

Summary
SIR2-HerA, a bacterial two-protein anti-phage defense system, induces bacterial death by depleting NAD+ upon phage infection. Biochemical reconstitution of SIR2, HerA, and the SIR2-HerA complex reveals a dynamic assembly process. Unlike other ATPases, HerA can form various oligomers, ranging from dimers to nanomers. When assembled with SIR2, HerA forms a hexamer and converts SIR2 from a nuclease to an NAD+ hydrolase, representing an unexpected regulatory mechanism mediated by protein assembly. Furthermore, high-concentrations of ATP can inhibit NAD+ hydrolysis by the SIR2-HerA complex. Cryo-EM structures of the SIR2-HerA complex reveal a giant supramolecular assembly up to 1 MDa, with SIR2 as a dodecamer and HerA as a hexamer, crucial for anti-phage defense. Unexpectedly, the HerA hexamer resembles a spiral staircase and exhibits helicase activities toward dual-forked DNA. Together, we reveal the supramolecular assembly of SIR2-HerA as a unique mechanism for switching enzymatic activities and bolstering anti-phage defense strategies.
Introduction
To combat phage infections, bacteria have evolved diverse anti-phage defense systems1–4. Recently, hundreds of novel anti-phage defense systems have been identified through bioinformatic analysis5–8. A few of the bacteria anti-phage defense systems have been well studied9–12, but most remain largely unexplored.
As a newly identified bacterial immune system, the SIR2-HerA system is composed of two components: HerA and SIR26,13. HerA, an ATPase identified in archaea and bacteria, has been shown to have DNA helicase activity14,15. In archaea or bacteria, NurA, a nuclease, and HerA form a 2:6 complex to process DNA double-strand breaks for subsequent DNA repair14,16. In contrast, SIR2 is homologous to sirtuin family members, which catalyze the removal of an acetyl moiety from modified lysine residues using NAD+ as a cofactor17–19. Previous studies have revealed that the activation of the SIR2-HerA system leads to the depletion of NAD+ in cells, thereby triggering bacterial cell death13. However, how HerA and SIR2, with contrasting enzymatic activities, assemble and cooperate to trigger bacteria cell death during phage infection remains unclear.
Here, we present a biochemical and structural analysis of E. coli HerA (EcHerA), SIR2 (EcSIR2), and the SIR2-HerA complex, revealing a unique mechanism of assembly-mediated activation that governs immune defense by the SIR2-HerA complex. Contrasting to its homologous proteins that tend to form a ring-like hexamer or heptamer, EcHerA assembles into various oligomers ranging from dimers to nanomers. Unexpectedly, EcSIR2 form a dodecamer, distinct from its homologs that function as a monomer. Moreover, dodecameric EcSIR2 serves as an organizer that assembles into a full complex with six protomers of EcHerA. Interestingly, the six protomers of EcHerA in the SIR2-HerA complex form an asymmetric hexamer with five protomers interacting with EcSIR2. Strikingly, we demonstrate that EcSir2 alone displays nuclease activity. When EcSIR2 forms a complex with EcHerA, the SIR2-HerA complex can efficiently hydrolyze NAD+ rather than digest nucleic acids. As such, the assembly of the SIR2-HerA complex converts the enzymatic activities of SIR2 from a nuclease to an NADase. Moreover, ATP can inhibit the NAD+ hydrolysis by the SIR2-HerA complex, while ATP hydrolysis by HerA can promote the NADase activity and helicase activity of the SIR2-HerA complex. The multiple enzymatic activities, together with the enzymatic activity switching and regulation by the assembly of the SIR2-HerA complex, constitute an intriguing mechanism for anti-phage defense by the supramolecular assembly of the SIR2-HerA complex.
Results
Assembly of SIR2, HerA, and the SIR2-HerA complex
To biochemically characterize the SIR2-HerA system, we first expressed and purified EcSIR2 and EcHerA, both of which appear to form large oligomers as estimated by the gel filtration profiles (Figures S1A–B). To further confirm the protein assembly, we collected cryo-EM datasets of EcSIR2 and EcHerA for structural analysis. Due to severe orientation preference problem, we did not obtain high-resolution structures of EcSIR2 and EcHerA (Figures 1A–C, S1C–D, & Table 1). The structure of EcSIR2 at 3.6 Å revealed that EcSIR2 alone assembles as a dodecamer, contrasting the well-studied SIR2 homologs that all exist as monomers17,18,20–22 (Figures 1A–B, S1D, & Table 1). Unexpectedly, EcHerA forms diverse oligomers, including dimers, trimers, tetramers, pentamers, hexamers, heptamers, octamers, and nonamers (Figure 1C). In addition, the oligomerization status of EcHerA appears to be concentration dependent, evidenced by the elution volumes on gel filtration at different concentrations (Figure S1E).
Figure 1. Structure and assembly of SIR2, HerA, and the SIR2-HerA complex.
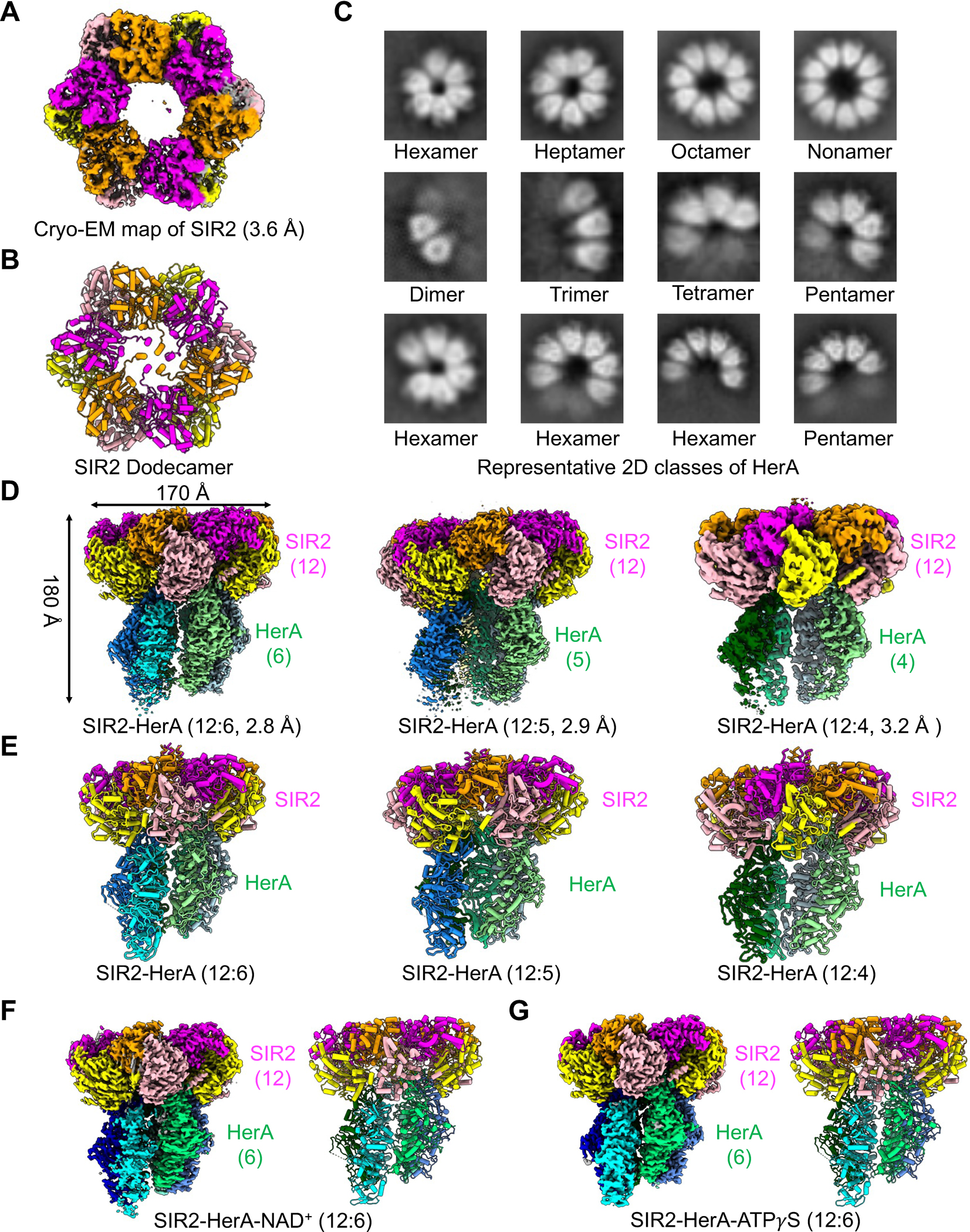
(A) Cryo-EM density map of SIR2 dodecamer with protomers color-coded.
