

Abstract
In recent years, the field of epitranscriptomics has witnessed significant breakthroughs with the identification of more than 150 different chemical modifications in different RNA species. It has become increasingly clear that these chemical modifications play an important role in the regulation of fundamental processes linked to cell fate and development. Further interest was sparked by the ability of the epitranscriptome to regulate pathogenesis. However, despite the involvement of macrophages in a multitude of diseases, a clear knowledge gap exists in the understanding of how RNA modifications regulate the phenotype of these cells. Here, we provide a comprehensive overview of the known roles of macrophage RNA modifications in the context of different diseases.
Keywords: MT: RNA and epigenetic editing Special Issue, macrophages, RNA modification, epitranscriptomics, m6A
Graphical abstract
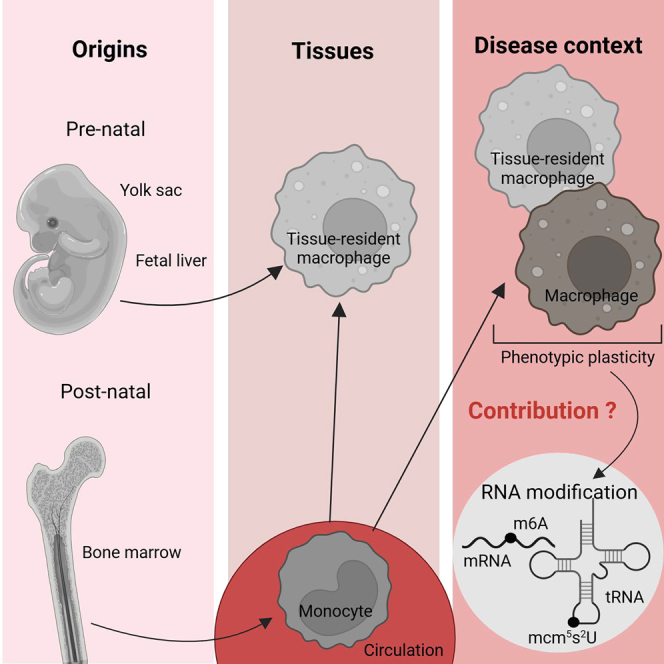
Macrophages are important contributors to a large variety of diseases, necessitating a thorough understanding of the mechanisms that regulate their phenotype. RNA modifications emerged as novel macrophage regulators. Here, current knowledge on resident macrophages in various tissues and the impact of RNA modifications in these cells during disease is reviewed.
Introduction
Macrophages play critical roles in maintaining tissue equilibrium during steady-state conditions, whereby tissue-resident macrophages execute specialized functions driven by cues from the microenvironment. Conversely, macrophages may contribute to inflammation and immunity during disease, taking part in the pathogenesis of various diseases such as cancer and inflammatory disorders.1 Consequently, a full understanding of the mechanisms that regulate these cells’ phenotype is required to fully grasp their impact during health and disease.
Over the past two decades, much attention was paid to the transcriptional plasticity of macrophages in response to a changing environment. Gene expression signatures were proposed for so-called classically and alternatively activated macrophages, first in a bimodal fashion referring to interferon (IFN)-γ versus interleukin (IL)-4/IL-13-stimulated macrophages,2 subsequently more fine-tuned to a variety of macrophage-modulating signals.3,4 Besides environmental factors, it also became clear that macrophage ontogeny can contribute to macrophage diversity. Embryonal progenitors from the yolk sac or the fetal liver give rise to long-lived tissue-resident macrophages, that, in some tissues, are gradually replaced by bone marrow (BM)-derived cells throughout life.5 Upon an inflammatory insult to the tissue, circulating monocytes usually contribute to the local macrophage pool, staying either temporarily to deal with the ongoing threat or incorporating in the long-term tissue-resident pool.6,7 With the advent of single cell RNA-sequencing and ATAC-sequencing technologies, the complexity of the macrophage compartment becomes even more evident, at least at the transcriptional and epigenetic level.
Recently, epitranscriptomics appeared as another level of cellular regulation. RNAs can be modified post-transcriptionally by more than 150 modifications,8 catalyzed by dedicated and typically highly conserved enzymatic machineries whose disruption can result in a range of diseases. Many of these modifications can be found on transfer RNAs (tRNAs), but these modifications can also decorate other RNA species, such as mRNAs, ribosomal RNAs (rRNAs), microRNAs, and long non-coding RNAs.9 N6-methyladenosine (m6A) is the most abundant internal modification associated with eukaryotic mRNAs and is also one of the most studied.10 It recently became clear that RNA modifications also affect the macrophage phenotype, with an important impact on these cells’ role during various diseases. In this review, we provide an overview of the current knowledge on the roles of RNA modifications in macrophages in the context of different diseases.
m6A RNA modifications
The role of the m6A RNA modification is currently being unveiled in immune cells in several pathologies.11 m6A was first identified in mammalian mRNA, and was later detected in nearly all types of RNA, including rRNA, small nuclear RNA and regulatory RNAs.12 m6A is deposited on mRNAs by m6A methyltransferases (also called writers), primarily by a complex composed of methyltransferase (METTL) 3 and METTL14. METTL3 is an S-adenosyl methionine-binding protein with a methyltransferase capacity, while METTL14 is an allosteric activator that binds to RNA.13 Remarkably, a variety of other proteins were reported to contribute to the writing of m6A on RNAs, including Wilms tumor 1-associated protein (WTAP),14 vir-like m6A methyltransferase-associated (originally known as KIAA1429),15 RNA-binding motif protein 15/15B (RBM15/15B),16 zinc-finger CCCH-type-containing 13 (ZC3H13),17 oncogene-like protein 1 (CBLL1),18 and METTL16.19 Zinc finger CCHC-type containing 4,20 METTL5,21 and tRNA methyltransferase activator subunit 11-222 regulate the methylation of rRNAs. m6A can also be removed from the RNA thanks to enzymes called erasers, which at the moment encompasses two members: fat mass and obesity-associated protein (FTO)11 and alkB homolog 5 (ALKBH5).23 Finally, enzymes capable of recognizing m6A-modified RNAs are referred to as m6A reader proteins. These readers regulate gene expression through several processes such as alternative splicing,24 mRNA export,25 mRNA stability,26 translation,27 mRNA decay,28 and the processing of pre-microRNA.29 m6A readers include YT521-B homology (YTH) domain-containing enzymes,30 such as YTHDF1, YTHDF2, YTHDF3,31 YTHDC1, and YTHDC2,28 as well as non-YTH-containing enzymes such as eukaryotic initiation factor 3,27 heterogeneous nuclear ribonucleoproteins,29 insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs),26 and fragile X mental retardation protein (FMRP).32 These actors are summarized in Figure 1.
Figure 1.

Schematic representation of the m6A RNA modification complex
The m6A methylation is catalyzed by the writer complex, including METTL3, METTL14, METTL16, WTAP, vir-like m6A methyltransferase-associated (VIRMA), RBM15/15B, zinc-finger CCCH-type-containing 13 (ZC3H13), zinc finger CCHC-type containing 4 (ZCCHC4), oncogene-like protein 1 (CBLL1), METTL5, and tRNA methyltransferase activator subunit 11-2 (TRMT112). The m6A modification is erased by demethylases, including FTO and ALKBH5. The m6A-modified RNA reader proteins include YTHDFs, YTHDCs, IGF2BPs, heterogeneous nuclear ribonucleoproteins (HNRNPs), and FMRP. The m6A modification modulates the following processes: microRNA biogenesis, RNA export, alternative splicing, RNA translation, RNA decay, and RNA stability.
The role of m6A in brain macrophages
Brain macrophages
In the brain, within different anatomical niches, distinct tissue-resident macrophage populations have been described. Microglia are found in the brain parenchyma, except for a unique microglial subset residing on the apical surface of the choroid plexus epithelium.33 Various barrier-associated macrophage (BAM) populations are located at interfaces between the brain and the periphery, such as the meninges, choroid plexus, and the perivascular space.33 During embryogenesis, microglia arise from yolk sac-derived progenitors. Once the microglia pool has been established, they are self-renewed by local proliferation, with virtually no input from BM-derived monocytes throughout life.5 BAMs residing in the dura mater, subdural meninges, and choroid plexus consist of distinct subsets, with tissue-specific transcriptional signatures, and, depending on the accessibility of the niche, a varying contribution of monocyte-derived macrophages in function of time.33
Several studies have identified a core homeostatic microglial gene signature, which includes Sall1, Hexb, Fcrls, Gpr43, Cx3cr1, Tmem119, Trem2, MerTK, Pros1, and Siglech.33,34,35 Homeostatic BAMs, in contrast, share a core transcriptional signature that is distinct from that of parenchymal microglia (except for core macrophage genes, such as Cx3cr1, Csf1r, and C1qb), including the expression of genes such as Ms4a7, Ms4a6c, Tgfbi, and Lyz2.36 BAM subtypes also exhibit compartment-specific transcriptional adaptations in the healthy mouse brain and can be subdivided in major histocompatibility complex (MHC)-IIhigh and MHC-IIlow subsets within the same compartment.33 Of note, in animal models of Alzheimer’s disease (AD), plaque-associated microglial activation states have been reported. These disease-associated microglial (DAM) clusters have been shown to express high levels of Lpl, Itgax, Apoe, and Cst7. In a model of toxic demyelination by cuprizone, the DAMs were found to exist in two different activation states, linked to demyelination and remyelination processes.37 These findings demonstrate the dynamic nature of the microglial activation states during disease.
The role of macrophage m6A in the context of brain disease
AD is a neurodegenerative disorder and a multifactorial disease, with an important regulatory role played by microglia.38 To understand the potential function of the m6A modification in myeloid cells associated with AD, wild-type (WT) mice were subjected to a WT or Mettl3−/− BM transplantation and subjected to amyloid beta (Aβ)-induced AD.39 Interestingly, Mettl3−/− BM-transplanted animals showed increased learning and decreased memory deficits. Similar results were obtained when monocytes isolated from Mettl3−/− animals were transferred into WT mice, highlighting a role for Mettl3 in monocyte-derived macrophages during AD. Indeed, inhibiting the recruitment of monocyte-derived macrophages into the brain abrogated the improvement in cognition. Mechanistically, the absence of Mettl3 led to a reduced expression of methyltransferase 3a (Dnmt3a), which in turn regulates the expression of tubulin acetyltransferase (Atat1). Lower Atat1 levels decreased microtubule acetylation, resulting in an enhanced mobility of macrophages. Consequently, an increase in monocyte-derived macrophages was observed in AD brains of Mettl3 conditional knock-out (cKO) (Lyz2Cre Mettl3fl/fl) mice compared with controls, confirming that the depletion of Mettl3 in myeloid cells enhances the transmigration of brain-infiltrating macrophages. Furthermore, these monocyte-derived macrophages showed an enhanced phagocytosis and clearance of Aβ. These results demonstrate that the m6A-Dnmt3a-Atat1 axis controls the progression of AD by regulating the migration and clearance of Aβ by monocyte-derived macrophages.39 It remains to be investigated if RNA modifications can also affect the phenotype of tissue-resident microglia and BAMs.
