

Abstract
The interest in transporter‐mediated drug interactions has been increasing in the field of drug development. In this study, we measured the plasma and urinary concentrations of coproporphyrin (CP) I and CP III as endogenous substrates for organic anion‐transporting polypeptide (OATP) using chimeric mice with human hepatocytes (PXB mice) and examined the influence of an OATP inhibitor, rifampicin (RIF). CP I and CP III were actively taken up intracellularly, and RIF inhibited the uptake in a concentration‐dependent manner for both CP I and CP III in human hepatocytes (PXB‐cells). Single doses of RIF at 10 and 30 mg/kg were orally or intravenously administered to PXB mice and wild‐type ICR mice. Plasma concentrations (AUC0‐8h) of CP I increased in both mice. However, a marked increase in CP III was only observed in ICR mice, after intravenous administration of RIF at 30 mg/kg. The IC50 values of RIF for intracellular CP I/III uptake and the unbound plasma concentrations of RIF suggested that the increase in plasma CP I is associated with the exposure of RIF to OATPs. The 24‐h cumulative urinary excretions of CP I and CP III increased in both mice, but more markedly in PXB mice. Thus, RIF increased the plasma and urinary concentrations of CP I and CP III in the mice, as reported in humans, and CP I may be a more sensitive biomarker of OATP‐mediated drug interactions in PXB mice.
Keywords: coproporphyrins, organic anion‐transporting polypeptide (OATP), PXB mice, rifampicin, transporter‐mediated drug interactions
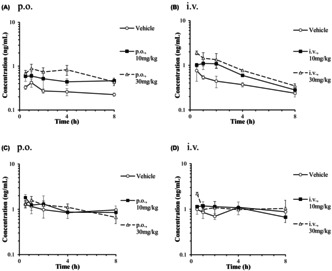
Plasma concentration‐time curves of CP I (A, B) and CP III (C, D) after a single oral or intravenous administration of rifampicin to male PXB mice. Plasma concentrations (AUC0‐8h) in CP I increased markedly with both administrations, but not in CP III.
Abbreviations
- AUC
Area under the plasma concentration‐time curve
- AUCR
AUC ratio
- C max
maximum plasma concentration
- CP
coproporphyrin
- CYP
cytochrome P450
- DDI
drug–drug interaction
- DMSO
dimethylsulfoxide
- HBSS
Hanks' balanced salt solution
- IC50
50% inhibitory concentration
- LC
liquid chromatography
- LLOQ
Lower limit of quantification
- MRP2
Multi‐drug resistance protein 2
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- OATP
Organic anion‐transporting polypeptide
- PBPK
physiologically‐based pharmacokinetics
- PBS
phosphate‐buffered saline
- RIF
rifampicin
- SD
standard deviation
- SCID
severe combined immunodeficiency
- t 1/2
elimination half‐life
- T max
time of maximum plasma concentration
- uPA
urokinase‐type plasminogen activator
- UPLC
ultra performance liquid chromatography
1. INTRODUCTION
Organic anion‐transporting polypeptide (OATP) 1B1 and OATP1B3 are primary solute carrier transporters responsible for hepatocellular drug uptake, appearing on the vascular side of hepatocytes. They are also known to affect the blood and liver concentrations of substrate drugs. The combination of OATP substrates (e.g., statins, repaglinide, and bosentan) with OATP inhibitors (e.g., cyclosporine A and rifampicin (RIF)) increases their blood concentration levels. For some drugs, the occurrence of serious adverse reactions has been reported in clinical cases. 1 In addition to drugs, OATP1B substrates include endogenous substances. 2 , 3
In recent years, clinical reports on transporter‐mediated drug–drug interactions (DDIs) have increased. Methodologies for assessing DDIs have been outlined in guidelines announced by the European Medicines Agency, Food, and Drug Administration (FDA), and Ministry of Health, Labour, and Welfare. 4 , 5 , 6 These guidelines state that transporter‐mediated inhibitory DDIs should be conducted using an adequate probe substrate, based on the expected or actual systemic exposure in humans. In the guidelines, the static model using I in,max,u is preferentially used for DDI risk assessment of OATP1B1 and 1B3, 7 , 8 which is suitable for screening but tends to produce false positives. 9 To avoid false positive results, a high‐accuracy dynamic model in which the kinetics of the inhibitor concentration are incorporated, such as a PBPK model with detailed human physiological conditions, is considered. However, it is difficult to accurately predict DDI risks non‐clinical models, even with information on the blood concentration of a test substance in clinical trials.
Attention is being focused on developing methods to directly predict clinical DDIs more accurately without conducting an additional clinical DDI trial. Studies using endogenous substrates for transporters as a biomarker have recently been conducted. 10 , 11 One major advantage of endogenous substrate biomarkers is the potential to quantitatively assess drug interactions in the early stages of clinical development, without a separate clinical DDI study. In particular, coproporphyrin (CP) I and CP III, intermediate products of the heme biosynthesis process, having high transporter selectivity, less diurnal variation, and abundant clinical DDI data, thus may be useful biomarkers of OATP1B. A study using transfected cells showed that OATP1B1 and 1B3 were responsible for CP I and CP III transport, OATP2B1 for CP III transport, and OCT1, OCT2, OAT1, and OAT3 were not involved in CP transport. 12 In humans and monkeys, administration of an OATP1B inhibitor (e.g., RIF) was found to increase blood and urine CP I/III levels. CPs, especially CP I, have emerged as important endogenous biomarkers to assess human hepatic OATP1B activities in vivo. 13 , 14 , 15 However, few studies have reported the usefulness of CPs as a biomarker in rodents.
Chimeric mice (i.e., PXB mice) were prepared by transplanting human hepatocytes into uPA/SCID mice, and ≥80% of the liver was substituted for human hepatocytes. 16 Previous studies have shown that the expressions of human drug‐metabolizing enzymes, including hepatic CYPs or non‐CYPs and human transporters, are similar in PXB mice and humans. 16 , 17 , 18 , 19 Furthermore, the metabolic pattern in PXB mice resembles that in humans. 20 , 21 , 22 , 23 , 24 , 25 PXB mice are increasingly being used as preclinical animal models to address DDIs in humans. 26 , 27 In addition, it was recently reported that the liver‐to‐plasma concentration ratio (K p,uu,liver) of OATP substrates in PXB mice resembled that in humans. 28 Accordingly, the measurement of CPs in PXB mice may directly contribute to the prediction of OATP1B activity in humans, suggesting that the PXB mouse is a valuable preclinical model for predicting human DDIs mediated by OATP1B transporters. In the present study, we measured the plasma concentration of RIF and plasma/urinary concentrations of CP I and CP III in PXB mice treated with RIF, of which the OATP1B‐mediated drug interactions were reported in clinical practice, and we compared the outcomes with those observed in wild‐type ICR mice. Based on the results, we considered the usefulness of CP I and CP III for clinical DDI risk assessment in PXB mice.
2. MATERIALS AND METHODS
2.1. Chemicals
RIF was obtained from Sigma‐Aldrich (St. Louis, MO, USA). Coproporphyrin I (CP I) and coproporphyrin III (CP III) reference standards were purchased from Frontier Scientific, Inc. (Logan, UT, USA). Isotopically labeled CP III sodium bisulfate salt (CP III‐15 N4) is used as an internal standard for CP I and CP III. All other chemicals and vehicles were of analytical grade or the highest commercially available quality.
