

Abstract
Background & Aims:
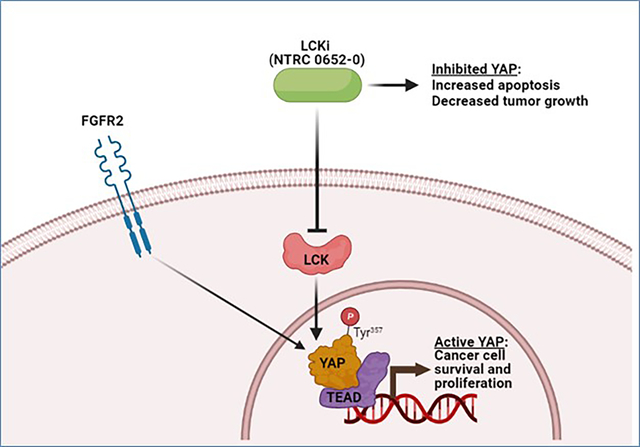
There is an unmet need to develop novel, effective medical therapies for cholangiocarcinoma (CCA). The Hippo pathway effector, YAP, is oncogenic in CCA, but has been historically difficult to target therapeutically. Recently, we described a novel role for the Src-family kinase LCK in activating YAP through tyrosine phosphorylation. This led to the hypothesis that LCK is a viable therapeutic target in CCA via regulation of YAP activity.
Methods:
A novel tyrosine kinase inhibitor with relative selectivity for LCK, NTRC 0652–0, was pharmacodynamically profiled in vitro and in CCA cells. A panel of eight CCA patient-derived organoids (PDO) were characterized and tested for sensitivity to NTRC 0652–0. Two patient-derived xenograft (PDX) models bearing FGFR2-rearrangements were utilized for in vivo assessment of pharmacokinetics, toxicity, and efficacy.
Results:
NTRC 0652–0 demonstrated selectivity for LCK inhibition in vitro and in CCA cells. LCK inhibition with NTRC 0652–0 led to decreased tyrosine phosphorylation, nuclear localization, and co-transcriptional activity of YAP, and resulted in apoptotic cell death in CCA cell lines. A subset of patient-derived organoids tested demonstrated sensitivity to NTRC 0652–0. CCA with FGFR2 fusions were identified as a potentially susceptible and clinically relevant genetic subset. In PDX models of FGFR2 fusion-positive CCA, daily oral treatment with NTRC 0652–0 resulted in stable plasma and tumor drug levels, acceptable toxicity, decreased YAP tyrosine phosphorylation, and significantly decreased tumor growth.
Conclusions:
A novel LCK inhibitor, NTRC 0652–0, inhibited YAP signaling and demonstrated preclinical efficacy in CCA cell-lines, and patient derived organoid and xenograft models.
Keywords: Hippo pathway, Src family kinase, tyrosine kinase inhibitor, bile duct tumors
Lay summary
Cholangiocarcinoma is an aggressive form of liver cancer arising in the biliary tract, which cannot be controlled by current chemotherapy or targeted therapy options. This paper describes a new targeted therapy approach, using an oral inhibitor of LCK. The data suggest this approach may be effective in cholangiocarcinoma with YAP dependence or FGFR2 fusions.
Graphical abstract
Introduction
Effective treatment options for advanced CCA are limited; with standard of care chemotherapy (GEMCIS, FOLFOX) producing modest response rates and poor overall survival.1 Targeted therapies based on recurrent genetic lesions in CCA are beginning to be clinically utilized with the availability of IDH and FGFR inhibitors.2 While promising, this approach is limited to genetically defined subsets of patients, and thus far has shown modest efficacy. Therefore, there is an ongoing clinical need for more effective CCA therapies.
YAP is a transcriptional co-activator and the effector protein of the Hippo signaling pathway. The Hippo pathway regulates organ size, tissue homeostasis, cell proliferation, and evasion of apoptosis by inhibiting YAP activity. Notably, YAP is aberrantly activated in many cancers.3 In the majority of human CCA, YAP is overexpressed and localized to the nucleus where it can drive an oncogenic gene expression program.4, 5 Mouse models of YAP activation in the liver have demonstrated that YAP is a bona fide oncogene in CCA.4, 6 However, less than 10% of CCAs have a defined genetic lesion in the canonical Hippo pathway, such as mutation or deletion of NF2 or SAV11, indicating that the molecular drivers of YAP activation in CCA are still poorly understood.
In contrast to the canonical regulatory paradigm, in which YAP activity is repressed by serine phosphorylation downstream of Hippo pathway kinases, our lab and others have recently identified YAP tyrosine phosphorylation as a novel activating mark and nuclear retention signal in CCA.7 We found that the Src family protein tyrosine kinase LCK phosphorylates YAP on tyrosine 357 (NCBI Reference Sequence: NM_001282098.2).7 Further, YAP tyrosine phosphorylation is required for its full activation, even in the absence of Hippo pathway repression, and in vivo growth of CCA patient-derived xenografts was inhibited by the Src-family kinase inhibitor dasatinib.7 These studies suggested that activation of YAP by tyrosine phosphorylation is a critical regulatory event that can be pharmacologically targeted. Therefore, in the present study we evaluated therapeutic targeting of LCK with a novel selective small molecule inhibitor, NTRC 0652–0, in preclinical models of CCA.
Materials and Methods
NTRC 0652–0 pharmacodynamics and pharmacokinetics assays
NTRC 0652–0 is a small molecule LCK inhibitor in development by Netherlands Translational Research Center B.V. (Oss, Netherlands). An in vitro screen of NTRC 0652–0 was performed against 152 kinases (Carna Biosciences, Kobe, Japan) at 1 mM ATP to identify targets. The half maximal inhibitory concentration (IC50), dissociation constant (KD), selectivity, and thermodynamic parameters of NTRC-0652 were determined for identified kinase targets using a surface plasmon resonance (SPR) biosensor (Biacore T200, GE Healthcare Life Sciences, Marlborough, MA, USA). For in vivo pharmacokinetic analyses, NTRC 0652–0 drug levels in paired tumor and plasma samples were measured by LC-MS/MS.
Total proteome and tyrosine phosphoproteome analysis
For stable isotope labeling with amino acid in cell culture (SILAC) experiments, parallel cultures of HuCCT-1 cells were grown in SILAC media containing light (control), medium (2H4-Lys and 13C6-Arg) (NTRC-treated) or heavy (13C615N2- Lys and 13C615N4-Arg) (sgLCK) amino acids to metabolically label the proteome. Once cells were >99% labeled, they were treated with vehicle (control), NTRC 0652–0 1 μM for 24 hours (NTRC), or doxycycline-induction of sgLCK expression for 72 hours (sgLCK). Cells were lysed and pooled for proteomic analysis by liquid chromatography tandem mass spectrometry. For tyrosine phosphoproteome analysis, tyrosine phosphorylated peptides were enriched on anti-phosphotyrosine antibody beads (pY1000, Cell Signaling Technology). Additional details in Supplemental Methods.