Ischemic stroke, which occurs when a vessel supplying blood to the brain is obstructed, is another common injury to the brain. In a rat model, initiated by a middle cerebral artery occlusion, the protein level of METTL14 was significantly increased in microglia identified via IBA1 staining.19 Invalidation of Mettl14 in HAPI cells (rat microglia) resulted in decreased levels of inflammatory macrophage markers (tumor necrosis factor [TNF]-α, inducible nitric oxide [iNOS], IL-1β, IL-6, and CD86) and increased anti-inflammatory markers (IL-10, Arg-1, and CD206) before and after oxygen-glucose deprivation/reperfusion. Mechanistically, METTL14 enhanced the expression of histone acetyltransferase P300 (Kat3b) by promoting the m6A modification of Kat3b mRNA. KAT3b controls the expression of STING, which in turn regulates the polarization of macrophages. Moreover, the absence of METTL14 also inhibited NLR family pyrin domain containing 3 (NLRP3) inflammasome activation. Overall, the invalidation of Mettl14 using siRNAs resulted in the alleviation of brain injury after ischemic stroke in rats.40 As the invalidation of Mettl14 was not specific to macrophages/microglia, it remains to be seen if similar results can be achieved when Mettl14 is invalidated only in those cells.
Autoimmune uveitis (AU) is an inflammatory process taking place at the uvea, in the interior of the eye, a region that is also populated by brain macrophages. In this disease, the expression of Fto in the retinal microglia was negatively correlated with the severity of the pathology.20 The authors also observed a decrease in the expression of Fto in a microglial cell line (HMC3 cells) treated with lipopolysaccharide (LPS) and IFN-ɣ to induce inflammation. Knocking down (KD) Fto in these cells led to an increased expression of inflammatory mediators, such as iNOS, IL-6, and TNF-α, in response to LPS and IFN-ɣ. Fto KD in HMC3 cells also resulted in a decreased proliferation, but an increased HMC3 cell migration after treatment with these inflammatory mediators. Bulk RNA sequencing revealed that the expression of Gpc4 was significantly upregulated after Fto KD in HMC3 cells due to an increased m6A content of its mRNA, leading to an increase in stability. GPC4 then regulates the TLR4/nuclear factor (NF)-κB signaling pathway. Finally, intravitreal injection of FB23-2, an inhibitor of FTO, resulted in higher clinical and histopathological scores after interphotoreceptor retinoid-binding protein immunization for the induction of experimental AU.41 Hence, FTO can significantly ameliorate the symptoms of AU, at least in part due to its effect in microglia. These findings are summarized in Figure 2A.
Figure 2.

m6A modifications and the mechanisms of their involvement in regulating disease-associated macrophages
(A) Mettl3, Mettl14, and Fto regulate the phenotype of brain macrophages through distinct response mechanisms. (B) Mettl3 regulates liver macrophages. (C) Igf2bp2, Ythdf2, and Mettl3 regulate the phenotype of lung macrophages. (D) Igf2bp2 regulates the phenotype of gastrointestinal macrophages. (E) Alkbh5 regulates the phenotype of cardiac macrophages.
The role of m6A in liver macrophages
Liver macrophages
The liver harbors multiple populations of macrophages, even in steady state. Kupffer cells (KCs) are first derived from yolk sac erythro-myeloid progenitors, but are subsequently replaced by fetal liver monocytes and then by blood monocytes in the post-natal period. KCs are found in the sinusoidal vessels,42 but can also reach into the perisinusoidal space of the liver, called the space of Disse, to be in contact with hepatic stellate cells and hepatocytes.43 KCs are implicated in lipid/cholesterol transport and metabolism,44 as well as in iron metabolism.45 These cells also play a key role in the removal of aged platelets.46
In addition to KCs, other hepatic macrophage populations have been described, representing less than 10% of all macrophages in the liver. Liver capsule macrophages (LCMs) have been described both in murine and human liver and were shown to be derived from monocytes accumulating in the capsule at weaning. LCMs specialize in mounting a bactericidal immune response against peritoneal bacteria that access the liver.47 Guilliams et al.48 identified a population of macrophages present at the central vein, referred to as the central vein macrophages. Aside from LCMs and central vein macrophages, Guilliams et al. also unveiled the presence of lipid-associated macrophages (LAMs) in proximity to the bile ducts in the healthy murine and human liver, leading to their annotation as bile-duct (BD)-LAMs.48 As of now, no functions for central vein macrophages nor BD-LAMs have been proposed during homeostasis.
CLEC4F was identified as a specific marker for murine KCs. However, CLEC4F is expressed rather late in the development of KCs, so the single use of CLEC4F is not sufficient to identify monocyte-derived KCs in the diseased liver. Moreover, CLEC4F does not permit the identification of human KCs. To circumvent this issue, a gene signature composed of CD5L, VSIG4, CD163, FOLR2, MARCO, and SLC40A1 was proposed to discriminate KCs from other macrophage populations.24 Moreover, the addition of the surface markers VSIG4 and FOLR2 to CLEC4F permit the identification of KCs in murine and human liver.48 There is also an ongoing debate regarding KC homogeneity in the healthy murine liver,49,50 with a recent proposal that KCs could be split into two populations based on the expression of CD206 and ESAM.51,52 In the murine liver, LCMs have been shown to express F4/80, CD64, CX3CR1, and CD207. Currently, a signature to specifically identify this population in humans is still missing. Of note, the transcriptional profile of LCMs is identical to that of central vein macrophages. The identification of BD-LAMs in the murine liver relies on negative gating strategies (CLEC4F−VSIG4−CLEC2−CD207−CD64+F4/80+). In the human liver, BD-LAMs can be studied based on the expression of the following surface markers: CD14, CD11a, CD26, CD141, and CD9. In addition, Gpnmb, Spp1, Trem2 and Cd9 are expressed by BD-LAMs at the mRNA level.43
The role of macrophage m6A in the context of liver disease
The function of METTL3 was investigated in hepatic macrophages in the context of liver fibrosis. By transiently overexpressing Mettl3 in isolated KCs, it was shown that an increase of m6A could upregulate the expression of Malat1, leading to an enhanced presence of inflammatory markers, such as IL-1β and iNOS, as well as NLRP3 inflammasome-induced pyroptosis. These features ultimately worsen carbon tetrachloride (CCl4)-induced liver fibrosis.53 Similar results were obtained by the overexpression of Mettl3 in THP-1 macrophages, which led to an upregulation of nuclear paraspeckle assembly transcript 1 (Neat1), while the invalidation of Mettl3 in these cells decreased Neat1. NEAT1 was found in exosomes from LPS-treated KCs, mediating the activation of the Sp1/transforming growth factor (TGF)-β1/Smad signaling pathway and causing the activation of hepatic stellate cells, which ultimately aggravates the progression of hepatic fibrosis.54 Together, these results suggest that the upregulation of m6A in KCs is contributing to the progression of liver fibrosis. Of note, both studies isolated KCs using the F4/80 marker, which is expressed on all liver macrophages. Thus, it remains to be determined if these results can be corroborated when using KC-specific markers for their isolation.
METTL3 was also found to play an important role in alcoholic steatohepatitis (ASH). It was shown that KCs, purified from the liver of an ASH mouse model, exhibited pyroptosis. Again, as a cautionary note, the KCs were isolated using Percoll density centrifugation and only F4/80 was used as a marker. The invalidation of Mettl3 in murine BM-derived macrophages and the Raw264.7 macrophage cell line resulted in decreased pyroptosis by influencing the splicing of pre-miR-34A. The authors hypothesized that targeting METTL3 could be a promising therapeutic strategy as some evidence emerged that liver cell pyroptosis could contribute to the progression of ASH.55
TGF-β1 secreted from activated KCs promotes the progression of non-alcoholic steatohepatitis (NASH) to liver fibrosis. NASH could be modeled by feeding a high-fat diet (HFD) to rats, which was accompanied by increased m6A and METTL3/METTL14 protein levels, but decreased FTO. Similar results were obtained when treating KCs, isolated via a Percoll gradient, with LPS. Using methylated RNA immunoprecipitation sequencing (MeRIP-seq), 2,859 differential m6A peaks were identified in KCs upon LPS treatment, including an LPS responsive m6A peak in the 5′UTR of TGF-β1 mRNA. siRNA against Mettl3 and Mettl14 abolished LPS-induced m6A hypermethylation and led to a decrease in the expression of TGF-β1. These results were verified in vivo by injecting LPS or CCl4 in cKO Mettl14 (Mettl14fl/fl Lys2Cre) and control mice. The absence of Mettl14 in myeloid cells completely abrogated the release of TGF-β1 by KCs and protected against CCl4-induced liver fibrosis. Interestingly, the decrease in TGF-β1 release by KCs cannot be recapitulated when invalidating Ythdf1 or Ythdf3,56 highlighting the key role of m6A writers in this process. As the Lys2 promoter is not specifically active in KCs, it remains to be investigated if similar conclusions can be drawn when using Clec4fCre mice.
Mettl3fl/fl LysMCre mice treated with a HFD showed a reduction in diet-induced non-alcoholic fatty liver disease and obesity. Bulk RNA sequencing and m6A epitranscriptomic microarray analyses were performed on splenic macrophages isolated from Mettl3 cKO and control mice, uncovering 37 genes that were both differentially m6A modified and differentially expressed. These unbiased analyses demonstrated that METTL3 regulates the expression of damage inducible transcript 4 (Ddit4), which was previously identified as a key regulator of energy homeostasis and metabolism. The absence of METTL3 in macrophages resulted in the downregulation of the mammalian target of rapamycin complex 1 (mTORC1), which led to increased autophagy. Furthermore, the decrease in m6A gave rise to a reduction of NF-κB signaling in macrophages. Validating these results in vivo, an activator of Ddit4 could inhibit D-galactosamine/LPS-induced acute hepatitis.57 Of note, these results are fully in line with the m6A effects seen in liver fibrosis. All these data are summarized in Figure 2B.