2.2. Animals
The present study was approved by the Ethics Committees of Toray Industries, Inc., and animal experiments were conducted according to the Guidelines for Animal Experiments of Toray Industries, Inc. PXB male mice were obtained from PhoenixBio Co., Ltd. (Hiroshima, Japan) at ages 17–18 weeks, and Crl:CD1 (ICR) male mice were purchased from Charles River Laboratories Japan, Inc. (Kanagawa, Japan) at ages 7 weeks. Replacement index (RI), repopulation ratio of human hepatocytes in host mouse liver, 16 was estimated to be 85%–91% by the concentration of human albumin in the blood of PXB mice. Each PXB mice was dosed three times (three doses) with two routes of administration (oral and intravenous) totaling six conditions with a seven‐day washout period between doses.
2.3. Human Hepatocytes Isolated from Chimeric Mice, PXB‐cells
Approximately, 1.5–1.7 × 108 human hepatocytes (PXB‐cells®) were obtained from a PXB mouse (repopulation ratio: 96%–101%) using the collagenase perfusion method. 29 Fresh human hepatocytes attached to 24‐well cell plates confluently at a density of 2.1 × 105 cells/cm2 (4.0 × 105 cells/well), and were maintained for two to four weeks with d‐HCGM medium consisting of Dulbecco's modified Eagle's medium. 30 PXB‐cells were used within 21 days after preparation, during which transporter expressions were confirmed. 31
2.4. In vitro concentration‐dependent uptake of CP I and CP III in PXB‐cells
Prior to the assay, cells were rinsed twice with pre‐warmed PBS (37°C) and equilibrated in HBSS for 2 min. Uptake studies were initiated by removing the equilibrating buffer and adding 0.50 mL of HBSS containing 0.1–3 μmol/L of CP I or CP III. Hepatocytes were then incubated at 37°C in a humidified 5% CO2 atmosphere. After 5 min, the incubation was terminated by removing the transport buffer, and cells were immediately washed with ice‐cold PBS three times. After adding 0.5 mL of acetonitrile containing 0.1% formic acid to each well, cells were scraped and recovered to another tube using a pipette. The concentrations of CP I and CP III were measured by LC–MS/MS. In order to assess the passive uptake of CP I and CP III in the hepatocytes, the same experiment was conducted on ice.
2.5. In vitro DDI for the uptake of CP I and CP III with RIF in PXB‐cells
The in vitro inhibitory effects of RIF on the uptake of CP I and CP III in PXB‐cells were examined using a similar method as the uptake studies on CP I and CP III described above. Uptake studies were initiated by removing the equilibrating buffer and adding 0.50 mL of HBSS containing between 0.01 and 100 μmol/L of RIF, and 0.3 μmol/L of CP I or CP III. Cells were incubated at 37°C for 10 min. In order to assess the passive uptake of CP I and CP III at the concentration of 0.3 μmol/L in the hepatocytes, the same experiment was conducted on ice.
2.6. Measurement of CP I, CP III, and RIF concentration in mouse plasma
Animals were fasted overnight (16 h) before dosing. RIF was dissolved in distilled water containing 10% dimethyl sulfoxide and 10% PEG400 for its oral and intravenous administration. RIF was administrated orally to mice by stomach tube or intravenously via tail vein at a dose of 10 or 30 mg/kg. In the PK analysis, serial blood samples were collected from the orbital vein at 0.5, 1, 2, 4, and 8 h. The plasma samples were obtained, followed by centrifugation at 1500g for 10 min, and stored at −30°C until analysis.
2.7. Measurement of CP I and CP III concentration in mouse urine
Animals were placed in metabolism cages and fasted overnight before dosing. After a single oral administration of RIF (30 mg/kg), urine was collected in a 0–24 h interval to evaluate urinary excretion of CP I and CP III. Collected urine was centrifuged to remove solid, diluted equal volume among samples with distilled water, and stored at −30°C until analysis.
2.8. Bioanalysis by liquid chromatography (LC) and Tandem Mass spectrometry (MS/MS)
All samples were kept in the dark and sample processes were carried out by protecting them from light as much as possible. A plasma or urine sample (20 μL) was mixed well with 180 μL of methanol/ acetonitrile (1:1) containing internal control and centrifuged (1500 × g, 4°C, 15 min). The supernatant was filtered and then applied to LC–MS/MS with the ACQUITY UPLC system (Waters Corporation, Massachusetts, USA) and the mass spectrometer API5000 (Applied Biosystems/MDS Sciex, Tokyo, Japan). Chromatographic separation was performed with Ace Excel C18 PFP 2.1 × 150 mm (particle size 3 μm) (Advanced Chromatography Technologies Limited, Aberdeen, Scotland) for CP I, CP III, and CP III‐15 N4, CAPCELL PAK C18 MGIII 2.0 × 50 mm (particle size 5 μm) (Shiseido Co., Ltd., Tokyo, Japan) for RIF. For CPs detection, mobile phase A consisted of 10 mmol/L ammonium formate, whereas mobile phase B consisted of 100% acetonitrile. A solvent gradient (flow rate of 0.5 mL/min) was adopted as follows for a total run time of 5 min: 0–2.0 min, isocratic at 45% phase B; 2.0–2.1 min, 98% B; 2.1–3.5 min, 98% B; 3.5–3.6 min, 45% B; 3.0–5.0 min 45% B. The mass spectrometer spray voltage was set at 5000 V, whereas the probe temperature was 650°C with positive ion polarity. For RIF detection, mobile phase A consisted of 10 mmol/L ammonium formate, whereas mobile phase B consisted of 0.1% formic acid in acetonitrile. The initial percentage of phase B was 0%. A linear gradient was applied from 0% to 50% phase B over 1 min, from 50% to 100% phase B over 2 min, maintained at 100% phase B for 1 min, and decreased to 0% phase B for equilibration. The mass spectrometer spray voltage was set at 5500 V, whereas the probe temperature was 500°C with positive ion polarity. The sample injection volume was 10 μL. The tandem mass spectrometer was operated in the multiple reaction monitoring mode (MRM) and Q1, Q3 quadrupoles were set at unit mass resolution, Mass transition, m/z 655.2 → 596.2 was selected to monitor CP I and CP III, while m/z 659.2 → 541.3 transition was used for CP III‐15 N4, and m/z 823.4 → 791.7 transition for RIF.
2.9. Data processing
2.9.1. In vitro transport activity
The in vitro uptake rates of CP I and CP III in PXB‐cells were calculated using the below Equation 1:
Uptake Rate
| (1) |
A kinetic analysis of the transporter‐mediated uptake of CP I and CP III in PXB‐cells was performed with the extended Michaelis–Menten equation, and K m and V max were calculated using the following Equation 2:
| (2) |
where S is the substrate concentration of CP I or CP III in the incubation buffer (μmol/L), K m is the Michaelis–Menten constant (μmol/L), and V max is the maximum uptake rate (pmol/min/106 cells). Fitting was performed by a non‐linear least‐squares regression method using Phoenix WinNonlin ver. 7.0 (Certara Inc., Prinston, NJ, USA).
