

Abstract
Bacteriophages are promising alternatives to traditional antimicrobial treatment of bacterial infections. To further increase the potential of phages, efficient engineering methods are needed. This study investigates an approach to phage engineering based on phage rebooting and compares selected methods of assembly and rebooting of phage genomes. GG assembly of phage genomes and subsequent rebooting by cell-free transcription–translation reactions yielded the most efficient phage engineering and allowed production of a proof-of-concept T7 phage library of 1.8 × 107 phages. We obtained 7.5 × 106 different phages, demonstrating generation of large and diverse libraries suitable for high-throughput screening of mutant phenotypes. Implementing versatile and high-throughput phage engineering methods allows vastly accelerated and improved phage engineering, bringing us closer to applying effective phages to treat infections in the clinic.
Keywords: phage, rebooting, DNA assembly, TXTL reaction, phage library
Graphical Abstract
1. Introduction
The antimicrobial resistance crisis will have drastic consequences if alternative treatments for bacterial infections are not found [1]. Infections by antimicrobial-resistant bacteria are already a major cause of death across the world and may soon entail that currently treatable infections routinely become life-threatening [2].
Phage therapy is a promising option to alleviate the consequences of widespread antimicrobial resistance [3–5]. Phage therapy harnesses bacteriophages (also called phages) to eliminate bacterial infections. While the interest in phage therapy diminished following the rise of the antibiotic era, phages are now reemerging as potential therapeutics [6]. Lytic phages are generally favored for this application, as the lytic life cycle ensures minimal bacterial survival [7]. Several clinical trials investigating the use of phage therapy have been conducted, and are currently being conducted, with results ranging from promising to inconclusive (reviewed in [4, 7]). Part of the inconsistent efficacy of phage therapy can be attributed to limited host range and resistance development, both common occurrences when using wildtype (wt) phages [8].
The effectiveness of phages can be enhanced through engineering, improving the potency of phage therapy. Phage engineering has been successfully applied to improve phages in vitro resulting in increased host range, more efficient killing of target cells, and reduced resistance development [9–11].
While phage engineering is very beneficial, the process is often slow and time-consuming which poses a barrier to developing phage-based medicines [9]. Engineering is frequently complicated by the lytic life cycle, a large genome size, phage gene toxicity, and difficulties when introducing multiple or large modifications [12]. Traditional methods for engineering lytic phages rely on homologues recombination between template DNA and phage DNA during infection of host cells; an approach that requires a rigorous screening process to select a correct recombinant phage [9, 10]. The efficacy of engineering by homologous recombination is low, as lytic phages do not reside long in the host cell and recombination occurs at low frequencies [10]. Although the success rate has been improved through various strategies, including co-introduction of selection markers, CRISPR-Cas-mediated counterselection, and expression of exogenous recombination genes, extensive screening of engineered phages is still necessary [9, 10].
Phage rebooting is an increasingly popular alternative engineering method [12–18] that can overcome some of the limitations of engineering strategies based on homologous recombination [12]. Phage rebooting is conducted by amplifying a phage genome in smaller fragments by PCR, introducing modifications, and re-assembling the fragments into a complete genome. The assembled genome is then reactivated (or ‘rebooted’), allowing production of phage particles from the engineered phage genome. Various methods for both assembling and rebooting phage genomes have been reported, each with unique advantages (reviewed in [9, 10]). In general, phage rebooting requires minimal screening of produced phages; any incorrectly assembled phage would likely lack essential elements and thus be unable to form viable phages during rebooting [19]. Another advantage is the ability to use synthesized DNA fragments in assemblies, mitigating the need for physically obtaining a wt phage of interest. Lastly, rebooting allows extensive engineering throughout the phage genome in a single round of engineering [12].
In this work, we investigated the applicability of two recent developments within phage rebooting: genome assembly by Golden Gate (GG) and rebooting using cell-free transcription–translation (TXTL) reactions. While both methods have previously been used for phage engineering and rebooting with promising results [18–22], the benefits of combining these methods have not been investigated in depth. In this study, we found that combining GG assembly and subsequent TXTL rebooting is advantageous compared to current state-of-the-art methods. We further demonstrated that diverse phage libraries of more than 107 phages can be created through GG assembly and TXTL rebooting.
2. Materials and methods
2.1. Growth conditions
Escherichia coli was grown at 37°C in lysogeny broth (LB) or on LB agar plates. A total of 10 mM MgCl2 and 5 mM CaCl2 were added when working with phages. Plaque-forming units (PFU)/ml was determined by phage titration, where diluted phage lysates were spotted in technical triplicates of 15 μl on overlays of LB top agar (0.4% agar) containing bacterial culture. Both wt and rebooted phages were propagated and titrated on the restriction-negative standard cloning strain NEB 10-beta (New England Biolabs, NEB). Yeast cells were grown in liquid media and on agar plates based on either yeast extract–peptone–dextrose media or synthetic complete dropout media lacking uracil.