(B) Ribbon diagram of the SIR2 dodecamer with protomers color-coded.
(C) Representative reference-free two-dimensional-class averages of the diverse HerA oligomerized complexes.
(D) Cryo-EM density maps of the SIR2-HerA complexes with different stoichiometries.
(E) Ribbon diagram of the SIR2-HerA complexes with stoichiometries indicated and protomers color-coded.
(F) Cryo-EM map and ribbon diagram of the SIR2-HerA complex with NAD+.
(G) Cryo-EM map and ribbon diagram of the SIR2-HerA complex with ATPγS.
See also Figures S1, S2, & Table S1.
Table 1.
Cryo-EM data collection, refinement, and validation statistics.
| SIR2_HerA complex 1 (12:6) EMD-40762 | SIR2_HerA complex 2 (12:5) EMD-40763 | SIR2_HerA complex 3 (12:4) EMD-40778 | SIR2 dodecamer EMD-40860 | SIR2_HerA complex 1 (12:6) bound with ATPγS EMD-42061 | SIR2_HerA complex 1 (12:6) bound with NAD+ MD-42062 | |
|---|---|---|---|---|---|---|
| Data collection and processing | ||||||
| Magnification | 81,000 | |||||
| Voltage (kV) | 300 | |||||
| Electron exposure (e−/Å2) | 50 | |||||
| Defocus range (μm) | 0.5 – 2.0 | |||||
| Pixel size (Å) | 1.11 | 0.4495 | 1.11 | 1.11 | ||
| Symmetry imposed | n/a | |||||
| Initial images (no.) | 7,541 | 4,788 | 2,314 | 3,102 | ||
| Initial particle images (no.) | 5,877,984 | 2,610,601 | 1,728,175 | 1,582,857 | ||
| Final particle images (no.) | 200,494 | 215,970 | 62,864 | 272,075 | 232,183 | 306,385 |
| Map resolution (Å) | 2.83 | 2.89 | 3.15 | 3.60 | 3.25 | 3.18 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map sharpening B factor (Å2) | −82.9 | −80.8 | −65.9 | −63.0 | −100.5 | −95.1 |
| Refinement | ||||||
| Initial model used | AlphaFold | |||||
| PDB ID | 8SU9 | 8SUB | 8SUW | 8SXX | 8UAE | 8UAF |
| Model resolution (Å) | 2.8 | 3.1 | 3.5 | 3.8 | 3.2 | 3.3 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Model composition | ||||||
| Non-hydrogen atoms | 65,385 | 61,226 | 56,577 | 37,940 | 64,630 | 64,015 |
| Protein residues | 8,134 | 7,611 | 7,035 | 4,674 | 8,033 | 8,033 |
| Ligands | ADPR: 12 ADP: 5 Mg2+: 5 |
ADPR: 12 ADP: 5 Mg2+: 5 |
ADPR: 12 ADP: 3 Mg2+: 3 |
NAD+: 12 | ADPR: 12 ATPγS: 5 Mg2+: 5 |
ADPR: 4 ADP: 5 NAD+: 8 Mg2+: 5 |
| B factors (Å2) | ||||||
| Protein | 63.77 | 74.47 | 52.06 | 66.42 | 69.12 | 79.64 |
| Ligand Nucleotide | 57.34 | 59.68 | 56.41 | 56.88 | 73.67 | 87.03 |
| R.m.s. deviations | ||||||
| Bond lengths (Å) | 0.003 | 0.005 | 0.002 | 0.003 | 0.004 | 0.004 |
| Bond angles (°) | 0.558 | 0.701 | 0.502 | 0.896 | 0.928 | 0.966 |
| Validation | ||||||
| MolProbity score | 1.27 | 1.47 | 1.40 | 1.83 | 1.37 | 1.57 |
| Clashscore | 2.16 | 2.94 | 3.16 | 7.75 | 2.78 | 4.50 |
| Poor rotamers (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 |
| Ramachandran plot | ||||||
| Favored (%) | 95.95 | 94.24 | 95.82 | 93.94 | 95.60 | 94.98 |
| Allowed (%) | 4.05 | 5.76 | 4.18 | 6.06 | 4.40 | 5.02 |
| Disallowed (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
We further determined the cryo-EM structures of the SIR2-HerA complex and identified three SIR2-HerA complexes with stoichiometries between SIR2 and HerA of 12:4, 12:5, and 12:6 (Figures 1D–E, S2, & Table 1). During our data processing, we also noticed dissociated SIR2 complexes (Figure S2B), indicating that the SIR2-HerA complex formation may be a dynamic process. As such, the SIR2-HerA with a stoichiometry of 12:6 may represent a fully assembled complex, while the other two may represent two intermediate states. Consistently, structures of SIR2-HerA in complex with NAD+ and ATPγS displayed a stoichiometry of 12:6 (Figures 1G–H, S2F–I & Table 1). Considering the diverse oligomeric status of EcHerA alone, we propose that SIR2 may function as a molecular organizer to lock HerA as a hexamer when assembling into a supramolecular complex.
SIR2 Dodecamer
Twelve EcSIR2 molecules assemble into a double-layered ring-like structure (Figure 2A). Each EcSIR2 protomer consists of 16 α-helices and 13 β-strands to form a small domain and a large domain, resembling the overall-fold of the well-studied Archaeoglobus fulgidus SIR2 (AfSIR2) 17 (Figures S3 & S4A–D). Compared to the AfSIR2 with two zinc coordinating sites17, the EcSIR2 lacks the typical zinc-binding motif and has no bound zinc ions (Figures S3 & S4A–D).
Figure 2. Assembly of SIR2 dodecamer.
