

Abstract

Herein we report progress toward a backup clinical candidate to the M1 positive allosteric modulator (PAM) VU319/ACP-319. Scaffold-hopping from the pyrrolo[2,3-b]pyridine-based M1 PAM VU6007477 to isomeric pyrrolo[3,2-b]pyridine and thieno[3,2-b]pyridine congeners identified several backup contenders. Ultimately, VU6007496, a pyrrolo[3,2-b]pyridine, advanced into late stage profiling, only to be plagued with unanticipated, species-specific metabolism and active/toxic metabolites which were identified in our phenotypic seizure liability in vivo screen, preventing further development. However, VU6007496 proved to be a highly selective and CNS penetrant M1 PAM, with minimal agonism, that displayed excellent multispecies IV/PO pharmacokinetics (PK), CNS penetration, no induction of long-term depression (or cholinergic toxicity) and robust efficacy in novel object recognition (minimum effective dose = 3 mg/kg p.o.). Thus, VU6007496 can serve as another valuable in vivo tool compound in rats and nonhuman primates, but not mouse, to study selective M1 activation.
Keywords: muscarinic acetylcholine receptor subtype 1 (M1), positive allosteric modulator (PAM), cognition, metabolism
Introduction
The resurgence of muscarinic acetylcholine receptors (mAChRs or M1–5) at the forefront of CNS drug discovery for a variety of neuropsychiatric disorders, driven by the pending FDA approval of xanomeline 1 (an M1/M4 agonist, combined with a peripheral muscarinic antagonist as KarXT),1−3 has focused attention on the development of selective M1 and M4 positive allosteric modulators (PAMs). While M4 PAMs have demonstrated preclinical and clinical efficacy for treatment of the positive symptoms of schizophrenia, M1 PAMs offer promise for treating cognitive dysfunction in schizophrenia, Alzheimer’s disease and other CNS disorders.4−7 Potent ago-PAMs, such as 2–5 (Figure 1),8−12 overstimulate the M1 receptor and lead to adverse events (AEs) and cholinergic toxicity, which has diminished enthusiasm for the mechanism. However, PAMs with minimal to no M1 agonism, such as 6–8,13−16 proved devoid of AEs and cholinergic toxicity in preclinical animal models. Interestingly, the M1 ago-PAM TAK-071 (9),17,18 displaying low cooperativity, was efficacious in a number of preclinical rodent models and has advanced into human clinical testing. From our efforts with VU319/ACP-319 (structure not disclosed at this time), an M1PAM with no detecable M1 agonsim proved devoid of AEs, cholinergic toxicity, and other toxicology findings in rat, dog, and nonhuman primate to support an open IND by the FDA. Recently, safety and pharmacokinetic data from a Phase I single ascending dose (SAD) study for ACP-319 have been reported.19,20 This M1 PAM was well tolerated, with no cholinergic side effects noted, at doses showing cognitive improvement and functional target engagement. Moreover, pharmacokinetic (PK) data demonstrated that ACP-319 demonstrated good absorption and bioavailability in man with a half-life supporting once daily dosing.19,20 Based on these data, the team was charged with developing a chemically orthogonal backup compound to ACP-319. Here, we disclose the chemical optimization of VU6007477 (8) and the challenges and tribulations that led to the discovery of not a backup clinical candidate, but a new in vivo tool compound, VU6007496.
Figure 1.
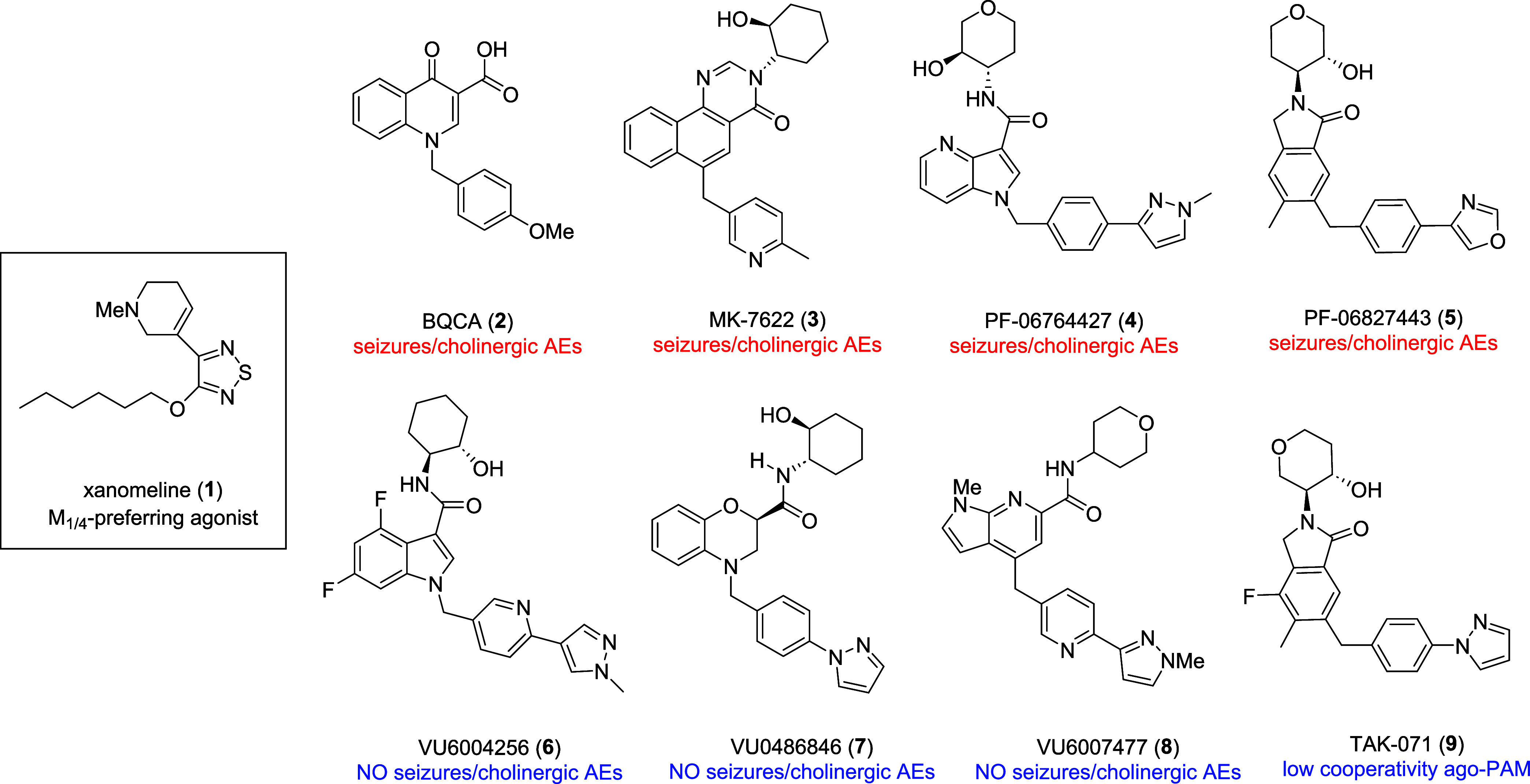
Structures of xanomeline (1), representative M1 ago-PAMs with cholinergic AEs 2–5, representative “pure” PAMs devoid of M1 agonism 6–8, and the low cooperative M1 ago-PAM TAK-071 (9).