2.2. DNA manipulation
PCRs were conducted using either Platinum SuperFi II DNA Polymerase (Thermo Fisher Scientific) or DreamTaq Green PCR Master Mix (2X) (Thermo Fisher Scientific). PCR products were visually examined after agarose gel electrophoresis and either purified directly by QIAquick PCR Purification Kit (Qiagen) or purified through gel extraction by use of QIAquick Gel Extraction Kit (Qiagen). DNA concentrations were determined by analysis via NanoDrop One. Sanger sequencing was conducted by Eurofins Genomics. All primers are listed in Supplementary Tables S1–S6. All phage DNA fragments were created using commercially available T7 DNA as a template (BIORON Life Science).
2.3. Yeast assembly
Phage assembly in yeast cells was achieved largely as described in Ando et al. [12] using the uracil auxotrophic Saccharomyces cerevisiae strain BY4741. The yeast artificial chromosome (YAC) backbone of 2.4 kb contained the metabolic marker (URA3), the CEN6 centromere, and the ARS4 replicon from pBK416 [23]. In all, 50 ng of YAC backbone and a molar ratio of 1:6 between backbone and phage fragments were used during assembly. Transformation of yeast cells was achieved by the lithium acetate-PEG4000-ssDNA method described by Gietz and Woods [24]. Cells were plated on selective plates lacking uracil followed by 2 days of incubation at 30°C. Assemblies were extracted using YeaStar™ Genomic DNA Kit (Zymo Research) following the manufacturer’s protocol I.
2.4. Golden Gate assembly
The T7 phage genome was split into fragments by PCR using primers tailored for scarless GG assembly by AarI. A total of 0.2 pmol of each fragment was used during assemblies which were conducted as described by Andreou and Nakayama [25], using 10–96 cycles of 37°C for 2.5 min and 16°C for 5 min, followed by 30 min incubation at 50°C and 5 min inactivation at 80°C. Fast GG engineering of phage genomes can be achieved using 10 cycles of restriction and ligation, but we used 96 cycles for quantification of rebooting efficiency and library creation as described in Pryor et al. [19].
The GG-assembled DNA was cleaned by isopropanol precipitation before DNA quantification and rebooting. The GG reaction was diluted to 100 μl in nuclease-free water and precipitated by 1 volume isopropanol and 0.1 volume 3 M sodium acetate. The mixture was incubated for 20 min at −20°C before pelleting, two washes in ethanol, and resuspension in nuclease-free water.
Control GG reactions with no addition of enzymes were included to ensure no contamination of intact template DNA in the assembled DNA. No phages were produced during rebooting by transformation or TXTL reactions, indicating that all rebooting phages originate from the assembly. Sequencing of random plaques was also used to ensure the presence of the mutations introduced during assembly.
2.5. Rebooting by transformation
Phage DNA was used to transform chemically competent E. coli cells created by rubidium chloride as described by Green and Rogers [26]. Different volumes of the mixture were added to 50 μl overnight culture of the host strain immediately after transformation, mixed with 3 ml top agar, and poured on LB plates for PFU determination. Transformation was conducted using chemically competent E. coli cells due to higher reliability and higher total PFU production when compared to transformation by electroporation (Supplementary Table S9).
2.6. Rebooting by TXTL reactions
In vitro rebooting of phage DNA was achieved using myTXTL Linear DNA Expression Kits (Arbor Biosciences). Reactions were conducted following the manufacturer’s recommendations and the description by Shin et al. [21]. No dNTPs were supplied to avoid replication of phage DNA. A total of 0.25 nM phage DNA was added to the TXTL reaction alongside PEG 8000 to a final concentration of 2%. Mixtures were incubated overnight at 29°C in 1.5 ml microcentrifuge tubes, after which the produced phages could be propagated or titrated as for wt phages.
2.7. Construction and sequencing of barcoded phage library
A barcode of 15 random nucleotides (see Supplementary data for barcode structure and insertion site) was added to a primer during amplification of DNA fragments for GG assembly. The barcoded T7 construct was assembled by GG using 96 cycles, cleaned, and subsequently rebooted by TXTL. The resulting 12 μl TXTL-rebooted phages were added to 6 ml media and 50 μl was removed for phage titration. The removed volume for titration corresponds to 0.8% of the undiluted TXTL reaction and entails minimal loss of barcoded phages. The remaining phages were amplified by overnight infection of NEB 10-beta. DNA was extracted from the entire phage lysate and sent for Illumina sequencing at BaseClear where the barcode-containing region was amplified in an Illumina 2-step PCR prep and sequenced on Illumina NovaSeq 6000.
