

Abstract
Immune effector cell (IEC) therapy is emerging as a promising approach in the field of cancer immunotherapy. Clinical IEC trials, predominantly using chimeric antigen receptor (CAR) T cells, have shown excellent responses in CD19+ B cell malignancies and multiple myeloma. In solid tumors, preclinical data are encouraging, but clinical data are in their infancy, and there are challenges in using CAR T therapy in this setting, including (1) on-target off-tumor toxicity, (2) optimal target identification, (3) effective trafficking into bulky tumor tissue, and (4) resistance to tumor immune evasion mechanisms.
Novel techniques and modifications are being explored in both the preclinical and clinical settings, aiming to improve treatment efficacy and address the aforementioned obstacles to successful CAR T therapy in solid tumors. Here we review these challenges in a clinically oriented approach and summarize published clinical trials using CAR T therapy in a variety of solid tumors.
Keywords: Immune effector cell therapy, Solid tumor, Cancer immunotherapy, Chimeric antigen receptor T cells, T cell receptor
INTRODUCTION
Over the past few years, adoptive cell therapy, also known as immune effector cell (IEC) therapy [1], has emerged as a leading technology, providing precise, immune-mediated antigen-directed therapy against cancer. Broadly, cellular immunotherapy strategies include tumor-infiltrating lymphocytes (TILs), activated tumor- and viral-specific T cells, and genetically modified T cell receptor (TCR) and chimeric antigen receptor (CAR) T cells. CAR T cells are an effective directed therapy, recently translated from the research phase of development to commercially available products for hematologic malignancies. Solid tumors are less susceptible than hematologic cancers to the classic CAR T cell therapy, and novel technologies are emerging to improve the efficacy of IEC in solid tumors.
CAR T cells are genetically modified autologous or allogeneic T cells, transfected with an engineered viral vector or electroporated with a plasmid that introduces a construct coding for a CAR into the T cell’s DNA [2]. On target recognition, the receptor activates the T cell’s cytotoxic capabilities while bypassing the costimulatory mechanisms that often inhibit the immune system’s killing of tumor cells [3]. The extracellular portion of the CAR is often composed of a single-chain variable fragment (scFv) of an antibody—the antigen recognition domain—that specifically targets the desired tumor-associated antigen (TAA). Other CAR constructs may include other antigen-recognition domains, with modified affinity to the target antigens, or targeting a spectrum of related antigens. CARs evolved from first-generation constructs that contained only the intracellular CD3 ζ moiety of the T cells, which activates the T cells through the antigen-recognition signaling pathway. However, T cells modified with first-generation CARs displayed limited in vivo proliferation and persistence [4]. Consequently, second-generation CARs were designed that added the CD28 or 4–1BB costimulatory domain into the construct, resulting in significantly improved in vivo CAR T proliferation and persistence and eventually leading to the Food and Drug Administration approval of second-generation CAR T cell therapy for leukemia and lymphoma [5–9]. Combining both CD28 and 4–1BB costimulatory domains into a third-generation CAR showed better expansion and longer persistence when infused simultaneously with second-generation CARs [10]. Although 1 patient death was reported with a third-generation CAR targeting erythroblastosis oncogene B (ERBB2) in solid tumors, presumably due to antigen recognition in the healthy lung tissue and a rapid cytokine storm [11], other third-generation CAR studies (mostly in hematologic malignancies) did not result in any patient mortality [12]. Further developments and modifications are being introduced to enhance CAR T activity while recognizing the need to avoid T cell exhaustion (Figure 1) [13].
Figure 1.
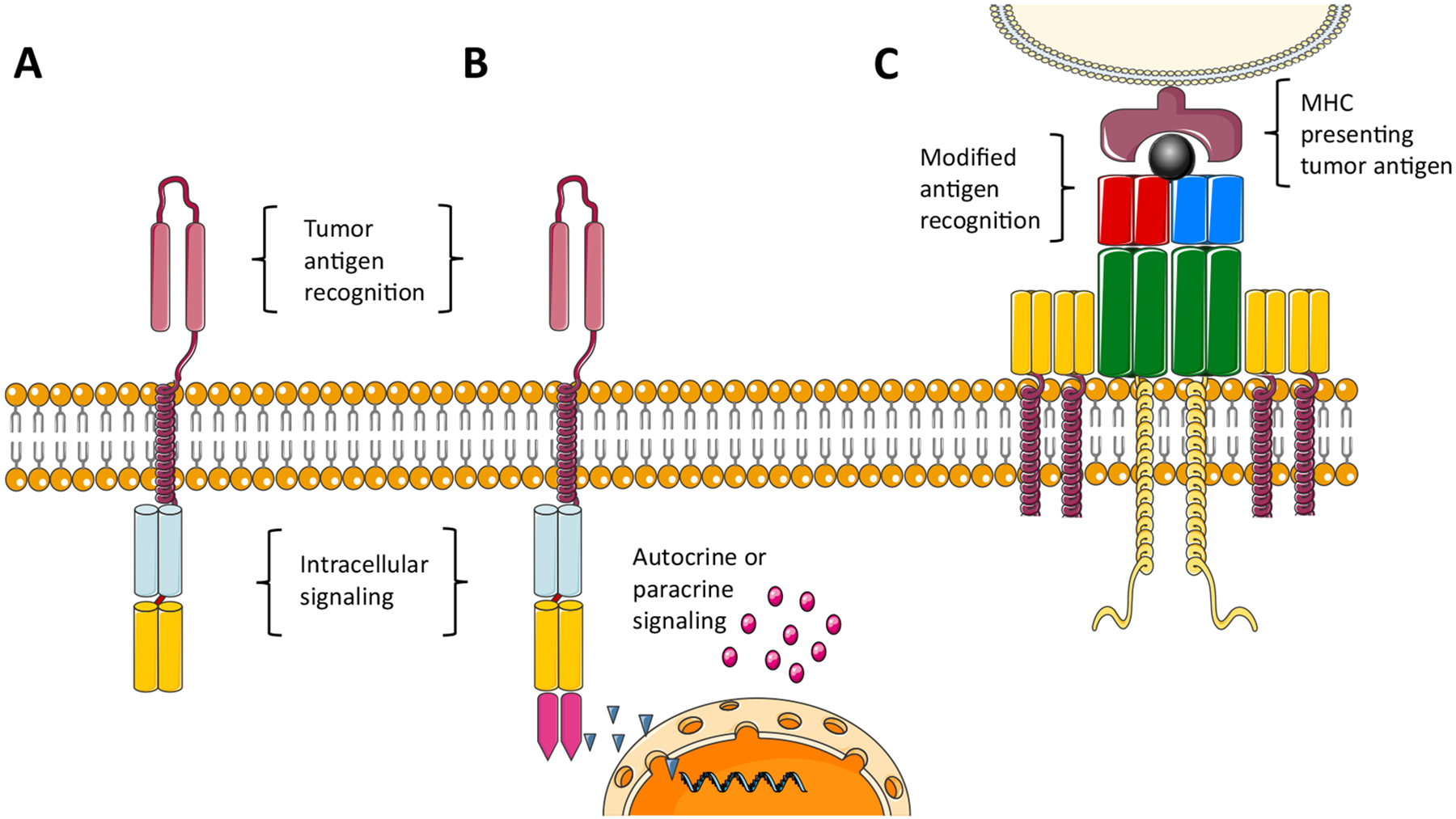
Adoptive cell therapy. (A) CAR. (B) Fourth-generation CAR coding for cytokine production. (C) Genetically modified T cell receptor targeting tumor MHC molecules presenting tumor-derived peptides. (Some elements of this figure are adapted from servier medical art under a CC 3.0 license.)
