

Abstract
Background and Aims:
Pathogenic variants in the NARS1 gene, which encodes for the asparaginyl-tRNA synthetase1 (NARS1) enzyme, were associated with complex central and peripheral nervous system phenotypes. Recently, Charcot–Marie–Tooth (CMT) disease has been linked to heterozygous pathogenic variants in NARS1 in nine patients. Here, we report two brothers and their mother from a French family with distal hereditary motor neuropathy (dHMN) carrying a previously unreported NARS1 variant.
Methods:
The NARS1 variant (c.1555G>C; p.(Gly519Arg)) was identified through whole-genome sequencing (WGS) performed on the family members. Clinical findings, nerve conduction studies (NCS), needle electromyography (EMG), and functional assays in yeast complementation assays are reported here.
Results:
The family members showed symptoms of dHMN, including distal weakness and osteoarticular deformities. They also exhibited brisk reflexes suggestive of upper motor neuron involvement. All patients were able to walk independently at the last follow-up. NCS and EMG confirmed pure motor neuropathy. Functional assays in yeast confirmed a loss-of-function effect of the variant on NARS1 activity.
Interpretation:
Our findings expand the clinical spectrum of NARS1-associated neuropathies, highlighting the association of NARS1 mutations with dHMN. The benign disease course observed in our patients suggests a slowly progressive phenotype. Further reports could contribute to a more comprehensive understanding of the spectrum of NARS1-associated neuropathies.
Keywords: asparaginyl-tRNA synthetase, Charcot–Marie–Tooth disease, distal hereditary motor neuropathy, NARS1
1 ∣. BACKGROUND AND AIMS
Recently, Beijer et al. (2024) reported that Charcot–Marie–Tooth (CMT) disease could be caused by heterozygous pathogenic variants in the NARS1 gene.1 CMT disease is a group of heterogeneous inherited neuropathies involving motor and sensory neurons, classified into three major subtypes based on nerve conduction study (NCS): a demyelinating form (CMT1 or CMT4, respectively for autosomal dominant or recessive inheritance), an axonal form (CMT2), and an intermediate form (CMT-I). When only motor neurons are involved, the term distal hereditary motor neuropathy (dHMN) is used. Asparaginyl-tRNA synthetase1 (NARS1) is a ubiquitously expressed enzyme that catalyzes the ligation of tRNA to asparagine in the cytoplasm, a crucial process for translation.2 Using whole-exome sequencing (WES), the authors identified three variants in the heterozygous state (p.-Met236del, p.Cys342Tyr, and p.Ser461Phe) in NARS1 in three unrelated CMT families.1 Nine patients belonging to these three families had late-onset sensorimotor neuropathy, responsible for walking impairment, foot deformities, and distal sensory loss.1 The NCS disclosed axonal features in seven patients (CMT2), and mildly reduced motor nerve conduction velocities compatible with an intermediate form in another one (CMT-I). The remaining patient did not undergo NCS.1 One of these seven individuals had associated central signs characterized by intellectual disability, spasticity, and cerebellar ataxia (CMT2+). Moreover, the authors provided functional evidence in yeast complementation assays and a Nars1 knock-in mouse, supporting the pathogenicity of these variants.1 Previously, NARS1 mutations had only been reported in patients with more complex phenotypes involving the central (microcephaly, seizures, ataxia) and peripheral (demyelinating neuropathy) nervous systems.2,3 Mutations in six other genes of the aminoacyl-tRNA synthetase family (AARS1, GARS1, HARS1, SARS1, WARS1, YARS1) had already been associated with dominant CMT.1 Here, we report two brothers and their mother from a French family who are affected by dHMN and who are heterozygous for a previously unreported NARS1 variant.
2 ∣. CASE REPORT
Whole-genome sequencing (WGS) analysis performed on parents and children revealed the following variant in NARS1 (NM_004539.4): c.1555G>C; p.(Gly519Arg). This variant was confirmed by Sanger sequencing and segregated with the disease in the family (Figure 1A). The variant, which was absent from the gnomAD v4, deCAFF, and Allo-fUS databases, affected a highly conserved amino acid residue (from baker's yeast to human) and was predicted to be probably pathogenic by bioinformatic tools (CADD Phred 26.3; REVEL 0.973; AlphaMissense 0.998). This amino acid maps to motif 3 involved in the NARS1 active site.3 No other pathogenic/probably pathogenic variants were identified. The patients gave their informed consents, and the study was carried out in accordance with the 1964 Helsinki declaration.
FIGURE 1.