2.8. Barcode extraction from sequenced phage library
Sequencing reads were converted to FASTA format without trimming due to high sequencing quality. The 10 bases flanking the barcodes on each side were referred to as starting and terminating sequences. Upon identification of a starting and terminating sequence in correct order, the region between these sequences was returned as a barcode. See Supplementary data and Fig. S1 for barcode extraction when no exact matches for starting and terminating sequences could be identified.
3. Results
3.1. Efficient phage genome assembly by Golden Gate
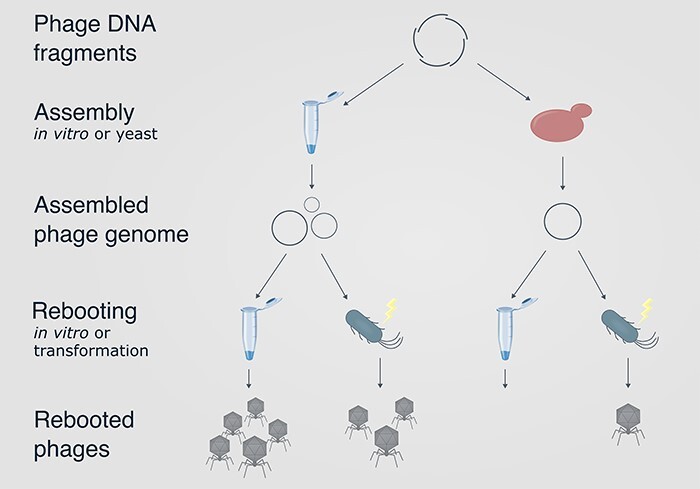
The lytic E. coli phage T7 was used as a model to evaluate the applicability of GG for phage genome assembly. DNA assembly by GG was compared to assembly in yeast cells, as yeast assembly has been extensively used for phage rebooting [9, 10]. A schematic overview of GG and yeast assembly followed by two methods of rebooting is shown in Fig. 1. Assembly by GG was conducted in a one-pot in vitro reaction, allowing creation of engineered rebooted phages in 1 day (Fig. 1). In contrast, rebooted phages were produced after 6 days when using yeast assembly, as a time-consuming process of culturing, transformation, and screening of yeast cells was required.
Figure 1.
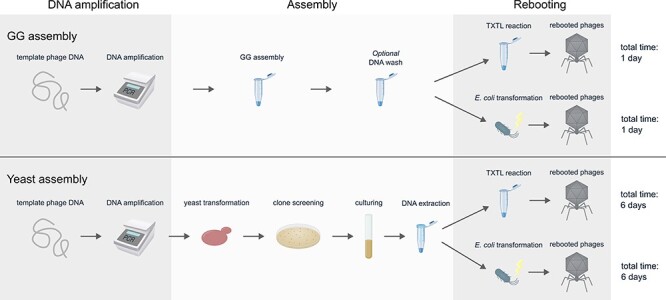
Schematic overview of the assembly process by GG (top) and yeast assembly (bottom) and subsequent rebooting. Two rebooting methods are illustrated: cell-free TXTL reactions and transformation of E. coli host cells. Microcentrifuge tubes illustrate in vitro reactions.
The GG assembly was conducted using the type IIS enzyme AarI. DNA fragments were amplified using primers containing AarI recognition sites for cloning, in addition to synonymous point mutations to remove native AarI recognition sites (Fig. 2 and Supplementary data). The T7 genome was assembled in a circular construct, despite the naturally linear topology of the phage genome, as circularized T7 phage DNA has been found to reboot more effectively than linear DNA [19].
Figure 2.

Overview of the DNA fragments used for assembly of T7 by GG and yeast cells. The wt T7 genome is shown from one direct terminal repeat to the other, indicated as black triangles. Genes of more than 500 bases are illustrated by gray boxes with points indicating orientation. Colored boxes below the wt T7 genome indicate the DNA fragments used in each assembly. Synonymous point mutations introduced in the genome during assembly are indicated by asterisks (*). The darker DNA fragment in the yeast assembly indicates the yeast artificial chromosome (YAC) element inserted between the terminal repeats. The point mutation in the yeast assembly was introduced for identification purposes. Both assemblies were created as circular constructs with fragment 1 joining the last fragment in the assembly (fragment 8 in the GG assembly and the YAC element in the yeast assembly).
Yeast assembly of T7 was achieved using DNA fragments with overlapping regions of 80 bases. To allow maintenance in yeast cells, the T7 genome was circularized and a YAC element was included, facilitating replication and selection. The YAC element was inserted between the two direct terminal repeats (Fig. 2). During maturation and packaging of the T7 genome, the phage genome is cleaved at the direct terminal repeats, and DNA is packaged from one terminal repeat to the next [27]. As a result, the YAC element placed between terminal repeats was excluded from the genome of the rebooted phages (Supplementary Fig. S2).
