

Abstract
Radical (R•) and R•-hole site-based interactions are comparatively studied, for the first time, using ab initio methods. In this regard, R•-bearing molecules •XO3 (where X = Cl, Br, and I) were subjected to direct interaction with NH3 within dimeric and trimeric forms in the form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 complexes, respectively. As confirmed by electrostatic potential analysis, the studied R•-bearing molecules •XO3 had the outstanding potentiality to interact as Lewis acid centers via two positive sites dubbed as R• and R•-hole sites. Such an observation proposed the potentiality of the considered •XO3 molecules to engage in unconventional R• and well-established R•-hole site-based interactions with Lewis bases. This was confirmed by negative interaction (Eint) energies, ranging from −4.93 to −19.89 kcal/mol, with higher favorability for R• site-based interactions over the R•-hole site-based ones. MP2 energetic features furnished higher preferability for the R• site-based interactions than the R•-hole site-based ones in the case of chlorine- and bromine-bearing complexes, and the reverse was true for the iodine-bearing complexes. Moreover, elevated Eint values were recorded for the NH3···•XO3···NH3 trimers over the NH3···•XO3 and •XO3···NH3 dimers, outlining the higher preference of the •XO3 molecules to engage in R• and R•-hole site-based interactions in the trimeric form over the dimeric one. These results might be considered a requisite linchpin for numerous forthcoming supramolecular chemistry and crystal engineering studies.
Introduction
Since the announcement of the crucial role of noncovalent interactions in sundry fields, including catalysis,1,2 biological systems,3−6 supramolecular chemistry,7−9 and molecular recognition,10−13 research societies began to strictly study the origin and nature of such crucial interactions. Astonishingly, σ-hole interactions are considered one of the most predominant noncovalent interactions as an upshot to their prevalent contributions in perceiving many vital applications.2,14−21 The σ-hole concept was first reported to describe an electron-deficient region that appeared along the extension of the σ-bond of the covalently bonded group IV–VII elements.15,16,22−26 Over the surface of the above-mentioned atoms, π- and lone pair (lp)-holes were afterward addressed27−30 by the presence of depletion in the electron density that is located nearly perpendicular to the framework of the molecular entity27,31 and in opposite to the lp,32−34 respectively.
In addition to the above-mentioned hole concept, in-depth systematic studies were recently conducted, with more efforts being directed toward clarifying the characteristics of radical (R•)-hole sites and their corresponding interactions. The term R•-hole is defined as an area with a deficiency of electron density that is nearly opposite to the single electron of the R•-bearing molecules.35 As a starting point, studying the R•-hole interactions of the •TF3 molecules (T = tetrel atom) with Lewis bases showed apparent preferability in coincidence with the atomic size of the radical centers (i.e., C < Si < Ge).35 Afterward, the strength of R•-hole interactions exhibited by group IV–VII R•-bearing molecules was disclosed, and it was found to increase as follows: •SiF3··· < •POF2··· < •SO2F··· < •ClO3···Lewis base complexes.36
According to the literature, the R•-bearing molecules revealed a prevalent potentiality to interact with Lewis acids via the unpaired electron (i.e., R• site), forming single-electron noncovalent interactions.37−41 In addition, various studies evinced the proton–acceptor behavior of R•-bearing molecules in hydrogen-bonded complexes of HF···•CH342−44 NH3···•CH3,45 OH2···•CH3,46−48 NCH···•CH3,49−51 and HCCH···•CH3.52 Consequently, R•-bearing molecules may exhibit an amphoteric nature, nucleophilic character through their R•-hole site and electrophilic character through their single-electron R• site. However, the nucleophilic character of R•-bearing molecules through the R•-site has not been systematically studied yet.
In this regard, the present work was devoted to examining the ability of sp2-hybridized group VII R•-bearing molecules to favorably engage in R• site-based interactions within the NH3···•XO3 dimers (where X = Cl, Br, and I). In a parallel manner, the R•-hole site-based interactions were comparatively investigated in the •XO3···NH3 dimers. For the first time, the potentiality of the investigated molecules to synchronously engage in R• and R•-hole site-based interactions was explored employing the NH3···•XO3···NH3 trimers (Figure 1). The obtained findings will provide a basis for forthcoming works relevant to the interactions of R•-bearing molecules with Lewis bases that would accordingly level up their admissible applications in supramolecular chemistry.
Figure 1.
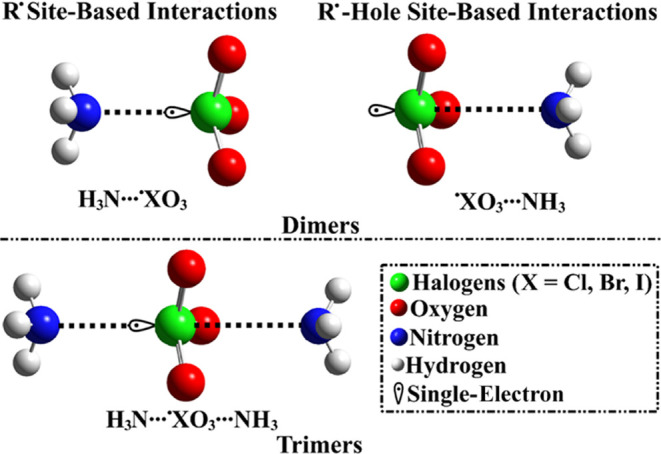
Illustrative representation of the R• and R•-hole site-based interactions within dimeric and trimeric forms in the form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 complexes, respectively.
Computational Methods
The current study was established to comparatively explore the potential of the sp2-hybridized group VII R•-bearing molecules (•XO3) to engage in R• and R•-hole site-based interactions via the NH3···•XO3 and •XO3···NH3 dimers, respectively. The trimeric form of the studied interactions was then considered in the form of NH3···•XO3···NH3 complexes to investigate the simultaneous potentiality of the examined systems to participate in R• and R•-hole site-based interactions.
All of the calculations were performed using Gaussian09 software53 by the MP2 method54 with the aug-cc-pVTZ basis set for H,55 N,55 O,55 and Cl56 atoms. The aug-cc-pVTZ(PP) basis set was devoted to the case of Br57 and I57,58 atoms. The adopted nonstandard basis sets were obtained from the EMSL Basis Set Exchange Library.59−61 First, geometry optimization calculations were carried out for the explored monomers, dimers, and trimers.
