, Juarez MA, Cooke CL, LaPointe L, Shavit JA, Yamaoka JS, and Lyons SE. 2007. Differential regulation of primitive myelopoiesis in the zebrafish by Spi-1/Pu.1 and C/ebp1. Zebrafish. 4: 187–199. [DOI] [PubMed] [Google Scholar]
- 49.Mesureur J, Feliciano JR, Wagner N, Gomes MC, Zhang L, Blanco-Gonzalez M, Vaart M, O’callaghan D, Meijer AH, and Vergunst AC. 2017. Macrophages, but not neutrophils, are critical for proliferation of Burkholderia cenocepacia and ensuing host-damaging inflammation. PLoS Pathogens. 13: e1006437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosowski EE, Deng Q, Keller NP, and Huttenlocher A. 2016. Rac2 functions in both neutrophils and macrophages to mediate motility and host defense in larval zebrafish. Journal of Immunology. 197: 4780–4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, and Valen E. 2019. CHOPCHOP V3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Research. 47: W171–W174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Labun K, Montague TG, Gagnon JA, Thyme SB, and Valen E. 2016. CHOPCHOP V2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Research. 44: W272–W276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Montague TG, Cruz JM, Gagnon JA, Church GM, and Valen E. 2014. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Research. 42: W401–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brinkman EK, Chen T, Amendola M, and van Steensel B. 2014. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research. 42: e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oliveira E, Casado M, Raldúa D, Soares A, Barata C, and Piña B. 2013. Retinoic acid receptors’ expression and function during zebrafish early development. The Journal of Steroid Biochemistry and Molecular Biology. 138: 143–151. [DOI] [PubMed] [Google Scholar]
- 56.Galindo-Villegas J, García-Moreno D, de Oliveira S, Meseguer J, and Mulero V. 2012. Regulation of immunity and disease resistance by commensal microbes and chromatin modifications during zebrafish development. Proceedings of the National Academy of Sciences. 109: E2605–E2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huemer K, Squirrell JM, Swader R, Pelkey K, LeBert DC, Huttenlocher A, and Eliceiri KW. 2017. Long-term live imaging device for improved experimental manipulation of zebrafish larvae. Journal of Visualized Experiments. 128:56340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huemer K, Squirrell JM, Swader R, LeBert DC, Huttenlocher A, and Eliceiri KW. 2017. zWEDGI: Wounding and entrapment device for imaging live zebrafish larvae. Zebrafish. 14: 42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J, White DJ, Hartenstein V, Eliceiri K, Tomancak P, and Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ibrahim-Granet O, Philippe B, Boleti H, Boisvieux-Ulrich E, Grenet D, Stern M, and Latge JP. 2003. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infection and Immunity. 71: 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rosowski EE, He J, Huisken J, Keller NP, and Huttenlocher A. 2020. Efficacy of voriconazole against Aspergillus fumigatus infection depends on host immune function. Antimicrobial Agents Chemotherapy. 64:e00917–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanner CD and Rosowski EE. 2024. Macrophages inhibit extracellular hyphal growth of A. fumigatus through Rac2 GTPase signaling. Infection and Immunity. 92: e0038023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Platanitis E and Decker T. 2018. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Frontiers in Immunology. 9: 2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan X, Zhao X, Huo R, and Xu T. 2020. IRF3 and IRF8 regulate NF-κB signaling by targeting MyD88 in teleost fish. Frontiers in Immunology. 11: 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kousha M, Tadi R, and Soubani AO. 2011. Pulmonary aspergillosis: a clinical review. European Respiratory Review. 20: 156–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Okaa UJ, Bertuzzi M, Fortune-Grant R, Thomson DD, Moyes DL, Naglik JR, and Bignell E. 2023. Aspergillus fumigatus drives tissue damage via iterative assaults upon mucosal integrity and immune homeostasis. Infection and Immunity. 91: e0033322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, and Schütz G. 1995. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes and Development. 9: 1608–1621. [DOI] [PubMed] [Google Scholar]
- 68.Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ, and Mak TW. 2000. Severe liver degeneration and lack of NF-kappaB activation in NEMO/IKKgamma-deficient mice. Genes and Development. 14: 854–862. [PMC free article] [PubMed] [Google Scholar]
- 69.Yang-Yen H, Chambard J, Sun Y, Smeal T, Schmidt TJ, Drouin J, and Karin M. 1990. Transcriptional interference between c-Jun and the glucocorticoid receptor: Mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 62: 1205–1215. [DOI] [PubMed] [Google Scholar]
- 70.Toyotome T, Adachi Y, Watanabe A, Ochiai E, Ohno N, and Kamei K. 2008. Activator protein 1 is triggered by Aspergillus fumigatus β-glucans surface-exposed during specific growth stages. Microbial Pathogenesis. 44: 141–150. [DOI] [PubMed] [Google Scholar]
