

Abstract
The angucycline group is the largest group of type II PKS-engineered natural products, rich in biological activities and chemical scaffolds. This stimulated synthetic creativity and biosynthetic inquisitiveness. The synthetic studies used five different strategies, involving Diels-Alder reactions, nucleophilic additions, electrophilic additions, transition-metal mediated cross-couplings and intramolecular cyclizations to generate the angucycline frames. Biosynthetic studies were particularly intriguing when unusual framework rearrangements by post-PKS tailoring oxidoreductases occurred, or when unusual glycosylation reactions were involved in decorating the benz[a]anthracene-derived cores. This review follows our previous reviews, which were published in 1992 and 1997, and covers new angucycline group antibiotics published between 1997 and 2010. However, in contrast to the previous reviews, the main focus of this article is on new synthetic approaches and biosynthetic investigations, most of which were published between 1997 and 2010, but go beyond, e.g. for some biosyntheses all the way back to the 1980s, to provide the necessary context of information.
1. Introduction
The angucycline group of natural products is the largest group of polycyclic aromatic polyketides, rich in chemical scaffolds and various biological activities, predominantly anticancer and antibacterial. Despite this chemical and biological diversity, none of these compounds have been developed into clinically applicable drugs, often due to toxicity or solubility issues. This review is the first in almost 15 years, and covers new examples of angucyclines and angucyclinones as well as intriguing new biological activities of angucyclines, which are quite diverse, ranging from enzyme inhibitory to antibacterial, anti-viral, anticancer and platelet-aggregation inhibition. However, the main focus of this article is on biosynthetic studies, most of which have emerged during the past decade, after the publication of our 1992 and 1997 reviews,1,2 or were not discussed in detail in the previous reviews. Another major focus of this article is to give an update on modern synthetic strategies applied to achieve the skeletons of the tetracyclic angucycline core, of complex angucyclines as well as of angucycline-derived natural products. The landomycins belong to the most active and most promising angucycline anticancer drugs, and most recently the first total synthesis of the complex, sugar-rich angucycline landomycin A, was reported.3
All angucyclin/one/s are polyketide derived, with type II polyketide synthases (PKS) responsible for the generation of the initial framework,4 and angucyclines form by far the largest group among type II PKS-decaketide-derived natural products. The biosyntheses of various typical angucyclines and angucyclinones were studied (e.g., urdamycins, landomycins, gaudimycins, simocyclinone, oviedomycin) whose angular benz[a] anthracene-derived polyketide backbones remained intact. However, the benz[a]anthracene frame typical for this group of natural products can either emerge directly or through a rearrangement of an anthracyclinone skeleton (Scheme 1). Considerable focus of the biosynthetic studies was given to unique angucyclinone-derived natural products, whose polyketide derived scaffold is drastically modified through post-PKS tailoring oxidative rearrangement reactions (e.g., jadomycins, gilvocarcins, kinamycins etc.). Thus, the past decade of research proved many natural product groups as angucyclinone-derived, a fact previously either undiscovered or just suggested from incorporation experiments with isotope-labeled precursors.
Scheme 1.

Two different pathways for biosynthesis of the typical benz[a]anthracene backbone of the angucycline group of natural products.
2. Biosynthesis
Angucycline group compounds retain, or are derived from, a benz[a]anthracene moiety. Type II polyketide synthase (PKS) enzymes utilize a starter unit, most commonly, acetyl CoA and extender units (9 malonyl CoA) to establish a decaketide backbone. Incorporation studies involving the feeding of [1-13C] acetate and [1,2-13C] acetate were carried out to identify the origin of the backbone carbons of this moiety. Such studies revealed two different routes for the formation of the benz[a] anthracene backbone. A typical example is the much studied urdamycin A (31), which derives from a decaketide intermediate (1) that folds in an angular fashion, facilitating ring closure reactions, thereby leading to the formation of the benz [a]anthracene moiety (route I, Scheme 1).5 This route has been suggested for the formation of the benz[a]anthracene intermediates and as the route to the biosynthesis of many other angucyline/angucyclinone-derived molecules, such as vineomycin A1, landomycin A (4), chrysomycin A (152), ravidomycin V (123), gilvocarcin V (122), and kinamycin D (205).6–11 However, in a few cases, the newly formed decaketide intermediate 1 follows a different folding and cyclization pattern (route II), leading to the formation of an anthracyclinone intermediate (3, route II). Oxidative disconnection of the bond between C10a–C11 of tetracyclic intermediate 3, possibly through a pathway specific Baeyer–Villiger oxygenase, and reconnection of the C3 and C11 establishes the benz[a]anthracene backbone. The latter pathway involving the rearrangement of an anthracycline type backbone was first discovered by Gould et al. in the biosynthesis of the angucyclinone PD116198, and more recently by Liu et al. in the biosynthesis of BE7585A (198).12,13
2.1. Biosynthesis of landomycins
The landomycins, discovered in 1990, form one of the best known families of the angucycline group.14 Landomycin A (4) and its congener landomycin C comprise hexasaccharidal chains, and were isolated along with the pentasaccharidal landomycin B, as well as the disaccharidal landomycin D from the culture broth of Streptomyces cyanogenus S136 (lan).7,14 The trisaccharidal variant of 4, namely landomycin E (14), was found later, isolated from Streptomyces globisporus. The biosynthetic origin of all the carbons and most of the oxygens of landomycinone, the aglycone of 4, were identified through the incorporation study of labeled precursors.7 Separate feeding experiments with [1-13C]-, [1,2-13C]- and [1-13C, 18O2]-acetate and fermentation of S. cyanogenus S136 under 18O2 atmosphere demonstrated that all of the carbons of landomycinone originated from acetate, and the biosynthesis involved a decaketide intermediate that underwent further cyclization and aromatization to establish the backbone of landomycinone (Scheme 2). The results also established that only the two oxygen atoms at C-1 and C-8 positions of landomycinone originated from acetate. Surprisingly, the oxygen atoms at C-7 and C-12, in addition to the expected C-11 position of 5,6-anhydrolandomycinone A (5), were found to originate from molecular oxygen. These results imply that the oxygen atoms at C-7 and C-12, originally derived from acetate during the course of decaketide backbone biosynthesis, must be removed and reinstalled later through the activity of other post-PKS tailoring enzymes. The origin of the oxygen atom at C-6 position still remains ambiguous.15
Scheme 2.

The incorporation of 18O-labeled precursor in landomycin A.
Genetic and biochemical studies of landomycin A (4) began with the cloning of a gene cluster from S. cyanogenus S136 (lan).16 Sequencing of a 35 kb region of the genomic DNA of S. cyanogenus revealed 32 putative open reading frames, including type II polyketide genes, post-polyketide tailoring enzymes (oxygenases, reductases), sugar biosynthetic genes, glycosyltransferases, regulatory and antibiotic transport genes (Fig. 1, Table 1). Similarly, the biosynthetic gene cluster of landomycin E (14) has been cloned and sequenced from S. globisporus (lnd). The two gene clusters appeared to be almost identical, except for three additional open reading frames (ORFs) lanK, lanGT3, and lanZ2 found only in the lan cluster, while a lndI homologue was missing in the lan cluster.15 Co-expression of the cosmid that harbors the whole biosynthetic gene cluster of landomycin A (4) with the regulatory gene lndI in the PKS enzymes deficient mutant of urdamycin producer S. fradiae resulted in the production of 4 and 5,6-anhydrolandomycinone A (5).17 These results provided clear proof that the isolated gene cluster contained all of the necessary genes that encode the biosynthetic machinery to generate 4.
Fig. 1.

Landomycin E and landomycin A biosynthetic gene clusters from S. globisporus 1912 (lnd genes, A) and S. cyanogenus S136 (lan genes, B), respectively.
Table 1.
Landomycin A biosynthetic genes and proposed functions of their products16
| Genes | Proposed function | Genes | Proposed function |
|---|---|---|---|
|
| |||
| lanE | Oxygenase | lanU | unknown |
| lanF | Cyclase (CYC) | lanV | reductase |
| lanA | ketoacyl synthase (KSα) | lanGT2 | glycosyltransferase |
| lanB | chain length factor (KSβ) | lanX | unknown |
| lanC | acyl carrier protein (ACP) | lanGT1 | glycosyltransferase |
| land | reductase | lanK | regulatory gene |
| lanL | Cyclase (CYC) | lanJ | transporter gene |
| lanM2 | oxidoreductase | lanZ1 | NDP-hexose 3,5-epimerase |
| lanO | reductase | lanGT3 | glycosyltransferase |
| lanP | decarboxylase | lanZ2 | NDP-hexose synthase |
| lanG | NDP-hexose synthase | lanZ3 | NDP-hexose 4-ketoreductase |
| lanH | NDP-hexose 4,6-dehydratase | lanGT4 | glycosyltransferase |
| lanQ | NDP-hexose 3,4-dehydratase | lanZ4 | reductase |
| lanR | NDP-hexose 4-ketoreductase | lanZ5 | oxygenase |
| lanS | NDP-hexose 2,3-dehydratase | lanZ6 | unknown |
| lanT | oxidoreductase | ||
2.1.1. Lan(Lnd)PKS enzymes.
The encoded products of lanA, lanB, lanC, and lanD represent ketoacyl synthase (KSα), chain length factor (CLF), acyl carrier protein, and a PKS associated C9-ketoreductase, respectively, which are typically found in aromatic polyketide biosynthetic gene clusters. The products of lanF and lanL are homologous to other cyclases found in type II polyketide gene clusters. Although there are no complete experimental data to assign the exact role of these PKS enzymes, it is anticipated that the products of lanABCDFL are sufficient for the biosynthesis of the earliest proposed intermediate, UWM6 (6). Through gene inactivation experiments in S. globisporus and subsequent analyses of products from the mutants, Fedorenko et al.18 suggested that LndL (homologue of LanL) controls the first cyclization–aromatization event during biosynthesis of landomycin E (14). Similarly, LndF (homologue of LanF) was proposed to catalyze the formation of the third and the fourth ring.
2.1.2. Post-PKS tailoring enzymes
2.1.2.1. Enzymes involved in biosynthesis of landomycinone/11-deoxylandomycinone.
Five oxygenases and reductases (Lan/LndZ4, Z5, E, M2, V) are proposed for the conversion of the Lan/LndPKS product UWM6 (6) to 11-deoxylandomycinone (13).15 Genes lanM and lanN, which were originally deposited as two separate oxygenase and reductase genes, were later revised to be a single oxidoreductase gene lanM2. A homologous enzyme LndM2, which shares 79% amino acid identity to LanM2, was also identified in the landomycin E producer S. globisporus 1912.15 Homology searches in the database revealed two clear domains of M2. The N-terminal 510 amino acids region shows similarity to a number of pyr_redox_superfamily enzymes, including both class I and class II oxidoreductases, NADH-oxidases and peroxidases. The C-terminal 233 amino acid region displays a Rossmann-fold NAD(P)H/NAD(P)(+) binding (NADB) domain, often found in a variety of dehydrogenases. Homologues of LanM2 are also found in several other angucycline biosynthetic pathways including the gaudimycin (PgaM)19 and urdamycin (UrdM).20 To identify the potential role of the encoded products of lanM2/lndM2 in landomycin A biosynthesis, Rohr et al.21 have expressed lndM2 in the urdamycin A (31) producer wild type strain S. fradiae, in the urdM disrupted mutant strain S. fradiae ΔurdM, in S. fradiae AX strain, which lacks three of the four urdamycin pathway glycosyltransferases (GTs) urdGT1a, urdGT1b and urdGT1c, and in S. fradiae A0, which lacks all of the GTs of urdamycin biosynthetic pathway. Despite a close resemblance of the urdamycin and landomycin biosynthetic pathways, expression of lndM2 in these mutants did not reveal any change in the production spectrum of the parent strain. These results suggested that neither urdamycin A (31) nor any of its intermediates can serve as substrates for LndM2.15
To further explore the exact role of LndM2 during the biosynthesis of landomycin E (14), the corresponfing gene was inactivated in S. globisporus 1912. The generated mutant strain completely abolished the production of 14 and accumulated tetrangomycin (9), 11-hydroxytetrangomycin (10), 4-hydroxytetrangomycin (7), and tetrangulol (8), all lacking sugar residues.15 Feeding of these isolated metabolites in the culture of the LndF (PKS-associated cyclase)-deficient mutant revealed that tetrangomycin (9) was the only compound that was converted to 14, thus is likely a pathway intermediate, while the others seem to be metabolic shunt products of the biosynthetic pathway. Interestingly, feeding of another structurally related compound, rabelomycin (11) also restored the production of landomycin E (14).15,18 These results together concluded in a hypothesis that tetrangomycin (9) can be converted to 11-deoxylandomycinone through at least two different routes (Scheme 3). In path I, LndM2 catalyzes hydroxylation at C-6 position of 9 to yield rabelomycin (11). The LndM2 catalyzed reduction of the C5–C6 double bond coupled with 2,3-dehydration of 11 generates 11-deoxylandomycinone (13). Alternatively, in path II, LndM2 may reduce the C5–C6 double bond of 9 to yield 5,6-dihydrotetrangomycin (12), and its oxygenation at C-6 position and aromatization of its A-ring generates 11-deoxylandomycinone (13).
Scheme 3.

Alternatives for the proposed biosynthetic pathway for landomycin E (14) via 11-deoxylandomycinone (13).
Among the remaining enzymes LanZ4/LndZ4 is a flavin reductase homologue while LanZ5/LndZ5 is an aromatic hydroxylase. Bechthold et al.22 inactivated both of lanZ4/lanZ5 together with the glycosyltransferase lanGT3. Thus, the generated mutant strain accumulated the disaccharidial derivative landomycin F (36), which lacked a hydroxy group at the 11-position. Expression of lanZ4/lanZ5 in this mutant strain led to the production of landomycin D (24), the hydroxylated analogue of landomycin F (36). This result demonstrated that the LanZ4–LanZ5 pair is responsible for the introduction of a hydroxyl group at the 11-position during the biosynthesis of landomycin A (4).23 Feeding experiments of Landomycin F (36) into the culture of a PKS mutant of S. cyanogenus S13618 produced 4, indicating that the hydroxylation at the 11-position is not dependent on the length of the sugar chain and can occur at different glycosylation stages during biosynthesis of 4. In contrast to these results, accumulation of 11-hydroxytetrangomycin (10) by the oxidoreductase LndM2-deficient mutant indicated that the Lan/LndZ4–Z5-catalyzed hydroxylation at the 11-position can occur prior to the first glycosylation.15 Thus, the exact timing of action and the preferred substrates for Lnd/LanZ4–Z5 are not fully understood, but it is clear that Lan/LndZ4–Z5 has broad substrate specificity. In addition to LndZ4–Z5, LanE was identified as another oxygenase in the biosynthetic pathway of landomycin E (14). Targeted inactivation of lndE gene in S. globisporus 1912 resulted in the complete abolishment of the production of 14.24 The mutant strain accumulated the intermediate prejadomycin (21) as the major metabolite instead (Scheme 5). This clearly showed that LndE is the earliest acting oxygenase, which incorporates an oxygen at the C-12 position. The enzyme was also proposed to catalyze the removal of the C-6 oxygen. The product of lanV displays the strongest amino acid identities (60–70%) with a number of NADB_Rossmann superfamily enzymes, including PgaM from gaudimycin A (185) and SaqN from saquayamycin Z (222) biosynthetic pathways.19,25 The product of this gene was proposed to catalyze the aromatization of ring A, which is necessary for the conversion of tetrangomycin (9) to 11-deoxylandomycinone (13)/landomycinone. The production of 9-C-olivosyltetrangulol (39), after complementation of an urdM-deletion mutant of S. fradiae (urdamycin A producer strain) with a lanV expression construct, suggested that LanV contributes to the aromatization of ring A as well as to the removal of the C-6 hydroxyl group.26 However, in a recent report, Lan/LndE was proposed to catalyze that deoxygenation step.24 Further experiments, including the inactivation of the lan/lndV gene and an in vitro activity assay involving Lan/LndV, would help assign the exact role of these enzymes in landomycin biosynthesis.
Scheme 5.

The proposed biosynthetic pathway for landomycins A and E.
2.1.3. Deoxysugar biosynthesis.
Landomycin A (4) comprises a hexasaccharidal side chain composed of a repeated sequence of a trisaccharidal unit of d-olivose-4–1-d-olivose-3–1-l-rhodinose. Thorough sequence analyses of the 4 biosynthetic gene cluster revealed 9 ORFs that could potentially be involved in the deoxysugar biosynthetic pathways.16 The genes lanG and lanZ2 encode NDP-hexose synthases, whereas lanH encodes an NDP-hexose-4,6-dehydratase. These three enzymes catalyze the conversion of d-glucose-1-phosphate (15) to TDP-4-keto-6-deoxy-d-glucose (17) (Scheme 4). The encoded products of lanS (NDP-hexose 2,3-dehydratase) and lanT (NDP-hexose-3-ketoreductase) were anticipated to catalyze a 2-deoxygenation reaction and 3-ketoreduction, respectively. Reduction of the 4-keto group is most likely catalyzed by the product of lanR, which generates NDP-d-olivose (19). The product of lanQ catalyzes C-3 deoxygenation of the 4-keto intermediate (18), whereas the proteins encoded by lanZ1 and lanZ3 were proposed to catalyze epimerization and 4-ketoreductions, respectively, during the biosynthesis of NDP-l-rhodinose (20) (Scheme 4).
Scheme 4.

The proposed pathway for biosynthesis of the NDP-d-olivose and NDP-l-rhodinose building blocks in context with landomycin biosynthesis.
2.1.4. Glycosylation.
Antibiotics containing oligosaccharide moieties are very promising regarding the engineering of new/novel analogues through glycodiversification. In this context, the pathway glycosyltransferases responsible for the construction of the hexasaccharidal side chain of landomycin A (4) have drawn considerable attention in the biosynthetic community. Only four glycosyltransferase genes (lanGT1, lanGT2, lanGT3, and lanGT4) were randomly distributed throughout the lan gene cluster, but responsible for 6 glycosyl transfer steps.16 The production of hexasaccharidal 4 through the expression of the cosmid that harbored all four glycosyltransferases (lanGT1, lanGT2, lanGT3, and lanGT4) and other biosynthetic genes clearly indicated that one or two of the encoded GTs has to work repeatedly.17 The functional roles of individual glycosyltransferases were identified through a series of experiments including gene inactivation and heterologous expression of these genes. Mutant strain (A-x) of the urdamycin A (31) producer S. fradiae Tü2717 that lacks all urdamycin glycosyltransferases, except for urdGT2 (responsible for the C-glycosidic attachment of a d-olivose at C-9 position in urdamycin A (31)) was used for complementation experiments, using various lanGT-expression constructs involving a single GT or a combination of multiple GTs.27 Expression of lanGT1 alone or in combination with other lanGTs in the A-x strain resulted in the production of new hybrid compounds, called ladamycins (ladamycin D3 (30), ladamycin B2 (28), ladamycin B3 (27), with an additional d-olivose attached to the C-glycosidically linked d-olivose (Fig. 2).
Fig. 2.

Engineered landomycin–urdamycin hybrid molecules.
Similarly, the expression of LanGT4 alone or along with other LanGTs generated molecules with a one-sugar (urdamycin G, 55), two-sugar (100–1, 49) and a three-sugar (ladamycin B3, 27) chain, respectively, with an l-rhodinose moiety at the end of these chains. These results indicated that lanGT1 and lanGT4 encode a d-olivosyltransferase and an l-rhodinosyltransferase, respectively (Scheme 5). The results also demonstrated a remarkably relaxed acceptor substrate flexibility of these glycosyltransferases. A follow up experiment involving the expression of lanGT4 alone in the urdamycin A (31) producer S. fradiae Tü2717 resulted in the production of new urdamycin A and B analogues (32, and 34, respectively, Scheme 6) with an extra l-rhodinose moiety linked to the terminal d-olivose through an α-(1,3)-linkage.28 Such an unusual flexibility of LanGT4 with regard to the saccharide length of the acceptor substrate provided a basis for the assumption that LanGT4 may catalyze the transfer of both l-rhodinose moieties of landomycin A (4). The mutant strain of S. cyanogenous S-136 that lacks a functional LanGT4 accumulated a biosynthetic pathway intermediate, landomycin D (24), which contains only two d-olivose moieties in the saccharide chain.22 This provided strong evidence that LanGT4 is responsible for the attachment of the rhodinose moiety as the third sugar of 4. Further experiments involving the feeding of landomycin E (14) into the LanGT4-deficient mutant revealed the production of the pentasaccharidal landomycin B (26).29 This proves the iterative role of LanGT4, which attaches the third and the sixth sugar (l-rhodinose) moiety during the biosynthesis of 4. These results do not only demonstrate a relaxed substrate specificity of these enzymes towards the sugar acceptor substrates but also highlight potential application of the enzymes towards glycodiversification of other natural products.28 Similar experiments involving the feeding of landomycin E (14) into the lanGT1 deficient mutant culture resulted in the production of the tetrasaccharidal congener landomycin I (23).29 These results combined with the previous heterologous expression results clearly showed that LanGT1 is responsible for the attachment of the second and the fifth sugar (d-olivose) moiety of landomycin A (4). Cross complementation experiments were conducted to explore the substrate specificities of LndGT4 and LndGT1. Complementation of the lanGT1-deficient mutant with a lndGT1-expression construct could not restore production of 4.30 This also revealed a non-iterative action of LndGT1, in contrast to the iteratively acting LanGT1. The complemented strain produced landomycin K (43) and landomycin L (38) instead (Fig. 3). This could be attributed to the relaxed sugar acceptor substrate specificity of the rhodinosyltransferase LanGT4. More recently, chimeric LndGT1s that are capable of functioning iteratively like LanGT1 have been engineered through the swapping of the N-terminal region of lndGT1 with lanGT1 nucleotide sequence.31 The expression of LndGT4 in LanGT4-deficient mutant fully restored the production of 4, showing a comparable flexibility of these enzymes towards acceptor substrates.
Scheme 6.

The generation of sugar chain extended urdamycin A and B analogues through the expression of lanGT4 in S. fradiae TU2717.
Fig. 3.

Landomycin analogues generated through the manipulation of biosynthetic pathways.
The fact that LanGT2 catalyzes the attachment of the first olivose moiety in the biosynthetic pathway of landomycin A (4) was confirmed through the accumulation of tetrangulol (8) devoid of a sugar moiety, after targeted disruption of lanGT2.32 Comparison of the landomycin A (4) and landomycin E (14) biosynthetic gene clusters revealed that the latter lacked lanGT3. This led to the speculation that LanGT3 might be responsible for the attachment of the fourth sugar (a d-olivose) moiety of 4.16,33 Production of 14 by the LanGT3-deficient mutant strain instead of the wild type product 4 provided proof for this hypothesis.34 Overexpression of lanGT3 in S. cyangenus S-136 led to the production of a new tetrasacchidal derivative, landomycin J (25, Scheme 4). Such an unusual production of metabolites was explained as being due to the unbalancing of the LanGTs.35
2.1.5. Regulation of landomycins production and their resistance.
Regulation and export of landomycin E (14) biosynthesis in S. globisporous 1912 have been extensively studied during the past several years. The gene lndI represents one of the key pathway specific positive regulatory (activator) genes for biosynthesis of 14 and is located at the end of the landomycin E gene cluster (Fig. 1).36 The encoded product of lndI binds with the promoter of the structural gene lndE and facilitates transcription. Although a weaker activation of lndI by lndI has been reported, biological significance of lndI-mediated autoregulation is not fully understood yet.36,37 Such an activating role of lndI was demonstrated through the heterologous production of landomycin A (4) utilizing the co-expression of the structural genes and lndI, while in the absence of the latter in a control experiment, 4 was not produced.17 Abrogation of landomycin E (14) production by the lndI-deficient mutant of S. globisporous 1912 further demonstrated its absolute necessity for the production of 14.33 Further sequencing of the 14 gene cluster revealed a putative proteinase gene homologue prx located next to the lndI gene. Targeted inactivation of prx in S. globisporus 1912 resulted in a significant loss in the production of 14 (~five-fold) compared to the parental strain, whereas introduction of additional copies of prx led to a 2.7-fold increase.38 These results clearly outline the positive regulatory role of Prx. However, a detailed mechanism concerning Prx-mediated regulation has yet to be elucidated.
The products of lanK and lanJ display their strongest amino acid sequence homologies to TetR-family regulator and efflux proteins, respectively. Overexpression of lanJ in S. cyanogenus S136 confers more resistance to the exogenously fed landomycin A (4) and landomycin D (24).39 The mutant strain also was found to increase the production of 24 with decreased production of landomycin A(4) and landomycin B (26) compared to the wild type strain. A dramatic decrease of landomycin production by the mutant S. cyanogenus strain, which harbors a lanK-overexpression construct indicated a repressor role for the gene product. Further studies revealed that LanK functions as an oligomer, and it negatively regulates the expression of lanJ and also some downstream genes encoding enzymes involved in the conversion of 24 to 4.40 Binding of landomycins (landomycin A (4), B (26), M (41), E (14), and G (37)) to lanK relieves its repressing activity and thus triggers the biosynthesis and export of landomycins. More recently, an ABC transporter homologue gene lndW has been identified at the end of the cluster (Fig. 1).41 Inactivation of lanW in S. globisporus 1912 did not reveal any significant change to the production of 14. However, overexpression of lndW in S. lividans TK24 conferred resistance to landomycin E (14), presumably through the export of the drug.
2.2. Urdamycin biosynthesis
Studies on the biosynthesis of urdamycins began shortly following the isolation of urdamycins A–H (31, 33, 65, 66, 67, 69, 55, 70) from the culture broth of S. fradiae Tü2717 in the mid 1980s.5,42–45 The presence of the C-glycosidically linked saccharide side chain and the enlarged aglycones of some of the urdamycins were particularly interesting for biosynthetic investigations. Rohr and coworkers conducted early studies on the biosynthesis of urdamycin A–D (31, 33, 65, 66) through feeding of isotope-labeled precursors [1-13C]acetate and [1,2-13C2] acetate. Like earlier studies on the vineomycins,6 the incorporation pattern revealed the benz[a]anthracene backbone of these natural products to be formed from a single decapolyketide chain.5 The additional structure elements of urdamycins C (65) and D (66) and E (67), which constitute unique chromophores, were found to be derived from tyrosine, tryptophan, and methionine, respectively.46–48 Tyrosine also served as a precursor for the (p-OH-phenyl)furan moiety of urdamycin H (70),49 while p-OH-phenylglycine did not incorporate into 70. Further experiments involving the feeding of [1-13C]glucose and [U-13C3] glycerol revealed that all of the sugar moieties of the urdamycins arise from glucose. The results also proved urdamycin A (31) to be a late intermediate of the biosynthesis of 65 and 66.1
2.2.1. Genetic and biochemical studies on biosynthesis of polyaromatic backbone of urdamycin A (31).
Decker and Haag cloned and sequenced five open reading frames (urdA, urdB, urdC, urdD, urdE and urdF) including the minimal polyketide synthase (PKS) genes involved in the biosynthesis of urdamycin A (31).50 Inactivation of urdA and urdB genes in S. fradiae Tü2717 completely abolished the production of urdamycins. Heterologous expression of these genes in tetracenomycin non-producer mutant strains that lack either ketoacyl synthase (TcmK) or chain length factor (TcmL) enzymes restored production of tetracenomycins. These results outlined the functional role of the UrdA and UrdB enzymes in urdamycin A (31) biosynthesis. Follow up sequencing of DNA spanning a ~31 kb region revealed a complete gene cluster for urdamycin A (31) (Fig. 4).20,51–53 The cluster comprises 27 putative genes, including PKS genes, post-PKS aglycone modifying genes, sugar biosynthetic genes, glycosyltransferases, regulatory genes and transporter genes (Table 2).
Fig. 4.

