

Abstract
Phagocytosis is an intensely physical process that depends on the mechanical properties of both the phagocytic cell and its chosen target. Here, we employed differentially deformable hydrogel microparticles to examine the role of cargo rigidity in the regulation of phagocytosis by macrophages. Whereas stiff cargos elicited canonical phagocytic cup formation and rapid engulfment, soft cargos induced an architecturally distinct response, characterized by filamentous actin protrusions at the center of the contact site, slower cup advancement, and frequent phagocytic stalling. Using phosphoproteomics, we identified β2 integrins as critical mediators of this mechanically regulated phagocytic switch. Macrophages lacking β2 integrins or their downstream effectors, Talin1 and Vinculin, exhibited specific defects in phagocytic cup architecture and selective suppression of stiff cargo uptake. We conclude that integrin signaling serves as a mechanical checkpoint during phagocytosis to pair cargo rigidity to the appropriate mode of engulfment.
Subject terms: Phagocytes, Biophysics, Actin, Integrins
Phagocytosis is regulated by the mechanical properties of both the phagocyte and its cargo. Here, the authors show that macrophages employ β2 integrins to sense the rigidity of phagocytic cargo and then mount the appropriate form of engulfment.
Introduction
Macrophages maintain homeostasis in multicellular organisms by phagocytosing microbes, cell corpses, and biomolecular debris. They also play a central role in emerging immunotherapies against infectious diseases and cancer, which has spawned intense interest in the mechanisms that control their activity1–4. Macrophages and other professional phagocytes, including neutrophils and dendritic cells, consume cargo that bears the molecular indices of immune targeting (antibodies and complement), microbial origin (e.g., lipopolysaccharide), or cellular distress (e.g., phosphatidylserine (PtdS)). Recognition of these ligands is mediated by phagocytic receptors specific for each ligand class: Fc receptors and β2 integrins bind to opsonizing antibodies and the iC3b subunit of complement, respectively, TLR4 binds to lipopolysaccharide, and a structurally diverse group of cell surface proteins recognizes PtdS. Receptor engagement induces dramatic reorganization of filamentous (F)-actin at the interface, which shapes the overlying plasma membrane into a phagocytic cup that engulfs the cargo, ultimately leading to its internalization in an acidifying phagosome.
Phagocytic cargo varies widely in its chemical composition, architecture, and mechanical properties, and phagocytes have developed multiple uptake strategies to accommodate this diversity5,6. The morphological features of phagocytosis have traditionally been linked to specific biochemical modes of target engagement. Thus, antibody-induced engulfment is thought to occur via a “reaching” mechanism in which F-actin rich pseudopods promote the sequential engagement (zippering) of Fc receptors, enabling the phagocyte to surround and then consume its target. Conversely, complement-dependent uptake has been proposed to proceed by a “sinking” mechanism in which cargo is pulled into the phagocyte with minimal formation of membrane pseudopods. These distinctions have been challenged, however, by studies highlighting architectural similarities between Fc- and complement-mediated phagocytosis, in particular the importance of protrusive F-actin for both processes7,8. More recent work has also revealed an alternative internalization response in which cargo is fragmented before consumption9–11. This “nibbling” process has been proposed to facilitate collaborative phagocytosis, which may be essential for the consumption of large cargos12. Deciphering how and why distinct uptake mechanisms are applied will require a comprehensive understanding of how macrophages sense and classify their cargo.
In considering this question, it is important to bear in mind that phagocytic cargos vary not only biochemically but also mechanically, ranging from hard, elongated bacterial cells to irregularly shaped and highly deformable apoptotic corpses13–15. The implications of this biophysical diversity for phagocytosis are not well understood6,16. Within the phagocytic cup, macrophages apply nanonewton scale protrusive forces to palpate the target surface17, potentially providing an avenue for cargo mechanosensing. Among the physical properties that could be detected in this manner, rigidity is particularly interesting because it varies substantially among cargos. Polystyrene beads and red blood cells, for instance, differ by six orders of magnitude in their Young’s modulus14. Prior studies have explored the effects of cargo rigidity on phagocytosis using hydrogel microparticles of differential stiffness as well as red blood cell targets treated with fixatives like glutaraldehyde. In general, increased rigidity was found to enhance uptake, indicating that phagocytes are indeed mechanosensitive and that they prefer stiffer cargo8,18–20. The molecular and mechanical bases that underlie this preference, however, remain largely unknown.
In the present study, we investigated the specific effects of cargo stiffness on macrophage phagocytosis. Central to our efforts was the application of microfabrication methodology to generate phagocytic targets of defined size, rigidity, and surface ligand composition20. Our results demonstrate that cargo mechanosensing is a general property of macrophages that is independent of cell lineage and mode of target recognition. We also found that increasing cargo stiffness is associated with stereotyped changes in both the speed and the morphology of phagocytic contacts, indicating that macrophages apply distinct phagocytic programs depending on the mechanical properties of their targets. Finally, we showed that the β2 integrin adhesion molecules serve as a mechanical checkpoint pairing cargo mechanics to the mechanism of uptake.
Results
Cargo rigidity promotes phagocytic uptake
To isolate the role of cargo rigidity on phagocytosis, we employed a recently developed method for generating deformable polyacrylamide acrylic acid micro (DAAM) particles of differential stiffness20 (Fig. 1a). For this study, we prepared four sets of particles, all 9–11 µm in diameter (Fig. 1b and Supplementary Fig. 1a), with stiffnesses (Young’s moduli) spanning 0.6–18 kPa (Fig. 1c), which encompasses the range between leukocytes (low rigidity) and yeast (high rigidity). Particles were loaded with phagocytic ligands (IgG or PtdS) via a streptavidin–biotin linkage in order to induce macrophage uptake. Using confocal microscopy, we confirmed that a coating ratio of 10 pmol IgG per million particles (pmol/M) affords particles of identical coating density (Supplementary Fig. 1b–d). Unless otherwise stated, this coating density was used for all phagocytosis assays.
Fig. 1. A hydrogel microparticle system to interrogate cargo mechanosensing.

a Schematic diagram for DAAM particle synthesis (see “Methods” section). Particle rigidity depends on the ratio of cross-linker to monomer. AIBN azobisisobutyronitrile. b Representative images of DAAM particles, prepared using the indicated bis-acrylamide contents and conjugated to LRB and FITC. Scale bars = 10 µm. c Atomic force microscopy measurements of stiffness (Young’s modulus) for particles prepared using the indicated bis-acrylamide contents. n = 36 (for 0.234% and 0.04%) or 34 (for 0.065% and 0.032%) DAAM particles. Error bars denote standard deviation (SD). d, e BMDMs were challenged with DAAM particles coated with different phagocytic ligands and particle uptake quantified by flow cytometry. d Top, schematic diagram for LRB/FITC dependent tracking of phagocytosis. Bottom, representative gating for flow cytometry-based quantification. e Uptake efficiency of DAAM particles coated with the indicated phagocytic ligands is graphed against particle stiffness. Columns and error bars denote mean and standard deviation (SD), respectively. n = 3 biological replicates. f–h BMDMs were imaged together with IgG-coated DAAM particles. f Representative brightfield and epifluorescence images of BMDMs phagocytosing DAAM particles of the indicated stiffness. Phagocytosed particles turn red, while unengulfed particles are yellow. Scale bars = 50 µm. g Phagocytosis quantified as the fraction of particles acidified per field of view after 2 h. Each point represents the mean of four fields of view in one technical replicate. Horizontal lines in each column denote the overall mean. P value determined by two-tailed Student’s t-test. h Histogram showing the number of 18 and 0.6 kPa particles phagocytosed per cell after 2 h. Schematics in (a), (d), and (e) created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en.
For most of our experiments, DAAM particles were conjugated to FITC, a pH-sensitive dye that fluoresces green, and Lissamine Rhodamine B (LRB), a pH insensitive red fluorophore. Uptake into an acidic phagolysosome quenches FITC fluorescence, enabling the identification of phagocytic macrophages by flow cytometry as LRB+FITClo cells (Fig. 1d and Supplementary Fig. 1e, f). In this manner, we quantified the phagocytic activity of primary murine bone marrow-derived macrophages (BMDMs) as a function of DAAM particle stiffness. These experiments revealed a distinct preference of macrophages for stiffer cargos; uptake increased monotonically with particle rigidity for both IgG-dependent and PtdS-dependent phagocytosis (Fig. 1e). We observed a similar pattern of results over a range of ligand coating densities (Supplementary Fig. 1g), indicating that mechanosensing occurs independently of target quality. We also performed live wide-field microscopy of BMDMs co-incubated with stiff (18 kPa) or soft (0.6 kPa) DAAM particles bearing IgG or PtdS (Supplementary Fig. 2a). Automated analysis of the imaging data indicated that the stiff cargo was more effectively engulfed and acidified on both a population level and a per-cell basis, with 24% of macrophages taking up two or more particles (Fig. 1f–h and Supplementary Fig. 2b–e). High stiffness also augmented the uptake of particles coated with streptavidin alone, BSA, or no protein at all, suggesting that this effect does not depend on a specific phagocytic ligand (Supplementary Fig. 2f). Collectively, these results demonstrate that stiffness promotes the phagocytosis of chemically diverse cargos, which is consistent with prior reports8,18–20.
Next, we assessed the generality of this behavior by measuring DAAM particle phagocytosis by different macrophage populations and in different contexts. Murine macrophages derived from HoxB8-ER-immortalized progenitor cells1,21 displayed a similar preference for stiffer cargo in both IgG and PtdS-dependent phagocytosis assays (Fig. 2a). We observed analogous results using human macrophages differentiated from induced pluripotent stem cells (hiPSCs) (Fig. 2b), indicating that cargo mechanosensing is not a species-specific phenomenon. We also examined microglia, the professional phagocytes of the central nervous system, which reside in an environment that is markedly softer than that of other macrophage subsets. Murine microglia consumed substantially more stiff (18 kPa) than soft (0.6 kPa) DAAM particles (Fig. 2c), strongly suggesting that this form of mechanosensing applies to most, if not all, macrophages. To assess whether the preference for stiff cargo also applies in vivo, we injected IgG-coated particles of differential rigidity into the peritonea of adult mice and measured phagocytosis by peritoneal macrophages after 2 h (Fig. 2d and Supplementary Fig. 3). In vivo uptake of stiff particles was robust, whereas uptake of soft particles was barely detectable (Fig. 2e). Importantly, macrophage recruitment to the peritoneum was comparable in both injection regimes (Fig. 2f), indicating that the observed preference for stiff particles did not result from differences in macrophage–particle encounter frequency. Together, these results suggest that cargo mechanosensing is a general phenomenon that applies to a wide range of macrophage cell types and environments.
Fig. 2. High cargo rigidity promotes phagocytic uptake.

a Phagocytic efficiency of HoxB8-ER derived macrophages challenged with IgG- and PtdS-coated DAAM particles of the indicated rigidities, quantified by flow cytometry. n = 3 replicate macrophage differentiations. hiPSC-derived macrophages (b) and murine microglia (c) were mixed with IgG- and PtdS-coated DAAM particles of the indicated rigidities, and phagocytosis monitored by video microscopy. Representative images of uptake of IgG-coated particles are shown to the left, with phagocytic index quantified on the right. Phagocytic index = number of particles phagocytosed per macrophage. In (b), n = 4 replicate macrophage differentiations, while in (c), n = 2 biological replicates (summary of 4 technical replicates/biological replicate). Scale bars in (b, c) = 30 µm. d–f DAAM particles of differential stiffness were injected into the mouse peritoneum to measure the phagocytic activity of elicited macrophages. d Schematic of macrophage elicitation and DAAM particle injection protocol (see “Methods” section). e Phagocytic efficiency (LRB+FITClo/all CD11b+F4/80+ cells) for particles of the indicated rigidities. n = 5 mice/condition. f Macrophage recruitment to the peritoneum, quantified as total F480+CD11b+ cells over all live cells in the peritoneal lavage. In (a–c) and (e, f), horizontal lines in columns denote mean values. All P values determined by two-tailed Student’s t-test. ns not significant. g–i BMDMs were challenged with Cypher5e-labeled E0771 cells expressing different levels of MRTF-A. g MRTF-A is expected to enhance phagocytosis by increasing cell stiffness. h Representative flow cytometry histogram showing Cypher5e signal in BMDMs challenged for 2 h with control E0771 cells or E0771 cells overexpressing MRTF-A. i Phagocytosis is quantified as %Cypher5e+ macrophages. Error bars indicate SD. n = 3 replicate macrophage differentiations. Schematics in (a–d), and (g) created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en.
