

Abstract
Nanofiber-based hydrogel delivery systems have recently shown great potential in biomedical applications, specifically due to their high surface-to-volume ratio of ultra-fine nanofibers and their ability to carry low solubility drugs. Herein, we introduce a visible light-triggered in situ-gelling drug vehicle (GAP Gel) composed of ascorbyl palmitate (AP) nanofibers and gelatin methacryloyl polymer. AP nanofibers form self-assembled structures through intermolecular interactions with a hydrophobic drug-loading core. We demonstrate that the hydrophilic periphery of AP nanofibers allows them to interact with other hydrophilic molecules via hydrogen bonds. The presence of AP nanofibers significantly enhances the viscoelasticity of GAP Gel in a concentration-dependent manner. Further, GAP Gel shows in vitro biocompatibility and sustained drug delivery efficacy when loaded with a hydrophobic antibiotic. Likewise, GAP Gel shows excellent in vivo biocompatibility when implanted in immunocompetent mice in various forms. Lastly, GAP Gels maintain cell viability when cultured in a 3D-environment over 7 days, establishing it as a promising and versatile hydrogel platform for the delivery of biotherapeutics.
Subject terms: Biomedical materials, Gels and hydrogels
Nanofiber-based hydrogels are useful delivery systems in biomedical applications due to their drug loading capability and controlled release. Here, a biocompatible visible light-triggered in situ-gelling drug delivery system is demonstrated consisting of ascorbyl palmitate nanofibers and gelatin methacryloyl polymer.
Introduction
The development of drug delivery systems has gained increasing popularity as a means to efficiently administer drugs and improve therapeutic outcomes. However, many carriers fail to address the delivery of hydrophobic molecules, limiting their use in clinical settings1. This limitation poses a major challenge as 40% of marketed drugs and 70–90% of drug candidates in the discovery pipeline are hydrophobic and lead to reduced therapeutic efficacy2,3. These drugs are often delivered without a suitable delivery vehicle, resulting in low bioavailability, low retention time in the systemic circulation, rapid metabolism, and a need for multiple dosages4. Consequently, there has been an upsurge in developing hydrophobic drug delivery vehicles; however, these carriers are often limited by their drug loading capacity.
To address this issue, nanofibers have emerged as a leading candidate for loading hydrophobic therapeutics due to their large surface area and high surface-to-volume ratio5,6. Natural (collagen7, chitosan8, hyaluronic acid9) and synthetic (polyvinyl alcohol10, polyimide11, polylactic acid12) polymers have been used to develop nanofibers for drug loading5. While these materials can successfully sustain the release of loaded molecules and can be synthesized with specifically tuned properties, they are often associated with lengthy, complex, costly, and energetically expensive syntheses12–14. In contrast, nanofibers with self-assembling properties can provide a greener and more sustainable synthesis by decreasing costs and energy associated with lengthy syntheses and purification processes15–19.
Ascorbyl Palmitate (AP), a vitamin C derivative made up of ascorbic acid and palmitic acid, has previously been shown to self-assemble and is classified as a Generally Recognized as Safe (GRAS) material by the United States Food and Drug Administration20–22. Notably, AP is an amphiphilic molecule that consists of a polyhydroxyl sugar hydrophilic head group and a polymethylene hydrocarbon hydrophobic chain. The presence of these functional groups allows for AP to be reinforced with two types of interactions: hydrogen bonding and van der Waals interactions20. As such, AP can self-assemble into an interdigitated bilayer to form a polymer-based nanofiber that can encapsulate and protect hydrophobic small molecules in the hydrophobic core of the polymer. Moreover, as a vitamin C derivative, AP retains the antioxidant bioactivity of its counterpart and can be exploited for its therapeutic effects related to oxidative stress and tissue and wound healing23.
Here, we will be exploring AP as a bioactive nanofiber to encapsulate a model hydrophobic drug, Erythromycin (Ery), a small molecule bacteriostatic antibiotic. To further increase its bioavailability and its efficacy as a mechanically resilient hydrophobic drug delivery vehicle, we will exploit the functional groups of AP nanofibers to interact with hydrophilic polymers. We hypothesize that AP nanofibers will be able to interact with gelatin methacryloyl (GelMA Gel) via hydrogen bonding. We have leveraged the benefits of GelMA-AP nanocomposite hydrogels (GAP Gel), including biocompatibility, biodegradability, ability to support cell attachment and retention, antioxidant activity, and mechanical resilience, to show its potential use in wound healing. Further, the flexibility in the fabrication of GAP Gels allows us to deliver the hydrogel precursor to a skin wound via injectable or spray-based methods, followed by visible light-triggered in situ gelation for less invasive treatment for soft tissues and ease of use.
Results and discussion
Ascorbyl palmitate self-assembles into fibrous nanofibers
AP is an amphiphilic molecule capable of forming multiple intermolecular interactions, allowing it to self-assemble. AP consists of a polyhydroxyl sugar hydrophilic head group and a polymethylene hydrocarbon hydrophobic chain20. These structural features allow AP nanofibers to self-assemble by forming hydrogen bonding networks between the hydrophilic head groups and van der Waals interactions between the hydrocarbon chains (Fig. 1A). Further, AP has a Krafft point of ~55–60 °C in a dimethyl sulfoxide (DMSO)/water mixture24,25. When heated in DMSO/water above the Krafft point, AP dissolves due to micelle formation. Upon cooling, AP forms liquid crystals in the form of self-assembled fibrous structures and a semi-solid viscous system20. During this self-assembly process, hydrophobic drugs can be introduced into the solution in order for the drug to be encapsulated within the hydrophobic core of the lamellar nanofibers. Here, we loaded a hydrophobic antibiotic, Ery, into AP nanofibers before the self-assembly process, such that Ery was encapsulated within the hydrophobic core of AP nanofibers. We confirmed the presence of Ery in the AP nanofibers via energy-dispersive X-ray spectroscopy (EDX) (Fig. 1B). The EDX spectra of AP-Ery nanofibers showed the corresponding peaks for carbon and oxygen which are constitutional elements found in AP nanofibers. The spectra also displayed an additional peak corresponding to nitrogen that can only be attributed to the nitrogen present in Ery. Moreover, transmission electron microscopy (TEM) confirmed the fibrous structure of AP-Ery nanofibers that were ~700 nm in length and 50 nm in thickness (Fig. 1C). However, upon loading of Ery in AP nanofibers, the viscosity of the nanofibers decreases (Supplementary Fig. 1).
Fig. 1. Preparation and characterization of Ascorbyl Palmitate (AP) nanofibers.