CAR T cell therapy has been more challenging for solid tumors than for hematologic malignancies. Emerging reports are showing lower response rates and few long-term remissions in preliminary IEC trials in solid tumors (Table 1) [14,15]. It appears that solid tumors pose more challenges and obstacles to the immune response, exhibiting tumor immune evasion, a hostile tumor microenvironment, and off-target toxicity due to less-specific tumor antigens. Numerous clinical trials of CARs and their efficacy in solid tumors are currently underway [16].
Table 1.
Results of Clinical Trials with CAR T Cells in Solid Tumors
| Trial | Year | Indication | Target Antigen | N* | Route | Distinctive Features | Clinical Results |
|---|---|---|---|---|---|---|---|
| Kershaw [4] | 2006 | Ovarian | α-folate receptor | 14 | IV | NA | No responses |
| Zhang [14] | 2017 | Colorectal | CEA | 10 | IV | NA | 2 patients with a PR and 7 with SD up to 30 wk |
| Thistlethwaite [15] | 2017 | CEA-positive tumors | CEA | 14 | IV | NA | No responses |
| O’Rourke [18] | 2017 | Glioblastoma | EGFR variant III | 10 | IV | NA | 1 patient with SD for 18 mo |
| Haas [19] | 2019 | Mesothelin-positive tumors | Mesothelin | 15 | IV | NA | 11 patients with SD |
| Lamers [20] | 2016 | Renal cell carcinoma | CAIX | 12 | IV | Repeated infusions | No responses |
| Feng [26] | 2018 | Biliary, pancreas | HER2 | 11 | IV | 1 or 2 infusions | 1 PR and 5 SD |
| Ahmed [27] | 2015 | Sarcoma | HER2 | 19 | IV | NA | 17 evaluable: 4 with SD for up to 14 mo |
| Ahmed [28]† | 2015 | Glioblastoma | HER2 | 16 | IV | CMV-directed cells | 1 PR and 4 SD for up to 24 mo |
| Zhan [29]† | 2019 | Gastric, pancreas | Claudin18.2 | 12 | IV | Up to 5 infusions | 11 evaluable: 1 CR, 3 PR, 5 SD |
| Specht [37]† | 2019 | Breast, triple-negative | ROR1 | 4 | IV | 1 or 2 infusions | 1 PR and 2 SD for up to 19 wk |
| Adusumilli [41]† | 2019 | Pleural tumors | Mesothelin | 20 | Intra-pleural | Local infusion; 14 patients + PD1 inhibitors | 14 with PD1 therapy: 2 CR, 5 PR, 4 SD |
| Beatty [43] | 2018 | Pancreas | Mesothelin | 6 | IV | Transient RNA expression, 9 infusions | 2 with SD for up to 5.4 mo |
| Yankelevich [44]† | 2019 | GD2-positive tumors | GD2 | 12 | IV | Chemically added surface antibodies | 1 with CR [for over 2.5 yr] and 2 PR |
| Becerra [45]† | 2019 | PSCA-positive tumors | PSCA | 15 | IV | Rimiducin-induced coactivation | 8 with SD of up to 30 wk |
| Wang [52] | 2018 | CD133-positive | CD 133 | 23 | IV | NA | 3 with PR, 14 with SD |
| Papa [64]† | 2018 | HNSCC | ErbB | 13 | Intra-tumor | Double CAR- ErbB and IL4 | Disease control rate, 69% |
| Brown [67]† | 2015 | Glioblastoma | IL13Rα2 | 3 | Intra-tumor | Up to 12 local infusions | Only transient responses were observed |
| Katz [68] | 2019 | CEA-positive tumors | CEA | 8 | Intra-arterial | Hepatic artery infusions | 2 patients with SD, good local responses |
| Goff [69] | 2019 | Glioblastoma | EGFR variant III | 18 | IV | NA | No responses |
| Heczey [73] | 2017 | Neuroblastoma | GD2 | 11 | IV | +\− PD 1 inhibitor | 5 with SD |
| Li [75]† | 2019 | Positive PD-L1 + MUC-1 tumors | MUC-1 | 13 | IV | CAR–NK + truncated PD-1 | 9 with SD |
ROR1 indicates Receptor Tyrosine Kinase Like Orphan Receptor 1; GD2, disialoganglioside; HNSCC, head and neck squamous cell carcinoma; ErbB, erythroblastosis oncogene B; MUC-1, Mucin1.
The number includes all patients reported, regardless of the dose level administered.
Abstract only.
Most CAR T trials target disease-specific antigens (Table 2), whereas others are aimed at a larger group of diseases with a shared tumor antigen (Table 3) [17]. Many of the CAR trials use enhanced CAR constructs that incorporate mechanisms for improved persistence, efficacy, or safety of the therapy. Finally, some trials use preselected T cells already specialized in viral recognition, which may be more cytotoxic when reprogrammed to target cancer cells.
Table 2.
Clinical Trials of Disease-Specific CARs Targeting Solid Tumors, as of February 5, 2020
CMV indicates cytomegalovirus.
Cells incorporating both CAR- and TCR-modified receptors.
Epstein-Barr virus/CMV/varicella zoster virus-specific T cells.
RNA electroporated autologous T cells.
Allogeneic CAR T cells.
CAR-NK cells.
Table 3.
Clinical Trials of Target-Specific CAR T Cells, as of February 5, 2020
| Cancer Type | Target Antigen | NCT Trials |
|---|---|---|
| Claudin18.2-positive solid tumors | Claudin18.2 | NCT03874897 |
| CD133-positive tumors | CD133 | NCT02541370 |
| CD70-positive solid tumors | CD70 | NCT02830724 |
| CEA-positive solid tumors | CEA | NCT02349724 |
| EGFR-positive solid tumors | EGFR | NCT03182816 |
| EPCAM-positive solid tumors | EpCAM | NCT03013712 |
| GD2-positive solid tumors | GD2 | NCT02992210 |
| HER-2-positive solid tumors | HER-2 | NCT01935843; NCT03740256; |
| NCT02713984; NCT00889954; | ||
| NCT00924287 | ||
| Lewis-Y-positive solid tumors | Lewis-Y | NCT03851146 |
| Mesothelin-positive solid tumors | Mesothelin | NCT03182803; NCT03054298; |
| NCT02580747; NCT02930993; | ||
| NCT02159716; NCT03545815; | ||
| NCT03747965; NCT03030001; | ||
| NCT01583686 | ||
| Relapsed or refractory solid tumors | NKG2DL | NCT04107142 |
| MUC1-positive solid tumors | MUC1 | NCT03179007; NCT02839954; |
| NCT04025216; NCT02617134; | ||
| NCT02587689 |
In this review, we focus on CAR T cell therapy for solid tumors and briefly touch on other cellular therapies currently under development. We report the results of recently published trials, review the present challenges to this therapy, and discuss the current efforts underway to mitigate barriers to successful therapies.