The urdamycin biosynthetic gene cluster.
Table 2.
Urdamycin biosynthetic genes and proposed functions
| Genes | Proposed function | Genes | Proposed function |
|---|---|---|---|
|
| |||
| urd-orf148 | regulatory gene | urdD | ketoreductase |
| urd-orf355 | unknown gene | urdL | cyclase |
| urdGT1a | glycosyltransferase | urdM | oxygenase |
| urdGT1b | glycosyltransferase | urdJ2 | transporter gene |
| urd-Int | integrase | urdZ1 | epimerase |
| urdGT1C | glycosyltransferase | urdGT2 | glycosyltransferase |
| urdK | regulatory gene | urdG | NDP-hexose synthase |
| urdJ | transporter gene | urdH | NDP-hexose4,6-dehydratase |
| urdO | NADPH:FMN reductase | urdZ3 | NDP-hexose-3-ketoreductase |
| urdE | oxygenase | urdQ | NDP-hexose 3,4-dehydratase |
| urdF | cyclase | urdR | NDP-hexose 4-ketoreductase |
| urdA | ketoacyl synthase (KSα) | urdS | NDP-hexose 2,3-dehydratase |
| urdB | chain length factor (KSβ) | urdT | oxidoreductase |
| urdC | acyl carrier protein (ACP) | ||
The polyketide-derived backbone requires a number of modifications, including oxygenation (at the 12b-position) and deoxygenation (at 6-position) to furnish the aglycone of urdamycin A (31) and aquayamycin (50). 18O-incorporation studies to identify the biosynthetic origin of oxygen atoms of aquayamycin showed the presence of isotopic oxygen in all of the oxygen atoms of the polyketide backbone except for the C-12- and C12b-positions when CH3-13C18O18OH was fed in the growing culture of S. fradiae Tü2717.54 Culturing under 18O2 atmosphere revealed labeling of the oxygen atom at the C12b-position. Although the isotopic oxygen was not incorporated at the C12-position, it is presumed that the oxygen atom at this position should be introduced by a pathway specific monooxygenase during an earlier stage of the biosynthesis.54
Bioinformatics studies of the urdamycin A (31) gene cluster revealed two candidate genes, urdE and urdM, whose encoded products could partake in oxidative modifications during the biosynthesis of aquayamycin (50). An in-frame deletion of the internal sequence of urdM resulted in the complete abolishment of production of 31.20 The urdM-impaired mutant strain predominantly accumulated rabelomycin (11) and a minimal amount of urdamycin L (45) as biosynthetic pathway shunt products.21 The production of 31 was restored when the urdM gene was expressed in the urdM-deficient mutant. These results indicated that UrdM is responsible for the oxygenation at the 12b-position of urdamycin A (31). Isolation of 45 also led to an assumption that UrdM catalyzes two distinct steps: a Baeyer–Villiger oxidation of UWM6 (6) and a subsequent base-assisted rearrangement of intermediate lactone 44 to establish an alkoxide ion at the C12b-position of 46 (Scheme 7). Alcoholate species 46 is reprotonated to form a pathway intermediate, 12b-hydroxy-UWM6 (47). According to this hypothesis, in-frame deleted-urdM still encodes a partly functional enzyme that retains some Baeyer–Villiger oxygenation activities but completely lacks the later rearrangement activities. As a result, UWM6 (6) and Baeyer–Villiger intermediate 44 undergo spontaneous dehydration/oxidation to generate rabelomyicin (11) and urdamycin L (45), respectively.21 Further in vitro studies involving isolated UrdM are necessary to verify these hypotheses.
Scheme 7.

The biosynthetic steps catalyzed by UrdM during urdamycin A (31) biosynthesis.
The encoded product of urdE displays strong amino acid sequence similarity with other flavin-dependent oxygenases involved in oxygenation of a variety of aromatic polyketides, including PgaE (77% amino acid sequence identity) and LanE (73% aa identity) from gaudimycin A (185) and landomycin a (4) biosynthetic pathways, respectively.24,55 Heterologous expression of the LanE protein in the tetracenomycin C-producer S. glaucescens GLA.O led to the production of a new derivative 6-hydroxy tetracenomycin C.50 This provided an early assumption that UrdE is responsible for the oxygenation at the 12b-position (Scheme 8). However, following the inactivation of the urdM gene (discussed above), a major revision has been made regarding the functional role of UrdE. According to the revised hypothesis, UrdE serves as an anthrone oxygenase, and catalyzes hydroxylation at the 12-position (para position of 7–OH group, which is comparable to the 6-hydroxylation in the para position of the 11-OH group of tetracenomycin C) rather than the 12b-position in urdamycins.
Scheme 8.

The proposed biosynthetic pathway of different urdamycins.
2.2.2. Deoxysugar biosynthesis.
Eight deoxysugar biosynthetic genes (urdG, urdH, urdS, urdT, urdQ, urdR, urdZ1 and urdZ3) have been identified in the urdamycin gene cluster.20,51,56 The products encoded by urdG and urdH catalyze the conversion of d-glucose-1-phosphate (15) to NDP-4-keto-6-deoxy-d-glucose (17). Enzymes UrdS and UrdT were anticipated to catalyze the deoxygenation at the C-2 position and C-3 ketoreduction to generate NDP-4-keto-2,6-dideoxy-d-glucose (18), which further undergoes UrdR-mediated ketoreduction to produce NDP-d-olivose (19) (Scheme 9). UrdQ catalyzes C-3 deoxygenation to generate NDP-d-cinerulose (57). The product then undergoes epimerization catalyzed by UrdZ1 and a final 4-ketoreduction step catalyzed by UrdZ3 to generate NDP-l-rhodinose (20). Inactivation of UrdR in S. fradiae Tü2717 led to the production of rhodinosyl containing urdamycin R (81), urdamycin S (82), 12b,4′-diderhodinosyl-urdamycin S (83), and urdamycin M (80).57 However, this mutant did not accumulate any d-olivosyl containing urdamycin. These results showed that UrdR is especially required for the biosynthesis of NDP-d-olivose and also that 3-deoxygenation occurs prior to the C-5 epimerization during l/d-rhodinose biosynthesis. The presence of a d-rhodinose moiety in urdamycin M also indicates an ability of UrdZ3 to reduce the keto group of 57 and 58.
Scheme 9.

The biosynthetic pathways for NDP-d-olivose and NDP-l-rhodinose.
Incorporation of a frame shift mutation in the urdS gene resulted in the complete abolishment of the urdamycin production.51 However, inactivation of urdQ, urdZ1, and urdZ3 led to the accumulation of urdamycinone B (48), showing that they are involved in the biosynthesis of NDP-l-rhodinose.51 The mutant strain (S. fradiaeΔurdQ/R) possesses mutations in the urdQ and urdR genes, which led to the production of an unexpected compound urdamycin X (79).56 A possibility of incorporating a polar effect was ruled out through the complementation of these genes in the mutant. These results together suggested a crucial role of the UrdQ–UrdR protein pair, which interacts/influences the other post-PKS tailoring enzymes to achieve the optimal transfer of the pathway intermediates from one enzyme to the other. Production of urdamycin I (71) and urdamycin J (73, Fig. 9) through the overexpression of l-oleandrose biosynthetic enzymes OleL (TDP-4-keto-2,6-dideoxy-d-glucose-3,5-epimerase) and OleU (TDP-4-keto-l-olivose-4-ketoreductase) further supports this hypothesis.56 urdQ, urdR or both have been utilized to construct a variety of plasmids (pFL845: generates NDP-d-amicetose, pFL844: generates NDP-l-amicetose, pLNR: generates NDP-d-olivose; pLNRHO: generates NDP-l-rhodinose), which direct the production of activated deoxysugars in vivo.58,59
Fig. 9.

Urdamycins isolated from natural producer S. fradiae and from engineered mutant strains.
2.2.3. Glycosylation.
Urdamycin A (31) comprises four sugar moieties, C-glycosidically linked d-olivose and O-glycosidically linked l-rhodinoses (2×) and d-olivose. Identification of four glycosyltransferases (GTs) (one C-GT, urdGT2, and three O-GTs, urdGT1a, urdGT1b and urdGT1c) in the biosynthetic gene cluster met a general requirement “one GT – one sugar” for sugar moieties of urdamycin A (31). Deletion of urdGT2 in S. fradiae Tü2717 resulted in the production of urdamycin I (71), J (73), and K (72) with all lacking the C-connected sugar. This clearly indicates that the GT-B family glycosyltransferase UrdGT2 is responsible for the attachment of d-olivose at the C-9 position of a benz[a]anthraquinone-derived polyketide, to yield the b-C-glycoside aquayamycin (Scheme 8).20,52 Presence of a C-12b-hydroxyl group in all of these accumulated metabolites also indicates that the hydroxyl group at the 12b-position is introduced prior to the C-glycosylation catalyzed by UrdGT2. However, production of prejadomycin-C-glycosides (59–61, Fig. 5) through the expression of the urdGT2 gene in the lndE-deficient mutant (accumulates prejadomycin predominantly) led to a revision regarding the substrate and the timing of C-glycosylation during urdamycin A (31) biosynthesis. According to the new hypothesis, UrdGT2 catalyzes the C-glycosylation of UWM6 (6) prior to the actions of the oxygenases UrdM and UrdE. C-glycosylation of methylnaphthazarine when fed to S. fradiae Tü2717 culture also indicated that UrdGT2 is less substrate specific, requiring only a minimal bicyclic structure of recognition.52 Production of urdamycin derivatives with C-glycosidically linked d- and l-configured rhodinoses in addition to the d-olivose and production of C-glycosylated premithramycins further confirmed the broad substrate specificity of UrdGT2, both regarding the NDP-sugar donor and acceptor substrates.57,60 Although UrdGT2 has a remarkable intrinsic flexibility towards both sugar-donor and acceptor substrates, it appeared to be strict regarding regioselectivity (a phenolic hydroxyl group is required to be ortho to the carbon being glycosylated).57,60
Fig. 5.

Prejadomycin C-glycosides.
Another experiment involving the feeding of the acceptor substrate mimic 1,2-dihydroxyanthraquinone (62) with the Urd-PKS deficient mutant strain resulted in the formation of an O-glycosylated product (63, Scheme 10).61 This unusual O-glycosylating ability of UrdGT2 in addition to its natural C-glycosylating activity drew much attention towards the elucidation of the molecular mechanism of its catalysis.61 Finally, Schulz and coworkers have crystalized UrdGT2 and have identified the key residues that could potentially be involved in binding the substrate and for the establishing of the C-glycosidic bond through computer modeling.62 Like other GT-B type glycosyltransferases, UrdGT2 has two domains that display Rossmanntype chain folds, and the putative active centers of the two domains are close to each other, which meet the general requirements for the binding of sugar donor substrate and the sugar acceptor substrate to catalyze glycosylation (Fig. 6). Through comparison of the UrdGT2 structure with other structurally related GTs and through the study of natural (6) or artificial substrate (62)-bound UrdGT2 models, putative sugar donor and acceptor binding sites were identified. According to this, 6 resides in a large non-polar binding pocket through the interaction of 7- and 8-hydroxyl groups with deeply buried Asp-137 via water molecules. Slightly different from the C-glycosylation mechanism suggested by Mittler et al.,62 we propose here a chemically more likely mechanism, which is equally supported by the UrdGT2 structure (Fig. 7): The interaction of the Asp-137 carboxylate group and the hydrogen bonding between the 7-hydroxyl and 6-keto-oxygen facilitates the abstraction of 8-OH, which in turn increases the electron density in the neighboring 9-position, thereby allowing the C-glycosylation to proceed via electrophilic, aromatic substitution and a σ-complex-like intermediate. The mobile loops made up of the 62–72 and 219–228 residues may cover the active center in an induced fit fashion. Similar binding interactions have been proposed for the O-glycosylation of the artificial substrate alizarin (62).62
Scheme 10.

O-glycosylation of alizarin catalyzed by UrdGT2.
Fig. 6.

The crystal structure of UrdGT2 (Mittler et al.)62
Fig. 7.

The aromatic substitution mechanism of the UrdGT2-catalyzed C-glycosylation.
To identify the exact role of the other three GTs (urdGT1a, urdGT1b and urdGT1c), each gene was inactivated alone or together with the other GTs and their effect on the production of urdamycin was compared with the wild type S. fradiae Tü2717.53 Inactivation of all of the three GTs in the triple mutant resulted in the production of aquayamycin (50) and urdamycinone B (48), both with a C-glycosidically linked d-olivose. Inactivation of urdGT1a resulted in the predominant production of urdamycin B (33) which lacks the l-rhodinose sugar moiety at 12b-position. Production of 12b-rhodinosylated compound 100–2 (29) through the expression of urdGT1a in the triple mutant clearly established that UrdGT1a catalyzes the attachment of l-rhodinose at 12b-position of aquayamycin during urdamycin A (31) biosynthesis.53 The deduced amino acid sequence of UrdGT1b and UrdGT1c are extremely similar (91% identity, different in only 33 amino acids). Expression of UrdGT1c in the triple mutant resulted in the production of disaccharidal urdamycin 100–1 (49). Co-expression of urdGT1b and urdGT1c in the triple mutant accumulated trisaccharidal derivative 12b-derhodinosyl-urdamycin A (52). However, the mutant strain lacking both GT1b and GT1c, but with a hybrid gene comprising 715 nucleotides from urdGT1b and 458 nucleotides from urdGT1c predominantly accumulated 100–2 (29). Summarizing these results, it is clear that UrdGT1c and UrdGT1b are responsible for the attachment of the second sugar, l-rhodinose, and the third sugar, d-olivose, moiety of the trisaccharide chain of urdamycin A (31).53 Incorporation of mutations in urdGT1b/urdGT1c, and subsequent study of their activities in vivo led to the identification of ten amino acid residues, which could potentially confer glycosylation specificity.63 A new metabolite urdamycin P (64) with an additional d-olivose attached at the 4′-OH group of the 12b-derhodinosyl-urdamycin G (51) was produced through the expression of chimeric GTs with novel functionality (Scheme 11). Screening of chimeras revealed some retained individual parental functionalities (UrdGT1b or UrdGT1c), some shared parental functionalities (UrdGTb and UrdGTc), and some novel and individual parental functionality (novel + UrdGTb or novel + UrdGTc), or the combination of all activities.63 Production of urdamycin A (31) through the co-expression of all three GTs in the triple mutant further showed that these GTs are sufficient for attaching all of the O-glycosidically linked sugars of urdamycin A (31). UrdGT2 catalyzes the first glycosylation at C-9 position to generate aquayamycin (50). The product undergoes second glycosylation catalyzed by UrdGT1a at 12–b position which followed by the second and the third glycosylation to complete urdamycin A (31) biosynthesis (Scheme 8). Similar sequences of glycosylations, except the 12b-rhodinosylation, have been proposed for urdamycin B (33) biosynthesis. Incorporation of amino acid-derived moieties in urdamycin C (65), urdamycin D (66), E (68) and urdamycin H (70, Fig. 8) are proposed to happen after the attachment of sugar moieties. These steps were shown to occur non-enzymatically.47–49
Scheme 11.

Generation of urdamycin P through the activity of engineered GTs.
Fig. 8.

Chromophore-modified urdamycin A congeners isolated from S. fradiae Tü2717.
2.3. Biosynthesis of jadomycin
Jadomycins were one of the earliest and most studied members of the angucycline group of antibiotics. Fermentation of Streptomyces venezuelae ISP5230 under heat shock or ethanol shock led to the initial isolation and characterization of jadomycins A (95) and B (96).64,65 Further attempts to optimize jadomycin production revealed phage infection as well as media components that could influence the expression of the cryptic jadomycin pathway.66
2.3.1. jadPKS Enzymes.
Biosynthetic studies of jadomycin B began with the isolation and sequence analysis of the jadomycin polyketide synthase genes jadA, jadB, jadC, jadE and jadD (Fig. 10) encoding a ketosynthase (KSα), chain length factor (CLF), acyl carrier protein (ACP), ketoreductase and cyclase/dehydratase, respectively.67 Two additional genes jadM and jadJ were identified as a putative phosphopantetheinyl transferase (PPTase) and acyl-coenzyme A carboxylase, respectively, and were found to be critical requirements for the biosynthesis of jadomycin B.68,69 The gene products of jadA, jadB, and jadC make up the minimal jadomycin PKS (jadPKS) and are the minimal genes required to produce the decaketide intermediate 1. Inactivation of jadA was found to abolish jadomycin B production, confirming the proposed ketosynthase functionality of JadA.70,71 Additional in vivo studies with jadE, jadD, and jadI established the first biosynthetic pathway to the production of UWM6 (6), the most minimal isolated angucyclinone, and proposed intermediate of many angucycline biosyntheses (Scheme 12).72 More recently, total enzymatic synthesis of rabelomycin (11), a spontaneous degradation product of 6, was achieved using a combination of jadomycin, gilvocarcin (see section 1.4), and ravidomycin (see section 1.4.5) PKS enzymes highlighting a conserved pathway to the production of 6.73
Fig. 10.

The biosynthetic gene cluster of jadomycin.
Scheme 12.

The proposed role of the polyketide synthase genes in jadomycin biosynthesis.
Through minimal jadPKS (jadA, jadB, and jadC) constructs containing different combinations of of jadE, jadD, and jadI, Hutchinson et al. discovered, by the formation of UWM4 (87) and SEK 43 (88), that jadE functions as a PKS-associated ketoreductase, while jadD clearly serves as the first ring cyclase catalyzing the aldol condensation between C7 and C12 of the decaketide intermediate 1.72 It is clear that the remaining cyclization events rely on jadI, as only the minimal jadPKS constructs containing jadE, jadD, and jadI were able to produce UWM6 (6).72 To date, the conversion of 84 to 6 remains ambiguous and it remains unclear which role jadD and jadE play in conjuction with jadI for the formation of 6 (Scheme 12).
2.3.2. jad Post-PKS enzymes.
The post-PKS reactions involved in jadomycin A (95) biosynthesis include two dehydration steps (2,3-dehydration and 4a,12b-dehydration), an enigmatic oxygenation cascade leading to oxidative C–C bond cleavage, incorporation of l-isoleucine and subsequent glycosylation (Scheme 13). Three genes (jadF, jadH and jadG) identified immediately upstream of jadD were proposed to partake in this oxidative cascade leading to ring opening based on their putative gene functions (Table 3).70,71,74 Inactivation experiments of jadF, jadG, and jadH resulted in the accumulation of shunt products rabelomycin (11) (ΔjadG)70,74 and l-digitoxosyl-dehydrorabelomycin (97) (ΔjadG) as well as pathway intermediates 2,3-dehydro-UWM6 (also called prejadomycin, 21) (ΔjadH)71 and dehydrorabelomycin (90) (ΔjadG).70 UWM6 (6), 21, and 90 were confirmed as pathway intermediates through bioconversion assays, where both substrates were converted to jadomycin A (95), B (96) or both depending on the assay conditions.71,75
Scheme 13.

Investigation of jadomycin A and B biosynthesis.
Table 3.
Jadomycin B biosynthetic genes and proposed functions
| Gene | Proposed Function | Gene | Proposed Function |
|---|---|---|---|
|
| |||
| jadW1 | Regulator | jadK | Hydrolase |
| jadW2 | Regulator | jadL | Efflux pump |
| jadW3 | Regulator | jadM | Phosphopantetheinyl transferase |
| jadR2 | Transcriptional regulator | jadN | Malonyl-coenzyme A decarboxylase |
| jadR1 | Transcriptional regulator | jadX | Unknown |
| jadJ | Acetyl-coenzyme A carboxylase | jadO | NDP-hexose-2,3-dehydratase |
| jadI | Cyclase | jadP | NDP-hexose-3-ketoreductase |
| jadA | Ketoacyl synthase | jadQ | NDP-glucose synthase |
| jadB | Chain length factor | jadS | Glycosyltransferase |
| jadC | Acyl carrier protein | jadT | NDP-hexose-4,6-dehydratase |
| jadE | Ketoreductase | jadU | NDP-hexose-5-epimerase |
| jadD | Cyclase/Dehydratase | jadV | NDP-hexose-4-ketoreductase |
| jadF | Oxygenase/Dehydratase | jadR* | tet R homologue |
| jadG | Anthrone oxygenase | jadY | Unknown |
| jadH | Oxygenase/Dehydratase | jadZ | Unknown |
The accumulation of rabelomycin (11) (retains 3-hydroxyl group) by ΔjadF and prejadomycin (21) (retains 4a-hydroxyl group) by ΔjadH indicate these proposed oxygenases also possess dehydratase activity where JadF and JadH catalyze 2,3-dehydration and 4a,12b-dehydration, respectively (Scheme 13).71,74 Interestingly, enzymatic studies of JadF found UWM6 (6) to be an unacceptable substrate for 2,3-dehydration; however, with the accumulation of prejadomycin (21) by ΔjadH, a 2,3-dehydrated product of 6, it is plausible that JadF activity partially depends on the presence of JadG.70,71 Invitro assays by Yang et al. clearly shows JadH possesses 4a,12b-dehydration activity towards 21, leading to the formation of dehydrorabelomycin (90). Moreover, a new intermediate, CR1 (89), was isolated from this reaction where in addition to 4a,12b-dehydration, JadH had catalyzed C-12 oxygenation (Scheme 13).75 JadH, an FAD- and NAD(P)H-dependant bifunctional hydroxylase/dehydratase, was the first characterized enzyme to catalyze angucycline C-12 oxygenation. JadG was initially proposed to function as the C-12 oxygenase as it has high sequence similarity to various anthrone-oxygenases; however, all metabolites produced by ΔjadG possess C-12 oxygenation.70 This was attributed to the spontaneous oxidation of UWM6 (6) to rabelomycin (11),72,76,77 and JadG remained the favored enzyme for C-12 oxygenation until the isolation of CR1 (89).
Further modification through C-5 oxygenation and subsequent C–C bond cleavage results in the proposed aldehyde/acid intermediate 93 (Scheme 13). Cross-complementation experiments with jadF in the gilOIV mutant strain (discussed in gilvocarcin biosynthesis below) reconstituted gilvocarcin production. Based on the proposed function of GilOI (initially as a C-5 hydroxylase), JadF was therefore hypothesized to catalyze the same reaction in jadomycin B (96) biosynthesis.78 Based on proposed pathway intermediates containing a C-6 carbonyl function in the ring to be cleaved, a Baeyer–Villiger-type mechanism was proposed to facilitate ring cleavage.70 As with expression of the jadF gene, cross-complementation of jadH in the gilOI mutant strain (discussed in gilvocarcin biosynthesis below) also reconstituted the gilvocarcin pathway and indirectly supported JadH as crucial for the oxidative C–C bond cleavage.78 The role of jadF, jadG, and jadH in the oxygenase cascade were further confirmed by the production of jadomycin A by the feeding of l-isoleucine and co-expressing jadF, jadG, and jadH together with pWHM1238, which alone affords the accumulation of UWM6 (6).78 Taken together this oxygenase cascade has been termed a “biosynthetic black box” and poses an extremely difficult challenge for researchers as inactivation experiments of jadF, jadG and jadH indicate their respective gene products form a co-dependent complex and the removal of an individual enzyme from this complex renders the remaining gene products unfunctional in terms of oxygenase activity.70,71,74 Despite rigorous attempts to delineate the oxygenase cascade leading to and including C–C bond cleavage, the conversion of 89 to 93 remained unclear.
2.3.3. Amino acid incorporation.
The incorporation of l-isoleucine during jadomycin biosynthesis was proposed to occur non-enzymatically by Rohr et al., who also fed structurally diverse natural amino acids to the jadomycin producer to generate a set of first generation jadomycin analogues with altered amino acid incorporation.2,79 Further feeding experiments by Jakeman et al. completed the incorporation of all 20 natural amino acids, and also expanded towards various non-proteogenic amino acids (Table 4).80–83 The ability to successfully include structurally diverse amino acids into the jadomycin skeleton in conjunction with the inability to assign an enzyme candidate from the jad pathway strongly supported the suggested nonenzymatic incorporation of l-isoleucine. Moreover, thorough NMR analysis of jadomycin B (96) surprisingly showed 96 to be an inseparable diastereomeric mixture of the 3a-S (67%) and 3a-R (33%) configurations, which exist as a dynamic mixture, where aldehyde intermediates are formed by the opening of the oxazolone ring upon nucleophilic attack (shown in Scheme 14 for jadomycin A (95)).79,82 It has been proposed that the preference for the 3a-S or 3a-R conformation depends on hydrogen bonding between the C-12 carbonyl group and the amino acid species being incorporated.79 Final proof for proposed non-enzymatic amino acid incorporation came from a recent biomimetic total synthesis of jadomycin A by O’Doherty et al. (see Section 5 below).84
Table 4.
Jadomycin analogues through non-enzymatic incorporation of various amino acids
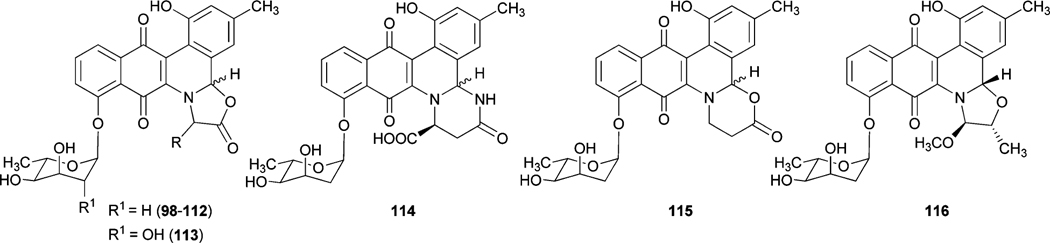
| |||||
|---|---|---|---|---|---|
|
| |||||
| Product | R | Amino Acid | Product | R | Amino Acid |
|
| |||||
| Jadomycin V (98)79 |
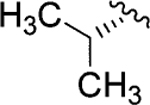
|
l-Val | Jadomycin DV (108) |
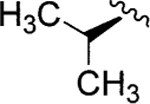
|
d-Val |
| Jadomycin M (99) |
|
l-Met | Jadmycin DM (109) |
|
d-Met |
| Jadomycin T (100)79 |
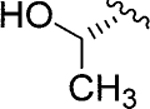
|
l-Thr | Jadomycin DT (110) |
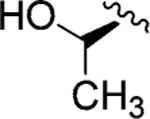
|
d-Val |
| Jadomycin G (101) |
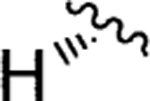
|
l-Gly | Jadomycin Ala (111)79 |
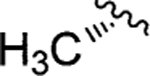
|
l-Ala |
| Jadomycin H (102) |
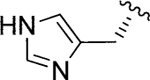
|
l-His | Jadomycin W (112) |
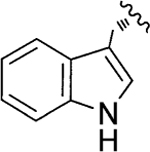
|
l-Trp |
| Jadomycin R-Phe (103) |
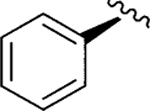
|
d-phenylglycine | ILEVS1080a (113)85 |
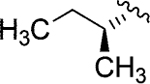
|
l-Ile |
| Jadomycin F (104)79 |
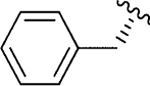
|
l-Phe | Jadomycin N (114) | — | l-Arg |
| Jadomycin S (105)79 |
|
l-Ser | Jadomycin β-ala (115) | — | β-Ala |
| Jadomycin Y (106) |
|
l-Tyr | Dalomycin T (116)82 | — | l-Thr |
| Jadomycin S-Phe (107) |
|
l-phenylglycine | |||
| Amino acid species with reported incorporation b | |||||
| d-Ile80 | l-Asp80 | l-Cys80 | l-Valine methyl ester81 | ||
| l-Leu80 | l-Glu80 | dl-4-Fluorophenylalanine81 | O-methoxy-l-Thr80 | ||
| l-Asn81 | l-Lys80 | 2-Aminoisobutyric acid81 | O-methoxy-methylene-l-Thr80 | ||
| l-Gln81 | l-Pro80 | dl-3-Aminoisobutyric acid81 | |||
Isolated from ΔJadO mutant.
Reported with only mass data.
Scheme 14.