The IC50 calculation was conducted in R (v. 4.1.2) environment with R package dr4pl. 32
2.10. PK analysis
Pharmacokinetic parameters were calculated by a non‐compartmental method using Phoenix WinNonlin ver 7.0. The AUC ratio (AUCR) was calculated using the following formula: AUCR = AUC0‐8h of the treatment group of interest/AUC0‐8 h of the vehicle control group.
2.11. Estimation of the maximum unbound concentration of RIF at the inlet to the liver
The unbound maximum hepatic inlet concentration of the RIF in blood (I in,max,u) was calculated using the following equations:
where I max,b is the maximum circulating blood concentration of the RIF, which is equal to C max; F a is the fraction of the RIF dose absorbed; F g is the fraction of the absorbed RIF dose escaping gut wall extraction; and dose is the RIF dose. f u,b is the unbound fraction of the RIF in blood, which was calculated from multiplying the unbound fraction of the RIF in plasma (f u,p) by the uptake rate of the RIF into blood cells, human f u,p was given 0.22 33 and mouse f u,p was given 0.033. 34 In PXB mice, fu,p was estimated 0.19, following previously reported that the unbound fraction in the mixed plasma of human and mouse plasma (85:15) was well correlated with f u,p in PXB mice. 35 In addition, the uptake rate of drug into blood cells was given 1, the absorption rate constant of the RIF (k a) was given 1.61, 36 the sum of the blood flow in hepatic artery and portal vein (Qh) was given 5.4 L/h/kg, 37 and an excreted rate of unchanged form of RIF in mouse urine (Ae) of was given 0.189. 38
2.12. Statistical analysis
The Student's t‐test was applied to assess the statistical significance of each parameter and value in the in vivo DDI study between CP I or CP III and RIF. In comparison with three groups, the Bonferroni method was employed. Difference was assessed with two‐sided test with a level of 0.01 or 0.05.
3. RESULTS
3.1. In vitro concentration‐dependent uptake of CP I and CP III in PXB‐cells
The potential ability to uptake each CP I and CP III in human hepatocytes (PXB‐cells®) derived from a PXB mouse was examined. CP I or CP III was incubated with PXB‐cells at a concentration of 0.1 to 3 μmol/L and temperature of 37°C or on ice. The concentrations of CP I and CP III in PXB‐cells without the addition of each CP I and CP III were below the LLOQ. The active uptake amount of CP I and CP III were calculated by subtracting each value obtained on ice from the corresponding value at 37°C. The Michaelis–Menten equation showed that K m of CP I and CP III were 1.11 and 1.46 μmol/L, and V max of CP I and CP III were 1.77 and 2.46 pmol/min/106 cells (0.708 and 0.984 pmol/min/well), respectively (Figure 1).
FIGURE 1.
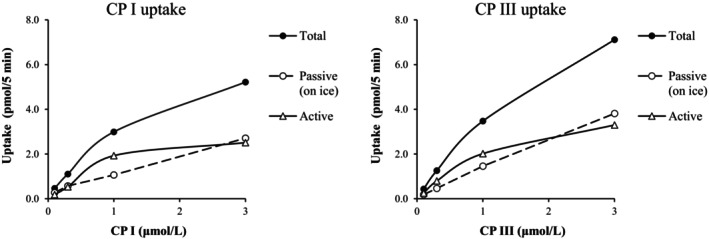
Concentration‐dependent accumulation of CP I and III in PXB‐cells. Active transport is calculated as the difference of total and passive uptake, passive uptake was determined on ice in this experiment. Each point is represented as mean (n = 2).
3.2. In vitro DDI for the uptake of CP I and CP III with RIF in PXB‐cells
The inhibitory effects of RIF (concentration: 0.02 to 200 μmol/L) on the uptake of each CP I and CP III (concentration: 0.3 μmol/L) in PXB‐cells were examined. The uptake of CP I and CP III were inhibited in the concentration‐dependent manner of RIF, and the IC50 were 1.67 and 2.62 μmol/L, respectively (Figure 2).
FIGURE 2.
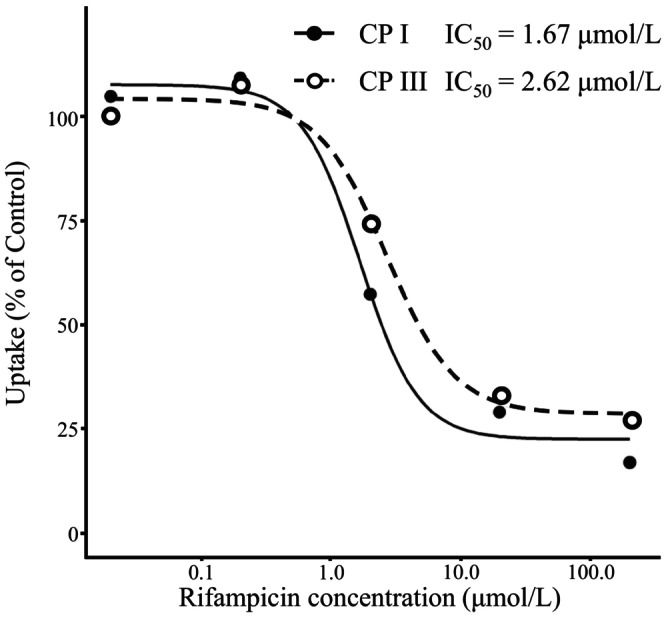
IC50 determination for the inhibition of CP I and III uptake by RIF in PXB‐cells. Intracellular CP I and III accumulation were determined after coincubation with 0.3 μmol/L CP I and III, and 0.02–200 μmol/L RIF for 5 min at 37°C. The RIF concentration causing half‐maximal inhibitory effect (IC50) on CP I and III accumulation in PXB‐cells was calculated. Each point is represented as mean (n = 2).
3.3. Effect of RIF on the concentration of CP I and CP III in ICR mice plasma
RIF was administrated orally and intravenously to each group consisting of three fasted ICR mice, at a dose of 10 or 30 mg/kg. The mean plasma concentration‐time curves of RIF after a single oral or intravenous administration of RIF to male ICR mice are presented in Figure 3. The pharmacokinetic parameters of RIF are presented in Table 1. Following a single oral dose of 10 and 30 mg/kg of RIF, the plasma concentrations of the unchanged form reached its maximum level (C max) of 5417 and 17 367 ng/mL at 2 h post‐dose and the elimination half‐life (t 1/2) were 3.96 and 3.64 h. Following a single intravenous dose of 10 and 30 mg/kg of RIF, the systemic plasma clearance (CLtot) was 100 and 59.8 mL/h/kg, and bioavailability (BA) was 40.7% and 33.2%, respectively. Both C max and AUC0‐24h were dose‐dependently increased. Considering that mouse plasma protein binding ratio of RIF was reported as 96.7%, 34 plasma concentrations of unbound RIF after oral and intravenous administration at 10 and 30 mg/kg to ICR mice was reached 573 ng/mL (0.70 μmol/L) and 1571 ng/mL (1.91 μmol/L), respectively. The unbound maximum hepatic inlet concentration of the RIF in blood (I in,max,u) after oral administration at 30 mg/kg to ICR mice was calculated as 672 ng/mL (0.82 μmol/L) (Table 2).
FIGURE 3.
