

ABSTRACT
A critical attribute of therapeutic antibodies is their ability to engage with humoral or cellular effector mechanisms, and this depends on the ability of the Fc region to bind to complement (C1q) or Fc receptors. Investigators have sought to optimize these effects by engineering the Fc region to bind to a greater or lesser extent to individual receptors. Different approaches have been used in the clinic, but they have not been systematically compared. We have now produced a matched set of anti-CD20 antibodies representing a range of variants and compared their activity in cell-based assays for complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity, and antibody-dependent phagocytosis using a range of individual Fc receptors. We have also compared the thermal stability of the variants by differential scanning fluorimetry (DSF). The results reveal a spectrum of activities which may be appropriate for different applications.
KEYWORDS: DSF, Fc region, Fc receptor, therapeutic antibody, antibody engineering, FcγRI, FcγRII, FcγRIII, C1q, CD16, CD32, CD64, antibody effector function
Introduction
Pharmacologic properties of immunoglobulins depend very much on their Fc region. Interaction with C1q initiates complement-dependent cytotoxicity (CDC). Binding to various Fc receptors on leucocytes induces antibody-dependent cell-mediated cytotoxicity (ADCC) or antibody-dependent cell-mediated phagocytosis (ADCP). Binding to the FcRn receptor is responsible for the comparatively long half-life of IgG. In a previous article, we cataloged the protein sequences of 819 antibodies and Fc fusion proteins which have been given international nonproprietary names (INNs).1 Within this dataset there are 756 human therapeutics which have an intact Fc region, of which 30 (about 4%) have been designed to enhance one or more effector functions. The most common method (at least 14 examples) is by modification of glycosylation to reduce the fucose content of the Fc-linked oligosaccharide.2–5 Antibodies lacking fucosylated structures have a substantially increased affinity for FcγRIII, resulting in a greater potency in ADCC, a phenomenon first observed when comparing alemtuzumab (Campath) produced from rat myeloma cells or Chinese hamster ovary (CHO) cells.6 In addition, numerous mutations have been discovered which modify the binding of IgG to one or more Fc receptors for the purpose of enhancing CDC, ADCC, and/or ADCP.7–12 Nevertheless, there have been few systematic comparisons of the activity of the different variants.
We have now constructed a matched set of IgG antibodies with identical Fab regions based on the anti-CD20 antibody rituximab and Fc regions representing all of the variants in our INN database as well as others described in the literature. We systematically compared their activity in cell-based assays and observed several different patterns of activity. We also analyzed the thermal stability of the different variants and found that many of them are impaired compared with wild-type antibodies.
Results
Catalog of immunoglobulin variants with enhanced effector function
Our INN dataset (Suplementary Tables S1 and S2 of reference 1) was interrogated to identify antibodies and Fc fusion proteins with an intact Fc designed to enhance effector function. There were 30 in total, all of them based on human IgG1 with 14 having low or no fucose and 16 having amino acid substitutions. One bispecific antibody, ivicentamab, had an asymmetric Fc containing F405L on one chain and K409R on the other to facilitate heterodimerisation. Both chains include E430G to enhance hexamerization and CDC. Two antibodies, tafasitamab (MOR00208, Xmab-5574) and talcotuzumab (INI-56022473), contained mutations additional to those reported in the literature. Whereas literature references13,14 mention only S239D/I332E as described by Lazar and colleagues15 for enhancement of ADCC, the INN listings show that they have six additional mutations K274Q/Y296F/Y300F/L309V/A339T/V397M. This agrees with a patent application which lists the sequence of MOR00208 heavy chain.16 The mutated residues are all found in wild-type IgG2. Their purpose in these antibodies is not explicit but, as we found, they likely contribute to modulation of Fc receptor binding even though they are located distant from the FcγR binding sites. We also considered the list of engineered variants assembled by the International ImMunoGeneTics information system (IMGT).17 Although this does not contain all of the variants found in the INN dataset, it includes some additional variants for which no therapeutic antibodies had been assigned an INN (as of April 2022). These variants, along with wild-type IgG1 and IgG3 and other variants were included in a panel of 24 antibodies to be synthesized and tested as listed in Table 1. This selection does not include every variant described in the literature. A review published after the initiation of this study lists 48 variants with some form of enhanced activity and does not include all of those found in the INN or IMGT lists.12
Table 1.
List of samples used in this study with the number of antibodies or fusion proteins that have been given an INN and the corresponding IMGT nomenclature (where available). So far as possible, citations are to the first description of the variant of which we are aware.