A Schematic of erythromycin-loaded ascorbyl palmitate nanofiber formation via self-assembly through hydrophobic and hydrophilic interactions, followed by the degradation of nanofibers and subsequent drug release. B EDX analysis of AP and AP-Ery nanofibers showing the absence or presence of Erythromycin molecules through the nitrogen peak in AP and drug-loaded AP nanofibers. C TEM images showing AP-Ery as long, fibrous structures ~700 nm in length and 50 nm in thickness. D The cumulative drug release profiles of varying concentrations of Erythromycin-loaded AP nanofibers in PBS over 96 h. E The scavenging ability of varying concentrations of AP nanofibers exposed to 0.4 mM DPPH.
After confirming the self-assembly of Ery-loaded AP nanofibers, we assessed the drug release profile of Ery from various concentrations of AP nanofibers to investigate whether there is potential to tailor the release of the drug towards a desired application. To determine the drug release profile of Ery from AP nanofibers, we encapsulated Ery into various concentrations of AP nanofibers (1.5%, 2.5%, 3.5%, 4.5% w/v) and measured the amount of drug encapsulated, followed by the cumulative drug release of loaded drug in phosphate buffered saline (PBS, pH 7.4) at 37 °C (Supplementary Fig. 2, Fig. 1D). We observed a slightly higher drug loading efficiency in lower concentrations of AP nanofibers; however slower release of Ery was observed with increasing concentrations of AP nanofibers. 1.5% (w/v) AP-Ery nanofibers showed ~80% release within 16 h, whereas 4.5% (w/v) AP-Ery nanofibers showed only ~20% release by the same time. This trend in the observed release kinetics can be attributed to the differences in molecular weight. As the molecular weight of the fibers increases, the mobility of the fibers decreases and thus, releases the drug more slowly26. This can be further confirmed when comparing AP nanofibers (2.5% w/v) to a bulk AP Gel (8% w/v). While AP nanofibers are able to steadily release Ery in the first 24 h, Ery-loaded AP Gel retains the drug much more strongly (Supplementary Fig. 3A). When assessing the bioactivity of the drug-loaded AP nanofibers or AP Gel loaded with 600 µg/mL Ery (for 2 mL culture), it was observed that AP nanofibers were able to release enough drug to prevent bacterial growth (Supplementary Figs. 3B–D, 4). However, the slow release of the drug from the AP Gel was not enough to prevent growth in 16 h. Therefore, drug delivery and release can be tailored to the desired application based on the concentration of AP nanofibers.
One potential application of AP is its role as an antioxidant to accelerate the wound healing process. As a vitamin C derivative, AP is a known antioxidant. It has previously been incorporated into an array of skin care products for being a free radical scavenger and plays a role in controlling wound oxidative stress in a tissue and wound healing environment23,27. To determine its scavenging ability, various concentrations of AP nanofibers were exposed to 0.4 mM of 2,2-diphenyl-1-picrylhydrazyl (DPPH), a stable free radical (Fig. 1E). As the concentration of AP nanofibers increases, the scavenging activity also increases. Subsequently, 2.5% AP nanofibers were exposed to varying concentrations of DPPH in order to determine the extent of antioxidant activity (Supplementary Fig. 5). As the concentration of DPPH increases, AP nanofibers show increased scavenging activity. This scavenging activity indicates that GAP Gels can aid in tissue and wound healing through their inherent ability to lower levels of oxidizing agents, such as reactive oxygen species, thereby initiating cell survival signaling and protecting tissues against infection28.
AP nanofibers are capable of hydrogen bonding with GelMA Gel networks
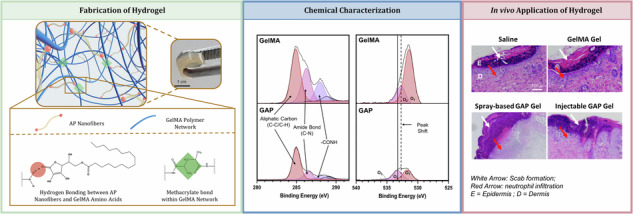
As mentioned previously, the structural features of AP give it the ability to form nanofibers through the presence of hydrophobic interactions and hydrogen bonding. We aimed to further exploit the structural features of AP nanofibers to investigate whether these nanofibers could be incorporated into GelMA Gels to further support its structure. Upon mixing, hydrogen bonds formed between the hydrophilic head groups of AP nanofibers and amino acids present within GelMA Gel (Fig. 2A).
Fig. 2. Fabrication of GAP Gels.

A Schematic of the formation of GAP Gels reinforced with hydrogen bonds by incorporating AP nanofibers and GelMA prepolymer. B XPS analyses of 1s C (left) and 1s O (right) spectra of GelMA and GAP Gels show characteristic peaks of GelMA Gel present in both samples as well a peak shift indicating hydrogen bonding between GelMA Gel and AP nanofibers.
Considering the structural features of AP, including hydroxyl functional groups, it becomes a reasonable hypothesis that hydrogen bonding may occur between AP nanofibers and GelMA Gel—a hydrogel composed of amino acids whose secondary structures are facilitated primarily based on their capacity to hydrogen bond. After mixing the AP nanofibers and GelMA Gel in a 1:3 ratio, we investigated the surface chemistry at the interface of a cross-section of GelMA and GAP Gels using X-ray photoelectron spectroscopy (XPS) (Fig. 2B). High-resolution C 1s spectra of GelMA and GAP Gel cross-sections show the presence of carbon peaks characteristic of GelMA Gel in both samples: (1) the presence of the aliphatic carbon (C-C/C-H) at 284.93 eV, (2) the amide bond (C-N) at 286.22 eV, and (3) the -CONH peak present at 289.03 eV29. At the same time, the O 1s spectra indicate a shift of the O2 peak towards higher ionization energy, where GelMA Gel shows a peak at 532.82 eV, and GAP Gel shows a peak at 533.47 eV. This shift towards higher ionization energy indicates the presence of hydrogen bonding between AP nanofibers and the GelMA Gel polymeric network30,31. Therefore, we can speculate that AP nanofibers do not interfere with the GelMA Gel polymeric network but instead, are dispersed within the sample and can act as reinforcements within the structure through intermolecular interactions.
Although there was an indicated presence of intermolecular interactions between GelMA Gel and AP nanofibers, the addition of nanofibers into a polymeric network can often interfere with the structure of that network. To ensure AP nanofibers were not interfering with the GelMA Gel polymer network, we used Fourier-transform infrared spectroscopy (FT-IR) to confirm the presence of the characteristic peaks of AP nanofibers and GelMA Gel within GAP Gels (Fig. 3A). To confirm the hydrogen bond interactions previously suggested by XPS, GAP Gel was further chemically characterized.