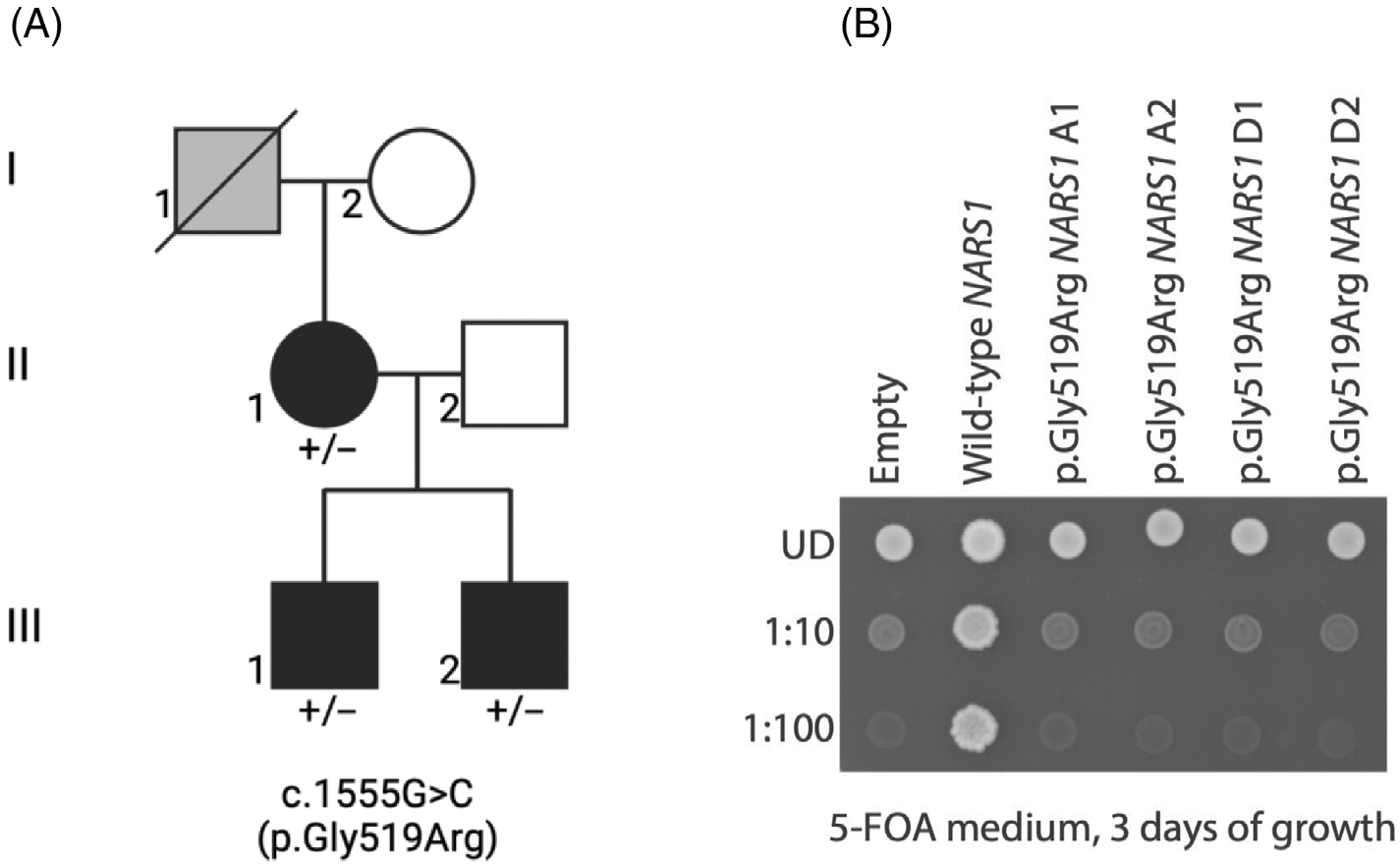
(A) Pedigree of the family showing co-segregation of the variant with the disease phenotype with affected individuals in black, unaffected individuals in white, and hearsay-affected individuals without clinical and genetic investigations in grey. (B) Haploid yeast lacking endogenous DED81 were transformed with plasmids expressing wild-type or human NARS1, or with a vector containing no NARS1 insert (“Empty”). Resultant yeast cultures were plated undiluted (UD) or diluted (1:10 or 1:100) on solid media containing 5-FOA and grown at 30°C for 3 days. Replicates “A” and “D” indicate transformations using two independently generated plasmid preparations, and “1” and “2” refer to individual colonies selected from each transformation.