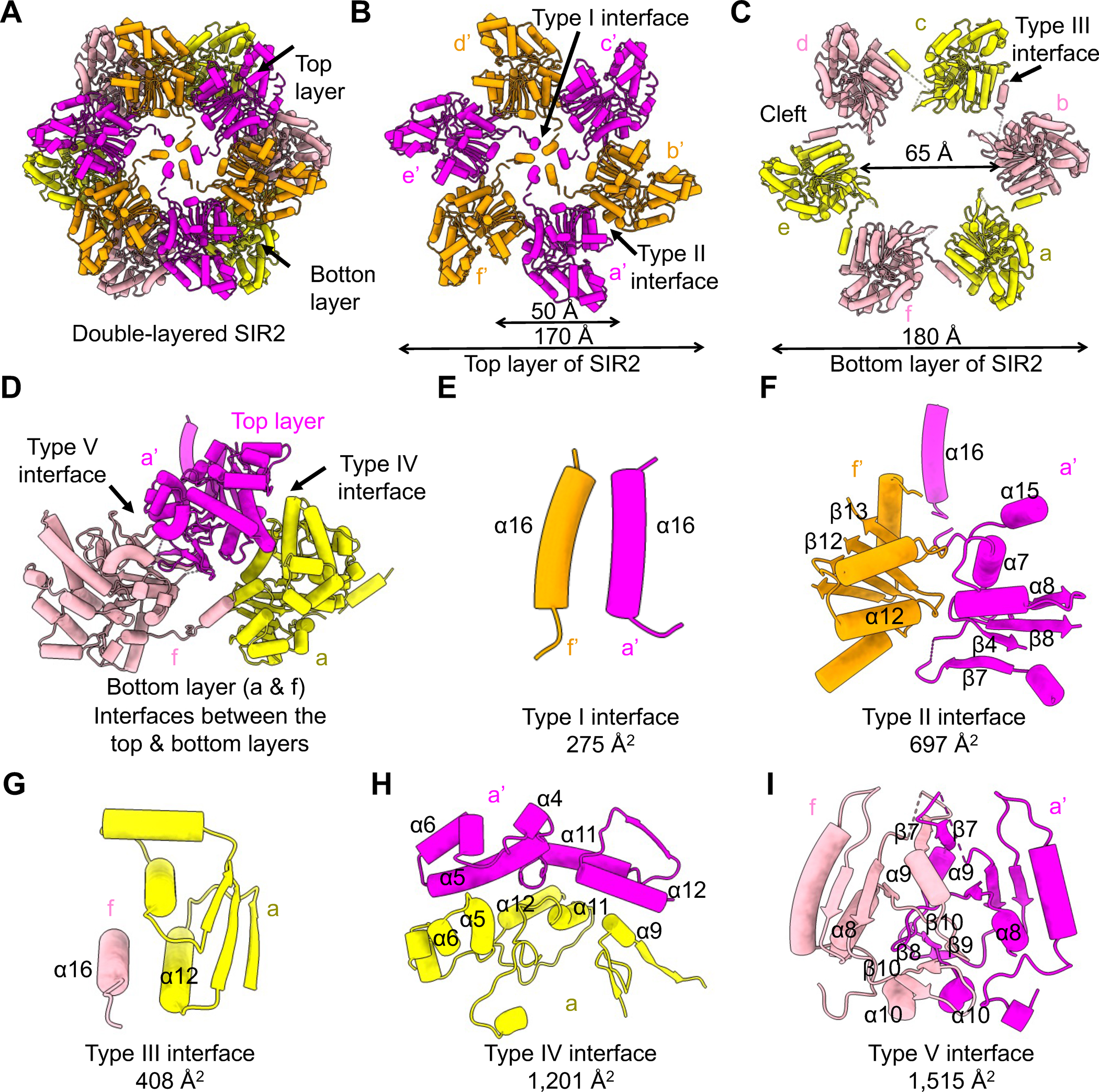
(A) Ribbon diagram of the double layered SIR2 dodecamer.
(B) The assembly of the SIR2 hexamer, mediated by the Type I and Type II interfaces, in the top layer. The six protomers are denoted as a’-f’, respectively.
(C) The assembly of the SIR2 hexamer mediated by the Type III interface in the bottom layer. The six protomers are denoted as a-f, respectively.
(D) Interactions between a protomer in the top layer and two protomers in the bottom layer, which are mediated by the Type IV and Type V interfaces.
(E-I) Expanded views of the type I-V interfaces, Type I (E), Type II (F), Type III (G), Type IV (H), and Type V (I), illustrating key elements involved.
See also Figures S3 and S4.
The small domain of EcSIR2 contains a two-stranded (β9–10) anti-parallel β-hairpin and six α-helices (α2–6 and α10). The small domain can be further divided into two modules, denoted as inserted module 1 and divergent module due to the differences with corresponding regions in the small domain of AfSIR217 (Figures S4B & S4D). Of note, α2–6 packed together to form the inserted module 1, while α10 and the β-hairpin constitute the divergent module (Figures S4B).
The large domain adopts a non-typical Rossmann-fold with seven β-strands (β1–3, β8, and β11–13) forming a central β sheet surrounded by eight α-helices (α1, α7–8, and α11–15). Compared to the AfSIR217, the large domain of EcSIR2 has two inserted modules, of which inserted module 2 contains β4–7, α9, and α16 while inserted module 3 is an α-hairpin formed by α13 and α14 (Figures S4B, S4D). Notably, the inserted module 1, the inserted module 2, and the divergent module in EcSIR2 are all involved in the assembly of the dodecamer (Figures 2B–I).
The SIR2 dodecamer can be divided into two layers, denoted as the upper layer (protomers a’-f’) and the bottom layer (protomers a-f), with six protomers in each layer (Figures 2A–C). The upper layer has an inner diameter of 50 Å and an outer diameter of 170 Å, while the bottom layer is slightly larger with an inner diameter of 65 Å and an outer diameter of 180 Å (Figures 2B–C). Protomers in the top and bottom layer resemble each other with obvious conformational changes in α5, α6, and α16, key elements for the oligomerization of SIR2 (Figures 2B–E, 2G–H, & S4E). In addition, SIR2 protomers at the bottom layer created large clefts in between, which accommodate protomers of the top layer (Figures 2B–D). As such, the upper layer sits on top of the bottom layer to form the double-layered dodecamer in a tail-to-tail manner (Figures 2A & 2D).
There are five different interfaces governing the assembly of the SIR2 dodecamer, denoted as Type I to V (Figures 2B–I & S4F–J). Type I and Type II interfaces are responsible for the assembly of the top layer, while the Type III interface dictates the formation of the bottom layer (Figures 2B–C). The Type I interface is mediated by hydrophobic interactions of α16, six of which come together to form a helical bundle in the middle of the top layer (Figures 2B, 2E, & S4F). The Type II interface consists of hydrophobic residues in α12 and β12–13 that packed against residues in α7, α8, α15, β4, and β7–8 (Figures 2B, 2F, & S4G). At the bottom layer, α16 of one protomer stretched out to engage α12 of a neighboring subunit, defining the Type III interface (Figures 2C, 2G, & S4H). Type I-III interactions, mediating the intra-layer assembly, are less extensive, with buried surface areas ranging from 275 Å2 to 697 Å2. In contrast, Type IV and Type V interfaces mediating inter-layer interactions are much more extensive, with buried surface areas of 1, 201 Å2 and 1, 515 Å2, respectively. A top protomer and a bottom protomer donate part of their inserted module 2 (α4–6) in the small domain and α9, α11–12 in the large domain to form the Type IV interface, whereas protomers of the bottom and top layer contributed their inserted module 2 in the large domain and divergent modules in the small domain to form the Type V interface (Figures 2D, 2H–I, & S4B). Both hydrophilic and hydrophobic interactions define the Type IV and Type V interfaces (Figures S4I–J). Together, these five interfaces cooperate to dictate the dodecameric assembly of EcSIR2.