| Sample number | Isotype | Mutations | Number of INNs | IMGT Code | Citation | doi |
|---|---|---|---|---|---|---|
| mAb-024 | IgG1 | 402 | ||||
| mAb-092 | IgG1 | afucose | 14 | Umana2 | 10.1038/6179 | |
| mAb-095 | IgG1 | L234Y L235Q G236W S239M H268D D270E S298A and D270E K326D A330M K334E | 0 | G1v10 and Gv11 | Mimoto34 | mabs.23452/mabs.23452 |
| mAb-080 | IgG1 | L235V F243L R292P Y300L P396L | 2 | Nordstrom28 | 10.1186/bcr3069 | |
| mAb-082 | IgG1 | G236A S239D A330L I332E | 1 | G1v12 | Smith26 | 10.1073/pnas.1203954109 |
| mAb-103 | IgG1 | G236A S239D I332E | 0 | G1v13 | Richards31 | 10.1158/1535-7163.MCT-08-0201 |
| mAb-100 | IgG1 | G236A S267E H268F S324T I332E | 0 | Moore24 | mabs.23452/mabs.2.2.11158 | |
| mAb-101 | IgG1 | G236A H268F S324T I332E | 0 | Moore24 | mabs.23452/mabs.2.2.11158 | |
| mAb-081 | IgG1 | G236A A330L I332E | 1 | G1v45 | Weitzenfeld30 | 10.1172/JCI128437 |
| mAb-083 | IgG1 | S239D K274Q Y296F Y300F L309V I332E A339T V397M | 2 | Foster16 | WO2015195498A1 | |
| mAb-084 | IgG1 | S239D A330L I332E | 1 | G1v8 | Lazar15 | 10.1073/pnas.0508123103 |
| mAb-085 | IgG1 | S239D I332E | 1 | G1v7 | Lazar15 | 10.1073/pnas.0508123103 |
| mAb-102 | IgG1 | F243L | 0 | Stewart21 | protein/protein/gzr015 | |
| mAb-094 | IgG1 | F243L R292P Y300L V305I P396L | 0 | G1v9 | Stavenhagen33 | 10.1158/0008-5472.CAN-07-0696 |
| mAb-086 | IgG1 | P247I A339Q | 1 | Bowles32 | 10.1182/blood-2006-04-020-057 | |
| mAb-087 | IgG1 | S267E | 1 | Chu25 | 10.1016/j.molimm.2008.06.027 | |
| mAb-088 | IgG1 | S267E L328F | 1 | Smith26 | 10.1073/pnas.1203954109 | |
| mAb-096 | IgG1 | D270E K326D A330M K334E | 0 | Mimoto34 | mabs.23452/mabs.23452 | |
| mAb-093 | IgG1 | S298A E333A K334A | 0 | G1v6 | Shields29 | 10.1074/jbc.M009483200 |
| mAb-089 | IgG1 | N325S L328F | 1 | Shang27 | 10.1074/jbc.M113.537936 | |
| mAb-099 | IgG1 | K326W E333S | 0 | Idusogie23 | 10.4049/jimmunol.166.4.2571 | |
| mAb-090 | IgG1 | T393A | 1 | Stewart21 | 10.1093/protein/gzr015 | |
| mAb-091 | IgG1 | E430G | 3 | de Jong22 | 10.1371/journal.pbio.1002344 | |
| mAb-022 | IgG3 | 1 |
Fc effector cell bioassays
The ability of anti-CD20 antibodies to engage with cellular Fcγ receptors was measured using the Promega Fc effector cell bioassay reporter system. This indicates the potential for ADCP or ADCC. Target cells were Raji, a CD20+ human B lymphocyte cell line and effector cells were Jurkat, a human T cell line with a luciferase gene under the control of a nuclear factor of activated T-cells (NFAT) promoter and transfected with the required human Fc receptor. Samples of variant antibodies were titrated 10-fold to give final concentrations from 3.33 to 0.0333 ng/mL (FcγRI) or from 333 to 3.33 ng/mL (FcγRII and FcγRIII). The activity of some samples was such that the calculated EC50 had to be extrapolated beyond the range of the titration. The numerical results, especially at the extremes, should therefore be treated with caution. EC50 values within three-fold of wild-type IgG1 are reported as ‘neutral’. Values which were more than three-fold lower are reported as ‘enhanced’ and values more than three-fold higher are reported as ‘reduced’. The results are summarized in Table 2.
Table 2.
Relative activity of CD20 antibodies in cell-based assays for ADCP potential, ADCC potential or CDC and thermal stability measured by DSF. The activity assays are normalized and expressed as a ratio of the EC50 of wild-type IgG1 to the EC50 of the sample. Cells are shaded to indicate the response range: green = within three-fold, blue = greater than three-fold, red = less than three-fold. For the DSF assays, the onset of denaturation (Ton) and first melting point (Tm) is reported. Cells are shaded to indicate the temperature compared with wild-type IgG1 (Ton = 63.1°C, Tm = 73.2°C): Ton green = greater than 61.9°C), yellow = 60°C to 61.9°C, orange = 56°C to 60°C, red = less than 56°C. Tm green = greater than 72.6°C), yellow = 70°C to 72.6°C, orange = 66°C to 70°C, red = less than 66°C. pd = point dropped due to technical failure.
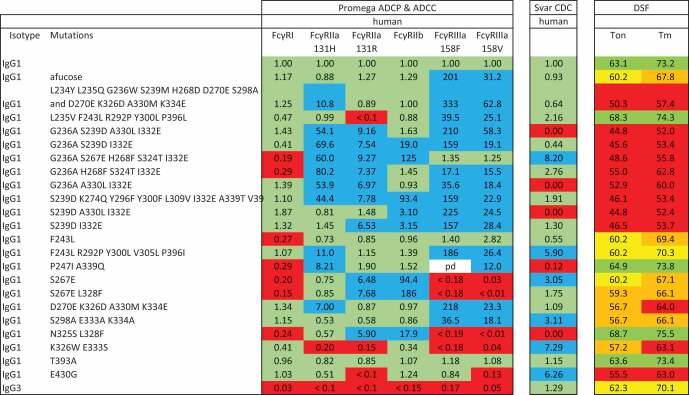 |
Consistent with our earlier observations,18 wild-type IgG3, despite binding strongly to all of the recombinant Fc receptors, gave very low levels of activity in all of the ADCP and ADCC assays. None of the variants enhanced ADCP with FcγRI and some were markedly reduced. Most (14/22) variants gave enhanced ADCC with both alleles of FcγRIIIa, always with greater enhancement for the 158F allele that normally gives lower levels of activity with wild-type IgG1. Meanwhile, 5 variants gave reduced activity and 3 were unchanged. The picture was mixed for FcγRII, with 7 variants giving no enhancement, 12 enhancing activity with only one isoform or one allele, and 3 enhancing with all three forms.