The role of m6A in lung macrophages
Lung macrophages
The two most prominent macrophage populations in the lung are alveolar macrophages (AMs) and interstitial macrophages (IMs), with the former being found at the luminal side of the alveolar niche, while IMs mainly reside in the interstitial space around the bronchovascular bundle. Consequently, AMs, which are the most abundant macrophage population in the lung, serve as the first defenders of the airways and the alveoli. IMs rather act as gatekeepers of the vasculature and the lung interstitium. The initial seeding of AMs is predominantly dependent on fetal monocytes, and AMs can self-maintain via proliferation without the input of circulating adult monocytes.58 IMs are also thought to originate from fetal progenitors, but they can be replenished by circulating adult monocytes.59 Interestingly, IMs are not specific to the lungs; they are present throughout the body and even share a similar transcriptional profile across the multiple organs where they reside.60,61
Human AMs are autofluorescent cells that express the surface markers HLA-DR, CD43, CD88, CD169, CD206, CD10 (MME), CD11c, CD36, CD141, CD64, and MARCO, as well as low amounts of CD14. In mice, AMs are identified by the expression of CD11c, SiglecF, CD64, F4/80, and MerTK. At the transcriptional level, human and mouse AMs both express FABP4, MARCO, and PPARG, with mouse AMs in addition being characterized by the expression of Car4, Chil3, Ear1, and Krt79.62,63 To identify IMs, Pf4, C1q, and Cd88 can be used in mice whereas, in humans, LGMN and MARCKS are selectively expressed by IMs.59
The role of macrophage m6A in the context of lung disease
The role of METTL3 was investigated in asthma. It was demonstrated that mice with a Mettl3-deficiency in myeloid cells (Mettl3fl/fl Lyz2Cre), that were exposed to cockroach extract (CRE) to induce allergic asthma, had aggravated airway inflammation compared with their control counterparts. In line with these findings, the expression of m6A writers (METTL3 and METTL14), erasers (FTO and ALKBH5), and readers (YTHDF3) was downregulated in peripheral blood mononuclear cells from human asthma sufferers compared with healthy controls. Mechanistically, the Mettl3 cKO AMs exposed to CRE demonstrated an exaggerated M2 activation profile. The increase in M2-associated markers was linked to a decrease in m6A, which impairs the m6A-Ythdf3-mediated degradation of pentraxin 3 (Ptx3) mRNA.64 Together, this led to enhanced Th2 responses, which drive the progression of allergic asthma.65 Using the same CRE-induced asthma model, the role of IGF2BP2, a m6A-binding protein, in macrophages was investigated. Mice with a hematopoietic IGF2BP2-deficiency showed decreased symptoms, correlating with a decreased expression of M2-associated genes, such as Arg1, Cd206, Chil3, Retnla, and Il10. It was suggested that the ability of IGF2BP2 to switch the phenotype of macrophages is linked to an increased stability of the tuberous sclerosis 1 (Tsc1) transcript. Indeed, in BM-derived macrophages (BMDMs), IGF2BP2 was shown to bind TSC1 to increase its stability.66 In turn, TSC1 is a well-known negative regulator of mTORC1 signaling, which prevents the expression of M2 markers.67,68 However, it remains to be determined if a similar conclusion can be drawn when IGF2BP2 invalidation occurs solely in macrophages.
Pulmonary hypertension (PH) is a cardiopulmonary disorder that leads to increased pulmonary arterial pressure, heart failure, and premature death. The absence of Ythdf2 in myeloid cells (Ythdf2fl/fl Lyz2Cre) was found to decrease SU5416/hypoxia-induced PH compared with control mice, as evidenced by an ameliorated pulmonary vascular remodeling (PVR) and decreased cardiac dysfunction. Mechanistically, the absence of Ythdf2 attenuated macrophage alternative activation at the early stage of hypoxia, as evidenced by a decrease in M2-associated marker mRNA levels. Conversely, heme oxygenase 1 (Hmox1) was found to be significantly upregulated in AMs from Ythdf2fl/fl Lyz2Cre mice, whereby Hmox1 mRNA can be recognized and bound by YTHDF2, resulting in a significant enrichment of the m6A modification in this mRNA. Subsequently, YTHDF2 is capable of targeting Hmox1 mRNA for degradation by reading the m6A modification sites. HMOX1 was indeed proven to be the crucial mediator in this disease, as its inhibition abrogated the anti-inflammatory and antioxidant effects observed in Ythdf2fl/fl Lyz2Cre mice under hypoxic exposure. Furthermore, the improvement of the PVR could be explained by the reduced proliferation and migration of pulmonary artery smooth muscle cells, which is seen after treatment with conditioned medium from Ythdf2fl/fl Lyz2Cre AMs.69 The effects of m6A on lung macrophages are summarized in Figure 2C.
The role of m6A in gastrointestinal macrophages
Gastrointestinal macrophages
Macrophages can be found throughout the different layers of the gastrointestinal tract.70,71 Lamina propria (LP) macrophages are monocyte-derived cells that are continuously replenished by progenitors. These cells play a role in the induction of tolerance to microbial stimuli. Furthermore, the LP macrophages are involved in the secretion of IL-10 promoting the maintenance of regulatory T cells.72 Besides LP macrophages, which constitute the most abundant population of gut macrophages, several other populations were described: the neuron-associated macrophages, the blood vessel-associated macrophages, and the crypt-associated macrophages, as well as the Peyer’s patch-associated macrophages. The neuron-associated macrophages are important for the survival of neurons within the myenteric and the submucosal plexus.73
LP macrophages can be identified using CD14hiCD11ChiCCR2+CX3CR1+. Interestingly, in humans, the expression of CX3CR1+ is decreased as monocytes differentiate into macrophages. The neuron-associated macrophages upregulate genes which have also been reported in microglia, such as Fcrls, Mef2a, Hexb, and Gpr34.72 The blood vessel-associated macrophages upregulate genes related to angiogenesis, such as Ecm, Tnfaip2, Anpep, Hif1a, Mmp2, and Mmp14. The crypt-associated macrophages have not yet been fully defined, but recent single-cell RNA sequencing data have shown that these macrophages upregulate the expression of typical Paneth cell markers like Itln1 and Defa. Remarkably, the Peyer’s patch-associated macrophages do not express typical macrophage markers like CD64, F4/80, and CD206, but are instead defined as Tim4+ CD4+ or Tim4− CD4+.74
The role of macrophage m6A in the context of gastrointestinal disease
In a mouse model of colonic inflammation (dextran sulfate sodium [DSS]-induced colitis), the absence of IGF2BP2 from myeloid cells is detrimental to the animals. Mice which received a BM transplantation from IGF2BP2 KO mice had increased DSS-induced colitis compared with mice which received BM transplantation from WT mice (Figure 2D). Body weight loss, shortened colon length, colon damage, and increased inflammation, as well as an elevated production of pro-inflammatory cytokines, was observed in mice with a hematopoietic IGF2BP2 deficiency. It was hypothesized that the protective effect of IGF2BP2 in the context of colitis is linked to the regulation of the macrophage phenotype via the Tsc1 transcript.66 However, this hypothesis needs to be substantiated by using macrophage-specific Igf2bp2 cKO mice.
The role of m6A in cardiac macrophages
Cardiac macrophages
Under steady-state conditions, resident cardiac macrophages remove senescent and dying cells and facilitate electrical conduction. Resident macrophages adopt a spindle-like shape and intermingle closely with myocytes and endothelial cells, as well as fibroblasts. It was shown that these cells originate from the embryonic yolk sac and from fetal liver progenitors.75 In the steady state, four populations of resident cardiac macrophage have been described, which can be distinguished based on the expression of Ly6C and the MHC class II in the mouse.75 Of note, Molawi et al.76 also proposed a classification to distinguish between four cardiac macrophage populations based on the expression of CX3CR1 and MHC-II. However, the distinctive roles of these four populations remain to be investigated.
The role of macrophage m6A in the context of cardiac disease
In a mouse model of hypertension-induced cardiac dysfunction, it was shown that hypertension increases the propensity of monocyte-derived macrophages, but not cardiac-resident macrophages, to transition toward myofibroblasts. Interestingly, Alkbh5 was significantly upregulated during the process of macrophage-to-myofibroblast transition (MMT). To investigate the role of ALKBH5 in macrophages in the context of MMT, Cx3cr1Cre, Rosa26Td, ALKBH5flox/flox mice were used, showing a decrease in the heart weight/body weight ratio and a lower left ventricular mass compared with the control animals. Furthermore, cardiac fibrosis was decreased in the cKO animals compared with controls, which could be explained by a diminished infiltration of circulating monocytes and a subsequently reduced MMT of the monocyte-derived macrophages. Mechanistically, the absence of ALKBH5 in macrophages led to an increase of m6A marks on the Il11 mRNA, resulting in a decreased stability of IL-11, which was identified as an important regulator of cardiovascular fibrosis77 (Figure 2E).
The role of macrophage m6A in the context of infection
Macrophages, as central players in the innate immune system and orchestraters of inflammation, often affect the course of an infectious disease, so an impact of macrophage m6A could be anticipated. Indeed, in a mouse model of sepsis caused by cecum ligation and puncture, the absence of Mettl14 in myeloid cells (Mettl14fl/fl Lyz2Cre mice) resulted in more severe symptoms than in control mice (Mettl14fl/fl). Similar results were obtained by using a LPS-induced sepsis model. After LPS stimulation, the gene expression of pro-inflammatory cytokines such as Tnf, Il1b, and Il6 was more pronounced in peritoneal macrophages as well as BMDMs of Mettl14 cKO mice compared with controls. This phenotype was mediated by a reduced expression of Socs1, a negative regulator of the LPS/TLR4 signaling pathway. Hence, m6A decoration of the Socs1 transcript is required for its stability, allowing SOCS1 to install an anti-inflammatory feedback loop.78 These data are corroborated by studying the effects of a Mettl3 deficiency in BMDMs. Indeed, LPS treatment of Mettl3 KO BMDMs resulted in an enhanced expression of Tnf, Il6, and Nos2.79 However, these findings are contradicted by another study, claiming that the absence of Mettl3 in macrophages led to a decreased expression of pro-inflammatory cytokines due to the induction of a negative regulator of the Toll-like receptor signaling pathway: IL-1 receptor associated kinase 3 (Irakm).80 Furthermore, the same research group showed that mice with a specific deletion of Mettl3 in myeloid cells (Mettl3fl/fl Lyz2Cre), upon infection with Salmonella typhimurium, had lower body weight and higher bacterial burden than control littermates (Mettl3fl/fl). It was suggested that the absence of Mettl3 impairs the anti-bacterial activity of macrophages, due to the reduced secretion of pro-inflammatory cytokines. How these contradictory findings, using the same cKO mice, can be reconciled will be a matter of future research.