Mechanism of NAD+ hydrolysis by SIR2
Previous studied EcSIR2 homologs catalyzed lysine deacetylation in a NAD+ dependent manner, while EcSIR2 simply catalyzed the hydrolysis of NAD+19. To better understand the catalytic mechanism of EcSIR2, we analyzed the cryo-EM structure of EcSIR2 alone and identified an NAD+ molecule in the active site of EcSIR2 (Figures 3A–B & S5A). In contrast, an ADPR molecule, the product of NAD+ hydrolysis, occupied the active site of EcSIR2 in the structure of the SIR2-HerA complex (Figures 3C & S5B). These observations suggested that the SIR2-HerA complex, not EcSIR2 alone, is capable of hydrolyzing NAD+, which was confirmed by subsequent enzymatic assay (Figure 3D). Therefore, the assembly of the SIR2-HerA complex is critical for the hydrolysis of NAD+.
Figure 3. Mechanism of SIR2 catalysis.
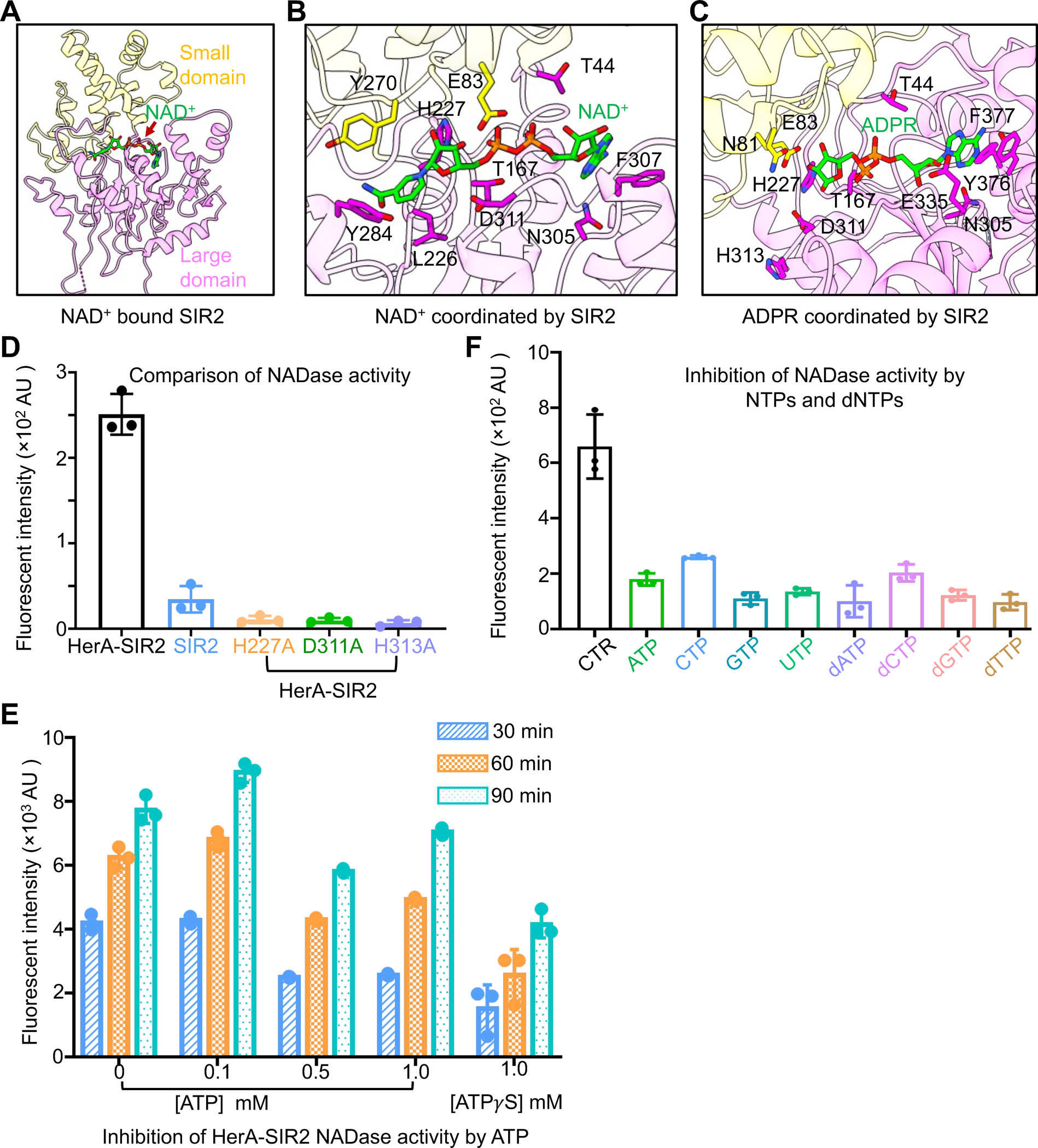
(A) NAD+ bins to the interface of the small domain and the large domain of SIR2.
(B) An expanded view of NAD+ coordination by key residues of SIR2.
(C) An expanded view of ADPR coordination by key residues of SIR2.
(D) Comparison of NAD+ hydrolase activity between HerA-SIR2 complex, HerA-SIR2H227A, HerA-SIR2D311A, HerA-SIR2H313A, and SIR2 apo dodecamer at 30 minutes. Mutations of residues impaired NAD+ hydrolysis. Data are mean ± SD from 3 replicates as indicated.
(E) Effects of ATP and ATPγS on NAD+ hydrolase activity of the HerA-SIR2 complex at different time points. Data are mean ± SD from 3 replicates as indicated.
(F) Effects of NTPs and dNTPs on NAD+ hydrolase activity of the HerA-SIR2 complex at 30 minutes. Data are mean ± SD from 3 replicates as indicated.
See also Figures S5.
Both NAD+ and ADPR molecules lay at the interface of the small domain and large domain of EcSIR2 (Figures 3A–C). As expected, aromatic residues including F377, Y376, and Y284 are responsible for coordinating the adenosine and nicotinamide groups while hydrophilic residues interact with the phosphate groups, and sugars (Figures 3B–C). Earlier studies on AfSIR2 revealed that residue H118 is critical for NAD+ hydrolysis17. The equivalent residue in EcSIR2, H227, is present in the active site in proximity to NAD+ or ADPR, indicating the functional significance of this residue (Figure S3). Substitution of H227 with alanine resulted in the loss of NAD+ hydrolytic activity of the SIR2-HerA complex, underscoring the critical role of H227 in catalysis (Figure 3D). Furthermore, substitutions of other residues in the catalytic center, including D311 and H313, also abolished the catalytic activity of the SIR2-HerA complex, highlighting the significance of these residues (Figure 3D).
Given that HerA contains an ATPase domain and the formation of the HerA-SIR2 complex is critical for SIR2 catalysis, we asked whether and how ATP molecules can affect NAD+ hydrolysis. A low concentration of ATP (0.1 mM) has an undetectable effect on NAD+ hydrolysis by the SIR2-HerA complex (Figure 3E). In contrast, higher concentrations of ATP (0.5 mM or 1mM) reduced 50% of the NADase activity of the SIR2-HerA complex at 30 minutes, while the inhibiting effects reduced at 60 minutes and 90 minutes, likely due to the hydrolysis of ATP (Figure 3E). To confirm this speculation, we tested the effect of ATPγS, an ATP analog with a much slower hydrolysis rate, and found that 1mM ATPγS significantly impaired the NADase activities at 30, 60, and 90 minutes. Additionally, other NTPs or dNTPs can also effectively inhibit the NADase activities of SIR2-HerA complex (Figure 3F). Together, these data suggested that NTP molecules at certain concentrations inhibit the NADase activities of the SIR2-HerA complex.