Upon optimization of •ClO3, •BrO3, and •IO3 systems, EP analysis was performed using a 0.002 au electron density, as recommended earlier, by dint of its worthy depiction for the surface of chemical molecules.62 Molecular electrostatic potential (MEP) maps were accordingly generated to envision the relative nucleophilicity and electrophilicity. Also, the surface electrostatic potential extrema (i.e., Vs,min/Vs,max) were quantified with the incorporation of Multiwfn 3.7 software.63
Upon optimization of NH3···•XO3/•XO3···NH3 dimers and NH3···•XO3···NH3 trimers, the interaction energy (Eint) was assessed as the variation in energy between the complex and the sum of its monomers in the complex form. Moreover, the basis set superposition error (BSSE) was eradicated from the computed interaction energies using the counterpoise correction procedure.64 The optimized complexes were not subjected to frequency calculations; therefore, it is feasible that these structures do not correspond to the true minima. For all investigated complexes, spin contamination was measured by the expectation value of the S2 operator. The S2 values were in the range of 0.7516–0.7589 after annihilation, confirming that spin contamination is negligible.
Benchmarking the MP2 interaction energies was done toward bestowing a more quantitative precision at the CCSD(T)/CBS level of theory based on eqs 1–3.65,66
| 1 |
where
| 2 |
| 3 |
Quantum theory of atoms in molecules (QTAIM)67 was accomplished to adequately elucidate the origin of the investigated interactions. Using QTAIM, we built the bond paths (BPs) and the bond critical points (BCPs) to indicate the origin of the investigated dimers and trimers. For a precise investigation of the origin of the considered interactions, various topological parameters, including the total energy density (Hb), electron density (ρb), and Laplacian (∇2ρb) were extracted. In addition to QTAIM, noncovalent interaction (NCI)6 index computations were conducted to precisely pinpoint and visualize the nature of noncovalent interactions exhibited by the R• and R•-hole sites. QTAIM and NCI index analyses were performed to generate the WFN files upon optimization of the complexes at the same level of theory with the help of Gaussian09 software. Based on the extracted WFN files, the QTAIM and NCI plots were built with the aid of the Multiwfn 3.7 software package63 and were plotted via Visual Molecular Dynamics software.68
Using frontier molecular orbital (FMO) theory, the electronic parameters were evaluated for the investigated systems after and before the complexation process. Pictorially, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) distributions were pictured for the monomers and complexes under study. Additionally, the energy of HOMO (EHOMO), energy of LUMO (ELUMO), Fermi-level energy (EFL), and the energy gap (Egap) were numerically assessed through eqs 4 and 5:
| 4 |
| 5 |
Moreover, the electronic properties, including global softness (S), chemical potential (μ), ionization potential (IP), electron affinity (EA), global hardness (η), work function (Φ), and electrophilicity index (ω) were assessed for the optimized monomers and complexes using eqs 6–12. It is worth mentioning that the vacuum-level electrostatic potential (VeL(+∞)) was set to nearly 0 within the work function estimation.
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
| 11 |
| 12 |
Results and Discussion
EP Calculations
EP analysis was devoted to inspecting the nucleophilic and electrophilic sites over the molecular entities according to previous recommendations.26,69 For the EP analysis, the MEP maps were generated for the •XO3 molecules using a 0.002 au electron density envelope. Moreover, Vs,min/Vs,max calculations were performed to numerically compute the magnitude of the positive R• and R•-hole sites. Figure 2 displays the MEP maps and Vs,min/Vs,max values.
Figure 2.
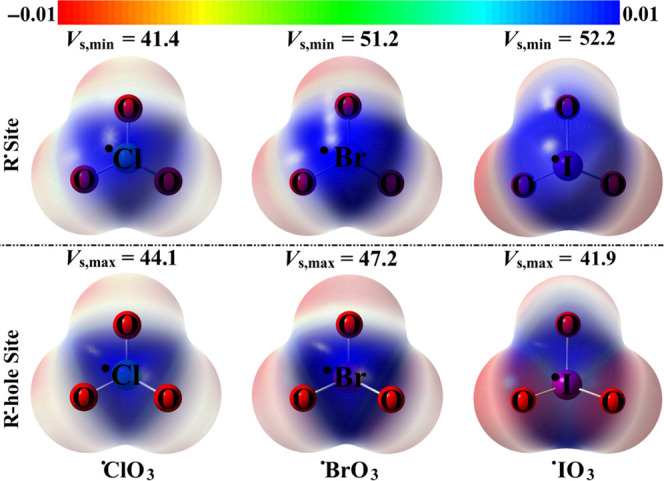
MEP maps of the R• and R•-hole sites over the optimized •XO3 molecules. The values of Vs,min/Vs,max are given in kcal/mol.
At first glance, Figure 2 unveils the distributions of the electron density over and opposite to the surface of radical location, namely, R• and R•-hole sites, respectively. Detailedly, an unconventional blue-colored R• site was observed over the surface of the single electron of the studied molecules. The aforementioned observation outlined the potentiality of the studied halogens to act as Lewis acid centers via the R• site and, accordingly, interact attractively with Lewis bases with disparate abilities (Figure 2). Notably, Vs,min values were found to increase with enlargement of the atomic size of the halogens in the sequence •ClO3 < •BrO3 < •IO3. Numerically, the positive EP magnitudes were 41.4, 51.2, and 52.2 kcal/mol for •ClO3, •BrO3, and •IO3 molecules, respectively.
On the other hand, a positive R•-hole site was detected mirroring that of the single electron, disclosing the potency of the considered R•-bearing molecules to engage in R•-hole site-based interactions. This observation was in line with the literature that reported the R•-hole site was located at the centroid of the three coplanar atoms.35 Regarding the R•-hole site, the Vs,max calculations highlighted the anomalous order in the R•-hole size where it increased as follows: •BrO3 < •ClO3 < •IO3. Overall, the forgoing manifestations would provide a proper corroboration for the potentiality of the inspected •XO3 molecules to simultaneous engage in R• and R•-hole site-based interactions.
Interaction Energy
To verify the efficacy of the investigated R•-bearing molecules to engage in R• and R•-hole site-based interactions, the NH3···•XO3 and •XO3···NH3 dimers were constructed, respectively. Furthermore, the propensity of •XO3 molecules to simultaneously interact via both sites was precisely examined within the NH3···•XO3···NH3 trimers. Geometrical optimization was performed for all of the studied complexes. The optimized geometries of the NH3···•XO3, •XO3···NH3, and NH3···•XO3···NH3 complexes along with the optimum distances are displayed in Figure 3. Upon optimization of the complexes, interaction energies (Eint) were evaluated. Table 1 gathers the complexation parameters along with energetic features of all of the optimized NH3···•XO3/•XO3···NH3 dimers and NH3···•XO3···NH3 trimers.