Proband II-1 had normal motor milestones. She complained of walking difficulties due to cramps in the lower limbs in her 30s. When she was assessed at the age of 36 years, pes cavus was noted. Hyperlaxity was also present in the upper limbs. Motor examination revealed a distal weakness affecting wrist flexion (Medical Research Council [MRC] grade 4) and fingers' abduction (grade 3). In the lower limbs, a motor weakness was found in toes dorsiflexion (grade 3), hallux dorsiflexion (grade 4), ankle dorsiflexion (grade 4), and plantar flexion (grade 4). She did not complain of sensory loss. Her deep tendon reflexes were brisk in the four limbs. Nerve conduction studies showed compound muscle action potentials (CMAP) of reduced amplitude in the lower limbs, while sensory nerve action potentials (SNAP) were normal in the four limbs. Needle electromyography (EMG) showed high-amplitude motor unit action potentials (MUAP) in the distal muscles of the four limbs. The recruitment pattern was reduced. Brain and spine MRI were normal. A cardiac workup including electrocardiogram, ambulatory electrocardiogram, and echocardiography was normal. At her last follow-up at 47 years of age, her neurological examination was stable. She was still ambulant, with no need for a walking aid. Her father (proband I-1) also presented pes cavus, but could not be evaluated as he was dead.
Proband III-1 had normal motor milestones. Before the age of 5, he began to walk on tiptoe. His neurological examination at 9 years revealed normal motor testing in the four limbs. His deep tendon reflexes were brisk in the four limbs. Ankles contractures and pes cavus were noted, as well as mild kyphosis. He was not able to walk on his heels due to these contractures. He complained of pain in the calf during running. His creatine kinase (CK) level was normal. He was able to run, climb stairs, and practice different sports activities (biking and football). His walk progressively improved and was considered normal at 18 years of age. At this age, the ankle contractures were still present, and a mild weakness was noted on toes extension (grade 4). Deep tendon reflexes were still brisk. The NCS was normal. Needle EMG showed rare fasciculations in the distal muscles of the lower limbs at rest, while MUAPs were found in these muscles during contraction. The recruitment pattern was reduced. At the last follow-up at 20 years of age, he was ambulant with no need for a walking aid and played rugby.
Proband III-2 had a disease course similar to that of his brother. He had normal motor milestones. From the age of 3, he walked on tiptoe. When he was assessed at 5 years of age, no motor weakness was found. His deep tendon reflexes were brisk in the four limbs, and ankle contractures were found. He had difficulty walking on his heels due to these contractures. A left Babinski sign was present. He was able to run and climb stairs. Brain and spinal cord MRI were normal. NCS was normal. His walk progressively improved during his early teenage years. At the last follow-up at 16 years of age, his walk was considered normal, and he was able to play football. His motor testing remained normal, but a mild atrophy of intrinsic foot muscles was observed along with kyphosis. NCS repeated at this time revealed reduced CMAP amplitudes in fibular nerves. Needle EMG showed high-amplitude MUAPs in the left tibialis anterior muscle, with a reduced recruitment pattern.
The clinical and paraclinical findings of the three patients are summarized in Table 1. None of them had seizures or intellectual disability.
TABLE 1.
Patients' clinical and paraclinical findings.
| Proband (gender) | II-1 (F) | III-1 (M) | III-2 (M) |
|---|---|---|---|
| Age at onset (years) | 30s | <5 | 3 |
| Muscle weakness | Distal UL + LL | Distal LL | − |
| Sensory loss | − | − | − |
| Contracture | − | + (ankles) | + (ankles) |
| Hyperlaxity | + | + | + |
| Pyramidal signs | Brisk DTR | Brisk DTR | Brisk DTR, Babinski sign |
| Osteoarticular deformities | Pes cavus | Pes cavus, kyphosis | Pes cavus, kyphosis |
| CNS signs | − | − | − |
| NCS | Decreased CMAP amplitudes in LL, normal SNAP amplitudes | Normal | Decreased CMAP amplitudes in LL, normal SNAP amplitudes |
| Needle EMG | High-amplitude MUAPs, reduced recruitment | High-amplitude MUAPs, reduced recruitment | High-amplitude MUAPs, reduced recruitment |
Abbreviations: CMAP, compound muscle action potential; CNS, central nervous system; DTR, deep tendon reflexes; EMG, electromyography; LL, lower limbs; MUAP, motor unit action potential; NCS, nerve conduction study; SNAP, sensory nerve action potential; UL, upper limbs.