HerA Hexamer
Similar to Sulfolobus solfataritus HerA (SfHerA) and Deinococcus radiodurans HerA (DrHerA) 14,15, each protomer of the EcHerA is composed of three domains: the N-terminal HAS (HerA and ATP synthase) β-barrel domain, the central RecA-like domain, and a four-helix bundle domain (Figures 4A & S5C–D). Compared to SfHerA and DrHerA14,15, both the HAS domain and the RecA-like domain of EcHerA have developed an additional β-hairpin. In contrast, the four-helix bundle domain of EcHerA contains a four-stranded β-sheet (Figures 4A & S5C–D).
Figure 4. Assembly of HerA hexamer.
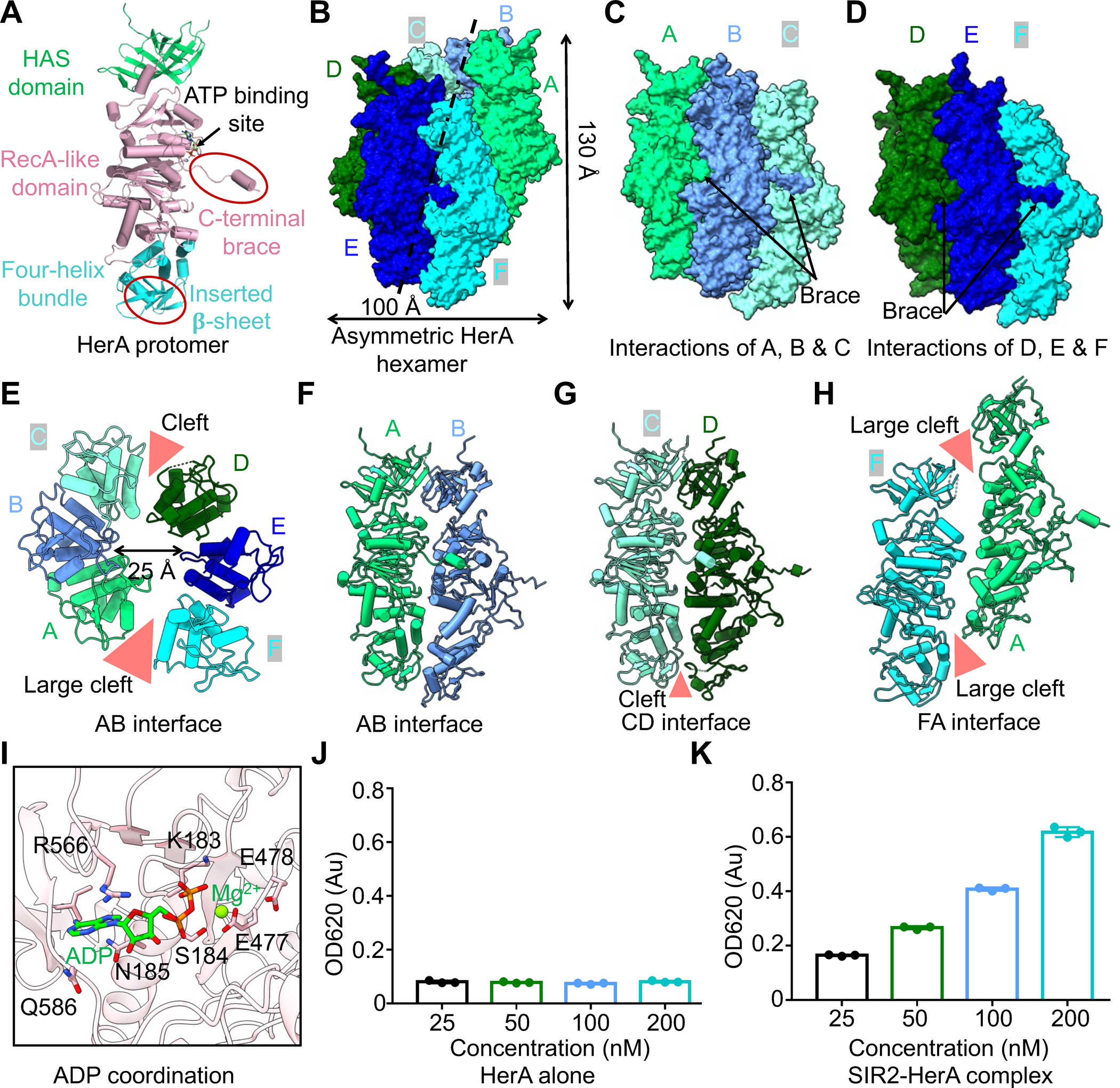
(A) Ribbon diagrams of HerA with domains color-coded. The C-terminal brace and the inserted four-stranded β-sheet are highlighted by oval circles.
(B) Asymmetric assembly of HerA hexamer with protomer A at the top and protomer F at the bottom. Each protomer is colored individually.
(C, D) Surface representations of HerA to illustrate interactions within trimers ABC (C) and DEF (D).
(E) Ribbon diagram of HerA hexamer viewed from the four-helical bundle domain with two clefts illustrated.
(F) Ribbon diagram of protomers A and B to illustrate their interacting interface.
(G) The interface between protomers C and D, revealing a cleft in between.
(H) The interface between protomers F and A, revealing a large cleft in between.
(I) ADP coordination by key residues of HerA.
(J) ATPase assay indicated that HerA alone has no ATPase activity. Data are mean ± SD from 3 replicates as indicated.
(K) The ATPase activities of the SIR2-HerA complex. Data are mean ± SD from 3 replicates as indicated.
See also Figure S5.
Different from the symmetric hexameric assembly of DrHerA14, EcHerA assembles as an asymmetric hexamer with dimensions of ~100 Å across and ~150 Å in height and with a central channel of ~25 Å (Figures 4B–E & S5E). The asymmetric EcHerA hexamer displayed a few unique features (Figures 4B–H & S5F–I).
First, the six protomers of EcHerA assembled into an asymmetric hexamer with a rise of ~6.7 Å and a rotation angle of ~60°, contrasting the symmetric hexamer of DrHerA14. Notably, the rise distance and the rotation angle between protomers C and D are ~4 Å and ~55°, smaller than other pairs. The six protomers of EcHerA assemble as a spiral staircase with promoter A on the top and protomer F at the bottom.
Second, the asymmetric hexamer of EcHerA is arranged in a dimer of trimers with two obvious clefts between the two pairs of trimers, in which protomers A, B and C form one trimer and the other three protomers assemble as another trimer (Figures 4C–H & S5F). Within each trimer, each protomer interacts with neighboring protomer via an extensive interface with a buried area ranging from ~2,000 Å2 to ~2,400 Å2 (Figures 4C–F & S5F). In contrast, protomers C and D, mediating trimer-timer interactions, form a less intensive interface with a buried area of ~1,500 Å2 (Figures 4G & S5F). More strikingly, the interface between protomers F and A is very limited, with a buried area of 432 Å2 (Figures 4H & S5F). As such, there is a large cleft between protomers F and A, making the asymmetric EcHerA form an open-ringed structure (Figures 4E & 4H).
Third, within each pair of EcHerA trimers, the three domains of each protomer interact with corresponding domains of a neighboring protomer to form intensive interfaces (Figures 4F & S5G–I). The HAS domains are packed against each other by forming an anti-paralleled β-sheet, while the helical-bundle domains mainly interact with each other via hydrophilic interactions (Figure S5G–H). Interestingly, the interactions between the RecA-like domain are mainly mediated by the very C-terminal motif, denoted as a brace motif (Figures 4A & S5I). This brace motif is amphipathic and extended into a pocket of a neighboring subunit to establish both hydrophobic and charge-charge interactions (Figure S5I). As such, this brace motif functions like a belt to link the six protomers of EcHerA (Figures 4B–D).