Results and Discussion
Design
Our initial backup campaign focused on the chemically orthogonal M1 PAM scaffold 7 (VU0486846), devoid of M1 agonism, AEs, and cholinergic toxicity, but which exhibited an unacceptable CYP450 profile.15 We elected to employ a scaffold-hopping approach to replace the benzomorpholine core while surveying alternate amides (deleting the agonism-prone hydroxyl moiety and incorporating heteroatoms) and various southern tail moieties (Figure 2). This exercise led to exceedingly steep structure–activity relationship (SAR) in terms of the generation of M1 ago-PAMs (displaying cholinergic side effects) versus M1 PAMs with minimal to no M1 agonism, with a single analog advancing as a new M1 PAM in vivo tool compound, 8 (VU6007477), with robust precognitive efficacy and no cholinergic toxicity or AEs.16 However, the overall profile of 8 did not warrant advancement as a backup candidate to VU-319/ACP-319.
Figure 2.
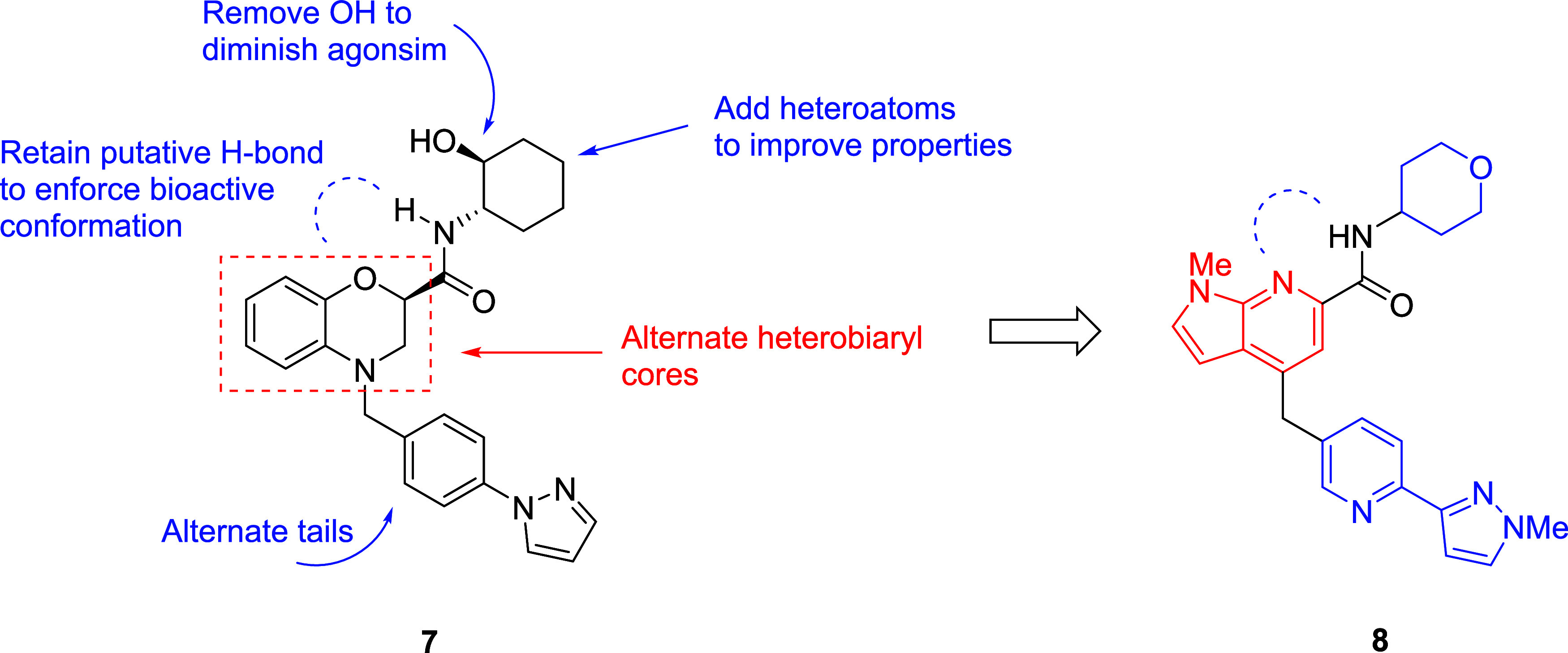
Initial scaffold-hopping exercise from 7 that led to 8, a novel M1 PAM devoid of cholinergic toxicities and AEs.
Far more dramatic departures in subsequent scaffold-hopping campaigns were met with resolute failure to provide M1 PAMs devoid of M1 agonism, so the team decided to revisit the pyrrolo[2,3-b]pyridine-based 8(16) and explore an alternate regioisomer, namely a pyrrolo[3,2-b]pyridine core 9 as well as an isosteric thieno[3,2-b]pyridine core 10 (Figure 3).21 Here again, SAR proved to be steep and failed to identify M1 PAMs with diminished M1 agonism worthy of further progression toward backup compounds. This exercise led to the discovery of two compounds that advanced into further profiling, VU6007496 (11, a pyrrolo[3,2-b]pyridine) and VU6006874 (12, a thieno[3,2-b]pyridine), with unexpected results and challenges.
Figure 3.

Scaffold-hopping from 8 to regioisomeric cores 9 and 10, which led to the discovery of VU6007496 (11) and VU6006874 (12) that were advanced into further profiling.
Synthesis
The synthesis of 11 and 12 (Scheme 1) first required the construction of key intermediates, namely a 7-chloro-1-methyl-1H-pyrrolo[3,2-b]pyridine-5-carbonitrile 13 and a 7-chlorothienyl[3,2-b]pyrdine-5-carbonitrile 14.21 Here, we envisioned that these key intermediates would be prepared from the commercially available chloropyridines 15 and 17 by application of a Reissert-Henze reaction sequence.22 Conversion of 15 to the pyridine N-oxide 16 by treatment with m-CPBA proceeded in 85% yield. Treatment of 16 with dimethyl sulfate and KCN facilitated the Reissert-Henze reaction followed by N-methylation affording the requisite cyanopyridine 13 in 98% over 3 steps. A similar sequence provided the analogous cyanopyridine 14, in albeit lower overall yield (∼53% over two steps).21
Scheme 1. Synthesis of 7-Chloro-1-methyl-1H-pyrrolo[3,2-b]pyridine-5-carbonitrile 13 and a 7-Chlorothienyl[3,2-b]pyrdine-5-carbonitrile 14.

Reagents and conditions: (a) m-CPBA, n-BuOAc:heptane (3:5), 0 °C—rt, 83%; (b) (MeO)2SO2, n-BuOAc, 75 °C, 16 h; (c) KCN, aq. NH4Cl, 50 °C, 2 h; quantitative yield (2 steps); (d) MeI, NaH, DMF, 0 °C—rt, 98%; (e) m-CPBA, DCM, 0 °C—rt, 54%; (f) TMSCN, (CH3)2NCOCl, DCM, rt, 16 h, 98%.
With intermediates 13 and 14 in hand, elaboration into the M1 PAMs 11 and 12 proved straightforward. Conversion of 13 to the boronate ester 19 proceeded smoothly (Scheme 2), followed by a Suzuki coupling with benzyl chloride 20 to provide 21 in 56% yield for the two steps. Acid-mediated hydrolysis of the nitrile to the carboxylic acid, and a HATU-facilitated coupling with tetrahydro-2H-pyran-4-amine delivered 11 in 71% yield for the two step sequence.21
Scheme 2. Synthesis of VU6007496 (11).