Fig. 3. Chemical characterization of GAP Gels.

A FTIR analyses of GAP Gel, GelMA Gel, and AP nanofibers highlight the characteristic peaks of GelMA Gel (orange) and AP nanofibers (purple). B NMR spectra of glycine (top), AP (middle), and a mix of glycine and AP (bottom) highlight the formation of hydrogen bonding between glycine and AP. C TGA derivative curves show the structural differences between GelMA and GAP Gels.
Since hydrogen bonding inherently involves a redistribution of electron density around hydrogen atoms, Nuclear Magnetic Resonance (NMR) emerges as an appropriate tool to ascertain the existence of such interactions (Fig. 3B). Glycine was chosen as the co-partner molecule for hydrogen bonding due to its (1) presence in the GelMA Gel polymeric network32, (2) simple NMR spectra, which minimizes interference with other spectral peaks, and (3) the versatility of its carboxylic acid functional group, capable of acting as both a hydrogen bond donor and a hydrogen bond acceptor33. The discernible broad peak observed at 8.61 ppm in the AP spectra is consistent with the hydroxyl group attached to the lactone moiety. Notably, after adding glycine to the solution, this peak at 8.61 ppm is no longer evident in the NMR spectra. This could be due to the extensive broadening of the peak after hydrogen bond formation, making it indistinguishable from the baseline34. Therefore, the disappearance of the peak at 8.61 ppm indicates that the hydroxyl groups attached to the lactone portion of AP can serve as hydrogen bond donors.
In agreement with NMR and XPS, the degradation of GAP and GelMA Gels also indicates the presence of hydrogen bonding in the GAP Gel, as measured by thermogravimetric analysis (TGA) (Fig. 3C). GAP and GelMA Gels show the gelation peaks at 50 °C and 330 °C. However, the GAP Gel shows continued degradation between 80–100 °C and ~160 °C. Intermolecular bonds linked through hydrogen bonds tend to release between 80–120 °C, whereas intermolecular bonds through interactions to carboxylate groups tend to release from 160 °C35,36. The thermogravimetric profile of GAP Gel is indicative of hydrogen bonds as the gel further degrades due to the intermolecular bonds released through weaker and stronger hydrogen bonds. Through FT-IR, NMR, XPS, and TGA, we have gained a good understanding of the intermolecular interactions between AP nanofibers and the GelMA Gel polymeric network within GAP Gels.
AP nanofibers reinforces the physical properties of GAP Gels
We aim to use GAP Gels as a topical antibacterial delivery vehicle. For this reason, hydrogels need to be elastic and should regain their structural stability when subjected to external deformative forces. We investigated the mechanical properties of the polymer network of GAP Gels to determine the potential of GAP Gels for topical applications. We hypothesized that hydrogels with increasing concentrations of AP nanofibers would result in increasing elasticity. To test this hypothesis, we subjected GAP Gels with increasing concentrations of AP nanofibers to shear stress applied by a rheometer (Fig. 4A–C, Supplementary Fig. 6). Results showed that as the concentration of AP nanofibers in the hydrogel increased, the values of storage modulus increased, indicating increased elasticity of the hydrogel, likely due to the intermolecular forces between the polymer network of GelMA Gel and the AP nanofibers. Further, results showed that for all gels, the storage modulus was higher than the loss modulus when the oscillation stress was smaller than 100 Pa, indicating the gel-like nature of the nanocomposite hydrogel. When the oscillation stress was larger than ~100 Pa, the values of the storage modulus decreased below that of the loss modulus, indicating that an increase in the oscillation stress broke the crosslinks of the hydrogel.
Fig. 4. Fabrication of GAP Gels through in situ gelation.

A Schematic displaying how the rheometer measures the translation of torque to shear stress. B, C Representative stress sweeps show that the hydrogels remain at the linear viscoelastic region within 10% of the applied strain and an increase in the storage modulus with increasing GAP Gel concentrations. D SEM images on the left display the pore sizes of GelMA and GAP Gels. Representative pictures of the hydrogels display the intact hydrogel being manually compressed. E Graph displaying the swelling profile of GelMA and GAP Gels. The swelling was reduced significantly with the introduction of AP nanofibers (Scale bar = 1 mm). F Degradation profile of GelMA and GAP Gels over 14 days.
Overall, the addition of AP nanofibers to GelMA Gel enhanced the elasticity of the hydrogel network in a concentration-dependent manner. This concentration-dependence is mainly attributed to hydrogen bonds between AP nanofibers and GelMA Gel.
We further demonstrated the elasticity of GAP Gels qualitatively by physically compressing the gels. GAP and GelMA Gels in their original shape are shown (Fig. 4D). When compressed, GelMA Gels broke under compression, while GAP Gel returned to its original shape when the compression was released, showing better structural integrity than GelMA Gel (Supplementary Movies 1, 2). Using scanning electron microscopy (SEM), we observed a porous structure of GAP Gel, which indicates suitable tissue integration and nutrient transfer37. Similarly, the degradability and swelling profiles can also indicate the tissue integration capabilities of the nanocomposite hydrogel. We measured the swelling ratio of GAP Gels to determine the water absorption capacity. In comparison to GelMA Gels, GAP Gels indicated a lower swelling ratio of 741.1% ± 23.7% compared to GelMA Gels, which swelled 822.5% ± 23.8% by 16 h in PBS (pH 7.4) (Fig. 4E, Supplementary Fig. 7). The water absorption capacity of GAP Gels could indicate increased cellular adhesion and tissue integration38,39. Taken together, we explored the possibility of GAP Gels’ ability to encapsulate and retain mammalian cells (Supplementary Fig. 8A). Over 7 days, we observed the 3D culture of NIH 3T3 cells within GAP Gels. Cells continued to grow and spread over time, and cells encapsulated within GAP Gel showed no visible difference in morphology or cell density compared to cells encapsulated within GelMA Gels (Supplementary Fig. 8B). After exploring the potential of cell encapsulation in GAP Gels due to their pore size and swelling capabilities, we investigated the in vitro degradation of GelMA and GAP Gels by incubating them in PBS (pH 7.4) for up to 14 days (Fig. 4F). GAP Gels were biodegradable and showed a higher degradation rate than GelMA Gels, with GAP Gels degrading ~8–10% higher at each time point. This degradation rate may be due to the added oxidative degradation of the gels by adding AP40. Overall, GAP Gels with lower swelling ratios than GelMA Gels demonstrated greater biodegradability under physiological conditions, which can be advantageous for soft tissue integration and cellular adhesion in vivo.