Fourth, ADP molecules were identified in the RecA-like domain of EcHerA, suggesting that EcHerA can hydrolyze ATP when forming a complex with SIR2 (Figures 4I & S6J). Further ATPase assay revealed that the SIR2-HerA complex, not HerA alone, can hydrolyze ATP, suggesting that the complex formation of the SIR2-HerA complex promotes the ATPase activities of HerA (Figures 4J–K). Linking back to the inhibition of the NADase activity by ATP (Figure 3E), we propose that HerA promotes the NADase activity of SIR2 by both forming a complex with SIR2 and hydrolyzing ATP molecules.
Interactions between SIR2 and HerA
Though EcHerA assembles as a hexamer in the HerA-SIR2 complex, five HerA protomers engage EcSIR2 dodecamer (Figures 5A–D & S6A–C). Protomers A, B, C, and D of EcHerA interact with EcSIR2 with similar buried surface areas ranging from 1,285 to 1,620 Å2 (Figures 5A & S6A–C). In contrast, the interface between EcHerA protomer E and EcSIR2 is less intensive, with a buried area of 976 Å2 (Figure 5B & 5D). Protomer F, sitting at the bottom of the EcHerA hexamer, has no interaction with any SIR2 molecules, partly due to the localization of protomer F distal from SIR2 (Figures 5C & S6D).
Figure 5. Interactions between HerA and SIR2.
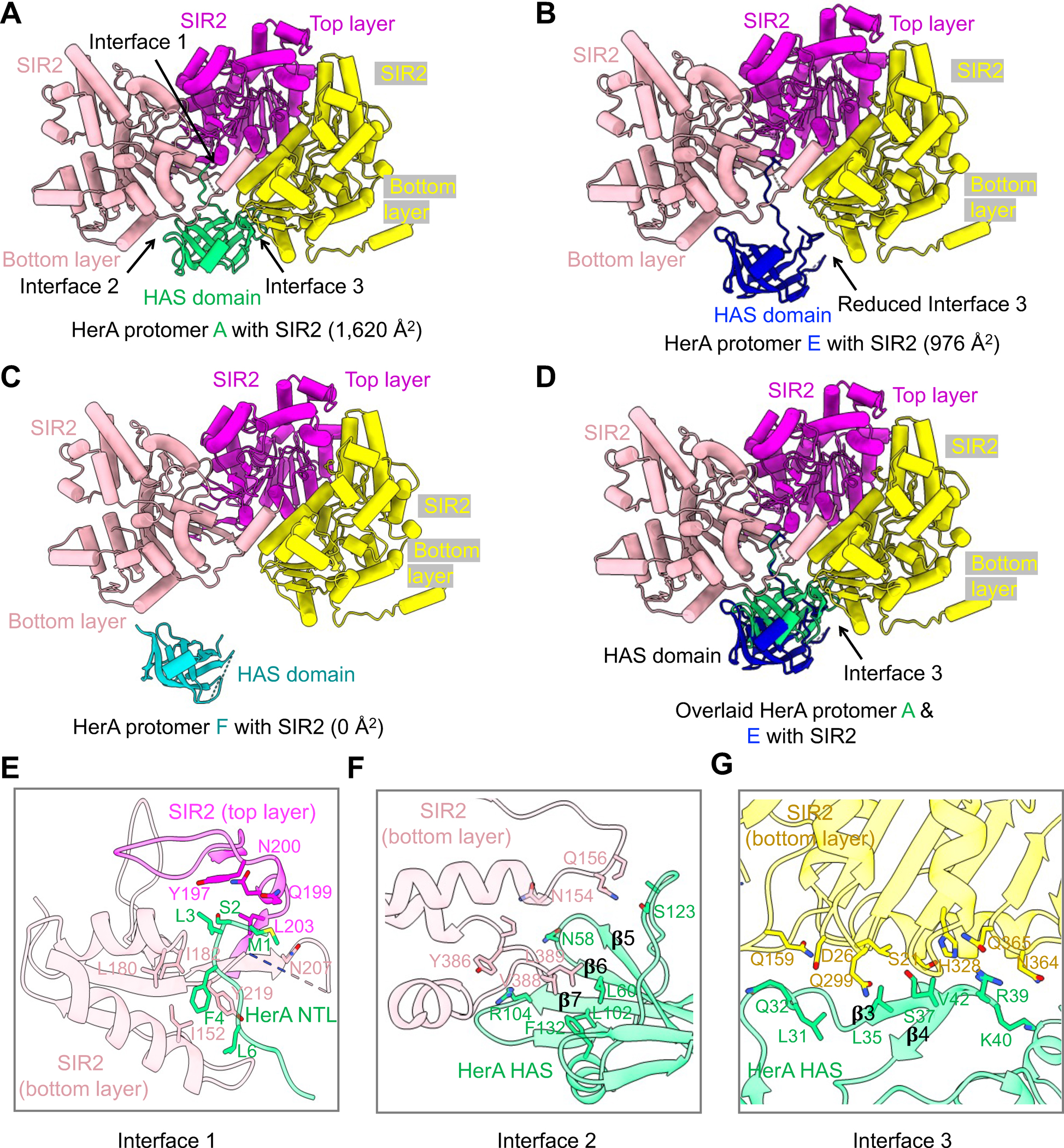
(A) Ribbon diagrams of the HAS domain of HerA protomer A in complex with three SIR2 protomers, illustrating three interfaces, interface 1, 2, and 3. SIR2 in the top layer is colored in magenta and protomers of SIR2 in the bottom layer are colored in yellow and pink.
(B) Ribbon diagrams of the HAS domain of HerA protomer E in complex with three SIR2 protomers, illustrating a reduced interface 3. SIR2 protomers in the top layer and in the bottom layer are color-coded.
(C) Ribbon diagrams of the HAS domain of HerA protomer F in complex with three SIR2 protomers, revealing that protomer F of HerA does not interact with SIR2. SIR2 protomers in the top layer and in the bottom layer are color-coded.
(D) Overlaid HerA protomer A and E with SIR2 interaction, revealing differences in interface 3.
(E, F, G) Expanded views of interface 1 (E), interface 2 (F) and interface 3 (G), revealing key residues on the interfaces.
See also Figure S6.
Compared to the DrHerA HAS domain that interacts with NurA mainly through an α-helix inserted between β6 and β714, EcHerA HAS domain developed more regions to engage SIR2 (Figures 5A, 5E–G, & S6E–G). Specifically, the HAS domain of HerA, a 9-stranded β barrel, form three interfaces with two neighboring SIR2 molecules at the bottom layer and a SIR2 molecule in the top layer (Figures 5A, 5E–G, & S6E). First, interface 1 is characterized by the very N-terminal loop of the HAS domain that established hydrophobic interactions with SIR2 protomers in the top and bottom layer (Figures 5E & S6E). Second, the HAS domain employed the β5-β6 hairpin and the β7 to form hydrophobic or π-cation interactions with a bottom SIR2, defining interface 2 (Figures 5F & S6E). Third, different from interfaces 1 and 2, interface 3 is mainly composed of hydrophilic interactions formed by the inserted β3-β4 hairpin of the EcHerA HAS domain and a bottom SIR2 (Figures 5G, S6E, & S6G). Interface 3 between EcHerA protomer E and SIR2 is less extensive than other pairs, accounting for the smaller overall buried interface areas in protomer E (Figure 5B & 5D).