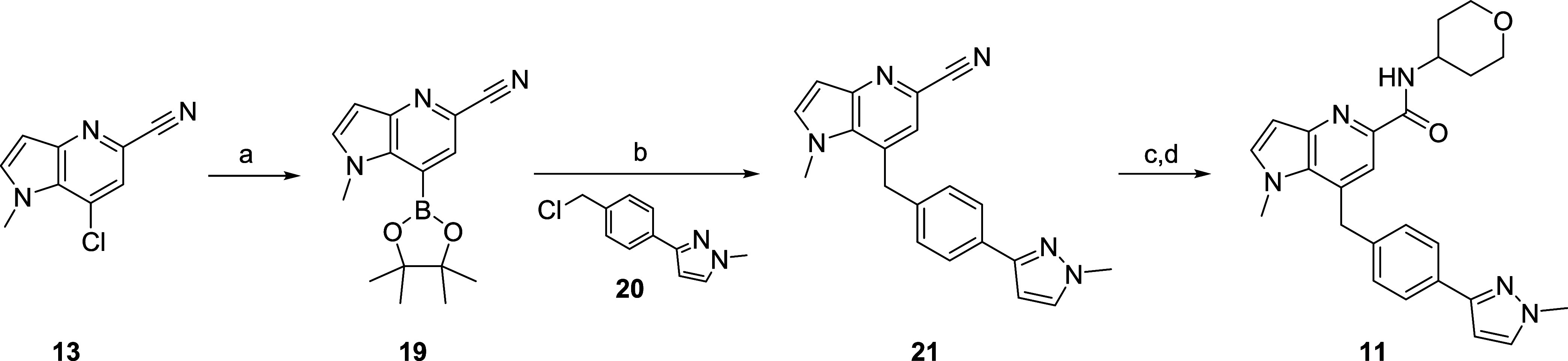
Reagents and conditions: (a) bis(pinacolato)diboron, KOAc, Pd(dppf)Cl2, 1,4-dioxane, 100 °C, 16 h; (b) benzyl chloride 20, Cs2CO3, Pd(dppf)Cl2, THF/H2O, 90 °C, 16 h; 56% (2 steps); (c) conc. HCl, reflux, 2 h; (d) tetrahydro-2H-pyran-4-amine, HATU, DIEA, DMF, rt, 20 min, 71% (2 steps).
Application of a similar reaction sequence with 7-chlorothienyl[3,2-b]pyridine-5-carbonitrile 14 provided 12 (Scheme 3). Here, conversion of 14 to the boronate ester 22 proceeded in excellent yield (95%). A Pd(dppf)Cl2-catalyzed Suzuki coupling with benzyl chloride 20 gave the elaborated nitrile 23 in 99% isolated yield. Finally, acid-mediated hydrolysis of the nitrile to the acid, and a HATU-facilitated coupling with tetrahydro-2H-pyran-4-amine delivered 12 in 61% yield for the two-step sequence.21
Scheme 3. Synthesis of VU6006874 (12).
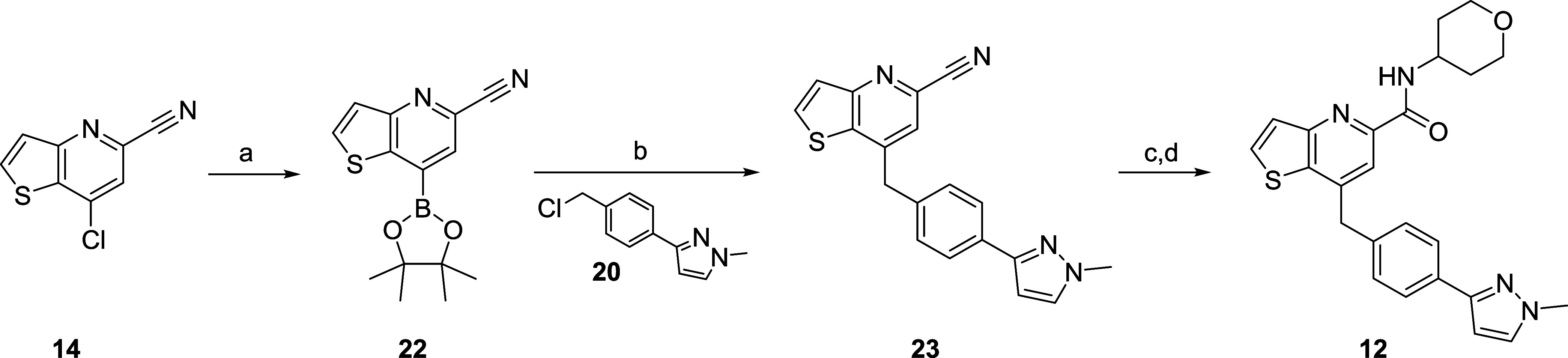
Reagents and conditions: (a) bis(pinacolato)diboron, KOAc, Pd(dppf)Cl2, 1,4-dioxane, 100 °C, 16 h, 95%; (b) benzyl chloride 20, Cs2CO3, Pd(dppf)Cl2, THF/H2O, 90 °C, 16 h, 99%; (c) conc. HCl, 100 °C, 3 h; (d) tetrahydro-2H-pyran-4-amine, HATU, DIEA, DMF, rt, 1 h, 61% (2 steps).
Molecular Pharmacology and In Vitro Drug Metabolism and Pharmacokinetics (DMPK)
With 11 and 12 in hand, both compounds were evaluated in a battery of molecular pharmacology and in vitro DMPK assays (Table 1) to assess their suitability to advance further down the testing cascade toward putative backup candidates.
Table 1. Pharmacology and In Vitro DMPK Profiles of 11 and 12a.