In light of these observations, we hypothesized that mechanically rigidifying cancer cells would promote their phagocytosis by macrophages. Overexpression of myocardin-related transcription factors (MRTFs) increases the Young’s modulus of multiple cell types without substantially altering their surface composition22 (Fig. 2g). Accordingly, we challenged BMDMs with E0771 breast cancer cells bearing an inducible MRTF-A expression cassette. These cells were stained with the pH-sensitive dye Cypher5e, and phagocytosis was measured in both the presence and the absence of anti-CD47 antibodies, which were used to promote Fc-dependent uptake and to block phagocytic “don’t eat me” signaling (Supplementary Fig. 2g). MRTF-A overexpression markedly increased E0771 uptake in these experiments, independently of anti-CD47 treatment (Fig. 2h, i). To determine whether this result depended specifically on the cytoskeletal effects of MRTF-A, target cells were incubated with mycalolide B (MycB), a small molecule toxin that depolymerizes F-actin in a sustained manner23. MycB pretreatment completely abrogated the enhanced phagocytosis of MRTF-A overexpressing cells (Fig. 2i), strongly suggesting that MRTF-induced rigidification is the critical determinant of this effect. Hence, cargo stiffness promotes the uptake of both live and inanimate phagocytic cargos.
Phagocytosis of soft cargo is slow and prone to stalling
To better understand the underlying mechanism of cargo mechanosensing, we analyzed phagocytic kinetics in wide-field videos of BMDMs engulfing stiff (18 kPa) and soft (0.6 kPa) DAAM particles (Fig. 3a, b). For each engulfment event, we scored three key steps: (1) contact, defined as the first frame the particle contacts the cell; (2) phagocytic cup initiation, visualized as an inversion in brightfield intensity around the particle; and (3) particle acidification, defined as the frame at which the FITC/LRB ratio reaches its minimum plateau (see “Methods” section). Whereas stiffness did not affect the time delay between particle contact and cup initiation (Fig. 3c and Supplementary Fig. 4a), the interval between initiation and particle acidification was significantly faster during stiff phagocytosis (Fig. 3d and Supplementary Fig. 4b). Given that the maximal rate of soft particle acidification was in fact higher than that of stiff particles (Supplementary Fig. 4c), we surmised that phagocytic mechanosensing occurs between cup initiation and closure.
Fig. 3. Phagocytosis of soft targets is slow and prone to stalling.

a–c BMDMs were challenged with 18 or 0.6 kPa IgG-coated DAAM particles and imaged by wide-field videomicroscopy. Above, time-lapse montages showing representative 18 kPa (a) and 0.6 kPa (b) uptake events. Fluorescence (LRB/FITC) and brightfield (BF) channels are shown individually and in merge. Particles undergoing phagocytosis are indicated by white arrows in the merged images. The BMDM of interest is outlined in yellow/black in the first image of each time-lapse. Time is indicated as M:SS in the top right of the fluorescence images, with time of first contact defined as t = 0. Scale bars = 30 µm. c Recognition time, defined as time difference between cup formation and first contact, n = 39 cells. ns not significant, two-tailed Welch’s t-test. d Total acidification time, defined as the difference between particle acidification and cup formation, n = 39 cells. P value determined by two-tailed Welch’s t-test. In (c) and (d), mean values and error bars (denoting SD) are indicated in red. e–g HoxB8-ER macrophages expressing F-tractin-mCherry were challenged with stiff (18 kPa) or soft (0.6 kPa) IgG-coated DAAM particles and imaged by spinning disk confocal microscopy. e Time-lapse montages of representative phagocytic events, visualized as side-views of 3D reconstructions. F-tractin is depicted in pseudocolor, with warmer colors indicating higher fluorescence, and the particle is shown in gray. A 40-min time point from the same 0.6 kPa particle conjugate is included for reference. Scale bars = 10 µm. f Mean phagocytic cup velocity (see “Methods” section and Supplementary Fig. 4d, e) on stiff and soft DAAM particles. Blue and red points denote cups that completed or failed to complete within 30 min, respectively. Statistical significance (p < 0.0001) includes all events, p = 0.0002 using only completed events, two-tailed Welch’s t-test. g Fraction of events that completed (blue) or did not complete (red) within 30 min. Completion defined as actin covering >95% of the particle at any time and persisting until the end of capture. h–i BMDMs were challenged with IgG-coated DAAM particles of different rigidities for various times, and then fixed and stained with phalloidin to visualize F-actin. h Left, Schematic for quantification of percent F-actin coverage on fixed phagocytic cups. Right, F-actin coverage for various particle rigidities, graphed against time. Values are sorted into five bins, with colors denoting the fraction of events falling within each bin. n = 15 events per time/stiffness condition (180 total events) in one representative experiment. i Quantification of stalling frequency, defined as the fraction of events that are 20–95% complete after 30 min co-incubation with BMDMs. n = 4 biological replicates (one mouse per replicate). P value determined by two-tailed paired t-test. j Representative en face maximum z-projections of F-actin in partial phagocytic cups formed on 18 and 0.6 kPa DAAM particles, taken from live imaging experiments. Color scale represents fluorescence intensity values divided by the maximum fluorescence intensity of each image. F-actin accumulation (k), defined as the mean phalloidin intensity at the particle interface divided by the mean phalloidin intensity over the entire cell, and F-actin clearance index (l), which compares F-actin signal at the periphery and the center of the phagocytic cup (see “Methods” section), determined in phagocytic cups formed with 18 kPa (5 min) and 0.6 kPa (5 or 30 min) DAAM particles. In (k) and (l), error bars denote SD. P values determined by two-tailed Welch’s t-test. n = 37, 48, 40 cells for 18 kPa, 0.6 kPa 5 min, and 0.6 kPa 30 min, respectively. Schematics in (h), (k), and (l) created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en.
Guided by these results, we turned to high speed (15 s interval) confocal microscopy to measure the rate of phagocytic cup closure more directly. These experiments utilized HoxB8-ER macrophages expressing F-tractin-mCherry, a probe for F-actin, which we mixed with fluorescently labeled DAAM particles just prior to imaging. F-actin rapidly accumulated at both stiff particle and soft particle interfaces. On stiff (18 kPa) particles, this accumulation promptly transitioned into a radially symmetric phagocytic cup containing a thick band of leading-edge F-actin, which advanced rapidly (~0.12 um/s) around the cargo, leading to full engulfment within 10 min of contact formation (Fig. 3e–g; Supplementary Fig. 4d; Supplementary Movie 1). Soft (600 Pa) particle contacts were markedly different: although macrophages appeared to form phagocytic cups on the target, most (10/15) failed to complete engulfment within 30 min, with little to no advancement of the phagocytic cup during that time (Fig. 3e–g; Supplementary Fig. 4d, e; Supplementary Movies 2, 3). This behavior, which we defined as “stalled phagocytosis”, was not observed in contacts with stiff particles (0/13) (Fig. 3g). Even in the minority of cases where soft particle engulfment did complete, phagocytic cup advancement was approximately threefold slower than the rate observed with stiff particles (Fig. 3f).
To confirm that phagocytic stalling is indeed a function of target rigidity, we imaged conjugates of BMDMs and IgG-coated DAAM particles, which had been fixed and stained for F-actin at various times after mixing (Fig. 3h). At early timepoints (5 and 10 min), samples containing stiff (18 kPa) particles exhibited both partially and fully internalized beads, indicative of ongoing and completed phagocytosis, respectively (Fig. 3h). After 30 min, all observed 18 kPa particles were fully internalized, implying that all phagocytic cups had fully enclosed their cargo by this time point. In contrast, samples containing softer DAAM particles (3, 1.5, 0.6 kPa) exhibited a substantial degree of incomplete engulfment at 30 min, consistent with the formation of stalled phagocytic cups (Fig. 3h, i). Notably, the fraction of stalled events at 30 min increased monotonically with decreasing particle rigidity, strongly suggesting that the likelihood to stall directly reflects target deformability. Hence, at the level of individual cells, phagocytic mechanosensing of DAAM particles manifests as a binary choice between the rapid “gulping” of cargo and an alternative stalling response.
Phagocytic cup maturation is typically associated with the clearance of F-actin from the center of the phagocyte–cargo interface24–26, which facilitates internalization of the newly formed phagosome into the cytoplasm. In videos of macrophages bound to soft DAAM particles, however, we typically observed a network of protrusive F-actin structures in the center of the contact for the entire duration of the experiment (~40 min) (Fig. 3j and Supplementary Movies 2, 3). This network was highly dynamic, with individual protrusions forming, moving, and dissolving on the timescale of minutes. The larger protrusions also physically distorted the cargo, creating micron-scale indentations on the particle surface (Supplementary Fig. 4f). This complex and heterogeneous F-actin architecture contrasted sharply with the rapidly progressing phagocytic cups seen during stiff cargo engulfment, which were characterized by the strong accumulation of F-actin at the leading edge and concomitant F-actin clearance from the central domain (Supplementary Movie 1).
To analyze these patterns in more detail, we mapped the F-actin signal of fixed, phalloidin-stained phagocytic cups onto the 3D rendered surfaces of their microparticle targets using a previously described analytical approach17,20,27 (Supplementary Fig. 4g). Our initial comparisons focused on early stage (<60% complete) interactions captured 5 min after BMDM-DAAM particle mixing. Contacts with stiff (18 kPa) and soft (0.6 kPa) particles both exhibited strong F-actin accumulation (Fig. 3k), which we measured by dividing the mean F-actin intensity within the phagocytic cup by the mean intensity over the whole cell. The morphologies of these accumulations, however, were markedly different. Stiff particle contacts displayed a ring-like configuration, with strong F-actin enrichment at the front and clearance from base of the phagocytic cup, whereas soft particle contacts were more disorganized, with a poorly defined front and punctate structures at the base (Supplementary Fig. 4g). To quantify these distinct patterns, we extracted two perpendicular intensity profiles through the center of the contact, then defined an actin clearance index as the ratio between mean F-actin intensity in the inner 50% of the contact and mean intensity in the outer 50%. By this metric, early phagocytic cups that formed on stiff particles were significantly more “ring-like” (lower clearance index) than their soft particle counterparts (Fig. 3l). We applied the same analysis to soft particle cups that remained incomplete after 30 min, presumably due to phagocytic stalling. These contacts were even less ring-like than early stage soft particle interactions (Fig. 3l), suggesting that stalling is caused, at least in part, by the failure to clear central F-actin.
β2 integrins respond differentially to stiff and soft cargo
We next sought to identify putative mediators of cargo mechanosensing. Reasoning that some of these molecules would be differentially phosphorylated in response to cargo rigidity, we performed phosphoproteomic analysis of BMDMs shortly after contact formation with IgG-coated DAAM particles (Fig. 4a), leading to the identification of proteins that were preferentially phosphorylated upon challenge with either soft (0.6 kPa) or stiff (18 kPa) targets (Supplementary Fig. 5a, b and Supplementary Data 1). Gene ontogeny analysis of the phosphosites enhanced by engagement with stiff particles revealed associations with the F-actin cytoskeleton, membrane reorganization, as well as both Rac and Rho GTPase signaling (Supplementary Fig. 5c). One of the most prominent of these sites was phosphothreonine 760 (pT760) of the β2 integrin subunit, which is also called CD18 (Fig. 4b, c and Supplementary Fig. 5a). This finding interested us because Fc receptor engagement drives the affinity maturation of integrins from a bent, autoinhibited conformation into an extended structure capable of ligand binding28,29. Furthermore, the β2 integrin Mac-1, a heterodimer composed of CD18 and CD11b (αM), is the established phagocytic receptor for complement. β2 integrins contribute to complement-independent forms of phagocytosis7,10, as well, although the basis for this involvement is poorly understood. Importantly, phosphorylation of human CD18 on T568, which is orthologous to mouse T760, has been associated with increased engagement of cytoskeletal adaptor molecules30,31, hinting at a role for this protein in mechanosensing.
Fig. 4. β2 integrins respond differently to stiff and soft targets.