Figure 3.

Optimized structures of the dimeric and trimeric forms of R• and R•-hole site-based interactions in the form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 complexes, respectively. The X···N optimum distances are evaluated in Å.
Table 1. Eint and ECCSD(T)/CBS of the Dimeric and Trimeric Forms of R• and R•-Hole Site-Based Interactions in the Form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 Complexes, Respectively, in kcal/mol and X···N Intermolecular Distances (d1 and d2) in Å.
| |
distance |
||||
|---|---|---|---|---|---|
| complex | d1 | d2 | Eint | ECCSD(T)/CBS | |
| dimers | R• Site-Based Interactions | ||||
| H3N···•ClO3 | 2.80 | –6.01 | –5.90 | ||
| H3N···•BrO3 | 2.83 | –7.59 | –7.29 | ||
| H3N···•IO3 | 2.91 | –9.05 | –8.55 | ||
| R•-Hole Site-Based Interactions | |||||
| •ClO3···NH3 | 2.90 | –4.93 | –5.05 | ||
| •BrO3···NH3 | 2.84 | –6.44 | –6.86 | ||
| •IO3···NH3 | 2.47 | –14.82 | –16.10 | ||
| trimers | R• and R•-Hole Site-Based Interactions | ||||
| H3N···•ClO3···NH3 | 2.93 | 2.99 | –9.52 | –9.55 | |
| H3N···•BrO3···NH3 | 2.98 | 2.94 | –12.02 | –12.32 | |
| H3N···•IO3···NH3 | 3.01 | 2.51 | –19.89 | –21.19 | |
At first glance, all of the studied R•-bearing molecules demonstrated their apparent ability to engage in unconventional R• site-based interactions and well-established R•-hole ones (Figure 3). Alongside, the optimum intermolecular distances were lower than the van der Waals (vdW) radii, ranging from 2.47 to 3.01 Å. Concerning NH3···•XO3 dimers, negative Eint values were noted, asserting the impressive potentiality of the studied R•-bearing molecules to participate in R• site-based interactions with a Lewis base. Obviously, the Eint was found to increase in line with growing positive EP size over the single-electron molecular surface (see Figure 2). For example, the Eint values were −6.01, −7.59, and −9.05 kcal/mol for the NH3···•ClO3, ···•BrO3, and ···•IO3 dimers versus Vs,max values of 41.4, 51.2, and 52.2 kcal/mol for the •ClO3, •BrO3, and •IO3 molecules, respectively.
Turning to the R•-hole site-based interactions, a direct proportion was noticed between the atomic size of the studied group VII R•-bearing molecules and the Eint values of the •XO3···NH3 dimers. This amplitude was in agreement with the previous findings of the tetrel-bearing molecules.35 Illustratively, the Eint were found to increase as follows: •ClO3··· < •BrO3··· < •IO3···NH3, amounting to −4.93, −6.44, and −14.82 kcal/mol, respectively. Such pattern reflected the imprecision perspective of Vs,max affirmations relevant to the irregular correlation of the positive ESP with the R• atomic size, as previously reported.36
Based upon the preceding observations, higher Eint values were perceived for the NH3···•XO3 complexes over the •XO3···NH3 ones when X = Cl and Br, highlighting the favorability of R• site-based interactions over the R•-hole site-based ones. On the other hand, contradictory findings were obtained for iodine-bearing complexes.
Turning to the NH3···•XO3···NH3 trimers, Eint was discerned to coincide with the energetic trends concerning the investigated dimers. Apparently, the Eint values were observed to follow the order: NH3···•IO3···NH3 > NH3···•BrO3···NH3 > NH3···•ClO3···NH3 trimers with values of −19.89, −12.02, and −9.52 kcal/mol, respectively. Undoubtedly, impressive Eint values were registered for the NH3···•XO3···NH3 trimers over those of the NH3···•XO3 and •XO3···NH3 dimers. For example, the Eint values of the NH3···•IO3, •IO3···NH3, and NH3···•IO3···NH3 complexes were −9.05, −14.82, and −19.89 kcal/mol, respectively. The earlier observations evidently demonstrated the salient propensity of the considered R•-bearing molecules to simultaneously participate in R• and R•-hole site-based interactions.
Moreover, the MP2 energetic features were benchmarked for the studied dimers and trimers by assessing Eint at the CCSD(T)/CBS level of theory. As listed in Table 1, the CCSD(T)/CBS energetic results were found to be consistent with the MP2 ones. Numerically, the Eint values of the R• site-based interactions were −5.90, −7.29, and −8.55 kcal/mol for the NH3···•ClO3, ···•BrO3, and ···•IO3 dimers at the CCSD(T)/CBS level of theory along with Eint values of −6.01, −7.59, and −9.05 at the MP2 counterparts.
QTAIM Analysis
For a better understanding of the nature of the studied R• and R•-hole site-based interactions, QTAIM67 was employed. QTAIM diagrams of the NH3···•XO3 and •XO3···NH3 dimers, along with the NH3···•XO3···NH3 trimers, are delineated in Figure 4. Topological parameters, including ρb, ∇2ρb, and Hb, are summarized in Table 2.
Figure 4.

QTAIM diagrams of dimeric and trimeric forms of R• and R•-hole site-based interactions in the form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 complexes, respectively. BCP locations are indicated by red dots. The symbols (a) and (b) refer to the generated BCPs within the R• and R•-hole site-based interactions, respectively.