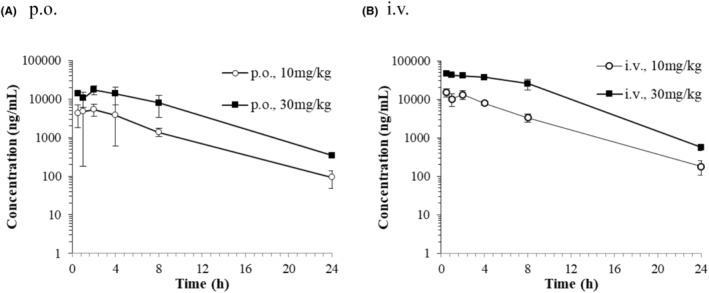
Plasma concentration‐time curves of unchanged form after a single oral (A) or intravenous (B) administration of RIF to male ICR mice. Each point and bar are represented as mean ± SD (n = 3).
TABLE 1.
Pharmacokinetic parameters of unchanged form after a single oral or intravenous administration of 10 and 30 mg/kg RIF to male ICR mice and PXB mice.
| Dose | (mg/kg) | Oral | Intravenous | |||
|---|---|---|---|---|---|---|
| 10 | 30 | 10 | 30 | |||
| ICR | C max | (ng/mL) | 5417 | 17 367 | — | — |
| C 0.5 h | (ng/mL) | — | — | 15 133 | 47 067 | |
| T max | (h) | 2.00 | 2.00 | — | — | |
| AUC0‐last | (h·ng/mL) | 40 124 | 164 672 | 98 842 | 498 982 | |
| CLtot | (mL/h/kg) | — | — | 100 | 59.8 | |
| V d,ss | (mL/kg) | — | — | 475 | 337 | |
| t 1/2 | (h) | 3.96 | 3.64 | 3.74 | 3.11 | |
| PXB | C max | (ng/mL) | 1277 | 4180 | — | — |
| C 0.5 h | (ng/mL) | — | — | 1840 | 5903 | |
| T max | (h) | 2.33 | 0.83 | — | — | |
| AUC0‐last | (h·ng/mL) | 6593 | 25 898 | 7989 | 24 388 | |
| CLtot | (mL/h/kg) | — | — | 1169 | 1161 | |
| V d,ss | (mL/kg) | — | — | 2432 | 3404 | |
| t 1/2 | (h) | 5.26 | 6.71 | 1.30 | 2.22 | |
Abbreviations: AUC0‐last, AUC from time zero to 8 h (intravenous administration in PXB mice) and time zero to 24 h (others); —, not applicable.
TABLE 2.
Estimated unbound RIF concentrations inlet to the liver and in systemic plasma in ICR and PXB mice at doses of 10 and 30 mg/kg.
| Dose (mg/kg) | Concentration (μmol/mL) | ||
|---|---|---|---|
| I in,max,u | Maximum observed systemic unbound in plasma | ||
| ICR | |||
| p.o. | 10 | 0.26 | 0.22 |
| 30 | 0.82 | 0.70 | |
| i.v. | 10 | — | 0.66 |
| 30 | — | 1.91 | |
| PXB | |||
| p.o. | 10 | 1.01 | 0.30 |
| 30 | 3.10 | 0.97 | |
| i.v. | 10 | — | 0.56 |
| 30 | — | 1.43 | |
Note: Data are expressed as mean (n = 3).
Plasma concentrations of CP I and CP III were determined in the ICR mice. The mean plasma concentration‐time curves of CP I and CP III are presented in Figure 4. Plasma concentrations of CP I and CP III in the vehicle‐dosed control samples were not constant but increased from 8 to 24 h after administrations. Besides, some samples in RIF‐treated groups also showed the increase of plasma concentrations of CP I and CP III in that time period despite very low RIF concentrations (data not shown). Implying that the tendency might have been caused by circadian rhythm and considering homeostasis of CP I and CP III, AUC of plasma CP I and CP III were calculated from samples collected until 8 h after administration in this study. There was no significant difference in CP I and CP III AUC0‐8h after an oral administration of RIF compared to the vehicle‐treated group, while significant 1.3‐fold and 2.2‐fold increases in CP I AUC0‐8h (p < 0.01) were observed in intravenously RIF administrated mice (Table 3). Plasma CP III AUC0‐8h following intravenous administration of 30 mg/kg RIF was significantly increased by 2.4‐fold compared with the vehicle‐treated group (p < 0.05). AUC0‐8h of CP III was higher than that of CP I in the orally and intravenously vehicle‐treated group (fold increases of 4.6 and 3.3, respectively). It has been reported that AUC0‐8h of CP III was higher than that of CP I by 4.5‐fold in non‐treated FVB/N mice. 14
FIGURE 4.
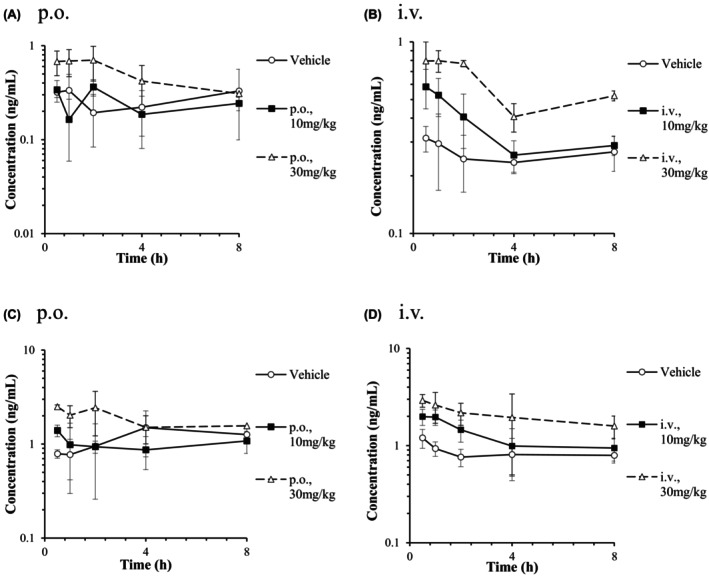
Plasma concentration‐time curves of CP I (A, B) and III (C, D) after a single oral or intravenous administration of RIF to male ICR mice. Each point and bar are represented as mean ± SD (n = 3).
TABLE 3.
Comparisons of AUC0‐8h of CP I and CP III following a single oral or intravenous administration of vehicle, 10 and 30 mg/kg RIF to male ICR mice and PXB mice.
| Dose (mg/kg) | AUC0‐8h (nmol/L·h) [fold change vs. vehicle] | ||||
|---|---|---|---|---|---|
| Oral | Intravenous | ||||
| CP I | CP III | CP I | CP III | ||
| ICR | Vehicle | 3.09 ± 1.30 [−] | 14.4 ± 1.3 [−] | 3.19 ± 0.35 [−] | 10.5 ± 2.0 [−] |
| RIF 10 | 2.87 ± 0.77 [0.9] | 11.6 ± 3.4 [0.8] | 4.28 ± 0.08* [1.3] | 15.4 ± 2.8 [1.5] | |
| RIF 30 | 4.83 ± 0.75 [1.6] | 18.1 ± 6.9 [1.3] | 7.09 ± 0.65** [2.2] | 25.3 ± 8.1 [2.4] | |
| PXB | Vehicle | 3.23 ± 0.25 [−] | 11.4 ± 1.3 [−] | 3.22 ± 0.85 [−] | 11.6 ± 1.9 [−] |
| RIF 10 | 5.75 ± 0.47** [1.8] | 12.4 ± 1.0 [1.1] | 6.42 ± 3.18 [2.0] | 9.16 ± 4.92 [0.8] | |
| RIF 30 | 8.34 ± 0.68** [2.6] | 12.8 ± 1.7 [1.1] | 12.6 ± 2.7* [3.9] | 14.8 ± 2.7 [1.3] | |
Notes: Data are expressed as mean ± SD (n = 3); **p < .01, *p < .05, statistically significant difference compared to the vehicle control group.