Complement-mediated cytotoxicity
The set of variants was tested for CDC using the Svar iLite assay system. This uses Ramos target cells which express CD20 and Svar luciferase (a modified form of Metridia longa luciferase). Upon lysis of the cells, this highly stabilized luciferase is released, accumulates in the cell medium over time, and can be measured with a suitable substrate. Samples of variant antibodies were titrated three-fold to give final concentrations from 1667 to 21 ng/mL. EC50 values which were within three-fold of wild-type IgG1 are reported as ‘neutral’. Values which are more than three-fold lower are reported as ‘enhanced’ and values more than three-fold higher are reported as ‘reduced’.
The results are summarized in Table 2.
In this assay, wild-type IgG3 gave similar activity to IgG1 as did 11 of the variants. Six variants gave enhanced activity and 5 were reduced or ablated.
Thermal stability
The thermal stability of the antibodies was assessed by differential scanning fluorimetry (DSF) using Protein Stable’s SUPR-DSF system. Intrinsic protein fluorescence was measured as an indicator of protein stability using the identical methodology as described previously.18 Comparing the first derivative melt curves, differences between samples were obvious (Figure 1). Particularly notable is the first transition, which is believed to be associated with the CH2 domain. The results are summarized in Table 2. For most of the variants, three temperature transitions could be resolved, but for the purpose of comparison, only the onset of melting (Ton) and the first transition point (Tm) are reported. Wild-type IgG1 gave a Ton of 63.1°C and Tm of 73.2°C. The mean fitting error of Ton was ± 0.4°C and of Tm ± 0.2°C. The Ton of the test samples was considered significantly lower if it was less than 61.9°C (i.e., lower by more than three times the fitting error). Similarly, the Tm was considered significantly lower if it was less than 72.6°C. Most (18/22) of the variants had a significantly lower Ton and Tm, sometimes reduced by more than 20°C. Only four variants showed a thermal profile similar to wild-type IgG1.
Figure 1.
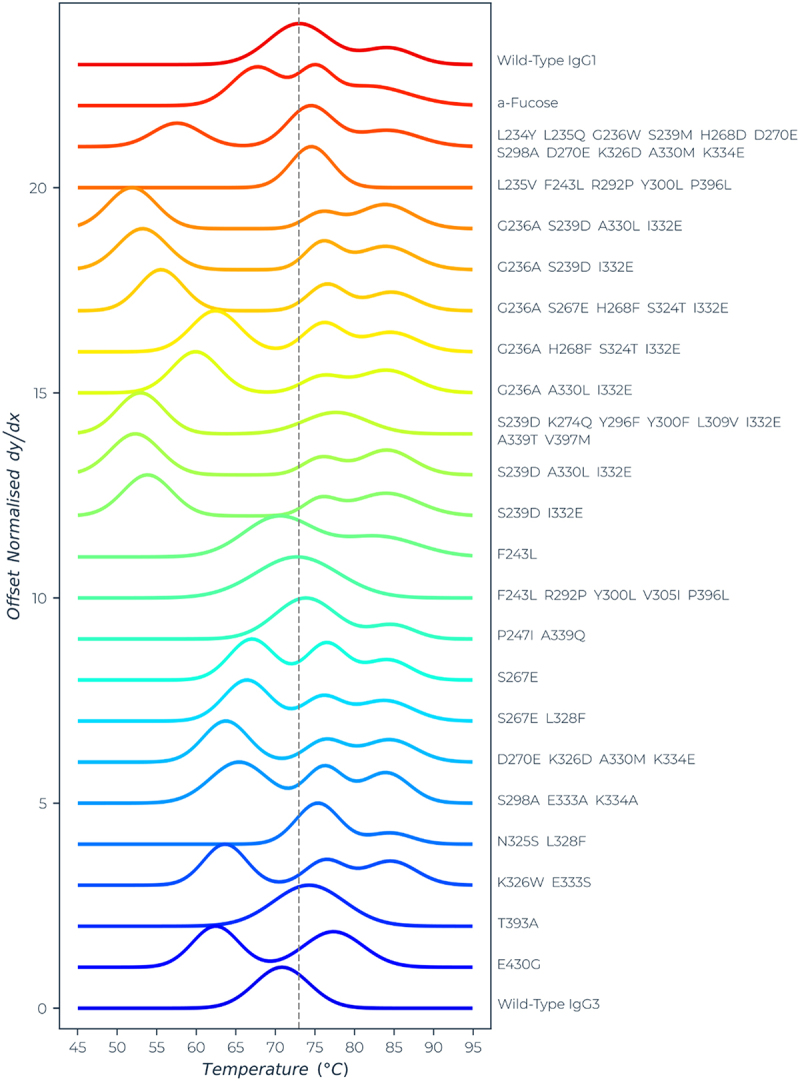
Thermal stability of CD20 antibodies measured by differential scanning fluorimetry. The melt curves (normalized, and offset, first differential) are shown for each sample. The dashed line is aligned with the Tm1 value of the wild-type IgG1 mAb. The corresponding Ton and Tm for the first transition are reported in Table 2.