Table 2. Topological Parameters, Including ∇2ρb, ρb, and Hb at the Extracted BCPs within the Dimeric and Trimeric Forms of R• and R•-Hole Site-Based Interactions in the Form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 Complexes, Respectivelya.
| complex | ρ (au) | ∇2ρb (au) | Hb (au) | |
|---|---|---|---|---|
| dimers | R• Site-Based Interactions | |||
| H3N···•ClO3 | 0.0236 | 0.0676 | 0.0006 | |
| H3N···•BrO3 | 0.0255 | 0.0657 | 0.0001 | |
| H3N···•IO3 | 0.0276 | 0.0589 | –0.0009 | |
| R•-Hole Site-Based Interactions | ||||
| •ClO3···NH3 | 0.0162 | 0.0592 | 0.0013 | |
| •BrO3···NH3 | 0.0217 | 0.0656 | 0.0006 | |
| •IO3···NH3 | 0.0573 | 0.0700 | –0.0153 | |
| trimers | R• and R•-Hole Site-Based Interactions | |||
| H3N···•ClO3···NH3 | 0.0177b/0.0134c | 0.0536b/0.0505c | 0.0012b/0.0014c | |
| H3N···•BrO3···NH3 | 0.0190b/0.0176c | 0.0517b/0.0561c | 0.0008b/0.0010c | |
| H3N···•IO3···NH3 | 0.0230b/0.0529c | 0.0517b/0.0706c | –0.0001b/–0.0128c | |
All of the computed parameters are given in au.
The QTAIM topological parameters of R• site-based interactions concerned with H3N···•XO3···NH3.
The QTAIM topological parameters of R•-hole site-based interactions concerned with H3N···•XO3···NH3.
As delineated in Figure 4, solo BPs and BCPs were observed within all of the NH3···•XO3 complexes, affirming the unconventional behavior exhibited by group VII elements toward efficiently engaging in the R• site-based interactions with Lewis bases. The same remarks were obtained for the R•-hole site-based interactions that align with preceding studies.36 Turning to the NH3···•XO3···NH3 trimers, the potency of the R•-bearing molecules to simultaneously engage in R• and R•-hole site-based interactions was emphasized via the presence of two BPs and BCPs (i.e., one at each site). These findings demonstrated the highly directional R• and R•-hole site-based interactions of the investigated halogens.
As listed in Table 2, the closed-shell nature of the R• and R•-hole site-based interactions was confirmed via the obtained positive values of ∇2ρb and Hb along with the low values of ρb for the considered complexes, except for the iodine-bearing complexes. For the latter complexes, negative Hb values were detected, highlighting their partially covalent nature. Eminently, a notable correlation was found between the ρb values with the atomic size of the studied halogens and MP2 energetic values. Illustratively, the ρb of the R• site-based interactions were 0.0236, 0.0255, and 0.0276 au of the NH3···•ClO3, ···•BrO3, and ···•IO3 dimers, respectively, whose corresponding Eint values were −6.01, −7.59, and −9.05 kcal/mol.
NCI Analysis
The NCI index was earlier documented as a reliable tool to unveil the nature of the interactions between the chemical species three-dimensionally.6 NCI isosurfaces were generated herein for the NH3···•XO3 and •XO3···NH3 dimers along with the NH3···•XO3···NH3 trimers and are pictured in Figure 5. These isosurfaces are pictured through a reduced density gradient (RDG) value of 0.50 au and colored from blue (sign (λ2)ρ = −0.035) to red (sign (λ2)ρ = 0.020).
Figure 5.

NCI isosurfaces of the dimeric and trimeric forms of R• and R•-hole site-based interactions in the form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 complexes, respectively.
As shown in Figure 5, the presence of the green-bluish-coded region between the interacting molecules evidently affirmed the occurrence of attractive forces within the complexes under study. Additionally, clear blue-coded regions were observed for the NH3···•IO3, •IO3···NH3, and NH3···•IO3···NH3 complexes, highlighting their partially covalent nature, which was in line with the aforementioned QTAIM topological parameters. For almost all R•-hole hole site-based interactions, the contributions of the coplanar substituents to the total forces beyond the formation of the studied complexes were assured via the resulting shamrock shape, consistent with the previous studies.36
Electronic Parameters
To trace the electronic features, the FMO theory was asserted. In the FMO theory, the energy of the EHOMO, ELUMO, and EFL were computed for the considered molecules and complexes. The Egap was then assessed as the variation between the energies of the HOMO and LUMO. In the case of the singly occupied molecular orbital (SOMO), the distributions of the HOMO levels could be labeled as the SOMO patterns. The HOMO/LUMO electron densities were plotted for the isolated systems and complexes (Figures 6 and 7, respectively). EHOMO, ELUMO, EFL, and Egap values are tabulated in Table 3.
Figure 6.
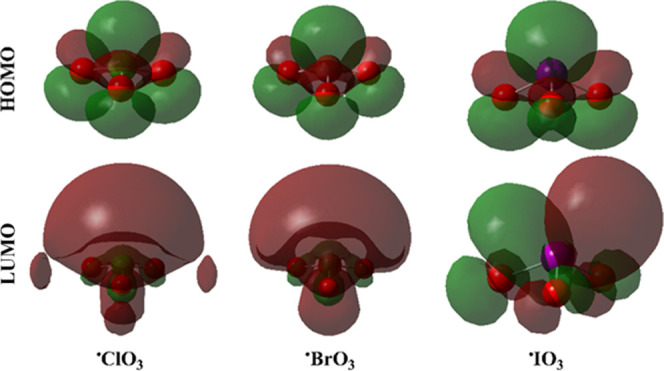
Plots of HOMO and LUMO distributions of the optimized •ClO3, •BrO3, and •IO3 monomers.
Figure 7.

Plots of HOMO and LUMO distributions within the dimeric and trimeric forms of R• and R•-hole site-based interactions in the form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 complexes, respectively.
Table 3. EHOMO, ELUMO, EFL, and Egap of the R•-Bearing Molecules and Dimeric and Trimeric Forms of R• and R•-Hole Site-Based Interactions in the Form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 Complexes, Respectively, Given in eV.
| molecule/complex | EHOMO (eV) | ELUMO (eV) | EFL (eV) | Egap (eV) | |
|---|---|---|---|---|---|
| monomers | R•-Bearing Molecules | ||||
| •ClO3 | –14.26 | –1.61 | –7.94 | 12.65 | |
| •BrO3 | –13.55 | –2.18 | –7.87 | 11.37 | |
| •IO3 | –12.60 | –0.67 | –6.63 | 11.93 | |
| dimers | R• Site-Based Interactions | ||||
| H3N···•ClO3 | –12.53 | –0.73 | –6.63 | 11.80 | |
| H3N···•BrO3 | –12.74 | –1.24 | –6.99 | 11.50 | |
| H3N···•IO3 | –12.46 | –1.20 | –6.83 | 11.26 | |
| R•-Hole Site-Based Interactions | |||||
| •ClO3···NH3 | –11.97 | –0.90 | –6.46 | 11.07 | |
| •BrO3···NH3 | –12.12 | –1.43 | –6.78 | 10.69 | |
| •IO3···NH3 | –0.12.27 | –0.98 | 6.62 | 11.29 | |
| trimers | R• and R•-Hole Site-Based Interactions | ||||
| H3N···•ClO3···NH3 | –11.58 | –0.18 | –5.88 | 11.40 | |
| H3N···•BrO3···NH3 | –11.62 | –0.71 | –6.17 | 10.91 | |
| H3N···•IO3···NH3 | –11.89 | –0.10 | –5.99 | 11.79 | |
As shown in Figure 6, the HOMO and LUMO distributions of the investigated systems were noticed over the nucleophilic and electrophilic sites, respectively. After complexation, the distributions of HOMO and LUMO were drastically changed (Figure 7).