Abbreviation: −, not applicable.
3.4. Effect of RIF on the concentration of CP I and CP III in PXB mice plasma
RIF was administrated orally and intravenously to each group consisting of three fasted PXB mice, at a dose of 10 or 30 mg/kg. The mean plasma concentration‐time curves of RIF after a single oral or intravenous administration of RIF to male PXB mice are presented in Figure 5. The pharmacokinetic parameters of RIF are presented in Table 1. Following a single oral dose of 10 and 30 mg/kg of RIF, the Cmax were 1277 and 4180 ng/mL at 2.33 and 0.83 h post‐dose and the t1/2 were 5.26 and 6.71 h. Following a single intravenous dose of 10 and 30 mg/kg of RIF, the plasma concentrations of the unchanged form at 0.5 h post‐dose (C0.5h) were 1840 and 5903 ng/mL and the CLtot were 1169 and 1161 mL/h/kg. Both C max and AUC were dose‐dependently increased. However, C max and AUC of RIF after oral administration in PXB mice were lower than those in ICR mice (by a fourth and by a sixth, respectively).
FIGURE 5.
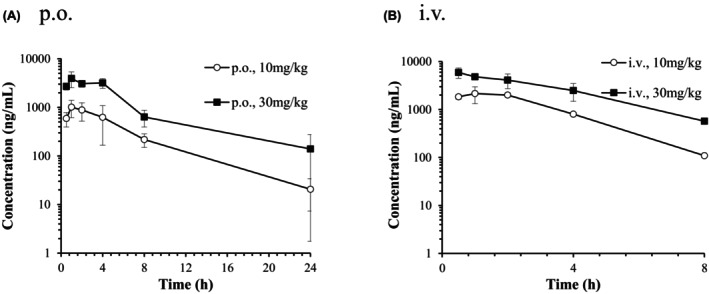
Plasma concentration‐time curves of unchanged form after a single oral (A) or intravenous (B) administration of RIF to male PXB mice. Each point and bar are represented as mean ± SD (n = 3). Some points are represented by mean of two (0.5 and 8 h after 10 mg/kg, i.v.; 4 h after 30 mg/kg, i.v.) or by one sample (4 h after 10 mg/kg, i.v.).
Considering that estimated f u,p of RIF in PXB mouse was 0.19, plasma unbound concentrations of RIF after oral and intravenous administration at 10 and 30 mg/kg to PXB mice was reached 929 ng/mL (1.13 μmol/L) and 1362 ng/mL (1.66 μmol/L) respectively. The I in,max,u after oral administration at 30 mg/kg was calculated as 2553 ng/mL (3.10 μmol/L) (Table 2).
Plasma samples for the determination of CP I and CP III concentrations were collected from PXB mice after a single oral or intravenous administration of vehicle or RIF (10 or 30 mg/kg). The mean plasma concentration‐time curves of CP I and CP III are presented in Figure 6. A significant 1.8‐fold and 2.6‐fold increases in CP I AUC0‐8h (p < 0.01) were observed in orally RIF (10 and 30 mg/kg) administrated PXB mice (Table 3). After a single intravenous administration of RIF (10 and 30 mg/kg), AUC0‐8h of CP I was increased by 2.0‐fold and 3.9‐fold respectively, whereas a significant difference was observed in 30 mg/kg RIF‐treated PXB mice (p < 0.01). There was no significant difference in CP III AUC0‐8h after RIF treatment compared to the vehicle‐treated group. AUC0‐8h of CP III was higher than that of CP I in the orally and intravenously vehicle‐treated group (fold increases of 4.5 and 3.3, respectively).
FIGURE 6.
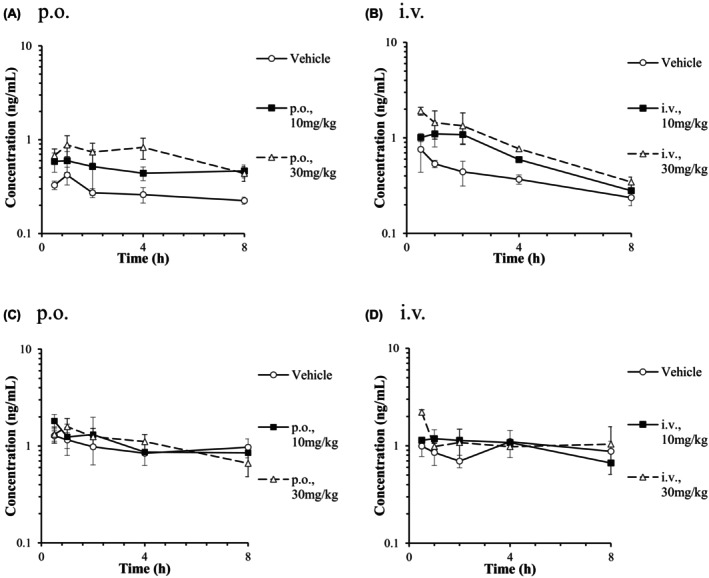
Plasma concentration‐time curves of CP I (A, B) and III (C, D) after a single oral or intravenous administration of RIF to male PXB mice. Each point and bar are represented as mean ± SD (n = 3). Some points are represented by mean of two (0.5 and 8 h after 10 mg/kg, i.v.; 4 h after 30 mg/kg, i.v.) or by one sample (4 h after 10 mg/kg, i.v.).
3.5. CP I/III levels in mouse urine following administration of RIF
Urine samples were collected from ICR and PXB mice orally administrated 30 mg/kg RIF (Figure 7). The amounts of CP I and CP III excreted in the urine for 24 h (X e(0‐24h)) were higher in RIF dosed ICR mice compared with the vehicle‐treated animals (fold increase of 1.4), and the difference observed in urinary CP I was statistically significant (p < 0.01). The amounts of CP I and III Xe(0‐24h) were significantly increased in RIF‐dosed PXB mice compared with the vehicle‐treated animals (fold increases of 3.2 (p < 0.01) and 2.4 (p < 0.05)). Among vehicle‐treated animals, the Xe(0‐24h) of CP III was higher than that of CP I in ICR and PXB mice urine (fold increase of 4.7 and 1.9).
FIGURE 7.