While phage T7 (40 kb) was used as a model in this study, both GG and yeast assembly were also applied to assemble phage lambda (46 kb) to validate the applicability of the assembly methods on other phage genomes (Supplementary data). Successful rebooting of GG- and yeast-assembled lambda genomes verified that assembly was achieved by both methods, allowing production of phages at a similar effectiveness as for T7 (Supplementary data and Tables S7 and S9). In contrast to the yeast assembly of T7, the YAC element was designed to remain within the genome of phage lambda after rebooting. No growth deficits were observed as an effect of the continued presence of the YAC element in the rebooted phages (Supplementary Table S8), indicating limited adverse effects of the inserted YAC element.
3.1.1. Comparison of the rebooting potential of DNA assembled by GG and yeast assembly.
Phage DNA assembled by GG and yeast assembly differed in rebooting frequency. T7 DNA assembled by GG required no screening of assemblies and was consistently rebooted by transformation of host cells at every attempt (data not shown). In contrast, T7 assemblies from yeast cells did not reboot reliably (Supplementary Table S9). Overall, only 5 out of 20 (25%) examined yeast colonies contained an assembly allowing production of rebooted phages, corresponding with previous reports of yeast assembly effectiveness [12]. A preliminary PCR screening for correct assembly improved the rebooting frequency to 5 out of 11 (45%, Supplementary Table S9).
To investigate how many phages were produced from the assembled phage genomes during rebooting, we rebooted (by transformation of an E. coli host) equal amounts of assembled T7 DNA and commercially available purified T7 DNA. The total number of produced phages from each reboot is shown with light gray squares in Fig. 3.
Figure 3.
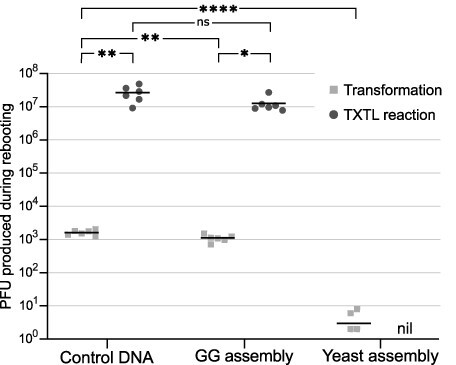
Number of functional phages (PFU) produced when rebooting by transformation (light gray squares) or cell-free TXTL reaction (dark gray dots). A total of 96 ng of commercial control T7 DNA, GG-assembled DNA, and yeast-assembled DNA were used for rebooting. Horizontal bars indicate the mean of six biological replicates; exact values are shown in Supplementary Tables S10 and S11. The limit of detection was 1 × 100 PFU. Rebooting of yeast-assembled DNA by TXTL reactions produced no phages, indicated by “nil.” Two of the replicates for TXTL rebooting of yeast-assembled DNA did not produce any phages. Significance was determined by unpaired t-test or Welch’s t-test depending on the variance between groups (Supplementary data). “ns” indicates no statistical significance, and *, **, and **** indicate statistical significance with P values below 0.05, 0.01, and 0.0001, respectively.
Rebooting of GG-assembled DNA by transformation was only 0.2 log10 (P value = .0085) less effective than rebooting of commercial T7 DNA (from 1.6 × 103 ± 0.3 × 103 PFU to 1.1 × 103 ± 0.3 × 103 PFU). The small decrease in rebooting effectiveness of GG assemblies compared to commercial DNA indicates a high prevalence of correctly assembled phage genomes in the GG assembly reaction.
In contrast, the rebooting effectiveness of yeast-assembled DNA was 2.7 log10 (P value < .0001) lower than for commercial DNA (from 1.6 × 103 ± 0.3 × 103 PFU to 3.0 × 100 ± 3.3 × 100 PFU). The poor rebooting of yeast-assembled DNA was likely caused by a low fraction of phage DNA in the DNA extracted from the yeast cells. Analysis by qPCR indicated that DNA extracted from yeast cells in general only contained 0.30 ± 0.01% phage DNA (Supplementary Fig. S3), in accordance with other reports in literature [28]. The majority of extracted DNA from the yeast cells thus originates from yeast genomic DNA and is likely the cause of the ineffective rebooting. Based on the rebooting effectiveness of DNA containing 0.3% phage DNA, we would expect to produce 1 × 103 PFU during rebooting if phage DNA constituted 100% of the extracted DNA. The theoretical effectiveness at 100% phage DNA is only 0.2 log10 lower than for commercial DNA; thus, optimizing the fraction of phage DNA in the yeast DNA extract would allow improved rebooting of yeast assemblies.