From Table 3, substantial changes in the EHOMO, ELUMO, EFL, and Egap values were prominently noted before and after interactions of the R•-bearing molecules with the Lewis base. For instance, the EHOMO/ELUMO/EFL/Egap values of the •ClO3 molecule were −14.26/–1.61/–7.94/12.65 eV that altered after complexation with NH3 to −12.53/–0.73/–6.63/11.80 and −11.97/–0.90/–6.46/11.07 eV for NH3···•ClO3 and •ClO3···NH3 complexes, respectively.
A resurgent electron-donating character was noted for the investigated R•-bearing molecules on going from •Cl to •Br and •I, which was evidently reported via diminishing the EHOMO values (i.e., less negative). For the inspected dimers, the higher preferability of R• site-based interactions over the R•-hole site-based ones was also affirmed via more positive Egap for the NH3···•XO3 complexes over the •XO3···NH3 counterparts when X = Cl and Br. The reverse was confirmed for the iodine-bearing complexes, which was consistent with the energetic claims. Quantitatively, for the NH3···•XO3 and •XO3···NH3 dimers where X = Cl/Br/I, the Egap were 11.80/11.50/11.26 and 11.07/10.69/11.29 accompanied by Eint of −6.01/–7.59/–9.05 and −4.93/–6.44/–14.82 kcal/mol, respectively. On the other hand, relatively less negative EHOMO/ELUMO/EFL and positive Egap values were noted upon the formation of H3N···•XO3···NH3 trimers compared to dimers. Such observations outlined the higher preferability of the trimeric form than that of the dimeric one for the studied systems.
Global Indices of Reactivity
In an effort to unveil the chemical reactivity of the radical species to participate in several noncovalent interactions, global indices of reactivity were assessed before and after the occurrence of the studied interactions. Various global reactivity indices, including EA, S, η, IP, ω, μ, and Φ, were assessed (Table 4).
Table 4. Global Indices of Reactivity of the R•-Bearing Molecules and Dimeric and Trimeric Forms of R• and R•-Hole Site-Based Interactions in the Form of NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 Complexes, Respectively.
| molecule/complex | system | IP (eV) | EA (eV) | μ (eV) | η (eV) | S (eV–1) | ω (eV) | Φ (eV) |
|---|---|---|---|---|---|---|---|---|
| monomers | R•-Bearing Molecules | |||||||
| •ClO3 | 14.26 | 1.61 | –7.94 | 6.32 | 0.16 | 4.98 | 7.94 | |
| •BrO3 | 13.55 | 2.18 | –7.87 | 5.69 | 0.18 | 5.44 | 7.87 | |
| •IO3 | 12.60 | 0.67 | –6.63 | 5.97 | 0.17 | 3.68 | 6.63 | |
| dimers | R• Site-Based Interactions | |||||||
| H3N···•ClO3 | 12.53 | 0.73 | –6.63 | 5.90 | 0.17 | 3.72 | 6.63 | |
| H3N···•BrO3 | 12.74 | 1.24 | –6.99 | 5.75 | 0.17 | 4.25 | 6.99 | |
| H3N···•IO3 | 12.46 | 1.20 | –6.83 | 5.63 | 0.18 | 4.14 | 6.83 | |
| R•-Hole Site-Based Interactions | ||||||||
| •ClO3···NH3 | 11.97 | 0.90 | –6.43 | 5.54 | 0.18 | 3.74 | 6.43 | |
| •BrO3···NH3 | 12.12 | 1.43 | –6.78 | 5.34 | 0.19 | 4.30 | 6.78 | |
| •IO3···NH3 | 12.27 | 0.98 | –6.62 | 5.65 | 0.18 | 3.88 | 6.62 | |
| trimers | R• and R•-Hole Site-Based Interactions | |||||||
| H3N···•ClO3···NH3 | 11.58 | 0.18 | –5.88 | 5.70 | 0.18 | 3.03 | 5.88 | |
| H3N···•BrO3···NH3 | 11.62 | 0.71 | –6.17 | 5.45 | 0.18 | 3.49 | 6.17 | |
| H3N···•IO3···NH3 | 11.89 | 0.10 | –5.99 | 5.89 | 0.17 | 3.05 | 5.99 | |
Upon the complexation of the investigated R•-bearing molecules with the NH3 Lewis base via the R• and R•-hole sites, drastic differences in the reactivity parameters were noted in the form of studied dimers and trimers (Table 4). Numerically, the •IO3 molecule had IP/EA/μ/η/S/ω/Φ values of 12.60/0.67/–6.63/5.97/0.17/3.68/6.63 eV that changed to 12.46/1.20/–6.83/5.63/0.18/4.14/6.83, 12.27/0.98/–6.62/5.65/0.18/3.88/6.62, and 11.89/0.10/–5.99/5.89/0.17/3.05/5.99 eV in the case of the H3N···•IO3, •IO3···H3N, and H3N···•IO3···H3N complexes, respectively. These findings assured the alteration of electronic parameters before and after the complexation, outlining the occurrence of the investigated interactions.
Conclusions
The predilection of R•-bearing molecules to engage in the unconventional R• site-based interactions and the well-established R•-hole site-based interactions was comparatively studied. Accordingly, various MP2 computations were performed and analyzed for the NH3···•XO3, •XO3···NH3 and NH3···•XO3···NH3 complexes. EP analyses enunciated the presence of positive blue-coded regions over and opposite the molecular surface of the single electron, namely, R• and R•-hole sites, respectively. MP2 energies also accentuated the noteworthy potentiality of the studied molecules to engage in R• and R•-hole site-based interactions with a higher preferability for the former interactions in the case of chlorine- and bromine-bearing complexes. The latter trend was reversed in the case of the iodine-bearing complexes. A direct correlation was also disclosed between the Eint values and the atomic sizes of the studied halogen (X) atoms. QTAIM and NCI affirmations outlined the closed-shell nature of the R• and R•-hole site-based interactions except for the iodine-bearing complexes that were detected with a partially covalent one. Overall, these results will support the forthcoming studies for the characterization and applications of R• and R•-hole site-based interactions.