a, b BMDMs were challenged with 18 and 0.6 kPa IgG-coated DAAM particles and then subjected to phosphoproteomic analysis. a Schematic diagram of phosphoproteomic sample preparation—BMDMs and microparticles were co-incubated for 10 min before transferring to ice, and cells were then lifted and pelleted at −80 °C. b Volcano plot of the resulting phosphopeptides, with negative and positive log fold-change (LogFC) denoting enrichment with 18 and 0.6 kPa particles, respectively. LogFC and significance were calculated using 8 independent replicates for each condition. c Above: sequence of Itgb2758–773, with phosphorylated threonine highlighted in red. Below left, normalized abundance values of the Itgb2758–773 phosphopeptide. Below right, normalized abundance of all Itgb2 peptides without phospho-enrichment. n = 8 replicates (independent macrophage differentiations from three pooled mice). Error bars denote SD. P values determined by two-tailed t-test, with Bonferroni adjustment. ns not significant. d–f BMDMs were challenged with IgG-coated DAAM particles of different rigidities for various times, fixed and stained for F-actin and CD18, and then imaged by confocal microscopy. d Representative images of DAAM particle–macrophage conjugates after 5 min incubation, stained with anti-CD18 antibody (cyan) and phalloidin (F-actin, magenta). Rows 1 and 3: top–down maximum intensity projections, rows 2 and 4: side-view maximum intensity projections. Scale bars = 10 µm. CD18 accumulation (e) defined as the ratio of CD18 at the particle interface divided by total CD18 in the cell, and CD18 clearance index (f) comparing CD18 signal at the periphery and the center of the phagocytic cup (see “Methods” section) determined in phagocytic cups formed with 18 kPa (5 min) and 0.6 kPa (5 and 30 min) DAAM particles. In (e) and (f), error bars denote SD. P values determined by two-tailed Welch’s t-test. n = 58, 53, and 15 cells for 18 kPa, 0.6 kPa 5 min, and 0.6 kPa 30 min, respectively. Schematics in (a), (e), and (f) created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en.
To investigate this possibility, we examined the localization of CD18 during phagocytosis. BMDMs were fixed 5 min after challenge with IgG-coated DAAM particles, stained for F-actin and CD18, and then imaged. CD18 strongly accumulated in phagocytic cups engaging both stiff (18 kPa) and soft (600 Pa) particles (Fig. 4d), which was notable given that β2 integrins are not known to bind directly to IgG or FcγR28. To quantify this behavior, we calculated a CD18 accumulation ratio by dividing the mean fluorescence intensity in direct contact with the particle by the intensity across the whole cell. This analysis revealed marked and largely rigidity-independent CD18 recruitment to the phagocytic cup (Fig. 4e), consistent with a role for β2 integrins in the direct recognition and mechanical interrogation of cargo.
Next, we assessed the organization of CD18 at the BMDM–cargo interface using the clearance ratio methodology applied above to profile F-actin (see Fig. 3l). Whereas CD18 formed ring-like structures on stiff particles, a more diffuse pattern prevailed during engagement with soft cargo, with appreciable accumulation at the center of the contact (Fig. 4f). Diffuse localization of CD18 was also observed in stalled phagocytic cups captured 30 min after mixing with soft particles (Fig. 4f). These data mirrored our results with F-actin, implying that CD18 and F-actin are recruited to at least some shared structures during phagocytosis. Consistent with this interpretation, we observed higher colocalization, determined by Pearson’s correlation, between F-actin and CD18 at the contact site compared to the whole macrophage (Supplementary Fig. 5d). This correspondence was not perfect, however, particularly in conjugates with stiff particles, where CD18 signal appeared to lag behind F-actin at the leading edge of the phagocytic cup. To quantify this behavior, we determined the distance between the F-actin and CD18 fronts in each contact, which we then normalized to the particle circumference. The resulting “Lag Distance” parameter was significantly larger in phagocytic cups containing 18 kPa particles (Supplementary Fig. 5e), indicating that an F-actin rich leading edge lacking β2 integrins is a characteristic feature of stiff cargo engulfment. Hence, similar to our observations with F-actin, β2 integrin organization, but not accumulation, depends on cargo rigidity.
β2 integrins are required for increased phagocytosis of stiff cargo
To better understand the role of β2 integrins during phagocytosis, we turned to genetic loss-of-function approaches. First, we applied CRISPR/Cas9 technology to target exon 2 of the Itgb2 locus (encoding CD18) in HoxB8-ER myeloid progenitors, which we then differentiated into CD18 deficient (CD18-KO) macrophages (Fig. 5a and Supplementary Fig. 6a). Control macrophages were prepared in parallel using a nontargeting (NT) guide RNA. Upon challenge with IgG-coated DAAM particles, CD18-KO macrophages exhibited two phagocytosis defects. The first was a significant reduction in the uptake of both stiff (18 kPa) and soft (0.6 kPa) particles, consistent with previous findings32. Second, and more intriguing, was a complete lack of preference for stiff over soft targets, indicating that CD18-KO macrophages are incapable of cargo mechanosensing (Fig. 5b). To examine this phenotype more closely, we derived and compared BMDMs from Itgb2−/− mice33 and Itgb2+/− littermate controls (Fig. 5c). Deletion of Itgb2 resulted in tenfold reduced expression of CD11b (αM), the predominant CD18 binding partner, but did not affect the levels of FcγRII/III and CD29 (β1 integrin) (Supplementary Fig. 7). CD61 (β3 integrin) expression was low on both Itgb2+/− and Itgb2−/− macrophages (Supplementary Fig. 7). In phagocytosis assays, Itgb2−/− BMDMs displayed both a global decrease in cargo uptake as well as reduced discrimination between stiff and soft targets (Fig. 5d and Supplementary Fig. 6b, c), similar to the behavior of CD18-KO HoxB8-ER macrophages. Interestingly, the stiffness independent part of this phagocytosis defect was rescued by increasing the concentration of IgG used for DAAM particle coating to 100 pmol/M from 10 pmol/M (Fig. 5e). High density IgG did not, however, restore mechanosensing; stiff and soft particles were still phagocytosed by Itgb2−/− BMDMs to the same extent (Fig. 5e). Hence, reduced cargo uptake and defective mechanosensing are separable aspects of the β2 integrin loss-of-function phenotype.
Fig. 5. β2 integrin signaling is necessary for phagocytic mechanosensing.

a Cas9+ HoxB8-ER conditionally immortalized progenitors were transduced with either a non-targeting sgRNA or sgRNA targeting exon 2 of CD18, and then differentiated into macrophages. b sgCD18 HoxB8-ER macrophages and sgNT controls were challenged with IgG-coated 18 and 0.6 kPa DAAM particles. Graph shows normalized phagocytic efficiency, defined as the ratio of percent phagocytic cells relative to the sgNT, 0.6 kPa sample. n = 3 biological replicates from independent differentiations. P values determined by two-tailed ratio paired t-test. ns not significant. c Diagram schematizing the preparation of BMDMs from Itgb2−/− mice. Heterozygous (Itgb2+/−) and homozygous knockout mice (Itgb2−/−) mice were bred and littermate F1 offspring of each genotype were compared in each biological replicate. Itgb2−/− BMDMs and Itgb2+/− controls were challenged with 18 and 0.6 kPa DAAM particles coated with 10 pmol/M (d) and 100 pmol/M (e) IgG. Graphs show phagocytic efficiency normalized against the Itgb2+/−, 0.6 kPa sample. n = 3 biological replicates for each genotype. P values determined by two-tailed ratio paired t-test. ns not significant. f Phagocytic efficiency of HoxB8-ER derived macrophages challenged with 100 pmol/M IgG-coated DAAM particles for 1 h, in the presence or absence of 500 µM MnCl2 added at the start of incubation. n = 3 independent macrophage differentiations. P values determined by two-tailed unpaired Student’s t-test. g F-actin coverage after 30 min incubation, determined for Itgb2−/− BMDMs and Itgb2+/− controls. Values are sorted into five bins, with colors denoting the fraction of events falling within each bin. Representative bars from one biological replicate are shown, from 20 events/condition. h Fraction of events that were 20–95% complete after 30 min co-incubation of DAAM particles with BMDMs. n = 3 biological replicates (litter-mate pairs, 20 events per condition/replicate). P values determined by two-tailed paired t-test. All error bars denote SD. Schematics in (a) and (c–e) created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en.
As a gain-of-function approach, we applied the divalent cation Mn2+, which promotes integrin affinity maturation by binding preferentially to the extended state34. Mn2+ treatment further increased the phagocytosis of stiff particles without altering the uptake of soft targets (Fig. 5f and Supplementary Fig. 6g), confirming that integrins play a key role in phagocytic mechanosensing. Mn2+ also enhanced stiff phagocytosis by CD18-KO macrophages, implying that the exogenous activation of other integrins can partially compensate for the absence of β2 isoforms.
Notably, the capacity of β2 integrins to augment stiff cargo uptake was independent of a specific phagocytic ligand. When challenged with particles coated with PtdS or streptavidin alone, macrophages lacking CD18 displayed significantly reduced overall phagocytosis and abrogation of cargo mechanosensing (Supplementary Fig. 6d–f), the same pattern of results we observed for IgG-coated targets. Given that certain β2 integrins can bind to denatured proteins35,36, we speculated that the CD18 dependent but ligand independent cargo mechanosensing we observed resulted from non-specific recognition of serum components adsorbed onto the DAAM particles. In fact, removing serum during particle coating or during the phagocytosis assay itself had no effect on the preferential uptake of stiff over soft IgG-coated cargo (Supplementary Fig. 6h). For particles bearing streptavidin alone, serum treatment actually dampened the mechanosensing response (Supplementary Fig. 6i). Collectively, these results suggest that β2 integrins bind directly either to streptavidin or to the polyacrylamide DAAM particle matrix. We favor the latter possibility, given that macrophages engulf stiff particles lacking even streptavidin coating (Supplementary Fig. 2f). Regardless of the actual ligand, our results clearly indicate that β2 integrins can mediate cargo mechanosensing via non-specific interactions with the target surface.
β2 integrins enable distinct responses to cargo stiffness
We reasoned that β2 integrin adhesion may act as a mechanical checkpoint to determine whether a phagocytic cup advances rapidly or stalls. To test this hypothesis, we challenged Itgb2−/− BMDMs and Itgb2+/− controls with IgG-coated DAAM particles and assessed phagocytic cup progress after 30 min by fixing and straining for F-actin. High concentration IgG coating was used in this experiment to increase conjugate formation in the Itgb2−/− samples. Stalling by Itgb2+/− BMDMs increased with decreasing cargo rigidity (Fig. 5g, h), mirroring the results we obtained using particles coated with low density IgG (see Fig. 3h, i). In striking contrast, Itgb2−/− BMDMs exhibited little to no stalling in any stiffness regime (Fig. 5g, h). With even the softest (0.6 kPa) microparticles, nearly all cell–particle conjugates had either detached or achieved complete engulfment by 30 min. The “all-or-nothing” behavior of Itgb2−/− macrophages raised the possibility that the phagocytic stalling induced by soft cargo does not simply represent a failure to gulp, but rather a second, distinct type of β2 integrin-dependent response. To investigate this idea, we compared F-actin architecture in phagocytic cups formed by Itgb2−/− BMDMs and Itgb2+/− controls 5 min after mixing with IgG-coated DAAM particles of differing stiffness (Fig. 6a–d). As expected, Itgb2+/− BMDMs exhibited high F-actin enrichment against both stiff (18 kPa) and soft (0.6 kPa) targets (Fig. 6a, b, e), with annular configuration predominating in stiff contacts and a more uniform accumulation on soft contacts (Fig. 6f). These characteristic patterns were almost completely abrogated in Itgb2−/− BMDMs, whose phagocytic cups contained F-actin but lacked any discernable F-actin enrichment compared to the rest of the cell (Fig. 6c–f). Together, these unexpected results indicate that β2 integrins play instructive roles in both the gulping phagocytosis of stiff cargo and the stalling response to soft targets.
Fig. 6. β2 integrin-mediated adhesion drives actin reinforcement at the phagocytic cup.

Itgb2−/− BMDMs and Itgb2+/− controls were challenged for various times with DAAM particles of differing rigidity coated with 100 pmol/M IgG. They were then fixed, stained for F-actin, and imaged by confocal microscopy. Representative maximum z-projections (top) and y-projections (bottom) of Itgb2+/− (a, b) and Itgb2−/− (c, d) BMDMs in the act of engulfing 18 kPa (a, c) and 0.6 kPa (b, d) DAAM particles (green). F-actin is visualized in magenta. Scale bars = 10 µm. e F-actin accumulation (see “Methods” section) for phagocytic cups, graphed as a function of particle stiffness and Itgb2 genotype. From left to right, n = 63, 57, 59, 58 cell–particle conjugates examined over three independent experiments. P values determined by two-tailed Welch’s t-test. f F-actin clearance index (see “Methods” section) for phagocytic cups graphed as a function of particle stiffness and Itgb2 genotype. From left to right, n = 55, 49, 50, 42 cell–particle conjugates examined over three independent experiments. P values determined by two-tailed Welch’s t-test. Columns and error bars in (e, f) denote mean and SD, respectively. Schematics in (e) and (f) created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en.