GAP Gels are cytocompatible with human stem cells in vitro
Recently, GelMA Gels have been used extensively for biomedical applications41,42, as has AP powder in the beauty industry43. However, one potential concern for GAP Gels was if the biocompatibility of the individual components would be maintained in the nanocomposite hydrogel. We evaluated the cytotoxicity of GAP Gels in vitro by investigating the mitochondrial activity of human mesenchymal stem cells (MSCs) exposed to GAP Gels for 24 h using MTS assay (Fig. 5A). The mitochondrial activity of the cells was not affected after 24 h exposure to GelMA and GAP Gels, indicating that GAP Gels did not invoke cytotoxic effects to MSCs. In order to further investigate the cytotoxicity of GAP Gels, we assessed the morphology of MSCs using Calcein fluorescent staining after 24 h of treatment (Fig. 5B). No morphological differences were observed between the control and treated groups, indicating the absence of intrinsic cytotoxicity of GAP Gels.
Fig. 5. Cytocompatibility of GelMA and GAP Gels in in vitro conditions.

A MTS mitochondrial activity assay, and (B) corresponding Calcein AM-stained MSCs exposed to GelMA and GAP Gels for 24 h show no cytotoxicity. C Plot displaying Annexin V-FITC/PI staining indicating % apoptotic cells, and (D) corresponding staining profiles of MSCs harvested after 24 h exposure to treatment groups show no significant increase in apoptosis when cells are exposed to GAP Gels.
After confirming that GAP Gels had no significant effect on mitochondrial activity or cell morphology, we aimed to show that GAP Gels were biocompatible with MSCs on a molecular level. To assess the effect of GAP Gels on the molecular level, we carried out flow cytometry analysis of MSCs exposed to GAP Gels for 24 h and measured the number of cells in apoptotic phases (Fig. 5C, D). MSCs exposed to hydrogels represent the treatment groups, untreated cells represent the negative control, and cells exposed to heat represent the positive control. Cells were classified into four subpopulations: Q1 indicates necrotic cells, Q2 indicates late apoptotic, Q3 indicates early apoptotic cells, and Q4 indicates viable cells. Exposure to GAP Gels shows a similar percentage of apoptotic cells compared to the untreated group (negative control). However, GAP Gel-treated cells show significantly fewer apoptotic cells than the heat-treated group (positive control), which showed ~75% apoptotic cells in the combined apoptotic phases. From these cytocompatibility assays, it can be concluded that the treatment groups did not reduce the number of MSCs or impact their morphology when treated with GAP Gels.
Investigating the in situ sol-gel transition of injectable and sprayable GAP Gels
In order to aid in limiting the progression of infections in the tissue and wound healing process, we developed Ery-loaded GAP Gels to sustainably delivery antibiotics. Previous clinical methods have relied on traditional dressings such as gauze to protect open wounds from infection. However, these often leave the wound bed dry and can further prolong the tissue and wound healing process when the dressing needs to be changed. This inconvenience results in decreased patient compliance and a subsequent burden on the healthcare system. Due to the flexibility in the fabrication of GAP Gels, we aimed to deliver GAP Gels directly to the skin wound via injectable or spray-based modes of delivery to increase patient compliance. Through the inversion test, we showed that GAP prepolymer is a free-floating substance, whereas once crosslinked, GAP Gel will stay in place (Fig. 6A). This flexibility gives GAP Gels the potential to be delivered through multiple modes of delivery, such as through injection using a syringe or spray-based methods using a medical spray bottle (Fig. 6B, Supplementary Movie 3). We then assessed the gelation performance of GAP Gels through both injectable and spray-based methods on a glass slide irradiated with blue light (405 nm) (Fig. 6C). Through both modes of delivery, GAP Gels undergo the sol-gel transition within a few seconds, and gelation is completed within 60 s. The ability of GAP Gels to undergo the sol-gel transition and adhere onto soft tissue as well as on glass was further assessed through the use of GAP Gels injected and sprayed onto porcine meat and chicken breast (Supplementary Movies 4–7, Supplementary Fig. 9). To confirm that the structural integrity of the hydrogel through these modes of delivery was maintained, we assessed the storage and loss modulus of the injectable and spray-based gels (Fig. 6D, E). Results showed that through both modes of delivery, GAP Gels maintained the high storage modulus observed earlier with larger gels. We also observed a similar gel-like nature of the hydrogel as the storage modulus was higher than the loss modulus when the oscillation stress was smaller than 100 Pa. Overall, the results showed the potential of GAP Gels to be delivered through injection or spray-based methods as the sol-gel transition can be precisely controlled by blue light at the location site.
Fig. 6. Injectability and sprayability of GAP Gels induced by in situ photocrosslinking.

A Inversion test of GAP Gel (200 µL) before and after exposure to 405 nm blue light. B Photographs of the GAP Gel loaded into and sprayed from a medical spray bottle. C Photographs of spray-based and injectable GAP Gels on a microscope slide with increasing blue light exposure, resulting in a final crosslinked hydrogel product. D, E Representative stress sweeps of spray-based and injectable GAP Gels show the hydrogel stiffness through multiple modes of delivery.
Assessing the in vitro antibacterial activity of GAP Gels
After confirming the ability of GAP Gels to undergo the sol-gel transition through multiple modes of delivery, we aimed to deliver Ery-loaded GAP Gel directly to skin wounds via injectable or spray-based applications, followed by blue light crosslinking (Fig. 7A). Overall, this allows us to deliver and sustain the release of Ery through an easy and less invasive treatment method. One significant advantage of using AP nanofibers in a gel system is its ability to deliver a hydrophobic drug in a hydrophilic system (Supplementary Fig. 10). E. coli was used as a representative pathogen to evaluate the antibacterial properties of Ery-loaded gels44. We incubated E. coli with injectable and spray-based GAP Gels loaded with varying concentrations of Ery (0, 64, 128, 256 µg/mL) at 37 °C in vitro for 24 h (Supplementary Fig. 3B, Fig. 7B, C). Compared with unloaded GAP Gels, bacterial growth with Ery-loaded hydrogels decreased as Ery concentration increased. The antibacterial activity of the hydrogels indicated that the drug’s bioactivity was sustained throughout the synthesis process of GAP Gels. Hydrogels loaded with 300 µg/mL of Ery prevented the growth of bacteria, which matches the concentration of free Ery needed to prevent the growth of the same number of bacteria (Supplementary Fig. 4). Further, no difference in bacterial growth was observed between injectable and spray-based hydrogels, indicating the successful release of the drug from both delivery systems. To reiterate, GAP Gels can deliver Ery without adversely affecting its bioactivity and successfully prevent the growth of E. coli.
Fig. 7. In vitro bioactivity and in vivo biocompatibility of spray-based and injectable GAP Gels.