The interconversion of jadomycin diastereomers.
2.3.4. Deoxysugar biosynthesis and glycosylation.
Genes for the biosynthesis and attachment of l-digitoxose, the 2,6-dideoxyhexose moiety found in jadomycin B (96), were found roughly 4.5 kb upstream of jadH, and included a proposed 2,3-dehydratase (jadO), 3-ketoreductase (jadP), NDP-glucose synthase (jadQ), glycosyltransferase (jadS), 4,6-dehydratase (jadT), 5-epimerase (jadU) and a 4-ketoreductase (jadV) (Fig. 10 and Table 3).86 Inactivation of the proposed deoxysugar biosynthetic genes, including the proposed glycosyltransferase, led to the accumulation of jadomycin A (95), therefore solidifying their role in the biosynthesis of 96 (Scheme 15).86 The attachment of l-digitoxose is believed to occur after amino acid incorporation and cyclization to form 95; however, the reported glycosylated aldimine containing the product from l-valine methyl ester feeding indicates the oxazolone ring is not an absolute requirement for JadS catalyzed glycosylation.81 Thus, JadS might also attach the sugar prior to the amino acid incorporation.4 If the glycosylation occurs as the last step, the library of glycosylated jadomycin B analogues created utilizing various amino acids illustrates a substantial inherent acceptor substrate flexibility of JadS. In an attempt to investigate the donor substrate flexibility of JadS, Jakeman et al. re-investigated the 2,3-dehydratase (jadO) mutant, S. venezuelae VS1080. With optimized culturing conditions the jadO mutant was found, contrary to previous reports, to produce a glycosylated product ILEVS1080 (113, Table 4). Structural characterization of this new product revealed the presence of 6-deoxy-l-altrose in lieu of l-digitoxose (Scheme 15).85 This finding proves JadS also exhibits donor acceptor substrate flexibility, and is the only example of a jadomycin analogue containing an altered deoxysugar moiety.
Scheme 15.

The biosynthetic pathway of l-digitoxose and 6-deoxy-l-altrose in the jadomycin pathway.
2.3.5. Regulation.
The requirement of combined nutritional and environmental stress, i.e. ethanol toxicity or heat shock, to elicit jadomycin B (96) biosynthesis in wild type S. venezuelae ISP5230 implies the presence of an atypical control mechanism for Streptomycete antibiotic production. The bioinformatical analysis of the sequence immediately downstream of jadI in the jadomcyin biosynthetic gene cluster identified five putative regulatory genes namely jadW1, jadW2, jadW3, jadR2 and jadR1. The jadW1–3 genes show strong amino acid sequence similarity to gene products associated with the regulation of morphological differentiation and secondary metabolism production in several streptomycetes through gamma-butyrolactone (GBL) autoregulators.87 The best studied case involves the γ-butyrolactone A-factor, which induces streptomycin biosynthesis in Streptomyces griseus.88 The inactivation of jadW1 caused slower growth rate and sporulation on solid media when compared to S. venezuelae ISP5230, and abolished antibiotic production except in galactose–isoleucine media, where jadomycin B production was restored to roughly half when compared to the wild type strain.87 Restoring a functional copy of the jadW1 gene improved growth rate and sporulation of the mutant (ΔjadW1), but did not return the strain to that of the wild type. Additionally, antibiotic production was increased up to 2.5 fold in the JadW1 restored strain. This was attributed to the presence of several copies of jadW1 in the complemented strain through multiple chromosomal integration events by the shuttle vector. These findings show the proposed GBL regulatory system in the jad gene cluster, particularly jadW1, plays a role in antibiotic regulation of jadomycin B (96) as well as cellular differentiation in S. venezuelae ISP5230.87
The proposed transcriptional regulators jadR2 and jadR1 are located immediately upstream of jadW3.89,90 Inactivation of jadR2, a putative GBL receptor, led to the production of jadomycin B without the additional stress of ethanol shock and overexpressed jadomycin B upon ethanol treatment when compared to wild type.89 These findings suggest jadR2 encodes a jad pathway repressor. A recent investigation of JadR2 confirmed its role as a repressor and a “pseudo” GBL receptor.91 JadR2 was found to directly repress the transcription of jadR1, a positive pathway regulator of jadomycin biosynthesis (see below). Additionally, jadomycin A and B were found to serve as ligands for JadR2 and upon binding successfully caused inhibition of the repressor signal, as determined by jadR1 transcription. Inactivation of JadR1 prevented jadomycin B biosynthesis entirely and therefore was concluded to be a positive jad pathway regulator.90 Furthermore, it was found that jadR1 encodes an “atypical” response regulator in which JadR1 binds and activates jadJ transcription.92 The ability of JadR1 to bind to the promoter region of jadJ was found to be inhibited by the binding of jadomycin B and early jadomycin intermediates (21, 90 and 95, Scheme 13) to the N-terminus of JadR1.
From these results, it is clear that jadR1 and jadR2 form an interacting regulatory system for jadomycin B biosynthesis where JadR1 activates jadJ transcription leading to the expression of jadomycin biosynthetic genes and JadR2 binds to and inhibits transcription of jadR1. Both gene products of jadR1 and jadR2 are inhibited by binding of jadomycin A and B, therefore allowing jadomycins to act as autoregulators of their own pathway. This regulatory system is complicated by the cross-regulation between the jadomycin B and chloramphenicol (naturally produced by S. venezuelae ISP5230) biosynthetic pathways. JadR1 was found not only to bind and activate jadJ transcription but also to directly repress chloramphenicol biosynthesis by binding the promoter region between the chloramphenicol biosynthetic genes cmlI–cmlJ.91 These findings continue to expand the understanding of an elaborate regulatory network that continues to offer interesting insights into antibiotic regulation. Despite intensive work on the regulatory mechanisms involved in jadomycin B biosynthesis it is still unknown how the two distinct autoregulatory systems (JadW1–3 and JadR1–2) interact with one another and how external environmental stress affect these regulatory circuits.
2.4. Gilvocarcin biosynthesis
Gilvocarcin V (122) was first published without complete structural characterization by Mizuno and coworkers as toromycin, the principal product of S. collinus (No. B-21085).93–95 Shortly thereafter, gilvocarcin V (122) and M (120) were fully characterized by Tomito and coworkers from the fermentation of S. gilvotanareus96,97 while gilvocarcin V, M, and gilvocarcin E (121) were reported by Balitz et al. from S. anandii (C-22437).98 Together 120, 121 and 122 comprise the gilvocarcins and are the most prominent member of a unique class of anticancer antibiotics, often referred to as gilvocarcin-type aryl-C-glycosides.
Initial biosynthetic studies began with feeding experiments to determine the biosynthetic origin of carbon atoms in the gilvocarcin chromophore. Through the addition of various 13C labeled acetate and propionate substrates to the fermentation broth of S. gilvotanareus, Takahashi et al. concluded that the gilvocarcin aglycon was synthesized via the polyketide pathway and involved subsequent oxidative cleavage and rearrangement to install the final backbone of gilvocarcins (Scheme 16). Additionally, it was hypothesized that the C8-side chains found in gilvocarcin M and V were the result of secondary alkylation steps with acetate or propionate, respectively.10 This hypothesis was later disproved by additional labeling experiments with ravidomycin (123) and chrysomycins (152 and 153), who share the same chromophore to that of gilvocarcins (Scheme 17).8,9,99,100 It was concluded that the C-8 side chains were the product of polyketide initiation with either acetate or propionate and not through secondary alkylation steps. Kingston and co-workers established with deuterium labeling experiments on 123 that the pro-S proton of the propionate starter unit is retained in the vinyl group101 (Scheme 16, box).
Scheme 16.

The originally suggested oxidative ring cleavage and vinyl group formation based on incorporation experiments with isotope-labeled precursors.
Scheme 17.

The biosynthetic pathway of gilvocarcins.
2.4.1. gilPKS enzymes.
Genetic and biochemical analysis of gilvocarcin V (122) biosynthesis started with the isolation and sequence analysis of its biosynthetic gene cluster from Streptomyces griseoflavus Gö 3592.99 The gilvocarcin gene (gil) cluster was found to contain 27 open reading frames (ORFs), including genes encoding the polyketide synthase (PKS) as well as several post-PKS tailoring and deoxysugar biosynthetic genes (Fig. 11 and Table 5). Heterologous expression of the gil cluster (cosG9B3) stimulated gilvocarcin production ensuring complete coverage of the biosynthetic gene cluster.99 As Streptomyces griseoflavus Gö 3592 is resistant to genetic modification, cosG9B3 was utilized as a suitable vehicle to further study the biosynthesis of gilvocarcin V (122).
Fig. 11.

The biosynthetic gene cluster of gilvocarcins.
Table 5.
Gilvocarcin biosynthetic genes and proposed functions
| Gene | Proposed Function | Gene | Proposed Function |
|---|---|---|---|
|
| |||
| gilS | Putative regulatory protein | gilH | Reductase |
| gilN | Unknown | gilOI | FAD-dependent oxygenase |
| gilL | Unknown | gilG | Cyclase |
| gilOIII | P-450 monooxygenase | gilA | Ketoacyl synthase |
| gilGT | Glycosyltransferase | gilB | Chain length factor |
| gilV | Unknown | gilC | Acyl carrier protein |
| gilM | Unknown | gilF | Ketoreductase |
| gilR | Oxidoreductase | gilK | Cyclase/dehydratase |
| gilOII | Anthrone monooxygenase | gilOIV | Cofactor-free oxygenase |
| gilMT | O-methyltransferase | gilP | Malonyl-CoA |
| acyltransferase | |||
| gilJ | Putatative efflux protein | gilQ | Acyltransferase |
| gilI | Repressor | gilT | Putative regulatory protein |
| gilE | NDP-glucose-4,6-dehydratase | gilU | NDP-hexose-4-ketoreductase |
| gilD | NDP-glucose-synthase | ||
The gilPKS, consisting of genes encoding a ketosynthase (gilA), chain length factor (gilB), acyl carrier protein (gilC), malonyl-CoA-acyltransferase (gilP) and acyltransferase (gilQ), catalyzes the formation of two individual C20 and C21 decaketide intermediates. The C20 decaketide (1) is formed through traditional acetate initiation of the polyketide backbone while the alternative C21 decaketide is formed through unusual propionate initiation by GilQ.102 It was hypothesized that further modification of both C20 and C21 decaketide intermediates by GilF (ketoreductase), GilK (cyclase/dehydratase) and GilG (cyclase) produce UWM6 (6) and homo-UWM6 (128), respectively.99
2.4.2. gil Post-PKS tailoring enzymes.
The initial post-PKS modification of UWM6 and homo-UWM6 is analogous to that observed in jadomycin B biosynthesis and involves an elusive “biosynthetic black box”, which results in the hypothetical ring opened intermediates 134–139 (Scheme 17). As with jadomycin B biosynthesis, this “biosynthetic black box” encompasses two dehydration steps (2,3-dehydration and 4a,12b-dehydration), two oxygenation steps (C-5 and C-12) as well as an oxidative ring opening reaction. Bioinformatic analysis of the gilvocarcin cluster revealed four oxygenase-encoding genes; gilOI, gilOII, gilOIII and gilOIV. Sequence comparisons showed gilOI, gilOII and gilOIV were closely related to the corresponding oxygenase-encoding genes from the jad cluster jadH, jadG and jadF, respectively.99
Inactivation of gilOIV was found to disrupt the gilvocarcin pathway and instead produced the shunt products rabelomycin (11) and homorabelomycin (124).100 Likewise, the inactivation of gilOI led to the accumulation of pathway intermediates prejadomycin (21) and homoprejadomycin (= homo-2,3-dehydro-UWM6, 129) as well as the unique shunt products pregilvocarcin M-o-quinone (125) and pregilvocarcin E-o-quinone (126).100 Lastly, the inactivation of gilOII caused the accumulation of mainly the shunt product dehydro-rabelomycin V (127), but also minor amounts of dehydrorabelomycin (90) and dehydrohomorabelomycin (130).78 In addition to inactivation experiments, these oxygenase encoding genes were co-expressed individually and in combination with pWHM1238, which when expressed alone accumulates UWM6 (6). Co-expression of gilOI with pWHM1238 accumulated rabelomycin (11), while replacement of gilOI with gilOIV afforded prejadomycin (21) and homodehydrorabelomycin (130). Alternatively, co-expression with gilOII did not alter the metabolic profile of pWHM1238. Only when gilOIV, gilOI and gilOII were co-expressed with pWHM1238 did the entire oxygenase cascade progress. This led to the completion of the oxidation reactions including the 5,6-bond breakage, and to the accumulation of jadomycin A (95), provided that the fermentation medium was supplemented with l-isoleucine.78 Collectively, these results indicated that gilOIV, gilOI, and gilOII form an oxygenase complex that converts UWM6 and homo-UWM6 into aldehyde–acid intermediates 134 and 135, respectively (Scheme 17).
The remaining oxygenase, gilOIII, belongs to the superfamily of cytochrome-P450 enzymes, and is not an absolute requirement of the oxygenase complex; however, it is required for the production of gilvocarcin V (122).78,103 The inactivation of gilOIII caused an accumulation of primarily gilvocarcin E (121), normally a very minor product of S. griseoflavus Gö 3592, as well as of gilvocarcin M (120). The substrate for GilOIII remains unknown as 121 fed to the gilOIV mutant (the earliest acting gilvocarcin mutant) did not convert gilvocarcin E to gilvocarcin V (122). Furthermore, in vitro studies also showed that gilvocarcin M and E were not suitable substrates for GilOIII (unpublished results). The results along with the isolation of vinyl containing shunt product 127 indicated that modification of the ethyl side chain occurred during early biosynthetic steps of gilvocarcin.78 Instead of gilvocarcin E (121), GilOIII may turn over the 4a,12b-dehydrated intermediate of the GilOI-reaction, which has not been characterized. The mechanism in which GilOIII converts the C-8 ethyl side chain to the vinyl functional group is also unknown. Cytochrome-P450 enzymes typically function as monooxygenases; however, it has been reported that some P450 enzymes catalyze very different reactions, including desaturation.104 GilOIII may catalyze hydroxylation of the aliphatic ethyl side chain and subsequent dehydration by an additional undetermined enzyme or – more likely – it may function as a simple desaturase, thereby directly installing the vinyl functional group.
The biosynthesis of gilvocarcin V therefore requires the activity of all four oxygenases, GilOIV, GilOI, GilOII and GilOIII to convert homo-UWM6 (128) to 136. Based on extensive in vivo and more recently also in vitro studies the oxygenase cascade has been hypothesized as shown in Scheme 17. Specifically, GilOIV is believed to catalyze 2,3-dehydration, forming 129. GilOIV is also believed to be involved in the decarboxylative release mechanism that may liberate 129 directly from the PKS–ACP, omitting 128. Subsequent 4a,12b-dehydration and the hydroxylation of C-12 by GilOI produces the proposed substrate for GilOIII, which then undergoes oxidative C–C bond cleavage, presumably catalyzed solely by GilOII, producing 136 via dioxetane 133 to complete the oxygenase cascade. Alternative to the dioxygenase mechanism shown in Scheme 17, GilOII also might act as twice as monooxygenase, introducing first a 5-OH group, and then inserting an O-atom between C5 and C6 in a Baeyer–Villiger fashion.
Additional post-PKS tailoring reactions, including the O-methylation of 10-OH and 12-OH groups, are believed to occur directly after C-12 hydroxylation. Two genes, gilM and gilMT, from the gil-cluster encode putative methyltransferases. Unlike gilMT, whose product clearly shows sequence similarity to various O-methyltransferases, the deduced gilM product only shows a clear S-adenosyl-l-methionine binding site and does not show homology to characterized enzymes with known functions. Recent experiments with synthetic model compounds closely related to 134 suggest that GilM is also responsible for the quinone reduction,105 and it is postulated that simultaneous O-methylation of the neighboring hydroquinone OH group will prevent spontaneous reoxidation to the quinone. This way, the reaction between the aldehyde group and the still free hydroquinone-OH group of 137/139 is facilitated and leads to the hemiacetal containing pre-defucogilvocarcins (140–142).106 Therefore it is hypothesized that C-12 O-methylation (gilvocarcin numbering, 122, Scheme 17) occurs prior to C-10 O-methylation. Unexpectedly, the accumulation of 12-demethyl-defucogilvocarcin (150) in the deoxysugar sugar knockout ΔgilU (discussed in gilvocarcin deoxysugar biosynthesis) seemed to suggest 10-OH methylation occurred before C-12-OH methylation.107 A possible explanation for the unanticipated formation of 150 can be attributed to formation of the lactone moiety (143 to 150) before the predicted O-methylation steps. The resulting didemethyl-pre-defucogilvocarcin shunt product may be further modified by a substrate flexible GilM or GilMT at the 10-OH and not the 12-OH position resulting in the formation of 150.
The formation of the shunt products defuco-gilvocarcins (147–149), like 150, are the product of premature lactone formation by a slightly substrate flexible oxidoreductase encoded by gilR.78,106 The initial inactivation of gilR led to the accumulation of pregilvocarcins (144–146) and showed GilR was directly involved in lactone formation.78 The hemiacetal functionality of the pregilvocarcins also proved that aldehyde-acids 134–136 rather than the earlier postulated diacids (Scheme 16) are the product of the above discussed 5,6-bond cleavage. Additionally, the formation of glycosylated hemiacetal intermediates 144–146 suggested that the glycosylation reaction occurred prior to lactone formation during gilvocarcin biosynthesis. Further in vitro characterization established GilR as a unique oxidoreductase which utilizes bicovalently bound FAD to directly dehydrogenate the hemiactal in 144–146 to form the lactone observed in gilvocarcins (120–122).106 The ability of GilR to accept pre-defucogilvocarcin V (142), albeit at a reduced rate when compared to pre-gilvocarcin V (146) (specific activity shows 4 fold preference for glycosylated substrate), explains the presence of the shunt products 147–149 and confirms that the glycosylation by GilGT occurs prior to the lactone formation.106
2.4.3. Deoxysugar biosynthesis and glycosylation.
Analysis of the gil cluster revealed three putative deoxysugar biosynthetic genes (gilD, gilE and gilU) as well as a putative glycosyltransferase encoding gene (gilGT) for the biosynthesis and attachment of d-fucofuranose to the aglycon of gilvocarcin.99 The attachment of a furanose sugar moiety, such as d-fucofuranose in natural products is rare as the vast majority of deoxysugar containing polyketides contain pyranose sugars. The genes gilD and gilE encode a TDP-glucose-synthase (unpublished results) and a putative TDP-glucose-4,6-dehydratase, respectively. Together, GilD and GilE form TDP-4-keto-6-deoxy-d-glucose (158), a common branching point in various deoxysugar biosyntheses (Scheme 18). The putative TDP-4-ketoreductase GilU is hypothesized to then regiospecifically install an axial 4-OH to produce TDP-d-fucose (156). This is supported by inactivation experiments with gilU (discussed below).107 An as of yet undetermined ring contraction event then creates the furanose sugar, TDP-d-fucofuranose (157) (Scheme 18). Recent elucidation of the biosynthetic pathway leading to TDP-d-fucofuranose in Escherichia coli 052 identified two unique enzymes Fcf1 and Fcf2 to be essential for 157 production.108 Fcf1 shows sequence homology with GilU and was found to catalyze the 4-ketoreduction step to form 156, while Fcf2 performed the ring contraction to form 157. The gil cluster lacks genes with sequence homology to Fcf2, and therefore ring contraction must depend on the activity of an uncharacterized enzyme in the gilvocarcin pathway or a homologous enzyme must be present in Streptomyces that is recruited by the gilvocarcin pathway during the biosynthesis of gilvocarcin V (122).
Scheme 18.

The biosynthetic pathway of natural (154, 157, 161 and 165) and engineered (155, 162 and 163) sugar moieties of gilvocarcins (157 and 155), chrysomycins (154), ravidomycins (165 and 161), olivosyl-gilvocarcins (162) and polycarcins (163).
Bacteria produce a wide variety of glycosylated natural products, most of which fall into the category of O-glycosides.109 Less common are C-glycosides, which are of particular interest as they are resistant to glycosidase activity. As gilvocarcins are C-glycosides containing an unusual sugar moiety, the biosynthetic investigation of the putative glycosyltransferase, GilGT, is significant. Inactivation of the gilGT gene afforded the accumulation of defucogilvocarcin E (148) and confirmed that GilGT plays an integral role in the C-4 glycosylation of the gilvocarcin aglycon.103 The expected shunt product 149 was not observed and was attributed to a polar effect on the downstream gilOIII gene due to the inactivation of gilGT. In context with the biosynthetic study of GilR, it is likely that GilGT glycosylates the gilvocarcin aglycone (140–142) producing 144–146 before the final lactone formation by GilR to produce gilvocarcins M,E and V (120–122, Scheme 17).
2.4.4. Engineering gilvocarcin derivatives with altered sugar residues.
While investigating the function of GilU as a TDP-4-ketoreductase, a mutant strain lacking gilU accumulated a glycosylated derivative of gilvocarcin V (122).107 The new compound, 4′-hydroxy-gilvocarcin V (168), contained 4-hydroxy-d-fucofuranose in lieu of d-fucofuranose (Fig. 12). The biosynthesis of TDP-4-hydroxy-d-fucofuranose (155) can be attributed to a loss in 4-ketoreductase activity as it is simply the ring contracted hydrated form of 158. The formation of 168, a more potent anticancer drug than the parent 122 (Section 2.1.3), provided direct evidence towards the flexibility of GilGT in installing modified sugars to the gilvocarcin aglycone.
Fig. 12.

Engineered gilvocarcin derivatives.
Further attempts to alter the sugar residue of gilvocarcins resulted in the formation of olivosyl-gilvocarcins (170–171) and polycarcin M (172) and E (173), which contain d-olivose (162) and l-rhamnose (163) in place of d-fucofuranose, respectively.110 Gilvocarcin derivatives with d-olivosyl attachment (170–171) were created by complementing the gilU-minus mutant strain with the plasmid pLN2, which contains deoxysugar biosynthetic genes from the oleandomycin gene cluster to produce TDP-l-olivose and TDP-d-olivose (Scheme 18).110 Polycarcin M (172) and E (173), containing l-rhamnose, were created similarly with the plasmid pRHAM which again utilizes deoxysugar genes from oleandomycin biosynthesis to create TDP-l-rhamnose (165, Scheme 18).111 Polycarcin V (234), which contains the vinyl C-8 side chain is a naturally occurring gilvocarcin analogue containing an l-rhamnose sugar moiety.112
2.4.5. Biosynthetic highlights of ravidomycins and chrysomycins.
Ravidomycin (123) and chrysomycin A (152) and B (153) share identical chromophores and differ only in the sugar attached at the C-4 position of their aglycon. Ravidomycin contains the amino pyranose sugar 4-O-acetyl-d-ravidosamine while chrysomycin contains the branched pyranose sugar d-virenose. The biosynthetic gene clusters leading to the formation of ravidomycin and chrysomycin A were isolated from S. ravidus and S. albaduncus, respectively.113 As expected, comparison of the gil, rav and chry clusters revealed homologous genes for the biosynthesis and tailoring of the gilvocarcin-type backbone. Interestingly, both the rav and chry clusters contain two genes with high sequence homology to acyl-carrier proteins (ACP) and initially led to the hypothesis that each ACP may show specificity for either initiation or elongation modules of their respective polyketide synthase, similar to that observed in R1128 biosynthesis.114 It was later found that the two ACP genes are likely functional repeats and do not show preference for either initiation or elongation module.102 The absence of gilN homologues in both rav and chry clusters suggested it may play a role in ring contraction; however, inactivation of gilN continued to produce gilvocarcin V (122, unpublished results). Another interesting feature conserved in both rav and chry clusters is the fusion of the homologous gilR and gilM genes producing the bimodular rav/chryRM.113
The main differences in the clusters were the genes encoding deoxysugar biosynthesis toward their respective sugar moiety. The NDP-glucose-synthase homologue (ChryD) and TDP-glucose-4,6-dehydratase (ChryE) form 158, as observed in gilvocarcin biosynthesis (Scheme 18). The putative C-methyltransferase (ChryCMT) is believed to install the equatorial C-3 methyl group and finally the putative 4-ketoreductase (ChryU) catalyzes the formation of the axial 4-OH seen in 154.
The biosynthesis of the sugar moiety in ravidomycin requires several steps and was investigated in detail by Kharel et al.115 Analogous to TDP-fucofuranose (157) and TDP-virenose (154) biosynthesis, RavE and RavD form the intermediate TDP-4-keto-6-deoxy-d-glucose (158). The 3,4-ketoisomerase, RavIM, then produces the 3-keto-isomer 159, which undergoes 3-transamination by RavAMT to form 160. Further modification by the N-methyltransferase RavNMT produces the N,N-dimethyl group found in TDP-d-ravidosamine (161).115 The final modification of 161, namely an O-acetylation at the 4-OH is believed to be catalyzed by RavW; however, the exact substrate of RavW is unclear. The most likely substrate is deacetyl-ravidomycin (151), a naturally occurring metabolite of S. ravidus containing d-ravidosamine in which RavW would act upon the sugar after glycosylation.116 Additionally, it is plausible that O-acetylation may occur prior to glycosylation where RavW acts directly on 161 to produce 165 or prior to methylation by RavNMT, RavW N-acetylates 160, which undergoes acyl migration to the 4-OH producing 164. Subsequent methylation by RavNMT would then create 165 which would serve as the donor substrate for glycosylation (Scheme 18).
Preliminary investigation into the substrate flexibility of the putative C-glycosyltransferases of ravidomycin (ravGT) and chrysomycin (chryGT) biosyntheses revealed ravGT, and not chryGT, could compliment the gilGT deficient mutant strain reconstituting gilvocarcin biosynthesis.113 This result is encouraging as several NDP-amino sugars were not accepted as GilGT donor substrates upon initial gilvocarcin glycodiversification studies (unpublished results).
2.5. Oviedomycin biosynthesis
An unusually rich oxygenated angucyclinone, oviedomycin (179), was produced in S. lividans TK24 and S. albus through the expression of its gene cluster, which was originally discovered in and cloned from the oleandomycin producer S. antibioticus ATCC 11891.117 Sequencing of the gene cluster revealed type II PKS genes (ovmPKST AC), post-PKS tailoring oxygenases (ovmOI, ovmOII and ovmOIII), malonyl-CoA biosynthetic genes and several other genes potentially involved in the regulation of oviedomycin production.118 The inability of the cluster to produce oviedomycin in the absence of the PKS-associated ketoreductase gene ovmT provided experimental proof that the cluster encoded oviedomycin biosynthesis.118 The functional roles of the pathway oxygenases were elucidated through the co-expression of the PKS genes with individual oxygenase genes, or with a combination of two or three of the oxygenase genes. OvmPKSTAC are sufficient for the production of fully cyclized angucyclinone intermediates UWM6 (6) and UWM6–2-carboxylate 174.119 Inclusion of three oxygenases (OvmOI, OII and OIII) in the PKS enzymes was necessary to fully constitute oviedomycin biosynthesis. The results also concluded that OvmI serves as an aromatic hydroxylase, which catalyzes the hydroxylation at the C5-position of 4a,12b-dehydro-UWM6 (22) (Scheme 19). OvmII might be a bifunctional (dehydratase/oxygenase) enzyme that catalyzes 2,3-dehydration prior to the OvmOIII mediated 2-hydroxylation as well as the oxygenation at 4-position at the last step of the oviedomycin biosynthetic pathway (Scheme 19). Production of 9-hydroxyrabelomycin (180) in the presence of OvmOIII also suggested that the enzyme has an inherent relaxed substrate specificity and is capable to catalyze hydroxylation at the 9-position of rabelomycin (11) in addition to the normal 2-hydroxylation in dehydrorabelomycin (90).119
Scheme 19.