Discussion
In a previous article, we described the analysis of a panel of variant antibodies with reduced binding to Fcγ receptors.18 Although investigators had aimed to eliminate binding to all of the receptors, we found a range of different levels of ‘silencing’ and in many cases, the mutations resulted in a decrease in thermal stability so that there were only a few variants which were completely silenced with regard to all Fc receptors and also retained the stability of wild-type immunoglobulin. However, strategies for the enhancement of FcγR binding are more complex. The overall aim is to increase the therapeutic effect of an antibody, typically to enhance the killing of tumor cells. But whether this is best achieved by enhancing ADCP (mediated by FcγI or FcγIIa), ADCC (mediated by FcγIIIa), or CDC, or by reduction in the inhibitory effects of engaging FcγIIb, or perhaps a combination of any of these, is not necessarily obvious. And is there an optimal level of enhancement of binding activity or functional activity? A considerable amount of research has been carried out, and many possible amino substitutions have been identified which modulate binding to Fcγ receptors and/or C1q, but the field is hard to navigate due to the lack of comparative studies and diversity of experimental methods. There are several recent reviews of the literature.7–12 One of the most comprehensive lists 48 variants with some level of enhanced binding to FcγR or C1q, ADCP, ADCC, or CDC.12 However, even this excellent resource does not include every variant actually used in the clinic, and the binding and functional data obtained from the cited literature are patchy and qualitative, making quantitative comparisons impossible.
We have made a start to addressing these problems by comparing a set of antibodies with identical V regions and allotypes using straightforward experimental methods which can be replicated in most laboratories. We used V regions from the CD20 antibody rituximab because cell-based assays for anti-CD20 antibodies are readily available. We focused on cell-based assays to measure the potential for ADCP, ADCC, and CDC, believing that these are more relevant for therapeutic applications than solution-phase binding assays, especially since aggregation of antibodies on the cell surface makes an important contribution to effector functions. It should be noted that these assay systems do not measure phagocytosis or cytotoxicity directly, but provide sensitive measures of upstream events, namely the activation of gene transcription through the pathway in the effector cells which is triggered by cross-linking of Fc receptors. We also measured the thermal stability of each sample using a high-throughput DSF method. Our panel of antibodies included all of the variants we previously identified from the World Health Organization’s INN lists (up till April 2022)1 with the addition of those listed by the IMGT17 and some others.
Wild-type IgG1 was used as the reference against which all other samples were compared. Wild-type IgG3 was included as a control. In our previous study, it gave higher binding than IgG1 to all of the Fcγ receptors by surface plasmon resonance,18 and higher CDC, but much lower activity in the ADCP and ADCC assays. The same results were seen here; the EC50s for ADCP with FcγRI and ADCC with FcγRIIIa were much lower than for IgG1, and ADCP with FcγRIIa was almost undetectable. We tested only one of the many allotypes of IgG3; it is possible that different results could be obtained with other allotypes.19
Few, if any, Fc mutations have been designed to enhance binding or ADCP with FcγRI, and most of the literature reports do not include data on FcγRI binding. We found comparatively little effect of most of the mutations. The greatest level of enhancement of ADCP with FcγRI was 1.87-fold (S239D/A330L/I332E). Seven variants gave more than 3-fold reduction.
The earliest, and most extensively used enhancement exploits the increased ADCC of antibodies with reduced levels of fucosylation.2–5 By the end of 2023, there were seven US Food and Drug Administration-approved therapeutics (amivantamab, belantamab mafodotin, benralizumab, inebilizumab, mogamulizumab, obinutuzumab, ublituximab) which had low or no fucose, typically by expression of the antibody in a cell line lacking fucosyltransferase. There have been few opportunities to directly compare fucosylated and afucosyl antibodies in the clinic, but one cross-over study of a patient with chronic lymphocytic leukemia showed a significant increase in clearance of tumor cells from the blood using a variant of alemtuzumab which was low in fucose.20 We observed an increase of 31-fold in the ADCC activity of the afucosyl sample using FcγRIIIa 158 V effector cells and 201-fold using FcγRIIIa 158F effector cells. However, there were no significant changes in the ADCP activity, either with FcγRI or FcγRII isoforms.
Two variants with single mutations, F243L and T393A, failed to show any significant increases in ADCC, ADCP, or CDC. They were originally identified as contributing to increased ADCC activity of a triple mutant F243L/T393A/H433P isolated by ribosome display selection.21 T393A is present in conbercept, an Fc fusion protein containing the extracellular domain of VEGF receptor 1, but no rationale for the mutation has been described.
Two variants, E430G22 and K326W/E333S23 had been specifically engineered to increase CDC; we confirmed increases of 6-fold and 7-fold. ADCC and ADCP with FcγRIIa were reduced, although the original report showed an increase in ADCC activity for E430G.22
One variant, G236A/S267E/H268F/S324T/I332E,24 gave increased ADCP activity with all isoforms of FcγRII and increased CDC, but no significant increase in ADCC, consistent with the published data for binding to the different Fc receptors. Notably, there was a greater increase in activity with the inhibitory receptor FcγRIIb than for the FcγRIIa isoforms, which probably accounts for why no net increase in ADCP was previously reported using macrophages as effector cells, as they express both FcγRIIa and FcγRIIb.