Blood from patients with sepsis demonstrated a decreased FTO expression in monocytes compared with healthy counterparts. The expression of FTO was shown to be correlated with the expression of IL1b, which plays a critical role in the inflammatory response. Furthermore, the in vivo silencing of Fto in macrophages via siRNA-loaded liposomes resulted in the protection of mice from a lethal dose of LPS, thanks to an inhibition of the NF-κB signaling pathway and consequently decreased levels of cytokines and a lowered expression of MHC-II, CD80, and CD86 in those macrophages.81 These findings are in line with the data described above,78,79 showing that the increase in macrophage m6A could protect mice from sepsis.
Infection of primary mouse peritoneal macrophages with an RNA virus, vesicular stomatitis virus (VSV), led to an increase in m6A levels at the beginning of the infection, which then returned to baseline at 16 h after infection. The change in m6A levels was accompanied by an increase of the viral load upon infection, followed by a decrease later on. An RNA interference-mediated knock-down of Mettl3, Mettl14, Wtap, Alkbh5, and Fto showed that the invalidation of Alkbh5 decreased the VSV viral load in mouse peritoneal macrophages. Similar results were reproduced in BMDMs and in the human THP-1 macrophage cell line. In vivo validation came from the observation that VSV-infected Alkbh5−/− mice survived longer than control mice. The authors demonstrated that Alkbh5-deficient macrophages produced fewer pro-inflammatory cytokines than WT macrophages. Mechanistically, m6A methylation was increased on α-ketoglutarate dehydrogenase (OGDH) mRNA, leading to its reduced stability. The decreased expression of OGDH resulted in a reduced production of itaconate, which is necessary for the replication of the virus.82
THP-1 macrophages infected with Treponema pallidum (TP), the causative agent of syphilis, were shown to upregulate the expression of YTHDF1. Interestingly, a knock-down of this gene in TP-infected macrophages led to an increased expression of M1 markers, such as CD86 and iNOS, while the expression of M2 markers like CD206 and Arg-1 were decreased. This phenotype was correlated with an enhanced level of m6A on the Socs3 mRNA, which was recognized by YTHDF1 to enhance its translation. SOCS3 negatively regulates the secretion of inflammatory cytokines, such as TNF-α and IL-1β, by TP-infected macrophages.83 These findings may thus explain why TP infection does not cause excessive inflammation, although it remains to be determined whether the expression of YTHDF1 in macrophages can modify the fate of TP-infected animals.
Altogether, it is clear that m6A RNA marks in myeloid cells strongly affect the susceptibility to bacterial and viral pathogens, as well as septic shock. These findings have been summarized in Figure 3. It will be important to determine whether these findings can be extended to a variety of human pathogens and whether common modes of action can be identified, which will be important for the future development of therapeutic strategies.
Figure 3.

The role of macrophage m6A in the context of infection
The absence of the writer complex (Mettl3/Mettl14) in macrophages increased bacterial burden (Salmonella typhimurium) as well as the symptoms linked to sepsis. The absence of the erasers (Fto and Alkbh5) in macrophages led to decreased symptoms of sepsis following the administration of a lethal LPS dose and upon viral infection (VSV). The absence of Ythdf1, an m6A reader, in macrophages decreased the expression of pro-inflammatory cytokines.
The role of macrophage m6A in the context of atherosclerosis
Atherosclerosis is characterized by the accumulation of fatty deposits called atheromatous plaques within the inner walls of the arteries. The plaques restrict blood flow, which can lead to several complications such as heart attack or stroke. Macrophages are active participants in the formation of the artery plaque, so therapeutic strategies that are based on macrophage targeting are currently being studied. In this context, RNA modifications could be considered as tools to shift the phenotype of macrophages toward an anti-atherosclerotic profile.
IFN-ɣ and LPS-treated macrophages that were silenced for Mettl3 demonstrated a reduced pro-atherosclerotic phenotype, with a decreased expression of M1-associated markers (iNOS, TNF-α, IL-1β, and Cxcl10), a lower accumulation of lipids and a diminished expression of the pro-atherosclerotic hepatoma-derived growth factor (HDGF). The importance of the latter is demonstrated by the fact that apolipoprotein E (ApoE)-KO mice with a myeloid deficiency for HDGF presented with a reduced development of atherosclerosis compared with ApoE KO mice.84 However, another study demonstrated that ApoE-KO mice fed with a western diet demonstrated decreased m6A levels in CD68-positive macrophages when compared with WT mice. Furthermore, ApoE-KO mice injected with lentiviral particles for the overexpression of matrin 3 (Matr3), which can promote the formation of the Mettl3-Mettl14 complex, led to smaller lesions in the aorta compared with the control group.85 The discrepancy between these studies may be linked to the mouse model. Indeed, overexpression of Matr3 does not significantly affect the m6A marks on RNAs.85 It can thus be hypothesized that a decrease in macrophage m6A levels may protect against atherosclerosis.
The role of FMRP, known as a m6A reader, was also evaluated in atherosclerosis. FMRP-deficient BMDMs showed an increased efferocytosis when compared with WT BMDMs, either under palmitate-induced ER stress conditions or without ER stress. It had been previously demonstrated that efferocytosis promotes the regression of an atherosclerotic plaque, allowing the authors to propose FMRP-targeting as a new therapeutic strategy to counteract atherosclerosis.86
The role of macrophage m6A in the context of cancer
Tumor-associated macrophages (TAMs) contribute to tumor growth, metastasis, and therapy resistance through a variety of mechanisms. Consequently, a greater density of TAMs is correlated with a worse prognosis in most cancer types. The advent of single-cell technologies resulted in a fine tuning of the TAM concept,35,62,87 with the identification of distinct TAM subsets playing distinct roles.
RNA epigenetics could be another level of TAM regulation. Indeed, the absence of Mettl3 in myeloid cells (Mettl3fl/fl Lyz2Cre) was found to promote the subcutaneous tumor growth of colon adenocarcinoma (MC38),80 melanoma (B16),88 and non-small cell lung carcinoma (LLC).88 This change in tumor growth was mainly attributed to a re-shaping of the tumor microenvironment, whereby TAMs adopted a more M2 activation state (increased CD206, decreased TNF-α and CD86) and both CD4+ and CD8+ T cells were exhausted, as evidenced by the increased expression of programmed cell death-1 (PD-1).80 Yin et al.88 reported a similar pro-tumoral effect of a myeloid Mettl3 deficiency, accompanied by an increased presence of macrophages as well as regulatory T cells, and a decrease in granulocytes and CD8+ T cells. These shifts in the tumor immune microenvironment could mainly be attributed to a change in translation induced by the absence of Mettl3 in myeloid cells. Indeed, in macrophages, the decrease in m6A downregulated the expression of Spred2, which was in turn shown to upregulate the expression of Erk, NF-κB, and Stat3. Especially an increased activity of Stat3 contributes to the creation of a pro-tumoral environment.88 Moreover, the absence of Mettl3 in myeloid cells enhanced lung metastasis. Indeed, tail vein injection of B16 cells or LLC cells led to a higher number of cancer cells in the lungs of Mettl3fl/fl LysMCre mice compared with WT controls.88
In line with the Mettl3-deficiency, also the absence of Mettl14 in myeloid cells (Mettl14fl/fl Lyz2Cre) could promote tumor growth in several models: B16-OZ, B16F10, LLC and MC38.89 Again, the tumor microenvironment was re-shaped, with the proportion of CD8+ T cells being significantly decreased in the Mettl14 cKO tumors due to a decrease in progenitor and effector CD8+ T cells. Interestingly, several m6A-marked immunoregulatory ligands showed an increased gene expression in Mettl14 cKO TAMs. This included Ebi3, a subunit of the heterodimeric cytokines IL-27 and IL-35, which have been demonstrated to induce T cell exhaustion. Interestingly, blocking Ebi3 with EBI3-neutralizing antibodies led to tumor reduction in Mettl14 cKO mice. These findings seem to translate to humans, as patients with low m6A levels showed lower effector T-cell signatures and higher exhausted T-cell signatures. Furthermore, using public scRNA-seq data from melanoma and non-small lung cancer patients, it was shown that the expression of EBI3 in macrophages was positively correlated with exhausted CD8+ T cells.89 To sum up, these results seem to strongly indicate that a reduction in m6A, either by targeting Mettl3 or Mettl14, is responsible for tumor growth in several models.