The proposed pathway for oviedomycin biosynthesis.
2.6. Gaudimycin biosynthesis
Angucyclinones gaudimycin A, B and C (185, 182, and 184) were generated through the reconstitution of enzymes from cryptic gene clusters. Gaudimycin B (182) was heterologously produced in Streptomyces lividans TK24 through the expression of the cryptic gene cluster (cabABCDEFJLMV1) isolated from Streptomyces sp. H021.19 Similarly, gaudimycin A was produced through the co-expression of nogalamycin minimal PKS (snoa123) and ketoreductase (snoaD) with the part of the cryptic gene cluster (pgaEFLM) isolated from Streptomyces sp. PGA64.19 UWM6 (6) was suggested as the earliest pathway intermediate for both the metabolites. The FAD-dependent monoxygenases PgaE/CabE catalyzes hydroxylation at the 12-position of 6, and the product of this reaction, 181, undergoes subsequent oxygenation, reduction and dehydration catalyzed by CabM (oxygenase) and CabV (reductase), respectively, to generate gaudimycin B (182) (Scheme 20). Recently, PgaE and CabE both have been crystallized and their structures have been solved.120 Structure analyses suggested that both enzymes are closely related homo-dimeric enzymes and are the members of the para-hydroxybenzoate hydroxylase (pHBH) fold family of hydroxylases. The exact active site residues and their role in catalysis have not been fully understood yet.120 In vitro results suggested that the oxidoreductase homologue PgaM catalyzes a series of reactions including 2,3-dehydration, isomerization to form the quinone-methide intermediate (183) and stereospecific reduction of the 6-keto group of 183 during gaudimycin C biosynthesis.55,121 The presence of both PgaE and PgaM was absolutely necessary to efficiently convert 6 to gaudimycin C (184). Endogenous enzymes of S. lividans TK24 might be contributing to the 5,6-dehydration and subsequent enoyl reduction of the PgaM products, which finish the production of gaudimycin A (185) in vivo (Scheme 20).19
Scheme 20.

The proposed pathway for gaudimycin biosynthesis.
2.7. Biosynthesis of azicemicins
Azicemicin A (188) comprises a unique aziridine moiety at the 3-position in addition to the fully decorated angucyclinone core structure.122,123 Feeding experiments conducted by Liu and co-workers have demonstrated that the aziridine moiety of azicemicin is originated from aspartic acid.124 Sequencing of a ~50 kb contiguous region of genomic DNA of the producer Kibdelosporangium sp. MJ126-NF4 revealed the azicemicin A gene cluster to contain angucyclinone core biosynthetic genes, aziridine moiety biosynthetic genes, and regulatory and resistance genes. An in vitro enzyme assay demonstrated that the NRPS adenylation-domain homologue AzicM preferentially activates the β-carboxyl group of either d- or l-aspartate. It is suggested that the adenylated product is then ligated to the carrier protein AzicP prior to the decarboxylation (catalyzed by the decarboxylase AzicN), hydroxylation (catalyzed by AzicOI, AzicO2 or AzicO6), and cyclization (catalyzed by yet unknown enzymes) to establish the aziridine carboxylate starter unit (187) (Scheme 21).
Scheme 21.

The proposed pathway for azicemicin A biosynthesis.
2.8. Biosynthesis of BE-7585A
The thymidylate synthase inhibitor BE-7585A (198) comprises a relatively unusual disaccharide 2-thiotrehalose moiety and an l-rhodinose linked to the angucyclinone core structure through the S- and O-glycosidic linkages, respectively. Formation of this thio-disaccharide and its attachment to the angucyclinone were particularly intriguing from a biosynthetic point of view. Incorporation studies involving singly and doubly labeled acetate suggested that the formation of the angucyclinone core of BE-7585A (198) proceeds through the linearly fused tetracyclic anthracyclinone intermediate 194 (Scheme 24).13 To provide more insights into the biosynthetic pathway, Liu and co-workers have cloned and sequenced the entire gene cluster (bex cluster) responsible for the biosynthesis of 198.13 Bioinformatic analyses of the cluster revealed 28 open reading frames covering type II PKS genes, post-PKS tailoring oxygenases, deoxysugar biosynthetic genes, glycosyltransferases, thiosugar biosynthetic genes, regulatory and resistance genes.
Scheme 24.

The proposed pathway for the biosynthesis of BE-7585A (198).
In vitro results demonstrated that the glycosyltransferase BexG2 is capable of producing 2-thiotrehalose 6-phosphate (192) when the enzyme was incubated with 2-thio-d-glucose-6-phosphate (190) and UDP-d-glucose (191) (Scheme 22). The pathway still requires a phosphatase that hydrolyses the phosphate of 192 prior to or after the attachment of the disaccharide to the angucyclinone. Although no such enzyme was identified in the cluster, the endogenous enzyme with such activity could be encoded elsewhere in the genome, or – more likely – the attachment of the disaccharide to the aglycone occurs non-enzymatically, similar to the SCH3-addition observed earlier in urdamycin E biosynthesis.125 A pathway analogous to the ThiG-catalzyed thiazole formation in thiamin biosynthesis has been proposed for the biosynthesis of 2-thio-d-glucose-6-phosphate 190 (Scheme 23). Recent in vitro results demonstrated that Lys110 of BexX (ThiG-homologue in the Bex cluster) reacts with the carbonyl group of glucose-6-phosphate specifically to generate a covalently linked adduct during the incubation with glucose-6-phosphate. This adduct still requires several isomerization steps prior to the incorporation of sulfur.13,125 However, the source of sulfur and the exact mechanism delineating the incorporation of sulfur in 2-thio-d-glucose-6-phosphate (190) still remains unclear.
Scheme 22.

The biosynthesis of 2-thiotrehalose-6-phosphate (192).
Scheme 23.

The proposed pathway for the biosynthesis of 2-thio-d-glucose-6-phosphate (190) and TDP-l-rhodinose (20).
PKS enzymes BexABCDLF are proposed to assemble the acetate units to form the tetracyclic intermediate 194. BexI serves as an anthrone oxygenase whereas BexE and BexM are proposed to catalyze Baeyer–Villiger oxidations during the conversion of 194 to the angucyclinone 195. The oxygenase homologue BexK could hydroxylate the 2-position of 195 to establish the fully functionalized angucyclinone core of BE-7585A (198) (Scheme 24). BgxG1 is proposed to catalyze the attachment of l-rhodinose at 12b-position of 198.
2.9. Kinamycin biosynthesis
Kinamycin D (205) was isolated from S. murayamaensis along with the other congener kinamycin A–C (206–208).126 More recently, kinamycins have been isolated from the spiramycin producer S. ambofaciens.127 Formation of a five membered ring (ring B) and a diazo group are the most biosynthetically intriguing features of the kinamycins. The biosynthetic pathway of the kinamycins has been largely established through feeding and incorporation experiments. Jadomycin A (92) biosynthesis is closely related (see section 1.3), as both pathways involve the common intermediate dehydrorabelomycin (90). Incorporation studies revealed that kinobscurinone (199),128 stealthin C (200)129 and kinamycin F (203) represent intermediates of the pathway (Scheme 25).130 The biosynthesis involves oxidative ring cleavage of the C5–C6 bond of dehydrorabelomycin and subsequent ring closure, to yield 199. Transamination of 199 followed by diazo group formation leads to the production of the proposed intermediate prekinamycin (201). Epoxidation of 201 and subsequent hydrolysis of the product results in the production of kinamycin F (203).131 An acetyltransferase sequentially acetylates 203 at the 4- and then at the 2-position to generate kinamycins E (204) and D (205), respectively.132 The kinamycin gene cluster has been recently cloned and sequenced (NCBI database accession AY228175). Production of 90, 199, and 200 through the expression of the cluster in S. lividans ZX7 indicated that the cluster covers most of the kinamycin D (205) biosynthetic genes.131 However, the exact role of these genes and the complete sequence of biosynthetic events are yet to be fully established.
Scheme 25.

The proposed pathway for the biosynthesis of kinamycin D (205).
2.10. Biosynthesis of hatomarubigins
Antitumor agents hatomarubigins A–D (211–213, 215) were isolated from Streptomyces sp. 2238-SVT4.133 Hatomarubigin D (215) is the most interesting molecule of these among other congeners as it comprises two identical angucyclinone moieties connected through a methylene bridge. Hayakawa and co-workers have cloned, sequenced and partially characterized the biosynthetic genes involved in the biosynthesis of 215.134 The cluster comprises 30 ORFs, including type II PKS genes, oxidoreductases, an O-methyltransferase, and oxygenases. Expression of a cosmid comprising the majority of the genes of the cluster, excluding the oxidoreductase gene hrbY, in S. lividans TK 23 led to the production of 211–213. Through in vitro assay, it is demonstrated that hatomarubigin C (213) is the last intermediate of the biosynthetic pathway of 215 where HrbY catalyzes the conversion 213 → 215 in the presence of NADPH and methylcobalamine (Scheme 26). Biosynthesis of hatomarubigin C can be achieved through two different routes (path I and path II, Scheme 26). Recent results demonstrated that conversion of 214 → 213 can be achieved through the activities of the methyltransferase HrbU in vitro.135 However, HrbU could also act earlier leading to the production of 211 and 212 through path II. Further experiments regarding the conversion of 209 → 213 are necessary to fully establish the biosynthesis of 215.
Scheme 26.

The proposed pathway for hatomarubigin D (215) biosynthesis.
2.11. Saquayamycin biosynthesis
Saquayamycin Z (222) and galtamycin B are produced in fermentations of Micromonospora sp. Tü6368, and both feature long saccharide side chains. Both saquayamycin Z and galtamycin possess a pentasaccharide at 3-O position consisting of l-rhodinose, d-olivose, l-rhodinose, d-olivose, and l-rhodinose. They also carry at the 9-position a C-glycosidically linked tetrasaccharide, comprised of d-olivose, 2-deoxy-l-fucose, l-rhodinose, and l-aculose. The elucidation of the saq biosynthetic gene cluster established six candidate glycosyltransferase genes encoding the enzymes responsible for the nine glycosyltransfer events.25 For the delineation of the biosynthetic pathway, five of these glycosyltransferase genes (saqGT1, saqGT2, saqGT3, saqGT5, and saqGT6) were inactivated (Scheme 27). The disruption of saqGT5 resulted in accumulation of 3-deoxyrabelomycin (216), and the successful complementation of this mutant strain with the urdamycin 9-C-d-olivosyltransferase encoding gene, urdGT2, suggested the encoded product of saqGT5 to be also a 9-C-d-olivosyltransferase. Inactivation of saqGT2 resulted in production of aquayamycin (50), and heterologous expression of saqGT2 in Streptomyces fradiae Ax, another aquayamycin producer, resulted in production of SaqAE3 (218). This showed that SaqGT2 is responsible for transferring the first l-rhodinose unit of the pentasaccharide. Disruption of saqGT3 resulted in accumulation of a new saquayamycin, SaqAE61 (219), which possessed a 9-C-d-olivose residue and a single 3-O-l-rhodinose residue. This showed that SaqGT3 possibly transfers the second and possibly also the fourth d-olivose units of this chain. SaqGT4 was hypothesized to transfer the third and fifth l-rhodinose units of the pentasaccharide. The saqGT6-disrupted strain produced SaqAE62 (220), which lacks the last three sugars of the tetrasaccharide chain, thereby characterizing SaqGT6 as the glycosyltransferase responsible for transferring the 2-deoxy-l-fucose unit. Disruption of saqGT1 resulted in production of SaqAE1 (221), which possessed an intact pentasaccharide and a 9-C-d-olivose-2-deoxy-l-fucose disaccharide, which revealed that it transfers the third and potentially the fourth sugars, both as l-rhodinoses, of the tetrasaccharide chain (the fourth sugar is possibly later converted to l-aculose by oxidoreductase SaqW, analogously to the AknOX reaction in aclacinomycin biosynthesis).136
Scheme 27.

The proposed glycosylation sequence of saquayamycin Z (222).
3. Biological activities of angucyclines
3.1. Cytotoxicity
Angucyclines have been attractive targets for synthetic and biosynthetic studies, in large part due to their promising biological activities. As such, a growing number of cellular targets and diverse antimicrobial and anticancer activities are being elucidated within the family of angucycline antibiotics. In recent years, novel angucycline analogues have been produced through traditional chemical synthesis and combinatorial biosynthetic methods, which affords a more thorough investigation of structure activity relationships (SAR). Therefore, it seems appropriate to discuss the cytotoxicity of angucyclines based on sub-groups of structure, and then to consider the enzymatic inhibition activities of angucycline antibiotics separately.
3.1.1. Angucyclinone N-heterocycles.
Recently, chemical syntheses by Valderrama et al. on angucyclinone derivatives have provided valuable insight into structure activity relationships.137 With an aim to alter the carbocyclic bioisostere of angucyclinone compounds, a series of angucyclinone derivatives were prepared with a nitrogen substitution at 5-position (Fig. 13). The biological activity of these derivatives was then assayed in an in vitro tetrazolium reduction assay against several human tumor cell lines (Fig. 13). 223 demonstrated 1.6 μg mL−1 activity against the AGS gastric adenocarcinoma cell line, whereas its congener with nitrogen at the 6-position (not shown) was 36-fold less active in the same cell line. Incorporation of the pyrimidopyridine moiety into the AB rings of 229 enhanced the cytotoxicity of the compound (IC50 5.9–9.2 μg mL−1) against several tested cell lines. Incorporation of a second nitrogen into ring D at the 8-position afforded the most promising compound, 231, which featured an IC50 range of 0.73–2.4 μg mL−1 against various tumor cell lines, comparable to the known topoisomerase II inhibitor etoposide (0.36–2.8 μg mL−1).
Fig. 13.

Angucycline N-heterocycles.
3.1.2. Jadomycins.
Rohr et al. and Jakeman et al. have recently produced a library of jadomycin B analogues by substituting various l- and d-amino acids into the oxazolone moiety.79–81,138 By exploiting the non-enzymatic nature of amino acid incorporation into the ring-cleaved jadomycin core, the resulting library of derivatives was assayed for their cytotoxicity in two breast cancer cell lines. Jadomycins B (96), and analogues 232 and 104 (jadomycin F) demonstrated inhibitory MIC’s of <2 μg mL−1, <1 μg mL−1, and 1 μg mL−1 when tested against a strain of methicillin resistant Staphylococcus aureus C623 (Fig. 14).138 This strain is of considerable clinical importance, as vancomycin is currently the only available drug for treatment of S. aureus C623. When these compounds were evaluated in two breast cancer cell lines, the jadomycins with l-serine and d/l-threonine moieties were shown to have the highest cytotoxicity (e.g. EC50 values in MDA-MB-435 cells 105 = 1.06 μM, 100 = 2.82 μM, 110 = 1.15 μM, 116 = 1.34 μM. In the breast cancer cell lines, the jadomycins with aromatic side chains were the least active; it is hypothesized that the hydrogen donor of the hydroxyl side chain is responsible for the excellent activity of 105, 100 and 110. Gel mobility assays were recently used to demonstrate that jadomycin derivatives might act as DNA cleaving agents.139
Fig. 14.

Bioactive jadomycins produced by mutasynthesis.
3.1.3. Gilvocarcins.
The gilvocarcin-type subclass of antitumor agents is an emerging family of oxidatively rearranged C-glycosylated angucyclines that exhibit promising biological activity while maintaining a low in vivo cytotoxicity.140–142 Gilvocarcin-type antibiotics with a characteristic 8-vinyl side chain (Fig. 15) have been shown to have a unique mode of action: photo-catalyzed [2 + 2] cycloaddition to thymidine residues in DNA.140,143–145 Additionally, crosslinking to histone H3 and heat-shock protein GRP78 was observed,143 for which the C-glycosidic moieties of these compounds are hypothesized to be responsible (Fig. 16). Similarly, desacetylravidomycin V (151), ravidomycin V (123), and the related chrysomycin A (152) maintains the same light-catalyzed photoactivity but differ from gilvocarcin V with respect to the C-glycoside (Fig. 15).113,146–148 The ketosugar containing chrysomycin derivatives Mer-1020dC (233) and Mer-1020dD (235) described in a patent by the Mercian Corporation have demonstrated better cytotoxicity towards solid tumor cell lines compared to chrysomycin A (152), with an IC50 0.10 and 0.15 μg mL−1 for 233 and 235 versus 0.84 μg mL−1 for 152 in human HT29 colic cancer cells.149 Polycarcin V (234) was recently accumulated in fermentations of Streptomyces polyformus112 and in a recombinant gilvocarcin producer strain.110 it exhibited IC70 values in human tumor cell lines ranging from <0.3 ng mL−1 to 0.4 ng mL−1 against several melanoma cell lines and <0.3 ng mL−1 against LXF 1211 L non-small-cell-lung cancer cells.112 4′-Hydroxygilvocarcin V (168), featuring an additional 4′-OH as compared to its parent drug gilvocarcin V (122), demonstrated similar activity against a murine lung cancer cell line (LL/2; 1.4% surviving treated cancer cells over untreated control cells (% T/C) at 100 μm concentration) and better activity in H460 human lung cancer cells (2.6% T/C at 100 μm for 168 versus 5.5% T/C for 122).107 The presence of an additional hydrogen donor could be responsible for the partially improved activity of 168.
Fig. 15.

Selected representatives of the gilvocarcin-class of antitumor antibiotics.
Fig. 16.

Depiction of gilvoarcin V 122 cross-linked with thymine residue and histone H3.
3.1.4. Kinamycins and marmycins.
The kinamycins have recently received attention for their antiproliferative and cell cycle disruption activities.150,151 Several studies have focused on the elucidation of the mechanism of action of the kinamycins (Fig. 17).152–154 Two competing mechanisms were proposed, one by Jebaratnam et al. suggesting oxidation of the C11-azide to a reactive radical species.154 The other, proposed by Dmitrienko et al.,155 involves nucleophilic attack upon the C11-azide and subsequent radical formation of isoprekinamycin (236). Eastman et al. developed a mechanism for kinamycin F (Scheme 28) via reduction to a semiquinone.153,154 Further resonance and subsequent intramolecular nucleophilic attack, thereby causing the diazide to leave as nitrogen gas, generates the reactive C11-radical species, 237. Kinamycins A (206) and C (208) demonstrated IC50 values in the ~10 nM range and 0.3 ±0.2 μM range, respectively, in Chinese hamster ovary cancer cells.150 Both demonstrated equal potency in abolishing topoisomerase IIα decatenation activity (7.6 μmol L−1 for kinamycin A and 9.2 μmol L−1 for kinamycin C), but when incubated with 500 μmol L−1 of reducing agent dithiothreitol (DTT), their activities were enhanced 7.5 fold and 11.4 fold. This may suggest a reductive mechanism of action of these antibiotics, affording IC50 values of 4.7 nM for kinamycin A against topoisomerase IIα activity and 330 nM for kinamycin C. Kinamycin F (203) was found to induce apoptosis and downregulate cyclin D3 in human leukemia K562 cells151 and induced single-stranded DNA breaks and inhibited topoisomerase IIα activity in the K562 cell line with an IC50 of 0.33 ± 0.02 μM.152
Fig. 17.

Representative members of the kinamycins (206, 208, and 203), and isoprekinamycin (236).
Scheme 28.

The proposed mechanism of action for the formation of activated radical species EN30 (237) for interaction with DNA.
The marmycins (marmycin A (238) and marmycin B (239), Fig. 18) were produced by a marine actinomycete. They have demonstrated marked cytotoxicity in human HCT-116 colon cancer cells (IC50 of 238 = 60.5 nM, 239 = 1.09 μM).156 It is noteworthy that the chlorinated derivative 239 was far less active than its non-chlorinated precursor 238, although halogenations usually enhance cytotoxicity of bioactive molecules.156
Fig. 18.

Marmycins A (238) and B (239).
3.1.5. Landomycins.
The landomycins have received much attention for their antitumoral activities, found against the NCI 60 human cancer cell line panel (Fig. 20).35 Landomycin A (4) has demonstrated cell cycle disruption activities, disrupts thymidine uptake, and has demonstrated activity against prostate cancer cell lines.157 Landomycin E (14) has shown promising activity in multiple drug resistant cell lines as an inducer of apoptosis (IC50 0.76 μg mL−1 for MDA-MB-231 human breast cancer cell line, mean GI50 at 1 μg mL−1 of 1μM), but still being less active than 4.157,158 Interestingly, landomycins with the intact hexasaccharide (landomycin A (4) and landomycin Y (240, Fig. 19) exhibited better activity in MCF-7 (estrogen responsive) and MDA-231 (estrogen refractory) breast cancer cell lines than landomycins with shorter saccharidal chains, when compared to a library of recently published new landomycins (IC50 of 2.2 μM and 2.0 μM for 4; 1.0 μM and 2.0 μM for 240 in MCF-7 and MDA-231 cell lines, respectively).159 Surprisingly, 11-deoxy-landomycinone (13) and landomycinone (241) demonstrate excellent activity in cancer cell lines as well, despite the absence of any saccharide chain (IC50 2.1 μM and 1.2 μM for 13 in MCF-7 and MDA-231 cell lines, respectively) and (IC50 2.9 μM and 2.9 μM in MCF-7 and LL/2 murine cancer cell lines, respectively).35,159 This might indicate two independent cytotoxic activity mechanisms for landomycin-type compounds: perhaps a thymidylate synthase uptake inhibition for hexasaccharidal landomycins, but some other activity for landomycinone type compounds that lack saccharide chains.35 Further investigations into the modes of action of landomycin-type compounds could prove valuable for overcoming multiple drug resistance in cancer treatment.
Fig. 20.

Urdamycin derivatives with xanthine oxidase inhibitory properties.
Fig. 19.

Representative landomycin-type antibiotics.
3.2. Enzyme inhibition and agonism
In addition to the cytotoxic activities of the angucyclines, diverse enzyme inhibitory and agonistic activities have been reported. As a result of their novel structural diversity, angucyclines are emerging as important ligands for inhibiting and activating an array of enzymatic targets, which is encouraging for their continued development as therapeutic agents. Some of these enzymatic inhibitions, such as JAK-STAT and PPAR-gamma antagonization, are particularly topical with the elucidation of these cellular pathways in recent years.
Some angucyclines with poor antitumoral and antibacterial activities possess strong enzyme inhibition. For example, ravidomycin M lacks the vinyl side chain, which serves as the warhead for the antitumoral ravidomycin V (123). Interestingly, the desacetylravidomycin M compound, the 8-methyl derivative of 123, despite having poor antimicrobial activity compared to 123, demonstrated a downregulation of interleukin-4 (IL-4, 0.05 ng mL−1 in C23-upregulated U937 cells). IL-4 is an important signal activator of the JAK-STAT pathway, and IL-4 inhibitors have been implicated as potential agents in preventing allergic reactions.160 Kirschning et al.161 reported several new glycomodified urdamycin-type angucyclines with poor antitumoral activity, but surprisingly, 242–245 demonstrate superior xanthine oxidase inhibition compared to allopurinol, one of the most potent inhibitors. Urdamycin diolivosyl isomers 242 and 243 demonstrate IC50 values of 0.42 μM to xanthine oxidase, and analogue 244 (α-glycosidic d-olivose connection) possess an IC50 of 0.46 μM.161 Note that these disaccharidal structural isomers differ from each other just by the region and nature of the glycosidic linkage of the O-glycosidically bound second d-olivose (Fig. 21).
Fig. 21.

Structures of structurally more complex new angucyclinones.
P371A1 and P371A2 have been shown to have antigastric protection properties. P371A1 demonstrated 61% pentagastric-induced acid secretions.162,163 It has been suggested that P371A1 serves as a CCK B/gastrin receptor antagonist. Furthermore, P371A1 inhibited the formation of lesions in the gastric mucosal tissue of rats when administered interperitoneally (a dose of 10 mg kg−1 provided 83.6% inhibition against HCl/ethanol (60% ethanol in 150 mL HCl) induced lesions and 72.8% against indomethacin (25 mg kg−1) induced lesions).
The bridged ether angucycline gephyromycin (285) demonstrates remarkable glutaminergic agonistic properties, for example, when incubated at a concentration of 3 μg mL−1 with neurons for five minutes, the intracellular Ca2+ concentration increased two-fold.164 The binding of gephyromycin has been shown to cause an influx of Ca2+ ions and perhaps a concomitant release of glutamate or aspartate into the extracellular space, typical with glutamate agonism. As such, it demonstrates a potency that is similar to the most potent glutamate receptor agonist, DCG-IV. Furthermore, due to its bridged ether, it does not exhibit any cytotoxicity or antibacterial properties as is typical for many angucyclinones, which further promotes its investigation as a potent glutamate receptor agonist.
For some known angucyclines, novel enzymatic targets are being identified. For example, recently Alvi and co-workers isolated vineomycin C, an A-ring cleaved derivative of the vineomycins and saquayamycins from fermentations of Streptomyces strain #AM1699. Vineomycin C and saquayamycin A1 were found to have weak inhibitory effects on inducible nitric oxide synthase (iNOS) with IC50s of 25.3 μM and 101.2 μM, respectively.
One emerging enzymatic target is PPAR-γ, an important activator for adipocyte formation and fluid retention. As such, its activation via the thiazolidinedione rosiglitazone (currently used in treatment of diabetes type II) results in weight-gain and other untoward side effects. PPAR-γ inhibitors are gaining attention for their attenuation of these effects, while still maintaining the positive therapeutic aspects of PPAR-γ activators. Recently, Potterat et al. have elaborated a strong PPAR-γ inhibition by the chlorocyclinone angucyclinone compounds.165 Chlorocyclinone C (266) inhibited rosiglitazone agonism at a concentration of 0.60 μM. All four chlorocyclinones were effective rosiglitazone antagonists, and they are the first angucyclinones to inhibit PPAR-λ.
4. New angucyclinones/angucyclines
This section contains new angucyclin(on)e or angucyclin(on)e derived compounds, which were discovered from 1997 until the end of 2010, and also includes some compounds missing in the earlier reviews. Occasionally, older structures were shown for comparison with new analogues. The newly isolated angucyclin (on)e or angucyclin(on)e derived compounds were divided into two categories: (i) new angucyclinones, (ii) new angucyclines (Table 6, Figs. 21–25).
Table 6.
Structures of newer members of the angucycline family
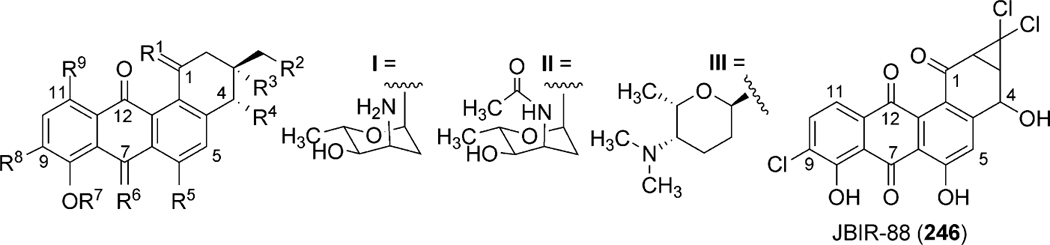
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| No | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | |
|
| ||||||||||
| 247 | O | CH3 | H | H | OH | O | I | H | H | Brasiliquinone A |
| 248 | O | CH3 | H | H | OH | O | H | H | H | Brasiliquinone B |
| 249 | O | CH3 | H | H | OH | O | CH3 | H | H | Brasiliquinone C |
| 250 | O | CH3 | H | H | OH | O | II | H | H | Brasiliquinone D |
| 251 | O | H | OH | H | H | O | CH3 | H | H | (—)-8-O-methyltetrangomycin |
| 252 | O | H | OH | H | H | H, OH | CH3 | H | H | 8-OMe-7-deoxo-7-OH-tetrangomycin |
| 253 | H, OH | H | OH | H | H | O | CH3 | H | H | 1-Deoxo-1-hydroxy-8-O-methylrabelomycin |
| 180 | O | H | OH | H | OH | O | H | OH | H | 9-Hydroxyrabelomycin |
| 254 | H, OH | H | OH | H | H | O | CH3 | H | OH | Seitomycin |
| 211 | O | H | H | H | OH | O | CH3 | H | H | Hatomarubigin A |
| 212 | O | H | H | H | H | O | CH3 | H | H | Hatomarubigin B |
| 213 | H, OH | H | H | H | H | O | H | H | OH | Hatomarubigin C |
| 214 | H, OH | H | H | H | H | O | CH3 | H | OH | Hatomarubigin E |
| 255 | H, OH | H | OH | H | H | O | CH3 | H | H | SM-196B |
| 256 | O | H | H | OH | H | O | H | H | H | Fujianmycin A |
| 257 | O | H | H | OH | H | O | CH3 | H | H | Fujianmycin B |
| 258 | O | OH | H | OH | H | O | CH3 | H | H | Fujianmycin C |
| 259 | O | H | OH | H | H | O | H | III | H | Frigocyclinone |
| 9 | O | H | OH | H | H | O | H | H | H | Tetrangomycin |
| 10 | O | H | OH | H | H | O | H | H | OH | 11-Hydroxytetrangomycin |
| 7 | O | H | OH | OH | H | O | H | H | H | 4-Hydroxytetrangomycin |
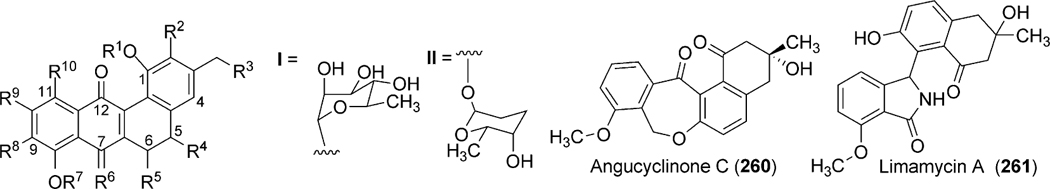
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| No | R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | R10 | |||
|
| |||||||||||||
| 262 | H | H | H | H | H | O | CH3 | H | H | H | Δ5,6 | 8-O-Methyltetrangulol | |
| 263 | I | H | H | H | H | H,OH | CH3 | H | H | H | Δ5,6 | 7-Deoxo-7-hydroxy-1-O-α-rhamnosyl-8-O-methyltetrangulol | |
| 177 | H | H | H | OH | OH | O | H | H | H | H | Δ5,6 | 5-Hydroxy-dehydrorabelomycin | |
| 264 | CH3 | Cl | H | H | OH | O | H | Et | COOMe | H | Δ5,6 | Chlorocyclinone A | |
| 265 | CH3 | Cl | H | H | OH | O | H | CH(OCOMe)Me | COOMe | H | Δ5,6 | Chlorocyclinone B | |
| 266 | CH3 | Cl | H | H | OH | O | H | CH(OCOCH2OH)Me | COOMe | H | Δ5,6 | Chlorocyclinone C | |
| 267 | H | H | OH | H | H | O | H | H | H | H | Δ5,6 | Kanglemycin M | |
| 268 | H | H | CH3 | β-H | α-OH | O | CH3 | H | H | H | — | PM070747 | |
| 269 | H | H | OH | β-OH | α-OH | O | CH3 | H | H | H | — | PD1167401 | |
| 270 | H | H | OH | β-OH | α-II | O | CH3 | H | H | H | — | TAN-1085 | |
| 271 | H | H | OH | H | H | O | CH3 | H | H | H | Δ5,6 | 19-Hydroxy-8-O-methyltetrangulol | |
| 272 | H | H | OH | OH | R4- R5 = β-O | O | CH3 | H | H | H | — | PD116740 | |
| 13 | H | H | H | H | β-OH | O | H | H | H | H | — | 11-Deoxy-landomycinone | |
| 241 | H | H | H | H | β-OH | O | H | H | H | OH | — | Landomycinone | |
| 273 | H | H | H | H | H | O | H | H | H | H | Δ5,6 | Landomycinone | |
| 274 | H | H | H | H | H | O | H | H | H | OH | Δ5,6 | Anhydrolandomycinone | |
Fig. 25.