CURRENT CHALLENGES OF CAR T CELL THERAPY IN SOLID TUMORS
Immune-Mediated Toxicity and Off-Tumor Effects
The limited clinical benefit of IEC therapy in solid tumors thus far stems from inherent difficulties, related both to the tumor biology itself, as well as to the tumor’s interaction with the immune system (Figure 2). In addition to tumor-related obstacles to successful IEC therapy, the toxicity of the treatment poses a risk to patients. IEC therapy has been associated with severe toxicity in hematologic malignancies, manifesting mostly as cytokine release syndrome (CRS) and/or immune effector cell-associated neurotoxicity syndrome (ICANS). However, only mild CRS and rare ICANS have been reported thus far in early, small series of IEC trials for solid tumors [18,19]. Distinguishing local, tumor-mediated toxicity from CRS may be challenging, complicating management.
Figure 2.
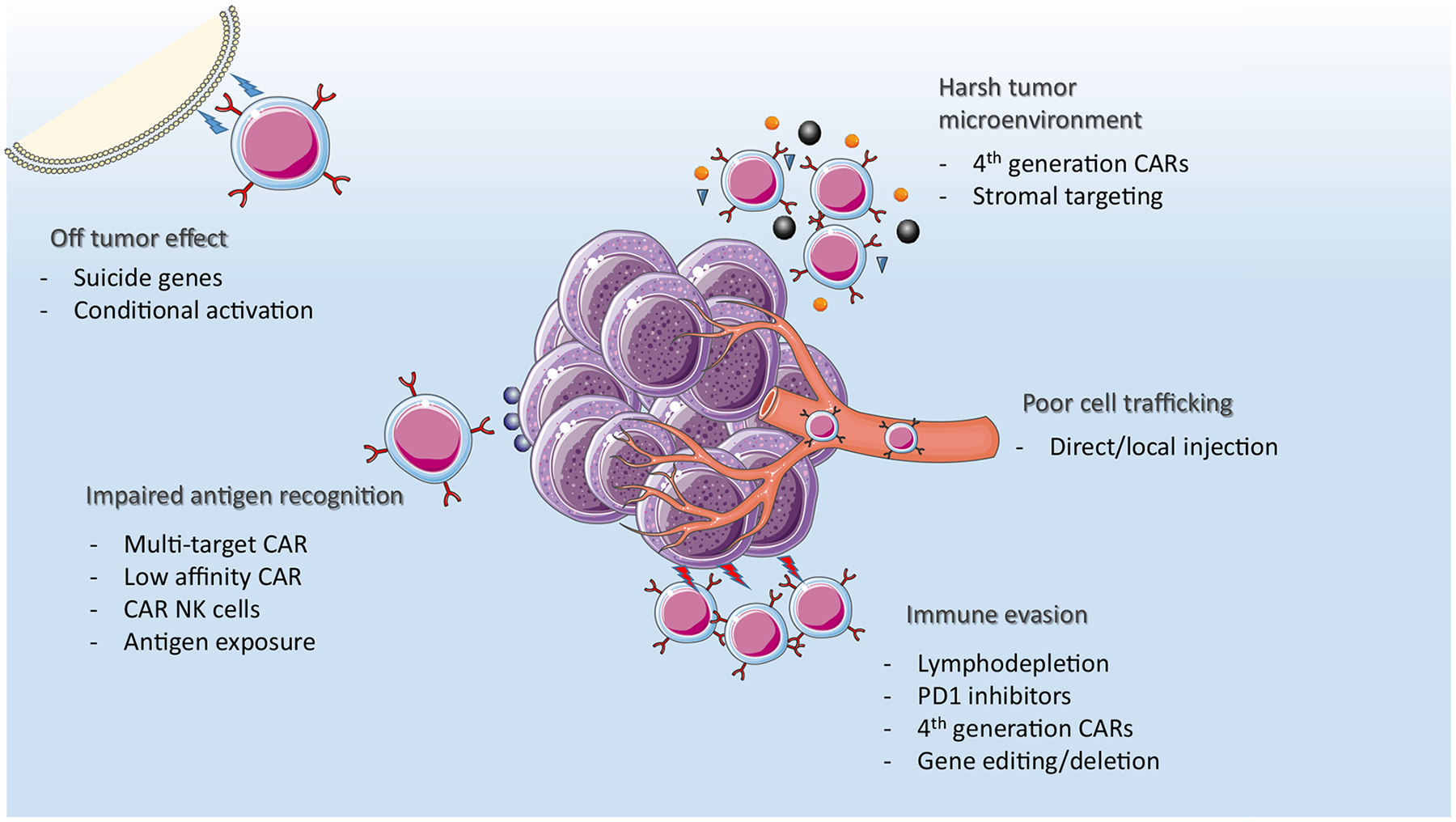
Challenges with cellular therapy in solid tumors and areas of investigation and improvements in future therapies and constructs. (Some elements of this figure are adapted from servier medical art under a CC 3.0 license.)
At times it may be more difficult to find a suitable target in solid tumors, as many of the tumor antigens are also present in healthy tissue. IEC directed at tumor antigens may present on-target off-tumor toxicity—that is, the targeting of antigens in healthy tissues that bear similarity to the tumor antigen. Serious off-tumor toxicities have been observed in solid tumors. In a study of CAR therapy targeting the carbonic anhydrase IX (CAIX) antigen in renal cell carcinoma, some patients developed severe liver toxicity, and a liver biopsy from 1 patient showed bile duct inflammation, likely caused by effector cells attacking healthy hepatic tissue expressing CAIX [20]. Another report described an immediate and fatal CRS reaction in a patient treated for lung cancer with a CAR targeting ERBB2, thought to be mediated by cytokine release owing to off-tumor target recognition in the patient’s lungs [11]. In a series of 9 patients treated with TCR-modified T cells targeting melanoma antigen gene (MAGE)-A3, 3 patients developed severe neurotoxicity, and 2 eventually succumbed to the neurologic damage. After further investigation using qRT-PCR and other methods on brain tissue samples, it was postulated that the toxicity was due to the presence of previously unrecognized MAGE family antigens expressed in the brain [21]. In a trial using an affinity-enhanced TCR against MAGE-A3 in patients with myeloma and melanoma, 2 patients had fatal cardiac toxicity caused by the T cells attacking titin, a protein found in cardiac myocytes that cross-reacts with MAGE-A3 [22]. The unexpected targeting of healthy tissues, which cannot always be detected in preclinical animal models underscores the challenge of identifying safe targets.