To examine the interplay between cargo stiffness, integrin engagement, and phagocytosis using bona fide biological targets, we compared the phagocytosis of control E0771 cells with the uptake of E0771 cells overexpressing MRTF-A. Target cell lines were stained with both plasma membrane and nuclear dyes and then imaged live together with either Itgb2+/− or Itgb2−/− BMDMs. Close inspection of the resulting time-lapse videos revealed two distinct forms of engulfment: whole cell gulping, which was characterized by nuclear uptake together with membrane (Supplementary Fig. 8a, white arrows), and “nibbling”, defined as the ingestion of small (~1 μm diameter) cell fragments containing the membrane stain alone (Supplementary Fig. 8c, yellow arrows). The nibbling behavior was similar to the “biting” or trogocytosis previously described in various immune cell subtypes, including macrophages and neutrophils37–39. Experiments were performed in both the presence and the absence of anti-CD47, which augmented phagocytosis somewhat but did not alter the overall pattern of responses. Relative to E0771 controls, E0771 cells overexpressing MRTF-A were more likely to be ingested via gulping and less likely to be subjected to nibbling (Supplementary Fig. 8b, d), in line with the interpretation that stiffness promotes the former uptake mechanism at the expense of the latter. Importantly, Itgb2−/− BMDMs exhibited reduced whole cell eating overall and no preference for consuming MRTF-A overexpressing cells by this mechanism (Supplementary Fig. 8b). These data, which mirror our results with stiff DAAM particles, lend further support to the idea that β2 integrins function as crucial activators of gulping. Notably, Itgb2 deficiency had no effect on phagocytic nibbling (Supplementary Fig. 8d), suggesting that this mode of uptake is independent of β2 integrin engagement, at least under these experimental conditions. We conclude that β2 integrins translate mechanical cues into distinct responses to cargo.
Mechanotransduction by Talin1 and Vinculin overcomes phagocytic stalling
Integrins mediate mechanosensing through the scaffolding proteins Talin1 and Vinculin, which form mechanically driven complexes that couple integrins to the F-actin cytoskeleton and initiate downstream signaling40,41. To evaluate the importance of Talin1 and Vinculin for cargo mechanosensing, we prepared CRISPR/Cas9 knockout macrophages lacking each molecule using the HoxB8-ER system (Fig. 7a and Supplementary Fig. 9a). As neither loss-of-function cell line expressed the related Talin isoform Talin2 (Supplementary Fig. 9b), we concluded that both lacked the machinery required to form canonical integrin adhesions. Importantly, depletion of either Talin1 or Vinculin significantly reduced the phagocytosis of stiff (18 kPa) DAAM particles without affecting soft (0.6 kPa) particle uptake (Fig. 7b). Taken together with the CD18-focused experiments described in the previous section, these data strongly suggest that β2 integrin adhesions drive the accelerated gulping of stiff cargo.
Fig. 7. Talin1 and Vinculin mediated mechanosensing is required for enhanced uptake of stiff cargo.

a, b Cas9+ HoxB8-ER conditionally immortalized progenitors were transduced with either a non-targeting sgRNA or sgRNA targeting Talin1 or Vinculin, and then differentiated into macrophages. These cells were then challenged with 10 pmol/M IgG-coated 18 kPa or 0.6 kPa DAAM particles. Graphs show phagocytic efficiency normalized against the sgNT, 0.6 kPa sample. n = 3 biological replicates for each genotype. P values determined by two-tailed paired ratio t-test. ns not significant. c F-actin coverage after 30 min incubation, determined for Talin1 and Vinculin CRISPR KOs (sgTalin1, sgVinculin) and non-targeting controls (sgNT). Values are sorted into five bins, with colors denoting the fraction of events falling within each bin. Representative bars from one biological replicate are shown, from 18 (sgVinculin) or 20 (sgNT and sgTalin1) events/condition. d Fraction of events that are 20–95% complete after 30 min co-incubation of 100 pmol/M IgG-coated DAAM particles with HoxB8-derived macrophages. n = 3 replicate differentiations (15–20 events/condition). P values determined by two-tailed paired t-test. e Representative images of BMDM-particle interactions showing Vinculin (yellow) and phalloidin (F-actin, magenta) staining, along with DAAM particles in green. Each panel contains top views (right) and side views (left). Scale bars = 5 µm. f Vinculin accumulation (see “Methods” section) at phagocytic cups, graphed as a function of particle stiffness and Itgb2 genotype. From left to right, n = 47, 78, 41, 47 cell–particle conjugates measured over three independent experiments. P values determined by Welch’s two-tailed t-test. ns not significant. All error bars denote SD. Schematics in (a) and (f) created with BioRender.com released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license https://creativecommons.org/licenses/by-nc-nd/4.0/deed.en.
To probe the basis for these phenotypes, we fixed Talin1-KO, Vinculin-KO, and NT control macrophages 30 min after challenge with IgG-coated DAAM particles, then assessed phagocytic progress by confocal microscopy. NT controls displayed pronounced phagocytic stalling on soft targets and little to none on stiff targets (Fig. 7c, d), similar to the behavior of WT and Itgb2+/− BMDMs shown above (see Figs. 3h and 5g). By contrast, Talin1-KO and Vinculin-KO macrophages exhibited substantial levels of stalling under all conditions (Fig. 7c, d). This was particularly notable in the case of stiff particles, where almost half of phagocytic cups remained unresolved after 30 min (compared to <10% for NT controls). Loss of Talin1 or Vinculin had no obvious effects on F-actin configuration at the interface, as all stalled contacts exhibited poor F-actin clearance regardless of target stiffness (Supplementary Fig. 9c). That being said, Talin1-KO and Vinculin-KO phagocytic cups were somewhat less enriched in F-actin than their NT control counterparts (Supplementary Fig. 9d), indicating that both adaptor molecules promote F-actin recruitment to the phagocyte–target interface. We conclude that Talin1 and Vinculin enhance F-actin accumulation to the cargo and are required for transition from phagocytic stalling to rapid gulping.
Previous studies have shown that force-dependent unfolding of Talin1 enables mechanosensitive Vinculin recruitment to focal adhesions42–44. To test whether differential target stiffness affects Vinculin recruitment, we challenged Itgb2+/− and Itgb2−/− BMDMs with DAAM particles for 5 min and then stained for Vinculin on the phagocytic cup (Fig. 7e). Vinculin accumulation at both soft (0.6 kPa) and stiff (18 kPa) interfaces was completely dependent on Itgb2, confirming the centrality of β2 integrins for Vinculin (and presumably Talin1) recruitment in this context. Surprisingly, Vinculin enrichment was more pronounced in soft cups than in stiff cups (Fig. 7f and Supplementary Fig. 9d), indicating that additional steps downstream of Vinculin association are required to trigger phagocytic gulping. These results highlight the importance of Talin1 and Vinculin for coupling mechanosensing to uptake mechanism.
Discussion
In the present study, we applied novel DAAM particle technology to study phagocytic mechanotransduction in macrophages independently of confounding factors such as cargo size and ligand presentation. Using this approach, we found that multiple macrophage subtypes respond to cargo rigidity and that they do so in the context of both IgG- and PtdS-induced uptake. Cargo mechanosensing manifested as a choice between rapid, “gulping” uptake, which was frequently observed on stiff DAAM particles, and phagocytic stalling, which was more prevalent with soft particles. These two forms of engagement differed not only in their dynamics but also in their underlying F-actin architectures: gulping interfaces were characterized by a strong peripheral ring of F-actin, whereas stalled interfaces exhibited F-actin accumulation both in the periphery and in punctate structures closer to the center of the contact. Similar stiffness-induced effects on cup morphology were observed in a previous study of “frustrated” phagocytosis on planar substrates45. Importantly, we found that β2 integrins are required for both gulping and stalling responses and for cargo mechanosensing in general. Finally, we identified Talin1 and Vinculin as critical mediators of stiff cargo gulping. Based on these results, we conclude that macrophages apply a β2 integrin-dependent mechanical checkpoint to tailor phagocytic responses to the deformability of the target (Fig. 8). Previous studies have documented integrin-dependent mechanosensing in the context of focal adhesions and podosomes46–49, both of which are F-actin rich assemblies that impart force against extracellular entities. Within these structures, the stress/strain relationship between force exertion and environmental deformation is interpreted via outside-in integrin signaling, ultimately resulting in stiffness dependent cellular responses. It is tempting to speculate that cargo mechanosensing by macrophages proceeds by an analogous mechanism in which β2 integrin mechanotransduction through Talin1 and Vinculin couples the degree of target deformation to the appropriate mode of uptake. Thus, the inability to deform cargo would induce phagocytic gulping, whereas strong distortion would prime an alternative approach, potentially embodied by the stalling response we observed on soft DAAM particles.
Fig. 8. β2 integrins, together with the downstream adaptors Talin1 and Vinculin, mediate a mechanosensitive checkpoint that dictates differential treatment of stiff and soft cargo.
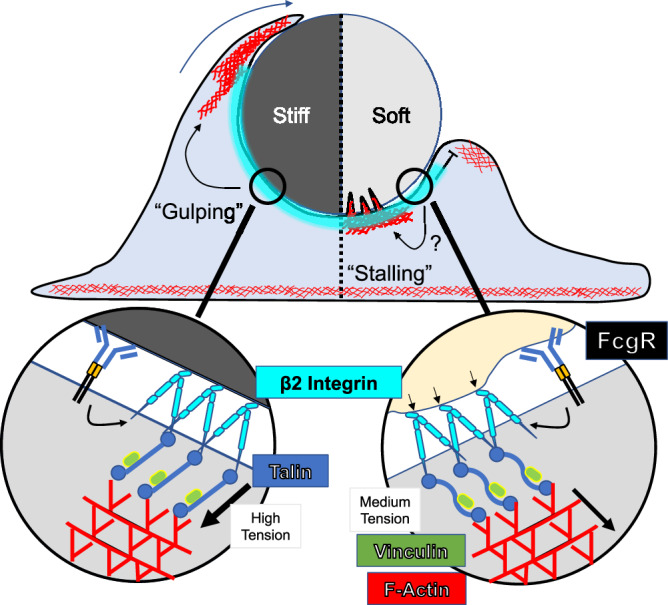
Stiff cargo fully engages β2 integrins and their downstream effectors, leading to rapid “gulping” phagocytosis (left). Conversely, soft cargo induces a distinct type of integrin contact, potentially involving a yet-to-be determined signaling component (denoted by the question mark), leading to the formation of central actin protrusions and a “stalling” response (right).
That Talin1-KO and Vinculin-KO macrophages stall on both stiff and soft DAAM particles suggests that stalling represents the failure to gulp. This interpretation, however, is inconsistent with the observation that CD18-KO macrophages do not stall on either stiff or soft cargos. We conclude instead that stalling is a distinct, β2 integrin-dependent response that occurs when integrins stably engage their ligands but signal only weakly through Talin1 and Vinculin. Precisely what stalling on DAAM particles might represent physiologically remains to be seen. The central F-actin protrusions that characterize stalled cups are reminiscent of the projections that mediate phagocytic nibbling9–11,37, a prevalent mode of uptake for large, soft cargos such as apoptotic corpses and tumor cells12,39,50–52. Indeed, one could imagine that nibbling might manifest as stalling on a soft but unfragmentable DAAM particle. Our results showing that MRTF-A dependent stiffening enhances the gulping uptake of E0771 cells at the expense of nibbling is consistent with this hypothesis. We also found, however, that the nibbling of E0771 cells was CD18-independent, in marked contrast to the requirement of CD18 for stalling. Determining whether other receptor-ligand interactions can functionally compensate for β2 integrins during target nibbling or, alternatively, whether stalling and nibbling are distinct processes, will require further investigation with live cell-targets and/or fragmentable synthetic cargo.
Given previous research indicating that Vinculin accumulation at focal adhesions increases with substrate stiffness42, it was surprising that we observed increased Vinculin recruitment to phagocytic cups containing soft cargo. This discrepancy likely reflects the functional dynamics of mature focal adhesions, which mediate stable anchorage, and gulping phagocytic cups, which surround and ingest cargo in <5 min. Indeed, the formation of sustained adhesions would be expected to impair rapid phagocytosis by delaying the clearance of interfacial F-actin required for cargo internalization. This is consistent with a previous finding that nascent integrin adhesions during the engulfment of latex beads are more akin to podosomes than focal adhesions26. Intriguingly, studies of force-dependent Vinculin binding to Talin1 have suggested that high levels of tension (~10–40 pN) promote Vinculin dissociation43. Stiff cargo may provide the high integrin tension necessary for rapid cycles of Vinculin recruitment and dissociation, thereby facilitating rapid engulfment, while moderate tension on a softer target could promote adhesion maturation (or stasis), which would inhibit phagocytic progression. Focused studies with molecular tension sensors could resolve this question.