Helicase and nuclease activities
Given that other well-studied HerA proteins function as helicases, we were curious whether EcHerA alone or the SIR2-HerA complex has helicase activity. We found that the SIR2-HerA complex, but not EcHerA alone, displayed helicase activities towards forked DNA substrates in an ATP-dependent manner (Figures 6A–B & S7A). This finding underscored the importance of the HerA-Sir2 complex formation in DNA unwinding.
Figure 6. Enzymatic characterization of SIR2-HerA and SIR2.
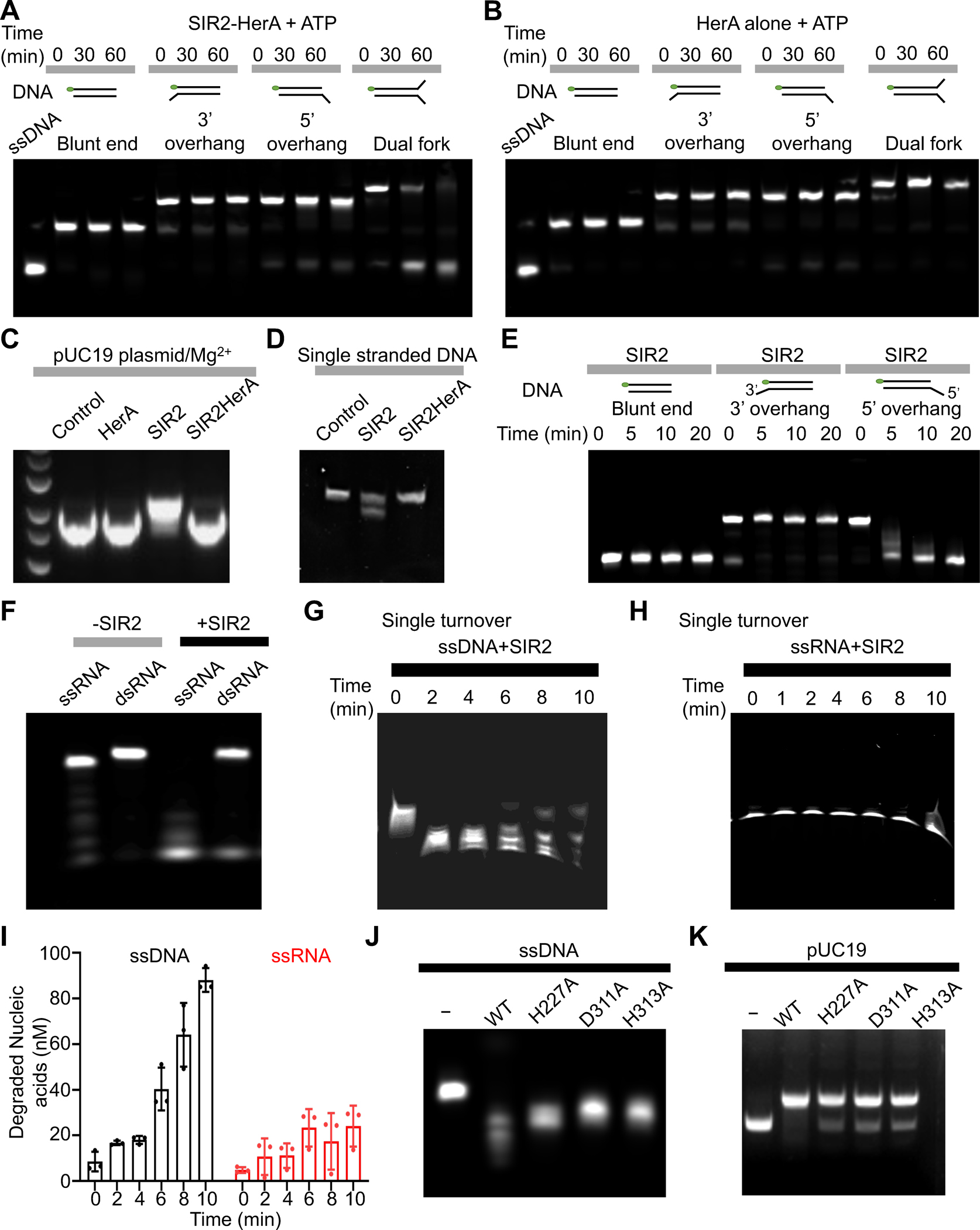
(A) In the presence of ATP, the SIR2-HerA complex can unwind dual fork DNA, but not blunt ended dsDNA, dsDNA with 3’ or 5’ overhang.
(B) In the presence of ATP, HerA alone cannot unwind dual fork DNA, blunt ended dsDNA, and dsDNA with 3’ or 5’ overhang.
(C) SIR2 alone, but not the SIR2-HerA complex, can generate nicks on the pUC19 plasmid in the presence of Mg2+, indicating SIR2 alone functions as a nuclease.
(D) SIR2, but not the SIR2-HerA complex, can cleave single-stranded DNA.
(E) SIR2 can process dsDNA with a 5’ overhang, but not blunt ended dsDNA and dsDNA with a 3’ overhang.
(F) SIR2 can cleave both single-stranded RNA and double-stranded RNA with a preference on ssRNA.
(G) Urea gel showing the processed single-stranded DNA by SIR2 in a single-turnover assay.
(H) Urea gel showing the processed single-stranded RNA by SIR2 in a single-turnover assay.
(I) Quantification of DNA and RNA cleavage by SIR2 in panel G and H. Data are mean ± SD from 3 replicates as indicated.
(J-K) Single-stranded DNA (J) or pUC19 plasmids (K) were processed by SIR2 or SIR2 mutants. All the experiments are repeated at least three times.
See also Figure S7.
Unexpectedly, we found that SIR2 alone can convert supercoiled plasmid pUC19 into relaxed forms probably via nicking the plasmids, indicating that SIR2 has nuclease activity (Figure 6C). Mg2+ is required for the nuclease activity of SIR2 because depletion of Mg2+ using EDTA abolished SIR2 nuclease activity (Figures 6C & S7B). Further analysis revealed that SIR2, not the SIR2-HerA complex, can effectively process single-stranded DNA, fork DNA, but not blunt-ended double-stranded DNA without obvious sequence preference (Figures 6D & S7C–D). Moreover, SIR2 can process the single-strand part of a double-stranded DNA with a 5’ overhang but not 3’ overhang, indicating SIR2 as an exonuclease (Figure 6E). In addition, SIR2 can digest single-stranded RNA more efficiently than double-stranded RNA (Figure 6F). Further single turnover nuclease assays revealed that SIR2 prefers to cleave single-stranded DNA over single-stranded RNA (Figure 6G–I). To test whether SIR2 uses the same catalytic center for nuclease and NADase activities, we further tested the nuclease activities of the three NAD+ catalytic dead mutants (H227A, D311A, and H313A). We found that the three mutants displayed slightly reduced nuclease activities towards single-stranded DNA, pUC19 plasmids, and fork DNA (Figures 6J–K & S7E), indicating that the nuclease catalytic center may be in proximity to the NAD+ hydrolysis site. Together, these data strongly support that SIR2 alone functions as a nuclease to non-specifically digest RNA or DNA molecules. As the SIR2-HerA complex can effectively hydrolyze NAD+, the assembly of the SIR2-HerA complex converts SIR2 from a nuclease to an NAD+ hydrolase.