Three variants, S267E,25,26 S267E/L328F26 and N325S/L328F27 gave no ADCC at all with FcγRIIIa, little change in ADCP with FcγRIIa-131 h, and significant increases in ADCP with FcγRIIa-131 R and FcγRIIb, consistent with previous literature. However, they differed in CDC activity, which was 3-fold increased for S267E, little changed for S267E/L328F and eliminated in N325S/L328F.
The remaining 14 variants all gave significantly increased ADCC, and consistently more so with FcγRIIIa-158F (17-fold to 333-fold) than with FcγRIIIa-158 V (12-fold to 62-fold). However, there were different patterns of activity in ADCP with FcγRII and in CDC. One variant, L235V/F243L/R292P/Y300L/P396L28 showed no ADCP activity with FcγRIIa-131 R but ADCP with FcγRIIa-131 h and FcγRIIb and CDC were not significantly affected. One variant, S298A/E333A/K334A29 showed no significant change in ADCP activity, but enhanced CDC. Two variants, G236A/A330L/I332E30 and G236A/S239D/A330L/I332E26 showed increased ADCP with both isoforms of FcγRIIa, unchanged with FcγRIIb and complete lack of CDC. One variant G236A/H268F/S324T/I332E24 showed a similar pattern of ADCP with a modest increase in CDC. Two variants, S239D/K274Q/Y296F/Y300F/L309V/I332E/A339T/V397M16 and G236A/S239D/I332E31 gave significant increases in ADCP with all isoforms of FcγRII, but little change in CDC. This is substantially different from the result with S239D/I332E,15 which gave much lower increases in ADCP with FcγRII. Two antibodies, tafasitamab and talacotuzumab, have consistently been described in the literature as having just the two mutations S239D/I332E, whereas they actually contain eight mutations S239D/K274Q/Y296F/Y300F/L309V/I332E/A339T/V397M and have higher activities with FcγRII than S239D/I332E alone. The variant S239D/A330L/I332E15 was similar to S239D/I332E except that ADCP with FcγRIIa-131 R was reduced to that of wild-type IgG1 and CDC was eliminated. Four variants, P247I/A339Q,32 F243L/R292P/Y300L/V305L/P396I,33 D270E/K326D/A330M/K334E,34 and the heterodimeric Fc L234Y/L235Q/G236W/S239M/H268D/D270E/S298A combined with D270E/K326D/A330M/K334E,34 all showed a similar pattern of ADCP activity, increased with FcγRIIa-131 h and unchanged with FcγRIIa-131 R and FcγRIIb. However, P247I/A339Q showed 8-fold reduction in CDC, F243L/R292P/Y300L/V305L/P396I a 6-fold increase and the other two had essentially unchanged CDC.
It can be appreciated that the range of possible enhancements is quite bewildering and it is not immediately obvious which would be the best suited for a particular purpose. For cancer therapy, it is generally believed that high levels of ADCC and a high activation:inhibition ratio of FcγRIIa:FcγRIIb activity is desirable. On this basis, G236A/S239D/A330L/I332E appears the best choice. However, the inclusion of A330L in this variant results in the complete loss of CDC. Removal of this mutation in G236A/S239D/I332E largely restores CDC at the cost of a significant increase in activity of FcγRIIb. It seems that the perfect combination of enhanced ADCC, ADCP, and CDC is yet to be achieved, but if CDC is a priority, then a good candidate might be F243L/R292P/Y300L/V305L/P396I.
For therapeutic applications, it is important that Fc modifications do not destabilize the antibody structure. We used thermal stability, measured by DSF as an indicator of the relative stability of the different antibody variants, but had insufficient material to carry out more detailed stability studies. None of the antibodies showed significant levels of aggregation by HPLC immediately after purification. However, others have shown that DSF correlates with stability-indicating measures such as differential scanning calorimetry and acid-induced aggregation.35,36
Most (18/22) of the variants showed reduced thermal stability, some with Tm as much as 21°C lower than wild-type IgG1. Variants with the highest ADCP and ADCC activities, such as G236A/S239D/A330L/I332E and G236A/S239D/I332E, were among the most severely affected, with a reduction in the Tm of the CH2 domain of about 20°C. We also found a reduction of about 16°C in the Tm of the heterodimeric antibody, although it had been engineered with a view to retaining thermal stability.33 The possible implications of reduced thermal stability for drug manufacture, storage, aggregation, and immunogenicity would need to be explored on a case-by-case basis, but these results raise a note of caution and show that there is still plenty of scope for optimization of Fc effector functions. The availability of the Protein Stable SUPR-DSF system makes it possible to screen large numbers of samples rapidly and economically (384 10 μg samples in 90 min), which is a huge improvement over the classic method of differential scanning calorimetry.
In contrast to the large number of therapeutic antibodies and fusion proteins which have been engineered to silence binding to Fc receptors for avoidance of inflammatory responses,18 comparatively few enhancing mutations have entered the clinic. This is despite a large amount of research over the past 25 years which has identified numerous different locations in the Fc region that can be mutated to fine-tune interactions with Fc receptors. The number of possible options may be part of the problem. The risk of introducing thermal instability may also have held back commercial development. Here we have attempted to assist newcomers to the field by providing comparative analyses of most, if not all, the clinically validated options to provide a database to enable them to identify a shortlist of mutations suitable for their particular application.