Tumor Target Identification
Finding a suitable target for T cell therapy is challenging in solid tumors, not only due to off-tumor effects, but also due to the intrinsic tumor biology. One approach is targeting TAAs that are produced by the cancer cells and not expressed by other healthy cells (Tables 2 and 3). These antigens are produced in cancer cells as a consequence of mutations formed during carcinogenesis causing formation of neoantigens (eg, KRAS mutations), or by viral oncogenes coding for viral proteins presented by the tumor cells (eg, E6 and E7 in human papillomavirus-associated tumors). Another mechanism for TAA formation is the abnormal transcription of silenced genes, such as MAGE antigens [23] and α-fetoprotein [24]. In addition, tumor-infiltrating T cells mount an immune response against these antigens, thus demonstrating their immunogenicity [25]. In a phase 1 study of 10 patients treated with CAR T cells directed at carcinoembryonic antigen (CEA), an antigen present in colorectal cancer cells, 2 patients had a partial response (PR) and 7 patients had stable disease (SD) [14]. In contrast, another study using a CAR aimed at CEA showed no clinical improvement, possibly owing to poor persistence of the CAR T cells [15]. ERBB2, also known as human epidermal growth factor receptor 2 (HER-2), is often expressed by pancreatic and biliary cancers and was targeted in a phase 1 CAR study, with 6 of 11 patients achieving PR or SD [26]. Another study testing a CAR targeting HER-2 in sarcoma achieved SD in 4 of 17 patients, lasting between 12 weeks and 14 months [27]. Finally, cytomegalovirus-specific cytotoxic T cells from patients with glioblastoma were transfected with a construct coding for a HER-2 CAR; among 15 patients treated, 5 demonstrated SD or a PR. Notably, these responses were durable, and 3 of these patients were reported to be alive at 18 months or longer postinfusion [28]. In a study of 11 patients treated with a CAR targeting the Claudin18.2 antigen, found in gastric and pancreatic cancers, 1 patient achieved a complete response (CR), 3 patients achieved a PR, and 5 patients had SD, with a median progression-free survival of 130 days [29].
Targeting tumor antigens can be further complicated by the fact that tumor cells often demonstrate intratumor heterogeneity in the expression of tumor-specific antigens [30]. These differences can sometimes be a phenotypic manifestation of subclones formed through mutations within the cancerous cells, due to either genetic instability or external pressure, resulting in loss of the target antigen. This has been noted following CD19-directed CAR T therapy in hematologic tumors [5] and in solid tumors, such as an epidermal growth factor receptor variant 3 (EGFRvIII) targeting CAR in glioblastomas [18]. In that study, 7 of the treated patients later underwent surgical tumor resection, and in 5 surgical specimens EGFRvIII antigen loss or decline in level of expression was observed. Moreover, some preclinical data show CAR T cells to be active only above a certain antigen density threshold, possibly deeming a heterogeneously expressing tumor cell population partially resistant to their effect [31]. In that study, approximately 200 antigen molecules per malignant cell were needed for lytic activity, whereas cytokine production by the T cells was achieved with an antigen density of approximately 2000 antigen molecules per cell. A study of EGFR expression on glioblastoma cells showed significant differences in expression levels before and after administration of chemotherapy [32], raising the question of the correct timing of cellular therapy after previous lines of treatment. Thus, the ideal target should be tumor-specific and uniformly expressed at a sufficient antigen density.
Finally, the TAA should be expressed on the tumor cell surface for optimal CAR T engagement. Although some cancer cell surface proteins are distinct and can serve as an identifiable target, other tumor-related proteins are mostly intracellular, and sometimes only displayed after cleavage inside the cell by major histocompatibility complex (MHC) molecules, making them “invisible” to chimeric receptors. The ability to identify intracellular proteins displayed only by MHC molecules may greatly enhance the IEC strategy.
Tumor Microenvironment and Cell Trafficking
Other factors that impair the immune response to IEC are inherent to the tumor microenvironment. Solid tumors often grow to a relatively large bulk. Some of the tumor regions feature harsh physiological conditions, impeding an effective immune reaction. Areas of necrosis impair immune cell growth and expansion, whereas the lack of sufficient blood supply makes the microenvironment hostile due to changes in pH, electrolytes, and, importantly, cytokines [33]. Oxygen levels are often markedly lower in tumors than in surrounding heathy tissue, yet during the initial T cell response, the cells adapt to such conditions with glycolysis, a non-oxygen-dependent metabolic pathway. However, if the microenvironment has persistent unfavorable conditions, T cells may become exhausted with mitochondrial dysfunction and up-regulated coinhibitory molecules which prevent their continued antitumor activity [34]. These conditions can also interfere with paracrine signaling to other cells that are required for efficient immune reactivity [35].
Likewise, tumor bulk and metastases, at times with relatively decreased blood supply, can impair the T cells’ ability to migrate into the tumor. In a study of the mechanisms driving the trafficking of effector cells into tumors, a lack of CD103+ dendritic cells reduced trafficking of immune cells, resulting in insufficient immune cell invasion and tumor immune escape [36].
Immune Evasion and Graft Rejection
Another obstacle hindering an effective immune response to effector cells is the tumor’s ability to evade the immune attack. T cells and other immune cells can infiltrate the tumor and even express specificity to the antigens on tumor cells, yet they may not display cytotoxic activity against them and are often dormant due to mechanisms blocking their activation. A study looking at CAR T cells targeting a surface receptor in triple-negative breast cancer found that after T cell expansion, there was an increase in inhibitory receptors on the cell surface [37]. These blocking mechanisms involve the Programmed Death 1 (PD1)-PD-L1 checkpoint pathway that is up-regulated in tumor cells and its interaction with T cells, as well as other immune-mediated tumor evasion mechanisms, such as TGF-β signaling [38]. Pancreatic cancer cells, for instance, produce IL-4, an inhibitory cytokine that interferes with immune surveillance while acting as a growth factor for tumor cells [39].
Similarly, the patients’ own immune system can attack the effector cells, causing graft rejection or cell inhibition via antibody production, effectively blocking the cells’ immune activity. This can be especially frequent for effector cells bearing chimeric receptors, specifically the murine scFv component. These are not recognized by the immune system as self and thus may stimulate an immune response against the effector cells. In a CAR T phase 1 study, T cells targeting mesothelin had good cell expansion but poor persistence, and anti-CAR antibodies were detected in 8 of 14 patients [19]. In another study, a CAR targeting tumor-associated glycoprotein (TAG)-72 in patients with liver cancer did not produce responses, and an antibody targeting the TAG-72-binding domain of the CAR was found in the patients’ blood [40]. Thus, immune rejection of the cells is also a potential limitation for allogeneic effector cells.