Yeast has proven to be an effective model system to study neuropathy-associated aminoacyl-tRNA synthetase mutations, with the vast majority of pathogenic variants causing loss-of-function and dominant negative effects.4,5 To determine the consequences of the p. (Gly519Arg) variant on NARS1 function, yeast complementation assays were performed. A haploid ΔDED81 yeast strain, the ortholog of human NARS1, was generated as previously described.1,6 Constructs to express wild-type or p.(Gly519Arg) human NARS1, or a construct with no NARS1 insert (“Empty”), were independently transformed into the yeast strains, which were then grown on media containing 5-FOA to select for loss of a maintenance vector that expresses wild-type DED81.1 After 3 days of incubation at 30°C, wild-type human NARS1 rescued yeast viability, whereas both the “Empty” vector and the constructs to express p. (Gly519Arg) human NARS1 did not allow any yeast growth (Figure 1B; a similar effect was seen after 5 days of growth [data not shown]). These data indicate that the p.(Gly519Arg) NARS1 variant causes a loss-of-function effect similar to other neuropathy-associated aminoacyl-tRNA synthetase alleles, including the previously reported NARS1 variants.1
3 ∣. INTERPRETATION
By analyzing WGS data in three members of a family presenting with a phenotype of dHMN, we identified a previously unreported c.1555G>C; p.(Gly519Arg) variant in NARS1. First, we confirm that hereditary neuropathy can be associated with NARS1 mutations by reporting this French family with three affected patients. These cases expand the clinical spectrum of NARS1-associated neuropathies, as the previously published patients had a sensorimotor phenotype, while our patients presented with pure motor axonal neuropathy corresponding to dHMN. Interestingly, these three patients exhibited pyramidal features suggestive of upper motor neuron involvement. Such a phenotype has already been described for other dHMN genes, such as BSCL2 and COQ7.7,8 Our patients share common features with the patients described by Beijer et al. Pes cavus was observed in two of three patients reported herein, and in eight of nine patients reported by Beijer et al.1 None of our patients had central signs such as seizures, intellectual disability, or cerebellar ataxia, which was the case in eight of nine patients in their study.1 No demyelinating abnormalities were found on NCS in our patients, and only one of the previously published patients had low motor nerve conduction velocity on the ulnar nerve and was classified as CMT-I.1 The remaining seven patients with available NCS findings were classified as CMT2.1 No walking aid was needed in our three patients, as in five of their nine patients. Thus, the disease course seems to be relatively benign, with a slowly progressive worsening.1 However, while the age at onset was late in the nine patients reported by Beijer et al. as in our proband II-1, probands III-1 and III-2 from our study developed motor signs in early childhood.1 The yeast model presented herein, which is a validated method, supports a loss-of-function effect of the p.(Gly519Arg) variant. Nevertheless, an associated gain-of-function mechanism cannot be ruled out, as several ARS genes responsible for dominant neuropathies have been reported to exert a gain-of-function effect.4
In summary, this work extends the clinical spectrum associated with pathogenic NARS1 variants, which now includes dHMN.
ACKNOWLEDGEMENTS
This research was made possible through access to the data generated by the France Genomic Medicine Plan 2025.
FUNDING INFORMATION
Sheila Marte is supported by the Michigan Pre-doctoral Training Program in Genetics (GM007544). Anthony Antonellis is supported by a grant from the National Institute of General Medical Sciences (GM136441).
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1.Beijer D, Marte S, Li JC, et al. Dominant NARS1 mutations causing axonal Charcot–Marie–Tooth disease expand NARS1-associated diseases. Brain Commun. 2024;6(2):fcae070. doi: 10.1093/braincomms/fcae070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang L, Li Z, Sievert D, et al. Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nat Commun. 2020;11(1):4038. doi: 10.1038/s41467-020-17454-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manole A, Efthymiou S, O'Connor E, et al. De novo and bi-allelic pathogenic variants in NARS1 cause neurodevelopmental delay due to toxic gain-of-function and partial loss-of-function effects. Am J Hum Genet. 2020;107(2):311–324. doi: 10.1016/j.ajhg.2020.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer-Schuman R, Antonellis A. Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum Mol Genet. 2017;26(R2):R114–R127. doi: 10.1093/hmg/ddx231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer-Schuman R, Marte S, Smith TJ, et al. A humanized yeast model reveals dominant-negative properties of neuropathy-associated alanyl-tRNA synthetase mutations. Hum Mol Genet. 2023;32(13):2177–2191. doi: 10.1093/hmg/ddad054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oprescu SN, Griffin LB, Beg AA, Antonellis A. Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods. 2017;113: 139–151. doi: 10.1016/j.ymeth.2016.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6–14. doi: 10.1136/jnnp-2011-300952 [DOI] [PubMed] [Google Scholar]
- 8.Jacquier A, Theuriet J, Fontaine F, et al. Homozygous COQ7 mutation: a new cause of potentially treatable distal hereditary motor neuropathy. Brain. 2023;146(8):3470–3483. doi: 10.1093/brain/awac453 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.