To provide fair comparisons of the different variants, we used the same anti-CD20 Fab region for all the samples. However, ADCP, ADCC, and CDC can all be influenced by the nature of the antigenic epitope. We believe that our results with anti-CD20 provide at least a reasonable indication of the rank order of different variants because, so far as comparisons are possible, our results correlate well with data reported for various different specificities in the original literature as summarized in Table 2 of reference 12. Nevertheless, it would be prudent for investigators to compare potential variants in the context of their own product. The cell-based reporter assays provide a convenient and high-throughput screening method for this purpose which allows dissection of interactions with individual alleles of Fc receptors, but more detailed characterization of lead candidates by ADCC and ADCP with a range of donor cells would be desirable. Thermal stability should also be evaluated in the context of individual products since it is possible that melting of Fab regions may precede the CH2 in some cases. To take our work further, it would be desirable to analyze other parameters such as different stability indicators or binding to FcRn. Researchers interested in such studies using this sample set are invited to contact us.
Since the completion of this work, Wang and coworkers described a new approach to express a large library of Fc variants in CHO cells, from which they identified novel variants with enhanced affinity and selectivity, and new mutation hotspots at P247 for FcγRIIIa, K290 for FcγRIIa and K334 for FcγRIIb.37 The biophysical stability of the new variants has not yet been reported and the authors acknowledge that this is not the end of the journey, but rather the dawn of a new era of screening high-quality large-diversity libraries which will no doubt incorporate machine learning so that Fc engineering can further advance therapeutic antibody development.
Materials and methods
Nomenclature
The EU numbering system38 is used throughout this article. Amino acid alterations are described thus: XnnnY, where X is the single letter code for the residue in the native amino acid sequence, nnn is the EU index position, and Y is the single letter code for the replacement amino acid residue.
Antibody design, expression, and purification
The amino acid sequences of the heavy chains of the variant panel were based on the variable region sequence of rituximab linked to the desired constant region based on human IgG1, IGHG*01 (G1m(za)). A control sample of human IgG3 (IGHG3*01, G3m(b*)) included the mutation R435H to facilitate purification using Protein A. A single light chain consisting of the variable region of rituximab with a kappa km1 constant region was used for all of the constructs. A set of 29 samples which included wild-type IgG1 and IgG3 and variants described in the literature but not appearing in the INN lists was made by Sanyou Biopharmaceuticals Co. (Shanghai, China) according to their standard procedures using synthetic genes expressed in ExpiCHO cells. Afucosylated antibody was prepared by expression of wild-type IgG1 in a variant of ExpiCHO in which the Fut8 gene encoding fucosyltransferase had been knocked out. Antibodies were purified by affinity chromatography on Protein A and formulated in phosphate-buffered saline (PBS). The antibodies were > 95% pure by SDS gel electrophoresis (reducing and non-reducing) and contained > 90% monomer and < 4% aggregates by size-exclusion chromatography. The yield of antibody from a 30 mL culture varied between 1.3 mg and 11.2 mg (mean 3.4 mg), but there was no particular correlation with antibody mutations. The variability in yield is largely attributed to losses in purification.
Fc effector cell bioassays
Antibodies were assessed for their ability to engage in ADCP or ADCC using Promega Fc effector bioassay systems as previously described.18 The assay kit contained CD20+ Raji target cells (Promega cat. no. G7016) and engineered Jurkat effector cells which stably express the desired Fc receptor and an NFAT response element to drive expression of firefly luciferase. Experiments were carried out according to the manufacturer’s instructions using the following effector cells:
| Receptor | Species | Supplier | Catalogue number |
|---|---|---|---|
| FcγRI (CD64) | human | Promega | GA133A |
| FcγRIIa (CD32a) 131H allele | human | Promega | G988A |
| FcγRIIa (CD32a) 131R allele | human | Promega | CS1781B11 |
| FcγRIIb (CD32b) | human | Promega | CS1781E01 |
| FcγRIIIa (CD16a) 158F allele | human | Promega | G979A |
| FcγRIIIa (CD16a) 158V allele | human | Promega | G701A |
Target cells, effector cells, and sample dilutions were all prepared in RPMI1640 culture medium containing 4% low IgG bovine serum. Samples were diluted in an off-line plate to give three 10-fold dilutions, and 25 µL was transferred to a white flat-bottomed assay plate. 25 µL of target cell suspension was added and mixed on a plate shaker. 25 µL of effector cell suspension was added and mixed and the plate was incubated at 37°C in 5% CO2 for 6 h. The final effector to target ratio was approximately 6:1. 50 μL of luciferase assay substrate was added, and luminescence was measured using a Glomax 96 luminometer (Promega). An estimate of the EC50 was calculated by non-linear regression to a four-parameter logistic curve using the Solver add-in of Excel. The upper asymptote was fixed at the maximum response of all the samples, the lower asymptote was fixed at the mean response of buffer alone and the slope was fixed at −2.0. The results were expressed relative to the response of wild-type IgG1, i.e., relative activity of test sample = EC50 (wild-type)/EC50 (test).