3.2. Cell-free transcription–translation reactions allow efficient rebooting of GG-assembled phage genomes
While host cell transformation is a common approach for rebooting engineered phage genomes [10], it was recently shown that engineered phages can also be rebooted in a cell-free manner using TXTL reactions [29]. These reactions contain all elements required for transcription and translation and thus also phage production. To investigate how well TXTL reactions reboot engineered phage genomes, assembled T7 phage DNA was rebooted by TXTL reactions and by transformation (Figs 1 and 3). Equal amounts of DNA were rebooted and absolute PFU counts were evaluated to allow comparison despite different volumes of rebooted phages (12 μl for TXTL reactions and 200 μl for transformations).
Rebooting of commercial T7 DNA and GG-assembled T7 DNA using TXTL yielded significantly higher phage production compared to rebooting by transformation (Fig. 3). Commercial DNA was rebooted by TXTL with an improvement of 4.2 log10 (P value = .0056) compared to rebooting by transformation (from 1.6 × 103 ± 0.3 × 103 PFU to 2.7 × 107 ± 1.4 × 107 PFU). Likewise, DNA assembled by GG was rebooted in TXTL reactions with an improvement of 4.1 log10 (P value = .043) compared to rebooting by transformation (from 1.1 × 103 ± 0.3 × 103 PFU to 1.3 × 107 ± 0.7 × 107 PFU), indicating that TXTL rebooting is more effective than rebooting by transformation for both assembled and commercial control DNA.
No significant difference was observed between TXTL rebooting of commercial T7 DNA and rebooting of GG-assembled T7 DNA. TXTL reactions are thus capable of rebooting GG-assembled phage genomes at high effectiveness comparable to commercial DNA.
In contrast, phages were not produced from the yeast assembly using TXTL reactions, while rebooting by transformation produced a small number of phages (0–8 PFU, Fig. 3). TXTL reactions function optimally when 0.25 nM template DNA with minimal chemical or DNA contaminants is used [29]; thus, the low phage DNA concentration in addition to the contaminating yeast DNA likely disrupts the TXTL reaction and results in impaired phage production.
3.3. Large diverse phage libraries can be created by Golden Gate assembly and cell-free transcription–translation rebooting
As GG assemblies were rebooted by TXTL reactions with high efficiency yielding more than 107 PFU (Fig. 3), we hypothesized that the GG–TXTL workflow could be used to generate libraries of engineered phages. To measure the diversity of libraries created by GG assembly and TXTL rebooting, we constructed a proof-of-concept library of T7 phages containing random DNA barcodes (Fig. 4 and Supplementary data).
Figure 4.
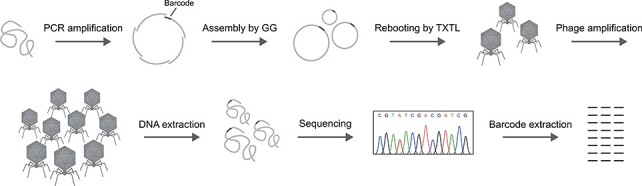
Process for constructing and sequencing the barcoded T7 library for diversity determination. T7 phage DNA was amplified in nine fragments by PCR for GG assembly. A barcode was incorporated in one of the fragments, indicated by a dark gray segment. See Supplementary data for details of barcode design. Assembled circular genomes were subsequently rebooted by cell-free TXTL reactions. The rebooted phages were amplified and DNA was extracted and sequenced by Illumina sequencing. Barcodes were extracted from the sequencing data.
To conduct an initial quality check, 20 plaques of barcoded phages rebooted by TXTL were Sanger sequenced. All examined phages were shown to contain unique barcodes (Supplementary Table S12); thus, we conducted Illumina sequencing to determine the total number of barcodes contained in the library. A small amount (<0.8%) of a TXTL reaction of barcoded T7 phages was titrated, showing 1.8 × 107 ± 0.9 × 106 PFU present in the examined TXTL reaction. The remaining TXTL reaction volume (>99.2%) was propagated overnight on an E. coli host to amplify the phage pool. This step potentially introduces some bias in the phage prevalence but reduces the risk of not detecting rare barcodes. Phage DNA was extracted from the resulting lysate and sequenced to establish the diversity of the phage library.
Illumina sequencing of the library identified 7.0 × 106 different barcodes distributed across 2.0 × 107 barcode-containing sequencing reads (Supplementary data). More than 75% of barcodes were observed three times or fewer (Fig. 5). However, a small subset of barcodes was identified at high frequencies, and 20 barcodes were observed more than 600 times (Supplementary data and Fig. S4). This overrepresentation of a small number of barcodes could have been caused by a combination of several factors: biases during either barcode synthesis, in phage amplification before DNA extraction, or PCR amplification during sequencing. These scenarios would result in an exponentially increased barcode prevalence, which could result in a false appearance of overabundance, which might not apply to other phage libraries.
Figure 5.