Organoid culture
Organoids were derived from primary human tumor samples in accordance with IRB 70703 at Mayo Clinic, Rochester, MN, USA. Freshly isolated human tumors were dissociated using a human tumor dissociation kit and MACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany). Dissociated cells were washed, strained (70 um), and resuspended in melted Matrigel domes (Corning, NY, USA). Organoids were cultured in complete media (supplemental methods), changed twice weekly and passaged by physical dissociation. Experiments were performed within 10 passages of generation.
Xenograft experiments
Mayo Clinic, Rochester, has an ongoing IRB-approved protocol for collection and xenotransplantation of resected biliary tract cancers, IRB 70703. In accordance with this protocol, ARRIVE guidelines for humane and reproducible animal research, and an established Institutional Animal Care and Use Committee protocol (A00003954–18-21), human CCA tumors were implanted in the flanks of female NOD/SCID mice, age 6–8 weeks. Treatment studies were initiated after tumors reached palpable size (>25 mm3).
See the CTAT table and supplemental methods for additional details.
Data availability
The mass spectrometry proteomics data are deposited to the ProteomeXchange Consortium via the PRIDE9 partner repository with the dataset identifier PXD026925. Other data generated in this study are available within the article and its supplementary files.
Results
NTRC 0652–0 preferentially inhibits LCK
NTRC 0652–0 was developed as a tyrosine kinase inhibitor selective for LCK (Figure 1A). To identify additional substrates of NTRC 0652–0 and define its selectivity, an in vitro screen of 150 kinases was performed. In addition to the top hit, LCK, eleven other kinases were identified with >50% activity inhibition by NTRC 0652–0, including other Src family kinases (SRC, BLK, LYN, YES, FYN, HCK, FRK, FGR), BRK, BTK, and SRM (Figure 1B, Supplemental table 1). Among the 5 hits with a dissociation constant (KD) <1 μM, NTRC 0652–0 showed increased potency and ~50 to 2000-fold selectivity for LCK, with an in vitro IC50 of 2 nM, KD of 0.08 nM, and residence time of approximately 10 hours for LCK (Figure 1C).
Fig. 1. NTRC is a novel TKI that preferentially inhibits LCK.
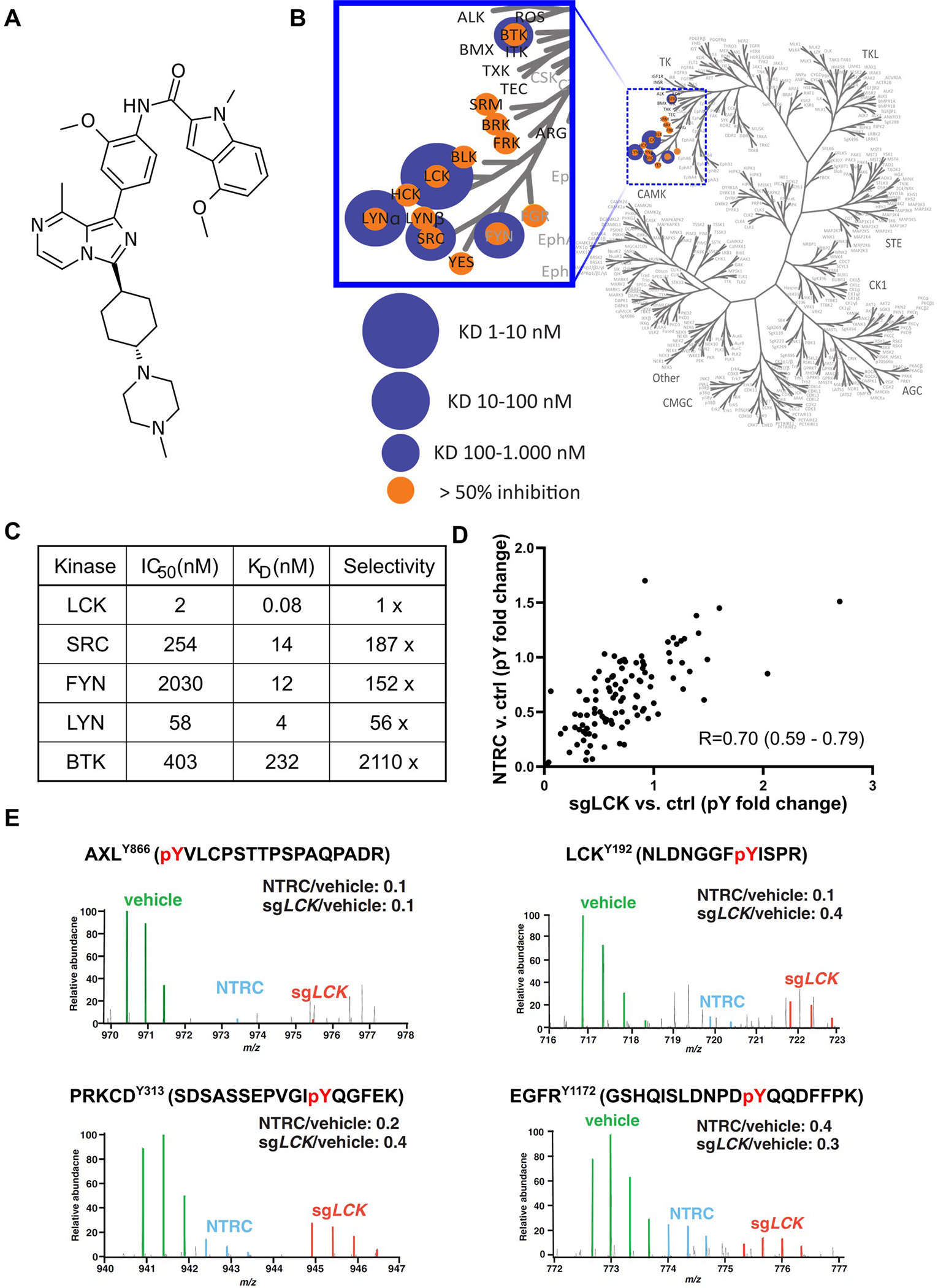
(A) Chemical structure of NTRC 0652–0. (B) An in vitro screen of 150 kinases identified 12 potential substrates. (C) Kinetics, affinity, and selectivity of top substrates. (D) Global changes in tyrosine phosphorylation (pY) were assessed in HuCCT-1 cells following pharmacologic LCK inhibition (NTRC, y-axis) or genetic deletion (sgLCK, x-axis) versus vehicle control. Changes in pY with NTRC and sgLCK were highly correlated (Spearman correlation, p<1e-15). (E) MS spectra for representative significantly hypophosphorylated proteins in both NTRC-treated and sgLCK conditions.