However, this working model was recently challenged by findings showing that the presence of lactate in the tumor microenvironment increased the expression of Mettl3 in tumor-infiltrating myeloid cells via histone lactylation, which is responsible for the promotion of tumor growth as shown using Mettl3fl/fl Lyz2Cre mice. These conflicting results were obtained despite using a similar strategy for the invalidation of Mettl3 in myeloid cells (Mettl3fl/fl Lyz2Cre) and even the same tumor models.90 Accordingly, MC38 cells co-injected with BMDMs treated with the METTL3-inhibitor STM245791 led to a decreased tumor growth compared with MC38 cells co-injected with vehicle-treated BMDMs.90 In this case, a decrease in PD-1+TIM-3+ exhausted CD8+ T cells and regulatory T cells, but an increase in tumor-infiltrating transcription factor 1 stem-like CD8+ T cells and IFN-ɣ-expressing effector CD8+ cytotoxic T lymphocytes was observed in cKO Mettl3 mice. In the myeloid compartment, a decrease in immunosuppressive CD14+Ly6G+ PMN-MDSCs was seen. The reprogramming of the tumor microenvironment was attributed to the decrease in m6A on Jak1 mRNA, which decreased its translation and prevents the phosphorylation of STAT3, thereby decreasing the transcription of pro-tumoral genes.90
The role of METTL3 was also investigated in KCs in the context of hepatocellular carcinoma. Treating Huh-7 and Li-7 cells with media from Mettl3-silenced KCs reduced their proliferation as well as migratory and invasive capacities. In vivo, nude mice co-injected with Huh-7 cells and Mettl3-silenced KCs showed a reduced tumor growth rate and tumor weight, as well as a lower lung metastatic burden compared with nude mice co-injected with Huh-7 cells and control KCs. Furthermore, the tumor microenvironment was enriched for M1 KCs, while the percentage of M2 KCs was decreased. However, the authors did not use KC-specific markers, so the identity of the involved liver macrophages remains uncertain. Mechanistically, the decrease in m6A methylation resulted in a lower expression of RBM14, which was, at least in part, responsible for controlling the polarization of macrophages.92
Beside the writers, some of the m6A readers have also been investigated in TAMs. It was first demonstrated that B16-OVA tumor growth was accelerated in Ythdf2 cKO (Ythfd2fl/fl Lyz2Cre) mice compared with control (Ythfd2fl/fl) mice, accompanied by a decrease in tumor-infiltrating CD8+ T cells.89 However, these results were also recently challenged by data showing that a specific deletion of Ythdf2 in macrophages is responsible for suppressing tumor growth as well as metastasis, again using the same cKO (Ythfd2fl/fl Lyz2Cre) mice and tumor models (B16-OVA and MC38). Furthermore, using a melanoma lung metastatic model, Ythdf2 cKO animals had a lower number of metastasis than controls. The absence of myeloid Ythdf2 led to an increase in tumor-infiltrating IFN-ɣ-producing CD8+ T cells and tumor-infiltrating CD62LlowCD44highCD8+ effector memory T cells and a switch in the phenotype of macrophages from pro-tumoral to anti-tumoral. Consequently, Ythdf2-deficient macrophages had a higher ability for antigen cross-presentation and T-cell activation. Mechanistically, the absence of Ythdf2 in macrophages resulted in an increased stability of Stat1 mRNA, which gave rise to the reprogramming of macrophages through the transcription of pro-inflammatory genes.93
Overall, these conflicting results, which have been summarized in Figure 4, seem to indicate that the role of RNA modifications in shaping the phenotype of TAMs is highly complex. Some of the discrepancies may be explained by non-genetic differences between the mouse strains used, for example, at the level of the microbiota of the animals. Moreover, most of these studies were conducted in non-orthotopic mouse models, and, at this point, we cannot exclude that the function of these RNA modifications might also differ based on macrophage ontogeny.
Figure 4.

The role of macrophage m6A in the context of cancer
The absence of the writer complex (Mettl3/Mettl14) in macrophages either has an anti-tumoral or a pro-tumoral role. The same holds true for the absence of Ythdf2, an m6A reader.
The role of macrophage non-m6A RNA modifications in the context of neurodegenerative disorders
C to U RNA editing is mediated by two proteins, namely APOBEC-1, the RNA-specific cytidine deaminase, and the cofactor Apobec-1 complementation factor. The absence of Apobec1 in microglia compromised the phagocytic capacities of these cells.94 Remarkably, these results might be dependent on the macrophage type as the absence of Apobec1 in BMDMs resulted in increased phagocytic abilities.95 In a full body KO Apobec1 mouse model, microglia were activated, which disrupted brain homeostasis. In addition, the loss of Apobec1 was associated with increased depression and anxiety, as well as recognition memory defects and an abnormal hindlimb reflex response in middle-aged mice.94 However, it remains to be investigated if similar results are observed if Apobec1 is invalidated only in microglia.
The role of macrophage non-m6A RNA modifications in the context of inflammation
RNA 5 methylcytosine (m5C) is an abundant modification that occurs on many different RNA species, such as tRNAs, mRNAs and rRNAs. DNA methyltransferase homolog 2 (Dnmt2) and the NOP2/Sun RNA methyltransferase (Nsun) family, composed of seven members (Nsun1-7), are implicated in the writing of the m5C modification, while AlkB homolog 1 and the ten-eleven translocation (Tet) family (Tet1, Tet2, and Tet3) facilitate the erasure of the m5C modification. Presently, two readers have been described as well: Aly/REF export factor and Y box binding protein 1 (Ybx1). The role of Ybx1 has been investigated in myeloid cells, using Ybx1fl/fl Lyz2Cre mice. In these mice, tubulointerstitial damage is enhanced in a model of renal interstitial fibrosis, i.e., unilateral ureteral obstruction. The authors suggested that the absence of Ybx1 results in an increase in the expression of CCL5, leading to the recruitment of immune cells into the kidney. The infiltrating macrophages cannot mount an appropriate anti-inflammatory response, and therefore these macrophages are unable to stop the inflammation leading to tissue damage, myofibroblast activation and fibrosis.96
Another example of an inflammatory insult that is regulated by RNA modifications is the impact of Elp3 on DSS-induced gut inflammation. Elongator (Elp) is a protein complex composed of Elp1 to 6, which is implicated in the formation of the 5-methoxycarbonylmethyl and 5-carbamoylmethyl side chains on uridines at the wobble position in tRNA. The absence of Elp3 in myeloid cells (Elp3fl/fl Lyz2Cre) resulted in a decreased survival of mice upon DSS administration.97 Further research will be needed to uncover the underlying mechanism.
The role of macrophage non-m6A RNA modifications in the context of cancer
The absence of Elp3 in myeloid cells also led to a decreased Wnt-driven intestinal tumor growth in Elp3fl/fl Lyz2Cre Apc+/− mice. A first hint of the underlying mechanism came from the in vitro observation that an Elp3-deficiency impairs M2 macrophage polarization, which is known to promote tumor growth. Furthermore, BMDMs treated with IL-13 alone or IL-13 in combination with IL-4 showed an increased expression of Elp3.97 However, more research is needed to fully elucidate the working mechanism.
Another RNA modification whose impact in TAMs was assessed is ac4C. This modification is found in most eukaryotic and prokaryotic RNAs. Currently, n-acetyltransferase 10 (NAT10) is the only known ac4C writer protein, producing ac4C marks on multiple RNA sites. To study the role of Nat10 in macrophages, remodelin hydrobromide (REM), an inhibitor of NAT10, was used. Bulk RNA sequencing performed on BMDMs treated with REM, as well as IFN-γ, demonstrated that REM treatment promoted BMDM survival, downregulated antigen processing and presentation, and upregulated genes linked to the HIF-1 signaling pathway. Furthermore, RIP sequencing of BMDMs treated with IFN-γ demonstrated the presence of ac4C modifications on the Cd274 (PD-L1) mRNA, which is known to suppress its protein expression. Hence, the inhibition of NAT10 via REM is responsible for upregulating PD-L1 on myeloid cells, resulting in a reduced tumor cell death.98
Outlook and future directions
Presently, targeting RNA modifications in macrophages as a treatment option remains in the theoretical stage, and there are no clinical application examples yet. RNA modifications may influence almost all types of RNA, and targeting these RNA modifications may cause a wide range of adverse effects. Therefore, a systematic analysis of the various RNA modifications that could play a role in macrophages during various diseases is mandatory. However, at present, the vast majority of the research is directed at unveiling the role of m6A in shaping the phenotype of disease-associated macrophages as m6A is considered as one of the most abundant RNA modifications in eukaryotic mRNAs. Moreover, the actors regulating m6A have been fairly well characterized, and thus, several fl/fl mice are available to study the role of these actors in specific cell types. The knowledge acquired on m6A was made possible thanks to transcriptome-wide m6A mapping, which was developed in 2012 and was called MeRIP-seq or m6A-seq.99,100 Nevertheless, the knowledge on m6A and its effects on the phenotype of macrophages is still rudimentary; many findings are contradictory as presented in this review. Furthermore, as novel technologies become more available, the importance of other RNA modifications in regulating the phenotype of macrophages should see the light of day. However, investigating the role of other RNA modifications is currently not straightforward due to the low prevalence of these modifications and the fact that the implicated enzymes are not specific to one RNA class. Indeed, many writers can modify both mRNA and tRNA, representing a serious challenge when studying the role of RNA modifications. However, this challenge can be overcome. Indeed, in the context of the m7G RNA modification, Zhang et al.101 demonstrated that METTL1 acted as a methyltransferase for both tRNAs and mRNAs, but the functions of METTL1-mediated internal m7G mRNA methylation could be investigated independently of the functions of METTL1-mediated m7G tRNA methylation. These results demonstrated that it is possible to study enzymes regulating RNA modifications, which are not specific to one RNA class, which should allow scientists to develop a better understanding of the role of other RNA modifications in macrophage biology.
Many questions are left to be addressed to fully understand the impact of RNA modifications on macrophage biology:
First, is the role of RNA modifications in macrophages dependent on the organ as well as the ontogeny of the macrophage? Indeed, most of the current research made use of the Lyz2-Cre strain to invalidate RNA-modifying genes, and most scientists are studying the role of one RNA-modifying gene independently of the ontogeny or the localization of macrophages. As more and more cre-deleter strains become available to target specific populations of (tissue-resident) macrophages, it will be necessary to go beyond Lyz2-Cre. For example, the targeting of brain-resident microglia is made possible thanks to constitutive and inducible Cx3cr1 promoter-driven Cre lines, with the caveat that these strains may display significant leakage in neurons.102 Similarly, Sall1-Cre and Cd11b-Cre strains can target microglia, but recombination in other brain cells was observed.103,104 Faust et al.103 compared five tamoxifen-inducible cre lines that are often used to study microglia: Cx3cr1-YFP-CreER, Cx3cr1-CreER, Tmem119-CreER, Hexb-CreER, and P2ry12-CreER. It was shown that Tmem119-CreER, P2ry12-CreER, and Hexb-CreER were less efficient deleters, but were more specific to microglia than the Cx3cr1-CreER lines.103 To solve these problems, the team of Steffen Jung developed a binary Cre approach (ncre and ccre, each driven by a different promoter, need to combine to yield a functional Cre recombinase), allowing the specific targeting of microglia using Sall1-ncre, Cx3cr1-ccre mice and the specific targeting of a subset of perivascular macrophages using Cx3cr1-ccre, and Lyve1-ncre mice.105 To unravel the role of RNA modifications in liver macrophages, the Clec4f-Cre line is available to target KCs.106 Clec4f expression is highly specific for mouse KCs, making this Cre line a highly suitable tool. There is also the possibility of investigating the role of AMs using the Itgax-Cre strain, although Itgax is also expressed by other cell types, such as dendritic cells.106 Hence, the identification of markers that are uniquely expressed by tissue-resident macrophages in the different organs is an ongoing effort that, together with novel binary Cre approaches (needing the identification of two markers that, combined, are unique to a certain tissue-resident macrophage), should enable the development of more specific Cre strains. Of note, beside differences between macrophages residing in distinct tissues, there could also be differences at the single cell level, even within the same tissue. Interestingly, at least in the context of m6A, the detection of m6A-modified transcripts is possible at single-cell and even single-molecule resolution.107 Second, it will be important to define the best strategy to target RNA modifications specifically in macrophages without affecting other cells. Third, it remains to be determined whether the writers, readers, and erasers regulating the same modification also affect the phenotype of macrophages in the same way or not. Fourth, it remains to be seen whether these discoveries in the mouse can be translated to humans. Initial proof could come from recapitulating the animal findings with human peripheral blood monocyte-derived macrophages, in which writers, readers or erasers could be invalidated by various approaches (CRISPR-Cas9, siRNA, etc.) or by using mice with a humanized immune system. Ultimate proof will come from investigating RNA modifications in macrophages from clinical samples. Fifth, once drugs that specifically target RNA-modifying proteins become available, combinations with standard-of-care treatments will have to be defined in various diseases. In that case, it will need to be assessed whether the systemic administration of such drugs can be tolerated, or whether a targeted delivery, for example, to macrophages, will be required. Current approaches, such as antibody or nanobody-drug conjugates, could be suitable to achieve this aim. Addressing these questions will certainly help to broaden the current understanding of RNA modifications and their role in macrophage biology, allowing for the development of new therapeutic drugs.