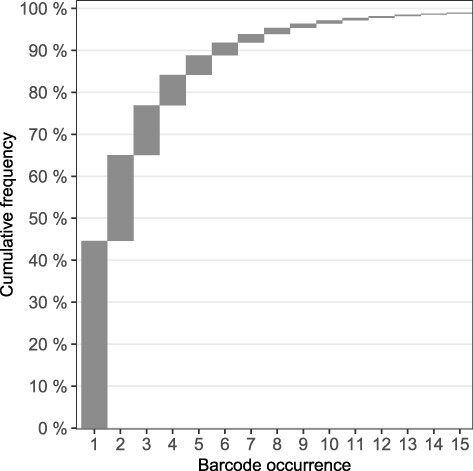
Frequency of barcode multiplicity in the barcoded T7 library. The cumulative frequency of the barcode multiplicity until 99% frequency is illustrated in a waterfall plot.
The number of identified barcodes increases slightly when accounting for the effect of the sequencing depth. Assuming that all barcodes are equally easy to find, we estimate to have detected approximately 93% of the total barcodes in the library given the sequencing depth (Supplementary data). The total pool of barcodes can thus be estimated to be 7.5 × 106 barcodes. While each phage in the library does not contain a unique barcode, the library overall contains a high diversity and low redundancy. Libraries produced by GG assembly and TXTL rebooting are thus suitable to deploy large screenings for example of tail fibers.
4. Discussion
Efficient methods of phage genome assembly and rebooting are necessary to fully utilize the advantages of rational phage engineering. In this study, the combination of GG assembly and TXTL rebooting of phage T7 was shown to outperform previous approaches based on assembly in yeast cells and rebooting by transformation. The GG–TXTL workflow was faster (Fig. 1), introduced fewer changes in the phage genome (Fig. 2), and produced a higher number of phages (Fig. 3).
GG is an effective DNA assembly method and has been shown to allow assembly of up to 52 fragments with minimal reduction in efficiency [19]. GG assembly is thus well-suited for phage genome assembly but requires removal of native recognition sites of the utilized type IIS restriction enzyme. In this study, recognition sites of the type IIS restriction enzyme used during GG assembly were removed by introducing synonymous mutation during PCR amplification of the DNA fragments for assembly. This approach was significantly faster and less intensive than removing recognition sites during DNA synthesis, which requires whole genome synthesis, cloning into both E. coli and yeast plasmids, sequencing, and amplification into fragments for GG assembly [30]. We selected the type IIS restriction enzyme AarI for GG assembly as this has only five native recognition sites in the T7 phage genome. All recognition sites were located in coding regions and were removed by synonymous mutations to minimize the effects on the phage. Recognition sites in vital areas such as packaging sites or regulatory regions require careful removal and should be avoided when possible. The ideal type IIS restriction enzyme used in the GG assembly should thus have minimal recognition sites in the phage genome, especially in vital or sensitive regions.
Currently, rebooting by TXTL reactions is limited by the narrow selection of commercially available TXTL reactions; some phages, such as lambda, are largely dependent on specific host factors that are missing the commercial reactions [21]. Creating TXTL reactions in-house from a suitable strain and/or supplying the required host factors to the reactions can mitigate the lack of commercial options [29, 31]. A recent study modified a gram-negative TXTL reaction and produced phages from gram-positive bacteria, demonstrating the large adaptability of TXTL reactions [32].
Implementing the GG–TXTL phage rebooting workflow will, in addition to improving rational phage engineering workflows, enable high-throughput work using phage libraries. We created a proof-of-concept barcoded library to fully investigate the diversity of produced libraries, as the benefit of using libraries of rebooted phages in screening for desired phenotypes has been demonstrated in recent studies [30, 33, 34]. The proof-of-concept library contained 1.8 × 107 phages, from which 7.5 × 106 different phages were identified. The diversity and size in the library created using our GG–TXTL workflow are similar to other libraries of rebooted phages [30, 33].
Alternative DNA assembly and rebooting methods have also been used for both individual phage engineering as well as creation of rebooted phage libraries. Higashi et al. [33] created a library of rebooted phages similar in size and diversity to the workflow described here. The newly developed PHEIGES method described by Levrier et al. [34] has also been used for efficient phage engineering and library creation. Overall, phage engineering based on phage rebooting is a growing field with many new methods developing [18–22, 30, 33, 34]. The most suitable assembly and rebooting method should be considered for individual experiments, as the efficiency of the methods may be greatly influenced by phage genome size, host characteristics, and other factors. For example, the T7 phage examined in this study had a relatively small genome of 40 kb and the host allowed transformation with the phage DNA. Transformation of host cells may not be possible for phages with large genome sizes or phages from hard-to-transform hosts, in which case TXTL rebooting or other approaches to rebooting should be considered.
In general, phage rebooting overcomes several limitations of traditional phage engineering, such as slow engineering and the requirement of extensive screening of phages. The GG–TXTL rebooting workflow further increases the engineering efficiency and applicability of phage rebooting, especially as it allows construction of large and diverse phage libraries. Implementing efficient and high-throughput methods based on engineered phage libraries results in significant time reduction when developing phages for phage therapy to combat antimicrobial resistance.