In our hands, CD18-KO macrophages employed a third form of phagocytosis characterized by the absence of both F-actin enrichment and mechanosensing. This rudimentary mode of uptake was particularly sensitive to ligand density, implying that abundant phagocytic receptor engagement can drive engulfment of large particles in the absence of integrin-induced adhesion and cytoskeletal remodeling. These results are intriguing in light of recent work showing that Itgb2−/− macrophages form aberrantly elongated phagocytic cups with IgG-coated red blood cells that are nevertheless capable of completing phagocytosis7. They are also consistent with prior work suggesting that integrin binding is critical for cup expansion only when phagocytic ligands are sparsely clustered24. Although further studies will be required to understand the mechanistic basis of this rudimentary uptake pathway, its existence strongly suggests that β2 integrins are not absolutely required for the phagocytosis of IgG-coated cargo, but rather allow for the application of distinct phagocytotic strategies that best match the specific mechanical features of that cargo.
β2 integrins are not known to interact directly with IgG and PtdS, raising the question of what these receptors recognized on the IgG- and PtdS-coated DAAM particles used in this study. It is formally possible that β2 integrins influence the mechanosensing of this cargo indirectly by establishing strong contact with underlying substrate ECM. The fact that CD18 strongly localized to both stiff and soft DAAM particles, however, argues against this hypothesis. Certain integrins, including Mac-1 and αXβ2, are notably promiscuous, exhibiting appreciable affinity for denatured proteins and even for uncoated plastic35,36. We investigated a potential role for adsorbed serum proteins as integrin ligands in our system, unexpectedly finding that serum coating actually dampened β2 integrin-dependent mechanosensing during DAAM particle phagocytosis. As such, we are left to conclude that β2 integrins engage DAAM particles via some other component. Although the balance of our results suggests direct recognition of the polyacrylamide matrix, we have not explicitly ruled out streptavidin or some other to-be-defined bridging molecule. It is important to note that these possibilities need not be mutually exclusive. Indeed, in physiological settings, the capacity of β2 integrins to bind many ligands, taken together with the molecular heterogeneity of biological targets, would be expected to facilitate cargo mechanosensing across a wide range of phagocyte–target pairings.
The specific CD18 phosphorylation event identified by our proteomic analysis (pT760 in mice, pT758 in humans) occurs in response to TCR ligands, phorbol ester, and chemokines53,54. This creates a recognition site for 14-3-3 proteins, which bind to the CD18 tail and thereby alter the composition of the adhesion complex. The functional consequences this process remain somewhat unclear; while some studies indicate that 14-3-3 binding promotes cell adhesion through the activation of Rho family GTPases, it has also been shown to disrupt interactions between CD18 and Talin130,31,55, which could attenuate integrin function. Here, we demonstrate that T760 phosphorylation is enhanced by target rigidity. This mechanoresponsiveness may simply reflect stronger engagement of β2 integrins, which would be expected to boost outside-in integrin signaling as well as any downstream feedback inhibition. It is also possible, however, that T760 phosphorylation plays a specific role in promoting the phagocytic gulping of stiff cargo. Targeted mechanistic studies will be required to resolve this issue.
In recent years, the potential of phagocyte-based immunotherapy has motivated intensive investigation of its governing mechanisms. Much attention has focused on the “eat” versus “don’t eat” choice point, no doubt reflecting the success of strategies targeting “don’t eat me” signals like CD4756. This critical decision, however, is just the first of many on the path to cargo uptake. Once the macrophage commits to phagocytosis, the key question becomes how to consume the cargo. Our results clearly demonstrate that this next stage is subject to complex biomechanical regulation. Decoding the molecular logic that controls this process could unlock new strategies for modulating phagocytic activity in translational settings.
Methods
The animal protocols used for this study were approved by the Institutional Animal Care and Use Committee at MSKCC. Use of the 731.2B-iPSC cell line was approved by the MSKCC Institutional Review Board.
Bone marrow-derived macrophage culture and characterization
BMDMs were differentiated from primary mouse bone marrow as previously described57. In brief, femurs from C57BL/6J mice (B6, Jackson #000664, 6–24 weeks old) were harvested, and bone marrow was extracted by centrifugation. After red blood cell lysis in ACK buffer (10 mM KHCO3, 150 mM NH4Cl, 100 µM EDTA), 3 × 106 bone marrow cells were plated in 10 cm non-TC-treated dishes with 10 mL DMEM supplemented with 25% M-CSF-conditioned media, which was obtained from L929 cells according to an established protocol57. Differentiated BMDMs were transferred to a new plate on day 6 and allowed to attach overnight for experiments on day 7. Day 6 BMDMs were assessed for purity flow cytometrically using anti-CD11b-APC (clone M1/70, Invitrogen 17-0112-82, 1:200), anti-CD11c-FITC (Clone N418, Invitrogen 11-0114-82, 1:200), and anti-F4/80-BV421 (clone T45-2342, BD Biosciences 565411, 1:200). For Itgb2−/− experiments, heterozygous mice were obtained from The Jackson Laboratory (B6.129S7-Itgb2tm2bay/J, #003329, 6–12 weeks old) and bred to generate Itgb2+/− × Itgb2−/− breeding pairs. Pups were genotyped by PCR and all experiments used gender-matched littermates for each biological replicate.
Plasmids and cell lines
For lentiviral transduction of F-tractin mCherry (pLVX-F-tractin-mCherry), the full F-tractin-mCherry coding sequence (from C1-F-tractin-mCherry, Addgene #155218) was subcloned into the lentiviral expression vector pLVX-M-puro (Takara 632164). For CRISPR-KO experiments, oligos for sequences targeting CD18 (CCTGTTCTAAGTCAGCGCCC), Talin1 (GCTTGGCTTGTGAGGCCAGT), Vinculin (GCACCTGGTGATTATGCACG), and non-targeting control (GCGAGGTATTCGGCTCCGCG) were annealed and cloned into lenti-sgRNA Blast (Addgene #104993). E0771 cells lines stably expressing a doxycycline-inducible MRTF-A construct were generated as previously described by sequential transduction with rTTA, TGL and either an MRTF-A expression construct or an empty control vector22. B16-GM-CSF cells were a gift from Michael Sykes. L929 cells were obtained from ATCC (CCL-1™).
HoxB8-ER conditionally immortalized progenitor culture and macrophage differentiation
HoxB8-ER progenitor cells were generated using an established protocol21 from bone marrow harvested from C57BL/6J (HoxB8-WT, 6–8 weeks old) and Cas9 knock-in mice (HoxB8-Cas9) (Jackson Labs #026179, 6–8 weeks old). After retroviral transduction of the HoxB8-ER oncogene, cells were cultured in HoxB8 Culture Media (RPMI supplemented with 1% pen–strep, 2 mM l-glutamine, 10% FBS, 1 µM β-estradiol, 5% GM-CSF conditioned media) for 1 month to select for clones expressing HoxB8-ER.
To generate CRISPR-KO lines, HoxB8-Cas9 cells were transduced with lentivirus containing targeting or non-targeting sgRNA by spinoculation (1400 × g, 2 h). After spinoculation, the viral media was replaced with HoxB8 Culture Media. After 24 h, half of the media was replaced with fresh HoxB8 Culture Media containing 5 µg/mL blasticidin-HCl (Thermo Fisher A1113903) for selection. Cells were kept under selection for 1 week, with daily exchange into fresh media. CD18 knockout progenitor cells were FACS-sorted after CD18 staining (BioLegend 101414) and recovered for 1 week. Knockout-efficiency of Talin1 and Vinculin was determined by western blot using the following antibody pairs: for Talin, Abcam ab157808, 1:1000 dilution followed by Goat anti-Mouse 800CW (LI-COR 925-32210), 1:15,000 dilution; for Vinculin, Abcam ab91459, 1:1000 dilution, followed by Goat anti-Rabbit 680RD (LI-COR 926-68071), 1:15,000 dilution. Talin2 expression was determined by western blot (Abcam ab105458, 1:1000; secondary: Goat anti-Mouse 800CW, LI-COR 925-32210, 1:15,000). Actin and GAPDH were detected using Sigma clone AC-15 (Sigma A2066, 1:1000) and clone 14C10 (Cell Signaling Technology 3683, 1:1000), respectively, followed by 1:15,000 Goat anti-Mouse 800CW and Goat anti-Rabbit 680RD, respectively.
To generate F-tractin mCherry reporter lines (HoxB8-F-tractin), HoxB8-WT cells were transduced with lentivirus containing pCMV-F-tractin-mCherry, following the same recovery protocol as above, with 5 µg/mL puromycin (Thermo Fisher A1113803) instead of blasticidin. After 1 week of selection, mCherry-positive cells were sorted by FACS and recovered for an additional week.
To generate HoxB8-ER macrophages, HoxB8-ER progenitor cells were washed three times in 10 mL complete RPMI without β-estradiol before transferring to complete BMDM differentiation media (see above), followed by 6 days of culture at 37 °C in non-TC-treated dishes (~5.3 × 104 cells/cm2). On day 6, cells were trypsinized and replated for downstream experiments.
Production of iPSC-derived human macrophages
Human macrophages were differentiated from 731.2B-iPSCs, which were generated from human fibroblasts (GM00731) purchased from the Coriell Institute58. 731.2B cells have been stocked and characterized by the Stem Cell Research Facility at MSKCC. They are mycoplasma free, express the expected levels of OCT4, SOX2, and NANOG, and have a normal karyotype. To generate macrophages, hiPSCs were seeded on CF1 mouse embryonic feeder cells (Thermo Fisher A34181) and maintained for 3 days in ESC medium (KO-DMEM (Thermo Fisher, 10829-018), 20% KO-Serum (Thermo Fisher, 10828-028), 2 mM l-glutamine (Thermo Fisher, 25030-024), 1% NEAA (Thermo Fisher, 11140-035), 0.2 % β-mercaptoethanol (Thermo Fisher, 31350-010), 1% Pen/Strep (Thermo Fisher, 15140163), with 10 ng/mL bFGF (Peprotech 100-18B)), and subsequently for 4 days in ESC media without bFGF. Seven days after seeding, hiPSC colonies were detached in clusters using a 13 min incubation with collagenase type IV (250 IU/mL final concentration) (Thermo Fisher Scientific [TFS]; 17104019) and transferred to 6-well low-adhesion plates in ESC media supplemented with 10 µM ROCK Inhibitor (Sigma; Y0503) to initiate differentiation. The plates were kept on an orbital shaker at 100 rpm for 6 days to allow for spontaneous formation of embryoid bodies (EBs) with hematopoietic potential. On day 6, 200–500 µm diameter EBs were picked under a dissecting microscope and transferred onto adherent tissue culture plates (2.5 EBs/cm2) for cultivation in hematopoietic differentiation (HD) medium (APEL2 (Stem Cell Tech 05270), with 0.5% protein free hybridoma medium (TFS; 12040077), 1% pen/strep, 25 ng/mL hIL-3 (Peprotech; 200-03), and 50 ng/mL hM-CSF (Peprotech, 300-25)). Starting from day 18 of the differentiation and then every week onwards for up to 2 months, macrophage progenitors produced by EBs were collected from suspension, filtered through a 100 µm mesh, plated at a density of 10,000 cells/cm2, and cultivated for 6 days in RPMI1640/GlutaMax (TFS; 61870036) medium supplemented with 10% FBS (EMD Millipore TMS-013-B) and 100 ng/mL human M-CSF before use in downstream experiments. Twenty-four hours before the start of an experiment, macrophages were harvested by trypsinization (0.25% with 1 mM EDTA, Thermo Fisher 25200056) and plated at a density of 25,000 macrophages per well in an 8-well imaging chamber (Ibidi 80821). Phagocytosis was assessed by live imaging as described below.