A Schematic displaying the versatility of GAP Gel applications as both spray-based and injectable applications for in vitro and in vivo analyses. B The plot demonstrates the OD600 of an E. coli bacterial culture after 24 h exposure to Erythromycin-loaded GAP Gels and (C) corresponding cultures plated onto LB-Agar. The concentrations displayed reflect the concentration of Erythromycin loaded into the hydrogels. Analysis of cutaneous wound after 3 days treatment of saline (control), GelMA Gel, spray-based GAP Gel, and injectable GAP Gel with (D) H&E staining, where white arrows indicate scab formation, red arrows indicate neutrophil infiltration; (E) Mason’s Trichrome Staining, and (F) CD68 staining indicative of pro-inflammatory monocytes and macrophages, where blue indicates DAPI staining, and red indicates CD68 staining.
Investigating the in vivo biocompatibility of GAP Gels
After evaluating the in vitro antibacterial activity of Ery-loaded GAP Gels, in vivo studies were carried out to assess the biocompatibility of GAP Gels (Fig. 7A). Through this study, we intend to develop a biocompatible hydrogel system for open wounds using injectable or spray-based delivery methods. Here, healthy mice were exposed to saline, GelMA Gel, injectable GAP Gel, and spray-based GAP Gel (Supplementary Movies 8, 9). A 4 mm wound was made on the dorsal of the mice before 100 µL of the treatment group was topically applied to the wound, followed by a 20-min exposure to 405 nm light to allow for suitable crosslinking. Three days post-treatment, the treated wound tissues were retrieved for H&E staining, Masson’s trichrome staining, and immunofluorescence staining using CD68 to investigate the biocompatibility of the GAP Gel. The H&E staining indicated little to no visual differences between the four groups (Fig. 7D). Clotting and scabbing occurred in all four groups with no visual increase in neutrophil infiltration in the GAP Gel group compared to the saline group. This was also confirmed by Masson’s trichrome staining (Fig. 7E), which showed no detrimental impact of the gel treatment towards collagen formation and the healing process compared to the saline treatment. We further assessed the biocompatibility by determining whether or not exposure to GAP Gels would illicit an immunotoxic response in comparison to control groups. We assessed pro-inflammatory macrophage infiltration through CD68 staining 3 days post-treatment (Fig. 7F, Supplementary Fig. 11). Few CD68+ cells (red) are observed in the GAP Gel treated groups and are comparable to the amount of CD68+ cells present in any open skin wound, as observed by the saline-treated group. These results demonstrate that GAP Gels do not negatively impact the early stages of wound healing or invoke immunotoxicity in vivo, showing great promise as a biocompatible dressing for future biomedical applications.
Conclusion and future outlook
We have demonstrated the diverse functionality of self-assembling AP nanofibers as a bioactive hydrophobic drug carrier (Fig. 8). In great contrast to emerging hydrophobic drug carriers, AP nanofibers provide a sustainable way to deliver hydrophobic therapeutics in a facile and inexpensive way, while carrying a high drug load. Further, we have taken advantage of the polar head groups of AP nanofibers to physcially crosslink them with GelMA via hydrogen bonding. The resulting biocompatible and biodegradable GAP Gel is a mechanically resilient and viscoelastic drug delivery system. The system developed here has shown sustained release of the loaded antibiotic, antioxidant activity, and in vitro and in vivo biocompatibility. Taken together, the presence of AP self-assembling nanofibers in GAP Gel maximized (1) the AP-drug interactions via van der Waals interactions and (2) AP-hydrogel interactions via intermolecular interactions, allowing us to deliver GAP Gel via injection and spray-based methods. To demonstrate the vast potential of GAP Gels, we have shown its capabilities in encapsulating and retaining mammalian cells. Further, we envision GAP Gels’ use can be extended to co-deliver cells within its hydrophilic regions and hydrophobic drugs within its hydrophobic core for 3D bioprinting applications. In summary, the in situ-forming nanocomposite hydrogel designed here offers a robust platform for biomedical applications while laying a foundation for a future in the co-delivery of cells and other therapeutics.
Fig. 8. Mechanically resilient GAP Gels reinforced with hydrogen bonding show potential in a vast array of biomedical applications.

GAP Gels have shown to be elastic, biodegradable, antioxidant, and antibacterial when loaded with antibiotics within the self-assembling AP nanofibers. Finally, GAP Gels can be delivered via spray-based or injection-based methods, and due to their biocompatibility, GAP Gels offer a great platform as wound dressings.
Methods
Fabrication of nanocomposite hydrogel
Fabrication of ascorbyl palmitate (AP) nanofibers and erythromycin-loaded AP
AP nanofibers (1–2.5%, w/v) were prepared using DMSO and H2O as a solvent pair (volume ratio, DMSO/H2O = 1:4)20. After dissolving AP in DMSO, 70 °C H2O was added dropwise. The glass scintillation vial was heated to 70 °C for 15 min or until AP was fully dissolved. Erythromycin was encapsulated by mixing 5 mg of Erythromycin with AP in DMSO before adding H2O.
Synthesis of GelMA foam
Gelatin (10%, w/v) was dissolved at 60 °C until fully dissolved. Methacrylic anhydride (0.8 mL/g) was added dropwise and mixed for 2 h at 60 °C41. 400 mL of PBS was added and mixed for 15 min at 60 °C to ensure dispersion. To purify the methacrylated gelatin, the solution was dialyzed (12–14 kDA molecular weight cut-off) with DI water for 7 days; the water was changed twice daily. The resulting GelMA was lyophilized and stored at room temperature for later use.
Fabrication of GelMA and GelMA-AP (GAP) Gels
To prepare GelMA and GAP Gels, GelMA prepolymer was mixed with AP nanofibers in a 1:3 volume ratio to a final concentration of 5% GelMA and 0%, 1.5%, 2.5%, 3.5%, or 4.5% AP nanofibers. GelMA prepolymer was fabricated by dissolving GelMA foam, 1% (w/v) lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), and 0.1% (w/v) tartrazine in PBS at 70 °C. The solutions were kept at 70 °C until ready for use. GelMA and GAP prepolymers were crosslinked using blue light (405 nm) for 40 min (20 min on each side).
Physicochemical characterization of nanocomposite hydrogel
Transmission electron microscopy (TEM)
TEM images were obtained using a Philips 420 Transmission Microscope. Samples were prepared by diluting AP nanofibers to a final volume of 0.5% using DI water.