- 71.Nicholson WJ, Slight J, and Donaldson K. 1996. Inhibition of the transcription factors NF-kappa B and AP-1 underlies loss of cytokine gene expression in rat alveolar macrophages treated with a diffusible product from the spores of Aspergillus fumigatus. American Journal of Respiratory Cell and Molecular Biology. 15: 88–96. [DOI] [PubMed] [Google Scholar]
- 72.Romani L 2011. Immunity to fungal infections. Nature Reviews Immunology. 11: 275–288. [DOI] [PubMed] [Google Scholar]
- 73.Bonnett CR, Cornish EJ, Harmsen AG, and Burritt JB. 2006. Early neutrophil recruitment and aggregation in the murine lung inhibit germination of Aspergillus fumigatus conidia. Infection and Immunity. 74: 6528–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lewis RE and Kontoyiannis DP. 2009. Invasive aspergillosis in glucocorticoid-treated patients. Medical Mycology. 47: S271–S281. [DOI] [PubMed] [Google Scholar]
- 75.Kyrmizi I, Gresnigt MS, Akoumianaki T, Samonis G, Sidiropoulos P, Boumpas D, Netea MG, van de Veerdonk FL, Kontoyiannis DP, and Chamilos G. 2013. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting Dectin-1/Syk kinase signaling. Journal of Immunology. 191: 1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luvanda MK, Posch W, Vosper J, Zaderer V, Noureen A, Lass-Flörl C, and Wilflingseder D. 2021. Dexamethasone promotes Aspergillus fumigatus growth in macrophages by triggering M2 repolarization via targeting PKM2. Journal of Fungi. 7: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Merkow L, Pardo M, Epstein SM, Verney E, and Sidransky H. 1968. Lysosomal stability during phagocytosis of Aspergillus flavus spores by alveolar macrophages of cortisone-treated mice. Science. 160: 79–81. [DOI] [PubMed] [Google Scholar]
- 78.Berenguer J, Allende MC, Lee JW, Garrett K, Lyman C, Ali NM, Bacher J, Pizzo PA, and Walsh TJ. 1995. Pathogenesis of pulmonary aspergillosis. Granulocytopenia versus cyclosporine and methylprednisolone-induced immunosuppression. American Journal of Respiratory and Critical Care Medicine. 152: 1079–1086. [DOI] [PubMed] [Google Scholar]
- 79.Segal AW 2005. How neutrophils kill microbes. Annual Review of Immunology. 23: 197–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J, Heesemann J, and Ebel F. 2010. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes and Infection. 12: 928–936. [DOI] [PubMed] [Google Scholar]
- 81.Röhm M, Grimm MJ, D’Auria AC, Almyroudis NG, Segal BH, and Urban CF. 2014. NADPH Oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infection and Immunity. 82: 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, Jeron A, Latgé J, Brakhage AA, and Gunzer M. 2010. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathogens. 6: e1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roilides E, Uhlig K, Venzon D, Pizzo PA, and Walsh TJ. 1993. Prevention of corticosteroid-induced suppression of human polymorphonuclear leukocyte-induced damage of Aspergillus fumigatus hyphae by granulocyte colony-stimulating factor and gamma interferon. Infection and Immunity. 61: 4870–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ellett F, Jorgensen J, Frydman GH, Jones CN, and Irimia D. 2017. Neutrophil interactions stimulate evasive hyphal branching by Aspergillus fumigatus. PLoS Pathogens. 13: e1006154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aimanianda V, Bayry J, Romani L, Latge J, Bozza S, Kniemeyer O, Perruccio K, Ramulu elluru S, Clavaud C, Paris S, Brakhage AA, and Kaveri SV. 2009. Surface hydrophobin prevents immune recognition of airborne fungal spores. Nature. 460: 1117–1121. [DOI] [PubMed] [Google Scholar]
- 86.Hohl TM, Van Epps HL, Rivera A, Morgan LA, Chen PL, Feldmesser M, and Pamer EG. 2005. Aspergillus fumigatus triggers inflammatory responses by stage-specific β-glucan display. PLoS Pathogens. 1: e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Caffrey-Carr AK, Kowalski CH, Beattie SR, Blaseg NA, Upshaw CR, Thammahong A, Lust HE, Tang Y, Hohl TM, Cramer RA, and Obar JJ. 2017. Interleukin 1α is critical for resistance against highly virulent Aspergillus fumigatus isolates. Infection and Immunity. 85:e00661–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Knox BP, Blachowicz A, Palmer JM, Romsdahl J, Huttenlocher A, Wang CCC, Keller NP, and Venkateswaran K. 2016. Characterization of Aspergillus fumigatus isolates from air and surfaces of the international space station. mSphere. 1: e00227–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lupiañez CB, Villaescusa MT, Carvalho A, Springer J, Lackner M, Sánchez-Maldonado JM, Canet LM, Cunha C, Segura-Catena J, Alcazar-Fuoli L, Solano C, Fianchi L, Pagano L, Potenza L, Aguado JM, Luppi M, Cuenca-Estrella M, Lass-Flörl C, Einsele H, Vázquez L, Ríos-Tamayo R, Loeffler J, Jurado M, and Sainz J. 2016. Common genetic polymorphisms within NFκB-related genes and the risk of developing invasive aspergillosis. Frontiers in Microbiology. 7: 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.