Acknowledgments
C.H., M.S.G., and J.V.G. are supported by FWO-EOS G0J5112N (EOS ID: 40007564).
Author contributions
The manuscript was written by C.H. and M.S.G. and revised by J.V.G. All authors contributed to the article and approved the submitted version.
Declaration of interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1.Watanabe S., Alexander M., Misharin A.V., Budinger G.R.S. The role of macrophages in the resolution of inflammation. J. Clin. Invest. 2019;129:2619–2628. doi: 10.1172/JCI124615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mills C.D., Kincaid K., Alt J.M., Heilman M.J., Hill A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000;164:6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 3.Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., Gordon S., Hamilton J.A., Ivashkiv L.B., Lawrence T., et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xue J., Schmidt S.V., Sander J., Draffehn A., Krebs W., Quester I., De Nardo D., Gohel T.D., Emde M., Schmidleithner L., et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ginhoux F., Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity. 2016;44:439–449. doi: 10.1016/j.immuni.2016.02.024. [DOI] [PubMed] [Google Scholar]
- 6.De Vlaminck K., Van Hove H., Kancheva D., Scheyltjens I., Pombo Antunes A.R., Bastos J., Vara-Perez M., Ali L., Mampay M., Deneyer L., et al. Differential plasticity and fate of brain-resident and recruited macrophages during the onset and resolution of neuroinflammation. Immunity. 2022;55:2085–2102.e9. doi: 10.1016/j.immuni.2022.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Musrati M.A., Stijlemans B., Azouz A., Kancheva D., Mesbahi S., Hadadi E., Lebegge E., Ali L., De Vlaminck K., Scheyltjens I., et al. Infection history imprints prolonged changes to the epigenome, transcriptome and function of Kupffer cells. J. Hepatol. 2024 doi: 10.1016/j.jhep.2024.07.007. [DOI] [PubMed] [Google Scholar]
- 8.Boccaletto P., Stefaniak F., Ray A., Cappannini A., Mukherjee S., Purta E., Kurkowska M., Shirvanizadeh N., Destefanis E., Groza P., et al. MODOMICS: a database of RNA modification pathways. 2021 update. Nucleic Acids Res. 2022;50:D231–D235. doi: 10.1093/nar/gkab1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delaunay S., Helm M., Frye M. RNA modifications in physiology and disease: towards clinical applications. Nat. Rev. Genet. 2024;25:104–122. doi: 10.1038/s41576-023-00645-2. [DOI] [PubMed] [Google Scholar]
- 10.Jiang X., Liu B., Nie Z., Duan L., Xiong Q., Jin Z., Yang C., Chen Y. The role of m6A modification in the biological functions and diseases. Signal Transduct. Targeted Ther. 2021;6:74. doi: 10.1038/s41392-020-00450-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gan L., Zhao Y., Fu Y., Chen Q. The potential role of m6A modifications on immune cells and immunotherapy. Biomed. Pharmacother. 2023;160 doi: 10.1016/j.biopha.2023.114343. [DOI] [PubMed] [Google Scholar]
- 12.Boulias K., Greer E.L. Biological roles of adenine methylation in RNA. Nat. Rev. Genet. 2023;24:143–160. doi: 10.1038/s41576-022-00534-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwala S.D., Blitzblau H.G., Hochwagen A., Fink G.R. RNA Methylation by the MIS Complex Regulates a Cell Fate Decision in Yeast. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwartz S., Mumbach M.R., Jovanovic M., Wang T., Maciag K., Bushkin G.G., Mertins P., Ter-Ovanesyan D., Habib N., Cacchiarelli D., et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Rep. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patil D.P., Chen C.-K., Pickering B.F., Chow A., Jackson C., Guttman M., Jaffrey S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–373. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen J., Lv R., Ma H., Shen H., He C., Wang J., Jiao F., Liu H., Yang P., Tan L., et al. Zc3h13 Regulates Nuclear RNA m6A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol. Cell. 2018;69:1028–1038.e6. doi: 10.1016/j.molcel.2018.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bawankar P., Lence T., Paolantoni C., Haussmann I.U., Kazlauskiene M., Jacob D., Heidelberger J.B., Richter F.M., Nallasivan M.P., Morin V., et al. Hakai is required for stabilization of core components of the m6A mRNA methylation machinery. Nat. Commun. 2021;12:3778. doi: 10.1038/s41467-021-23892-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pendleton K.E., Chen B., Liu K., Hunter O.V., Xie Y., Tu B.P., Conrad N.K. The U6 snRNA m 6 A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 2017;169:824–835.e14. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma H., Wang X., Cai J., Dai Q., Natchiar S.K., Lv R., Chen K., Lu Z., Chen H., Shi Y.G., et al. N6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 2019;15:88–94. doi: 10.1038/s41589-018-0184-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ignatova V.V., Stolz P., Kaiser S., Gustafsson T.H., Lastres P.R., Sanz-Moreno A., Cho Y.-L., Amarie O.V., Aguilar-Pimentel A., Klein-Rodewald T., et al. The rRNA m 6 A methyltransferase METTL5 is involved in pluripotency and developmental programs. Genes Dev. 2020;34:715–729. doi: 10.1101/gad.333369.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Tran N., Ernst F.G.M., Hawley B.R., Zorbas C., Ulryck N., Hackert P., Bohnsack K.E., Bohnsack M.T., Jaffrey S.R., Graille M., Lafontaine D.L.J. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47:7719–7733. doi: 10.1093/nar/gkz619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.-M., Li C.J., Vågbø C.B., Shi Y., Wang W.-L., Song S.-H., et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao W., Adhikari S., Dahal U., Chen Y.-S., Hao Y.-J., Sun B.-F., Sun H.-Y., Li A., Ping X.-L., Lai W.-Y., et al. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Roundtree I.A., Luo G.-Z., Zhang Z., Wang X., Zhou T., Cui Y., Sha J., Huang X., Guerrero L., Xie P., et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. Elife. 2017;6 doi: 10.7554/eLife.31311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H., Weng H., Sun W., Qin X., Shi H., Wu H., Zhao B.S., Mesquita A., Liu C., Yuan C.L., et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018;20:285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O., Pestova T.V., Qian S.-B., Jaffrey S.R. 5′ UTR m6A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu P.J., Zhu Y., Ma H., Guo Y., Shi X., Liu Y., Qi M., Lu Z., Shi H., Wang J., et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–1127. doi: 10.1038/cr.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alarcón C.R., Goodarzi H., Lee H., Liu X., Tavazoie S., Tavazoie S.F. HNRNPA2B1 Is a Mediator of m6A-Dependent Nuclear RNA Processing Events. Cell. 2015;162:1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lasman L., Krupalnik V., Viukov S., Mor N., Aguilera-Castrejon A., Schneir D., Bayerl J., Mizrahi O., Peles S., Tawil S., et al. Context-dependent functional compensation between Ythdf m 6 A reader proteins. Genes Dev. 2020;34:1373–1391. doi: 10.1101/gad.340695.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi H., Wang X., Lu Z., Zhao B.S., Ma H., Hsu P.J., Liu C., He C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017;27:315–328. doi: 10.1038/cr.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang F., Kang Y., Wang M., Li Y., Xu T., Yang W., Song H., Wu H., Shu Q., Jin P. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 2018;27:3936–3950. doi: 10.1093/hmg/ddy292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Hove H., Martens L., Scheyltjens I., De Vlaminck K., Pombo Antunes A.R., De Prijck S., Vandamme N., De Schepper S., Van Isterdael G., Scott C.L., et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat. Neurosci. 2019;22:1021–1035. doi: 10.1038/s41593-019-0393-4. [DOI] [PubMed] [Google Scholar]
- 34.Jordão M.J.C., Sankowski R., Brendecke S.M., Sagar, Locatelli G., Tai Y.-H., Tay T.L., Schramm E., Armbruster S., Hagemeyer N., et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 2019;363 doi: 10.1126/science.aat7554. [DOI] [PubMed] [Google Scholar]
- 35.Pombo Antunes A.R., Scheyltjens I., Lodi F., Messiaen J., Antoranz A., Duerinck J., Kancheva D., Martens L., De Vlaminck K., Van Hove H., et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021;24:595–610. doi: 10.1038/s41593-020-00789-y. [DOI] [PubMed] [Google Scholar]
- 36.Munro D.A.D., Movahedi K., Priller J. Macrophage compartmentalization in the brain and cerebrospinal fluid system. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abk0391. [DOI] [PubMed] [Google Scholar]
- 37.Masuda T., Sankowski R., Staszewski O., Böttcher C., Amann L., Sagar, Scheiwe C., Nessler S., Kunz P., van Loo G., et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–392. doi: 10.1038/s41586-019-0924-x. [DOI] [PubMed] [Google Scholar]
- 38.Prater K.E., Green K.J., Mamde S., Sun W., Cochoit A., Smith C.L., Chiou K.L., Heath L., Rose S.E., Wiley J., et al. Human microglia show unique transcriptional changes in Alzheimer’s disease. Nat. Aging. 2023;3:894–907. doi: 10.1038/s43587-023-00424-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yin H., Ju Z., Zheng M., Zhang X., Zuo W., Wang Y., Ding X., Zhang X., Peng Y., Li J., et al. Loss of the m6A methyltransferase METTL3 in monocyte-derived macrophages ameliorates Alzheimer’s disease pathology in mice. PLoS Biol. 2023;21 doi: 10.1371/journal.pbio.3002017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y., Li J., Yu Q., Ji L., Peng B. METTL14 regulates microglia/macrophage polarization and NLRP3 inflammasome activation after ischemic stroke by the KAT3B-STING axis. Neurobiol. Dis. 2023;185 doi: 10.1016/j.nbd.2023.106253. [DOI] [PubMed] [Google Scholar]