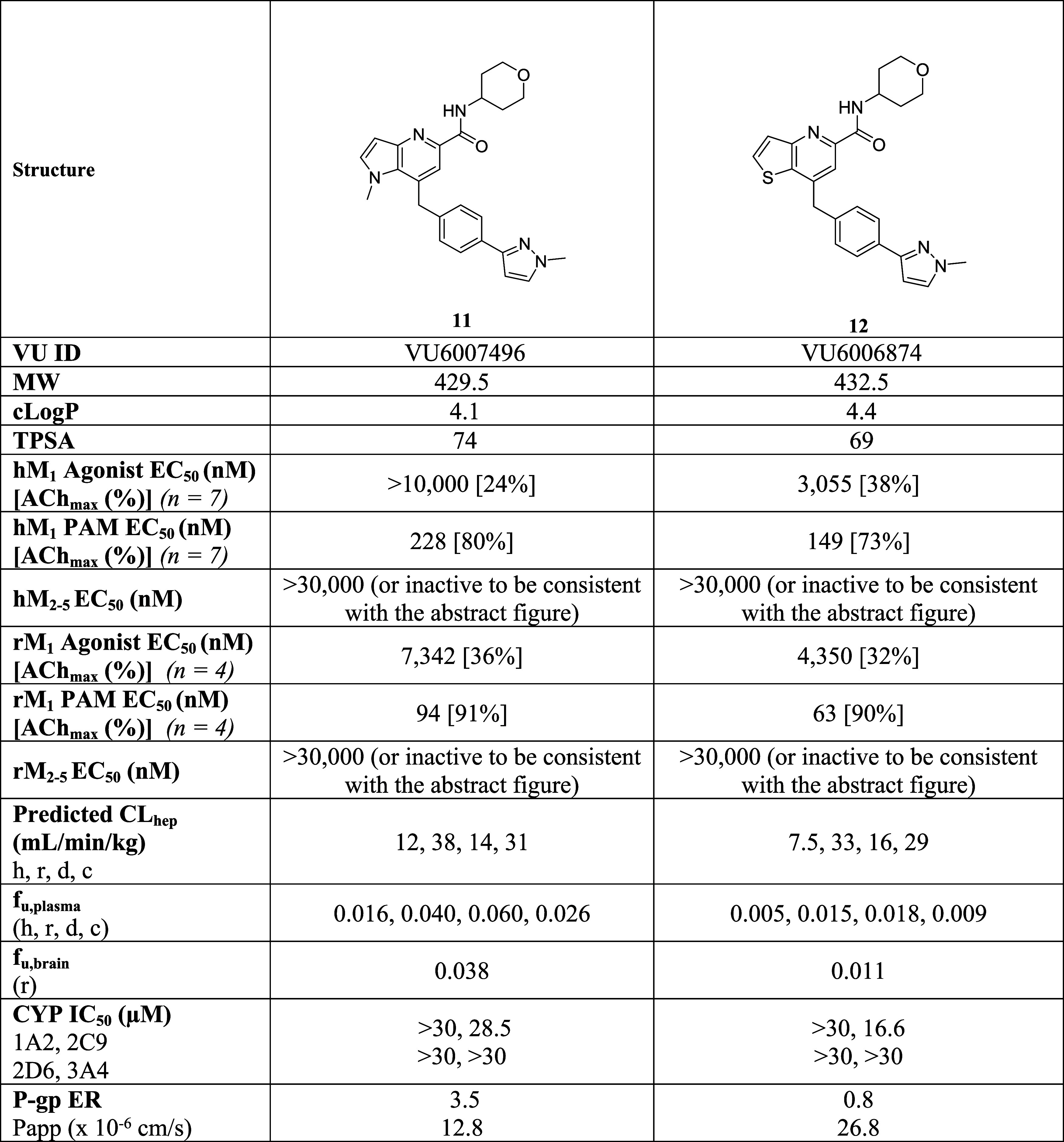
c Log P and TPSA were calculated using ChemDraw Professional 22.2.
On human M1, 11 was found to be an M1 PAM (EC50 = 228 nM, 80% ACh max, pEC50 = 6.78 ± 0.11) with minimal M1 agonism (EC50 > 10 μM, 24% ACh Max), whereas 12 was a more potent M1 PAM (EC50 = 149 nM, 73% ACh max, pEC50 = 6.88 ± 0.08), but displayed moderate M1 agonism (EC50 = 3.1 μM, 38% ACh Max).21 Both 11 and 12 were potent on rat M1 with PAM potencies of 94 nM (91% ACh Max) and 63 nM (90% ACh Max), respectively, and both showed moderate rat M1 agonism. Importantly, both 11 and 12 were inactive on human and rat M2–5 (EC50s > 30 μM). PAMs 11 and 12 displayed moderate predicted hepatic clearance across human, rat and dog; however, both PAMs possessed high predicted hepatic clearance in cyno (≥30 mL/min/kg). Both PAMs showed acceptable plasma and brain homogenate binding profiles, but 12 uniformly showed less fraction unbound. Similarly, both 11 and 12 demonstrated acceptable CYP450 profiles against CYP3A4, CYP1A2, CYP2C9 and CYP2D6 (IC50s > 16 μM). In terms of predicted CNS penetration in human, 11 had an MDCK-MDR1 ER ratio of 3.5 with a Papp of 12.8, and, based on controls in this assay, was acceptable to move forward, whereas 12 was not a P-gp substrate (ER = 0.8) and had good permeability (Papp = 26.8). In a Lead Profiling Screen of 68 GPCRs, ion channels and transporters employing radioligand displacement to assess ancillary pharmacology, 11 had no significant results (<50% displacement at 10 μM), whereas 12 had a single significant hit (melatonin MT1, 70% at 10 μM).21 In a 4-strain Ames assay, with and without S9, both 11 and 12 were negative. A GSH trapping study in human liver microsomes to test for reactive intermediates showed similar negative results for the two PAMs. Finally, an electrophysiology panel of cardiac ion channels was clean (<17% inhibition at 10 μM) for both PAMs.21 Thus, both PAMs possessed acceptable overall in vitro profiles to advance into the tier of the development workflow; however, in the team’s opinion, PAM 11 was the leading contender due to higher fraction unbound and less M1 agonism.
In Vivo Behavior and DMPK
PAMs 11 and 12 were next evaluated in our rat IV plasma: brain level cassette paradigm (0.25 mg/kg compound, 1 mg/kg total dose, 10.1% EtOH:40.4% PEG400:49.5% DMSO) sampled at a set 15 min time point to assess CNS penetration. 11 had a Kp (the partitioning coefficient between plasma and brain) of 0.42 (plasma, 92.2 ng/mL: brain, 39 ng/g) and a Kp,uu of 0.36; in contrast, 12 had a Kp of 1.1 (plasma, 105 ng/mL: brain, 120 ng/g) and a Kp,uu of 0.7.20 These data were in alignment with the human predicted CNS penetration from the MDCK-MDR1 P-gp in vitro assay discussed earlier and supported continued advancement. As mice are the most sensitive species to cholinergic mechanisms,11,13,23,24 we placed a high-throughput phenotypic seizure liability assay into the work-flow in which potent ago-PAMs, such as 2–5,8−15 display robust Racine scale 4/5 behavioral convulsions that develop within minutes of dosing and last for the 3 h duration of the study. Here, a high dose (100 mg/kg intraperitoneal (i.p.)) of either 11 or 12 in mice did not induce seizure liability, akin to 6–9 and VU-319/ACP-319, for the 3-h duration of the study (Figure 4A).21 For 11 in particular, the 100 mg/kg i.p. study afforded total brain exposure of 1.04 μM, and the mouse M1 PAM EC50 was determined to be 92 nM (66% ACh max). In parallel, we performed electrophysiology studies in mouse native tissue layer V medial prefrontal cortex (mPFC). While ago-PAMs such as 2–5 induce substantial long-term depression that correlates with a lack of robust pro-cognitive efficacy, PAM 11 did not induce significant changes in field excitatory post synaptic potentials (fEPSPs) recorded from layer V and evoked by electrical stimulation in layer II/III at 3 μM concentration (∼30x above the functional mouse EC50), and, therefore, maintain activity dependence of PFC function (Figure 4B).21 Thus, both compounds cleared the major hurdles of CNS penetration and the liability of M1 overstimulation.
Figure 4.

(A) Modified Racine Score test in mice with M1 PAMs. Pretreatment with M1 PAMs (100 mg/kg, i.p., 10 mL/kg, 180 min) BQCA (2), MK-7622 (3), PF-0674427 (4), resulted in robust behavioral convulsions at 3 h post administration, while VU6007496 (11) and VU6006874 (12) did not cause any observed adverse effects. N = 3/group of male C57Bl/6 mice. ANOVA p < 0.0001; ****p < 0.0001 as compared to vehicle control. (B) Time course graph showing that bath application of 3 μM VU6007496 (11) for 20 min led to no significant change in fEPSP slope. N = 8 brain slices from 3 different male C57Bl/6 mice.