Supplementary Material
Contributor Information
Camilla S Kristensen, SNIPR Biome, Copenhagen 2100, Denmark; The Novo Nordisk Foundation Center for Biosustainability, DTU Biosustain, Technical University of Denmark, Kongens Lyngby 2800, Denmark.
Anders Ø Petersen, SNIPR Biome, Copenhagen 2100, Denmark.
Mogens Kilstrup, DTU Bioengineering, Technical University of Denmark, Kongens Lyngby 2800, Denmark.
Eric van der Helm, SNIPR Biome, Copenhagen 2100, Denmark.
Adam Takos, SNIPR Biome, Copenhagen 2100, Denmark.
Supplementary data
Supplementary data are available at SYNBIO online.
Conflict of interest:
All authors affiliated with SNIPR Biome are present or past employees of SNIPR Biome and may be share and/or warrant holders.
Funding
This work is partly funded by the Innovation Fund Denmark (IFD) under File No. 0153–00229B.
Data Availability
Sequencing data and barcode frequencies are available at Zenodo (https://zenodo.org/record/8366567). The scripts for barcode extraction are available at GitHub (https://github.com/sniprbiome/phage_rebooting_barcode_analysis).
References
- 1. World Health Organization . Antimicrobial Resistance: Global Report on Surveillance. Geneva, Switzerland: WHO, 2014. [Google Scholar]
- 2. Antimicrobial Resistance Collaborators . Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 2022;399:629–55. doi: 10.1016/S0140-6736(21)02724-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hesse S, Adhya S. Phage therapy in the twenty-first century: facing the decline of the antibiotic era; is it finally time for the age of the phage? Annu Rev Microbiol 2019;73:155–74. doi: 10.1146/annurev-micro-090817-062535 [DOI] [PubMed] [Google Scholar]
- 4. Abedon ST, Danis-Wlodarczyk KM, Alves DR. Phage therapy in the 21st century: is there modern, clinical evidence of phage-mediated efficacy? Pharmaceuticals 2021;14:1157. doi: 10.3390/ph14111157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Czaplewski L, Bax R, Clokie M et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect Dis 2016;16:239–51. doi: 10.1016/S1473-3099(15)00466-1 [DOI] [PubMed] [Google Scholar]
- 6. Międzybrodzki R, Hoyle N, Zhvaniya F et al. Current updates from the long-standing phage research centers in Georgia, Poland, and Russia. Bacteriophages 2021;1:921–51. [Google Scholar]
- 7. Uyttebroek S, Chen B, Onsea J et al. Safety and efficacy of phage therapy in difficult-to-treat infections: a systematic review. Lancet Infect Dis 2022;22:e208–20. doi: 10.1016/S1473-3099(21)00612-5 [DOI] [PubMed] [Google Scholar]
- 8. Lin J, Du F, Long M et al. Limitations of phage therapy and corresponding optimization strategies: a review. Molecules 2022;27:1857. doi: 10.3390/molecules27061857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lenneman BR, Fernbach J, Loessner MJ et al. Enhancing phage therapy through synthetic biology and genome engineering. Curr Opin Biotechnol 2021;68:151–59. doi: 10.1016/j.copbio.2020.11.003 [DOI] [PubMed] [Google Scholar]
- 10. Pires DP, Cleto S, Sillankorva S et al. Genetically engineered phages: a review of advances over the last decade. Microbiol Mol Biol Rev 2016;80:523–43. doi: 10.1128/MMBR.00069-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gencay YE, Jasinskytė D, Robert C et al. Engineered phage with antibacterial CRISPR–Cas selectively reduce E. coli burden in mice. Nat Biotechnol 2023;2023:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ando H, Lemire S, Pires DP et al. Engineering modular viral scaffolds for targeted bacterial population editing. Cell Syst 2015;1:187–96. doi: 10.1016/j.cels.2015.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meile S, Sarbach A, Du J et al. Engineered reporter phages for rapid bioluminescence-based detection and differentiation of viable listeria cells. Appl Environ Microbiol 2020;86:e00442–20. doi: 10.1128/AEM.00442-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pires DP, Monteiro R, Mil-Homens D et al. Designing P. aeruginosa synthetic phages with reduced genomes. Sci Rep 2021;11:2164. doi: 10.1038/s41598-021-81580-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dunne M, Rupf B, Tala M et al. Reprogramming bacteriophage host range through structure-guided design of chimeric receptor binding proteins. Cell Rep 2019;29:1336–50.e4. doi: 10.1016/j.celrep.2019.09.062 [DOI] [PubMed] [Google Scholar]