NOVEL STRATEGIES IN IEC THERAPY TO IMPROVE SOLID TUMOR RESPONSE
Owing to the somewhat discouraging results reported in preliminary studies of CAR T cell therapy for solid tumors, other trials are trying to mitigate some of the challenges and obstacles discussed above. Novel constructs, sometimes referred to as “next-generation” CAR T cells, include mechanisms to enhance cell activity or to mitigate factors that impair cell activity. These include genes coding for cytokines or cytokine receptors, which improve expansion and persistence. Other additions to CAR T cells target immune checkpoints, with blockade of immune inhibition or enhancement of immune interactions as mechanisms built into the CAR constructs. Although these novel constructs are mostly in preclinical stages of development, they appear to hold substantial promise for improving CAR T cell therapy in solid tumors. We present some preliminary clinical data below.
Other improvements, such as new modes of delivery and systemic adjuncts to IEC, are being explored as ways to improve the safety of treatment, as well as enhance the response to treatment and overcome the obstacles hindering T cell activity (Figure 2). Different IEC modalities, such as TCR-engineered T cells, are being developed in an effort to better target TAAs and improve the immune response against the tumors.
Toxicity Management: Suicide Genes and Conditional Activation
The main toxicities of CAR T therapy in hematologic malignancies, CRS and ICANS, are usually not observed with solid tumor IEC. The harmful and sometimes fatal complications of IEC, especially if arising from on-target off-tumor toxicity, have driven the development of novel CAR T designs incorporating “suicide genes” that can be triggered to immediately stop the cytotoxic activity and cause CAR T cell apoptosis. For example, a CAR directed at mesothelin had a I-caspase-9 safety gene incorporated in the construct, allowing for caspase-induced apoptosis in the event of severe toxicity [41]. Another method for T cell elimination is to include a truncated non-functioning receptor, such as EGFRt, that can be targeted with monoclonal antibodies administered in cases of toxicity [42]. Thus far, these techniques have yet to be used in a clinical setting, due to a lack of life-threatening toxicity.
Because CAR T cells can potentially cause prolonged suppression of cells bearing a targeted antigen, some have tried to avoid incorporating the construct into the DNA by having the cells transiently express the chimeric receptors. In a study using mRNA molecules, CARs directed at mesothelin were produced in T cells without viral transfection into the DNA. Multiple infusions were required owing to the short duration of cell persistence, and 2 out of 6 patients showed a stable disease response [43]. In another study, anti-GD2 monoclonal antibodies were chemically conjugated to autologous T cells and administered as 8 biweekly infusions. Three of 12 patients in this dose escalation study showed a response, with 1 patient in CR more than 2 years after the treatment [44].
Studies are currently underway to refine the control of T cell activity and proliferation using conditional activation, thus improving its safety. Some constructs have “on-switches,” in which a molecule given after the infusion of the cells triggers the cells to produce cytokines, proliferate, and activate. In a study targeting prostate stem cell antigen (PSCA), the construct included a rimiducin-inducible coactivation switch [45]. Seven days after cell infusion, in the absence of major toxicity, a single dose of rimiducin was administered, which caused the dimerization of a costimulatory domain, resulting in further expansion and persistence of the T cells. In a preliminary phase 1 dose expansion study, patients with PSCA-expressing tumors (mainly pancreatic, gastric, and prostate cancers) received PSCA-directed CAR T cells with a rimiducin coactivation domain. The best responses observed were SD in 8 out of 15 patients. Four of the patients remained free of further treatment for up to 30.1 weeks [45].
Some researchers have proposed preclinical models using tumor microenvironment factors as conditional “on-switches” for T cell activation, enhancing their activation within the tumor and limiting off-tumor toxicity. For instance, a preclinical model of a conditional activation CAR was designed to activate only in the low-pH tumor microenvironment, where a conformational change in the receptor renders it active [46].
Improved Tumor and Stem Cell Antigen Recognition and Targeting
Tumor heterogeneity, in both antigen expression and variation due to antigenic shift, presents another challenge to the efficacy of CAR T cells in solid tumors. Some investigators have constructed CARs that can target a range of closely related antigens, and others have designed CARs that target more than 1 defined antigen in an effort to maintain activity even in the case of antigen loss [47]. A preclinical model proposed low-dose radiation as a modality to presensitize tumor cells to immune clearance by effector cells [48].
Some have proposed using natural killer (NK) cells to attack cancer cells missing MHC domains, which are less dependent on a specific antigen and more dependent on the aberrant MHC expression of the tumor [49]. Preclinical data also hint at the possibility that NK cells may be able to target cancer stem cells, thereby preventing relapse and cancer spread [50]. Importantly, because NK cells do not recognize MHC and can be obtained from an allogeneic source, they can be offered as an “off-the-shelf” drug avoiding the burden of producing autologous product [51].
A study of CAR T cells aimed at CD133 expressed on stem cells of epithelial cancers did not show a reduction in tumor size but did seem to stabilize the disease, with a progression-free survival of 8 to 22 weeks [52].
TCR-Modified T Cell Therapy
CAR T cells can only detect antigens present on cell surfaces, and CARs often target cluster-of-differentiation molecules or receptors displayed on the cells. However, many of the aberrant cancer proteins are intracellular and expressed within the MHC complex only after proteasome cleavage inside the cells. Unlike the interaction of CAR T cells with the target cells, which involves the receptor’s scFv domain, TCR-engineered T cells interact with their target through the more physiological peptide-MHC complex formed by the target cell (Figure 1). Because cancer-specific proteins are formed in tumor cells, they are presented by this cell as part of the MHC complex; however, the patient’s cytotoxic T cells fail to recognize them. Through transfection of the patient’s T cells with a constructed TCR with high affinity to the specific cancer-associated peptide-MHC complex, the engineered T cells recognize and activate against tumor cells. Thus, TCR therapies can target both cell surface and intracellular TAAs. It is important to note, however, that these cells are restricted to the specific HLA subtype that comprises the patient’s MHC molecules.
T cells transfected with a construct coding for a TCR identifying these MHC-peptide complexes have been generated, and preliminary TCR clinical trials are currently in progress [53,54]. For instance, cells bearing an engineered TCR recognizing MAGE-A3 was given in a dose-escalation study. One patient in the dose- escalation cohort had a CR lasting 29 months, and 3 patients out of a 9-patient cohort receiving the full dose had a PR, 1 of which lasted more than 19 months [55]. A clinical phase 1 study used a vector that also coded for small interfering RNA production, silencing the endogenous TCR [56]. Silencing of the native TCR promotes improves the efficacy of the cells and avoids T cell receptor mismatching between the novel TCR and native TCRs in an effort to avoid autoimmune toxicity. In a study of 9 patients with endometrial cancer (n = 1), ovarian cancer (n = 1), melanoma (n = 3), or synovial sarcoma (n = 4) receiving TCR- modified cells targeting NY-ESO-1, 2 patients achieved a PR and 5 patients had SD [57]. A novel mechanism to silence the native TCR using the CRISPR CAS 9 editing system was recently introduced in a preclinical model. It has shown higher TCR antigen affinity than TCR-transduced cells without deletion of the native TCR [58].