To evaluate the effect and selectivity of NTRC 0652–0 in CCA cells in an unbiased manner, we quantified changes in the phosphotyrosine proteome in HuCCT-1 cells following treatment with NTRC 0652–0 or knockout of LCK using a previously reported doxycycline-inducible CRISPR/Cas9 system (HuCCT-1-sgLCK).7 Relative quantitation of total and phosphotyrosine proteome was carried out using high resolution mass spectrometry. We employed three state stable isotope labeling with amino acids in cell culture (SILAC)8 of HuCCT-1 cells following LCK pharmacologic inhibition (NTRC 0652–0 at 1 μM for 24 hours) or genetic deletion (HuCCT-1-sgLCK cells, induced 72 hours) compared to control HuCCT-1 cells. Phosphotyrosine peptides were enriched using an anti-phosphotyrosine antibody and their relative abundance was measured by mass spectrometry (Supplemental figure 1A). Proteins with decreased tyrosine phosphorylation (log2fold change <−1) but stable total protein level (log2 fold change −1 to 1) in an experimental condition compared to control HuCCT-1 cells were considered significantly hypophosphorylated (Supplemental figure 1C and D). Following NTRC 0652–0 treatment, 36 tyrosine sites on 31 proteins were hypophosphorylated. Upon induction of LCK knockout, 30 tyrosine sites on 27 proteins were hypophosphorylated. Importantly, 21 sites on 20 proteins were significantly hypophosphorylated under both conditions, representing a highly significant overlap in the effects of pharmacologic inhibition and genetic deletion (chi square, p <1e-54, Supplemental table 2). Analysis of the phosphotyrosine fold-change compared to control as a continuous variable also showed a highly significant correlation between the effects of NTRC 0652–0 treatment and LCK knockout (Spearman correlation, p<1e-15, Figure 1D). Representative hypophosphorylated peptides are depicted in Figure 1E with the expression level in each condition evaluated from mass spectrometry. Notably, an LCK-specific peptide containing phospho-LCKY192 was significantly decreased in both NTRC 0652–0-treated and LCK knockout conditions. Tyrosine phosphorylation was significantly decreased on additional, functionally important peptides common to LCK and other Src-family kinases, including phospho-LCKY394 which is a conserved autophosphorylation site required for LCK activation (Supplemental figure 1B). Downstream of LCK, protein kinase C delta type (PRKCD) was significantly hypophosphorylated on Y313, a residue known to be phosphorylated by LCK and other Src-family kinases.9 AXLY866 is also a known target of LCK10 that was hypophosphorylated in both conditions. Other significantly hypophosphorylated proteins of interest include Epidermal Growth Factor Receptor (EGFR) at residue Y1172 which contributes to EGFR activation but is not known to be regulated downstream of LCK, and downstream targets of EGFR, including EPHA2Y772, raising the possibility of a broader role for LCK signaling upstream of receptor tyrosine kinases in CCA. Overall, these findings demonstrate on-target effect of NTRC 0652–0 with a high degree of overlap in the effects of NTRC 0652–0 treatment and LCK knockout, as well as hypophosphorylation of known LCK targets. In addition, proteomic analysis provided several less investigated phosphorylation sites that are potentially regulated by LCK, which may germinate future studies on the role of LCK in CCA signaling (Supplemental table 2).
NTRC 0652–0 inhibits YAP tyrosine phosphorylation and activity and induces cell death by apoptosis in CCA cells
We next examined YAP tyrosine phosphorylation and activity in cells treated with NTRC 0652–0. We previously demonstrated that inducible knockout of LCK in HuCCT-1 cells decreased YAPY357 phosphorylation, nuclear localization, and co-transcriptional activity.7 Herein, we identified that pharmacologic LCK inhibition with NTRC 0652–0 in HuCCT-1 cells led to decreased YAPY357 phosphorylation without changing the level of YAPS127 phosphorylation or total YAP protein level (Figure 2A). This was associated with redistribution of YAP from the nucleus to the cytoplasm (Figure 2B), and decreased YAP co-transcriptional activity measured by both a luminescent reporter of TEAD-dependent transcription (Figure 2C) and mRNA expression of canonical YAP target genes including the anti-apoptotic gene MCL1 (Figure 2D).
Fig. 2. NTRC 0652–0 inhibits YAP tyrosine phosphorylation and activity and induces apoptotic cell death.
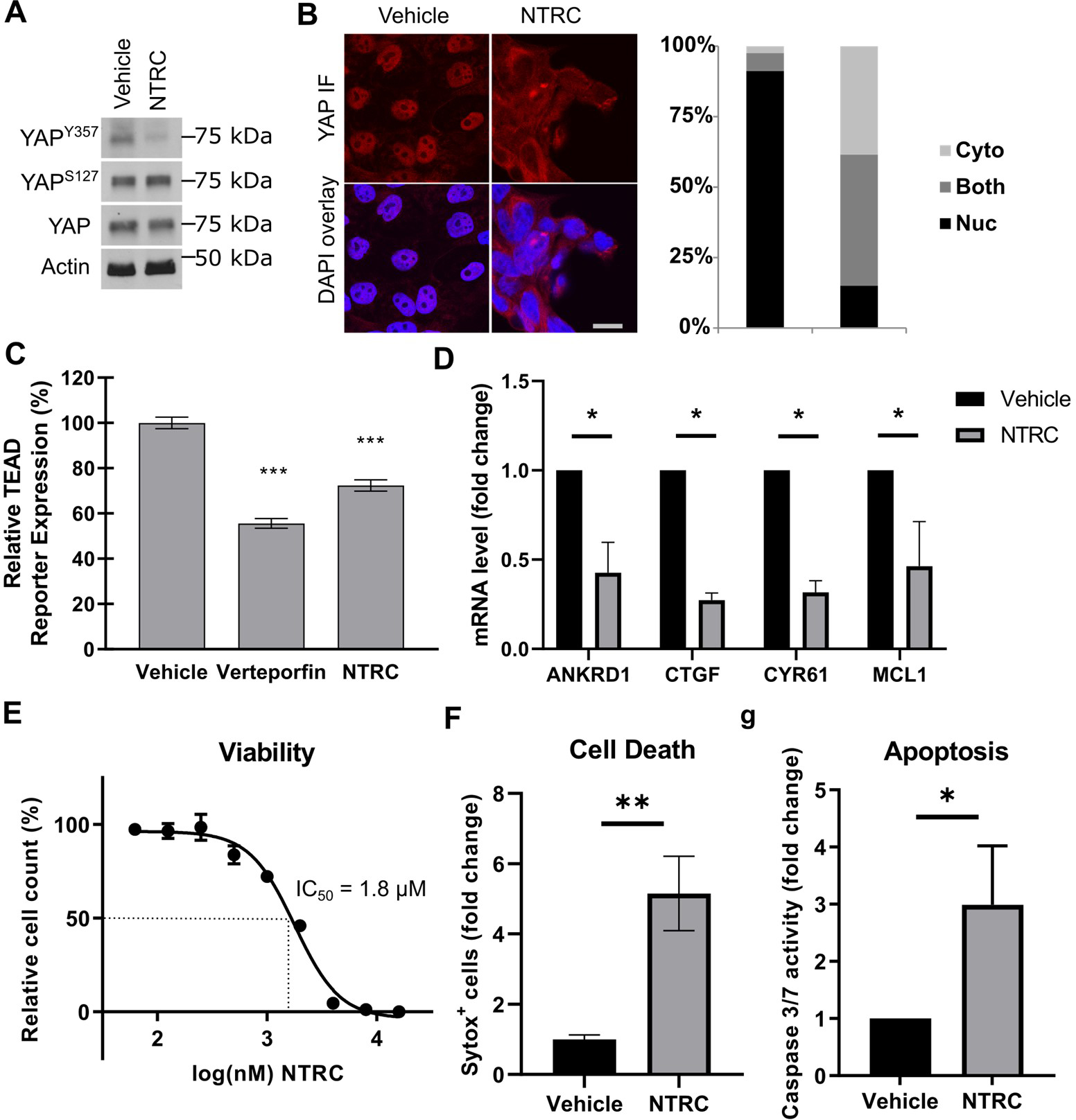
(A) Immunoblot analysis of HuCCT-1 cells showed decreased YAP Y357 phosphorylation following NTRC 0652–0 treatment. (B) Immunofluorescence in HuCCT-1 cells showed nuclear to cytoplasmic redistribution of YAP following NTRC 0652–0 treatment, scale 10 μm. (C) Global TEAD-dependent transcription was decreased in HuCCT-1 reporter cells treated with Verteporfin or NTRC 0652–0. (D) Expression of canonical YAP target genes by qRT-PCR was significantly decreased following NTRC 0652–0 treatment. (E) Viability dose-response curve and calculated IC50 of HuCCT-1 cells treated with NTRC 0652–0. (F) Cell death and (G) caspase 3/7 activity were increased in HuCCT-1 cells treated with NTRC 0652–0. (All panels represent N≥3 replicate experiments, significance by unpaired t-test: * p<0.05).