Complement-mediated cytotoxicity
Antibodies were tested for CDC using the iLite reporter system of Svar Life Science (Malmo, Sweden). Experiments were carried out in accordance with the manufacturer’s instructions. Low IgG fetal bovine serum (FBS) (Promega cat no G7110) was heat-inactivated by incubation at 56°C for 40 min. Dilution medium was prepared by adding the heat-inactivated FBS to the RPMI culture medium (Promega cat no G708A) to give a final concentration of 9%. Samples were diluted in an off-line plate to give five three-fold dilutions, and 20 μL was added to white flat-bottomed microplates, followed by 20 μL of target cell suspension (iLite CD20+ Svar Luc cat. no. BM5028) and 10 μL of 25% human serum (Svar cat. no. 5980). The plates were incubated at 37°C and 5% CO2 for 4 h. 50 μL of Quanti-Luc 4 substrate (Invivogen cat. no. rep-qlc4lg1) was added, and luminescence was read after 10 min. An estimate of the EC50 was calculated by non-linear regression to a four-parameter logistic curve using the Solver add-in of Excel. The upper asymptote was calculated from the maximum response of positive samples, the lower asymptote was fixed at the mean response of buffer alone and the slope was fixed at −1.82. The results were expressed relative to the response of wild-type IgG1, i.e., relative activity of test sample = EC50 (wild-type)/EC50 (test).
Thermal stability by DSF
Antibodies were tested using the SUPR-DSF system (Protein Stable Ltd, Leatherhead, UK) in accordance with the manufacturer’s instructions. 10 μL of each test sample at 1 mg/mL in PBS was pipetted in triplicate into a black 384-well microplate (BioRad cat. no. HSP 3866). The plate was sealed using pressure-activated adhesive qPCR seals (Azenta Cat No 4ti-0560). The plate was placed in the SUPR-DSF and subjected to a thermal ramp from 20°C to 105°C at 1°C/min with a 25 ms integration time. The fluorescence data were analyzed using the Protein Stable software package to determine the first onset of melting (Ton) and the midpoint of each melting phase (Tm) exactly as described previously.18 The mean fitting error for Ton was 0.4°C and for Tm was 0.2°C.
Funding Statement
The author(s) reported that there is no funding associated with the work featured in this article.
Abbreviations
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ADCP
antibody-dependent cell-mediated phagocytosis
- CDC
complement-dependent cytotoxicity
- CHO
Chinese hamster ovary
- DSF
differential scanning fluorimetry
- C50
the effective concentration which gives half maximal activity
- Fab
fragment antigen binding
- Fc
fragment crystallizable
- FcγR
Fc gamma receptor
- FcRn
neonatal Fc receptor
- NFAT
nuclear factor of activated T-cells
- IMGT
International ImMunoGeneTics information system
- INN
International nonproprietary name.
Disclosure statement
This research was sponsored by mAbsolve Limited. Geoff Hale and Ian Wilkinson have financial interests in mAbsolve.
References
- 1.Wilkinson I, Hale G.. Systematic analysis of the varied designs of 819 therapeutic antibodies and Fc fusion proteins assigned international nonproprietary names. mAbs. 2022;14(1):2123299. doi: 10.1080/19420862.2022.2123299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Umana P, Jean-Mairet J, Moudry R, Amstrutz H, Bailey JE. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nature. 1999;17(2):176–8. doi: 10.1038/6179. [DOI] [PubMed] [Google Scholar]
- 3.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, Weikert SHA, Presta LG. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human FcγRIII and antibody-dependent cellular toxicity. J Biol Chem. 2002;277(30):26733–26740. doi: 10.1074/jbc.M20206920. [DOI] [PubMed] [Google Scholar]
- 4.Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, Uchida K, Anazawa H, Satoh M, Yamasaki M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278(5):3466–3473. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- 5.Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, et al. Unique carbohydrate–carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc Natl Acad Sci USA. 2011;108(31):12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lifely MR, Hale C, Boyce S, Keen MJ, Phillips J. Glycosylation and biological activity of CAMPATH-1H expressed in different cell lines and grown under different conditions. Glycobiology. 1995;5(8):813–822. doi: 10.1093/glycob/5.8.813. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Mathieu M, Brezski RJ. IgG Fc engineering to modulate antibody effector functions. Protein Cell. 2018;9(1):63–73. doi: 10.1007/s13238-017-0473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang TN, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp Mol Med. 2019;51(11):1–9. doi: 10.1038/s12276-019-0345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saunders K. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10:1296. doi: 10.3389/fimmu.2019.01296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdeldaim DT, Schindowski K. Fc-engineered therapeutic antibodies: recent advances and future directions. Pharmaceutics. 2023;15(10):2402. doi: 10.3390/pharmaceutics15102402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damelang T, Brinkhaus M, van Oschl TLJ, Schuurman J, Labrijn AF, Rispens T, Vidarsson G. Impact of structural modifications of IgG antibodies on effector functions. Front Immunol. 2023;14:1304365. doi: 10.3389/fimmu.2023.1304365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kellner C, Otta A, Cappuzzello E, Klausz K, Peipp M. Modulating cytotoxic effector functions by Fc engineering to improve cancer therapy. Transfus Med Hemother. 2017;44(5):327–336. doi: 10.1159/000479980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van der Horst HJ, Nijhof IS, Mutis T, Chamuleau ED. Fc-engineered antibodies with enhanced Fc-effector function for the treatment of B-cell malignancies. Cancers. 2020;12(10):3041. doi: 10.3390/cancers12103041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. 2006;103(11):4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foster P, Byrd J. WO2015195498A1: treatment for chronic lymphocytic leukemia (CLL). Geneva, Switzerland: World Intellectual Property Organization Internal Bureau; 2015. p. 1–30. [Google Scholar]