- 16. Faber MS, Van Leuven JT, Ederer MM et al. Saturation mutagenesis genome engineering of infective φx174 bacteriophage via unamplified oligo pools and golden gate assembly. ACS Synth Biol 2020;9:125–31. doi: 10.1021/acssynbio.9b00411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kilcher S, Studer P, Muessner C et al. Cross-genus rebooting of custom-made, synthetic bacteriophage genomes in L-form bacteria. Proc Natl Acad Sci USA 2018;115:567–72. doi: 10.1073/pnas.1714658115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitsunaka S, Yamazaki K, Pramono AK et al. Synthetic engineering and biological containment of bacteriophages. Proc Natl Acad Sci USA 2022;119:e2206739119. doi: 10.1073/pnas.2206739119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pryor JM, Potapov V, Bilotti K et al. Rapid 40 kb genome construction from 52 parts through data-optimized assembly design. ACS Synth Biol 2022;11:2036–42. doi: 10.1021/acssynbio.1c00525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rustad M, Eastlund A, Jardine P et al. Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth Biol 2018;3:ysy002. doi: 10.1093/synbio/ysy002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shin J, Jardine P, Noireaux V. Genome replication, synthesis, and assembly of the bacteriophage T7 in a single cell-free reaction. ACS Synth Biol 2012;1:408–13. doi: 10.1021/sb300049p [DOI] [PubMed] [Google Scholar]
- 22. Garamella J, Marshall R, Rustad M et al. The All E. coli TX-TL toolbox 2.0: a platform for cell-free synthetic biology. ACS Synth Biol 2016;5:344–55. doi: 10.1021/acssynbio.5b00296 [DOI] [PubMed] [Google Scholar]
- 23. Frazer LAN, O’Keefe RT. A new series of yeast shuttle vectors for the recovery and identification of multiple plasmids from Saccharomyces cerevisiae. Yeast 2007;24:777–89. doi: 10.1002/yea.1509 [DOI] [PubMed] [Google Scholar]
- 24. Gietz RD, Woods RA. Genetic transformation of yeast. Biotechniques 2001;30:816–31. doi: 10.2144/01304rv0 [DOI] [PubMed] [Google Scholar]
- 25. Andreou AI, Nakayama N, Isalan M. Mobius assembly: a versatile golden-gate framework towards universal DNA assembly. PLoS One 2018;13:e0189892. doi: 10.1371/journal.pone.0189892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Green R, Rogers EJ. Transformation of chemically competent E. coli. Methods Enzymol 2013;529:329–36. doi: 10.1016/B978-0-12-418687-3.00028-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Langman L, Paetkau V, Scraba D et al. The structure and maturation of intermediates in bacteriophage T7 DNA replication. Can J Biochem 1978;56:508–16. doi: 10.1139/o78-078 [DOI] [PubMed] [Google Scholar]
- 28. Bodner K, Melkonian AL, Covert MW. A protocol to engineer bacteriophages for live-cell imaging of bacterial prophage induction inside mammalian cells. STAR Protoc 2020;1:100084. doi: 10.1016/j.xpro.2020.100084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liyanagedera SBW, Williams J, Wheatley JP et al. SpyPhage: a cell-free TXTL platform for rapid engineering of targeted phage therapies. ACS Synth Biol 2022;11:3330–42. doi: 10.1021/acssynbio.2c00244 [DOI] [PubMed] [Google Scholar]
- 30. Liang J, Zhang H, Tan YL et al. Directed evolution of replication-competent double-stranded DNA bacteriophage toward new host specificity. ACS Synth Biol 2022;11:634–43. doi: 10.1021/acssynbio.1c00319 [DOI] [PubMed] [Google Scholar]
- 31. Noireaux V, Liu AP. The new age of cell-free biology. Annu Rev Biomed Eng 2020;22:51–77. doi: 10.1146/annurev-bioeng-092019-111110 [DOI] [PubMed] [Google Scholar]
- 32. Emslander Q, Vogele K, Braun P et al. Cell-free production of personalized therapeutic phages targeting multidrug-resistant bacteria. Cell Chem Biol 2022;29:1434–45.e7. [DOI] [PubMed] [Google Scholar]
- 33. Higashi K, Oda S, Fujii M et al. Construction of a T7 phage random peptide library by combining seamless cloning with in vitro translation. J Biochem 2024;175:85–93. doi: 10.1093/jb/mvad077 [DOI] [PubMed] [Google Scholar]
- 34. Levrier A, Karpathakis I, Nash B et al. PHEIGES: all-cell-free phage synthesis and selection from engineered genomes. Nat Commun 2024;15:2223. doi: 10.1038/s41467-024-46585-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data and barcode frequencies are available at Zenodo (https://zenodo.org/record/8366567). The scripts for barcode extraction are available at GitHub (https://github.com/sniprbiome/phage_rebooting_barcode_analysis).