- 41.He S., Li W., Wang G., Wang X., Fan W., Zhang Z., Li N., Hou S. FTO-mediated m6A modification alleviates autoimmune uveitis by regulating microglia phenotypes via the GPC4/TLR4/NF-κB signaling axis. Genes Dis. 2023;10:2179–2193. doi: 10.1016/j.gendis.2022.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musrati M.A., De Baetselier P., Movahedi K., Van Ginderachter J.A. Ontogeny, functions and reprogramming of Kupffer cells upon infectious disease. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1238452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guilliams M., Scott C.L. Liver macrophages in health and disease. Immunity. 2022;55:1515–1529. doi: 10.1016/j.immuni.2022.08.002. [DOI] [PubMed] [Google Scholar]
- 44.Remmerie A., Scott C.L. Macrophages and lipid metabolism. Cell. Immunol. 2018;330:27–42. doi: 10.1016/j.cellimm.2018.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scott C.L., Guilliams M. The role of Kupffer cells in hepatic iron and lipid metabolism. J. Hepatol. 2018;69:1197–1199. doi: 10.1016/j.jhep.2018.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deppermann C., Kratofil R.M., Peiseler M., David B.A., Zindel J., Castanheira F.V.E.S., van der Wal F., Carestia A., Jenne C.N., Marth J.D., Kubes P. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 2020;217 doi: 10.1084/jem.20190723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sierro F., Evrard M., Rizzetto S., Melino M., Mitchell A.J., Florido M., Beattie L., Walters S.B., Tay S.S., Lu B., et al. A Liver Capsular Network of Monocyte-Derived Macrophages Restricts Hepatic Dissemination of Intraperitoneal Bacteria by Neutrophil Recruitment. Immunity. 2017;47:374–388.e6. doi: 10.1016/j.immuni.2017.07.018. [DOI] [PubMed] [Google Scholar]
- 48.Guilliams M., Bonnardel J., Haest B., Vanderborght B., Wagner C., Remmerie A., Bujko A., Martens L., Thoné T., Browaeys R., et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. 2022;185:379–396.e38. doi: 10.1016/j.cell.2021.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hume D.A., Offermanns S., Bonnavion R. Contamination of isolated mouse Kupffer cells with liver sinusoidal endothelial cells. Immunity. 2022;55:1139–1140. doi: 10.1016/j.immuni.2022.06.010. [DOI] [PubMed] [Google Scholar]
- 50.Iannacone M., Blériot C., Andreata F., Ficht X., Laura C., Garcia-Manteiga J.M., Uderhardt S., Ginhoux F. Response to contamination of isolated mouse Kupffer cells with liver sinusoidal endothelial cells. Immunity. 2022;55:1141–1142. doi: 10.1016/j.immuni.2022.06.012. [DOI] [PubMed] [Google Scholar]
- 51.Blériot C., Barreby E., Dunsmore G., Ballaire R., Chakarov S., Ficht X., De Simone G., Andreata F., Fumagalli V., Guo W., et al. A subset of Kupffer cells regulates metabolism through the expression of CD36. Immunity. 2021;54:2101–2116.e6. doi: 10.1016/j.immuni.2021.08.006. [DOI] [PubMed] [Google Scholar]
- 52.De Simone G., Andreata F., Bleriot C., Fumagalli V., Laura C., Garcia-Manteiga J.M., Di Lucia P., Gilotto S., Ficht X., De Ponti F.F., et al. Identification of a Kupffer cell subset capable of reverting the T cell dysfunction induced by hepatocellular priming. Immunity. 2021;54:2089–2100.e8. doi: 10.1016/j.immuni.2021.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shu B., Zhou Y.-X., Li H., Zhang R.-Z., He C., Yang X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death Dis. 2021;7:368. doi: 10.1038/s41420-021-00756-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shu B., Zhang R.-Z., Zhou Y.-X., He C., Yang X. METTL3-mediated macrophage exosomal NEAT1 contributes to hepatic fibrosis progression through Sp1/TGF-β1/Smad signaling pathway. Cell Death Dis. 2022;8:266. doi: 10.1038/s41420-022-01036-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan X.S., Li B.-W., Wang L.-L., Li N., Lin H.-M., Zhang J., Du N., Zhu Y.-Q., Wu X., Hu C.-M., et al. Kupffer cell pyroptosis mediated by METTL3 contributes to the progression of alcoholic steatohepatitis. Faseb. J. 2023;37 doi: 10.1096/fj.202300059RR. [DOI] [PubMed] [Google Scholar]
- 56.Feng Y., Dong H., Sun B., Hu Y., Yang Y., Jia Y., Jia L., Zhong X., Zhao R. METTL3/METTL14 Transactivation and m6A-Dependent TGF-β1 Translation in Activated Kupffer Cells. Cell. Mol. Gastroenterol. Hepatol. 2021;12:839–856. doi: 10.1016/j.jcmgh.2021.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin Y., Li B., Arumugam S., Lu Q., Mankash S.M., Li J., Sun B., Li J., Flavell R.A., Li H.-B., Ouyang X. m6A mRNA methylation-directed myeloid cell activation controls progression of NAFLD and obesity. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Evren E., Ringqvist E., Doisne J.-M., Thaller A., Sleiers N., Flavell R.A., Di Santo J.P., Willinger T. CD116+ fetal precursors migrate to the perinatal lung and give rise to human alveolar macrophages. J. Exp. Med. 2022;219 doi: 10.1084/jem.20210987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aegerter H., Lambrecht B.N., Jakubzick C.V. Biology of lung macrophages in health and disease. Immunity. 2022;55:1564–1580. doi: 10.1016/j.immuni.2022.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gibbings S.L., Thomas S.M., Atif S.M., McCubbrey A.L., Desch A.N., Danhorn T., Leach S.M., Bratton D.L., Henson P.M., Janssen W.J., Jakubzick C.V. Three Unique Interstitial Macrophages in the Murine Lung at Steady State. Am. J. Respir. Cell Mol. Biol. 2017;57:66–76. doi: 10.1165/rcmb.2016-0361OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dick S.A., Wong A., Hamidzada H., Nejat S., Nechanitzky R., Vohra S., Mueller B., Zaman R., Kantores C., Aronoff L., et al. Three tissue resident macrophage subsets coexist across organs with conserved origins and life cycles. Sci. Immunol. 2022;7 doi: 10.1126/sciimmunol.abf7777. [DOI] [PubMed] [Google Scholar]
- 62.Zilionis R., Engblom C., Pfirschke C., Savova V., Zemmour D., Saatcioglu H.D., Krishnan I., Maroni G., Meyerovitz C.V., Kerwin C.M., et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity. 2019;50:1317–1334.e10. doi: 10.1016/j.immuni.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma R.-Y., Black A., Qian B.-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43:546–563. doi: 10.1016/j.it.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 64.Chen F.-W., Wu Y.-L., Cheng C.-C., Hsiao Y.-W., Chi J.-Y., Hung L.-Y., Chang C.-P., Lai M.-D., Wang J.-M. Inactivation of pentraxin 3 suppresses M2-like macrophage activity and immunosuppression in colon cancer. J. Biomed. Sci. 2024;31:10. doi: 10.1186/s12929-023-00991-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han X., Liu L., Huang S., Xiao W., Gao Y., Zhou W., Zhang C., Zheng H., Yang L., Xie X., et al. RNA m6A methylation modulates airway inflammation in allergic asthma via PTX3-dependent macrophage homeostasis. Nat. Commun. 2023;14:7328. doi: 10.1038/s41467-023-43219-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang X., Ji Y., Feng P., Liu R., Li G., Zheng J., Xue Y., Wei Y., Ji C., Chen D., Li J. The m6A Reader IGF2BP2 Regulates Macrophage Phenotypic Activation and Inflammatory Diseases by Stabilizing TSC1 and PPARγ. Adv. Sci. 2021;8 doi: 10.1002/advs.202100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Byles V., Covarrubias A.J., Ben-Sahra I., Lamming D.W., Sabatini D.M., Manning B.D., Horng T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013;4:2834. doi: 10.1038/ncomms3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu L., Yang T., Li L., Sun L., Hou Y., Hu X., Zhang L., Tian H., Zhao Q., Peng J., et al. TSC1 controls macrophage polarization to prevent inflammatory disease. Nat. Commun. 2014;5:4696. doi: 10.1038/ncomms5696. [DOI] [PubMed] [Google Scholar]
- 69.Hu L., Yu Y., Shen Y., Huang H., Lin D., Wang K., Yu Y., Li K., Cao Y., Wang Q., et al. Ythdf2 promotes pulmonary hypertension by suppressing Hmox1-dependent anti-inflammatory and antioxidant function in alveolar macrophages. Redox Biol. 2023;61 doi: 10.1016/j.redox.2023.102638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yip J.L.K., Balasuriya G.K., Spencer S.J., Hill-Yardin E.L. The Role of Intestinal Macrophages in Gastrointestinal Homeostasis: Heterogeneity and Implications in Disease. Cell. Mol. Gastroenterol. Hepatol. 2021;12:1701–1718. doi: 10.1016/j.jcmgh.2021.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delfini M., Stakenborg N., Viola M.F., Boeckxstaens G. Macrophages in the gut: Masters in multitasking. Immunity. 2022;55:1530–1548. doi: 10.1016/j.immuni.2022.08.005. [DOI] [PubMed] [Google Scholar]
- 72.De Schepper S., Verheijden S., Aguilera-Lizarraga J., Viola M.F., Boesmans W., Stakenborg N., Voytyuk I., Schmidt I., Boeckx B., Dierckx De Casterlé I., et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell. 2018;175:400–415.e13. doi: 10.1016/j.cell.2018.07.048. [DOI] [PubMed] [Google Scholar]
- 73.Viola M.F., Boeckxstaens G. Niche-specific functional heterogeneity of intestinal resident macrophages. Gut. 2021;70:1383–1395. doi: 10.1136/gutjnl-2020-323121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bonnardel J., Da Silva C., Henri S., Tamoutounour S., Chasson L., Montañana-Sanchis F., Gorvel J.-P., Lelouard H. Innate and Adaptive Immune Functions of Peyer’s Patch Monocyte-Derived Cells. Cell Rep. 2015;11:770–784. doi: 10.1016/j.celrep.2015.03.067. [DOI] [PubMed] [Google Scholar]