Next, HuCCT-1 cells were treated with a range of NTRC 0652–0 drug concentrations to determine the effects on cell viability (62.5 nM – 16 μM, Figure 2E). The IC50 at 72 hours was 1.8 μM, and cell viability approached 0% at maximum drug concentrations. Cell death was confirmed by sytox staining (Figure 2F), and was associated with apoptosis, as demonstrated by increased caspase-3 and -7 activity (Figure 2G).
NTRC 0652–0 is cytotoxic in a subset of patient-derived organoids
We next sought to confirm the cytotoxic effect of NTRC 0652–0 in a broader panel of clinically relevant CCA models. Patient-derived organoid (PDO) models were derived from primary CCA tumor samples at surgical resection and cultured in matrigel with complete organoid media (Figure 3A, Table 1). PDOs were histologically evaluated and confirmed to grow as spherical structures with open lumens (Figure 3A and Supplemental figure 2). Expression of the CCA phenotype marker cytokeratin 19 (CK19) was confirmed by immunofluorescence. Organoids did not express phenotypic markers of fibroblasts (alpha-smooth muscle actin [SMA]) or hepatocytes (HNF4-a, Figure 3A, and Supplemental figure 2).
Fig. 3. NTRC is cytotoxic in a subset of patient-derived organoids (PDO) with basal YAP tyrosine phosphorylation and drug-induced YAP inhibition.
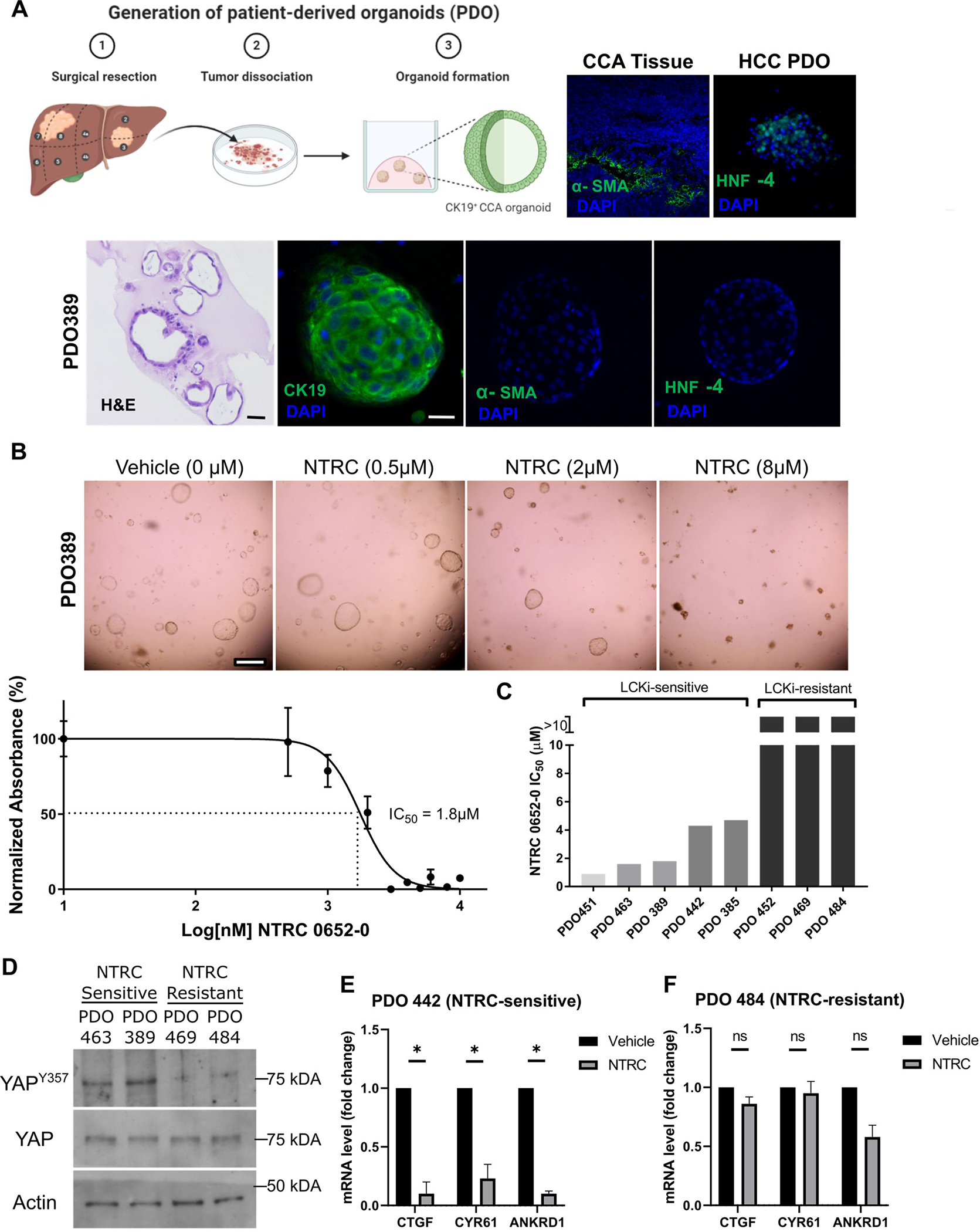
(A) Derivation of PDO from primary human CCA (schema). PDO histology and phenotypic marker immunofluorescence, with expected CCA pattern (CK19+/HNF4a−/α-SMA−, positive controls for HNF4a and α-SMA staining included, original magnification 200x.) (B) Photomicrographs and viability dose-response curve of a representative NTRC-sensitive organoid, PDO389 (scale bar 500 μm). (C) Calculated IC50 of NTRC 0652–0 in a panel of PDO models. (D) Immunoblot showed hypophosphorylation of YAP Y357 in NTRC-resistant PDOs. (E) YAP target gene expression was reduced in the NTRC-sensitive PDO442 following NTRC treatment (2 μM, 24 hr; N=3, unpaired t-tests, p< 0.05). (F) No change in YAP target gene expression was seen in the NTRC-resistant PDO484 (4 μM, 24 hr; N=3, unpaired t-tests, p = ns).