TILs and Unconventional T Cells
TILs are one of the first adoptive cell therapies and have shown efficacy in solid tumors, especially melanoma [59,60]. TILs are isolated from tumors, ex vivo expanded, and returned to the patient after lymphodepleting chemotherapy. Because these cells are already partially primed against the tumor, their stimulation, usually with IL-2, leads to greater activity against the tumor. Recently, this modality has been explored in combination with checkpoint inhibition, with some patients experiencing prolonged responses [61].
Other T cells, termed unconventional T cells, are known to attack cancer cells. Unlike conventional T cells, whose TCR recognizes peptide-MHC complexes, unconventional T cells recognize other molecules on target cells, and some, such as mucosal-associated invariant T cells, have been shown to recognize and kill tumor cells [62]. Recently, a subtype of unconventional T cells recognizing MHC-related-protein 1 (MR1) has been shown to target many types of cancer cells, sparing cells from healthy tissues, and adoptive transfer of these cells’ TCR to naïve T cells grants them activity against tumor cells [63]. However, the exact peptide presented on the MR1 target is not yet known. These novel mechanisms of tumor immune targeting can lead to innovative therapeutic strategies, harnessing the immune system’s ability to recognize and kill cancer cells.
Tumor Microenvironment and Cell Trafficking
Direct infusion of modified T cells into the tumor cavity, or into involved body cavities for pleural malignancies, has been performed in an effort to achieve direct cell migration to the tumor [41,64]. In a reported case of a patient with gastric cancer, 7-methylguanosine (MG7)-directed CAR T cells were administered both systematically and into different metastatic sites, with more than 80% of the tumor mass in the metastases undergoing necrosis after 40 days of treatment [65]. In a case report of a patient with metastatic glioblastoma, CAR T cells aimed at IL-13Rα2 were injected directly into the tumor cavity and cerebral ventricles, after which the investigators noted a significant reduction in tumor size and improvement in the patient’s clinical condition that persisted for longer than 7 months [66]. However, other patients showed only transient responses to this therapy, and pretreatment and post-treatment biopsy specimens showed reduced expression of the target antigen [67]. CAR T cells injected directly into the hepatic artery, followed by local radiation, caused shrinkage of CEA-positive liver metastases, with a prolonged CR of >30 months in 1 patient [68]. However, in a study of 18 patients with glioblastoma, targeting the glioblastoma deletion variant of the EGFRvIII via direct injection into the tumor did not produce a clinical benefit [69].
Further strategies to circumvent the hostile tumor microenvironment include “fourth-generation” CAR T cells that produce interleukins and other cytokines that enhance T cell proliferation and trafficking [13,70]. These novel constructs, also called TRUCK T cells (“T cells redirected for antigen–unrestricted cytokine–initiated killing”) produce cytokines only on CAR activation, thus directing their effect on the tumor environment and avoiding the adverse effects of systemic cytokine therapy. Through production of cytokines like IL–7, IL–12, IL–15, and IL–18, they can enhance their own activity with autocrine signaling and recruit more immune cells for a more robust paracrine response [71]. Other CAR T constructs target stromal elements found in the tumor microenvironment and lead indirectly to tumor shrinkage [72].
Immune Evasion and Graft Rejection
To mitigate graft rejection, it is now common practice to administer lymphodepleting chemotherapy before cell infusion, reducing the immune rejection of the cells and prolonging their persistence. A study of PSCA-targeted CAR T cells showed improved persistence and expansion after fludarabine and cyclophosphamide lymphodepletion versus cyclophosphamide alone, and this regimen is being increasingly used in solid tumors, although the optimal dosage and administration schedule remain to be determined [45].
Tumor immune evasion is being addressed in clinical trials of strategies to disrupt the mechanisms leading to inefficient immune responses. The use of PD-1 pathway inhibitors was evaluated in a CAR T cell trial targeting GD2 on neuroblastoma cells, which found that PD-1 blockade did not increase cell expansion or persistence [73]. However, in a recent study investigating a CAR construct targeting mesothelin and containing I-caspase-9, a cohort of 14 patients received a PD-1 inhibitor after the CAR T cell therapy. Of the 14 patients receiving both CAR T cells and anti-PD-1, 2 patients achieved a CR as the best response, 5 had a PR, and 4 had SD [41]. A preclinical model has proposed gene editing to delete PD-1 molecules from the CAR T cells, making them resistant to PD-1-mediated blockade [74]. A phase 1 study using a CAR targeting Mucin-1 with a PD-1 truncated peptide found SD in 9 of 13 enrolled patients [75].
Other immune evasion mechanisms involving immune suppressing cytokines are also being explored as in the setting of IEC [13]. A CAR construct blocking the transforming growth factor β (TGF-β) pathway is currently being tested in a study with PSCA-directed CAR T cells [76], and another trial is recruiting patients with glypican 3 (GPC3)-positive tumors, where the CAR construct also includes IL-15 and IL-21 coexpression [77]. Whereas pancreatic tumors were associated with IL-4 elevation, repressing the immune reaction, a CAR was constructed to also code for a receptor with an IL-4 exodomain and an IL-7 endodomain. Thus, exposure to IL-4 causes CAR T cell expansion in response to elevated IL-4 levels, instead of the usual immune repression [39,64]. In this study, besides the IL-4-responsive receptor, the CAR T cells were constructed to target a variety of ERBB antigens, thus minimizing the possibility of cells escaping due to heterogeneous antigen expression. Furthermore, to enhance direct trafficking of cells to the tumors or the metastases, the cells were injected directly into the tumor. Disease control, with stable disease, was achieved in 69% of the cases [64].
FUTURE DIRECTIONS IN SOLID TUMOR IEC THERAPY
Numerous preclinical and early-phase trials of CAR T therapy for solid tumor malignancies are currently underway. As described above, the “classic” CAR construct used in hematologic malignancies shows limited response in solid tumors (Table 1). Modified CAR constructs developed to address some of the issues unique to solid tumors show great promise in preliminary studies, with improved CAR T persistence and tumor trafficking. However, the financial burden of CAR T cell and IEC therapy remains very high, given the high costs of development and production of the cells for each individual patient. Toward this end, “off-the-shelf” cells, such as CAR NK cells, allogeneic CAR T cells [78], and other cellular platforms, are being developed for use as allogeneic cell grafts [79]. CAR T cell and IEC therapies are in their infancy in solid tumors, but novel and promising approaches are driving the field forward.
ACKNOWLEDGMENTS
Financial disclosure:
U.G. is the recipient of a Fellowship Grant from the American Physicians Fellowship for Medicine in Israel.