We next conducted multispecies IV/PO PK in parallel as a potential discerning data set. PAM 11 possessed an attractive PK profile across rat, dog and nonhuman primate (cynomologus monkey (cyno)) (Table 2). In rat, 11 showed low-to-moderate clearance (26 mL/min/kg) with a 6.1 h half-life and 66% oral bioavailability. The PK of 11 in dog and cyno was characterized by very low clearance (2.4 and 5.9 mL/min/kg, respectively), long half-life in dog (12.8 h) and a short half-life in cyno (1 h), driven by a low volume (0.39). Oral bioavailability for dog was 35% and cyno was 59%. These data were generated prior to any significant formulation of vehicle screens; thus, we were very pleased with the profile of 11. PAM 12 was similar in disposition, with moderate clearance (33 mL/min/kg) with a 5.3 h half-life and 100% oral bioavailability in rat. Clearance was low in dog (1.3 mL/min/kg) and cyno (2.3 mL/min/kg), and, once again, a long half-life in dog (36.6 h) and moderate in cyno (4.5 h). However, while oral bioavailability was excellent in rat (100%) and cyno (79%), but it was poor in dog (9%).21 With a strong desire to employ dog as the nonrodent safety species, the challenge to address the low %F in dog was enough to triage PAM 12 and focus on the advancement of 11 (Table 3).
Table 2. Pharmacokinetic Parameters of 11.
| parameter | rat (SD) | dog (beagle or mongrel) | NHP (cyno) |
|---|---|---|---|
| dose (mg/kg) iv/po | 1/10 | 1/5 | 1/5 |
| CLp (mL/min/kg) | 26 | 2.4 | 5.9 |
| Vss (L/kg) | 3.2 | 2.2 | 0.39 |
| elimination t1/2 (h) | 6.1 | 12.8 | 1.0 |
| F (%) po | 66 | 35 | 52 |
| Kp | 0.42 | ||
| Kp,uu | 0.36 |
Table 3. Pharmacokinetic Parameters of 12.
| parameter | rat Sprague–Dawley (SD) | dog (beagle or mongrel) | NHP (cyno) |
|---|---|---|---|
| dose (mg/kg) iv/po | 1/10 | 1/3 | 1/3 |
| CLp (mL/min/kg) | 33 | 1.3 | 2.3 |
| Vss (L/kg) | 8.2 | 4.0 | 0.7 |
| elimination t1/2 (h) | 5.3 | 36.6 | 4.5 |
| F (%) po | 100 | 9 | 79 |
| Kp | 1.1 | ||
| Kp,uu | 0.7 |
Previously, we have shown that potent M1 ago-PAMs with agonist activity in the PFC showed little efficacy in novel object recognition (NOR); in contrast,15,16 M1 PAMs with no to minimal agonist activity in the PFC displayed robust dose-dependent enhancement of NOR. As illustrated in Figure 5, PAM 11 dose-dependently enhanced recognition memory in rats with a minimum effective dose (MED) of 3 mg/kg p.o., which was in-line with data obtained with previous M1 PAMs. Interestingly, a 3 mg/kg p.o. rat satellite PK study demonstrated total brain concentration of 990 nM and an unbound brain concentration of 39.8 nM (rat M1 PAM EC50 = 94 nM (91% ACh max)). Historically, our PK/PD for M1 PAMs in NOR has a strong correlation with total brain as opposed to unbound levels, with MEDs typically 0.25 to 0.4 of the rat EC50.21 This may be due to higher endogenous cholinergic tone in healthy animals, and the in vitro PAM EC50s being derived from an arbitrarily selected EC20 concentration of ACh as a subthreshold value.
Figure 5.

Novel object recognition (NOR) test in rats with VU6007496 (11). PAM 11 dose-dependently enhanced recognition memory in rats. Pretreatment with 0.1, 0.3, 1, and 3 mg/kg VU6007496 (p.o, 0.5% natrosol/0.015% Tween 80 in water, 30 min) prior to exposure to identical objects significantly enhanced recognition memory assessed 24 h later. N = 15–18/group of male Sprague–Dawley rats. ANOVA p = 0.0283; **p < 0.01.
In-Depth DMPK Profiling
To advance 11 as a candidate and into IND-enabling studies, we next needed to better understand its CYP profile to minimize drug–drug interactions in the clinic. Here, a substrate depletion approach employing recombinant human cytochrome P450s (rCYPs) indicated that rCYP2J2 and rCYP3A4 are responsible for 2.4 and 97.6% of the hepatic CYP-mediated clearance of PAM 11, respectively. We were pleased to see the contribution of another CYP beyond 3A4 for the metabolism of 11.21 In parallel, we investigated the ability of PAM 11 to induce the expression of CYP450s (measuring mRNA) in cryopreserved human hepatocytes from three separate donors. Weak induction liability was noted for CYP1A2 (∼3-fold) and CYP2B6 (∼8-fold), while more concerning induction liability was reported for CYP3A4 (∼21-fold), and would require subsequent evaluation using the assessment of protein expression instead of mRNA.21
In parallel to these CYP studies, we performed metabolite identification (MET ID) studies in rat and human S9 liver microsomes (Figure 6) and observed comparable coverage of metabolites across human and rat. The PAM 11 proved to be reasonably stable, with the extent of metabolism being 36.8% in rat and 58.1% in human (based on MS peak areas). Eight oxidative metabolites were identified, with the major metabolite in both rat and human being Metabolite A (VU6036463, 24), the result of oxidative N-demethylation of the southern pyrazole (Scheme 4). The majority of other oxidative metabolism occurred on the tetrahydropyran moiety and included ring opening, but there was no amide hydrolysis observed.21
Figure 6.

Metabolism of VU6007496 (11) in rat and human liver S9, with the major metabolite, VU6036463 (24), an N-demethylation product, exemplified.
Scheme 4. Synthesis of Metabolite VU6036463 (24).
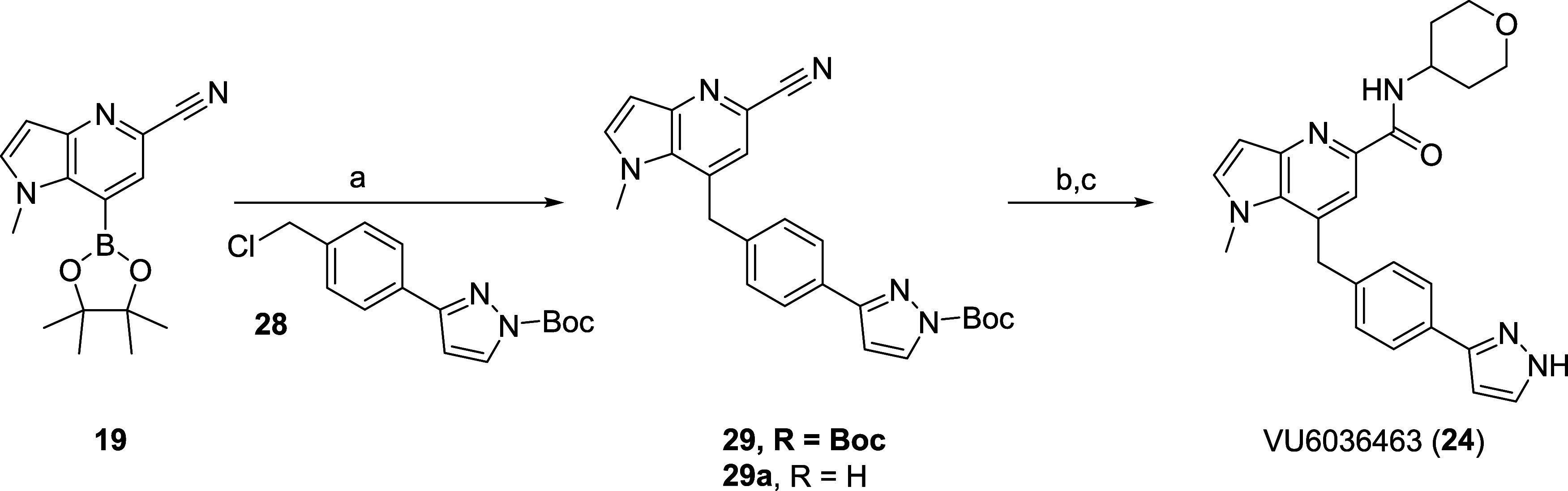
Reagents and conditions: (a) benzyl chloride 28, Cs2CO3, Pd(dppf)Cl2, THF/H2O, 90 °C, 16 h; 58%; (b) conc. HCl, reflux, 2 h; (c) tetrahydro-2H-pyran-4-amine, HATU, DIEA, DMF, rt, 20 min, 81% (2 steps).