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R743), King Saud University, Riyadh, Saudi Arabia, for funding this work. The computational work was completed with resources provided by the CompChem Lab (Minia University, Egypt, hpc.compchem.net), Center for High-Performance Computing (Cape Town, South Africa, http://www.chpc.ac.za), Bibliotheca Alexandrina (http://hpc.bibalex.org), and the American University in Cairo.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c04620.
Cartesian atomic coordinates and expectation value of the S2 operator value of the optimized NH3···•XO3/•XO3···NH3 and NH3···•XO3···NH3 complexes (where X = Cl, Br, and I) (PDF)
Author Contributions
M.A.A.I.: Conceptualization, methodology, software, resources, project administration, supervision, writing—review and editing. H.S.M.A.E.: Data curation, formal analysis, investigation, visualization, writing—original draft. M.N.I.S.: Methodology, investigation, project administration, writing—review and editing. N.A.M.M.: Methodology, investigation, project administration, writing—review and editing. S.R.M.S.: Resources, writing—review and editing. M.E.S.S.: Resources, writing—review and editing. M.N.A.: Visualization, writing—review and editing. M.K.A.E.-R.: Writing—review and editing. T.S.: Conceptualization, resources, methodology, writing—review and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Mahmudov K. T.; Gurbanov A. V.; Guseinov F. I.; da Silva M. F. C. G. Noncovalent interactions in metal complex catalysis. Coord. Chem. Rev. 2019, 387, 32–46. 10.1016/j.ccr.2019.02.011. [DOI] [Google Scholar]
- Jiang S.; Zhang L.; Cui D.; Yao Z.; Gao B.; Lin J.; Wei D. The Important Role of Halogen Bond in Substrate Selectivity of Enzymatic Catalysis. Sci. Rep. 2016, 6, 34750 10.1038/srep34750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Liu Y.; Xu Z.; Li H.; Liu H.; Zhu W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discovery 2012, 7, 375–383. 10.1517/17460441.2012.678829. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Wang Y.; Zhu W. Nonbonding interactions of organic halogens in biological systems: Implications for drug discovery and biomolecular design. Phys. Chem. Chem. Phys. 2010, 12, 4543–4551. 10.1039/b926326h. [DOI] [PubMed] [Google Scholar]
- Mendez L.; Henriquez G.; Sirimulla S.; Narayan M. Looking back, looking forward at halogen bonding in drug discovery. Molecules 2017, 22, 1397. 10.3390/molecules22091397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson E. R.; Keinan S.; Mori-Sanchez P.; Contreras-Garcia J.; Cohen A. J.; Yang W. Revealing noncovalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. 10.1021/ja100936w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Qin K.; Deng G.; Wu G.; Bai Y.; Xu J. F.; Wang Z.; Yu Z.; Scherman O. A.; Zhang X. Supramolecular chemistry of cucurbiturils: Tuning cooperativity with multiple noncovalent interactions from positive to negative. Langmuir 2016, 32, 12352–12360. 10.1021/acs.langmuir.6b01709. [DOI] [PubMed] [Google Scholar]
- Bartkowski M.; Giordani S. Supramolecular chemistry of carbon nano-onions. Nanoscale 2020, 12, 9352–9358. 10.1039/D0NR01713B. [DOI] [PubMed] [Google Scholar]
- Li Z.-T.; Wu L.-Z.. Hydrogen Bonded Supramolecular Structures; Springer, 2015; Vol. 87. [Google Scholar]
- Davis A. P.; Wareham R. S. Carbohydrate recognition through noncovalent interactions: A challenge for biomimetic and supramolecular chemistry. Angew. Chem., Int. Ed. 1999, 38, 2978–2996. . [DOI] [PubMed] [Google Scholar]
- Mazik M. Molecular recognition of carbohydrates by acyclic receptors employing noncovalent interactions. Chem. Soc. Rev. 2009, 38, 935–956. 10.1039/b710910p. [DOI] [PubMed] [Google Scholar]
- Schalley C. A. Supramolecular chemistry goes gas phase: the mass spectrometric examination of noncovalent interactions in host–guest chemistry and molecular recognition. Int. J. Mass Spectrom. 2000, 194, 11–39. 10.1016/S1387-3806(99)00243-2. [DOI] [Google Scholar]
- Fish R. H.; Jaouen G. Bioorganometallic chemistry: Structural diversity of organometallic complexes with bioligands and molecular recognition studies of several supramolecular hosts with biomolecules, alkali-metal ions, and organometallic pharmaceuticals. Organometallics 2003, 22, 2166–2177. 10.1021/om0300777. [DOI] [Google Scholar]
- Mó O. Some interesting features of the rich chemistry around electron-deficient systems. Pure Appl. Chem. 2020, 92, 773–787. 10.1515/pac-2019-0814. [DOI] [Google Scholar]
- Politzer P.; Lane P.; Concha M. C.; Ma Y.; Murray J. S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. 10.1007/s00894-006-0154-7. [DOI] [PubMed] [Google Scholar]
- Clark T.; Hennemann M.; Murray J. S.; Politzer P. Halogen bonding: the sigma-hole. J. Mol. Model. 2007, 13, 291–296. 10.1007/s00894-006-0130-2. [DOI] [PubMed] [Google Scholar]
- Angarov V.; Kozuch S. On the σ, π and δ hole interactions: A molecular orbital overview. New J. Chem. 2018, 42, 1413–1422. 10.1039/C7NJ03632A. [DOI] [Google Scholar]
- Varadwaj P. R.; Varadwaj A.; Marques H.; Yamashita K. Can combined electrostatic and polarization effects alone explain the F···F negative-negative bonding in simple fluoro-substituted benzene derivatives? A first-principles perspective. Computation 2018, 6, 51–84. 10.3390/computation6040051. [DOI] [Google Scholar]