Conflict of interest statement:
E.J.S. serves on the scientific advisory boards of Magenta, Novartis, Celgene, Adaptimmune, and Zelluna. K.R. has a licensing agreement with Takeda; has received educational grants from Affymed and Pharmacyclics; and serves on scientific advisory boards of Virogen, Adicet Bio, Formula Pharma, and GemoAb. G.B. reports grants from Adaptimmune, Elelixis, GlaxoSmithKline, Immatics, Immunocore, Incyte, Kite Pharma, Macrogenics, and Torque; personal fees from Clovis Oncology, Abbvie, Adicet, Amgen, Ariad, Virogin Biotech, Johnson & Johnson/Janssen, and Maverick Therapeutics; and grants and personal fees from Novartis, Bayer, AstraZeneca, Bristol-Myers Squibb, Celgene, Genetech, MedImmune, Merck, Roche, and Xcovery. D.S.H. reports research/grant funding from AbbVie, Adaptimmune, Aldi-Norte, Amgen, Astra-Zeneca, Bayer, BMS, Daiichi-Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, GSK, Ignyta, Infinity, Kite, Kyowa, Lilly, LOXO, Merck, MedImmune, Mirati, miRNA, Molecular Templates, Mologen, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, Takeda, Turning Point Therapeutics; has received reimbursement for travel, accommodations, and expenses from Bayer, Genmab, AACR, ASCO, SITC. Consulting or Advisory Role: Alpha Insights, Amgen, Axiom, Adaptimmune, Baxter, Bayer, eCancer, Genentech, GLG, Group H, Guidepoint, Infinity, Medscape, Numab, Oncology Education Project Association, Pfizer, Prime Oncology, Takeda, Trieza Therapeutics, and WebMD; and has other ownership interests in Molecular Match (advisor), OncoResponse (founder), and Presagia (advisor). P.K. has received research support from Amgen and Ziopharm; has served on advisory boards for Pfizer, Kite, and Novartis; and has received consulting fees from Jazz Pharmaceuticals.
REFERENCES
- 1.Maus MV, Nikiforow S. The why, what, and how of the new FACT standards for immune effector cells. J Immunother Cancer. 2017;5:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine BL. Performance-enhancing drugs: design and production of redirected chimeric antigen receptor (CAR) T cells. Cancer Gene Ther. 2015;22:79–84. [DOI] [PubMed] [Google Scholar]
- 3.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immuneglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster SJ, Bishop MR, Tam CS, et al. Long-term follow-up of tisagenlecleucel in adult patients with relapsed or refractory diffuse large B-cell lymphoma: updated analysis of Juliet study. Biol Blood Marrow Transplant. 2019;25:S20–S21. [Google Scholar]
- 7.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang M, Munoz J, Goy A, et al. KTE-X19, an anti-CD19 chimeric antigen receptor (CAR) T cell therapy, in patients (Pts) with relapsed/refractory mantle cell lymphoma (R/R MCL): results of the phase 2 ZUMA-2 study. Biol Blood Marrow Transplant. 2020;26:S1. [Google Scholar]
- 9.Gauthier J, Hirayama AV, Hay KA, et al. Comparison of efficacy and toxicity of CD19-specific chimeric antigen receptor T-cells alone or in combination with ibrutinib for relapsed and/or refractory CLL. Blood. 2018;132(suppl 1). 299. [Google Scholar]
- 10.Ramos CA, Rouce R, Robertson CS, et al. In vivo fate and activity of second-versus third-generation CD19-specific CAR-T cells in B cell non-Hodgkin’s lymphomas. Mol Ther. 2018;26:2727–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enblad G, Karlsson H, Gammelgård G, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. 2018;24:6185–6194. [DOI] [PubMed] [Google Scholar]
- 13.Chmielewski M, Abken H. TRUCKs with IL–18 payload: toward shaping the immune landscape for a more efficacious CAR T–cell therapy of solid cancer. Adv Cell Gene Ther. 2018;1:e7. [Google Scholar]
- 14.Zhang C, Wang Z, Yang Z, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA+ metastatic colorectal cancers. Mol Ther. 2017;25:1248–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thistlethwaite FC, Gilham DE, Guest RD, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother. 2017;66:1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/results?cond=&term=chimeric+antigen+receptor&cntry=&state=&city=&dist=]. Accessed February5, 2020.
- 17.MacKay M, Afshinnekoo E, Rub J, et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020;38:233–244. [DOI] [PubMed] [Google Scholar]
- 18.O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9. eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haas AR, Tanyi JL, O’Hara MH, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther. 2019;27:1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lamers CH, Klaver Y, Gratama JW, Sleijfer S, Debets R. Treatment of meta-static renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells—a completed study overview. Biochem Soc Trans. 2016;44:951–959. [DOI] [PubMed] [Google Scholar]
- 21.Morgan RA, Chinnasamy N, Abate-Daga DD, et al. Cancer regression and neurologic toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linette GP, Stadtmauer EA, Maus MV, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weon JL, Potts PR. The MAGE protein family and cancer. Curr Opin Cell Biol. 2015;37:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017;17:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tran E, Ahmadzadeh M, Lu YC, et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng K, Liu Y, Guo Y, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor–modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33:1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed NM, Brawley VS, Diouf O, et al. Autologous HER2 CMV bispecific CAR T cells for progressive glioblastoma: results from a phase I clinical trial. J Clin Oncol. 2015;33(15 suppl). 3008.26304901 [Google Scholar]
- 29.Zhan X, Wang B, Li Z, et al. Phase I trial of Claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. J Clin Oncol. 2019;37(15 suppl). 2509. [Google Scholar]
- 30.Chen N, Li X, Chintala NK, Tano ZE, Adusumilli PS. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Curr Opin Immunol. 2018;51:103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe K, Terakura S, Martens AC, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. 2015;194:911–920. [DOI] [PubMed] [Google Scholar]
- 32.van den Bent MJ, Gao Y, Kerkhof M, et al. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro Oncol. 2015;17:935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schurich A, Magalhaes I, Mattsson J. Metabolic regulation of CAR T cell function by the hypoxic microenvironment in solid tumors. Immunotherapy. 2019;11:335–345. [DOI] [PubMed] [Google Scholar]
- 34.Scharping NE, Delgoffe GM. Tumor microenvironment metabolism: a new checkpoint for anti-tumor immunity. Vaccines (Basel). 2016;4:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology. 2014;3: e970027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spranger S, Dai D, Horton B, Gajewski T. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31:711–723e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Specht J, Lee S, Turtle C, et al. Abstract P2-09-13: a phase I study of adoptive immunotherapy for ROR1+ advanced triple negative breast cancer (TNBC) with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res. 2019;79(4 suppl). P2-09-13. [Google Scholar]