Ex vivo mouse microglia culture
Primary mouse microglia were harvested from P2-P4 C57BL/6 mice by first dissociating brains using the Miltenyi gentleMACS Octo Dissociator with Heaters, brain dissociation kits (Miltenyi Biotec 130-096-427, 130-107-677, 130-092-628), and gentleMACS C Tubes (Miltenyi Biotech 130-093-237), following the manufacturer’s instructions. Microglia were purified by magnetic separation using CD11b MicroBeads (Miltenyi Biotec 130-097-142), LS Columns (Miltenyi Biotech 130-042-401), and the QuadroMACS separator (Miltenyi Biotech 130-091-051), following the manufacturer’s instructions. Isolated primary microglia were then cultured in complete DMEM, and phagocytosis was assessed by live imaging in 8-well chambers (Nunc 155409) coated with collagen IV (Corning 354233) at a density of 100,000 cells/well.
Microparticle synthesis, functionalization, and characterization
Microparticles were synthesized as previously described20,27, with modifications. Aqueous acrylamide solutions were prepared containing 150 mM MOPS (pH 7.4), 0.3% (v/v) tetramethylethylenediamine (TEMED), 150 mM NaOH, and 10% (v/v) acrylic acid. The mass fraction of total acrylamide was kept constant at 10% and the cross-linker mass fraction was altered to adjust Young’s modulus (Table 1). The mixture was sparged with N2 gas for 15 min and kept under N2 pressure. Hydrophobic Shirasu porous glass (SPG) filters (pore size specific to each batch, see Table 1) were sonicated under vacuum in n-dodecane, mounted on an internal pressure micro kit extruder (SPG Technology Co.), and submerged into the organic phase, consisting of 99% hexanes and 1% (v/v) Span-80 (Sigma-Aldrich, S6760), with continuous stirring at 300 rpm. Ten milliliters of the acrylamide mixture was then loaded into the apparatus and extruded under N2 pressure into 250 mL of the hexane mixture. To initiate polymerization, the resulting emulsion was heated to 60 °C and 2,2′-azobisisobutyronitrile (2 mg/mL, Sigma-Aldrich) was added. The reaction was kept at 60 °C for 3 h and then at 40 °C overnight. Polymerized microparticles were harvested by washing three times in hexanes and two times in ethanol (30 min, 1000 × g), followed by resuspension in 100 mL phosphate buffered saline (PBS). For some batches, excess ethanol caused incomplete partition of particles into aqueous solution, so additional PBS was added until the particles formed one pellet under centrifugation (1000 × g, 5 min). Particles were washed two more times in PBS and reduced to an appropriate volume (108 particles/mL). See Table 1 for exact extrusion conditions and formulations for each batch of particles used in this study.
Table 1.
DAAM particle formulations and properties
| Batch # | Young’s modulus (kPa) | Diameter mean (µm) | Mono: total acrylamide ratio | % Bis-arylamide (by mass) | Membrane pore size (µm) | Membrane lot # | Extrusion pressure (psi) | Span-80 conc. (v/V) |
|---|---|---|---|---|---|---|---|---|
| 0.31 | 2.54 (±0.4) | 8.31 (±0.8) | 154:1 | 0.065 | 2.4 | PJU15E21 | 3.51 | 1% |
| 0.32 | 1.28 (±0.2) | 9.31 (±1.2) | 250:1 | 0.040 | 2.4 | PJU15E21 | 3.60 | 1% |
| 0.33 | 0.598 (±0.12) | 9.66 (±0.8) | 308:1 | 0.032 | 2.4 | PJU15E21 | 3.42 | 1% |
| 0.34 | 18.0 (±4.1) | 9.65 (±0.8) | 42.7:1 | 0.234 | 3.0 | PJU13A28 | 1.67 | 1% |
For labeling, the particles were streptavidinated using amine coupling as previously described27: briefly, 4.0 × 107 beads were activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, TFS 22980) and N-hydroxysuccinimide (NHS, TFS 24500) in MES buffer (pH 6.0) with 0.1 % (v/v) Tween 20. Then, the buffer was exchanged into basic PBS (pH 8.0) with 0.1 % Tween 20 for a 2 h conjugation with 10 μM streptavidin (Prozyme, SA10) and 10 μM fluorescent dye (LRB ethylenediamine, FITC ethylenediamine, or Alexa Fluor 488 Cadaverine). Unreacted carboxy-NHS was quenched with excess ethanolamine for 30 min, and the resulting DAAM particles exchanged into PBS (pH 7.4). To functionalize particles for phagocytosis assays, streptavidinated particles were incubated for 2 h in PBS with Biotin-SP-Whole IgG (Jackson Immuno Research 015060003) or Biotin-Phosphatidylserine (Echelon 31B16), followed by three washes in PBS pH 7.4 (10,000 × g, 2 min).
Diameter and Young’s moduli were measured on particles conjugated to streptavidin, LRB, and FITC, and then coated with Biotin-IgG (10 pmol/M). Young’s modulus was measured by atomic force microscopy using large radius hemispherical tips (LRCH-15-750, Team NanoTec). Particles were indented three times each and the apparent Young’s modulus was taken as the mean of the 3 measurements. Thirty particles were measured for each batch. Particle size was assessed using an inverted fluorescence microscope (Olympus IX-81) fitted with a 20× objective lens (0.75, Olympus). Images of particles (N ≥ 39) were taken at the focal plane of maximum diameter, and diameters were subsequently measured using Fiji 2.0.
In vitro phagocytosis of DAAM particles
For flow cytometry-based detection, BMDMs were cultured for 6 days on non-TC-treated dishes, trypsinized, and plated in a 24-well non-TC-treated plate with 105 cells in 1 mL of BMDM differentiation media per well. On the following day, IgG- or PtdS-coated DAAM particles were vortexed and added into each well, followed by centrifugation at 100 × g for 3 min to accelerate particle settling. BMDMs and particles were co-incubated for the specified amount of time (usually 1 h), then washed twice with PBS and removed from the well by trypsinization and scraping. The resulting cells were collected and transferred to a 96-well plate for washing and staining. Cells were washed 3× with 200 µL FACS buffer. In experiments where CD11b or CD18 were measured, cells were stained for 1 h at 4 °C in 1:200 conjugated antibody (anti-CD11b-APC (M1/70), Invitrogen 0112-82 or anti-CD18-AF647(M18/2), BioLegend 101414), followed by two washes with FACS buffer. Finally, 3 µM DAPI was added 5 min prior to flow cytometric analysis. Measurements were taken on a CytoFLEX benchtop flow cytometer (Beckman Coulter). Live cells were identified using FlowJo 10.10 software by examining the forward scatter, side scatter, and DAPI channels. Non-conjugate beads were used to define the LRB+FITClo gate. Phagocytosis was measured as LRB+FITCloDAPI− cells divided by the total DAPI− population.
For microscopy-based phagocytosis assays, macrophages were plated at a density of 2.5 × 104 cells/well in an 8-well chamber slide (Ibidi 80821) and labeled with 1 µg/mL Hoechst 3342 (Thermo Fisher H1399). DAAM particles were added and centrifuged at 100 × g for 3 min to encourage contact formation. Imaging was started within 5 min of centrifugation using a ZEISS Axio Observer.Z1 epifluorescence microscope fitted with an X-Cite exacte light source and a 10×/0.45 numerical aperture (NA) objective. Four channels (FITC, TRITC, DAPI, and Brightfield) and 4 fields of view/well were captured at 2 min intervals for 2 h.
A custom MATLAB (2022b) script was used to derive phagocytic efficiency and phagocytic index from time-lapse data. Still images were thresholded and segmented by LRB signal to label particles in each field of view. For each labeled particle, mean FITC and LRB intensity values were obtained and the FITC/LRB ratio computed. Every tenth frame (20 min) was processed independently. To compute the threshold for acidified targets, FITC/LRB ratios for all labeled particles across all processed frames were pooled to generate a distribution of represented ratios and the MATLAB functions ksdensity and findpeaks were used to identify one FITChi peak and one FITClo peak as the two largest peaks in the distribution. The threshold was set as the midpoint between the peaks. In some experiments, the number of phagocytosed particles was low, and only one peak could be identified. In these cases, the threshold was set at 0.5× the peak ratio value. Cells were counted in each well by thresholding and segmenting using the DAPI channel. Phagocytosis efficiency for each time point was calculated as the percentage of labeled particles below the calculated threshold. Phagocytic index was calculated as the number of labeled particles below the threshold divided by the number of cells in the field of view.
In vivo phagocytosis of DAAM particles
One milliliter of aged thioglycolate (TG) was injected intraperitoneally into B6 mice. Following 24 h of elicitation, 107 DAAM particles (coated with 10 pmol/M IgG) were injected intraperitoneally (volume 200 µL in 1× PBS). After 2 h, mice were sacrificed, and cells were isolated by peritoneal lavage as previously described59. Samples were then stained with fluorescently conjugated antibodies against CD11b-APC (Invitrogen 0112-82), F4/80-BV421 (BD 565411), and Ly6G-BV650 (BD 740554). Peritoneal macrophages were identified as the F4/80+CD11b+Ly6G− cells. Within this population, LRB+FITClo events were identified in FlowJo based on a non-conjugate bead control and phagocytic efficiency was measured as the percentage of CD11b+F4/80+Ly6G− cells in the LRB+FITClo gate.
In vitro phagocytosis of cancer cells
E0771 cells transduced with doxycycline-inducible MRTF-A or empty vector control were cultured in complete DMEM and grown to ~80% confluence in 10 cm TC-treated dishes. Twenty-four hours before experiments, E0771 cells were harvested by trypsinization and cultured at a density of 106 cells/well in 6-well Nunclon™ Sphera™ low-adhesion plates (Thermo Fisher, 174932) to prevent substrate attachment. Five hundred nanograms per milliliter doxycycline (Sigma, D5207) was then added for 24 h to induce overexpression of MRTF-A. D7 BMDMs stained with CellTrace™ CFSE according to manufacturer’s instructions and were plated in 24-well non-TC dishes at 5 × 104 cells/well. On the next day, E0771 target cells were pre-treated with 100 nM MycB or DMSO for 1 h at 37 °C, followed by staining with 1 µM Cypher5e (Sigma GEPA15405) and 1 µg/mL Hoechst 3342 (Thermo Fisher H1399) according to manufacturer’s instructions. They were then purified by centrifugation over Histopaque® (Millipore Sigma 10771). 1 × 105 target cells were added to the 24-well BMDM plate and incubated with 1 mg/mL anti-mouse Isotype or anti-CD47 antibodies (BioXCell BE0083 & BE02830). After 2 h, macrophage–target conjugates were harvested by trypsinization and analyzed by flow cytometry. Phagocytosis was measured as the ratio of CFSE+Cypher5e+ events over all CFSE+ events. The Cypher5e+ threshold was established using CFSE+Hoechst− events in the isotype control sample.
For imaging-based analysis of whole-cell uptake and nibbling, BMDMs were stained with CellTrace™ CFSE as described above and plated in 8-well imaging chambers (iBidi) at a density of 2.5 × 104 cells/well. Target cells were generated as described above, then stained with CellMask™ Deep Red (Thermo Fisher C10046) and 1 µg/mL Hoechst 3342 according to manufacturer’s instructions. 2.5 × 104 target cells were added to the imaging chambers and imaged using a ZEISS Axio Observer.Z1 epifluorescence microscope fitted with an X-Cite exacte light source and a 10×/0.45 NA objective. Four channels (FITC, Far Red, DAPI, and Brightfield) were captured at 1 min intervals for 2 h. To blind the samples for manual analysis, a custom MATLAB script was used to split each time-lapse movie into 150 µm × 150 µm sub-sections with randomly barcoded labels. Each sub-section was analyzed as follows. Macrophages in frame were counted using FITC and Brightfield channels, target cells were counted and cell membranes identified by CellMask-Deep Red signal, and nuclei were identified by Hoechst signal. Dead target cells were identified by high Hoechst in the nucleus (~5–10-fold increase over mode) and removed from analysis. Whole-cell uptake events were defined as complete internalization of both membrane and nucleus within a single cytoplasmic void in the macrophage (see Supplementary Fig. 8 for examples). Nibbling uptake events were defined as internalization of CellMask+ material without simultaneous internalization of the Hoechst+ nucleus. Any event captured during the 2 h imaging window was counted. Sub-sections were then unblinded and each sample was quantified by the number of occurrences of each type of uptake event as a percentage of all live target cells examined. To enable manual analysis within a reasonable timeframe, ~15% of sub-sections from each sample were analyzed, constituting 50–100 target cells analyzed per condition/replicate.