- Brammer L. Halogen bonding, chalcogen bonding, pnictogen bonding, tetrel bonding: origins, current status and discussion. Faraday Discuss. 2017, 203, 485–507. 10.1039/C7FD00199A. [DOI] [PubMed] [Google Scholar]
- Varadwaj A.; Marques H. M.; Varadwaj P. R. Is the fluorine in molecules dispersive? Is molecular electrostatic potential a valid property to explore fluorine-centered non-covalent interactions?. Molecules 2019, 24, 379–407. 10.3390/molecules24030379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkorta I.; Elguero J.; Frontera A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180–208. 10.3390/cryst10030180. [DOI] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T.; Resnati G. The sigma-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. 10.1039/C7CP06793C. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. A. A.; Saeed R. R. A.; Shehata M. N. I.; Ahmed M. N.; Shawky A. M.; Khowdiary M. M.; Elkaeed E. B.; Soliman M. E. S.; Moussa N. A. M. Type I–IV halogen···halogen interactions: A comparative theoretical study in halobenzene···halobenzene homodimers. Int. J. Mol. Sci. 2022, 23, 3114. 10.3390/ijms23063114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. A. A.; Shehata M. N. I.; Rady A. S. M.; Abuelliel H. A. A.; Abd Elhafez H. S. M.; Shawky A. M.; Oraby H. F.; Hasanin T. H. A.; Soliman M. E. S.; Moussa N. A. M. Effects of Lewis basicity and acidity on sigma-hole interactions in carbon-bearing complexes: A comparative ab initio study. Int. J. Mol. Sci. 2022, 23, 13023. 10.3390/ijms232113023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. A. A.; Shehata M. N. I.; Soliman M. E. S.; Moustafa M. F.; El-Mageed H. R. A.; Moussa N. A. M. Unusual chalcogen···chalcogen interactions in like···like and unlike Y = C=Y···Y = C=Y complexes (Y = O, S, and Se). Phys. Chem. Chem. Phys. 2022, 24, 3386–3399. 10.1039/D1CP02706A. [DOI] [PubMed] [Google Scholar]
- Murray J. S.; Politzer P. The electrostatic potential: An overview. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 153–163. 10.1002/wcms.19. [DOI] [Google Scholar]
- Frontera A. σ- and π-Hole Interactions. Crystals 2020, 10, 721. 10.3390/cryst10090721. [DOI] [Google Scholar]
- Ibrahim M. A. A.; Saeed R. R. A.; Shehata M. N. I.; Mohamed E. E. B.; Soliman M. E. S.; Al-Fahemi J. H.; El-Mageed H. R. A.; Ahmed M. N.; Shawky A. M.; Moussa N. A. M. Unexplored σ-hole and π-hole interactions in (X2CY)2 complexes (X = F, Cl; Y = O, S). J. Mol. Struct. 2022, 1265, 133232 10.1016/j.molstruc.2022.133232. [DOI] [Google Scholar]
- Politzer P.; Murray J. S.; Clark T. The pi-hole revisited. Phys. Chem. Chem. Phys. 2021, 23, 16458–16468. 10.1039/D1CP02602J. [DOI] [PubMed] [Google Scholar]
- Bauza A.; Frontera A.; Mooibroek T. J. pi-Hole interactions involving nitro aromatic ligands in protein structures. Chemistry 2019, 25, 13436–13443. 10.1002/chem.201903404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. A. A.; Saeed R. R. A.; Shehata M. N. I.; Moussa N. A. M.; Tawfeek A. M.; Ahmed M. N.; Abd El-Rahman M. K.; Shoeib T. Sigma-hole and lone-pair-hole site-based interactions of seesaw tetravalent chalcogen-bearing molecules with Lewis bases. ACS Omega 2023, 8, 32828–32837. 10.1021/acsomega.3c03981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauza A.; Mooibroek T. J.; Frontera A. Sigma-hole opposite to a lone pair: Unconventional pnicogen bonding interactions between ZF3 (Z = N, P, As, and Sb) compounds and several donors. ChemPhysChem 2016, 17, 1608–1614. 10.1002/cphc.201600073. [DOI] [PubMed] [Google Scholar]
- Shukla R.; Yu D.; Mu T.; Kozuch S. Yet another perspective on hole interactions, part II: lp-hole vs. lp-hole interactions. Phys. Chem. Chem. Phys. 2023, 25, 12641–12649. 10.1039/D3CP00225J. [DOI] [PubMed] [Google Scholar]
- Del Bene J. E.; Alkorta I.; Elguero J.; Sanchez-Sanz G. Lone-pair hole on P: P···N pnicogen bonds assisted by halogen bonds. J. Phys. Chem. A 2017, 121, 1362–1370. 10.1021/acs.jpca.6b12553. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. A. A.; Telb E. M. Z. Comparison of ± σ-hole and ± R-hole interactions formed by tetrel-containing complexes: a computational study. RSC Adv. 2021, 11, 4011–4021. 10.1039/D0RA09564H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim M. A. A.; Mohamed Y. A. M.; Abd Elhafez H. S. M.; Shehata M. N. I.; Soliman M. E. S.; Ahmed M. N.; Abd El-Mageed H. R.; Moussa N. A. M. R•-hole interactions of group IV-VII radical-containing molecules: A comparative study. J. Mol. Graph. Model. 2022, 111, 108097 10.1016/j.jmgm.2021.108097. [DOI] [PubMed] [Google Scholar]
- Wang Y.-H.; Zou J.-W.; Lu Y.-X.; Yu Q.-S.; Xu H.-Y. Single-electron halogen bond: Ab initio study. Int. J. Quantum Chem. 2007, 107, 501–506. 10.1002/qua.21097. [DOI] [Google Scholar]
- Alkorta I.; Elguero J.; Solimannejad M. Single electron pnicogen bonded complexes. J. Phys. Chem. A 2014, 118, 947–953. 10.1021/jp412144r. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wu D.; Li Z. R.; Chen W.; Sun C. C. Do single-electron lithium bonds exist? Prediction and characterization of the H3C···Li-Y (Y = H, F, OH, CN, NC, and CCH) complexes. J. Chem. Phys. 2006, 125, 084317 10.1063/1.2339020. [DOI] [PubMed] [Google Scholar]
- Li Z. F.; Yang X. P.; DeYonker N. J.; Xu X. Y.; Guo Z.; Zhao C. Y. Binding energies and interaction origins between nonclassical single-electron hydrogen, sodium and lithium bonds and neutral boron-containing radicals: a theoretical investigation. China Sci. Bull. 2014, 59, 2597–2607. 10.1007/s11434-014-0361-z. [DOI] [Google Scholar]