Unique new angucyclines.
4.1. New angucyclinones
The halogen-rich JBIR-88 (246)166 was isolated from the two lichen-derived actinomycetes RI104-LiC106 and RI104-LiB101. It is a 1,1-dichlorocyclopropane-contaning angucyclinone whose structure was elucidated on the basis of 1D and 2D NMR spectroscopy and MS analyses. It is reported to show cytotoxic activities against cancer cells and antimicrobial activity against Micrococcus luteus.
The brasiliquinones A–C (247–249) are three cytotoxic benz[a] anthraquinones with an ethyl group attached at position C-3, indicating a rarely found for non-rearranged angucyclinones propionate biosynthetic starter unit (the other exception is MM2002, see below). They were isolated from the actinomycete Nocardia brasiliensis IFM 0089.167 Structures 247–249 were elucidated on the basis of spectroscopic data and by chemical means. Brasiliquinones A–C (247–249) are active against Gram-positive bacteria including Mycobacterium sp., but not active against Gram-negative bacteria or fungi. They were also active against multiple drug-resistant P388/ADRtumor cells.168 Brasiliquinone A(247) was reported as the first benz[a]anthraquinone possessing ristosamine as an O-glycosidic moiety, while brasiliquinones Band C (248 and 249) correspond to the aglycon of 247 and its 8-O-Me derivative, respectively. The antitumor, antibacterial, and kinase-inhibiting activities of 247–249 were also reported and discussed. The fourth compound, brasiliquinone D (250), was isolated from the actinomycete Nocardia brasiliensis IFM 0667. Brasiliquinone D (250) exhibited cytotoxicity and antibacterial activity.169 Brasiliquinone B (248) was also produced by the two lichen-derived actinomycetes RI104-LiC106 and RI104-LiB101.166
The three angucylinones; (–)-8-O-methyltetrangomycin (251), 8-O-methyltetrangulol (262) and 8-O-methyl-7-deoxo-7-hydroxytetrangomycin (252) were isolated by cultivation in an arginine–glycerol medium from the new isolated strain Streptomyces sp. AC113. The Streptomyces sp. AC113 is 99% identical with the Streptomyces flavidofuscus according to 16S ribosomal DNA-based.170 Compounds 251, 252, and 262 have antimicrobial activity against Bacillus cereus and Listeria monocytogenes. The cytotoxic activities of angucyclinones 251, 252, and 262 were evaluated against four cell lines (B16, HT-29 and non-tumor lines V79, L929), and 8-O-methyl-7-deoxo-7-hydroxytetrangomycin (252) was the most potent anticancer agent with IC50 0.054 μg mL−1 against cell line B16.
Six new angucyclinones (252–253, 263, 260–261) were reported and identified from two Indonesian Streptomyces strains, ICBB8309 and ICBB8415. The angucyclinones 7-deoxo-6-deoxy-7-hydroxy-8-O-methylrabelomycin (252), 1-deoxo-1-hydroxy-8-O-methylrabelomycin (253), and the angucycline 7-deoxo-7-hydroxy-1-O-α-rhamnosyl-8-O-methyltetrangulol (263) have common angular backbones, whereas angucyclinone C (260), limamycin A (261), and limamycin B (275) seem to be rearranged angucyclinones. Compound 180 has weak activity against S. aureus, P. aeruginosa and Mucor miehei; compound 253 has weak activity against S. aureus and Mycobacterium smegmatis; while compound 260 inhibited S. aureus and Mucor miehei. For the other compounds 260, 261 and 275, no antibiotic activity was reported. Furthermore, compounds 252 and 260 were reported to have moderate antiproliferative activity against PC3-cells (androgen independent prostate cancer cells).171
The unusual angucyclinone, oviedomycin (179), is a novel natural product encoded by genes of the oleandomycin producer Streptomyces antibioticus ATCC 11891. Compound 179 was produced using S. albus as a transformation host for heterologous expression of the oviedomycin biosynthetic gene cluster. The structure of 179 was elucidated by NMR spectroscopy and mass spectrometry. Besides weak cytostatic activity, no other biological activities were reported.117,118 However, the studies of its biosynthesis allowed for the elucidation of novel angucyclinone intermediates, particularly during the elucidation of the oxygenation steps (see above), which revealed several new angucyclinone derivatives. Prejadomycin-2-carboxylate (175), 4a,12b-dehydro-UWM6 (22), 5-hydroxy-dehydrorabelomycin (177), and 9-hydroxyrabelomycin (180), were isolated and characterized for the first time. Compounds 175 and 22 showed a significant increase in their in vitro antitumor activity relative to oviedomycin (179).119
Chlorinated angucyclinones are relatively rare in nature. The fermentation of the Streptomyces sp. strain DSM 17045 led to the isolation of four new chlorinated angucyclinones, the chlorocyclinones A–D (264–266, 276). Compounds 264–266 and 276 are reported to antagonize rosiglitazone-induced peroxisome proliferator-activated receptor gamma (PPAR-γ) activation with IC50’s < 0.4 μM in vitro using an AlphaScreen assay and are able to displace rosiglitazone from the PPAR-γ ligand-binding domain (LBD) in a scintillation proximity assay (SPA). These compounds also proved to be active in a cell-based reporter gene assay as well, antagonizing rosiglitazone-induced PPAR-γ activity with IC50 values between 0.60 and 7.0 μM. Chlorocyclinone C (266) has the most potent activity in all assays. Several natural products have been found to activate PPAR-γ. However, chlorocyclinones A–D (264–266, 276) were reported to be the first angucyclinones to modulate the activity of the nuclear hormone receptor PPAR-γ in a dose dependent manner. Furthermore, chlorocyclinones A–D (264–266, 276) were reported to be the first PPAR-γ antagonists of natural origin.165
The panglimycins A–F (277–282) are six new angucyclinone polyketides isolated from the bioassay-guided fractionation of the extract of the Indonesian Streptomyces strain ICBB8230. These were isolated together along with the three previously known metabolites fujianmycin A (256), ochromycinone, and emycin C. The new angucyclinones are highly oxygenated angucyclinones that appear to be biosynthetically derived from ochromycinone or fujianmycin (256). Their structures were determined by X-ray crystallography, interpretation of 1D- and 2D-NMR spectra, and comparison of the data with those of structurally known natural products. In spite of structural similarities to angucyclinones with antibiotic activities, the panglimycins A–F (277–282) did not exhibit any growth inhibition against several bacteria and fungi.172 Very recently, Abdalla et al. reported the highly related fujianmycin C (258) along with the known fujianmycins A (256) and B (257). Fujianmycin C (258) has weak antibacterial activity against Streptomyces viridochromogenes (Tü57).173
The angucyclinone chemomicin A (283)174 was obtained from the culture broth of Nocardia mediterranei subsp. kanglensis 1747–64, along with kanglemycin C, kanglemycin M (267) and SF2315B (284). Compound 283 had antimicrobial activity against Bacillus subtilis and Enterococcus faecium with MIC values of 10.2 and 20.4 μM, respectively, and showed cytotoxicity against human colorectal cancer HCT116 cells and human esophageal carcinoma YES-2 cells with IC50 values of 127 and 153 μM, respectively. Kanglemycin M (267) was isolated from Nocardia mediterranei var kanglensis 1747–64 and was not mentioned in our earlier reviews in 1992 and 1997. Compound 267 inhibited the growth of Gram-positive bacteria.
Antibiotic seitomycin (254) was isolated by Laatsch and co-workers and was produced by the two terrestrial Streptomyces spp. GW19/1251, and GW10/1118. This was isolated along with the other previously known angucyclinones hatomarubigin C (213), SM-196 B (255), and tetrangulol methyl ether (262). Compound 254 had moderate antimicrobial and weak phytotoxic activity, comparable to tetrangulol methyl ether (262).175
Gephyromycin (285)164 is a structurally unique angucyclinone isolated from the Streptomyces griseus NTK 14 by Bringmann et al. It is highly oxygenated, with an unprecedented ether-bridge between C-3 and C-12a. The structure was confirmed by single crystal X-ray analysis, and its absolute configuration was deduced by quantum chemical CD calculations. Gephyromycin (285) was reported to exhibits glutaminergic activity towards neuronal cells.
The marine Saccharopolyspora taberi PEM-06-F23–019B is the producer of the cytotoxic angucyclinone PM070747 (268).176 This was isolated along with the known PD1167401 (269), to which it is structurally related,1 and TAN-1085 (270).1,2 PD1167401 (269) was isolated from the unknown actinomycete, WP4669, and it has antitumor properties against leukemia and adenocarcinoma cell lines.
Urdamycin L (45) was isolated as a shunt product of the urdamycin A biosynthetic pathway of Streptomyces fradiae Tü2717.21 Its structure is similar to rabelomycin, however, it has a seven-membered lactone A-ring instead of the normal six-membered carbocycle. The isolation of urdamycin L (45) provided evidence for the hypothesis that the 12b-hydroxylation catalyzed by oxygenase UrdM enzyme involves a Baeyer–Villiger monooxygenation reaction, followed by rearrangement into the six-membered carbocycle and the generation of the 12b-hydroxy group, which are observed in the biosynthetic pathways of many angucycline-group antibiotics (see above, also for alternative mechanisms for this oxygenation step).
The simocyclinones D4 and D8 (286, 287) are two angucycli-none-type antibiotics isolated from the mycelium of Streptomyces antibioticus Tü6040.177–180 Compounds 286 and 287 contain four structural elements: the angucyclinone backbone, a C-glycosidically linked deoxysugar, and octatetraene dicarboxylate linker and an aminocoumarin unit. There structures were elucidated based on NMR techniques and with incorporation experiments using 13C labeled precursors. Simocyclinones D4 and D8 (286, 287) were reported to have antibiotic activities against Gram-positive bacteria and cytostatic effects on various tumor cell lines.
Frigocyclinone (259) was isolated from Streptomyces griseus NTK 97 and it consists of a C-glycosidic linkage with the rare aminodeoxysugar ossamine attached to C-9 of the tetrangomycin moiety. Compound 259 has antibacterial activities against Gram-positive bacteria.181
The cytotoxic pentacyclic C-glycosides, marmycins A and B (238 and 239),156 were isolated by Fenical and co-workers from the culture broth of a marine sediment-derived actinomycete, which is related to the genus Streptomyces. Their structures and absolute configurations were determined on the basis of spectroscopic and crystallographic techniques. Compound 238 has significant cytotoxicity against several cancer cell lines, some at nanomolar concentrations; whereas the chloro-analogue of 238, marmycin B (239) is less potent.
The very recently published antibiotic R2 (288)182 was described as a highly oxygenated angucyclinone obtained from the newly isolated strain Streptosporangium sp. Sg3, which was collected from an Algerian soil sample.183 However, the compound is not an angucyclinone by definition (not benz[a] anthracene derived),1 but belongs to the class of pentangular polyphenols.4 Compound 288 has strong antibacterial activity against the Gram-positive Micrococcus luteus ATCC 9314, Bacillus subtilis ATCC 6633, and moderate activity against Staphylococcus aureus CIP 7625. However, it showed no activity against Gram-negative bacteria or fungi and yeasts.
The four hatomarubigins A-D (211–213, 215) were produced by Streptomyces sp. 2238-SVT4, which enhanced the cytotoxicity of colchicine in multidrug-resistant tumor cells. The fifth hatomarubigin analogue, hatomarubigin E (214)135 was recently identified as an intermediate of the pathway. Bioinformatic analyses and in vitro experiments indicated that HrbU is able to convert structure 214 to hatomarubigin C (212).
Tetrangomycin (9) was re-isolated and characterized with corrected 13C-NMR assignments, by Shaaban et al.184 along with many other angucyclin(on)es. Compound 9 was shown to possess cytotoxic activity against MDA 231, and estrogen refractory breast cancer cell line (IC50 = 1.55 μM). Similarly, the structurally simple 11-deoxylandomycinone (13)159 was recently isolated from the culture broth of a Streptomyces cyanogenus K62 mutant, along with many other angucyclin(on)e analogues. Compound 13 showed a high cytotoxic activity against both MCF-7 (estrogen responsive) and MDA 231 (estrogen refractory) human breast cancer cell lines comparable to the hexasaccharidal landomycin A (4), indicating that both compounds may have different mechanisms of actions.
The saccharothrixins A-C (289–291) are three new angucyclinone isomers that were isolated and reported from the actinomycete Saccharothrix espanaensis An 113, associated with the marine mollusk Anadara broughtoni.185 These compounds were isolated along with the previously reported X-14881 A, X-14881 B, X-14881 C and X-14881 E, and ochromycinone.1 Compounds 289–291 have moderate activity against B. subtilis, E. faecium, and Xanthomonas sp. pv. badrii.
MM2002 (292)76 is another novel angucyclinone containing a rarely observed ethyl side chain in its A-ring, and was obtained by heterologously expressing a fourth-ring cyclase gene, pgaF, which was isolated from a silent antibiotic biosynthesis gene cluster. PgaF, and when co-expressed with the early biosynthesis genes of the nogalamycin biosynthetic pathway resulted in accumulation of the angucyclinone 292. This effectively steered the polyketide framework away from an anthracycline pathway towards a C4–C17-cyclized angucyclinone.
11-Hydroxytetrangomycin (10) and 4-hydroxytetrangomycin (7) are two novel shunt products isolated along with tetrangomycin (9) and tetrangulol (8) from Streptomyces globisporus mutant M12.15 The isolation of compounds 271 and 6 from this mutant strain confirm the involvement of lndM2 in the early biosynthetic pathway of landomycin E, in particular in the formation of the alicyclic 6-hydroxy function of the landomycin aglycon.
UWM6 (6), structurally somewhat related to MM2002 (292), was isolated and fully characterized by Hutchinson and co-workers.72 Compound 6 was obtained after expression of a jadomycin (jad) gene cassette (e.g. pWHM1238) composed of the minimal polyketide synthase (PKS, jadABC), a cyclase (jadD), a ketoreductase (jadE), and jadI in Streptomyces lividans. Expression of pWHM1238 and the resultant accumulation of 6 confirmed the role of JadI as a C4–C17 fourth ring cyclase.
Through the cloning and heterologous expression of the entire gene clusters for PD 116740 (272) from Streptomyces strain WP 4669 and tetrangulol (8) and tetrangomycin (9) from Streptomyces rimosus NRRL 3016, the angucyclinone analogues 19-hydroxytetrangulol; kanglemycin M (267); 8-O-methyltetrangulol (262); 19-hydroxy-8-O-methyltetrangulol (271); and PD 116740 (272) were reported.186 Compounds 262, 267, 271 and 272 were obtained from the WP 4669 cluster (pksWP-2) and characterized by 1H NMR spectroscopy and HR-mass spectrometry. Prejadomycin (21) is a novel angucyclinone metabolite obtained in a jadH-deficient mutant of Streptomyces venezuelae ISP5230,71 and an important intermediate of the biosynthesis of various members of the angucycline family.
As mentioned above, the gaudimycins A (185) and B (182) are two new angucyclinones reported by Palmu et al. from reconstitution of the gene clusters from Streptomyces spp. PGA64 and H021.19 Compounds 185 and 182 are very similar to WP 3688–2 (293), both of which undergo C2–C3 dehydration.
4.2. New angucyclines
As defined in the earlier reviews,1,2 the term angucycline refers to compounds with hydrolyzable sugar moieties, i.e. compounds combining an angucyclinone (benz[a] anthracene) aglycone with an appended N-, O-, or S-saccharide side chain. This part will be divided into three subsections, (i) landomycins, (ii) saquayamycins and urdamycin analogues, and (iii) all other angucyclines.
4.2.1. Landomycins.
Landomycins, along with the urdamycins, became the largest sub-families of angucycline antibiotics. The landomycins are characterized by the presence of a single deoxyoligosaccharide chain of varying lengths attached at the 8-position of the aglycone. The isolation of landomycins A–D14 by Rohr et al.,7,14 from Streptomyces cyanogenus S-136 and later of the trisaccharidal landomycin E (14)187,188 from Streptomyces globisporus 1912 opened the door for many other new analogues. The major congener, landomycin A (4), contains the longest sugar side chain, a hexasaccharide constructed from two repeating trisaccharide patterns (d-olivose-4–1-d-olivose-3–1-l-rhodinose).
The increased knowledge regarding the details of landomycin biosynthesis (see above) led to several new landomycins generated by genetic engineering (Fig. 22), leading to variants with respect to the saccharide moiety. The three landomycins F–H (35–37)23,29 were generated by deletion of the lndGT4 gene of the landomycin E cluster in S. globisporus. Compounds (35–37) contains di-, tri- and mono-saccharidal side chains, and they have no hydroxyl group attached at C-11 of the aglycon. Landomycins I (23) and J (25)35 were isolated in 2007 by Zhu et al., from a strain of S. cyanogenus that overexpressed glycosyltransferase lanGT3. Landomycin I (23) contains only one sugar moiety (d-olivose), and landomycin J (25) was the first compound found with a tetrasaccharide side chain. Recently, two new landomycins K (43) and L (38) were isolated by Erb et al. in 2009.30 It is interesting that the last sugar moiety in the pentassacharidal landomycin K (43) as well as in the disaccharidal landomycin L (38) is l-rhodinose instead of d-olivose normally found at this position of other penta- and di-saccharidal landomycins, caused by the acceptor substrate flexibility of rhodinosyltransferase LanGT4.
Fig. 22.

New natural or genetically engineered landomycins.
The generation of new landomycins with changes in the angucyclinone moiety can give further data for structure–activity-relationships. Landomycins M (41) and O (40)22 are compounds that have a tetrangulol (8) aglycon instead of landomycinone (241). Compounds 38, 40 and 41 are inactive against common lung and breast cancer cell lines, indicating that the absence of 11- and 6-OH groups in landomycins M (41) and O (40) reduces the activity of the molecules. Thus, the hydroxyl groups in the 6- and 11-positions seem to be important for the antitumor activity exhibited by landomycin B (26) and D (24).
More recently, Shaaban et al.184 isolated eight new landomycins namely, landomycin P–W (295–302), upon re-examining the fermentation of Streptomyces cyanogenus S-136. Another three congeners, landomycins X–Z (303–305),159 were isolated from the Streptomyces cyanogenus K62 mutant strain along with several known angucyclin(on)es. Together, these new derivatives provide more insight into the structure–activity-relationship (SAR) of landomycins, with respect to the oxygenation patterns of the aglycone and the length of attached sugar chains. Interestingly, the sugar chains of all previously reported landomycins consist solely of l-rhodinose and d-olivose units. Landomycin C (294) was the only analogue that bears an additional, different sugar, namely d-amicetose. The fourth sugar moiety in the three new landomycins X–Z (303–305) was also found to be d-amicetose, as in landomycin C (294), instead of d-olivose, which is usually found in this position in all the other landomycins.
4.2.2. New saquayamycin and urdamycin analogues.
Saquayamycins and urdamycins are well known members of the growing group of angucycline antitumor antibiotics. The structures of both saquayamycins and urdamycins contain aquayamycin (50) as aglycone. Saquayamycins A–D (306–309) were first isolated from Streptomyces nodosus MH190–16F3 in 1985 by Uchida et al.189 and were reported as platelet aggregation inhibitors. Saquayamycins A and B (306–307, Fig. 23) contain three O-glycosidically bounded deoxysugars, one l-rhodinose, and two l-aculoses, in the case of 307 the aculose (sugar B) is converted to l-cinerulose. Compound 306 was reported to be unstable to acids even by contact with silica gel during chromatography leading to its conversion to 307. Saquayamycins A1, B1 and C1 (310–312) were generated by partial acid hydrolysis of saquayamycins A (306), B (307) and C (308), respectively. Two new isomers, saquayamycins E (313) and F (314) were reported by Sekizawa et al., and produced by Actinomyces strain MK290-AF1.190 They differ slightly from the earlier saquayamycins A (306) and C (308) with respect of their sugar moieties. Compounds 313–314 were reported to inhibit the EPTase from bovine brain with IC50 values of 1.8 and 2.0 μM, respectively. A-7884 (315) was isolated from the Streptomyces sp. #AM1699 along with saquayamycin A1 (310) by Alvi et al.191 Compound 315 contains an additional l-rhodinose sugar moiety inserted between the first C-glycosidic sugar d-olivose and l-aculose of saquayamycin A1 (310), affording a trisaccharidal side chain connected at C-9 of the angucyclinone part. Compounds 310 and 315 have potent inhibitory activity in the inducible nitric oxide synthase (iNOS) assay with IC50 values of 101.2 and 43.5 μM, respectively.
Fig. 23.

New members of the saquamycin family.
Recently, Ströch et al. reported an even larger saquayamycin derivative, saquayamycin Z (222, Fig. 23) from Micromonospora sp. strain Tü6368,192 which was biosynthetically investigated four years later by Erb et al.25 Saquayamycin Z (222) contains tetra- and pentasaccharidal side chains linked at 3- and 9-positions of the benz[a]anthracene core. The tetrasaccharide side chain of saquayamycin Z is striking due to the presence of an l-oliose, which is an unprecedented sugar constituent of angucyclines.
More recently, the two anticancer antibiotics moromycins A (316) and B (317, Fig. 23), were isolated from the culture broth of Streptomyces sp. KY002. The attachment and composition of the sugar chains of moromycins A and B are like those of saquayamycins B (307) and B1 (311), respectively, however, their tetracyclic angucyclinone core has an aromatic ring B, missing the angular hydroxy groups at 4a- and 12b-positions. Moromycin B (317) with only one saccharide side chain linked at 9-position has superior antitumor activity against the human lung (H-460) and human breast cancer cell lines (MCF-7) compared to moromycin A (316) and saquayamycin B (307).193
Urdamycins are closely related to the saquayamycins, however, they differ in their saccharide patterns, which are attached at the 9- and 12b-positions. In addition to the earlier reported natural urdamycins A–H (31, 33, 65, 66, 67, 69, 55, 70)5,42,43,45,194 several new urdamycin analogues were generated by combinatorial biosynthesis (Figs. 8 and 24), typically through gene inactivation or gene overexpression experiments. The three angucyclin(on)es; urdamycins I (71), J (72), and K (73) were isolated from the urdamycin A (31) producer Streptomyces fradiae Tü2717 after inactivation of urdGT2, which encodes a glycosyltransferase responsible for the C-glycosyltransfer of activated d-olivose.52,20 The most recent reported analogues were urdamycins M (80),51 R (81) and S (82). Both urdamycins R (81) and S (82) contain a C-glycosidically linked rhodinose moiety at the 9-position, but differ with respect to the configuration of this sugar moiety.57 Trefzer et al. studied the function of glycosyltransferases involved in urdamycin A biosynthesis and found that overexpression of urdGT1c and urdGT1a in a triple mutant led to the formation of compounds 100–1 (49) and 100–2 (29), respectively.53 Both compounds were already known as pathway intermediates from earlier randomly generated block mutants.195
Fig. 24.