- 38.Massagué J TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. [DOI] [PubMed] [Google Scholar]
- 39.Mohammed S, Sukumaran S, Bajgain P, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25:249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hege KM, Bergsland EK, Fisher GA, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adusumilli PS, Zauderer MG, Rusch VW, et al. Regional delivery of mesothelin-targeted CAR T cells for pleural cancers: safety and preliminary efficacy in combination with anti-PD-1 agent. J Clin Oncol. 2019;37(15 suppl):2511.31154918 [Google Scholar]
- 42.Specht JM, Lee S, Turtle C, et al. Phase I study of immunotherapy for advanced ROR1+ malignancies with autologous ROR1-specific chimeric antigen receptor-modified (CAR)-T cells. J Clin Oncol. 2018;36(5 suppl). TPS79. [Google Scholar]
- 43.Beatty GL, O’Hara MH, Lacey SF, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155:29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yankelevich M, Modak S, Chu R, et al. Phase I study of OKT3 × hu3F8 bispecific antibody (GD2Bi) armed T cells (GD2BATs) in GD2-positive tumors. J Clin Oncol. 2019;37(15 suppl). 2533. [Google Scholar]
- 45.Becerra CR, Manji GA, Kim DW, et al. Ligand-inducible, prostate stem cell antigen (PSCA)-directed GoCAR-T cells in advanced solid tumors: preliminary results with cyclophosphamide (Cy) fludarabine (Flu) lymphodepletion (LD). J Clin Oncol. 2019;37(15 suppl). 2536. [Google Scholar]
- 46.Hu J, Lopez B, Lam T, et al. Abstract 3189: CAB-CAR-T: a novel conditionally active biologics approach to minimize on-target off-tumor effects in adoptive immunotherapy. Cancer Res. 2019;79(13 suppl). 3189. [Google Scholar]
- 47.Shah NN, Zhu F, Schneider D, et al. Results of a phase I study of bispecific anti-CD19, anti-CD20 chimeric antigen receptor (CAR) modified T cells for relapsed, refractory, non-Hodgkin lymphoma. J Clin Oncol. 2019;37(15 suppl). 2510. [Google Scholar]
- 48.DeSelm C, Palomba ML, Yahalom J, et al. Low-dose radiation conditioning enables CAR T cells to mitigate antigen escape. Mol Ther. 2018;26:2542–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Souza-Fonseca-Guimaraes F, Cursons J, Huntington ND. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol. 2019;40:142–158. [DOI] [PubMed] [Google Scholar]
- 50.Tallerico R, Garofalo C, Carbone E. A new biological feature of natural killer cells: the recognition of solid tumor-derived cancer stem cells. Front Immunol. 2016;7:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Chen M, Wu Z, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7: e1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lam VK, Hong DS, Heymach J, et al. Initial safety assessment of MAGE-A10c796TCR T-cells in two clinical trials. J Clin Oncol. 2018;36(15 suppl). 3056.30183464 [Google Scholar]
- 54.Goyal L, Frigault M, Meyer T, et al. Abstract 3183: Initial safety of AFP SPEAR T-cells in patients with advanced hepatocellular carcinoma. Cancer Res. 2019;79(13 suppl):3183. [Google Scholar]
- 55.Lu YC, Parker LL, Lu T, et al. Treatment of patients with metastatic cancer using a major histocompatibility complex class II-restricted T-cell receptor targeting the cancer germline antigen MAGE-A3. J Clin Oncol. 2017;35:3322–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun Q, Zhang X, Wang L, et al. T-cell receptor gene therapy targeting melanoma-associated antigen-A4 by silencing of endogenous TCR inhibits tumor growth in mice and human. Cell Death Dis. 2019;10:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Butler MO, Saibil S, Bonilla L, et al. Effect of minimal lymphodepletion prior to ACT with TBI-1301, NY-ESO-1 specific gene-engineered TCR-T cells, on clinical responses and CRS. J Clin Oncol. 2019;37(15 suppl). 2537.31386610 [Google Scholar]
- 58.Legut M, Dolton G, Mian A, Ottmann OG, Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood. 2018;131:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. N Engl J Med. 1988;319:1676–1680. [DOI] [PubMed] [Google Scholar]
- 60.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mullinax JE, Hall M, Prabhakaran S, et al. Combination of ipilimumab and adoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic melanoma. Front Oncol. 2018;8:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gherardin NA, Loh L, Admojo L, et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci Rep. 2018;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crowther MD, Dolton G, Legut M, et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol. 2020;21:178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Papa S, Adami A, Metoudi M, et al. A phase I trial of T4 CAR T-cell immunotherapy in head and neck squamous cancer (HNSCC). J Clin Oncol. 2018;36 (15 suppl). 3046. [Google Scholar]
- 65.Yuan J CT056 Abstract: The first-in-human clinical trial of MG7-CART for metastasis colon cancer. Cancer Res. 2018;78(13 suppl). CT056. [Google Scholar]
- 66.Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375:2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Katz SC, Hardaway J, Prince E, et al. HITM-SIR: phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA+ liver metastases. Cancer Gene Ther. 2020;27:341–355. [DOI] [PubMed] [Google Scholar]
- 69.Goff SL, Morgan RA, Yang JC, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42:126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yeku OO, Purdon T, Spriggs DR, Brentjens RJ. Interleukin-12 armored chimeric antigen receptor (CAR) T cells for heterogeneous antigen-expressing ovarian cancer. J Clin Oncol. 2018;36(5 suppl). 12. [Google Scholar]
- 71.Chmielewski M, Abken H. TRUCKS, the fourth–generation CAR T cells: current developments and clinical translation. Adv Cell Gene Ther. 2020:e84. [Google Scholar]
- 72.Lo A, Wang LS, Scholler J, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75:2800–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Heczey A, Louis CU, Savoldo B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25:2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rupp LJ, Schumann K, Roybal KT, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li Q, Wang Y, Lin M, et al. Abstract A014: Phase I clinical trial with PD-1/MUC1 CAR-pNK92 immunotherapy. Cancer Immunol Res. 2019;7(2 suppl). A014. [Google Scholar]
- 76.Kloss CC, Lee J, Zhang A, et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26:1855–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Steffin DHM, Batra SA, Rathi P, et al. A phase I clinical trial using armored GPC3 CAR T cells for children with relapsed/refractory liver tumors. J Clin Oncol. 2019;37(15 suppl). TPS2647. [Google Scholar]
- 78.Van Cutsem E, Machiels J, Van den Eynde M, et al. Phase 1 studies assessing the safety and clinical activity of autologous and allogeneic NKG2D-based CAR-T therapy in metastatic colorectal cancer. Ann Oncol. 2019;30. 10.1093/annonc/mdz157.008. iv124–iv125. [DOI] [Google Scholar]
- 79.Montagner IM, Penna A, Fracasso G, et al. Anti-PSMA CAR-engineered NK-92 Cells: An Off-the-shelf Cell Therapy for Prostate Cancer. Cells. 2020;9(6):1382. 10.3390/cells9061382. [DOI] [PMC free article] [PubMed] [Google Scholar]