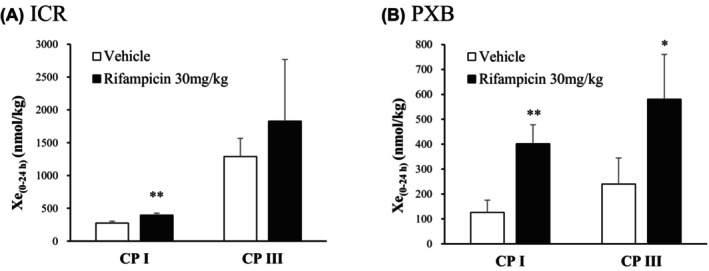
Effects of RIF on urinary excretion of CP I and CP III in ICR (A) and PXB (B) mice. The 24‐h urinary excretion rates of CP I and CP III were determined in mice after a single oral administration of RIF. Data are expressed as mean ± SD (n = 3); *p < 0.05 and **p < 0.01, statistically significant difference compared to the vehicle control group.
4. DISCUSSION
In human hepatocytes isolated from the chimeric PXB mice, CP I and III were taken up into the cells in a substrate concentration‐dependent manner, with K m values of 1.11 and 1.46 μmol/L and V max values of 1.77 and 2.46 pmol/min/106 cells, respectively (Figure 1). Bednarczyk reported that the K m values of CP I were 0.13 (OATP1B1) and 3.25 (1B3) μmol/L and the K m values of CP III were 0.22 (1B1), 4.61 (1B3), and 0.034 (2B1) μmol/L using transporter‐expressing cells. CP I was not a substrate for 2B1. 12 Based on previous studies, the K m values of CP I and CP III obtained in the present study were considered reasonable. RIF showed a concentration‐dependent inhibition on the cellular uptake of CP I and CP III with IC50 values of 1.67 and 2.62 μmol/L, respectively (Figure 2). In studies using transporter‐expressing cells, the IC50 values of RIF for CP I and CP III uptake ranged from 0.66 to 4.61 μmol/L using OATP 1B1, from 0.25 to 1.61 μmol/L using OATP1B3, and only CP III uptake was observed using OATP2B1, with an IC50 of 48.4 μmol/L to N/A. 12 , 15 The rate of uptake of CP III using OATP2B1 was approximately 0.8 to 1.3 times faster than that using OATP1B1 or 1B3, and the amounts of CP III uptake using each transporter were similar. 12 In addition, Hirano et al. examined the expression rates of OATP1B1, 1B3, and 2B1 in human hepatocytes from various lots and reported that 2B1 is expressed 0.07‐ to 0.16‐fold more than 1B1 and 1B3. 7 Wang et al. reported similar expression levels of OATP1B1, 1B3, and 2B1 in human hepatocytes (each around 10% of the total transporter expression). 39 Therefore, OATP2B1 may be responsible for approximately 20% to 30% of the overall clearance in the uptake of CP III induced by OATPs in human hepatocytes. The effect of OATP2B1 on CP III uptake and the difference in IC50 between CP I and CP III were not examined in this study.
Next, we measured the plasma concentrations of CP I and CP III in mice in vivo. The plasma concentration of CP III in the vehicle group of wild‐type ICR mice was similar to that reported in FVB/N and Oatp1a/1b‐null mice 14 and was higher than the plasma concentration of CP I (ratio of CP I/CP III: 0.30 ± 0.07) (Figure 4). In PXB mice, the plasma CP III was also higher than that of CP I (ratio of CP I/CP III: 0.29 ± 0.03) (Figure 6). Notably, the plasma CP I/CP III ratio reported in humans is 5.7 to 6.5, 13 and that in monkeys is 3.1. 40 CPs are by‐products of heme biosynthesis. Bone marrow erythroblasts (75%–80%) and the liver (15%–20%) are primarily responsible for heme biosynthesis in humans. 41 Even though the greater portion of the PXB mouse liver used in the present study was substituted for human hepatocytes, the plasma CP I/CP III ratio was similar to that in ICR mice, but not in humans, possibly because the greater portion of CPs is derived from the bone marrow.
Subsequently, we confirmed the influence of RIF administration in mice on plasma CP I and CP III concentrations. In ICR mice, oral administration of RIF did not significantly increase CP I or CP III in plasma. However, intravenous administration showed a dose‐dependent increase in plasma CP I and CP III, with a significant 2.2‐ (p < .01) and 2.4‐fold (p < .05) increase using 30 mg/kg RIF, respectively (Table 3). There are no reports on the IC50 of RIF for CP I or CP III transports mediated by mouse OATP proteins (translated by Oatp1a1, Oatp1a4, Oatp1b2, and Oatp2b1), owing to the difficulty in obtaining the appropriate tools. The RIF concentrations after oral administration in ICR mice may be insufficient to inhibit CP I and CP III uptake induced by mouse OATPs (Table 2). In PXB mice, the systemic clearance of RIF was 11 to 19 times greater than that in ICR mice, and the plasma concentrations of RIF in ICR mice were lower. RIF is primarily metabolized in the liver, and the formation of deacetyl and formyl metabolites has been well‐characterized, but no reports have mentioned that significant differences appeared in the RIF metabolic profiles between humans and mice. 42 Meanwhile, the plasma protein binding rate in mice is reported to be 96.7%, 34 whereas that in humans is 77.7%, 33 suggesting that there may be no significant difference in systemic unbound plasma RIF concentrations between PXB and ICR mice (Table 2). In PXB mice, both oral and intravenous administrations of RIF showed a dose‐dependent increase in plasma CP I, with a significant 2.6‐ (p < .01) and 3.9‐fold (p < .01) increase using 30 mg/kg RIF, respectively (Table 3). However, unlike ICR mice, no CP III increase in plasma was observed in PXB mice. Considering the unbound plasma RIF concentrations and in vitro IC50 values (CP I: 1.67 μmol/L; CP III: 2.62 μmol/L), the plasma RIF concentrations may be insufficient to quantitatively inhibit OATP‐mediated CP III uptake in the liver in PXB mice. Additional experiments using more than 30 mg/kg RIF are necessary.
Mori et al. studied plasma endogenous substrate concentrations, such as CP I and CP III, in humans after oral administration of RIF and reported a dose‐dependent increase in the AUC0‐24h of CP I in plasma. 43 In their study, the C max of unbound RIF ranged from 1.06 to 5.06 μmol/L, and the AUCR of CP I ranged from 1.5 to 3.7. Thus, the unbound RIF in PXB mice was similar to or slightly lower than that reported in humans (Table 2), and the increases in plasma CP I were not significantly different from those observed in humans (Table 3). The extent of the increase in CPIII levels in plasma after administration of RIF to PXB mice was smaller than that of CPI. The same tendency was reported in human plasma after OTAP inhibition by RIF. 13
Both CP I and CP III are excreted from the body through urinary excretion in the kidneys and bile excretion in the liver. The bile extraction rates of CPs up to 30 min after intravenous administration of the isomers in rats were reported to be 72% of the dose for CP I and 35% of the dose for CP III. 44 The renal clearance of CP III in monkeys and humans at a steady state is greater than that of CP I 13 , 14 ; therefore, CP I is primarily excreted in bile, and CP III is primarily excreted in urine. Neither CP I nor CP III is influenced by enterohepatic circulation. 27 CP I and CP III transport is mediated by OATP1B1/B3, but not by OAT1/2/3/4, OCT1/2, MATE1/2‐K, or NTCP. 12 , 15 CP I is primarily excreted through passive transport, and CP III is considered to be excreted through active transport, but no reports have identified specific transport carriers. 11 OATP2B1 expression is detected in the proximal kidney tubule, and its activity may influence the concentration of CP III in circulating blood. 45 In the present study, urinary excretion of CP III was higher than that of CP I in both ICR and PXB mice, which is similar to monkeys and humans. The elevated urinary CP I and CP III levels in the RIF‐treated group in PXB mice [CP I: 3.2‐fold (p < .01), CP III: 2.4‐fold (p < .05)] were more pronounced compared with those in ICR mice [CP I: 1.4‐fold (p < .01), CP III: 1.4‐fold (no significant difference)] (Figure 7). A previous study on the human urinary CP levels (Xe(0‐24h)) after RIF administration showed a 3.6‐fold increase in CP I and 1.6‐fold increase in CP III. 13 In another study on cynomolgus monkeys, a single oral administration of RIF increased the urinary CP I and CP III levels (Xe(0‐48h)) 4.3‐ and 3.1‐fold, respectively. 14 In particular, significant changes in urinary concentrations were observed in PXB mice for CP III, whereas no changes were observed in plasma. This is important considering that OATP2B1 is an uptake transporter for CP III from urine in the renal proximal tubules. Additionally, the urinary excretion of CP III was lower in PXB mice than in ICR mice in the vehicle group. Therefore, the renal tissue expression of transporters, including OATP2B1 in ICR and PXB mice, should be investigated in the future.