While awaiting more definitive MET ID studies in multispecies hepatocytes, we elected to perform a rat dose escalation study to assess exposures in a standard, tox-friendly oral vehicle (30% captisol) to lay the foundation for dose range finding/maximum tolerated dose (DRF/MTD) studies. Male Sprague–Dawley rats were dosed with VU6007496 (11) at doses of 3, 10, 30, 100, and 300 mg/kg p.o. in 30% captisol. At all doses, PAM 11 was rapidly absorbed with tight Tmax values under 2 h (Figure 7). Linear dose escalation was noted from 3 to 30 mg/kg. However, severe adverse cholinergic events were noted after 4.5 h in the 100 and 300 mg/kg dose groups which was not expected due to the clean mouse phenotypic assay and our long history with other M1 PAMs.20 Recall, exposures in rat at the NOR MED were 990 nM total and 39.8 nM unbound brain concentrations, respectively. At Cmax, the 100 mg/kg dose achieved 9.1 μM total and 298 nM unbound brain concentrations (∼9-fold over the NOR MED exposure), while the 300 mg/kg dose afforded 17.2 μM total and 569 nM unbound brain concentrations (∼17-fold over the NOR MED exposure).21 There was a clear, yet unanticipated disconnect.
Figure 7.

Rat dose escalation study with VU6007496 (11).
More concerning were the results generated via outsourced multispecies hepatocyte MET ID, which were drastically different than the liver S9MET ID. The extent of metabolism for VU6007496 with rat, dog, monkey and human hepatocytes was 64.7, 51.5, 63.9 and 99.5%, respectively (Figure 8). Twelve metabolites were observed in the 4-h hepatocytes samples, and as opposed to the S9 study, there was no parent 11 remaining in human hepatocytes. In human hepatocytes, PAM 11 was metabolized primarily to Metabolite A (24), the result of oxidative N-demethylation of the southern pyrazole. The other minor metabolites were oxidation on the tetrahydropyran moiety and included ring opening (Metabolite F, 25).21 With these data, PAM 11 was no longer a backup candidate, but the disconnects and paradigm changing cholinergic toxicity profile warranted further investigation to inform the next backup campaign.
Figure 8.

Metabolism of VU6007496 (11) in rat, dog, primate and human hepatocytes, with the major metabolite, VU6036463 (24), an N-demethylation product. Unexpectedly, no parent PAM 11 remained in human after the 4 h incubation.
We typically would not examine mouse hepatocyte MET ID, as mouse was not a safety species for the IND-enabling toxicology package, but with the unusual metabolism profile for human, and the lack of predictive cholinergic tox in the mouse phenotypic assay, we felt this was worthy of exploration. As shown in Figure 9, PAM 11 rapidly undergoes extensive metabolism (95.4%) on the tetrahydropyran moiety and almost complete ring opening to the hydroxy acid Metabolite F (25).21 Clearly, based on the hepatocyte data, there is a tremendous difference in the concentration of parent 11 and metabolite composition, which sheds light on the mouse-rat disconnect for observed cholinergic toxicity in rat versus the more cholinergic sensitive mouse.
Figure 9.

Extensive metabolism (95.4%) of VU6007496 (11) in mouse hepatocytes, with metabolite F (25) produced in high abundance.
The team was still puzzled by the severe cholinergic adverse events observed in rats at the 100 and 300 mg/kg arms of the dose escalation study, as this was not consistent with the past 20 years of M1 PAM research. Could the N-demethylated metabolite A (24) be responsible? Due to the disconnect between liver S9 and hepatocytes, we felt it was prudent to examine in vivo rat MET ID at doses of 100 and 300 mg/kg to ensure 24 is produced upon in vivo oral dosing after 5–30 min and 160–240 min, and to determine if any other putative metabolites are generated at detectable levels (Figure 10). Across both doses and time points, 73–81% of the parent 11 remained. Metabolite 24 was produced in vivo at 2.2 to 5.6% relative abundance and lower than from in vitro incubations, along with two oxidative metabolites, a dioxygenated species M461 (26) and a mono-oxygenated species M445 (27) on the tetrahydropyranyl moiety. These were produced in higher relative abundance than 24, representing 7.7 to 13.8% (26) and 8.3 to 11.1% (27), respectively.21 A species such as 27 would resemble the hydroxy pyranyl congeners known to be potent ago-PAMs and prone to severe cholinergic side effects, and the potential for 24 to elicit similar cholinergic toxicity was unknown. However, it was clear from these data that PAM 11 rapidly generated stable metabolites with strong potential to be “active” metabolites.
Figure 10.

In vivo MET ID and metabolic pathways of VU6007496 (11) in mouse rat plasma at 100 mg/kg and 300 mg/kg p.o. at 5–30 min and 160–240 min at each dose. Three metabolites are produced in vivo: the N-demethylated 24, and two oxidative metabolites, a dioxygenated species M461 (26) and a mono-oxygenated species M445 (27) on the tetrahydropyranyl moiety.
Synthesis and Characterization of Metabolites
Due to the prevalence of metabolite A (VU6036463, 24) in both in vitro preparations and in vivo, we first synthesized and characterized 24. Utilizing intermediate 19 (Scheme 3), a Suzuki coupling with benzyl chloride 28 afforded a mixture of N-Boc protected analog 29 and N-H analog 29a in moderate isolated yield. Hydrolysis of the nitriles to the carboxylic acid, followed by a HATU-mediated coupling with tetrahydro-2H-pyran-4-amine delivered 24 in 81% yield over the two steps.21 In our kinetic assays, 24 was an “active” metabolite, with the profile of an M1 ago-PAM. Metabolite 24 was a potent M1 PAM on both rat (EC50 = 56 nM, 89% ACh max) and human (EC50 = 144 nM, 73% ACh max); in fact, more potent than the parent 11. PAM 24 also displayed M1 agonism at both the rat (EC50 = 3.1 μM, 48% ACh Max) and human receptors (EC50 = 2.1 μM, 36% ACh Max). As with the parent 11, metabolite 24 was inactive on rat and human M2–5. In our tier 1 in vitro DMPK panel, 24 displayed an acceptable profile with moderate predicted hepatic clearance (hCLhep = 12.8 mL/min/kg and rCLhep = 44 mL/min/kg), good unbound fraction in human (fu = 0.047), rat (fu = 0.134) and rat brain homogenate binding (fu = 0.025), and an acceptable CYP profile (>30 μM @ 1A2, 2C9, 2D6 and 5.7 μM @ 3A4). To ascertain if 24 could be the source of the adverse cholinergic events in rats, we dosed 11 and 24, in parallel, at a dose of 100 mg/kg i.p. in 30% captisol and prepared plasma and brain samples at a 3-h time point to determine brain exposure of the M1 ago-PAM 24. In this study, 11 achieved a total plasma concentration of 3.6 μM and a total brain concentration of 0.93 μM (Kp = 0.26). In contrast, and again—unexpectedly—the metabolite 24 displayed lower exposure in plasma (1.2 μM) and very low (0.08 μM) brain exposure (Kp = 0.07).21 These data suggest that the adverse events in rats was unlikely due to CNS activity of the “active” metabolite 24. To further confirm this in vivo finding, 24 was found to be a P-gp substrate (MDR1-MDCK ER = 31.9, Papp = 2.0 × 10–6 cm/s) rendering 24 a nonparticipant in the observed toxicity of 11. At the same time, we explored an N-CD3 congener of 11 to determine if the kinetic isotope effect would engender metabolic stability to the alkylated pyrazole and avoid the production of 24, but the metabolism proved to be identical to the N-CH3.