Table 1:
Clinical characteristics of patient derived PDO and PDX models.
| Model | Patient Sex | Subtype | Primary vs. Recurrent | Neoadjuvant chemotherapy | FGFR2 fusion status | Other molecular testing |
|---|---|---|---|---|---|---|
| PDO385 | M | ICCA | Primary | None | negative | None |
| PDO389 | M | ICCA | Primary | GEMCIS | negative | None |
| PDO442 | M | HCCA | Primary | GEMCIS | negative | pMMR (IHC) |
| PDO451 | M | ICCA | Recurrent | GEMCIS and FOLFIRINOX | Detected (BAP FISH) | None |
| PDO463 | M | DCCA | Primary | None | negative | pMMR (ICH); AR mutation (Tempus) |
| PDO484 | M | HCCA | Primary | None | negative | dMMR: Loss of MLH1/PMS2 No significant alterations (targeted panel) |
| PDO469 | M | ICCA | Primary | None | negative | pMMR (IHC), ARID1A mutation (Tempus) |
| PDO452 | F | ICCA | Primary | GEMOX | negative | pMMR (IHC) |
| PDX283 | F | ICCA | Primary | GEMCIS | Detected (FGFR2-AFF4) | MSS, FGFR2-AFF4 fusion, BAP1 mutation, MCL1 amplification (FoundationOne) |
| Liv31 | F | ICCA | Metastatic | GEMCIS, CAPOX, and multiple others | Detected (FGFR2-CCDC6) | FGFR2-CCDC6 Fusion, PTPRB mutation (Tempus) |
| PDX42 | M | ICCA | Primary | None | negative | No clinical genetic testing |
PDO were treated with vehicle or NTRC 0652–0 at a range of concentrations (500 nM – 10 μM) for 72 hours to identify the IC50 (Figure 3B and C). Of the PDO models tested, 5 of 8 (62.5%) were sensitive to NTRC 0652–0 with an IC50 ranging from 0.9 to 4.7 μM, while 3 models (37.5%) had no change in viability with NTRC treatment up to 10 μM, indicating there is a subset with inherent resistance to treatment.
NTRC 0652–0 inhibits YAP cotranscriptional activity in sensitive organoids
To understand the observed differences in PDO sensitivity to NTRC 0652–0, we initially examined the PDOs for expression of the drug target, LCK. Of note, analysis of The Cancer Genome Atlas data showed that LCK RNA levels were increased in CCA compared to normal tissue controls, suggesting that LCK may be upregulated in CCA (Supplemental figure 3A, https://www.cancer.gov/tcga). We characterized the basal level of LCK expression and phosphorylation by immunoblot in 7 of 8 PDOs for which adequate material was available. LCK was expressed in all PDO tested, and expression level did not correlate with NTRC 0652–0 sensitivity or resistance, indicating the mechanism of resistance was not lack of drug target expression (Supplemental figure 3B). LCKY394 phosphorylation level as a marker of LCK activity also did not correlate with NTRC sensitivity or resistance. Antibodies to this conserved site cross-react with other SFKs and are not a specific readout of LCK activation. However, this data suggests that overall SFK activation does not predict sensitivity to NTRC 0652–0, and likely LCK activation alone is not a strong predictor of response (Supplemental figure 3B).
Rather, we found that while total YAP levels were consistent across models, the subset of NTRC-sensitive models profiled had a higher level of YAPY357 phosphorylation under basal conditions (Figure 3D). However, YAP co-transcriptional activity was not significantly different in NTRC-sensitive versus resistant models, suggesting alternative routes of YAP activation in NTRC-resistant models (Supplemental figure 3C). PDO treatment with NTRC 0652–0 led to decreased expression of canonical YAP target genes in sensitive but not resistant organoids (Figure 3E and F). These results further support a correlation between YAP activation by tyrosine phosphorylation and sensitivity to LCK inhibition.
NTRC 0652–0 is therapeutic in multiple in vivo models of FGFR2-fusion CCA
We next sought to evaluate the efficacy and toxicity of NTRC 0652–0 treatment in vivo. In selecting a preclinical model for testing, we considered both YAP-dependence and clinically relevant genetic subtypes. Of the PDO profiled, the most sensitive model, PDO451, was found to have FGFR2 rearrangement by clinical break-apart probe analysis (Table 1), leading to the hypothesis that FGFR2-fusion CCA may be sensitive to LCK inhibition. FGFR2 rearrangements occur in 10–15% of intrahepatic CCA and are thought to be oncogenic via increased downstream MAPK/ERK and AKT/PI3K signalling.2 In prior work we found that YAP can be activated downstream of FGFR211, suggesting that this genetic subset may be YAP-dependent as well. We analyzed expression of YAP target genes in CCA with or without an FGFR2 rearrangement, using the TCGA dataset.12 In CCA with FGFR2 rearrangement, overexpression of the YAP target gene set was enriched 2.73-fold (p=0.034), indicating that FGFR2 rearrangement is associated with elevated expression of YAP target genes (Supplemental table 3).
PDX283 is a patient-derived xenograft model from a surgically resected intrahepatic CCA. Genetic analysis of the primary tumor revealed presence of an FGFR2-AFF4 fusion, BAP1 frameshift mutation, and MCL1 amplification (Table 1). The FGFR2-AFF4 fusion has been previously reported in CCA and is predicted to be functional.2 To test whether PDX283 tumors depend on YAP for tumor growth, we treated mice bearing PDX283 tumors with CA3, a small molecule inhibitor of YAP.13 Mice bearing PDX283 tumors were treated with vehicle or CA3 at 1 mg/kg/mouse by intraperitoneal injection 3 times per week for 3 weeks. CA3 treatment significantly decreased the growth of PDX283 tumors (Supplemental figure 4A). Inhibition of YAP target gene expression in tumors from CA3-treated mice was confirmed by qRT-PCR (Supplemental figure 4B). These data support the YAP-dependence of FGFR2-altered cholangiocarcinoma in an in vivo patient-derived model.
Unexpectedly, cells isolated from PDX283 tumor and propagated in vitro showed resistance to pemigatinib, an FGFR inhibitor clinically used in patients with FGFR2 alterations, at concentrations up to 16 μM (Supplemental figure 4C). In contrast, cells from PDX283 were sensitive to NTRC 0652–0 in vitro with an IC50 of 2.5 μM (Supplemental figure 4D), suggesting LCK activity as a potential resistance mechanism for FGFR inhibition. Therefore, this model was selected for further testing in vivo.
We treated NOD/SCID mice bearing PDX283 flank tumors with NTRC 0652–0 at a dose of 30 or 45 mg/kg by daily oral gavage for 2 weeks. Mice were sacrificed at multiple time points (1, 6, 12, and 24 hours) following the final dose of drug to establish plasma and tumor pharmacokinetics by LC-MS/MS. Drug levels in both the plasma and tumor were high and remained stable over 24 hours (Figure 4A). Given the established target affinity and in vitro cytotoxicity data, these levels were predicted to provide continuous LCK target coverage. Indeed, treatment with either 30 or 45 mg/kg of NTRC 0652–0 significantly decreased xenograft tumor growth (Figure 4B).
Fig. 4. NTRC 0652–0 is therapeutic in an in vivo model of FGFR2-fusion CCA.