This is the first report on plasma and urinary concentrations of CP I and CP III in chimeric mice with human hepatocytes, namely PXB mice. This study showed that the treatment of PXB mice with an OATP1B inhibitor, namely RIF, significantly increased the plasma and urinary CP I concentrations. Notably, we obtained information on the relationship between the unbound plasma RIF concentration and changes in CP I in ICR and PXB mice over the range of RIF exposure that has been reported to alter plasma CP I in humans. Furthermore, changes in CP III levels were different from those in CP I. Particularly, CP III levels in urinary excretion differed between ICR and PXB mice. For OATP‐mediated DDI assessment, plasma and urinary CP I and/or CP III levels in these mice can serve as biomarkers.
AUTHOR CONTRIBUTIONS
Participation in research design: Uchida and Shishido. Conducted experiments: Shishido, Uchida, and Yoshida. Performed data analysis: Shishido, and Uchida. Wrote or contributed to the writing of the manuscript: Shishido, Uchida, and Oshida.
ETHICS STATEMENT
We obtained approvals from the Animal Care and Use Committee of Toray Industries, Inc. (Approval number: AC2018‐136, AC2019‐43, AC2019‐51) and performed experiments in accordance with the Guideline for the Animal Experiments, Research & Development Division, Toray Industries, Inc. This study was performed according to procedures approved by the Epidemic Prevention Committee, Toray Industries, Inc. (Approval number: 2019‐1025).
ACKNOWLEDGMENTS
The authors thank Masakazu Kakuni, and other members in PhoenixBio Co. Ltd. for providing PXB‐cells, PXB mice, and other materials.
Shishido Y, Yoshida T, Oshida K, Uchida M. Plasma and urinary CP I and CP III concentrations in chimeric mice with human hepatocytes after rifampicin administration. Pharmacol Res Perspect. 2024;12:e70017. doi: 10.1002/prp2.70017
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Mück W, Mai I, Fritsche L, et al. Increase in cerivastatin systemic exposure after single and multiple dosing in cyclosporine‐treated kidney transplant recipients. Clin Pharmacol Ther. 1999;65:251‐261. [DOI] [PubMed] [Google Scholar]
- 2. Li Y, Talebi Z, Chen X, Sparreboom A and, Hu S (2021) Endogenous biomarkers for SLC transporter‐mediated drug‐drug interaction evaluation. Molecules 26:5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ciută AD, Nosol K, Kowal J, et al. Structure of human drug transporters OATP1B1 and OATP1B3. Nat Commun. 2023;14:5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. European Medicines Agency, Committee for Human Medicinal Products . Guideline on the Investigation of Drug Interactions [Internet]. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf
- 5. US Department of Health and Human Services , Food and Drug Administration , Center for Drug Evaluation and Research (CDER) . Vitro Drug Interaction Studies — Cytochrome P450 Enzyme‐ and Transporter‐Mediated Drug Interactions Guidance for Industry [Internet]. 2020. https://www.fda.gov/media/134582/download
- 6. Ministry of Health. Labour and Welfare, Japan . Guideline of drug interaction studies for drug development and appropriate provision of information [Internet]. 2018. https://www.pmda.go.jp/files/000228122.pdf
- 7. Hirano M, Maeda K, Shitara Y, Sugiyama Y. Drug‐drug interaction between pitavastatin and various drugs via OATP1B1. Drug Metab Dispos. 2006;34:1229‐1236. [DOI] [PubMed] [Google Scholar]
- 8. Sharma P, Butters CJ, Smith V, Elsby R, Surryet D. Prediction of the in vivo OATP1B1‐mediated drug‐drug interaction potential of an investigational drug against a range of statins. Eur J Pharm Sci. 2012;47:244‐255. [DOI] [PubMed] [Google Scholar]
- 9. Yoshida K, Maeda K, Sugiyama Y. Transporter‐mediated drug–drug interactions involving OATP substrates: predictions based on in vitro inhibition studies. Clin Pharmacol Ther. 2012;91:1053‐1064. [DOI] [PubMed] [Google Scholar]
- 10. Chu X, Chan GH, Evers R. Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter‐mediated drug‐drug interactions. J Pharm Sci. 2017;106:2357‐2367. [DOI] [PubMed] [Google Scholar]
- 11. Müller F, Sharma A, König J, Fromm MF. Biomarkers for in vivo assessment of transporter function. Pharmacol Rev. 2018;70:246‐277.12. [DOI] [PubMed] [Google Scholar]
- 12. Bednarczyk D, Boiselle C. Organic anion transporting polypeptide (OATP)‐mediated transport of coproporphyrins I and III. Xenobiotica. 2016;46:457‐466. [DOI] [PubMed] [Google Scholar]
- 13. Lai Y, Mandlekar S, Shen H, et al. Coproporphyrins in plasma and urine can Be appropriate clinical biomarkers to recapitulate drug‐drug interactions mediated by organic anion transporting polypeptide inhibition. J Pharmacol Exp Ther. 2016;358:397‐404. [DOI] [PubMed] [Google Scholar]
- 14. Shen H, Dai J, Liu T, et al. Coproporphyrins I and III as functional markers of OATP1B activity: in vitro and in vivo evaluation in preclinical species. J Pharmacol Exp Ther. 2016;357:382‐393. [DOI] [PubMed] [Google Scholar]
- 15. Shen H, Chen W, Drexler DM, et al. Comparative evaluation of plasma bile acids, Dehydroepiandrosterone sulfate, Hexadecanedioate, and Tetradecanedioate with Coproporphyrins I and III as markers of OATP inhibition in healthy subjects. Drug Metab Dispos. 2017;45:908‐919. [DOI] [PubMed] [Google Scholar]
- 16. Tateno C, Yoshizane Y, Saito N, et al. Near completely humanized liver in mice shows human‐type metabolic responses to drugs. Am J Pathol. 2004;165:901‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nishimura M, Yoshitsugu H, Yokoi T, et al. Evaluation of mRNA expression of human drug‐metabolizing enzymes and transporters in chimeric mouse with humanized liver. Xenobiotica. 2005;35:877‐890. [DOI] [PubMed] [Google Scholar]
- 18. Okumura H, Katoh M, Sawada T, et al. Humanization of excretory pathway in chimeric mice with humanized liver. Toxicol Sci. 2007;97:533‐538. [DOI] [PubMed] [Google Scholar]