New urdamycin analogues.
4.3. Other new angucyclines
In addition to the larger groups described above, various new angucyclines were described during the past 13 years (Fig. 25), some of which contained rare aminosugars, including two different tetradeoxyaminosugars. P371A1 (318) was isolated from Streptomyces sp. P371 by Uesato et al. in 1998.163 Later, its congener P371A2 (319) from the same Streptomyces sp. P371 was described by the same group.162 The absolute stereochemistry of compounds 318–319 was determined by NMR spectroscopy, acid degradation, and by application of the modified Mosher method to produce P371A2 C-glycoside MTPA esters. Exciton chirality studies on P371A2 C-glycoside benzoate derivatives confirmed the absolute stereochemistry. Unique about compounds 318 and 319 is the presence of an ureido group connected at C-4C of the last sugar moiety of the trisaccharide side chain, a tetradeoxyaminosugar. The compounds also show unusual stereochemistry and a rich oxygenation pattern in the ring A/B regions. P371A1 (318) and its congener 319 were reported to have inhibitory activity against pentagastrin-stimulated acid secretion as well as protective activities against HCl/ethanol- and indomethacin-induced gastric lesions.
Very recently, mayamycin (320) was reported by Schneeman et al. from Streptomyces sp. HB202, which was isolated from the marine sponge Halichondria panacea by varying the culture conditions. This strain was also the producer of streptophenazines A–H. Structure 320 consists of an angucyclinone skeleton which identical with dehydrorabelomycin and a unique C-glycosidically linked aminosugar attached at 5-position of the benz[a]anthracenone core. Compound 320 showed antibacterial activity, including antibiotic-resistant strains, and exhibited potent cytotoxic activity against eight human cancer lines; HepG2 (hepatocellular carcinoma), HT-29 (colon adenocarcinoma), GXF251L (gastric cancer), LXF529L (non-small-cell lung cancer), MAXF401NL (mammary cancer), MEXF462NL (melanoma cancer), PAXF1657L (pancreatic cancer) and RXF486L (renal cancer) with IC50 range of 0.33–0.13 μM.196
The grecocyclines A and B (321, 322), along with the C12a–C12b ring B-opened grecocycline C, (323) were very recently isolated from Streptomyces acta 1262. Compound 321 has cytotoxic activity while grecocycline B (322) inhibits the protein tyrosine phosphatase 1B with an IC50 of 0.52 ± 0.17 μM. Grecocycline A (321) is structurally characterized by the presence of an epoxide ring between C6a and C12a, while grecocycline B (322) shows a 12a-OH along with a 6a-SH group.197 A sulfur substituent at C-6a was earlier reported for the potent endothelin receptor antagonists WS009A and B (FR901366 and FR901367), where an N-acetylcysteine is present at C-6a. Structurally most unique is grecocyclin C (323), in which an unprecedented oxidative 12a,12b-bond cleavage was observed.
Only very few thiosugar-containing angucyclines are known. These include BE-7585A (198) and rhodonocarcin A (324). BE-7585A (198) inhibits thymidylate synthase and was produced by Amycolatopsis orientalis subsp. vinearia BA-07585. Its biosynthetic pathway was recently studied by Liu and co-workers.13 The saccharothrixmicines A (325) and B (326) are two new angucycline antibiotics isolated from the actinomycete Saccharothrix espanaensis An 113 associated with the marine mollusk Anadara broughtoni.198 Structures 325 and 326 differ in the aglycone and glycosidic bond attachment. Both compounds 325 and 326 contain the same sugar moiety O-α-l-6-deoxyaltrose, attached at C-8 and C-7, respectively.198
9-C-d-Olivosyl-tetrangulol (327) is a genetically engineered angucyclinone, for which the urdamycin glycosyltransferase urdGT2 was heterologously expressed in a lanGT2-minus mutant strain, resulting in the first landomycin C-glycoside analogue.32
5. Synthesis of angucyclines and angucycline derived natural products
Due to their diverse range of biological activities and attractive structural features, a number of different strategies have been developed for the synthesis of angucyclines and angucycline derived natural products. The methodologies described for the synthesis of angucyclines until 2005 were summarized in the two earlier reviews by Krohn and Rohr2 and Carreno and Urbano.199 The syntheses of gilvocarcins were also already reviewed by Hua and Shankar.200 Thus, in this section, we will mostly discuss recent reports added to this topic, from 2005 on for the synthesis of angucyclines, and from 2000 on for angucycline-derived natural products.
The first ever reported synthesis of an angucycline group natural product is that of tetrangulol (8),201 roughly 10 years after this compound was published as the very first angucycline group natural product (together with tetrangomycin).202,203 However, a synthesis of a benz[a]anthraquinone core was reported earlier.204 While most of the earlier syntheses used classical reactions like the Diels–Alder or a nucleophilic/electrophilic addition reaction as the key step to form the angucycline backbone,2 a variety of newer reactions and variants have been evolved in recent years. Depending upon the key steps used for the synthesis of the tetracyclic core structure, the here highlighted synthetic strategies have been divided into the following five sections: (i) Diels–Alder, (ii) nucleophilic addition, (iii) electrophilic addition, (iv) transition metal catalyzed cross coupling, and (v) intramolecular cyclization strategies.
5.1. Diels–Alder strategies
The Diels–Alder reaction between two suitably substituted diene and dienophile components is the most widely used technique for the synthesis of the tetracyclic condensed ring skeletons of angucyclines. Because of the possibility of variation in the structure of dienes and dienophiles, the reaction can be utilized for the regio- and stereoselective construction of different rings of the angucycline structures. The synthesis of ochromycinone (343) by Guingant and Barreto205 was the first example of application of this methodology in the total synthesis of an angucyclinone natural product.
More recently, Kaliappan and Ravikumar206 used an intermolecular Diels–Alder reaction between bromonaphthoquinone 332 and chiral vinyl cyclohexenes (e.g., 331, 335, 336) for the enantioselective synthesis of five closely related natural products namely (+)-ochromycinone (343), (+)-rubiginone B2 (344), (—)-tetrangomycin (9), MM-47755 (345), and YM-181741 (334) (Schemes 29 and 30). The key starting materials, vinyl cyclohexenes, were prepared through an enyne metathesis reaction using Grubbs 1st or 2nd generation catalyst. The regiochemical course of the cycloaddition reaction was steered using bromojuglone 332. Thus the reaction of the chiral vinylcyclohexene 331, synthesized from hexenoic acid in five steps, with the bromojuglone 332 in toluene at 80 °C afforded an intermediate cycloadduct, which on aromatization with K2CO3 in MeOH provided the angucyclinone 333. Introduction of the angular carbonyl by a well-known regioselective photo oxidation207 and subsequent deprotection of the silyl protection group completed the total synthesis of YM-181741 (334) in 22% overall yield (Scheme 29). A similar reaction sequence starting from vinyl cyclohexene 335 and 336 allowed them to complete the enantioselective total synthesis of the other four natural products 343, 344, 9, and 345 (Scheme 30).
Scheme 29.

Kaliappan’s total synthesis of YM181741 (334).206
Scheme 30.

Kaliappan’s total synthesis of (+)-ochromycinone (343), (+)-rubiginone B2 (344),(-)-tetrangomycin (9), MM-47755 (345).206
Based on an identical strategy, Maugel and Snider208 used vinyl cyclohexene and naphthoquinone for a Diels–Alder reaction, to synthesize the 8-aminobenz[a]anthraquinone core of the marmycins (238, 239), cytotoxic natural products isolated from a marine sediment-derived actinomycete (Scheme 31).156 The vinyl cyclohexene 347 was prepared along with 348 as a mixture of regioisomers by the reaction of a vinyl Grignard reagent with 3-methylcyclohexanone (346) followed by dehydration. Without purification, the mixture of 347 and 348 was allowed to react with naphthoquinone 349 and the corresponding Diels–Alder adducts were oxidized to obtain angucyclinone compounds 350 and 351. Surprisingly, none of the regioisomers of 352 was observed, although the electron withdrawing substituent at C-5 of the naphthoquinone mostly favors the Diels–Alder reaction at the nucleophilic end of the diene through C-2. The anomaly was explained by the steric preference of the corresponding transition state over the others during the formation of 351. A similar steric factor also explains the meager production of 350 (7%) compared to the 351 (63%). Subsequent aromatization of the A-ring and reduction of the nitro group produced the desired aminoquinone 353. The authors also synthesized a diastereomeric mixture of the 3′-desmethyl-4′-acetyl-marmycin A analogues 355 and 356 using a In(OTf)3 catalyzed glycosidation with protected sugar 354, which was essentially a modification of the process developed earlier by Ding et al. (discussed later in Section 5.4).209
Scheme 31.

The synthesis of 3′-desmethyl-4′-acetyl-marmycin A analogues.209,208
Using azadienes (e.g., 359) in place of vinyl cyclohexenes Guingant et al.210,211 succeeded to synthesize different, non-natural 5-aza-analogues of angucyclinones (e.g., 362). The azadiene 359 was prepared from cyclohexane-1,3-dione (357) in two steps, which included the preparation of enaminone 358 by the treatment with NH4OAc in refluxing toluene followed by imine formation with N-(dimethoxymethyl)-N,N-dimethylamine. The reaction actually produced a 1 : 3 mixture of Z,E & E,E isomers 360 & 359, which could be completely converted to the more stable E,E-isomer by refluxing with toluene (Scheme 32). They used the reaction with differently substituted bromojuglones for the synthesis of a series of 5-aza-angucyclines. More recently, the same group prepared different chiral azadienes and successfully utilized them to synthesize chiral analogues of 5-aza-angucyclinones.212
Scheme 32.

The Guingant synthesis of 5-aza-angucyclinones.211
Another example of synthesis of 5-aza-angucyclinones was found in the work of Valderrama et al.213 They used the Diels–Alder reaction between a 1,3-diene and a substituted 7,10-phenanthrenedione to synthesize the d-ring of the angucyclinone core. Thus treatment of phenanthridine-1,7,10-trione 365, a Michael addition-heterocyclization product of acyl benzoquinone 363 and cyclic enamine 364, with diene 366 produced the nitrogen containing angucyclinone skeleton 367, which could be transferred to the corresponding 8-hydroxy or 8-non hydroxyl angucyclinone analogue 368 and 369, respectively, using selective deprotection–oxidation techniques (Scheme 33). Applying a similar synthetic strategy they have also reported the synthesis of different angucylinone AB-pyrido[2,3-d]pyrimidine analogues.137,214
Scheme 33.

An alternative strategy for the synthesis of 5-aza angucyclinones by Valderrama et al.213
Ishikawa and co-workers215,216 used the Diels–Alder reaction between a benz[f]indenone and a 1,3-diene to construct the benzo [b]fluorenone skeleton of the angucyclinone-derived kinamycin natural products for their stereoselective total synthesis of (±)-methyl-kinamycin C (380) (Scheme 34). The synthesis starts with the Lewis acid catalyzed Diels–Alder reaction between indenone 370, obtained in 5 steps starting from a substituted bromojuglone, and a Danishefsky-type diene 371. The tetracyclic adduct of the Diels–Alder reaction 372 was subjected to hydrolysis and oxygenation to give racemic enone 373. Diastereoselective dihydroxylation using OsO4 and TMEDA followed by protection of the hydroxyl groups as silyl ethers yielded 374, then oxidation with MCPBA afforded 1β-silyloxyketone 375, which was converted to the desired α-isomer 376 by aerial oxidation. Removal of TMS groups and subsequent selective acetylation provided the diacetate 377. Diastereoselective reduction with tetramethylammonium triacetoxyborohydride produced 378, possibly via a chelation between ketone and 3-OH, which favored the reduction at the vacant β-site. Diol protection followed by dehydration afforded 379. A subsequent deketalization and partial acetylation followed by treatment with tosylhydrazine yielded the hydrazone, which was finally exposed to CAN for hydroquinone oxidation along with desulfination to give (±)-methyl-kinamycin C (380).
Scheme 34.

Ishikawa’s total synthesis of (±)-methyl-kinamycin C (380).216
5.2. Nucleophilic addition strategies
Starting from the first synthesis of tetrangulol (8),201 the inter- or intra-molecular nucleophilic addition–cyclization reactions were being used for the regioselective construction of the tetracyclic benz[a]anthraquinone core of angucyclines. In recent years the mostly used version of this kind of reaction is the anionic [4 + 2]-cycloaddition commonly termed as Hauser annulation217 or phthalide218 annulation. The reaction is an intermolecular Michael-type addition followed by cyclization between a base-generated, stabilized anion from substituted phthalides with α,β-unsaturated carbonyl compounds. The process can be exemplified with the total synthesis of BE-23254 (387), a chlorinated angucyclinone by Mal and Dey (Scheme 35).219 The key step of the synthesis was a reaction between an anion, generated by the treatment of phthalide 383 with lithium tert-butoxide, and quinone monoketal 384 to provide the preliminary angucyclinone backbone 385. Aromatization of the A-ring with DDQ produced 386, which on subsequent demethylation and deesterification reaction yielded the natural product 387. The chloro-substituted cyanophthalide 383 was obtained from cyclohexenone 381 using a six step reaction sequence, which included a SO2Cl2 mediated regioselective chlorination reaction as the key step. On the other hand, the acceptor enone was prepared from tetrahydro-2-naphthol derivative by using a hypervalent iodine oxidation.220,221
Scheme 35.

Mal’s total synthesis of BE-23254 (387).219
The Hauser annulation as key step for the synthesis of angucyclinone-derived natural products was also applied in the synthesis of defucogilvocarcin M derivatives by Mal and his group (Scheme 36).222 A styryl sulfone 390 was used as an acceptor in the annulation reaction, which on reaction with cyanophthalide 391 provided the tetracyclic gilvocarcin skeleton 392. Radical initiated desulfonation followed by aromatization led to the synthesis of defucogilvocarcin derivative 393. The unusual starting styryl sulfone 390 was prepared in four steps from 2-methoxy-4-methyl aniline using a Heck-coupling with vinyl sulfone.
Scheme 36.

Synthesis of 1-O-methyl-defucogilvocarcin M (393) by Mal et al.222
Mal and coworkers223,224 also showed that, with the variation of the acceptor substrate, a similar Hauser annulation strategy could be utilized for the synthesis of the benzonaphthopyranone skeletons of the chartreusins and hayumicins. Coumarine derivative 395, generated from 5-hydroxy-2-methyl benzoate (394) using a Wittig reaction cyclization sequence, was allowed to react with the anion generated from the cyanophthalide 396 to produce the methyl chartarin derivative 397. Demethylation with HBr–AcOH completed the total synthesis of chartarin (398), the aglycon of the chartreusins (Scheme 37). An identical reaction, when performed with ortho-naphthoquinone monoketal 402, produced the pentacyclic compound 403 in one step. Final deketalization with acid yielded the O-methylhayumicinone (404), the O-methyl aglycon of the hayumicins (Scheme 38). The required ortho-naphthoquinone monoketal 402 was prepared in eight steps starting from the benzyl derivative of 6-hydroxy tetralone 399.
Scheme 37.

Mal’s total synthesis of chartarin (398).224
Scheme 38.

The synthesis of O-methylhayumicinone (404).224
Finally, Mal et al.225 also described the total synthesis of arnottin I (409), a gilvocarcin type natural product, using a variation of the conventional Hauser annulation (Scheme 39). In the synthesis, they used a 2-aryl acrylic acid ester (e.g., 408) as an acceptor, which underwent an unusual in situ decarboxylation process to give easy access to the benz[d]naphthopyranone skeleton. Thus, starting from phthalide 406 and acrylic ester 408, total synthesis of arnottin I (409) was achieved in a one-step reaction along with subtle amounts of the ester containing side product 410.
Scheme 39.

Mal’s total synthesis of arnottin I (409).225
Besides Hauser annulation, the directed remote metalation strategy has also been used for the construction of angucyclinone-derived natural product skeletons.226 A recent example is the total synthesis of defucogilvocarcin V (149), M (147), and E (148) and arnottin I (409) by Snieckus and James (Scheme 40).227 Starting from the biarylcarbamate 413, prepared by a palladium catalyzed Suzuki–Miyaura cross-coupling between iodonaphthalene 411 and aryl boronic acid 412, the benzonaphthopyranone skeleton of defucogilvocarcin was constructed using an LDA-induced directed remote metalation–carbamoyl migration followed by standard acid hydrolysis. Protection of 8-OH with triflic anhydride produced 414, in which the 8-triflate served as a handle for the introduction of vinyl, ethyl and methyl side chains through Stille, Suzuki and Negishi cross-coupling, respectively. A final removal of the isopropyl group provided defucogilvocarcin V (149), E (148), and M (147) in overall 15–16% yields. The synthesis of arnottin I (409) was also achieved following an identical reaction sequence (Scheme 41). Here, the Suzuki–Miyaura cross-coupling between iodocarbamate 415 and phenyl boronic acid 416 produced the carbamate-containing biaryl derivative 417. After installing the right choice of the protecting groups, compound 419 was subjected to a directed remote metalation–carbamoyl migration and acid hydrolysis reaction sequence to obtain the desired arnottin skeleton 420. Finally, methylenation of the catechol functionality completed the total synthesis of arnottin I (409).
Scheme 40.

Snieckus’s total synthesis of defucogilvocarcin M (147), E (148), and V (149).227
Scheme 41.

Snieckus’s total synthesis of arnottin I (409).227
An example of a different type of nucleophilic addition reaction was found in the enantioselective total syntheses of kinamycins C (208), F (203) and J (428) by Nicolaou and co-workers.228 They have used an intramolecular benzoin-type condensation for the construction of the tetracyclic ring system of the kinamycins (Scheme 42). The synthesis started with the Ullmann-type cross-coupling between a bromo-aldehyde 421 and iodo-enone 422. The starting materials 421 and 422 were prepared in five steps from bromojuglone and TBS-protected 4-hydroxycyclohexenone, respectively. The benzoin-type reaction of the coupling product 425 in presence of a Rovis catalyst (424) provided a 3 : 1 diastereomeric mixture of the tetracyclic hydroxy ketone 425. Temporary acylation followed by removal of the acetate by exposure to SmI2/MeOH and migration of the double bond in conjugation provided the fluorenone framework, which was subjected to a stereoselective allylic oxidation with SeO2. Removal of the TBS and acetonide groups, selective protection of secondary alcohols and Pd/C-mediated debenzylation provided 426. Protection of free hydroxyl group as TBS, introduction of azide functionality with tosyl hydrazine and protected hydroquinone oxidation with CAN lead to TBS-protected kinamycin C 427, from which the natural products were obtained using selective protection/deprotection techniques.
Scheme 42.

Nicolaou’s total synthesis of kinamycin C (208), F (203), and J (428).228
The final example of this section to be discussed is the total synthesis of landomycinone (241) by Roush and Neitz (Scheme 43),229 and the first total synthesis of the complex angucycline landomycin A (4) by Yu et al. (Scheme 44).3 In their synthesis, Roush and Neitz used a base mediated intramolecular nucleophilic addition-cyclization reaction of 431 to construct the tetracyclic angucyclinone skeleton 432. The starting naphthoquinone 431 was prepared using a Dötz benzannulation reaction between alkyne 430 and chromium carbene 429. The compound 430, in turn, was prepared from m-cresol using a 13-step reaction sequence, which included a Corey–Fuchs reaction to introduce the alkyne moiety. The chromium carbene 429 was prepared in three steps from a MOM-protected hydroquinone. The nucleophilic addition-cyclization reaction sequence was initiated by NaOEt and the unstable intermediate hydroquinone was oxidized to the quinone using aerial oxidation, to give 432. Removal of the protecting groups provided landomycinone (241), the stereochemistry of the C-6 chiral center was confirmed by the crystal structure data. Due to misinterpretation of the reported NMR data of the natural product the authors mistakenly concluded that the synthetic compound was not the actual natural product.
Scheme 43.

The synthesis of landomycinone (241) by Roush and Neitz.229
Scheme 44.

The total synthesis of landomycin A (4) by Yu and co-workers.3
Very recently Yu and co-workers3 reported the first total synthesis of hexasaccharidal natural product landomycin A (4), for which they have used a similar intramolecular nucleophilic–addition cyclization reaction sequence to construct the landomycinone aglycone. The synthesis started with the preparation of a naphthalene 435 using a Hauser annulation between cyanophthalide 433, synthesized in 8 steps from 2,5-dihydroxybenzoic acid, and aryl enone 434, obtained from 3,5-dimethylphenol. The annulation product 435 was converted to naphthoquinone 436 by stereoselective reduction of the C-6 carbonyl and oxidation of the hydroquinone. Intramolecular nucleophilic addition–cyclization followed by protection of 1-OH and deprotection of the 8-OH produced 437. The six sugar units of the natural product were introduced using a two step reaction sequence, a clever strategy to overcome the difficulty of glycosylating a naphthazarine OH group. The first glycosylation was achieved using a SN2-type substitution with in situ generated iodide of 438 to form the monoglycosylated derivative 439. Compound 438 was prepared in six steps starting from triacetyl d-glucal. For the second glycosylation step the trifluoroacetamide derivative of pentasaccharide 440 was prepared from d-glucal and d-xylose derivatives in 10 linear steps. Coupling of the monoglycoside 439 after deprotection of the TBS with pentasaccharide 440 produced the protected hexasaccharidal landomycin precursor 441. Global deprotection and reductive removal of the iodide functional groups furnished the total synthesis of landomycin A (4), in overall 63 steps and with 0.34% overall yield. The total synthesis of landomycin A is a highlight, since it completed by far the most complex angucycline group natural product to date, and put to rest any doubts regarding the natural landomycinone (241) skeleton, which might still linger in the synthetic community after Roush’s inaccurate conclusion (see above).229
5.3. Electrophilic addition strategies
The electrophilic addition or Friedel–Crafts type reactions have been extensively used for the synthesis of angucyclinone skeletons. One of the recent applications of the methodology in the synthesis of angucycline derived natural product was observed during the first enantioselective total synthesis of kinamycin C (208) by Lei and Porco (Scheme 45).230 The synthesis started with the successful construction of substituted naphthalene derivative 444 through Pd-catalyzed Stille cross-coupling between vinyl bromide 443 and MOM-protected naphthylstannane 442. The vinyl bromide 443 was synthesized from known starting material 2-bromo-3,6-dihydroxybenzaldehyde in 11 steps, while the naphthylstanane counterpart 442 was synthesized in 3 steps from bromojuglone. The carbonyl group of the coupling product 444 was subjected to diastereoselective reduction with Super-hydride and a subsequent epoxide-opening using Ti(OiPr)4/nBu4NOAc. Acetyl protection of the hydroxyl groups and TBS deprotection produced primary alcohol 445, which was oxidized to the corresponding carboxylic acid 446 in a two-step reaction sequence. An intramolecular Friedel–Crafts cyclization with trifluoroacetic acid constructed the cyclopentane ring to provide the tetracyclic core structure 447. A subsequent oxidation of hydroquinone and installation of diazo functionality completed the total synthesis of kinamycin C (208).
Scheme 45.

Porco’s total synthesis of kinamycin C (208).230
During their synthesis of prekinamycin (201), Kimura et al.231 also used a Friedel–Crafts acylation for the synthesis of the tetracyclic benzofluorenone ring system (Scheme 46). The key step of the synthesis was a palladium-catalyzed Suzuki cross-coupling between naphthyl boronic acid 448 and bromo aldehyde 449. The resulting naphthyl-fused benzaldehyde 450 was oxidized to acid 451. Subsequent acid chloride formation and an intramolecular Friedel–Crafts reaction in presence of AlCl3 produced the tetracyclic compound 452. Demethylation with BBr3, followed by hydrazone formation with TsNHNH2 and oxidation with Fetizone’s reagent afforded prekinamycin (201) in 7% overall yield.
Scheme 46.

The synthesis of prekinamaycin (201).231
5.4. Transition metal catalyzed cross-coupling strategies
In recent years, transition metal catalyzed C–C bond forming reactions have turned into a very useful tool for the synthesis of complex natural products, including angucyclines and their derivatives. A recent example is the synthesis of C-3′ desmethyl analogues of marmycin A (238). In their synthesis Ding et al.209 followed a modification of the synthetic strategy used by Brown and Thomson201 during the synthesis of tetrangulol (8), for the construction of core angucyclinone structure. The synthesis started with a AgNO3/(NH4)2S2O8 initiated decarboxylative radical alkylation of 453 with 454 to yield 455 (Scheme 47). But, unlike Brown and Thomson, the authors used a Pd(OAc)2-catalyzed intramolecular Heck-coupling to construct the B-ring of the angucyclinone skeleton. The reaction produced 8-methoxy-3-methylbenzo[a]anthraquinone 456 as the single regio-isomer, which on aromatization of the B-ring provided 457. The methoxy group was transformed into an amino functionality of 8-amino derivative 353 by demethylation followed by triflation and Pd-catalyzed transamination. A Lewis acid (InBr3) catalyzed simultaneous C- and N-glycosidation with di-O-acetyl-l-rhamnal 354 followed by hydrolysis of the acetate group produced 3-desmethylmarmycin A 459 and its diasteromer 460 in a 2 : 1 ratio. Although the authors were able to prepare five different derivatives of 3-desmethylmarmycin A by attaching different sugars, the extreme instability of the required sugar unit eluded the possibility of the total synthesis of marmycin A (238).
Scheme 47.

The synthesis C-3′ desmethyl analogues of marmycin A.209
A similar Michael addition–elimination and Heck coupling reaction sequence was also adopted by Herzon and co-workers232 for the construction of the diazofluorene moiety during the convergent and enantioselective total synthesis of kinamycin F (203) (Scheme 48). The synthesis started with the tris(diethylamino)sulfonium trimethyldifluorosilicate [TASF(Et)] mediated desilylative Michael addition between β-(trimethylsilylmethyl)-cyclohexenone 462 and naphthoquinone derivative 461. The product 463 was then subjected to Heck coupling by heating with a mixture of Pd(OAc)2, PPh3, and AgNO3 to afford the tetracyclic benzofluorene 464. Introduction of the diazo functionality with trifluoromethanesulfonyl azide, followed by α-oxidation, carbonyl reduction and global deprotection produced kinamycin F (203).
Scheme 48.

Herzon’s total synthesis of kinamycin F (203).232
In their total synthesis of ravidomycin V (123), Suzuki and his group233 used an intramolecular Pd-catalyzed cross-coupling reaction for the construction of benzonaphthopyranone ring skeleton (Scheme 49). The key starting material, the equatorially connected C-glycoside 465, was prepared in 8 steps from a previously known lactone. The presence of bulky TBDPS group in the C-3′ favored the equatorial disposition of the compound during the glycosylation reaction. A regioselective [4 + 2]-cycloaddition between 465 and 2-methoxyfuran (466) provided the naphthyl glycoside 467, after protection of the phenol with MOM-group. Introduction of an azide group at C-3′ through inversion of configuration and subsequent reduction and methylation to N,N-dimethylamino functionality produced 468. A Mitsunobu reaction between the phenol, produced by the removal of the MOM group, and the benzoic acid derivative 469 yielded 470. Finally, the tetracyclic benzonaphthopyranone skeleton 471 was constructed by a Pd-catalyzed intramolecular cross-coupling reaction. A series of protection/deprotection reactions and an elimination to introduce the vinyl side chain completed the total synthesis of ravidomycin V (123).
Scheme 49.

Suzuki’s total synthesis of ravidomycin V (123).233
Using the Buchwald protocol, Ishikawa and co-workers234 coupled o-bromobenzoate with α-tetralone for the construction of the tetracyclic core structure of arnottin I (409) (Scheme 50). A Pd-catalyzed arylation of tetralone 473 with 472 produced dihydro-arnottin 474, which on DDQ-promoted aromatization furnished arnottin I (409). More recently, the same group used an analogous path for the synthesis of dimethyl-jadomycin A 481.235 The synthesis started with a similar Pd-catalyzed cross coupling between bromo benzoate 475 and tetralone 476 to produce the bicyclic enol-lactone 477. 477 underwent an oxidative ring contraction to form the spirocyclic compound 478, which was converted to masked benzoquinone 479 using a bromination, hydroxylation and oxidation reaction sequence. Treatment of methyl isoleucinate opened up the spirocyclic ring to form a diastereomeric mixture of the conjugate addition product 480. Finally, reduction of the acid functionality to an aldehyde formed the oxazolo-phenanthridine ring skeleton of dimethyljadomycin A 481 (Scheme 50). The authors failed to complete the total synthesis of jadomycin A (95) because of the inability to remove the methoxy protecting group.
Scheme 50.

Ishikawa’s synthesis of arnottin I (409) and dimethyl jadomycin A 481.235
Madan and Cheng236 reported a Ni-catalyzed one-step Negishi type cross coupling cyclization reaction sequence for the regioselective synthesis of arnottin I (409) (Scheme 51). Thus Ni-catalyzed cross-coupling between a Zn-complex of o-iodobenzoate 483 and oxabenzonorbornadiene 482 followed by in situ ring opening and cyclization through transesterification produced the tetracyclic benzonaphthopyranone 484. Removal of the silyl protecting groups and methenylation using CH2Br2 completed a six step total synthesis of 409 with 21% overall yield.
Scheme 51.