Based on the known cholinergic adverse events with hydroxy pyranyl amides such as 2–5,8−12 and the rat in vivo MET ID suggesting, by mass, that M445 (27) was a similar species, we prepared analog 30 (VU6007519) following Scheme 2. As shown in Figure 11, this putative metabolite was an extremely potent human M1 ago-PAM (PAM EC50 = 4.9 nM, 71% ACh Max; agonist EC50 = 176 nM, 60%), and this profile would likely give rise to the cholinergic adverse events seen in rats at doses over 100 mg/kg. Importantly, 30 was also brain penetrant in rat at 100 mg/kg i.p. (Kp = 0.38) and comparable to 11 (Kp = 0.26). To provide additional evidence that this “active” metabolite was responsible for the overactivation of M1 and the observed toxicity, coinjection of 30 with the rat in vivo MET ID proved that 30 was not in fact the metabolite 445 (27) despite identical masses.21 Thus, 27 was possibly another stereo- or regio-isomer of 30, but quite likely a potent M1 ago-PAM. While a valuable and interesting line of investigation, the project team had to refocus on ligands with the potential to advance as backups to our clinical asset, VU319/ACP-319.
Figure 11.

Structure and human M1 pharmacology of 30, an extremely potent M1 ago-PAM with CNS penetration in rat.
Conclusions
In summary, a lead optimization campaign to identify a suitable backup for the clinical compound, VU319/ACP-319, focused on scaffold-hopping from the pyrrolo[2,3-b]pyridine-based M1 PAM, VU6007477, to isomeric pyrrolo[3,2-b]pyridine and thieno[3,2-b]pyridine congeners. From this effort, VU6007496, a pyrrolo[3,2-b]pyridine, advanced into late stage profiling, only to be plagued with unanticipated, species-specific metabolism, in vitro/in vivo disconnects and “active” and potentially toxic metabolites. For this program, in vitro liver S9, hepatocyte and in vivo MET ID were critical to identify putative “active” metabolites. The unexpected and species-specific mouse metabolism thwarted our phenotypic cholinergic seizure liability in vivo screen as a stage gate, and thus prevented further development of VU6007496 as a backup clinical candidate. However, VU6007496 proved to be a highly selective and CNS penetrant M1 PAM (with minimal agonism), with excellent multispecies IV/PO PK, CNS penetration, no impact on long-term depression (or cholinergic toxicity) and robust efficacy in novel object recognition (MED = 3 mg/kg p.o.). Cholinergic toxicity was not observed in rats at 30 mg/kg p.o., providing a 10-fold window from the MED, and suggesting that VU6007496 can serve as another valuable in vivo rat tool compound, but not mouse, to study selective M1 activation in vivo.
Acknowledgments
The authors thank William K. Warren, Jr., and the William K. Warren Foundation for support of our programs and endowing both the Warren Center for Neuroscience Drug Discovery and the William K. Warren, Jr., Chair in Medicine (C.W.L.).
Glossary
Abbreviations
- PAM
positive allosteric modulator
- PBL
plasma/brain level
- DMPK
drug metabolism and pharmacokinetics
- AE
adverse event
- M1
muscarinic acetylcholine receptor subtype 1
- MED
minimum effective dose
- NOR
novel object recognition
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.4c00508.
Additional experimental details; methods for the synthesis and characterization of all compounds; in vitro and in vivo DMPK protocols; eurofins lead profiling screen data; synthesis of VU6006874 (12) (PDF)
Author Contributions
C.W.L., D.W.E., P.J.C., C.M.N., J.K.R., A.L.B., O.B. A.L.R. and H.P.C. oversaw the medicinal chemistry, target selection and interpreted biological/DMPK data. C.W.L. wrote the manuscript. J.L.E., K.A.B., R.A.C., M.F.L., A.M.B., D.W.E. performed chemical synthesis. C.C.P. performed and analyzed HRMS reports. H.P.C., A.L.R, and C.M.N. performed and analyzed in vitro pharmacology assays. S.P.M. and Z.X. and S.P.M. performed slice electrophysiology studies. J.W.D., W.P. and J.M.R. performed in vivo behavior pharmacology assays and in vivo DMPK. A.L.B. and O.B. performed in vitro and in vivo DMPK studies. All authors have given approval to the final version of the manuscript.
Studies were supported by NIH (NIMH, MH082867, MH073676 and MH108498) and Acadia Pharmaceuticals (UNIV61505).
The authors declare the following competing financial interest(s): We hold U.S. patents on M1 PAMs (the chemical series in this article are no longer under development) and are working with Acadia Pharmaceuticals on new, distinct chemical matter.