Drug loading and release
AP nanofibers (1.5%, 2.5%, 3.5%, 4.5%) were loaded with 5 mg of erythromycin for cumulative drug release (n = 3). To determine drug loading efficiency, AP nanofibers were washed 3 times in PBS to isolate unloaded drug. The absorbance of collected washes were measured at 268 nm. Drug loading efficiency was calculated using the following equation:
Samples were then incubated in 1.5 mL of PBS at 37 °C on an orbital shaker. Drug release was measured by centrifuging samples at 14,000 × g for 20 min and measuring the absorbance of the supernatant at 267 nm.
Antioxidant activity
To study the antioxidant activity of AP nanofibers, 2,2-Diphenyl-1-picrylhydrazyl (DPPH) assay was performed45. 20 µL of AP nanofibers were pipetted into a 96-well plate containing 180 µL of DPPH to a final concentration of 0.4 mM DPPH and 1.5%, 2.5%, 3.5%, or 4.5% (w/v). The resulting mixture was incubated at 37 °C for 30 min before measuring the absorbance at 517 nm. The percentage scavenging activity was calculated using the following equation:
Where A0 = Absorbance at t = 0 min
Where A1 = Absorbance at t = 30 min
1H Nuclear magnetic resonance (NMR)
Glycine, ascorbyl palmitate, and a glycine-AP mixture were characterized by 1H NMR using a Bruker 400 MHz NMR spectrometer with deuterated DMSO as the solvent46.
X-ray photoelectron spectroscopy (XPS)
GelMA and GAP Gels were cross-sectioned and lyophilized. XPS analysis was done on the interface of the cross-sections using a Kratos AXIS Supra spectrometer at Surface Science Western.
Thermogravimetric analysis (TGA)
GelMA and GAP Gels were prepared and lyophilized for thermogravimetric analysis (TGA) using the TGA 5500 (TA Instruments). Samples were held under nitrogen (flow rate of 60 mL/min) at a heating rate of 10 °C/min from 20 °C to 800 °C.
Fourier-transform Infrared spectroscopy (FTIR)
FTIR was performed using a Nicolet Sumit LITE FTIR Spectrometer (Fisher Scientific, USA). All samples were lyophilized before data acquisition.
Rheological analysis
All rheological analyses were performed using a HAAKE Modular Advanced Rheometer System (MARS) (Fisher Scientific, USA). Hydrogels were tested using a P20/Ti titanium plate. Stress sweep tests were performed at 37 °C in the range of 0.1 to 104 Pa47.
Scanning electron microscopy/electron dispersive X-ray analysis
Samples were analyzed by SEM and EDX using a Hitachi SU3500 variable pressure SEM combined with an Oxford X-Max50 SDD X-ray detector. SEM was used to image the surface topography of GelMA and GAP Gel samples. EDX detected carbon, nitrogen, and oxygen in AP and AP-Ery, with a minimum detection limit of ~0.1 weight %. The samples were coated with a thin layer of gold to minimize sample charging artifacts.
In vitro and in vivo assessment of nanocomposite hydrogel
Swelling
GelMA and GAP Gels were prepared in 96-well plates and lyophilized (n = 3). Samples were placed in 2 mL of PBS at 37 °C on an orbital shaker48. Samples were taken out of PBS, weighed, and returned to PBS at each time point. Swelling was calculated based on the following equation:
Where w0 = initial dried sample weight
Where w = swollen weight
Degradation
GelMA and GAP Gels were prepared in 96-well plates and lyophilized (n = 3). Samples were degraded in 1 mL of PBS containing 1% v/v Pen-Strep (replaced every 48 h) at 37 °C on an orbital shaker for 3, 7, 10, 14, and 21 days49. Samples were lyophilized and weighed before measuring degradation based on the following equation:
Where w0 = initial dried sample weight
Where w1 = dried sample weight after degradation
Cell culture
Human MSCs were purchased from Lonza (Cat. No. PT-5006) and cultured in MSC basal media (Lonza, Cat. No. PT-3273) supplemented with the ASC-GM Singlequots™ Supplement Kit (Lonza, Cat. No. PT-4503). Cells were incubated at 37 °C with 5% CO2.
Cytocompatibility of hydrogels on MSCs
GelMA and GAP Gels were prepared in a 96-well plate and washed with PBS containing 1% v/v Pen-Strep overnight. The next day, wells were washed with three 10-min washes of PBS. MSCs were then seeded into the wells with a seeding density of 10,000 cells/well and cultured for 24 h. Fresh media was added and followed by addition of 3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)−2H-tetrazolium (MTS; Promega, USA) assay to check for cell cytocompatibility49. Cell viability was also confirmed with the above groups using Calcein AM staining (Thermo Fisher Scientific, Cat. No. C1430).
Cellular apoptosis assessment of MSCs
GelMA and GAP Gels were prepared in 6-well plates and washed with PBS containing 1% v/v Pen-Strep overnight. The next day, wells were washed with three 10-min washes of PBS. MSCs were then seeded into the wells with a seeding density of 500,000 cells/well and cultured for 24 h. Cells were then harvested via trypsinization and centrifugation (as per the centrifugation conditions of MSCs collected during normal passaging) before being subjected to the manufacturer’s protocol from the purchased Annexin V and PI apoptosis kit (Biotium, Cat. No. 30061), and samples were passed through the flow cytometer (Cytoflex, Beckman Coulter, Canada) and data was recorded for analysis50. Untreated cells acted as the negative control, while cells subjected to a 20-min incubation period at 55 °C, followed by a 24 h recovery at 37 °C acted as the positive control51.
Antibacterial activity of hydrogel against E. coli
Overnight cultures of E. coli DH5 were grown in LB medium (37 °C, 210 RPM). Cultures were pelleted at 13,000 g for 3 min and washed two times with PBS, pH 7.0, and resuspended in PBS. The optical density at 600 nm (OD600) of this bacterial suspension was determined, and the cells were normalized to an OD600 equal to 0.1 in PBS52. A 2 mL suspension of LB and the experimental group was inoculated with 20 μL E. coli (equal to OD600 of 0.01 in 13 mL tubes at 37 °C with 210 RPM shaking for 24 h). The OD600 values of bacterial cultures were measured before being serially diluted and plated onto LB-agar to enumerate CFUs at 24 h post-inoculation.
In vivo biocompatibility
All animal experiments were conducted under the approval of The University of Western Ontario Animal Ethics Committee (AUP #: 2020-110) and in accordance with the Canadian code of practice. Five C57BL/6 male mice were purchased and held at the Animal Care and Veterinary Services facility. Prior to biocompatibility experimentation, the animals were allowed to acclimatize for 1 week.