- 19. Ohtsuki S, Kawakami H, Inoue T, et al. Validation of uPA/SCID mouse with humanized liver as a human liver model: protein quantification of transporters, cytochromes P450, and UDP‐glucuronosyltransferases by LC‐MS/MS. Drug Metab Dispos. 2014;42:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 20. Inoue T, Nitta K, Sugihara K, Horie T, Kitamura S, Ohta S. CYP2C9‐catalyzed metabolism of S‐warfarin to 7‐hydroxywarfarin in vivo and in vitro in chimeric mice with humanized liver. Drug Metab Dispos. 2008;36:2429‐2433. [DOI] [PubMed] [Google Scholar]
- 21. Sanoh S, Horiguchi A, Sugihara K, et al. Predictability of metabolism of ibuprofen and naproxen using chimeric mice with human hepatocytes. Drug Metab Dispos. 2012;40:2267‐2272. [DOI] [PubMed] [Google Scholar]
- 22. Sanoh S, Nozaki K, Murai H, Terashita S, Teramura T, Ohta S. Prediction of human metabolism of FK3453 by aldehyde oxidase using chimeric mice transplanted with human or rat hepatocytes. Drug Metab Dispos. 2012;40:76‐82. [DOI] [PubMed] [Google Scholar]
- 23. Schulz‐Utermoehl T, Sarda S, Foster JR, et al. Evaluation of the pharmacokinetics, biotransformation and hepatic transporter effects of troglitazone in mice with humanized livers. Xenobiotica. 2012;42:503‐517. [DOI] [PubMed] [Google Scholar]
- 24. Tanoue C, Sugihara K, Uramaru N, et al. Prediction of human metabolism of the sedative‐hypnotic zaleplon using chimeric mice transplanted with human hepatocytes. Xenobiotica. 2013;43:956‐962. [DOI] [PubMed] [Google Scholar]
- 25. Nakada N, Oda K. Identification and characterization of metabolites of ASP015K, a novel oral Janus kinase inhibitor, in rats, chimeric mice with humanized liver, and humans. Xenobiotica. 2015;45:757‐765. [DOI] [PubMed] [Google Scholar]
- 26. Kakuni M, Yamasaki C, Tachibana A, Yoshizane Y, Ishida Y, Tateno C. Chimeric mice with humanized livers: a unique tool for in vivo and in vitro enzyme induction studies. Int J Mol Sci. 2013;15:58‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takehara I, Watanabe N, Mori D, Ando O, Kusuhara H. Effect of rifampicin on the plasma concentrations of bile acid‐O‐sulfates in monkeys and human liver‐transplanted chimeric mice with or without bile flow diversion. J Pharm Sci. 2019;108:2756‐2764. [DOI] [PubMed] [Google Scholar]
- 28. Feng B, Pemberton R, Dworakowski W, et al. Evaluation of the utility of PXB chimeric mice for predicting human liver partitioning of hepatic organic anion‐transporting polypeptide transporter substrates. Drug Metab Dispos. 2021;49:254‐264. [DOI] [PubMed] [Google Scholar]
- 29. Yamasaki C, Kataoka M, Kato Y, et al. In vitro evaluation of cytochrome P450 and glucuronidation activities in hepatocytes isolated from liver‐humanized mice. Drug Metab Pharmacokinet. 2010;25:539‐550. [DOI] [PubMed] [Google Scholar]
- 30. Yamasaki C, Tateno C, Aratani A, et al. Growth and differentiation of colony‐forming human hepatocytes in vitro. J Hepatol. 2006;44:749‐757. [DOI] [PubMed] [Google Scholar]
- 31. Yamasaki C, Ishida Y, Yanagi A, et al. Culture density contributes to hepatic functions of fresh human hepatocytes isolated from chimeric mice with humanized livers: novel, long‐term, functional two‐dimensional in vitro tool for developing new drugs. PLoS One. 2020;11(15):e0237809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. An H, Landis JT, Bailey AG, Marron JS, Dittmer DP. dr4pl: a stable convergence algorithm for the 4 parameter logistic model. R I Dent J. 2019;11:171. [Google Scholar]
- 33. Polasa K, Murthy J, Krishnaswamy K. Rifampicin kinetics in undernutrition. Br J Clin Pharmacol. 1984;17:481‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steenwinkel JEM, Aarnoutse RE, Knegt GJ, et al. Optimization of the rifampin dosage to improve the therapeutic efficacy in tuberculosis treatment using a murine model. Am J Respir Crit Care Med. 2013;187:1127‐1134. [DOI] [PubMed] [Google Scholar]
- 35. Miyamoto M, Kosugi Y, Iwasaki S, et al. Characterization of plasma protein binding in two mouse models of humanized liver, PXB mouse and humanized TK‐NOG mouse. Xenobiotica. 2021;51:51‐60. [DOI] [PubMed] [Google Scholar]
- 36. Lyons MA, Reisfeld B, Yang RSH, Lenaerts AJ. A physiologically based pharmacokinetic model of rifampin in mice. Antimicrob Agents Chemother. 2013;57:1763‐1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993;10:1093‐1095. [DOI] [PubMed] [Google Scholar]
- 38. Binda G, Domenichini E, Gottardi A, et al. Rifampicin, a general review. Arzneimittelforschung. 1971;21:1907‐1977. [PubMed] [Google Scholar]
- 39. Wang L, Prasad B, Salphati L, et al. Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab Dispos. 2015;43:367‐374. [DOI] [PubMed] [Google Scholar]
- 40. Gu X, Wang L, Gan J, et al. Absorption and disposition of Coproporphyrin I (CPI) in Cynomolgus monkeys and mice: pharmacokinetic evidence to support the use of CPI to inform the potential for organic anion‐transporting polypeptide inhibition. Drug Metab Dispos. 2020;48:724‐734. [DOI] [PubMed] [Google Scholar]
- 41. Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute hepatic Porphyrias: review and recent Progress. Hepatol Commun. 2018;3:193‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. National Center for Biotechnology Information . PubChem Compound Summary for CID 135398735, Rifampin. Accessed April 18, 2024. https://pubchem.ncbi.nlm.nih.gov/compound/Rifampin
- 43. Mori D, Kimoto E, Rago B, et al. Dose‐dependent inhibition of OATP1B by rifampicin in healthy volunteers: comprehensive evaluation of candidate biomarkers and OATP1B probe drugs. Clin Pharmacol Ther. 2020;107:1004‐1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaplowitz N, Javitt N, Kappaset A. Coproporphyrin I and 3 excretion in bile and urine. J Clin Invest. 1972;51:2895‐2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Medwid S, Price HR, Taylor DP, et al. Organic anion transporting polypeptide 2B1 (OATP2B1) genetic variants: in vitro functional characterization and association with circulating concentrations of endogenous substrates. Front Pharmacol. 2021;12:713567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.