- 75.Ma Y., Mouton A.J., Lindsey M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018;191:15–28. doi: 10.1016/j.trsl.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Molawi K., Wolf Y., Kandalla P.K., Favret J., Hagemeyer N., Frenzel K., Pinto A.R., Klapproth K., Henri S., Malissen B., et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014;211:2151–2158. doi: 10.1084/jem.20140639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhuang T., Chen M.-H., Wu R.-X., Wang J., Hu X.-D., Meng T., Wu A.-H., Li Y., Yang Y.-F., Lei Y., et al. ALKBH5-mediated m6A modification of IL-11 drives macrophage-to-myofibroblast transition and pathological cardiac fibrosis in mice. Nat. Commun. 2024;15:1995. doi: 10.1038/s41467-024-46357-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Du J., Liao W., Liu W., Deb D.K., He L., Hsu P.J., Nguyen T., Zhang L., Bissonnette M., He C., Li Y.C. N6-Adenosine Methylation of Socs1 mRNA Is Required to Sustain the Negative Feedback Control of Macrophage Activation. Dev. Cell. 2020;55:737–753.e7. doi: 10.1016/j.devcel.2020.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cai Y., Yu R., Kong Y., Feng Z., Xu Q. METTL3 regulates LPS-induced inflammatory response via the NOD1 signaling pathway. Cell. Signal. 2022;93 doi: 10.1016/j.cellsig.2022.110283. [DOI] [PubMed] [Google Scholar]
- 80.Tong J., Wang X., Liu Y., Ren X., Wang A., Chen Z., Yao J., Mao K., Liu T., Meng F.-L., et al. Pooled CRISPR screening identifies m 6 A as a positive regulator of macrophage activation. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abd4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luo J., Wang F., Sun F., Yue T., Zhou Q., Yang C., Rong S., Yang P., Xiong F., Yu Q., et al. Targeted Inhibition of FTO Demethylase Protects Mice Against LPS-Induced Septic Shock by Suppressing NLRP3 Inflammasome. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.663295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Y., You Y., Lu Z., Yang J., Li P., Liu L., Xu H., Niu Y., Cao X. N6 -methyladenosine RNA modification–mediated cellular metabolism rewiring inhibits viral replication. Science. 2019;365:1171–1176. doi: 10.1126/science.aax4468. [DOI] [PubMed] [Google Scholar]
- 83.Li Z., Teng M., Jiang Y., Zhang L., Luo X., Liao Y., Yang B. YTHDF1 Negatively Regulates Treponema pallidum-Induced Inflammation in THP-1 Macrophages by Promoting SOCS3 Translation in an m6A-Dependent Manner. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.857727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng L., Chen X., Yin Q., Gu J., Chen J., Chen M., Zhang Y., Dong M., Jiang H., Yin N., et al. RNA-m6A modification of HDGF mediated by Mettl3 aggravates the progression of atherosclerosis by regulating macrophages polarization via energy metabolism reprogramming. Biochem. Biophys. Res. Commun. 2022;635:120–127. doi: 10.1016/j.bbrc.2022.10.032. [DOI] [PubMed] [Google Scholar]
- 85.Sun Z., Chen W., Wang Z., Wang S., Zan J., Zheng L., Zhao W. 2022. Matr3 Reshapes m6A Modification Complex to Alleviate Macrophage Inflammation during Atherosclerosis. [DOI] [PubMed] [Google Scholar]
- 86.Yildirim Z., Baboo S., Hamid S.M., Dogan A.E., Tufanli O., Robichaud S., Emerton C., Diedrich J.K., Vatandaslar H., Nikolos F., et al. Intercepting IRE1 kinase-FMRP signaling prevents atherosclerosis progression. EMBO Mol. Med. 2022;14 doi: 10.15252/emmm.202115344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Casanova-Acebes M., Dalla E., Leader A.M., LeBerichel J., Nikolic J., Morales B.M., Brown M., Chang C., Troncoso L., Chen S.T., et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;595:578–584. doi: 10.1038/s41586-021-03651-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yin H., Zhang X., Yang P., Zhang X., Peng Y., Li D., Yu Y., Wu Y., Wang Y., Zhang J., et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat. Commun. 2021;12:1394. doi: 10.1038/s41467-021-21514-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dong L., Chen C., Zhang Y., Guo P., Wang Z., Li J., Liu Y., Liu J., Chang R., Li Y., et al. The loss of RNA N6-adenosine methyltransferase Mettl14 in tumor-associated macrophages promotes CD8+ T cell dysfunction and tumor growth. Cancer Cell. 2021;39:945–957.e10. doi: 10.1016/j.ccell.2021.04.016. [DOI] [PubMed] [Google Scholar]
- 90.Xiong J., He J., Zhu J., Pan J., Liao W., Ye H., Wang H., Song Y., Du Y., Cui B., et al. Lactylation-driven METTL3-mediated RNA m6A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol. Cell. 2022;82:1660–1677.e10. doi: 10.1016/j.molcel.2022.02.033. [DOI] [PubMed] [Google Scholar]
- 91.Yankova E., Blackaby W., Albertella M., Rak J., De Braekeleer E., Tsagkogeorga G., Pilka E.S., Aspris D., Leggate D., Hendrick A.G., et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601. doi: 10.1038/s41586-021-03536-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hu J., Yang L., Peng X., Mao M., Liu X., Song J., Li H., Chen F. METTL3 promotes m6A hypermethylation of RBM14 via YTHDF1 leading to the progression of hepatocellular carcinoma. Hum. Cell. 2022;35:1838–1855. doi: 10.1007/s13577-022-00769-3. [DOI] [PubMed] [Google Scholar]
- 93.Ma S., Sun B., Duan S., Han J., Barr T., Zhang J., Bissonnette M.B., Kortylewski M., He C., Chen J., et al. YTHDF2 orchestrates tumor-associated macrophage reprogramming and controls antitumor immunity through CD8+ T cells. Nat. Immunol. 2023;24:255–266. doi: 10.1038/s41590-022-01398-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cole D.C., Chung Y., Gagnidze K., Hajdarovic K.H., Rayon-Estrada V., Harjanto D., Bigio B., Gal-Toth J., Milner T.A., McEwen B.S., et al. Loss of APOBEC1 RNA-editing function in microglia exacerbates age-related CNS pathophysiology. Proc. Natl. Acad. Sci. USA. 2017;114:13272–13277. doi: 10.1073/pnas.1710493114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rayon-Estrada V., Harjanto D., Hamilton C.E., Berchiche Y.A., Gantman E.C., Sakmar T.P., Bulloch K., Gagnidze K., Harroch S., McEwen B.S., Papavasiliou F.N. Epitranscriptomic profiling across cell types reveals associations between APOBEC1-mediated RNA editing, gene expression outcomes, and cellular function. Proc. Natl. Acad. Sci. USA. 2017;114:13296–13301. doi: 10.1073/pnas.1714227114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bernhardt A., Fehr A., Brandt S., Jerchel S., Ballhause T.M., Philipsen L., Stolze S., Geffers R., Weng H., Fischer K.-D., et al. Inflammatory cell infiltration and resolution of kidney inflammation is orchestrated by the cold-shock protein Y-box binding protein-1. Kidney Int. 2017;92:1157–1177. doi: 10.1016/j.kint.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 97.Chen D., Nemazanyy I., Peulen O., Shostak K., Xu X., Tang S.C., Wathieu C., Turchetto S., Tielens S., Nguyen L., et al. Elp3-mediated codon-dependent translation promotes mTORC2 activation and regulates macrophage polarization. EMBO J. 2022;41 doi: 10.15252/embj.2021109353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xu N., Zhuo J., Chen Y., Su R., Chen H., Zhang Z., Lian Z., Lu D., Wei X., Zheng S., et al. Downregulation of N4-acetylcytidine modification in myeloid cells attenuates immunotherapy and exacerbates hepatocellular carcinoma progression. Br. J. Cancer. 2024;130:201–212. doi: 10.1038/s41416-023-02510-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 100.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang L.-S., Liu C., Ma H., Dai Q., Sun H.-L., Luo G., Zhang Z., Zhang L., Hu L., Dong X., He C. Transcriptome-wide Mapping of Internal N7-Methylguanosine Methylome in Mammalian mRNA. Mol. Cell. 2019;74:1304–1316.e8. doi: 10.1016/j.molcel.2019.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhao X.-F., Alam M.M., Liao Y., Huang T., Mathur R., Zhu X., Huang Y. Targeting Microglia Using Cx3cr1-Cre Lines: Revisiting the Specificity. eNeuro. 2019;6 doi: 10.1523/ENEURO.0114-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Faust T.E., Feinberg P.A., O'Connor C., Kawaguchi R., Chan A., Strasburger H., Frosch M., Boyle M.A., Masuda T., Amann L., et al. A comparative analysis of microglial inducible Cre lines. Cell Rep. 2023;42 doi: 10.1016/j.celrep.2023.113031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chappell-Maor L., Kolesnikov M., Kim J.-S., Shemer A., Haimon Z., Grozovski J., Boura-Halfon S., Masuda T., Prinz M., Jung S. Comparative analysis of CreER transgenic mice for the study of brain macrophages: A case study. Eur. J. Immunol. 2020;50:353–362. doi: 10.1002/eji.201948342. [DOI] [PubMed] [Google Scholar]
- 105.Kim J.-S., Kolesnikov M., Peled-Hajaj S., Scheyltjens I., Xia Y., Trzebanski S., Haimon Z., Shemer A., Lubart A., Van Hove H., et al. A Binary Cre Transgenic Approach Dissects Microglia and CNS Border-Associated Macrophages. Immunity. 2021;54:176–190.e7. doi: 10.1016/j.immuni.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 106.Scott C.L., T'Jonck W., Martens L., Todorov H., Sichien D., Soen B., Bonnardel J., De Prijck S., Vandamme N., Cannoodt R., et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity. 2018;49:312–325.e5. doi: 10.1016/j.immuni.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kim K.L., van Galen P., Hovestadt V., Rahme G.J., Andreishcheva E.N., Shinde A., Gaskell E., Jones D.R., Shema E., Bernstein B.E. Systematic detection of m6A-modified transcripts at single-molecule and single-cell resolution. Cell Rep. Methods. 2021;1 doi: 10.1016/j.crmeth.2021.100061. [DOI] [PMC free article] [PubMed] [Google Scholar]