All animals were anesthetized before performing the experiment by isofluorane. Then, the skin of the dorsal area was shaved clearly, and four wounds were created on each mouse using a 4 mm diameter biopsy punch. GelMA Gel, spray-based GAP Gel, injectable GAP Gel, and saline were applied to each wound before being exposed to 405 nm blue light for 20 min. Wounds were covered with Tegaderm adhesive bandages. On day 3, all mice were sacrificed, and the wound surface was cut and fixed in 10% formalin to perform H&E staining, Masson’s Trichrome Staining, and immunofluorescent staining using CD6853.
Statistical analysis
To determine whether a significant difference existed between experimental groups (n = 3), one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison tests were performed. Results are shown as mean ± standard deviation with n = 3 (* = p < 0.05, ** = p < 0.01, *** = p < 0.001). GraphPad Prism 9 was used for all statistical analyses.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
Arghya Paul is thankful to the following funding agencies for providing support: New Frontiers in Research Fund (NFRF)—Exploration Stream (NFRFE-2019-00743), Early Research Award (ERA) from the Province of Ontario, Canada Research Chairs Program of the Natural Sciences and Engineering Research Council (NSERC) of Canada (CRC-2018-00028), Wolfe-Western Fellowship At-Large for Outstanding Newly Recruited Research Scholar, and Canadian Institutes of Health Research Operating Grant (CIHR – IMHA, Grant no: 185629). Yasmeen Shamiya would like to acknowledge the funding and support from NSERC Canada Graduate Scholarship—Doctoral Program (CGS D). Aishik Chakraborty is supported in part by a Transdisciplinary Award from the Bone and Joint Institute, The University of Western Ontario, Canada. The authors would also like to acknowledge Reza Khazee from the Biotron Facility (TEM) and Surface Science Western (SEM, XPS) at The University of Western Ontario. The authors would also like to thank BioRender, as some of the images and illustrations were created with BioRender.com.
Author contributions
A.P. and Y.S. developed the idea and A.P. supervised the work. A.P., Y.S., and A.C. designed the outline and methodology. Y.S. prepared all the samples, performed the experiments, and analyzed the work. Y.S. wrote the manuscript, with contributions from A.C. Y.S. and A.C. took all pictures and supplementary movies. A.A.Z. performed flow cytometry experiments and analysis. N.B. performed NMR spectroscopy. J.G. and D.P. performed TGA experiments. Y.S., A.C., B.F., and S.C. performed in vivo studies and analysis. All authors commented on the manuscript.
Peer review
Peer review information
Communications Materials thanks Praveen Kumar Vemula and the other anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Youn Soo Kim and Jet-Sing Lee.
Data availability
The raw data in support of most of the quantitative figures reported in this work are reported in the Supplementary Information. All of the other data are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
10/8/2024
A Correction to this paper has been published: 10.1038/s43246-024-00663-5
Supplementary information
The online version contains supplementary material available at 10.1038/s43246-024-00641-x.
References
- 1.Ghosh, S. & Banerjee, M. A smart viral vector for targeted delivery of hydrophobic drugs. Sci. Rep.11, 7030 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalepu, S. & Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B5, 442–453 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larrañeta, E., Stewart, S., Ervine, M., Al-Kasasbeh, R. & Donnelly, R. F. Hydrogels for hydrophobic drug delivery. Classification, synthesis and applications. J. Funct. Biomater.9, 13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wen, H., Jung, H. & Li, X. Drug delivery approaches in addressing clinical pharmacology-related issues: opportunities and challenges. AAPS J.17, 1327–1340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hiwrale, A., Bharati, S., Pingale, P. & Rajput, A. Nanofibers: a current era in drug delivery system. Heliyon9, e18917 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laha, A., Yadav, S., Majumdar, S. & Sharma, C. S. In-vitro release study of hydrophobic drug using electrospun cross-linked gelatin nanofibers. Biochem. Eng. J.105, 481–488 (2016). [Google Scholar]
- 7.Huo, P. et al. Electrospun nanofibers of polycaprolactone/collagen as a sustained-release drug delivery system for artemisinin. Pharmaceutics13, 1228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fathollahipour, S., Abouei Mehrizi, A., Ghaee, A. & Koosha, M. Electrospinning of PVA/chitosan nanocomposite nanofibers containing gelatin nanoparticles as a dual drug delivery system. J. Biomed. Mater. Res. A103, 3852–3862 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Grimaudo, M. A., Concheiro, A. & Alvarez-Lorenzo, C. Crosslinked hyaluronan electrospun nanofibers for ferulic acid ocular delivery. Pharmaceutics12, 274 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kharaghani, D. et al. Design and characterization of dual drug delivery based on in-situ assembled PVA/PAN core-shell nanofibers for wound dressing application. Sci. Rep.9, 12640 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serbezeanu, D. et al. Functional polyimide-based electrospun fibers for biomedical application. Materials12, 3201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mao, Z. et al. Preparation of poly(lactic acid)/graphene oxide nanofiber membranes with different structures by electrospinning for drug delivery. RSC Adv.8, 16619–16625 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shamiya, Y., Ravi, S. P., Coyle, A., Chakrabarti, S. & Paul, A. Engineering nanoparticle therapeutics for impaired wound healing in diabetes. Drug Discov. Today27, 1156–1166 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Dubey, R., Dutta, D., Sarkar, A. & Chattopadhyay, P. Functionalized carbon nanotubes: synthesis, properties and applications in water purification, drug delivery, and material and biomedical sciences. Nanoscale Adv.3, 5722–5744 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Al-Zarah, H., Serag, M. F., Abadi, M. & Habuchi, S. Self-assembly of geometry-based DNA origami-histone protein hybrid nanostructures for constructing rationally-designed higher-order structures. ACS Appl. Nano Mater.6, 9515–9522 (2023). [Google Scholar]
- 16.Xu, Z. Y. et al. Self-assembled nanoparticles based on supramolecular-organic frameworks and temoporfin for an enhanced photodynamic therapy in vitro and in vivo. J. Mater. Chem. B10, 899–908 (2022). [DOI] [PubMed] [Google Scholar]
- 17.Sivagnanam, S. et al. Self-assembled dipeptide based fluorescent nanoparticles as a platform for developing cellular imaging probes and targeted drug delivery chaperones. Nanoscale Adv.4, 1694–1706 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu, C. et al. His-mediated reversible self-assembly of ferritin nanocages through two different switches for encapsulation of cargo molecules. ACS Nano14, 17080–17090 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Loureiro, J., Miguel, S. P., Seabra, I. J., Ribeiro, M. P. & Coutinho, P. Single-step self-assembly of zein–honey–chitosan nanoparticles for hydrophilic drug incorporation by flash nanoprecipitation. Pharmaceutics14, 920 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vemula, P. K. et al. On-demand drug delivery from self-assembled nanofibrous gels: a new approach for treatment of proteolytic disease. J. Biomed. Mater. Res. A97A, 103–110 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Zhang, S. et al. An inflammation-targeting hydrogel for local drug delivery in inflammatory bowel disease. Sci. Transl. Med.7, 300ra128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.FDA. Substances Generally Recognized as Safe. Code of Federal Regulations Title 21 (Canadian Journal of Chemisty, 2015).