Supplementary Material
References
- Bodick N. C.; Offen W. W.; Levey A. I.; Cutler N. R.; Gauther S. G.; Satlin A.; Shannon H. E.; Tollefson G. D.; Rasmussen K.; Bymaster F. P.; Hurley D. J.; et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch. Neurol. 1997, 54 (4), 465–473. 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- Shekhar A.; Potter W. Z.; Lightfoot J.; Lienemann D.; Dube S.; Mallinckrodt C.; Bymaster F. P.; McKinzie D. L.; Felder C. C. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry. 2008, 165, 1033–1039. 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Brannan S. K.; Sawchak S.; Miller A. C.; Lieberman J. A.; Paul S. M.; Brier A. Muscarinic Cholinergic Receptor Agonist and Peripheral Antagonist for Schizophrenia. N. Engl. J. Med. 2021, 384 (8), 717–726. 10.1056/NEJMoa2017015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melancon B. J.; Tarr J. C.; Panarese J. D.; Wood M. R.; Lindsley C. W. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer’s disease. Drug Discovery Today 2013, 18 (23–24), 1185–1199. 10.1016/j.drudis.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges T. M.; LeBois E. P.; Hopkins C. R.; Wood M. R.; Jones J. K.; Conn P. J.; Lindsley C. W. Antipsychotic potential of muscarinic allosteric modulation. Drug News Perspect. 2010, 23, 229–240. 10.1358/dnp.2010.23.4.1416977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H. T. M.; van der Westhuizen E. T.; Langmead C. J.; Tobin A. B.; Sexton P. M.; Christopoulos A.; Valant C. Opportunities and challenges for the development of M1 muscarinic receptor positive allosteric modulators in the treatment for neurocognitive deficits. Br. J. Pharmacol. 2024, 181 (14), 2114–2142. 10.1111/bph.15982. [DOI] [PubMed] [Google Scholar]
- Dwomoh L.; Rossi M.; Scarpa M.; Khajehali E.; Molloy C.; Herzyk P.; Bottrill A. R.; Sexton P. M.; Christopoulos A.; Conn P. J.; Lindsley C. W.; Bradley S. J.; Tobin A. B. M1 muscarinic receptor activation reduces pathology and slows the progression of prion-mediated neurodegenerative diseases. Sci. Signaling 2022, 15, eabm3720 10.1126/scisignal.abm3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Seager M.; Wittman M.; Bickel N.; Burno M.; Jones K.; Graufelds V. K.; Xu G.; Pearson M.; McCampbell A.; Gaspar R.; Shughrue P.; Danzinger A.; Regan C.; Garson S.; Doran S.; Kreatsoulas C.; Veng L.; Lindsley C. W.; Shipe W.; Kuduk S.; Jacobson M.; Sur C.; Kinney G.; Seabrook G. R.; Ray W. J.; et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 15950–15955. 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey J. K.; Brady A. E.; Jones P. J.; Davis A. A.; Bridges T. M.; Jadhav S. B.; Menon U.; Christain E. P.; Doherty J. J.; Quirk M. C.; Snyder D. H.; Levey A. I.; Watson M. L.; Nicolle M. M.; Lindsley C. W.; Conn P. J. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and can restore impairments of reversal learning. J. Neurosci. 2009, 29, 14271–14286. 10.1523/jneurosci.3930-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beshore D. C.; Di Marco C. N.; Chang R. K.; Greshock T. J.; Ma L.; Wittman M.; Seager M. A.; Koeplinger K. A.; Thompson C. D.; Fuerst J.; Hartman G. D.; Bilodeau M. T.; Ray W. J.; Kuduk S. D. MK-7622: A first-in-class M1 positive allosteric modulator development candidate. ACS Med. Chem. Lett. 2018, 9, 652–656. 10.1021/acsmedchemlett.8b00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S. P.; Dickerson J. W.; Plumley H. C.; Xiang Z.; Maksymetz J.; Remke D. H.; Doyle C. A.; Niswender C. M.; Engers D. W.; Lindsley C. W.; Rook J. M.; Conn P. J.; et al. M1 positive lacking agonist activity provide the optimal profile for enhancing cognition. Neuropsychopharmacology 2018, 43, 1763–1771. 10.1038/s41386-018-0033-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoren J. E.; Garnsey M.; Pettersen B.; Brodeney M. A.; Edgerton J. R.; Fortin J.-P.; Grimwood S.; Harris A. R.; Jenkison S.; Kenakin T.; Lazzaro J. T.; Lee C.-W.; Lotarski S. M.; Nottebaum L.; O’Neil S. V.; Popiolek M.; Ramsey S.; Steyn S. J.; Thorn C. A.; Zhang L.; Webb D. Design and synthesis of γ- and δ-lactam M1 positive allosteric modulators (PAMs): convulsion and cholinergic toxicity of M1-selective PAM with weak agonist activity. J. Med. Chem. 2017, 60, 6649–6663. 10.1021/acs.jmedchem.7b00597. [DOI] [PubMed] [Google Scholar]
- Moran S. P.; Cho H. P.; Maksymetz J.; Remke D.; Hanson R.; Niswender C. M.; Lindsley C. W.; Rook J. M.; Conn P. J. PF-06827443 displays robust allosteric agonist and positive allosteric modulator activity in high receptor reserve and native systems. ACS Chem. Neurosci. 2018, 9, 1572–1581. 10.1021/acschemneuro.8b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook J. M.; Abe M.; Cho H. P.; Nance K. D.; Luscombe V. B.; Adams J. J.; Dickerson J. W.; Remke D. H.; Garcia-Barrantes P. M.; Engers D. W.; Engers J. L.; Chang S.; Foster J. J.; Blobaum A. L.; Niswender C. M.; Jones C. K.; Conn P. J.; Lindsley C. W. Diverse Effects on M1 Signaling and Adverse Effect Liability within a Series of M1 Ago-PAMs. ACS Chem. Neurosci. 2017, 8, 866–883. 10.1021/acschemneuro.6b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rook J. M.; Berton J. L.; Cho H. P.; Gracia-Barrantes P. M.; Moran S. P.; Maksymetz J. T.; Nance K. D.; Dickerosn J. W.; Remke D. H.; Chang S.; Harp J. M.; Blobaum A. L.; Niswender C. M.; Jones C. K.; Stauffer S. R.; Conn P. J.; Lindsley C. W. A novel M1 PAM VU0486846 exerts efficacy in cognition models without displaying agonist activity or cholinergic toxicity. ACS Chem. Neurosci. 2018, 9, 2274–2285. 10.1021/acschemneuro.8b00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engers J. L.; Childress E. S.; Long M. F.; Capstick R. A.; Luscombe V. B.; Cho H. P.; Dickerson J. W.; Rook J. M.; Blobaum A. L.; Niswender C. M.; Conn P. J.; Lindsley C. W. VU6007477, a novel M1 PAM based on a pyrrolo[2,3-b]pyridine carboxamide core devoid of cholinergic side effects. ACS Med. Chem. Lett. 2018, 9, 2641–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako Y.; Kurimoto E.; Mandai T.; Suzuki A.; Tanaka M.; Suzuki M.; Shimizu Y.; Yamada M.; Kiruma H. TAK-071, a novel M1 positive allosteric modulator with low cooperativity, improves cognitive function in rodents with few cholinergic side effects. Neuropsychopharmacology 2019, 44, 950–960. 10.1038/s41386-018-0168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W.; Mamashli F.; Buhl F. M. D. L.; Buhl D. L.; Khudyakov P.; Khudyakov P.; Volfson D.; Volfson D.; Martenyi F.; Martenyi F.; Gevorkyan H.; Gevorkyan H.; Rosen L.; Rosen L.; Simen A. A. Safety, pharmacokinetics and quantitative EEG modulation of TAK-071, a novel muscarinic M1 receptor positive allosteric modulator, in healthy subjects. Br. J. Pharmacol. 2022, 88, 600–612. 10.1111/bcp.14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley A. C.; Key A. P.; Blackford J. U.; Rook J. M.; Conn P. J.; Lindsley C. W.; Jones C. K.; Newhouse P. A. Cognitive performance effects following a single dose of the M1 muscarinic positive allosteric modulator VU319. Alzheimer’s Dementia 2020, 16, e045339 10.1002/alz.045339. [DOI] [Google Scholar]
- Conley A. C.; Key A. P.; Blackford J. U.; Rook J. M.; Conn P. J.; Lindsley C. W.; Jones C. K.; Newhouse P. A. Functional activity of the muscarinic positive allosteric modulator VU319 during a Phase 1 single ascending dose study. Am. J. Geriatr. Psychiatry 2021, 29, S43 10.1016/j.jagp.2021.01.038. [DOI] [Google Scholar]
- See Supporting Information for full details.
- Storz T.; Bartberger M. D.; Sukits S.; Wilde C.; Soukup T. The first practical and efficient one-pot synthesis of 6-substituted 7-azaindoles via a Reissert-Henze reaction. Synthesis 2008, 2008, 201–214. 10.1055/s-2007-1000853. [DOI] [Google Scholar]
- Hamilton S. E.; Loose M. D.; Qi M.; Levey A. I.; Hille B.; McKnight G. S.; Idzerda R. L.; Nathanson N. M. Disruption of the M1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 13311–13316. 10.1073/pnas.94.24.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löscher W.; Ferland J.; Ferraro T. N. The relevance of inter- and intrastrain differences in mice and rats and their implications for models of seizures and epilepsy. Epilepsy Behav. 2017, 73, 214–235. 10.1016/j.yebeh.2017.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.