- 17.Lefranc M-O, Lefranc G. IMGT® nomenclature of engineered IGHG variants involved in antibody effector properties and formats. Antibodies. 2022;11(4):65. doi: 10.3390/antib11040065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hale G, De Vos J, Davy AD, Wilkinson I. Systematic analysis of antibodies with Fc mutations designed to reduce binding to Fc-gamma receptors. mAbs. 2024;16(1). in press. doi: 10.1080/19420862.2024.2402701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Taeye SW, Bentlage AEH, Mebius MM, Meesters JI, Lissenberg-Thunnissen S, Falck D, Senard T, Salehi N, Wuhrer M, Schuurman J, et al. FcγR binding and ADCC activity of human IgG allotypes. Front Immunol. 2020;11:740. doi: 10.3389/fimmu.2020.00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dyer MJS, Moser S, Brunker P, Bird P, Almond N, Puentener U, Wheat LMW, Bolam E, Berrie E, Gratt R, et al. Enhanced potency of glycoengineered anti-CD52 monoclonal antibodies (MAbs). Blood. 2005;106(11):2958. doi: 10.1182/blood.V106.11.2958.2958. [DOI] [Google Scholar]
- 21.Stewart R, Thom G, Levens M, Guler-Gane G, Holgate R, Rudd PM, Webster C, Jermutus L, Lund J. A variant human IgG1-Fc mediates improved ADCC. Prot Eng Des Sel. 2011;24(9):671–678. doi: 10.1093/protein/gzr015. [DOI] [PubMed] [Google Scholar]
- 22.de Jong RN, Beurskens FJ, Verploegen S, Strumane K, van Kampen MD, Voorhorst M, Horstman W, Engleberts PJ, Oostindie SC, Wang G, et al. A novel platform for the potentiation of therapeutic antibodies based on antigen-dependent formation of IgG hexamers at the cell surface. PLOS Biol. 2016;14(1):e1002344. doi: 10.1371/journal.pbio.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, Mulkerrin MG. Engineered antibodies with increased activity to recruit complement. J Immunol. 2001;166(4):2571–2575. doi: 10.4049/jimmunol.166.4.2571. [DOI] [PubMed] [Google Scholar]
- 24.Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. 2010;2(2):181–189. doi: 10.4161/mabs.2.2.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, Joyce PF, Szymkowski DE, Desjarlais JD. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcγRIIb with Fc-engineered antibodies. Mol Immunol. 2008;45(15):3926–3933. doi: 10.1016/j.molimm.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 26.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc Natl Acad Sci USA. 2012;109(16):6181–6186. doi: 10.1073/pnas.1203954109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shang L, Daubeuf B, Triantafilou M, Olden R, Depis F, Raby A-C, Herren S, Dos Santos A, Malinge P, Dunn-Siegrist I, et al. Selective antibody intervention of Toll-like receptor 4 activation through Fcγ receptor tethering. J Biol Chem. 2014;289(22):15309–15318. doi: 10.1074/jbc.M113.537936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nordstrom JL, Gorlativ S, Zhang W, Yang Y, Huang L, Burke S, Li H, Ciccarone V, Zhang T, Stavenhagen J, et al. Anti-tumor activity and toxicokinetics analysis of MGAH22, an anti-HER2 monoclonal antibody with enhanced Fcγ receptor binding properties. Breast Cancer Res. 2011;13(6):R123. doi: 10.1186/bcr3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shields RL, Namenuk AK, Hong K, Meng G, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for FcγRI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J Biol Chem. 2001;276(9):6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 30.Weitzenfeld P, Bournazos S, Ravetch JV. Antibodies targeting sialyl Lewis a mediate tumor clearance through distinct effector pathways. J Clin Invest. 2019;129(9):3952–3962. doi: 10.1172/JCI128437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcγRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther. 2008;7(8):2517–2527. doi: 10.1158/1535-7163.MCT-08-0201. [DOI] [PubMed] [Google Scholar]
- 32.Bowles JA, Wang S-Y, Link BK, Allan B, Beuerlein G, CAmpbell M-A, Marquis D, Ondek B, Wooldridge JE, Smith BJ, et al. Anti-CD20 monoclonal antibody with enhanced affinity for CD16 activates NK cells at lower concentrations and more effectively than rituximab. Blood. 2006;108(8):2648–2654. doi: 10.1182/blood-2006-04-020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, Huang L, Vijh S, Johnson S, Bonvini E, et al. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcγ receptors. Cancer Res. 2007;67(18):8882–8890. doi: 10.1158/0008-5472.CAN-07-0696. [DOI] [PubMed] [Google Scholar]
- 34.Mimoto F, Igawa T, Kuramochi T, Katada H, Kadono S, Kamikawa T, Shida-Kawazoe M, Hattori K. Novel asymmetrically engineered antibody Fc variant with superior FcγR binding affinity and specificity compared with afucosylated Fc variant. mAbs. 2013;5(2):229–236. doi: 10.4161/mabs.23452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pejchal R, Cooper AB, Brown ME, Vasquez M, Krauland EM. Profiling the biophysical developability properties of common IgG1 Fc effector silencing variants. Antibodies. 2023;12:54. doi: 10.3390/antibod12030054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ito T, Tsumoto K. Effects of subclass change on the structural stability of chimeric, humanized and human antibodies under thermal stress. Protein Science. 2013;22:1542–1551. doi: 10.1002/pro.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Kang M, Ebrahimpour A, Chen C, Ge X. Fc engineering by monoclonal mammalian cell display for improved affinity and selectivity towards FcγRs. Antibody Ther. 2024;7(3):209–220. doi: 10.1093/abt/tbae017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest. 5th ed. Bethesda (MD): National Institutes of Health; 1991. [Google Scholar]