- 23.Fitzmaurice, S. D., Sivamani, R. K. & Isseroff, R. R. Antioxidant therapies for wound healing: a clinical guide to currently commercially available products. Ski. Pharmacol. Physiol.24, 113–126 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Joshi, N. et al. Towards an arthritis flare-responsive drug delivery system. Nat. Commun.9, 1275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sapper, H., Cameron, D. G. & Mantsch, H. H. The thermotropic phase behavior of ascorbyl palmitate: an infrared spectroscopic study. Can. J. Chem. 59, 2543–2549 (1981).
- 26.Hrib, J. et al. Nanofibers for drug delivery—incorporation and release of model molecules, influence of molecular weight and polymer structure. Beilstein J. Nanotechnol.6, 1939–1945 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meves, A., Stock, S. N., Beyerle, A., Pittelkow, M. R. & Peus, D. Vitamin C derivative ascorbyl palmitate promotes ultraviolet-B-induced lipid peroxidation and cytotoxicity in keratinocytes. J. Investig. Dermatol.119, 1103–1108 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Comino-Sanz, I. M., López-Franco, M. D., Castro, B. & Pancorbo-Hidalgo, P. L. The role of antioxidants on wound healing: a review of the current evidence. J. Clin. Med.10, 3558 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang, L. et al. A double network strategy to improve epithelization of a poly(2-hydroxyethyl methacrylate) hydrogel for corneal repair application. RSC Adv.6, 1194–1202 (2016). [Google Scholar]
- 30.Flamia, R., Lanza, G., Salvi, A. M., Castle, J. E. & Tamburro, A. M. Conformational study and hydrogen bonds detection on elastin-related polypeptides using x-ray photoelectron spectroscopy. Biomacromolecules6, 1299–1309 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Kerber, S. J. et al. The nature of hydrogen in x-ray photoelectron spectroscopy: General patterns from hydroxides to hydrogen bonding. J. Vac. Sci. Technol. A Vac. Surf. Film.14, 1314–1320 (1996). [Google Scholar]
- 32.Zhu, M. et al. Gelatin methacryloyl and its hydrogels with an exceptional degree of controllability and batch-to-batch consistency. Sci. Rep.9, 6863 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin, J., Pozharski, E. & Wilson, M. A. Short carboxylic acid-carboxylate hydrogen bonds can have fully localized protons. Biochemistry56, 391–402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charisiadis, P. et al. 1H-NMR as a structural and analytical tool of intra- and intermolecular hydrogen bonds of phenol-containing natural products and model compounds. Molecules19, 13643–13682 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Velasco‐ Rodriguez, B. et al. Hybrid methacrylated gelatin and hyaluronic acid hydrogel scaffolds. Preparation and systematic characterization for prospective tissue engineering applications. Int. J. Mol. Sci.22, 6758 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ostrowska-Czubenko, J., Pierõg, M. & Gierszewska-Druzyńska, M. Water state in chemically and physically crosslinked chitosan membranes. J. Appl. Polym. Sci.130, 1707–1715 (2013). [Google Scholar]
- 37.Fan, C. & Wang, D. A. Macroporous hydrogel scaffolds for three-dimensional cell culture and tissue engineering. Tissue Eng. - Part B Rev.23, 451–461 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Shin, S. R. et al. Reduced graphene oxide-GelMA hybrid hydrogels as scaffolds for cardiac tissue engineering. Small12, 3677–3689 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Augustine, R. et al. Chitosan ascorbate hydrogel improves water uptake capacity and cell adhesion of electrospun poly(epsilon-caprolactone) membranes. Int. J. Pharm.559, 420–426 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Špiclin, P., Gašperlin, M. & Kmetec, V. Stability of ascorbyl palmitate in topical microemulsions. Int. J. Pharm.222, 271–279 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Yue, K. et al. Synthesis, properties, and biomedical applications of gelatin methacryloyl (GelMA) hydrogels. Biomaterials73, 254–271 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bessot, A. et al. GelMA and biomimetic culture allow the engineering of mineralized, adipose, and tumor tissue human microenvironments for the study of advanced prostate cancer in vitro and in vivo. Adv. Healthc. Mater.12, 2201701 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ravetti, S. et al. Ascorbic acid in skin health. Cosmetics6, 58 (2019). [Google Scholar]
- 44.Arbab, S., Ullah, H., Wang, W. & Zhang, J. Antimicrobial drug resistance against Escherichia coli and its harmful effect on animal health. Vet. Med. Sci.8, 1780–1786 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mukherjee, S. et al. Evaluation of free-radical quenching properties of standard Ayurvedic formulation Vayasthapana Rasayana. BMC Complement. Altern. Med.11, 38 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han, Z. et al. A versatile hydrogel network–repairing strategy achieved by the covalent-like hydrogen bond interaction. Sci. Adv.8, eabl5066 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basu, S. et al. Harnessing the noncovalent interactions of DNA backbone with 2D silicate nanodisks to fabricate injectable therapeutic hydrogels. ACS Nano12, 9866–9880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu, F., Pang, Y. & Liu, J. Swelling-strengthening hydrogels by embedding with deformable nanobarriers. Nat. Commun.11, 4502 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paul, A. et al. Nanoengineered biomimetic hydrogels for guiding human stem cell osteogenesis in three dimensional microenvironments. J. Mater. Chem. B4, 3544–3554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu, K. et al. Injectable hyaluronic acid-tyramine hydrogels incorporating interferon-α2a for liver cancer therapy. J. Control. Release166, 203–210 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Leber, B. et al. Impact of temperature on cell death in a cell-culture model of hepatocellular carcinoma. Anticancer Res.32, 915–921 (2012). [PubMed] [Google Scholar]
- 52.Laçin, N. T. Development of biodegradable antibacterial cellulose based hydrogel membranes for wound healing. Int. J. Biol. Macromol.67, 22–27 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Khalaf, A. A., Hassanen, E. I., Zaki, A. R., Tohamy, A. F. & Ibrahim, M. A. Histopathological, immunohistochemical, and molecular studies for determination of wound age and vitality in rats. Int. Wound J.16, 1416–1425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The raw data in support of most of the quantitative figures reported in this work are reported in the Supplementary Information. All of the other data are available from the corresponding author upon request.