- Yu D.; Wu D.; Li Y.; Li S. Y. On the formation of beryllium bonds where radicals act as electron donors. Theor. Chem. Acc. 2016, 135, 112. 10.1007/s00214-016-1877-x. [DOI] [Google Scholar]
- Merritt J. M.; Rudic S.; Miller R. E. Infrared laser spectroscopy of CH3···HF in helium nanodroplets: The exit-channel complex of the F + CH4 reaction. J. Chem. Phys. 2006, 124, 084301 10.1063/1.2168450. [DOI] [PubMed] [Google Scholar]
- Tachikawa H. A direct ab initio dynamics study on the finite temperature effects on the hyperfine coupling constant of a weakly bonded complex. J. Phys. Chem. A 1998, 102, 7065–7069. 10.1021/jp981733x. [DOI] [Google Scholar]
- Misochko E. Y.; Benderskii V. A.; Goldschleger A. U.; Akimov A. V.; Shestakov A. F. Formation of the CH3-HF complex in reaction of thermal F atoms with CH4 in solid Ar. J. Am. Chem. Soc. 1995, 117, 11997–11998. 10.1021/ja00153a023. [DOI] [Google Scholar]
- Alkorta I.; Rozas I.; Elguero J. Radicals as hydrogen bond acceptors. Ber. Bunsen-Ges. Phys. Chem. 1998, 102, 429–435. 10.1002/bbpc.19981020323. [DOI] [Google Scholar]
- An X.; Liu H.; Li Q.; Gong B.; Cheng J. Influence of substitution, hybridization, and solvent on the properties of C-HO single-electron hydrogen bond in CH3-H2O complex. J. Phys. Chem. A 2008, 112, 5258–5263. 10.1021/jp710414g. [DOI] [PubMed] [Google Scholar]
- Hammerum S. Alkyl radicals as hydrogen bond acceptors: computational evidence. J. Am. Chem. Soc. 2009, 131, 8627–8635. 10.1021/ja901854t. [DOI] [PubMed] [Google Scholar]
- Rudić S.; Merritt J. M.; Miller R. E. Study of the CH3 H2O radical complex stabilized in helium nanodroplets. Phys. Chem. Chem. Phys. 2009, 11, 5345–5352. 10.1039/b817484a. [DOI] [PubMed] [Google Scholar]
- Li Q.; An X.; Luan F.; Li W.; Gong B.; Cheng J.; Sun J. Cooperativity between two types of hydrogen bond in H(3)C-HCN-HCN and H(3)C-HNC-HNC complexes. J. Chem. Phys. 2008, 128, 154102 10.1063/1.2898499. [DOI] [PubMed] [Google Scholar]
- Rudić S.; Merritt J. M.; Miller R. E. Infrared laser spectroscopy of the CH3-HCN radical complex stabilized in helium nanodroplets. J. Chem. Phys. 2006, 124, 104305 10.1063/1.2170087. [DOI] [PubMed] [Google Scholar]
- Solimannejad M.; Alikhani M. E. Theoretical study of the HCN-CH and HNC-CH radicals: Hydrogen and covalent bonding. Chem. Phys. Lett. 2005, 406, 351–354. 10.1016/j.cplett.2005.03.025. [DOI] [Google Scholar]
- Wang B.-Q.; Li Z.-R.; Wu D.; Hao X.-Y.; Li R.-J.; Sun C.-C. Single-electron hydrogen bonds in the methyl radical complexes H3C···HF and H3C···HCCH: an ab initio study. Chem. Phys. Lett. 2003, 375, 91–95. 10.1016/S0009-2614(03)00822-4. [DOI] [Google Scholar]
- Frisch J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.; et al. Gaussian 09, revision E.01; Gaussian, Inc.: Wallingford CT, 2009.
- Møller C.; Plesset M. S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. 10.1103/PhysRev.46.618. [DOI] [Google Scholar]
- Kendall R. A.; Dunning T. H.; Harrison R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. 10.1063/1.462569. [DOI] [Google Scholar]
- Woon D. E.; Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. 10.1063/1.464303. [DOI] [Google Scholar]
- Peterson K. A.; Figgen D.; Goll E.; Stoll H.; Dolg M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. 10.1063/1.1622924. [DOI] [Google Scholar]
- Peterson K. A.; Shepler B. C.; Figgen D.; Stoll H. On the Spectroscopic and Thermochemical Properties of ClO, BrO, IO, and Their Anions. J. Phys. Chem. A 2006, 110, 13877–13883. 10.1021/jp065887l. [DOI] [PubMed] [Google Scholar]
- Pritchard B. P.; Altarawy D.; Didier B.; Gibson T. D.; Windus T. L. New basis set exchange: An open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model. 2019, 59, 4814–4820. 10.1021/acs.jcim.9b00725. [DOI] [PubMed] [Google Scholar]
- Feller D. The role of databases in support of computational chemistry calculations. J. Comput. Chem. 1996, 17, 1571–1586. . [DOI] [Google Scholar]
- Schuchardt K. L.; Didier B. T.; Elsethagen T.; Sun L.; Gurumoorthi V.; Chase J.; Li J.; Windus T. L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. 10.1021/ci600510j. [DOI] [PubMed] [Google Scholar]
- Ibrahim M. A. A. Molecular mechanical perspective on halogen bonding. J. Mol. Model. 2012, 18, 4625–4638. 10.1007/s00894-012-1454-8. [DOI] [PubMed] [Google Scholar]
- Lu T.; Chen F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Boys S. F.; Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. 10.1080/00268977000101561. [DOI] [Google Scholar]
- Mishra B. K.; Karthikeyan S.; Ramanathan V. Tuning the C-H···Pi interaction by different substitutions in benzene-acetylene complexes. J. Chem. Theory Comput. 2012, 8, 1935–1942. 10.1021/ct300100h. [DOI] [PubMed] [Google Scholar]
- Helgaker T.; Klopper W.; Koch H.; Noga J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639–9646. 10.1063/1.473863. [DOI] [Google Scholar]
- Bader R. F. W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. 10.1021/ar00109a003. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Weiner P. K.; Langridge R.; Blaney J. M.; Schaefer R.; Kollman P. A. Electrostatic potential molecular surfaces. Proc. Natl. Acad. Sci. U.S.A. 1982, 79, 3754–3758. 10.1073/pnas.79.12.3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.