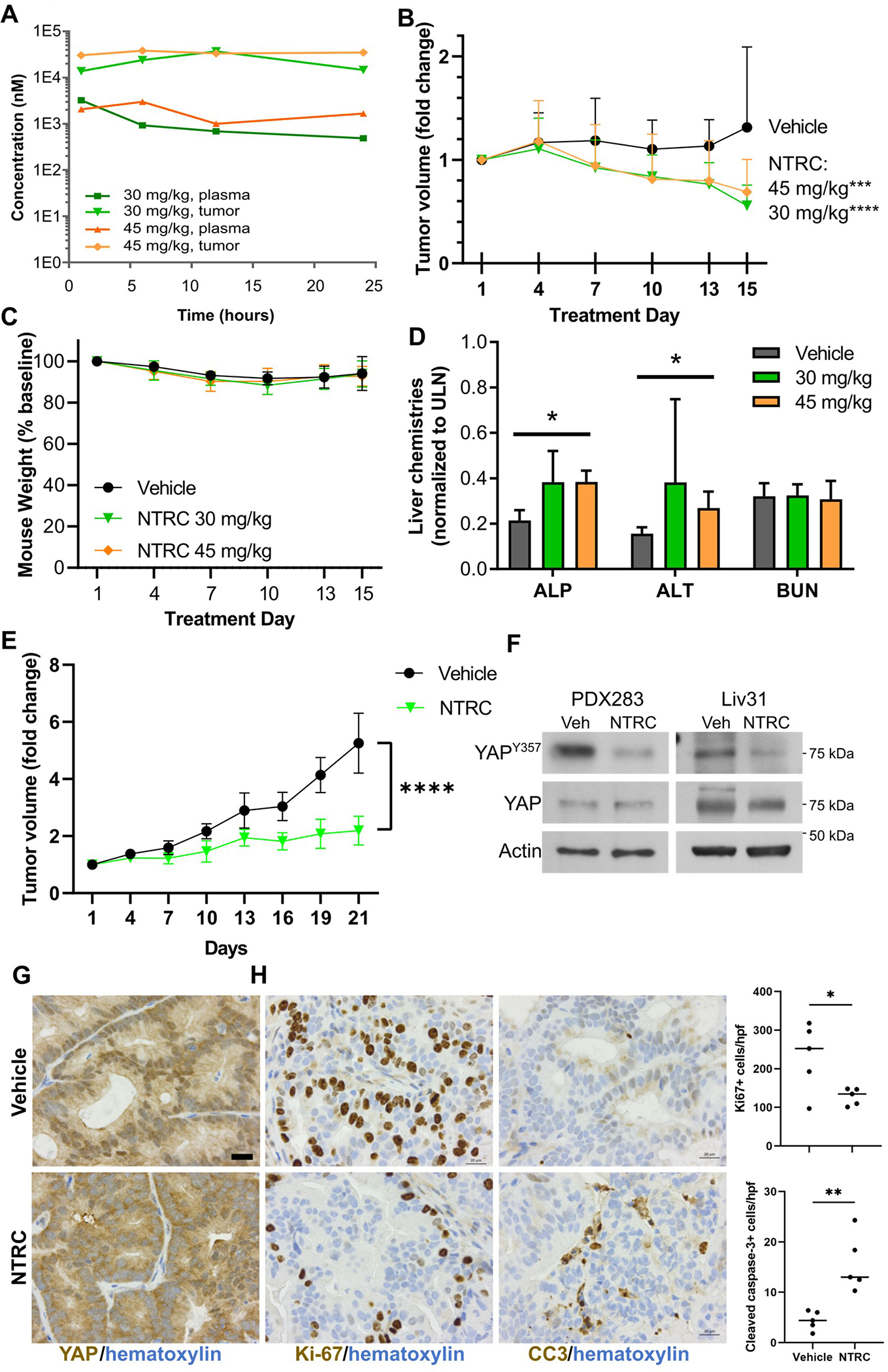
(A) Pharmacokinetic analysis of NTRC 0652–0 drug level in plasma and tumor from PDX283 mice (N=2 per dose, per time point). (B) PDX283 tumor volume was decreased in mice treated with NTRC 0652–0 at 30 or 45 mg/kg/day for 15 days versus vehicle control (N=8 per group, 2way ANOVA, multiple comparisons for each dose versus vehicle. adjusted p <0.0001 [30 mg/kg], <0.0006 [45 mg/kg]). (C) Mouse body weight (2way ANOVA, multiple comparisons, adjusted p= ns). (D) Serum liver and renal chemistries. Elevation of ALP and ALT remained below the upper limit of normal, with no change in BUN (2way ANOVA, multiple comparisons, * P <0.05). (E) Mice bearing Liv31 tumors were treated with vehicle or 30 mg/kg of NTRC 0652–0 for 21 days (N=5 per group). NTRC 0652–0 treatment significantly decreased tumor growth. (Mixed effects analysis due to missing data. One vehicle-treated mouse was sacrificed on day 16 for excess tumor growth per animal care protocols. Adjusted P-value = 0.0002). (F) Immunoblot of phospho-YAPY357, total YAP, and actin in PDX283 and Liv31 tumors from representative mice treated with vehicle or NTRC 0652–0 at 30 mg/kg/day. (G) YAP immunohistochemistry in Liv31 tumors (40x images, 20 μm scale bar). (H) Ki-67 and cleaved caspase 3 immunohistochemistry in Liv31 tumors (40x images, 20 μm scale bar. Positive cells per 20x field, unpaired t-test, P <0.05.)
NTRC 0652–0 treatment was well tolerated overall, with no difference in weight loss between vehicle- and drug-treated groups (Figure 4C), and clinically insignificant changes in liver chemistries within the normal range (Figure 4D).
YAPY357 phosphorylation was decreased in tumors from NTRC 0652–0-treated mice (Figure 4F), confirming the effect on YAP tyrosine phosphorylation in vivo. To assess proteome-wide drug effects and specificity, global changes in the total and phosphotyrosine proteome were assessed in PDX283 flank tumors from a parallel set of NOD/SCID mice treated with a short course of NTRC 0652–0 at 30 mg/kg/day or vehicle for 5 days (Supplemental table 4, Supplemental figure 6). Separate tumors were utilized for this experiment to provide adequate tissue for proteomic analyses and to assess early changes in protein phosphorylation. This analysis confirmed hypophosphorylation of several phosphotyrosine sites noted to be regulated by NTRC 0652–0 treatment and LCK inducible knockout in HuCCT-1 cells, including AXLY866 and PRKCDY313 (Supplemental figure 6), supporting the specificity of NTRC 0652–0 as an inhibitor of LCK and the relevance of these targets in multiple tumor models.