Live confocal microscopy and quantification
HoxB8-F-tractin progenitor cells were differentiated into macrophages as described above and transferred to 8-well chamber slides at a density of 1.5 × 104 cells/well 24 h prior to the start of the experiment. The chamber slides were imaged using a SoRa spinning disk confocal microscope (Nikon) fitted with a 63×/1.49 NA objective. To capture images, chamber slides were first placed onto the microscope to focus on the macrophage plane. Fifty microliters (2.5 × 104) DAAM particles were then pipetted into the center of the well. After ~5 min, the region was scanned to identify potential initiating events (particle and cell in contact) and image capture was initiated. Image stacks encompassing the entire particle and macrophage (~60–100 slices/stack, 0.3 µm slice depth) were recorded at the maximum rate obtainable by the system (10–15 s interval/image stack). Time-lapses were captured for a minimum of 10 min, up to 40 min if phagocytosis did not complete. Phagocytic events that did not complete within 30 min were considered “stalled.” Supplementary Movies were generated using Imaris 9.9.0 (Oxford Instruments).
To calculate mean phagocytic cup velocity, we first identified the time points at which phagocytosis was initiated (start of contact) and completed (particle fully encompassed in F-tractin). If phagocytosis did not complete, the final timepoint of the movie was used as the end time point. Then, for each time point, using Fiji measurement tools, an effective engulfed diameter (dn) was measured as the distance between the phagocytic cup base and the intersection of the plane of maximum advancement (see Supplementary Fig. 4 for schematic diagram). Instantaneous phagocytic cup velocity was calculated as the distance traveled by actin during the measured period of time:
| 1 |
And the mean phagocytic cup velocity was calculated using the total distance traveled during the full engulfment progress:
| 2 |
or, if phagocytosis did not complete, the total distance traveled over the entire movie was used:
| 3 |
Fixed confocal microscopy and quantification
To quantify phagocytic cup progress, 5 × 104 macrophages were plated in each well of an 8-well chamber slide (Ibidi 80821), and 105 DAAM particles (IgG-coated, AF488 labeled) were added and centrifuged at 100 × g for 5 min at 4 °C to synchronize contact formation. After centrifugation, plates were incubated at 37 °C for 5 min, 10 min, or 30 min and fixed by adding an equal volume of 4% pre-warmed paraformaldehyde (Electron Microscopy Sciences, final concentration 2%), followed by 20 min incubation at 37 °C. Each well was then neutralized by removing 200 µL of PFA/media waste and adding 500 µL complete media (DMEM, 10% FBS). Neuralization was completed by three washes with 500 µL RPMI/well, followed by three washes with 500 µL PBS. Cell–particle conjugates were then permeabilized by 5 min incubation in 0.5% Triton X-100 in PBS (PBST), followed by a 1 h incubation in PBST with 1% Goat Serum to block. Samples were then stained for 1 h with Phalloidin-AF594 or Phalloidin-AF568 (1:400 dilution). Following staining, samples were washed 3× with 500 µL PBST and 5× with 500 µL PBS. At each washing step, care was taken to not allow the wells to dry or to disturb particles at the bottom of the well.
Samples were imaged using a Leica Stellaris 8 point scanning confocal microscope fitted with a 63×/1.4 NA oil objective. Particle–cell conjugates were identified morphologically by first focusing on particles and then confirming by phalloidin signal that a cell was in contact or nearby. Image stacks were captured using 0.3-μm z-sectioning (~80 slices/stack). Phagocytic cup progress was quantified using a custom MATLAB GUI script: each TIFF stack is first presented to the user using the sliceViewer MATLAB function, which prompts the user to click the center of the particle. This center is then used to present an X–Z slice through the center of the particle. The user then outlines the phagocytic cup in the phalloidin channel using the MATLAB drawpolygon function. This enables the particle to be segmented using the particle fluorescence channel and converted into a series of equally spaces points around its circumference. Percent actin coverage is then calculated as the percentage of particle edge coordinates that fall within the user-defined polygon. To avoid user bias, images from a given experiment were loaded randomly, and no identifying information about each image was given to the user. After processing, incidental/non-phagocytic macrophage–particle contacts (<20% coverage area) were removed from the dataset.
For analysis of CD18-localization, permeabilized cells were blocked with 1% Goat Serum and 1:50 Fc-Block (Tonbo 70-0161-U500), then incubated overnight with 1:200 anti-CD18-AF647 (BioLegend 101414) in the presence of 1% Goat Serum and 1:50 Fc-Block. The following day, they were washed once with PBST, stained with 1:400 Phalloidin-AF568, processed, and imaged as described above. To analyze F-actin and CD18 accumulation at the phagocytic cup, image stacks were first thresholded and binarized based on AF488 signal to identify the particle volume and boundaries. This mask was then dilated by 4 pixels in all directions to create a “shell” around the particle. Phalloidin and CD18 signal was also thresholded and segmented to identify the cell volume and boundary. Enrichment of each signal at the cup was then calculated as the ratio of the mean fluorescence intensity within the intersection between the particle shell and cell volume divided by the mean fluorescence intensity in the entire cell volume. For analysis of Vinculin localization, permeabilized cells were blocked as described above and then incubated with anti-1:100 Vinculin Monoclonal Rabbit antibody (Invitrogen 42H89L44) overnight. The following day, they were washed three times with PBST and stained for 1 h with Phalloidin-AF568 and 1:2000 goat anti-rabbit AF647 secondary antibody (Invitrogen A21244). Samples were then processed, imaged, and analyzed as described above.
To calculate actin and CD18 clearance ratios, we first pre-processed the data using a previously established MATLAB pipeline to render the particles as surface coordinates in 3D cartesian and polar space20,27. Phalloidin and CD18 signal intensity was localized to the particles using a 0.3 µm search distance. Fully engulfed particles were removed from this analysis, and particles were rotated such that the base of the phagocytic cup aligned with the bottom of the particle in XYZ coordinates. The rendered particle was transformed to 2D by stereographic projection to a point below the particle in the Z direction (see Supplementary Fig. 4), and two perpendicular line profiles were then measured through the center of the projected interface. To identify the edge of the contact, a custom MATLAB GUI script was used and the peaks of F-actin or CD18 intensity furthest from the contact center in either direction were selected. Clearance ratio was calculated as the mean fluorescence intensity of points in the central 50% of the contact surface area divided by the mean fluorescence intensity of points in the outer 50%. CD18-F-actin lag distance was calculated as the distance between the phagocytic cup edge, as defined by the CD18 signal or the F-actin signal (whichever one was more advanced), and the lagging marker.
Phosphoproteomic analysis
Bone marrow from three mice was pooled and differentiated into BMDMs as described above. Twenty-four hours before the experiment, BMDMs were trypsinized and replated into 6-well non-TC-treated culture plates. IgG-coated (10 pmol/M) DAAM particles were overlayed onto the cells, centrifuged for 3 min at 300 × g, and then incubated at 37 °C. After 10 min, plates were quickly transferred to ice, trypsinized, and snap frozen with liquid nitrogen. One replicate consisted of pooled material from all 6 wells of one plate per stiffness condition. To ensure rapid processing after the addition of DAAM particles, two plates (one with 18 kPa particles, one with 0.6 kPa) were processed at a time, and all eight replicates were completed in batches over the course of 4 h.
Samples were lysed in buffer containing 8 M urea and 200 mM EPPS (pH at 8.5) with protease inhibitor (Roche) and phosphatase inhibitor cocktails 2 and 3 (Sigma). Benzonase (Millipore) was added to a concentration of 50 U/mL and incubated for 15 min and room temperature, followed by water bath sonication. Samples were then centrifuged at 4 °C, 14,000 × g for 10 min and the supernatant extracted. The Pierce bicinchoninic acid (BCA) assay was used to determine protein concentration. Protein disulfide bonds were reduced with 5 mM tris(2-carboxyethyl)phosphine (room temperature, 15 min), then alkylated with 10 mM iodoacetamide (RT, 30 min, dark). The reaction was quenched with 10 mM dithiothreitol (RT, 15 min). Aliquots of 100 μg were taken for each sample and diluted to ~100 μL with lysis buffer. Samples were then subjected to chloroform/methanol precipitation as previously described1. Pellets were reconstituted in 50 μL of 200 mM EPPS buffer and digested with Lys-C (1:50 enzyme-to-protein ratio) at 37 °C for 4 h. Trypsin was then added (1:50 enzyme-to-protein ratio) and the samples incubated overnight at 37 °C.
Anhydrous acetonitrile (ACN) was added to a final concentration of 30%. Peptides were TMT-labeled as described60. Briefly, peptides were TMT-tagged by adding 10 μL (28 μg/μL) TMTPro reagents (16plex) for each respective sample and incubated for 1 h (RT). A ratio check was performed by taking a 1 μL aliquot from each sample and desalted using the StageTip method61. TMT-tags were then quenched with hydroxylamine to a final concentration of 0.3% for 15 min (RT). Samples were pooled 1:1 based on the ratio check and vacuum-centrifuged to dryness. Dried peptides were reconstituted in 1 mL of 3% ACN/1% TFA, desalted using a 100 mg tC18 SepPak (Waters), and vacuum-centrifuged overnight.
Phosphopeptides were enriched using the Thermo High-Select Fe-NTA Phosphopeptide Enrichment Kit (Cat. No.: A32992). Unbound peptides and washes were saved and dried down for further analysis of the non-phosphorylated (phospho-depleted) peptides. The phosphopeptide eluate was vacuum-centrifuged to dryness and reconstituted in 100 μL of 1% ACN/25 mM ammonium bicarbonate (ABC). A StageTip was constructed by placing two plugs with a narrow bore syringe of a C18 disk (3M Empore Solid Phase Extraction Disk, #2315) into a 200 μL tip (VWR, Cat. #89079-458). StageTips were conditioned with 100 μL of 100% ACN, 70% ACN/25 mM ABC, then 1% ACN/25 mM ABC. Phospho-enriched sample was loaded onto the StageTip and eluted into six fractions of 3, 5, 8, 10, 12, and 70% ACN/25 mM ABC with 100 μL each. Fractions were immediately dried down by vacuum-centrifugation and reconstituted in 0.1% formic acid (FA) for LC–MS/MS.
Phospho-depleted peptides were reconstituted in 300 μL 0.1% TFA then fractionated by Pierce High pH Reversed-Phase Peptide Fractionation (Cat. No.: 84868) into eight fractions (10, 12.5, 15, 17.5, 20, 22.5, 25, and 70% ACN). Fractions were dried by vacuum-centrifugation and reconstituted in 0.1% FA for LC–MS/MS.
Phosphopeptide-enriched and phospho-depleted peptide fractions were analyzed by LC–MS/MS using a Thermo Easy-nLC 1200 (TFS) with a 50 cm (inner diameter 75 µm) EASY-Spray Column (PepMap RSLC, C18, 2 µm, 100 Å) heated to 60 °C and coupled to an Orbitrap Fusion Lumos Tribrid Mass Spectrometer (TFS). Peptides were separated by direct inject at a flow rate of 300 nL/min using a gradient of 5–30% ACN (0.1% FA) in water (0.1% FA) over 3 h, then to 50% ACN in 30 min, and analyzed by SPS-MS3. MS1 scans were acquired over an m/z range of 375–1500, 120 K resolution, AGC target (standard), and maximum IT of 50 ms. MS2 scans were acquired on MS1 scans of charge 2-7 using an isolation of 0.5 m/z, collision induced dissociation with activation of 32%, turbo scan and max IT of 120 ms. MS3 scans were acquired using specific precursor selection (SPS) of 10 isolation notches, m/z range 110–1000, 50 K resolution, AGC target (custom, 200%), HCD activation of 65%, max IT of 150 ms, and dynamic exclusion of 60 s.
Raw data files were processed using Proteome Discoverer (PD) version 2.4.1.15 (Thermo Scientific). For each of the TMT experiments, raw files from all fractions were merged and searched with the SEQUEST HT search engine using a mouse UniProt protein database downloaded on 2019/12/13 (92,249 entries). Cysteine carbamidomethylation was specified as a fixed modification, while methionine oxidation, acetylation of the protein N-terminus, TMTpro (K) and/TMTpro (N-term), phosphorylation (STY), and deamidation (NQ) were set as variable modifications. The precursor and fragment mass tolerances were 10 ppm and 0.6 Da, respectively. A maximum of two trypsin missed cleavages were permitted. Searches used a reversed sequence decoy strategy to control peptide false discovery rate (FDR) and 1% FDR was set as the threshold for identification. Phosphosite localization was assigned by PD IMP-ptmRS node. Phosphopeptides measured in all replicates were log2-transformed. Unpaired t-test was used to calculate p values in differential analysis, and volcano plots were generated based on log2FC and p value.