Cheng’s total synthesis of arnottin I (409).236
5.5. Intramolecular cyclization reaction strategies
Intramolecular [2 + 2 + 2] or [4 + 2]-cycloaddition reactions were also used for the one-step synthesis of the core structures of angucyclinones and angucyclinone-derived natural product skeletons. The reactions are generally catalyzed by transition metals like Co or Au, although thermal electrocyclization reactions are also reported.199,237
In this context, an example to be discussed is the stereoselective total synthesis of (—)-tetrangomycin (9) by Groth et al. (Scheme 52).238 The key step of the synthesis was a cobalt mediated [2 + 2 + 2]-cycloaddition of a chiral triyne 488, which constructed the angular tetracyclic skeleton 489 in one step. The required triyne was prepared by the addition of lithiated octadiyne 486, obtained from geraniol in 10 steps, to the substituted benzaldehyde 485 followed by K2CO3 mediated TMS removal and TBS protection of the newly generated secondary alcohol. Oxidation of 489 by 1 : 2 mixture of Ag(Py)2MnO4 and silica gel followed by deprotection of the MOM protection group afforded the angucyclinone compound 490. The total synthesis of 9 was completed by TBS deprotection and regioselective photooxidation at C-1. Using an identical synthetic methodology the authors were also able to synthesize (+)-rubiginone B2 (344).
Scheme 52.

Groth’s total synthesis of (−)-tetrangomycin (9).238
Roulland and co-workers239 used a Rh(I)-catalyzed intra-molecular [2 + 2 + 2]-cycloaddition to construct the B and C rings of the landomycinone (241) skeleton. The final D ring was made using a 2nd generation Grubb’s catalyzed ring closing metathesis reaction. The synthesis started with propargyl alcohol 491 prepared in nine steps from commercially available starting material (Scheme 53). A Mitsunobu coupling with propargylic acid 492 produced triyne 493, which in the presence of Wilkinson’s catalyst underwent a [2 + 2 + 2]-cycloaddition to construct the tetracyclic lactone 494. Selective addition of vinyllithium to the lactone formed the acid sensitive intermediate diene 495, which on treatment with a 2nd generation Grubbs catalyst produced a mixture of the tetracyclic angucycline compounds 496 and 497. Unfortunately, the authors could not oxidize the C ring to a quinone, which precluded the possibility of a total synthesis of landomycinone (241).
Scheme 53.

Roulland’s attempt toward the total synthesis of landomycinone.239
Sato et al.240 used an Au(III) mediated [4 + 2]-cycloaddition strategy for the stereoselective total synthesis of (+)-ochromycinone (343) and (+)-rubiginone B2 (344) (Scheme 54). The key starting material alkynyl enynal 500 was prepared by the Stille coupling between a chiral alkynyl stannane (499) and a bromo substituted naphthalene aldehyde derivative (498), followed by removal of TMS. AuCl3-mediated intramolecular benzannulation produced the tetracyclic compound 502, which could be transformed to rubiginone B2 (344) via oxidation with CAN. Enantiopure (ee 99%) (+)-ochromycinone (343) was obtained from 344 by BCl3 mediated demethylation.
Scheme 54.

Asao’s total synthesis of (+)-ochromycinone (343) and (+)-rubiginone B2 (344).240
Recently, O’Doherty and his group84 used a regioselective 6π-electrocylization reaction to construct the phenanthridine ring skeleton of the jadomycins during their total synthesis of jadomycin A (95) (Scheme 55). The synthesis started with a Pd-catalyzed Stille cross coupling between aryl stannane 505 and bromojuglone 503 to produce biaryl compound 506. Removal of the MOM and acetal protecting groups yielded aldehyde 508, which on exposure with protected isoleucine 509 produced imine intermediate 510. In situ electrocyclization in toluene at room temperature produced hemiaminal 512, possibly via water addition and oxidation of the unstable intermediate 511. Final cyclization and benzyl deprotection by treatment with neat TFA resulted the total synthesis of jadomycin A (95) in 17% overall yield. The author’s attempt to complete the total synthesis of jadomycin B (96), the natural product with a digitoxose moiety in 8-OH position, failed because of the extreme sensitivity of the glycosylated product towards the mild acidic conditions required for the glycosylation reaction. To prove the high instability of the digitoxosyl sugar containing compound and to have an analogue for structure–activity relationship studies, compound 515 was synthesized, which is the carbasugar analogue of jadomycin B, following an identical reaction sequence starting from quinone aldehyde 514. The carbasugar containing compound 514 was prepared by connecting cyclohexane 513 (carba-analogue of digitoxose) to 8-OH of 507 using a Mitsunobu reaction.
Scheme 55.

O’Doherty’s total synthesis of jadomycin A (95) and of carbasugar analogue 515 of jadomycin B (96).84
A different type of thermal 6π-electrocyclization reaction was applied in the work of Suzuki et al.241 during a short total synthesis of defucogilvocarcin M (147). The group used a thermal benzocyclobutenone ring opening followed by an electrocyclic ring closure reaction sequence for an efficient synthesis of the 2-phenylnaphthalene skeleton of 514 (Scheme 56). The key starting material 513 was prepared by connecting a benzyl protected benzocyclobutenone 512 and a bromostyrene 509 by treatment with t-BuLi. The benzocyclobutenone 512 was known to be formed from 510 and 511 by a benzyne-alkene [2 + 2]-cycloaddition. The bromostyrene building block 509 was prepared from benzoic ester 507 in 6 steps. The one-step cyclobutenone ring opening electrocyclization was achieved by refluxing 513 in toluene. Subsequent acetylation of the phenolic hydroxyl group produced 2-phenylnaphthalene 514. Finally, the lactone ring of the natural product was introduced by an oxidation of the liberated aldehyde to the acid, followed by methanolysis and acidification.
Scheme 56.

Suzuki’s total synthesis of defucogilvocarcin M (147).241
Suzuki and coworkers242 also applied an identical cyclobutenone ring opening electrocyclization strategy (Scheme 56) in their total synthesis of the angucyclinone natural product PD-116740 (272, Scheme 57). The synthesis started with vinylstannane 525, which was prepared by a Pd-catalyzed hydrostannylation of arylalkyne 524. The E-geometry of its double bond was necessary to hinder an unwanted [1,7]-H shift during the electrocyclic reaction. MeLi-mediated coupling of 525 with cyclobutenone 526 produced 527, which on refluxing in toluene underwent 6π-electrocyclization to produce 2-phenyl naphthalene 528. Deprotection of the hydroxymethyl side chain followed by Swern oxidation gave dialdehyde 529. A SmI2-mediated pinacol reaction of 529 constructed the remaining B-ring of the angucyclinone, and produced mostly the trans- isomer of 530 (trans/cis = 10 : 1). To prove the absolute configuration of the natural product, the racemic 530 was reacted with (—)-camphanic chloride (531) to yield diastereomeric 532. Separation of the diastereomers and removal of camphanoyl group yielded both (S,S)- and the (R,R)-enantiomers of 533. Both isomers were subsequently converted to the natural product structure 272 using a 5-step reaction sequence, which included triflation of C-3 hydroxyl group followed by Pd-catalyzed carbonyl insertion, reduction of the formed carbonyl and standard deprotection. Comparison of the optical rotations of both synthetic compounds with that of the natural product 272 confirmed that the absolute stereochemistry of the natural product, being S-configured at both C-5 and C-6.
Scheme 57.

Suzuki’s total synthesis of PD116740 (272).242
6. Conclusions
The angucycline group of natural products has grown significantly during the past 14 years, and yielded interesting new molecules, with unsual amino sugars and striking biological activites. Some of these were isolated from marine organisms, but the majority of new natural products of this class were derived from terrestrial strains. Most significantly, modern synthetic methodology was applied to synthesize artificial angucyclinone analogues not existing in nature and to improve yields of classical angucyclinones, such as ochromycinone, rubiginone or tetrangomycin. Compared to the syntheses described in our previous review, much more complex molecules, such as kinamycins, ravidomycin, landomycin A or the carba analogue of jadomycin B were achieved. Extensive biosynthetic investigations not only revealed previously not recognized or not proven relationships between typical angucycline and various groups of natural products that were now shown to be angucyclinone-derived, but also paved the way for the exploitation of biosynthetic methodology, such as combinatorial biosynthesis, to yield many new analogues of angucyclinones and angucyclines. The studies also revealed intriguing enzymology, although this is just the tip of an iceberg.
Biographies

Madan K: Kharel
Dr. Madan Kharel was born in Jhapa, Nepal. After completing a Masters from the Tribhuvan University, Nepal, he joined the department of chemistry, Sun Moon University, South Korea, and received a PhD degree under the supervision of Prof. Jae Kyung Sohng. In 2004, he joined Prof. Jürgen Rohr’s lab where he completed his second PhD in the department of Pharmaceutical Sciences, University of Kentucky, USA. During doctoral and post-doctoral research work (2000–2010), he studied biosynthesis of aminoglycoside antibiotics and angucycline anticancer drugs. Currently, he is an Assistant Professor of Medicinal Chemistry in Midway College School of Pharmacy, KY, USA. His research interests include elucidation of biosynthetic pathways, enzymatic synthesis of natural products and isolation of new bioactive microbial natural products.

Pallab Pahari
Dr. Pallab Pahari was born in Midnapur (W), a district of West Bengal, India. He obtained his PhD from the Indian Institute of Technology Kharagpur, India, in 2008 under the supervision of Prof. Dipakranjan Mal with a thesis on the synthetic studies of angucycline and isobenzofuranone natural products. Since then, he is continuing his research at Prof. Jürgen Rohr’s laboratory, University of Kentucky, USA, as a post-doctoral fellow. He is currently working on elucidation of biosynthetic pathways of angucycline natural products and nucleoside antibiotics. His research interests also include isolation, characterization, synthesis and structure–activity relationship studies of furanocoumarine type natural products.

Micah D: Shepherd
Dr. Micah Shepherd was born in Stanton, Kentucky, USA. He studied Biology at the University of Kentucky (Lexington, KY), where he graduated with a BS degree in 2006. He went on to join the graduate program of Pharmaceutical Sciences at the University of Kentucky and the research group of Prof. Jürgen Rohr in early 2007. His PhD research applied combinatorial biosynthetic techniques to generate glycodiversity among gilvocarcin-like anticancer antibiotics through deoxysugar pathway and glycosyltransferase engineering as well as post-glycosylation modification. After completion of the PhD program in 2011, he joined the research and development team at zuChem (Peoria, IL) where he continues to work in the field of glycobiology.

Nidhi Tibrewal
Nidhi Tibrewal is a fourth year graduate student in the lab of Prof. Jürgen Rohr. She was born and brought up in Delhi, India and completed her undergrad in Chemistry from Delhi University. After completing her Masters in Pharmaceutical Chemistry, she worked in industrial labs before she joined the graduate program of the College of Pharmacy at the University of Kentucky in 2008. Her interests are organic synthesis and enzymology. She is currently investigating the gilvocarcin V biosynthetic pathway.

S: Eric Nybo
Eric Nybo was born in Nashville, Tennessee. He studied Biology and Classics at Transylvania University in Lexington, Kentucky where he received his Baccalaureate degree in 2006. He was accepted as a graduate student at the University of Kentucky in the Department of Pharmaceutical Sciences in the fall semester of 2006, where he soon joined the lab of Prof. Jürgen Rohr. In his PhD work, he focused on the study of the biosynthesis of chrysomycin, and he also focused on the use of combinatorial biosynthesis to diversify deoxysugar substituents of type II polyketides. His PhD graduation is anticipated for fall 2011.