To confirm the sensitivity of FGFR2-altered CCA to LCK inhibition, we identified a second PDX model, Liv31, driven by an FGFR2-CCDC6 gene fusion and previously found to be sensitive to FGFR inhibition.14 NOD/SCID mice bearing Liv31 flank tumors were treated with NTRC 0652–0 at a dose of 30 mg/kg by daily oral gavage for 3 weeks, resulting in significantly decreased tumor growth (Figure 4E). Weight loss was not significantly different between drug- and vehicle-treated mice (Supplemental figure 5A) and histologic analysis of liver tissue from treated mice showed no overt toxicity (Supplemental figure 5B). YAPY357 phosphorylation was decreased in NTRC 0652–0 treated tumors (Figure 4F). Concordantly, YAP localization shifted to the cytoplasm by immunohistochemistry (Figure 4G.) Ki-67 was significantly decreased and cleaved-caspase 3 was significantly increased in tumors from mice treated with NTRC 0652–0 (Figure 4G). These results indicate that tumor growth inhibition by NTRC 0652–0 is due to a combination of decreased proliferation and increased apoptosis, consistent with the known biology of YAP as a regulator of proliferative and cytoprotective pathways.3
Discussion
This study supports targeting LCK as a therapeutic approach in CCA, by demonstrating: 1) NTRC 0652–0 is a selective LCK inhibitor with favorable pharmacokinetic and toxicity profiles, 2) targeting LCK decreases CCA viability both in vitro and in vivo, 3) sensitivity to LCK inhibition may be correlated with YAP tyrosine phosphorylation, and 4) CCA bearing FGFR2 fusions may represent a clinically relevant genetic subset that is sensitive to LCK inhibition.
LCK is a member of the Src family of protein tyrosine kinases, which functions in T-cell receptor signaling.15 In hematologic malignancies, LCK is a protooncogene and potential therapeutic target.16 A recent study identified a subset of T-ALL sensitive to LCK inhibition with dasatinib, due to dependence on pre-T cell receptor activation of LCK.17
In solid tumors, the expression and function of LCK is less well defined. In a model of endometrioid cancer, LCK signaling was implicated in chemotherapy resistance and LCK knockdown or inhibition promoted cisplatin sensitivity through regulation of DNA repair proteins.18 In glioblastoma, the expression of LCK and abundance of activated pLCKY394 were increased in high-grade tumors.19 Moreover, LCK was found to regulate tumor cell migration and cancer stem cell self-renewal, and LCK inhibitor treatment decreased tumor growth in an orthotopic glioblastoma xenograft model.19 Our data suggest that LCK may also be a viable drug target in CCA. However, questions remain regarding the mechanistic basis of LCK dependence in tumorigenesis. In contrast to prior studies, we did not observe significant regulation of DNA repair proteins or a cancer stem cell signature. Rather, we identified that CCA models with sensitivity to LCK inhibition exhibited higher levels of YAP tyrosine phosphorylation, and YAP-dependent transcription was decreased by LCK inhibitor treatment.
Historically, YAP has been difficult to target therapeutically, due to its canonical negative regulation by the Hippo pathway, and the inherent difficulty of pharmacologically activating a tumor suppressor pathway. While compounds interrupting the YAP/TEAD interaction or downstream targets of YAP are in development, no YAP inhibitors are yet clinically available.13, 20 Therefore, our finding that YAP activity is inhibited by NTRC 0652–0 represent an advance with potentially significant therapeutic implications.
In addition to inhibiting YAP, we observed that NTRC 0652–0 decreased tyrosine phosphorylation of multiple other proteins in a mass spectrometry-based, unbiased analysis of the tyrosine phosphoproteome. We confirmed the specificity of these effects with an inducible genetic knock-out of LCK, suggesting that the majority of identified proteins are bona fide LCK targets in CCA. Interestingly, the list of targets includes a small number of additional receptor tyrosine kinases, including EGFR. This suggests that LCK acts not only as a signal transducer downstream of receptor tyrosine kinases but can be an upstream regulator as well. Whether these additional targets are required for the cytotoxicity of LCK targeted therapy or represent biomarkers for sensitivity to LCK inhibition remains to be determined. Additionally, while NTRC 0652–0 is selective for LCK versus other kinase targets, inhibition of other identified kinases may contribute to the observed signaling changes, cytotoxicity, and tumor growth inhibition.
Regarding the translational potential of LCK inhibitors, Dasatinib is a multi-kinase inhibitor which can inhibit LCK in addition to other targets. Dasatinib is already in clinical use for hematologic malignancies driven by BCR-ABL rearrangement, and for metastatic gastrointestinal stromal tumors due to inhibition of KIT and PDGFR. Profiling suggests that dasatinib inhibits over 30 different kinases, including LCK and other SFKs. Although this polypharmacology has allowed dasatinib to be repurposed for multiple cancer indications, the lack of selectivity likely also contributes to toxicity. A more selective inhibitor of LCK, such as NTRC 0652–0, could potentially offer decreased toxicity.
Given the role of LCK in T-cell receptor signaling15, and effects of YAP on tumor immunology3, questions remain regarding the effect of LCK inhibition on the tumor immune microenvironment and response to LCK inhibition in immunocompetent settings. In future work, detailed examination of those dynamics including profiling and functional assessment of the immune repertoire and tumor growth phenotypes in syngeneic models of cholangiocarcinoma will be required to translate this approach.
In this study, LCK inhibition with NTRC 0652–0 demonstrated efficacy in PDO and PDX models harboring FGFR2 fusions. This is a clinically relevant genetic subset of CCA, for which FGFR inhibitors, pemigatinib and infigratinib, have accelerated FDA-approval in the advanced, previously treated setting.21, 22 The objective response rate of these agents was limited (23% - 36%) indicating that there is a significant proportion of FGFR2-altered CCA which is primarily resistant to FGFR targeted therapy.21, 22 Therefore, development of alternative therapeutic strategies that may be effective in this genetic subset is imperative.
The role of YAP in mediating oncogenesis downstream of FGFR2 has been investigated previously. Multiple receptor tyrosine kinases, including FGFR2, have been shown to phosphorylate YAP on tyrosine residues to promote its oncogenic functionality.23 We previously found that YAP can be activated downstream of FGFR2 in CCA specifically.11 Here we confirmed that an FGFR2-altered PDX model was sensitive to the YAP inhibitor CA3.
In gastric cancer, FGFR2 was shown to activate YAP via c-Jun, and co-targeting FGFR2 and YAP was therapeutic in preclinical models.24 In triple negative breast cancer, epigenetic YAP activation was identified as a resistance mechanism to FGFR2 inhibition, and co-targeting FGFR2 and YAP was efficatious.25 Given these results, combining FGFR and LCK inhibition could be a rational approach in future work.
Ultimately, successful implementation of LCK-targeted therapy for CCA patients will require identification of predictive biomarkers for sensitivity and resistance. While FGFR2 fusion is a relevant biomarker that is routinely assessed in current clinical care of cholangiocarcinoma patients, further work will be needed to define the predictive utility of YAPY357 phosphorylation and other phosphoproteins regulated by LCK inhibition.
Overall, these results demonstrate preclinical efficacy of a novel small molecule LCK inhibitor, NTRC 0652–0, in models of CCA with YAP dependence and FGFR2-rearranged CCA. These data support further development of LCK-targeted therapy as a novel approach in CCA.
Supplementary Material
Highlights.