Schematic diagrams created with BioRender.com
The following diagrams were created with BioRender.com: Figs. 1a, d, e, 2a–d, g, 3h, k, l, 4a, e, f, 5a, c–e, 6e, f, and 7a, f. Supplementary Figs. 1e, 2a, 4d, e, and 5e.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
We thank the staff of the Research Animal Resource Center, the Flow Cytometry and Cell Sorting Core, the Molecular Cytology Core, and the Proteomics Core at MSKCC for their assistance. We also thank the members of the Perry and Huse labs for advice and technical assistance. We gratefully acknowledge David B. Sykes for the generous gift of HoxB8-ER plasmids and cell lines. Supported in part by the NIH (R01-AI087644 to M.H., P30-CA008748 to MSKCC), the MSKCC Basic Research Innovation Award Program (M.H.), the National Science Foundation Graduate Research Fellowship program (A.H.S.), and the Center for Experimental Immuno-Oncology at MSKCC (A.H.S.).
Author contributions
A.H.S., D.V., J.S.A.P, and M.H. conceived of the project. A.H.S and M.H. analyzed data, generated all figures, and wrote the manuscript. A.H.S., B.Y.W, M.M.D.J., L.S., Z.W., Z.L., and M.M.M. performed experiments and/or analysis for the manuscript. B.Y.W., M.M.D.J., E.C., Y.R., R.C.H., D.V., and J.S.A.P provided key expertise in experimental design and interpretation.
Peer review
Peer review information
Nature Communications thanks Stefan Linder, Patrick Oakes, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
Raw proteomic LC/MS data are available on ProteomXchange (PXD049434). Raw flow cytometry output has been deposited on the Zenodo Repository (https://zenodo.org/records/13351483). Imaging files are available upon request from the lead author (M.H., husem@mskcc.org). Source data are provided with this paper.
Code availability
All custom MATLAB scripts used for data analysis in this study can be found on the Zenodo repository (https://zenodo.org/records/13313112).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52453-9.
References
- 1.Accarias, S. et al. Genetic engineering of Hoxb8-immortalized hematopoietic progenitors - a potent tool to study macrophage tissue migration. J. Cell Sci.133, jcs236703 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Klichinsky, M. et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol.38, 947–953 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov.21, 799–820 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sly, L. M. & McKay, D. M. Macrophage immunotherapy: overcoming impediments to realize promise. Trends Immunol.43, 959–968 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Swanson, J. A. & Baer, S. C. Phagocytosis by zippers and triggers. Trends Cell Biol.5, 89–93 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Jaumouillé, V. & Waterman, C. M. Physical constraints and forces involved in phagocytosis. Front. Immunol.11, 1097 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walbaum, S. et al. Complement receptor 3 mediates both sinking phagocytosis and phagocytic cup formation via distinct mechanisms. J. Biol. Chem.296, 100256 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jaumouille, V., Cartagena-Rivera, A. X. & Waterman, C. M. Coupling of beta2 integrins to actin by a mechanosensitive molecular clutch drives complement receptor-mediated phagocytosis. Nat. Cell Biol.21, 1357–1369 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Velmurugan, R., Challa, D. K., Ram, S., Ober, R. J. & Ward, E. S. Macrophage-mediated trogocytosis leads to death of antibody-opsonized tumor cells. Mol. Cancer Ther.15, 1879–1889 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vorselen, D. et al. Cell surface receptors TREM2, CD14 and integrin αMβ2 drive sinking engulfment in phosphatidylserine-mediated phagocytosis. Preprint at bioRxiv10.1101/2022.07.30.502145 (2022).
- 11.Zhao, S. Y. et al. Gnawing between cells and cells in the immune system: friend or foe? A review of trogocytosis. Front. Immunol.13, 791006 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dooling, L. J. et al. Cooperative phagocytosis of solid tumours by macrophages triggers durable anti-tumour responses. Nat. Biomed. Eng.7, 1081–1096 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eaton, P., Fernandes, J. C., Pereira, E., Pintado, M. E. & Xavier Malcata, F. Atomic force microscopy study of the antibacterial effects of chitosans on Escherichia coli and Staphylococcus aureus. Ultramicroscopy108, 1128–1134 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Bufi, N. et al. Human primary immune cells exhibit distinct mechanical properties that are modified by inflammation. Biophys. J.108, 2181–2190 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Planade, J. et al. Mechanical stiffness of reconstituted actin patches correlates tightly with endocytosis efficiency. PLoS Biol.17, e3000500 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vorselen, D., Labitigan, R. L. D. & Theriot, J. A. A mechanical perspective on phagocytic cup formation. Curr. Opin. Cell Biol.66, 112–122 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Vorselen, D. et al. Phagocytic ‘teeth’ and myosin-II ‘jaw’ power target constriction during phagocytosis. Elife10, e68627 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sosale, N. G. et al. Cell rigidity and shape override CD47’s “self”-signaling in phagocytosis by hyperactivating myosin-II. Blood125, 542–552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beningo, K. A. & Wang, Y. L. Fc-receptor-mediated phagocytosis is regulated by mechanical properties of the target. J. Cell Sci.115, 849–856 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Vorselen, D. et al. Microparticle traction force microscopy reveals subcellular force exertion patterns in immune cell-target interactions. Nat. Commun.11, 20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang, G. G. et al. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods3, 287–293 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Tello-Lafoz, M. et al. Cytotoxic lymphocytes target characteristic biophysical vulnerabilities in cancer. Immunity54, 1037–1054.e1037 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito, S., Watabe, S., Ozaki, H., Fusetani, N. & Karaki, H. Mycalolide B, a novel actin depolymerizing agent. J. Biol. Chem.269, 29710–29714 (1994). [PubMed] [Google Scholar]
- 24.Freeman, S. A. et al. Integrins form an expanding diffusional barrier that coordinates phagocytosis. Cell164, 128–140 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox, D., Tseng, C.-C., Bjekic, G. & Greenberg, S. A requirement for phosphatidylinositol 3-kinase in pseudopod extension*. J. Biol. Chem.274, 1240–1247 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Ostrowski, P. P., Freeman, S. A., Fairn, G. & Grinstein, S. Dynamic podosome-like structures in nascent phagosomes are coordinated by phosphoinositides. Dev. Cell50, 397–410.e393 (2019). [DOI] [PubMed] [Google Scholar]
- 27.de Jesus, M. M. et al. Topographical analysis of immune cell interactions reveals a biomechanical signature for immune cytolysis. Sci. Immunol.9, eadj2898 (2024). [DOI] [PubMed]
- 28.Jongstra-Bilen, J., Harrison, R. & Grinstein, S. Fcγ-receptors induce Mac-1 (CD11b/CD18) mobilization and accumulation in the phagocytic cup for optimal phagocytosis*. J. Biol. Chem.278, 45720–45729 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Chung, J. et al. Rap1 activation is required for Fc gamma receptor-dependent phagocytosis. J. Immunol.181, 5501–5509 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fagerholm, S. C., Hilden, T. J., Nurmi, S. M. & Gahmberg, C. G. Specific integrin alpha and beta chain phosphorylations regulate LFA-1 activation through affinity-dependent and -independent mechanisms. J. Cell Biol.171, 705–715 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nurmi, S. M., Autero, M., Raunio, A. K., Gahmberg, C. G. & Fagerholm, S. C. Phosphorylation of the LFA-1 integrin beta2-chain on Thr-758 leads to adhesion, Rac-1/Cdc42 activation, and stimulation of CD69 expression in human T cells. J. Biol. Chem.282, 968–975 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Morrissey, M. A., Kern, N. & Vale, R. D. CD47 ligation repositions the inhibitory receptor SIRPA to suppress integrin activation and phagocytosis. Immunity53, 290–302.e296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson, R. W. et al. Gene targeting yields a CD18-mutant mouse for study of inflammation. J. Immunol.151, 1571–1578 (1993). [PubMed] [Google Scholar]
- 34.Ni, H., Li, A., Simonsen, N. & Wilkins, J. A. Integrin activation by dithiothreitol or Mn2+ induces a ligand-occupied conformation and exposure of a novel NH2-terminal regulatory site on the beta1 integrin chain. J. Biol. Chem.273, 7981–7987 (1998). [DOI] [PubMed] [Google Scholar]
- 35.Yakubenko, V. P., Lishko, V. K., Lam, S. C. & Ugarova, T. P. A molecular basis for integrin alphaMbeta 2 ligand binding promiscuity. J. Biol. Chem.277, 48635–48642 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Lamers, C., Plüss, C. J. & Ricklin, D. The promiscuous profile of complement receptor 3 in ligand binding, immune modulation, and pathophysiology. Front. Immunol.12, 662164 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrissey, M. A. et al. Chimeric antigen receptors that trigger phagocytosis. Elife7, e36688 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schriek, P. et al. Marginal zone B cells acquire dendritic cell functions by trogocytosis. Science375, eabf7470 (2022). [DOI] [PubMed] [Google Scholar]
- 39.Matlung, H. L. et al. Neutrophils kill antibody-opsonized cancer cells by trogoptosis. Cell Rep.23, 3946–3959.e3946 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Kim, C., Ye, F. & Ginsberg, M. H. Regulation of integrin activation. Annu. Rev. Cell Dev. Biol.27, 321–345 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Bays, J. L. & DeMali, K. A. Vinculin in cell-cell and cell-matrix adhesions. Cell. Mol. Life Sci.74, 2999–3009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou, D. W., Lee, T. T., Weng, S., Fu, J. & García, A. J. Effects of substrate stiffness and actomyosin contractility on coupling between force transmission and vinculin-paxillin recruitment at single focal adhesions. Mol. Biol. Cell28, 1901–1911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yao, M. et al. Mechanical activation of vinculin binding to talin locks talin in an unfolded conformation. Sci. Rep.4, 4610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar, A. et al. Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J. Cell Biol.213, 371–383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu, Y. et al. Molecular force imaging reveals that integrin-dependent mechanical checkpoint regulates Fcγ-receptor-mediated phagocytosis in macrophages. Nano Lett.23, 5562–5572 (2023). [DOI] [PubMed] [Google Scholar]
- 46.Case, L. B. & Waterman, C. M. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol.17, 955–963 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Driscoll, T. P. et al. Integrin-based mechanosensing through conformational deformation. Biophys. J.120, 4349–4359 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Collin, O. et al. Self-organized podosomes are dynamic mechanosensors. Curr. Biol.18, 1288–1294 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friedland, J. C., Lee, M. H. & Boettiger, D. Mechanically activated integrin switch controls α5β1 function. Science323, 642–644 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Brouckaert, G. et al. Phagocytosis of necrotic cells by macrophages is phosphatidylserine dependent and does not induce inflammatory cytokine production. Mol. Biol. Cell15, 1089–1100 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weinhard, L. et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun.9, 1228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim, T. K. & Ruthazer, E. S. Microglial trogocytosis and the complement system regulate axonal pruning in vivo. Elife10, e62167 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fagerholm, S. C., Varis, M., Stefanidakis, M., Hilden, T. J. & Gahmberg, C. G. alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation. Blood108, 3379–3386 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Jahan, F. et al. Phosphorylation of the α-chain in the integrin LFA-1 enables β2-chain phosphorylation and α-actinin binding required for cell adhesion. J. Biol. Chem.293, 12318–12330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takala, H. et al. β2 integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14-3-3 and filamin binding. Blood112, 1853–1862 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Maute, R., Xu, J. & Weissman, I. L. CD47-SIRPalpha-targeted therapeutics: status and prospects. Immunooncol. Technol.13, 100070 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weischenfeldt, J. & Porse, B. Bone marrow-derived macrophages (BMM): isolation and applications. Cold Spring Harb. Protoc.2008, pdb.prot5080 (2008). [DOI] [PubMed]
- 58.Miller, J. D. et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell13, 691–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ray, A. & Dittel, B. N. Isolation of mouse peritoneal cavity cells. J. Vis. Exp.35, e1488 (2010). [DOI] [PMC free article] [PubMed]
- 60.Navarrete-Perea, J., Yu, Q., Gygi, S. P. & Paulo, J. A. Streamlined tandem mass tag (SL-TMT) protocol: an efficient strategy for quantitative (phospho)proteome profiling using tandem mass tag-synchronous precursor selection-MS3. J. Proteome Res.17, 2226–2236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc.2, 1896–1906 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Raw proteomic LC/MS data are available on ProteomXchange (PXD049434). Raw flow cytometry output has been deposited on the Zenodo Repository (https://zenodo.org/records/13351483). Imaging files are available upon request from the lead author (M.H., husem@mskcc.org). Source data are provided with this paper.
All custom MATLAB scripts used for data analysis in this study can be found on the Zenodo repository (https://zenodo.org/records/13313112).