Khaled A: Shaaban
Dr. Khaled A. Shaaban was born in Karam, El-Mansoura, Dakahlia (Egypt) in 1978. He received his MSc and PhD in Chemistry at the University of Göttingen, Germany, in 2005 and 2009, respectively under the supervision of Prof. Hartmut Laatsch. Throughout his PhD studies, Dr. Shaaban isolated more than 60 new compounds from Streptomyces sp., macroalgae, marine coral, and medicinal plants, many of these with novel skeletons. He won five awards for his contributions to natural products chemistry. In April 2009, Dr. Shaaban joined the College of Pharmacy at University of Kentucky (USA) as a postdoctoral fellow in the group of Prof. Jürgen Rohr. He is currently working on isolation, structure elucidation, and biosynthesis of new pharmaceutical drugs from Streptomycetes and plant extracts. His research interests include isolation of bacterial strains, prescreening, and searching for new bioactive compounds.
7 References
- 1.Rohr J and Thiericke R, Nat. Prod. Rep, 1992, 9, 103–137. [DOI] [PubMed] [Google Scholar]
- 2.Krohn K and Rohr J, Top. Curr. Chem, 1997, 188, 127–195. [Google Scholar]
- 3.Yang X, Fu B and Yu B, J. Am. Chem. Soc, 2011, 133, 12433–12435. [DOI] [PubMed] [Google Scholar]
- 4.Rohr J and Hertweck C, in Comprehensive Natural Products II – Chemistry and Biology, ed. Mander L and Liu H.-w., Elsevier, Oxford, 2010, pp. 227–303. [Google Scholar]
- 5.Rohr J, Beale JM and Floss HG, J. Antibiot, 1989, 42, 1151–1157. [DOI] [PubMed] [Google Scholar]
- 6.Imamura N, Kakinuma K, Ikekawa N, Tanaka H and Omura S, J. Antibiot, 1982, 35, 602–608. [DOI] [PubMed] [Google Scholar]
- 7.Weber S, Zolke C, Rohr J and Beale JM, J. Org. Chem, 1994, 59, 4211–4214. [Google Scholar]
- 8.Carter GT, Fantini AA, James JC, Borders DB and White RJ, J. Antibiot, 1985, 38, 242–248. [DOI] [PubMed] [Google Scholar]
- 9.Carter GT, Fantini AA, James JC, Borders DB and White RJ, Tetrahedron Lett., 1984, 25, 255–258. [Google Scholar]
- 10.Takahashi K and Tomita F, J. Antibiot, 1983, 36, 1531–1535. [DOI] [PubMed] [Google Scholar]
- 11.Gould SJ, Chem. Rev, 1997, 97, 2499–2510. [DOI] [PubMed] [Google Scholar]
- 12.Gould SJ, Cheng XC and Halley KA, J. Am. Chem. Soc, 1992, 114, 10066–10068. [Google Scholar]
- 13.Sasaki E, Ogasawara Y and Liu HW, J. Am. Chem. Soc, 2010, 132, 7405–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henkel T, Rohr J, Beale JM and Schwenen L, J. Antibiot, 1990, 43, 492–503. [DOI] [PubMed] [Google Scholar]
- 15.Zhu LL, Ostash B, Rix U, Nur-e-Alam M, Mayers A, Luzhetskyy A, Mendez C, Salas JA, Bechthold A, Fedorenko V and Rohr J, J. Org. Chem, 2005, 70, 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westrich L, Domann S, Faust B, Bedford D, Hopwood DA and Bechthold A, FEMS Microbiol. Lett, 1999, 170, 381–387. [DOI] [PubMed] [Google Scholar]
- 17.von Mülert U, Luzhetskyy A, Hofmann C, Mayer A and Bechthold A, FEMS Microbiol. Lett, 2004, 230, 91–97. [DOI] [PubMed] [Google Scholar]
- 18.Ostash B, Rebets Y, Yuskevich V, Luzhetskyy A, Tkachenko V and Fedorenko V, Folia Microbiol., 2003, 48, 484–488. [DOI] [PubMed] [Google Scholar]
- 19.Palmu K, Ishida K, Mantsälä P, Hertweck C and Metsä-Ketelä M, ChemBioChem, 2007, 8, 1577–1584. [DOI] [PubMed] [Google Scholar]
- 20.Faust B, Hoffmeister D, Weitnauer G, Westrich L, Haag S, Schneider P, Decker H, Künzel E, Rohr J and Bechthold A, Microbiology, 2000, 146, 147–154. [DOI] [PubMed] [Google Scholar]
- 21.Rix U, Remsing LL, Hoffmeister D, Bechthold A and Rohr J, ChemBioChem, 2003, 4, 109–111. [DOI] [PubMed] [Google Scholar]
- 22.Luzhetskyy A, Zhu L, Gibson M, Fedoryshyn M, Dürr C, Hofmann C, Hoffmeister D, Ostash B, Mattingly C, Adams V, Fedorenko V, Rohr J and Bechthold A, ChemBioChem, 2005, 6, 675–678. [DOI] [PubMed] [Google Scholar]
- 23.Ostash B, Rix U, Rix LL, Liu T, Lombó F, Luzhetskyy A, Gromyko O, Wang C, Braña AF, Méndez C, Salas JA, Fedorenko V and Rohr J, Chem. Biol, 2004, 11, 547–555. [DOI] [PubMed] [Google Scholar]
- 24.Baig I, Kharel M, Kobylyanskyy A, Zhu LL, Rebets Y, Ostash B, Luzhetskyy A, Bechthold A, Fedorenko VA and Rohr J, Angew. Chem., Int. Ed, 2006, 45, 7842–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erb A, Luzhetskyy A, Hardter U and Bechthold A, ChemBioChem, 2009, 10, 1392–1401. [DOI] [PubMed] [Google Scholar]
- 26.Mayer A, Taguchi T, Linnenbrink A, Hofmann C, Luzhetskyy A and Bechthold A, ChemBioChem, 2005, 6, 2312–2315. [DOI] [PubMed] [Google Scholar]
- 27.Trefzer A, Fischer C, Stockert S, Westrich L, Künzel E, Girreser U, Rohr J and Bechthold A, Chem. Biol, 2001, 8, 1239–1252. [DOI] [PubMed] [Google Scholar]
- 28.Hoffmeister D, Weber M, Dräger G, Ichinose K, Durr C and Bechthold A, ChemBioChem, 2004, 5, 369–371. [DOI] [PubMed] [Google Scholar]
- 29.Luzhetskyy A, Fedoryshyn M, Durr C, Taguchi T, Novikov V and Bechthold A, Chem. Biol, 2005, 12, 725–729. [DOI] [PubMed] [Google Scholar]
- 30.Erb A, Krauth C, Luzhetskyy A and Bechthold A, Appl. Microbiol. Biotechnol, 2009, 83, 1067–1076. [DOI] [PubMed] [Google Scholar]
- 31.Krauth C, Fedoryshyn M, Schleberger C, Luzhetskyy A and Bechthold, Chem. Biol, 2009, 16, 28–35. [DOI] [PubMed] [Google Scholar]
- 32.Luzhetskyy A, Taguchi T, Fedoryshyn M, Dürr C, Wohlert SE, Novikov V and Bechthold A, ChemBioChem, 2005, 6, 1406–1410. [DOI] [PubMed] [Google Scholar]
- 33.Rebets Y, Ostash B, Luzhetskyy A, Hoffmeister D, Brana A, Mendez C, Salas JA, Bechthold A and Fedorenko V, FEMS Microbiol. Lett, 2003, 222, 149–153. [DOI] [PubMed] [Google Scholar]
- 34.Luzhetskyy A, Liu T, Fedoryshyn M, Ostash B, Fedorenko V, Rohr J and Bechthold A, ChemBioChem, 2004, 5, 1567–1570. [DOI] [PubMed] [Google Scholar]
- 35.Zhu L, Luzhetskyy A, Luzhetska M, Mattingly C, Adams V, Bechthold A and Rohr J, ChemBioChem, 2007, 8, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rebets YV, Ostash BO, Fukuhara M, Nakamura T and Fedorenko VO, FEMS Microbiol. Lett, 2006, 256, 30–37. [DOI] [PubMed] [Google Scholar]
- 37.Rebets Y, Ostash B, Luzhetskyy A, Kushnir S, Fukuhara M, Bechthold A, Nashimoto M, Nakamura T and Fedorenko V, Microbiology, 2005, 151, 281–290. [DOI] [PubMed] [Google Scholar]
- 38.Dutko L, Rebets Y, Ostash B, Luzhetskyy A, Bechthold A, Nakamura T and Fedorenko V, FEMS Microbiol. Lett, 2006, 255, 280–285. [DOI] [PubMed] [Google Scholar]
- 39.Ostash I, Ostash B, Luzhetskyy A, Bechthold A, Walker S and Fedorenko V, FEMS Microbiol. Lett, 2008, 285, 195–202. [DOI] [PubMed] [Google Scholar]
- 40.Ostash B, Ostash I, Zhu L, Kharel MK, Luzhetskyy A, Bechthold A, Walker S, Rohr J and Fedorenko V, Russ. J. Genet, 2010, 46, 530–535. [Google Scholar]
- 41.Ostash I, Rebets Y, Ostash B, Kobylyanskyy A, Myronovskyy M, Nakamura T, Walker S and Fedorenko V, Arch. Microbiol, 2008, 190, 105–109. [DOI] [PubMed] [Google Scholar]
- 42.Drautz H, Zahner H, Rohr J and Zeeck A, J. Antibiot, 1986, 39, 1657–1669. [DOI] [PubMed] [Google Scholar]
- 43.Rohr J and Zeeck A, J. Antibiot, 1987, 40, 459–467. [DOI] [PubMed] [Google Scholar]
- 44.Rohr J, Zeeck A and Floss HG, J. Antibiot, 1988, 41, 126–129. [DOI] [PubMed] [Google Scholar]
- 45.Rohr J, J. Antibiot, 1989, 42, 1482–1488. [DOI] [PubMed] [Google Scholar]
- 46.Henkel T, Ciesiolka T, Rohr J and Zeeck A, J. Antibiot, 1989, 42, 299–311. [DOI] [PubMed] [Google Scholar]
- 47.Rohr J, J. Chem. Soc., Chem. Commun, 1989, 492–493. [Google Scholar]
- 48.Rohr J, J. Chem. Soc., Chem. Commun, 1990, 113–114. [Google Scholar]
- 49.Rohr J, Angew. Chem., Int. Ed. Engl, 1990, 29, 1051–1053. [Google Scholar]
- 50.Decker H and Haag S, J. Bacteriol, 1995, 177, 6126–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hoffmeister D, Ichinose K, Domann S, Faust B, Trefzer A, Dräger G, Kirschning A, Fischer C, Künzel E, Bearden D, Rohr J and Bechthold A, Chem. Biol, 2000, 7, 821–831. [DOI] [PubMed] [Google Scholar]
- 52.Künzel E, Faust B, Oelkers C, Weissbach U, Bearden DW, Weitnauer G, Westrich L, Bechthold A and Rohr J, J. Am. Chem. Soc, 1999, 121, 11058–11062. [Google Scholar]
- 53.Trefzer A, Hoffmeister D, Kunzel E, Stockert S, Weitnauer G, Westrich L, Rix U, Fuchser J, Bindseil KU, Rohr J and Bechthold A, Chem. Biol, 2000, 7, 133–142. [DOI] [PubMed] [Google Scholar]
- 54.Udvarnoki G, Henkel T, Machinek R and Rohr J, J. Org. Chem, 1992, 57, 1274–1276. [Google Scholar]
- 55.Kallio P, Liu ZL, Mantsälä P, Niemi J and Metsä-Ketelä M, Chem. Biol, 2008, 15, 157–166. [DOI] [PubMed] [Google Scholar]
- 56.Fedoryshyn M, Nur-e-Alam M, Zhu L, Luzhetskyy A, Rohr J and Bechthold A, J. Biotechnol, 2007, 130, 32–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hoffmeister D, Draäger G, Ichinose K, Rohr J and Bechthold A, J. Am. Chem. Soc, 2003, 125, 4678–4679. [DOI] [PubMed] [Google Scholar]
- 58.Perez M, Lombó F, Zhu LL, Gibson M, Braña AF, Rohr R, Salas JA and Méndez C, Chem. Commun, 2005, 1604–1606. [DOI] [PubMed] [Google Scholar]
- 59.Rodriguez L, Aguirrezabalaga I, Allende N, Braña AF, Méndez C and Salas JA, Chem. Biol, 2002, 9, 721–729. [DOI] [PubMed] [Google Scholar]
- 60.Trefzer A, Blanco G, Remsing L, Künzel E, Rix U, Lipata F, Braña AF, Méndez C, Rohr J, Bechthold A and Salas JA, J. Am. Chem. Soc, 2002, 124, 6056–6062. [DOI] [PubMed] [Google Scholar]
- 61.Dürr C, Hoffmeister D, Wohlert SE, Ichinose K, Weber M, von Mülert U, Thorson JS and Bechthold A, Angew. Chem., Int. Ed, 2004, 43, 2962–2965. [DOI] [PubMed] [Google Scholar]
- 62.Mittler M, Bechthold A and Schulz GE, J. Mol. Biol, 2007, 372, 67–76. [DOI] [PubMed] [Google Scholar]
- 63.Hoffmeister D, Wilkinson B, Foster G, Sidebottom PJ, Ichinose K and Bechthold A, Chem. Biol, 2002, 9, 287–295. [DOI] [PubMed] [Google Scholar]
- 64.Ayer SW, Mcinnes AG, Thibault P, Walter JA, Doull JL, Parnell T and Vining LC, Tetrahedron Lett., 1991, 32, 6301–6304. [Google Scholar]
- 65.Doull JL, Ayer SW, Singh AK and Thibault P, J. Antibiot, 1993, 46, 869–871. [DOI] [PubMed] [Google Scholar]
- 66.Doull JL, Singh AK, Hoare M and Ayer SW, J. Ind. Microbiol, 1994, 13, 120–125. [DOI] [PubMed] [Google Scholar]
- 67.Han L, Yang KQ, Ramalingam E, Mosher RH and Vining LC, Microbiology, 1994, 140, 3379–3389. [DOI] [PubMed] [Google Scholar]
- 68.Han L, Yang K, Kulowski K, Wendt-Pienkowski E, Hutchinson CR and Vining LC, Microbiology, 2000, 146(Pt 4), 903–910. [DOI] [PubMed] [Google Scholar]
- 69.Wang L, McVey J and Vining LC, Microbiology, 2001, 147, 1535–1545. [DOI] [PubMed] [Google Scholar]
- 70.Rix U, Wang CC, Chen YH, Lipata FM, Rix LLR, Greenwell LM, Vining LC, Yang KQ and Rohr J, ChemBioChem, 2005, 6, 838–835. [DOI] [PubMed] [Google Scholar]
- 71.Chen YH, Wang CC, Greenwell L, Rix U, Hoffmeister D, Vining LC, Rohr J and Yang KQ, J. Biol. Chem, 2005, 280, 22508–22514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kulowski K, Wendt-Pienkowski E, Han L, Yang KQ, Vining LC and Hutchinson CR, J. Am. Chem. Soc, 1999, 121, 1786–1794. [Google Scholar]
- 73.Kharel MK, Pahari P, Lian H and Rohr J, Org. Lett, 2010, 12, 2814–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang KQ, Han L, Ayer SW and Vining LC, Microbiology, 1996, 142, 123–132. [DOI] [PubMed] [Google Scholar]
- 75.Chen Y, Fan K, He Y, Xu X, Peng Y, Yu T, Jia C and Yang K, ChemBioChem, 2010, 11, 1055–1060. [DOI] [PubMed] [Google Scholar]
- 76.Metsä-Ketelä M, Palmu K, Kunnari T, Ylihonko K and Mäntsälä P, Antimicrob. Agents Chemother, 2003, 47, 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Metsä-Ketelä M, Ylihonko K and Mäntsälä P, J. Antibiot, 2004, 57, 502–510. [DOI] [PubMed] [Google Scholar]
- 78.Kharel MK, Zhu LL, Liu T and Rohr J, J. Am. Chem. Soc, 2007, 129, 3780–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rix U, Zheng JT, Rix LLR, Greenwell L, Yang KQ and Rohr J, J. Am. Chem. Soc, 2004, 126, 4496–4497. [DOI] [PubMed] [Google Scholar]
- 80.Jakeman DL, Farrell S, Young W, Doucet RJ and Timmons SC, Bioorg. Med. Chem. Lett, 2005, 15, 1447–1449. [DOI] [PubMed] [Google Scholar]
- 81.Jakeman DL, Graham CL and Reid TR, Bioorg. Med. Chem. Lett, 2005, 15, 5280–5283. [DOI] [PubMed] [Google Scholar]
- 82.Syvitski RT, Borissow CN, Graham CL and Jakeman DL, Org. Lett, 2006, 8, 697–700. [DOI] [PubMed] [Google Scholar]
- 83.Jakeman DL, Borissow CN, Graham CL, Syvitski RT, Reid TR and Blay J, ChemBioChem, 2007, 8, 1198–1203. [DOI] [PubMed] [Google Scholar]
- 84.Shan MD, Sharif EU and O’Doherty GA, Angew. Chem., Int. Ed, 2010, 49, 9492–9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jakeman DL, Borissow CN, Graham CL, Timmons SC, Reid TR and Syvitski RT, Chem. Commun, 2006, 3738–3740. [DOI] [PubMed] [Google Scholar]
- 86.Wang L, White RL and Vining LC, Microbiology, 2002, 148, 1091–1103. [DOI] [PubMed] [Google Scholar]
- 87.Wang L and Vining LC, Microbiology, 2003, 149, 1991–2004. [DOI] [PubMed] [Google Scholar]
- 88.Kato JY, Funa N, Watanabe H, Ohnishi Y and Horinouchi S, Proc. Natl. Acad. Sci. U. S. A, 2007, 104, 2378–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang K, Han L and Vining LC, J. Bacteriol, 1995, 177, 6111–6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang K, Han L, He J, Wang L and Vining LC, Gene, 2001, 279, 165–173. [DOI] [PubMed] [Google Scholar]
- 91.Xu G, Wang J, Wang L, Tian X, Yang H, Fan K, Yang K and Tan H, J. Biol. Chem, 2010, 285, 27440–27448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang L, Tian X, Wang J, Yang H, Fan K, Xu G, Yang K and Tan H, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 8617–8622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hatano K, Higashide E, Shibata M, Kameda Y, Horii S and Mizuno K, Agric. Biol. Chem, 1980, 44, 1157–1163. [Google Scholar]
- 94.Horii S, Fukase H, Mizuta E, Hatano K and Mizuno K, Chem. Pharm. Bull, 1980, 28, 3601–3611. [Google Scholar]
- 95.Jain TC, Simolike GC and Jackman LM, Tetrahedron, 1983, 39, 599–605. [Google Scholar]
- 96.Nakano H, Matsuda Y, Ito K, Ohkubo S, Morimoto M and Tomita F, J. Antibiot, 1981, 34, 266–270. [DOI] [PubMed] [Google Scholar]
- 97.Takahashi KY, Tomita M,F and Shirahata K, J. Antibiot, 1981, 43, 271–275. [DOI] [PubMed] [Google Scholar]
- 98.Balitz DM, O’Herron FA, Bush J, Vyas DM, Nettleton DE, Grulich RE, Bradner WT, Doyle TW, Arnold E and Clardy J, J. Antibiot, 1981, 34, 1544–1555. [DOI] [PubMed] [Google Scholar]
- 99.Fischer C, Lipata F and Rohr J, J. Am. Chem. Soc, 2003, 125, 7818–7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu T, Fischer C, Beninga C and Rohr J, J. Am. Chem. Soc, 2004, 126, 12262–12263. [DOI] [PubMed] [Google Scholar]
- 101.Keyes RF and Kingston DGI, J. Org. Chem, 1989, 54, 6127–6129. [Google Scholar]
- 102.Shepherd MD, Kharel MK, Zhu LL, van Lanen SG and Rohr J, Org. Biomol. Chem, 2010, 8, 3851–3856. [DOI] [PubMed] [Google Scholar]
- 103.Liu T, Kharel MK, Fischer C, McCormick A and Rohr J, ChemBioChem, 2006, 7, 1070–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Guengerich FP, Chem. Res. Toxicol, 2001, 14, 611–650. [DOI] [PubMed] [Google Scholar]
- 105.Tibrewal N, Pahari P, Kharel M, O’Doherty G and Rohr J, 2011, unpublished. [Google Scholar]
- 106.Kharel MK, Pahari P, Lian H and Rohr J, ChemBioChem, 2009, 10, 1305–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu T, Kharel MK, Zhu L, Bright S and Rohr J, ChemBioChem, 2009, 10, 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Q, Ding P, Perepelov AV, Xu YL, Wang Y, Knirel YA, Wang L and Feng L, Mol. Microbiol, 2008, 70, 1358–1367. [DOI] [PubMed] [Google Scholar]
- 109.Thibodeaux CJ, Melancon CE and Liu HW, Angew. Chem., Int. Ed, 2008, 47, 9814–9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shepherd MD, Liu T, Méndez C, Salas JA and Rohr J, Appl. Environ. Microbiol, 2011, 77, 435–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rodriguez L, Oelkers C, Aguirrezabalaga I, Braña AF, Rohr J, Méndez C and Salas JA, J. Mol. Microbiol. Biotechnol, 2000, 2, 271–276. [PubMed] [Google Scholar]
- 112.Li YQ, Huang XS, Ishida K, Maier A, Kelter G, Jiang Y, Peschel G, Menzel KD, Li MG, Wen ML, Xu LH, Grabley S, Fiebig HH, Jiang CL, Hertweck C and Sattler I, Org. Biomol. Chem, 2008, 6, 3601–3605. [DOI] [PubMed] [Google Scholar]
- 113.Kharel MK, Nybo SE, Shepherd MD and Rohr J, ChemBioChem, 2010, 11, 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Meadows ES and Khosla C, Biochemistry, 2001, 40, 14855–14861. [DOI] [PubMed] [Google Scholar]
- 115.Kharel MK, Lian H and Rohr J, Org. Biomol. Chem, 2011, 9, 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Narita T, Matsumoto M, Mogi K, Kukita K, Kawahara R and Nakashima T, J. Antibiot, 1989, 42, 347–356. [DOI] [PubMed] [Google Scholar]
- 117.Méndez C, Künzel E, Lipata F, Lombo F, Cotham W, Walla M, Bearden DW, Braña AF, Salas JA and Rohr J, J. Nat. Prod, 2002, 65, 779–782. [DOI] [PubMed] [Google Scholar]
- 118.Lombo F, Braña AF, Salas JA and Méndez C, ChemBioChem, 2004, 5, 1181–1187. [DOI] [PubMed] [Google Scholar]
- 119.Lombo F, Abdelfattah MS, Braña AF, Salas JA, Rohr J and Méndez C, ChemBioChem, 2009, 10, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Koskiniemi H, Metsä-Ketelä M, Dobritzsch D, Kallio P, Korhonen H, Mäntsälä P, Schneider G and Niemi J, J. Mol. Biol, 2007, 372, 633–648. [DOI] [PubMed] [Google Scholar]
- 121.Kallio P, Liu Z, Mäntsälä P, Niemi J and Metsä-Ketelä M, J. Mol. Biol, 2008, 375, 1212–1221. [DOI] [PubMed] [Google Scholar]
- 122.Tsuchida T, Iinuma H, Kinoshita N, Ikeda T, Sawa R, Takahashi Y, Naganawa H, Sawa T, Hamada M and Takeuchi T, J. Antibiot, 1993, 46, 1772–1774. [DOI] [PubMed] [Google Scholar]
- 123.Tsuchida T, Sawa R, Takahashi Y, Iinuma H, Sawa T, Naganawa H and Takeuchi T, J. Antibiot, 1995, 48, 1148–1152. [DOI] [PubMed] [Google Scholar]
- 124.Ogasawara Y and Liu HW, J. Am. Chem. Soc, 2009, 131, 18066–18068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sasaki E and Liu HW, J. Am. Chem. Soc, 2010, 132, 15544–15546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Omura S, Nakagawa A, Yamada H, Hata T and Furusaki A, Chem. Pharm. Bull, 1973, 21, 931–940. [DOI] [PubMed] [Google Scholar]
- 127.Bunet R, Song L, Mendes MV, Corre C, Hotel L, Rouhier N, Framboisier X, Leblond P, Challis GL and Aigle B, J. Bacteriol, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gould SJ and Melville CR, Bioorg. Med. Chem. Lett, 1995, 5, 51–54. [Google Scholar]
- 129.Gould SJ, Melville CR, Cone MC, Chen J and Carney JR, J. Org. Chem, 1997, 62, 320–324. [DOI] [PubMed] [Google Scholar]
- 130.Seaton PJ and Gould SJ, J. Antibiot, 1989, 42, 189–197. [DOI] [PubMed] [Google Scholar]
- 131.Gould SJ, Hong ST and Carney JR, J. Antibiot, 1998, 51, 50–57. [DOI] [PubMed] [Google Scholar]
- 132.Gould SJ, O’Hare T, Seaton P, Soodsma J and Tang Z, Bioorg. Med. Chem, 1996, 4, 987–994. [DOI] [PubMed] [Google Scholar]
- 133.Hayakawa Y, Ha SC, Kim YJ, Furihata K and Seto H, J. Antibiot, 1991, 44, 1179–1186. [DOI] [PubMed] [Google Scholar]
- 134.Kawasaki T, Hirashima R, Maruta T, Sato H, Maeda A, Yamada Y, Takeda M and Hayakawa Y, Appl. Environ. Microbiol, 2010, 76, 4201–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kawasaki T, Yamada Y, Maruta T, Maeda A and Hayakawa Y, J. Antibiot, 2010, 63, 725–727. [DOI] [PubMed] [Google Scholar]
- 136.Alexeev I, Sultana A, Mäntsälä P, Niemi J and Schneider G, Proc. Natl. Acad. Sci. USA, 2007, 104, 6170–6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Valderrama JA, Colonelli P, Vasquez D, Gonzalez MF, Rodriguez JA and Theoduloz C, Bioorg. Med. Chem, 2008, 16, 10172–10181. [DOI] [PubMed] [Google Scholar]
- 138.Jakeman DL, Bandi S, Graham CL, Reid TR, Wentzell JR and Douglas SE, Antimicrob. Agents Chemother, 2009, 53, 1245–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cottreau KM, Spencer C, Wentzell JR, Graham CL, Borissow CN, Jakeman DL and McFarland SA, Org. Lett, 2010, 12, 1172–1175. [DOI] [PubMed] [Google Scholar]
- 140.Lorico A and Long BH, Eur. J. Cancer, 1993, 29A, 1985–1991. [DOI] [PubMed] [Google Scholar]
- 141.Morimoto M, Okobu S, Tomita F and Marumo H, J. Antibiot, 1981, 34, 701–707. [DOI] [PubMed] [Google Scholar]
- 142.Nakano H, Matsuda Y, Ito K, Ohkubo S, Morimoto M and Tomita F, J. Antibiot, 1981, 34, 266–270. [DOI] [PubMed] [Google Scholar]
- 143.Matsumoto A and Hanawalt PC, Cancer Res., 2000, 60, 3921–3926. [PubMed] [Google Scholar]
- 144.Oyola R, Arce R, Alegria AE and Garcia C, Photochem. Photobiol, 1997, 65, 802–810. [DOI] [PubMed] [Google Scholar]
- 145.Elespuru RK and Gonda SK, Science, 1984, 223, 69–71. [DOI] [PubMed] [Google Scholar]
- 146.Findlay JA, Liu JS, Radics L and Rakhit S, Can. J. Chem, 1981, 59, 3018–3020. [Google Scholar]
- 147.Singh K, J. Antibiot, 1984, 37, 71–73. [DOI] [PubMed] [Google Scholar]
- 148.Greenstein M, Monji T, Yeung R, Maiese WM and White RJ, Antimicrob. Agents Chemother, 1986, 29, 861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nakashima T, Tadashi F, Kazuya S, Tomohiro S, Hiroyuki K and Takeo Y, United States Patent 09/308710, 1999. [Google Scholar]
- 150.Hasinoff BB, Wu X, Yalowich JC, Goodfellow V, Laufer RS, Adedayo O and Dmitrienko GI, Anticancer Drugs, 2006, 17, 825–837. [DOI] [PubMed] [Google Scholar]
- 151.O’Hara KA, Dmitrienko GI and Hasinoff BB, Chem.–Biol. Interact., 2010, 184, 396–402. [DOI] [PubMed] [Google Scholar]
- 152.O’Hara KA, Wu X, Patel D, Liang H, Yalowich JC, Chen N, Goodfellow V, Adedayo O, Dmitrienko GI and Hasinoff BB, Free Radic. Biol. Med, 2007, 43, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Feldman KS and Eastman KJ, J. Am. Chem. Soc, 2006, 128, 12562–12573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Feldman KS and Eastman KJ, J. Am. Chem. Soc, 2005, 127, 15344–15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Laufer RS and Dmitrienko GI, J. Am. Chem. Soc, 2002, 124, 1854–1855. [DOI] [PubMed] [Google Scholar]
- 156.Martin GDA, Tan LT, Jensen PR, Dimayuga RE, Fairchild CR, Raventos-Suarez C and Fenical W, J. Nat. Prod, 2007, 70, 1406–1409. [DOI] [PubMed] [Google Scholar]
- 157.Luzhetskyy A, Vente A and Bechthold A, Mol. BioSyst, 2005, 1, 117–126. [DOI] [PubMed] [Google Scholar]
- 158.Korynevska A, Heffeter P, Matselyukh B, Elbling L, Micksche M, Stoika R and Berger W, Biochem. Pharmacol, 2007, 74, 1713–1726. [DOI] [PubMed] [Google Scholar]
- 159.Shaaban KA, Stamatkin C, Damodaran C and Rohr J, J. Antibiot, 2011, 64, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Arai M, Tomoda H, Matsumoto A, Takahashi Y, Woodruff BH, Ishiguro N, Kobayashi S and Omura S, J. Antibiot, 2001, 54, 554–561. [DOI] [PubMed] [Google Scholar]
- 161.Kirschning A, Chen GW, Dräager G, Schuberth I and Tietze LF, Bioorg. Med. Chem, 2000, 8, 2347–2354. [DOI] [PubMed] [Google Scholar]
- 162.Uesato S, Tokunaga T, Mizuno Y, Fujioka H, Kada S and Kuwajima H, J. Nat. Prod, 2000, 63, 787–792. [DOI] [PubMed] [Google Scholar]
- 163.Uesato S, Tokunaga T and Takeuchi K, Bioorg. Med. Chem. Lett, 1998, 8, 1969–1972. [DOI] [PubMed] [Google Scholar]
- 164.Bringmann G, Lang G, Maksimenka K, Hamm A, Gulder TA, Dieter A, Bull AT, Stach JE, Kocher N, Muller WE and Fiedler HP, Phytochemistry, 2005, 66, 1366–1373. [DOI] [PubMed] [Google Scholar]
- 165.Potterat O, Puder C, Wagner K, Bolek W, Vettermann R and Kauschke SG, J. Nat. Prod, 2007, 70, 1934–1938. [DOI] [PubMed] [Google Scholar]
- 166.Motohashi K, Takagi M, Yamamura H, Hayakawa M and Shin-ya K, J. Antibiot, 2010, 63, 545–548. [DOI] [PubMed] [Google Scholar]
- 167.Nemoto A, Tanaka Y, Karasaki Y, Komaki H, Yazawa K, Mikami Y, Tojo T, Kadowaki K, Tsuda M and Kobayashi J, J. Antibiot, 1997, 50, 18–21. [DOI] [PubMed] [Google Scholar]
- 168.Tsuda M, Sato H, Tanaka Y, Yazawa G, Mikami Y, Sasaki T and Kobayashi J, J. Chem. Soc., Perkin Trans 1, 1996, 1773–1775. [Google Scholar]
- 169.Tsuda M, Nemoto A, Komaki H, Tanaka Y, Yazawa K, Mikami Y and Kobayashi J, J. Nat. Prod, 1999, 62, 1640–1642. [DOI] [PubMed] [Google Scholar]
- 170.Maruna M, Sturdikova M, Liptaj T, Godany A, Muckova M, Certik M, Pronayova N and Proksa B, J. Basic. Microbiol, 2010, 50, 135–142. [DOI] [PubMed] [Google Scholar]
- 171.Fotso S, Mahmud T, Zabriskie TM, Santosa DA and Proteau PJ, J. Antibiot, 2008, 61, 449–456. [DOI] [PubMed] [Google Scholar]
- 172.Fotso S, Mahmud T, Zabriskie TM, Santosa DA, Sulastri and Proteau PJ, J. Nat. Prod, 2008, 71, 61–65. [DOI] [PubMed] [Google Scholar]
- 173.Abdalla MA, Helmke E and Laatsch H, Nat. Prod. Commun, 2010, 5, 1917–1920. [PubMed] [Google Scholar]
- 174.Sun CH, Wang Y, Wang Z, Zhou JQ, Jin WZ, You XF, Gao H, Zhao LX, Si SY and Li X, J. Antibiot, 2007, 60, 211–215. [DOI] [PubMed] [Google Scholar]
- 175.Abdelfattah M, Maskey RP, Asolkar RN, Grun-Wollny I and Laatsch H, J. Antibiot, 2003, 56, 539–542. [DOI] [PubMed] [Google Scholar]
- 176.Perez M, Schleissner C, Rodriguez P, Zuniga P, Benedit G, Sanchez-Sancho F and de la Calle F, J. Antibiot, 2009, 62, 167–169. [DOI] [PubMed] [Google Scholar]
- 177.Holzenkämpfer M, Walker M, Zeeck A, Schimana J and Fiedler HP, J. Antibiot, 2002, 55, 301–317. [DOI] [PubMed] [Google Scholar]
- 178.Schimana J, Fiedler HP, Groth I, Su€ssmuth R, Beil W, Walker M and Zeeck A, J. Antibiot, 2000, 53, 779–787. [DOI] [PubMed] [Google Scholar]
- 179.Schimana J, Walker M, Zeeck A and Fiedler HP, J. Ind. Microbiol. Biotechnol, 2001, 27, 144–148. [DOI] [PubMed] [Google Scholar]
- 180.Galm U, Schimana J, Fiedler HP, Schmidt J, Li SM and Heide L, Arch. Microbiol, 2002, 178, 102–114. [DOI] [PubMed] [Google Scholar]
- 181.Bruntner C, Binder T, Pathom-aree W, Goodfellow M, Bull AT, Potterat O, Puder C, Horer S, Schmid A, Bolek W, Wagner K, Mihm G and Fiedler HP, J. Antibiot, 2005, 58, 346–349. [DOI] [PubMed] [Google Scholar]
- 182.Boudjella H, Zitouni A, Coppel Y, Mathieu F, Monje MC, Sabaou N and Lebrihi A, J. Antibiot, 2010, 63, 709–711. [DOI] [PubMed] [Google Scholar]
- 183.Boudjella H, Bouti K, Zitouni A, Mathieu F, Lebrihi A and Sabaou N, J. Appl. Microbiol, 2007, 103, 228–236. [DOI] [PubMed] [Google Scholar]
- 184.Shaaban KA, Srinivasan S, Kumar R, Damodaran C and Rohr J, J. Nat. Prod, 2011, 74, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Kalinovskaya NI, Kalinovsky AI, Romanenko LA, Pushilin MA, Dmitrenok PS and Kuznetsova TA, Nat. Prod. Commun, 2008, 3, 1611–1616. [Google Scholar]
- 186.Hong ST, Carney JR and Gould SJ, J. Bacteriol, 1997, 179, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Luzhetskii AN, Ostash BE and Fedorenko VA, Genetika, 2001, 37, 1340–1347. [PubMed] [Google Scholar]
- 188.Matseliukh BP and Lavrinchuk V, Mikrobiol. Z, 1999, 61, 22–27. [PubMed] [Google Scholar]
- 189.Uchida T, Imoto M, Watanabe Y, Miura K, Dobashi T, Matsuda N, Sawa T, Naganawa H, Hamada M and Takeuchi T, J. Antibiot, 1985, 38, 1171–1181. [DOI] [PubMed] [Google Scholar]
- 190.Sekizawa R, Iinuma H, Naganawa H, Hamada M, Takeuchi T, Yamaizumi J and Umezawa K, J. Antibiot, 1996, 49, 487–490. [DOI] [PubMed] [Google Scholar]
- 191.Alvi KA, Baker DD, Stienecker V, Hosken M and Nair BG, J. Antibiot, 2000, 53, 496–501. [PubMed] [Google Scholar]
- 192.Stro€ch K, Zeeck A, Antal N and Fiedler HP, J. Antibiot, 2005, 58, 103–110. [DOI] [PubMed] [Google Scholar]
- 193.Abdelfattah MS, Kharel MK, Hitron JA, Baig I and Rohr J, J. Nat. Prod, 2008, 71, 1569–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zeeck A, Rohr J, Sheldrick GM, Jones PG and Paulus EF, J. Chem. Res.-S, 1986, 104–105. [Google Scholar]
- 195.Rohr J, Scho€newolf M, Udvarnoki G, Eckardt K, Schumann G, Wagner C, Beale JM and Sorey SD, J. Org. Chem, 1993, 58, 2547–2551. [Google Scholar]
- 196.Schneemann I, Kajahn I, Ohlendorf B, Zinecker H, Erhard A, Nagel K, Wiese J and Imhoff JF, J. Nat. Prod, 2010, 73, 1309–1312. [DOI] [PubMed] [Google Scholar]
- 197.Paululat T, Kulik A, Hausmann H, Karagouni AD, Zinecker H, Imhoff JF and Fiedler HP, Eur. J. Org. Chem, 2010, 2344–2350. [Google Scholar]
- 198.Kalinovskaya NI, Kalinovsky AI, Romanenko LA, Dmitrenok PS and Kuznetsova TA, Nat. Prod. Commun, 2010, 5, 597–602. [PubMed] [Google Scholar]
- 199.Carreno MC and Urbano A, Synlett, 2005, 1–25. [Google Scholar]
- 200.Hua DH and Saha S, Recl. Trav. Chim. Pays-Bas, 1995, 114, 341–355. [Google Scholar]
- 201.Brown PM and Thomson RH, J. Chem. Soc., Perkin Trans. 1, 1976, 997–1000. [Google Scholar]
- 202.Dann M, Lefemine DV, Barbatschi F, Shu P, Kunstmann MP, Mitscher LA and Bohonos N, Antimicrob. Agents Chemother, 1965, 5, 832–835. [PubMed] [Google Scholar]
- 203.Kuntsmann MP and Mitscher LA, J. Org. Chem, 1966, 31, 2920–2925. [DOI] [PubMed] [Google Scholar]
- 204.Carothers WH and Coffman DD, J. Am. Chem. Soc, 1932, 54, 4071–4076. [Google Scholar]
- 205.Guingant A and Barreto MM, Tetrahedron Lett., 1987, 28, 3107–3110. [Google Scholar]
- 206.Kaliappan KP and Ravikumar V, J. Org. Chem, 2007, 72, 6116–6126. [DOI] [PubMed] [Google Scholar]
- 207.Krohn K, Ballwanz F and Baltus W, Liebigs Ann. Chem, 1993, 911–913. [Google Scholar]
- 208.Maugel N and Snider BB, Org. Lett, 2009, 11, 4926–4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ding CY, Tu SH, Li FY, Wang YX, Yao QZ, Hu WX, Xie H, Meng LH and Zhang A, J. Org. Chem, 2009, 74, 6111–6119. [DOI] [PubMed] [Google Scholar]
- 210.Collet SC, Remi JF, Cariou C, Laib S, Guingant AY, Vu NQ and Dujardin G, Tetrahedron Lett., 2004, 45, 4911–4915. [Google Scholar]
- 211.Vu NQ, Dujardin G, Collet SC, Raiber EA, Guingant AY and Evain M, Tetrahedron Lett., 2005, 46, 7669–7673. [Google Scholar]
- 212.Sissouma D, Dequirez G, Collet S and Guingant A, Tetrahedron Lett., 2011, 52, 2336–2339. [Google Scholar]
- 213.Valderrama JA, Gonzalez MF, Colonelli P and Vasquez D, Synlett, 2006, 2777–2780. [Google Scholar]
- 214.Valderrama JA and Vasquez D, Tetrahedron Lett., 2008, 49, 703–706. [Google Scholar]
- 215.Kitani Y, Morita A, Kumamoto T and Ishikawa T, Helv. Chim. Acta, 2002, 85, 1186–1195. [Google Scholar]
- 216.Kumamoto T, Kitani Y, Tsuchiya H, Yamaguchi K, Seki H and Ishikawa T, Tetrahedron, 2007, 63, 5189–5199. [Google Scholar]
- 217.Mal D and Pahari P, Chem. Rev, 2007, 107, 1892–1918. [DOI] [PubMed] [Google Scholar]
- 218.Rathwell K and Brimble MA, Synthesis, 2007, 643–662. [Google Scholar]
- 219.Mal D and Dey S, Tetrahedron, 2006, 62, 9589–9602. [Google Scholar]
- 220.Mal D, Roy HN, Hazra NK and Adhikari S, Tetrahedron, 1997, 53, 2177–2184. [Google Scholar]
- 221.Mal D and Roy HN, J. Chem. Soc., Perkin Trans. 1, 1999, 3167–3171. [Google Scholar]
- 222.Patra A, Pahari P, Ray S and Mal D, J. Org. Chem, 2005, 70, 9017–9020. [DOI] [PubMed] [Google Scholar]
- 223.Mal D, Patra A and Roy H, Tetrahedron Lett., 2004, 45, 7895–7898. [Google Scholar]
- 224.Ray S, Patra A and Mal D, Tetrahedron, 2008, 64, 3253–3267. [Google Scholar]
- 225.Mal D, Jana AK, Mitra P and Ghosh K, J. Org. Chem, 2011, 76, 3392–3398. [DOI] [PubMed] [Google Scholar]
- 226.Anctil EJG and Snieckus V, J. Organomet. Chem, 2002, 653, 150–160. [Google Scholar]
- 227.James CA and Snieckus V, J. Org. Chem, 2009, 74, 4080–4093. [DOI] [PubMed] [Google Scholar]
- 228.Nicolaou KC, Li HM, Nold AL, Pappo D and Lenzen A, J. Am. Chem. Soc, 2007, 129, 10356–10357. [DOI] [PubMed] [Google Scholar]
- 229.Roush WR and Neitz RJ, J. Org. Chem, 2004, 69, 4906–4912. [DOI] [PubMed] [Google Scholar]
- 230.Lei XG and Porco JA, J. Am. Chem. Soc, 2006, 128, 14790–14791. [DOI] [PubMed] [Google Scholar]
- 231.Kimura S, Kobayashi S, Kumamoto T, Akagi A, Sato N and Ishikawa T, Helv. Chim. Acta, 2011, 94, 578–591. [Google Scholar]
- 232.Woo CM, Lu L, Gholap SL, Smith DR and Herzon SB, J. Am. Chem. Soc, 2010, 132, 2540–2541. [DOI] [PubMed] [Google Scholar]
- 233.Futagami S, Ohashi Y, Imura K, Hosoya T, Ohmori K, Matsumoto T and Suzuki K, Tetrahedron Lett., 2000, 41, 1063–1067. [Google Scholar]
- 234.Konno F, Ishikawa T, Kawahata M and Yamaguchi K, J. Org. Chem, 2006, 71, 9818–9823. [DOI] [PubMed] [Google Scholar]
- 235.Akagi Y, Yamada S, Etomi N, Kumamoto T, Nakanishi W and Ishikawa T, Tetrahedron Lett., 2010, 51, 1338–1340. [Google Scholar]
- 236.Madan S and Cheng CH, J. Org. Chem, 2006, 71, 8312–8315. [DOI] [PubMed] [Google Scholar]
- 237.Asao N, Synlett, 2006, 1645–1656. [Google Scholar]
- 238.Kesenheimer C, Kalogerakis A, Meissner A and Groth U, Chem.–Eur. J., 2010, 16, 8805–8821. [DOI] [PubMed] [Google Scholar]
- 239.Bugaut X, Guinchard X and Roulland E, J. Org. Chem, 2010, 75, 8190–8198. [DOI] [PubMed] [Google Scholar]
- 240.Sato K, Asao N and Yamamoto Y, J. Org. Chem, 2005, 70, 8977–8981. [DOI] [PubMed] [Google Scholar]
- 241.Takemura I, Imura K, Matsumoto T and Suzuki K, Org. Lett, 2004, 6, 2503–2505. [DOI] [PubMed] [Google Scholar]
- 242.Mori K, Tanaka Y, Ohmori K and Suzuki K, Chem. Lett, 2008, 37, 470–471. [Google Scholar]