LCK is a novel therapeutic target in cholangiocarcinoma
Cholangiocarcinoma organoid and xenograft tumor models respond to LCK inhibition
FGFR2-altered cholangiocarcinomas have enriched YAP activity and are sensitive to LCK inhibition
Acknowledgements
We thank Chantal McCabe for bioinformatics support. This work was supported by a Department of Defense Career Development Grant W81XWH-18-1-0297 (R.L.S), Eagles Cancer Research Fund Fellowship grant (C.B.C.), the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), a supplement for liver cancer infrastructure (NCI Cancer Center Support Grant 5P30 CA15083 -43C1), the Mayo Clinic Hepatobiliary SPORE from NCI (P50 CA210964), the Satter Family Fellowship in Liver Cancer Research, the Mayo Clinic Department of Surgery, and the Mayo Clinic. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The results are in part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Schematic diagram figures were created with BioRender.com.
Financial disclosure:
This work was supported by a Department of Defense Career Development Grant W81XWH-18-1-0297 (R.L.S), Eagles Cancer Research Fund Fellowship grant (C.B.C.), the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), a supplement for liver cancer infrastructure (NCI Cancer Center Support Grant 5P30 CA15083-43C1), the Mayo Clinic Hepatobiliary SPORE form the NCI (P50 CA210964), the Satter Family Fellowship in Liver Cancer Research, the Mayo Clinic Department of Surgery, and the Mayo Clinic.
These funding sources had no role in the conduct of the research or preparation of the article.
Abbreviations
- BAP
break apart probes
- CCA
cholangiocarcinoma
- CAPOX
capecitabine and oxaliplatin
- FISH
fluorescence in situ hybridization
- FOLFIRINOX
5-FU, leucovorin, irinotecan, oxaliplatin
- GEMCIS
Gemcitabine and cisplatin
- GEMOX
gemcitabine and oxaliplatin
- IC50
half maximal inhibitory concentration
- IHC
immunohistochemistry
- MMR
Mismatch repair
Footnotes
Competing interests: R.C. Buijsman is managing director and shareholder of Netherlands Translational Research Center B.V. Other authors declare no potential conflicts of interest.
References
Author names in bold designate shared co-first authorship.
- [1].Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nature Reviews Gastroenterology & Hepatology 2020;17:557–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Silverman IM, Hollebecque A, Friboulet L, Owens S, Newton RC, Zhen H, et al. Clinicogenomic Analysis of FGFR2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov 2021;11:326–339. [DOI] [PubMed] [Google Scholar]
- [3].Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer cell 2016;29:783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li X, Tao J, Cigliano A, Sini M, Calderaro J, Azoulay D, et al. Co-activation of PIK3CA and Yap promotes development of hepatocellular and cholangiocellular tumors in mouse and human liver. Oncotarget 2015;6:10102–10115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015;6:17206–17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yamada D, Rizvi S, Razumilava N, Bronk SF, Davila JI, Champion MD, et al. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology (Baltimore, Md) 2015;61:1627–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sugihara T, Werneburg NW, Hernandez MC, Yang L, Kabashima A, Hirsova P, et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Molecular cancer research : MCR 2018;16:1556–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harsha HC, Molina H, Pandey A. Quantitative proteomics using stable isotope labeling with amino acids in cell culture. Nat Protoc 2008;3:505–516. [DOI] [PubMed] [Google Scholar]
- [9].Konishi H, Yamauchi E, Taniguchi H, Yamamoto T, Matsuzaki H, Takemura Y, et al. Phosphorylation sites of protein kinase C δ in H<sub>2</sub>O<sub>2</sub>-treated cells and its activation by tyrosine kinase <em>in</em> <em>vitro</em>. Proceedings of the National Academy of Sciences 2001;98:6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Braunger J, Schleithoff L, Schulz AS, Kessler H, Lammers R, Ullrich A, et al. Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 1997;14:2619–2631. [DOI] [PubMed] [Google Scholar]
- [11].Rizvi S, Yamada D, Hirsova P, Bronk SF, Werneburg NW, Krishnan A, et al. A Hippo and Fibroblast Growth Factor Receptor Autocrine Pathway in Cholangiocarcinoma. The Journal of biological chemistry 2016;291:8031–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Farshidfar F, Zheng S, Gingras M-C, Newton Y, Shih J, Robertson AG, et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Reports 2017;18:2780–2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Song S, Xie M, Scott AW, Jin J, Ma L, Dong X, et al. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Molecular Cancer Therapeutics 2018;17:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wang Y, Ding X, Wang S, Moser CD, Shaleh HM, Mohamed EA, et al. Antitumor effect of FGFR inhibitors on a novel cholangiocarcinoma patient derived xenograft mouse model endogenously expressing an FGFR2-CCDC6 fusion protein. Cancer Letters 2016;380:163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nature Reviews Immunology 2018;18:485–497. [DOI] [PubMed] [Google Scholar]
- [16].Serafin V, Capuzzo G, Milani G, Minuzzo SA, Pinazza M, Bortolozzi R, et al. Glucocorticoid resistance is reverted by LCK inhibition in pediatric T-cell acute lymphoblastic leukemia. Blood 2017;130:2750–2761. [DOI] [PubMed] [Google Scholar]
- [17].Gocho Y, Liu J, Hu J, Yang W, Dharia NV, Zhang J, et al. Network-based systems pharmacology reveals heterogeneity in LCK and BCL2 signaling and therapeutic sensitivity of T-cell acute lymphoblastic leukemia. Nature Cancer 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Saygin C, Wiechert A, Rao VS, Alluri R, Connor E, Thiagarajan PS, et al. CD55 regulates self-renewal and cisplatin resistance in endometrioid tumors. Journal of Experimental Medicine 2017;214:2715–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zepecki JP, Snyder KM, Moreno MM, Fajardo E, Fiser A, Ness J, et al. Regulation of human glioma cell migration, tumor growth, and stemness gene expression using a Lck targeted inhibitor. Oncogene 2019;38:1734–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dey A, Varelas X, Guan KL. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat Rev Drug Discov 2020;19:480–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol 2020;21:671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Javle MM, Roychowdhury S, Kelley RK, Sadeghi S, Macarulla T, Waldschmidt DT, et al. Final results from a phase II study of infigratinib (BGJ398), an FGFR-selective tyrosine kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma harboring an FGFR2 gene fusion or rearrangement. Journal of Clinical Oncology 2021;39:265–265.33503392 [Google Scholar]
- [23].Azad T, Nouri K, Janse van Rensburg HJ, Maritan SM, Wu L, Hao Y, et al. A gain-of-functional screen identifies the Hippo pathway as a central mediator of receptor tyrosine kinases during tumorigenesis. Oncogene 2020;39:334–355. [DOI] [PubMed] [Google Scholar]
- [24].Zhang J, Wong CC, Leung KT, Wu F, Zhou Y, Tong JHM, et al. FGF18-FGFR2 signaling triggers the activation of c-Jun-YAP1 axis to promote carcinogenesis in a subgroup of gastric cancer patients and indicates translational potential. Oncogene 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li Y, Qiu X, Wang X, Liu H, Geck RC, Tewari AK, et al. FGFR-inhibitor-mediated dismissal of SWI/SNF complexes from YAP-dependent enhancers induces adaptive therapeutic resistance. Nature Cell Biology 2021;23:1187–1198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data are deposited to the ProteomeXchange Consortium via the PRIDE9 partner repository with the dataset identifier PXD026925. Other data generated in this study are available within the article and its supplementary files.