Mechanism of Anti-phage defense by the SIR2-HerA complex
To further test whether the assembly of the SIR2-HerA complex is required for anti-phage defense, we performed bacteria growth assay. Upon T5 phage infection at low MOI, bacteria with EcSIR2 or EcHerA alone died of phage infection, while bacteria with the SIR2-HerA complex can grow up (Figures 7A–C). Upon T5 phage infection, in the presence of the SIR2-HerA complex, the concentration of NAD+ in bacteria plummeted to levels that were nearly undetectable within 30 minutes. This discovery supports that the SIR2-HerA complex exerts its anti-phage defense through hydrolyzing NAD+ in bacteria (Figure S7F). Consistently, EcSIR2 H227A, a defective mutant for NAD+ hydrolysis, impairs the anti-phage defense activity of the SIR2-HerA complex (Figures 3D & 7D). Together, these data suggested that both the assembly of the SIR2-HerA complex and the NAD+ hydrolase activity of SIR2 are critical for anti-phage defense.
Figure 7. Mechanism of anti-phage defense by SIR2-HerA.
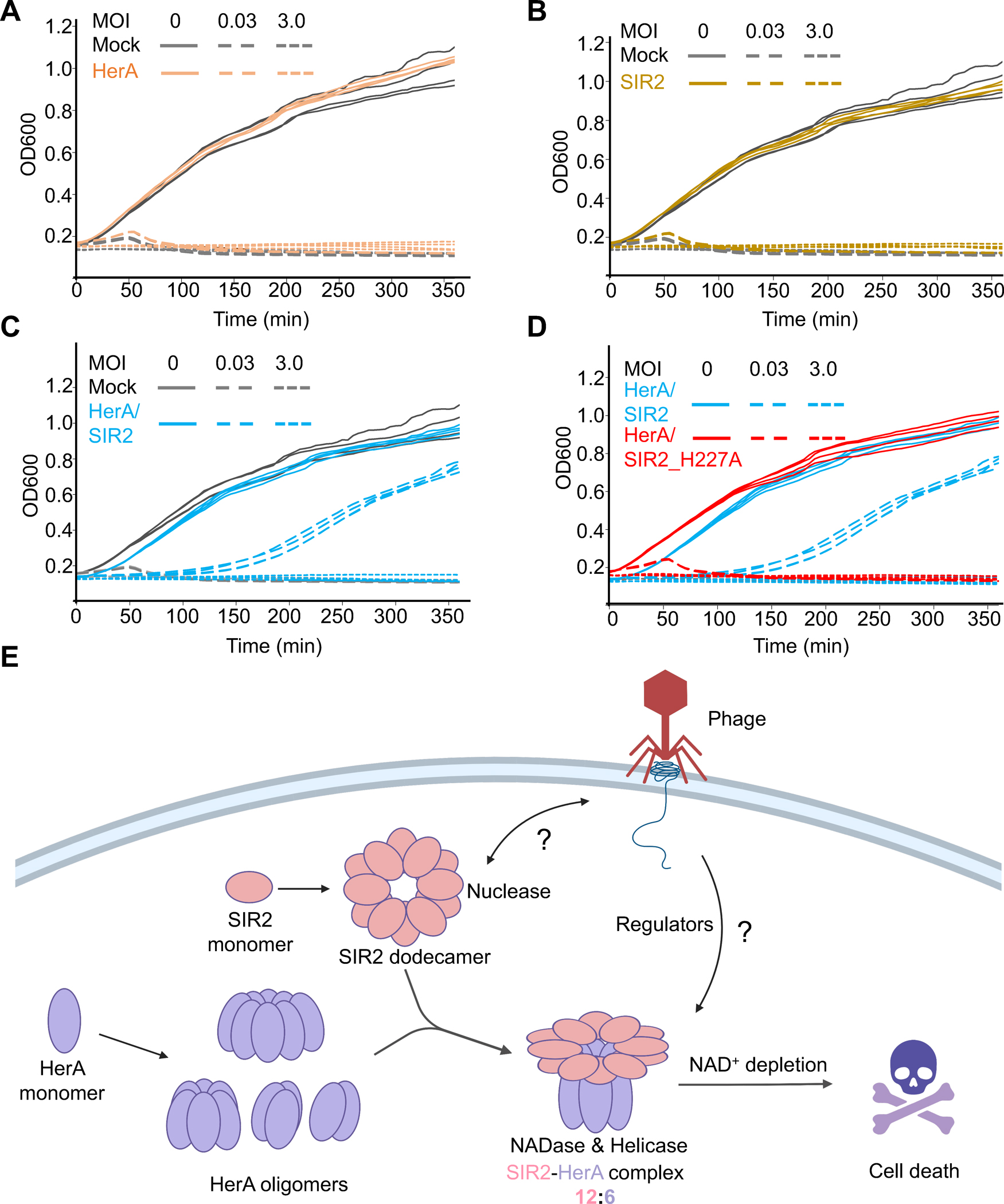
(A-D) Bacterial growth curves in the presence of HerA, SIR2, the HerA-SIR2 complex, or the HerA-SIR2_H227A mutant upon infection by T5 phage at different MOI as indicated. (E) Assembly and mechanism of the SIR2-HerA complex in anti-phage defense.
See also Figure S7.
Upon T5 phage infection at high MOI (MOI equal to 5), all the bacteria were infected, and the SIR2-HerA complex was activated in all the bacteria for depleting NAD+, leading to bacterial cell death (Figure 7C & S7F). As such, the SIR2-HerA complex protects bacteria from phage infection using a mechanism of abortive infection in which bacteria undergo suicide upon infection to protect the safety of the community.
Discussion
Our study has revealed a fascinating mechanism of assembly-mediated activation of the SIR2-HerA complex with multiple enzymatic activities for anti-phage immune defense (Figure 7E).
First, our study revealed fascinating mechanism of the assembly of HerA, SIR2, and the SIR2-HerA complex. EcHerA can form variable oligomers ranging from dimers to nanomers dependent on concentrations, quite distinct from other known ATPases that tend to form an oligomer with a fixed stoichiometry. Unexpectedly, SIR2, a dodecamer, serves as an organizer to form a supramolecular complex with HerA with a stoichiometry of 12:6.
Second, a striking discovery from our study is the multiple enzymatic activities in the HerA-SIR2 system. SIR2 alone functions as a nuclease that can effectively process DNA and RNA. In contrast, the SIR2-HerA complex display ATPase, helicase, and NADase activities. In particular, all these enzymatic activities are dependent on the assembly of the SIR2-HerA complex.
Third, more intriguingly, we revealed that the assembly of the SIR2-HerA complex serves as a switching mechanism to convert SIR2 from a nuclease to an NAD+ hydrolase. Perhaps, large conformational changes in the catalytic center and changes of the NAD+ binding mode upon SIR2-HerA complex formation may partially explain the NADase activity in the complex (Figure S7G). Currently, we have no evidence to show whether the nuclease and NADase share the same active site. Mutations of those residues that are critical for NADases slightly reduced the nuclease activity, indicating that the nuclease active center is in proximity to the NAD+ catalytic site.
Fourth, elegantly, HerA promotes the NADase activities of SIR2 not only by assembly but also through hydrolyzing ATP. ATP can effectively inhibit the NADase activities of the SIR2-HerA complex at concentrations higher than 0.5 mM. Under physiological conditions, the ATP concentration is about 2–3 mM in bacteria23. As such, the NADase activity of the SIR2-HerA complex is arrested at the basal level to avoid cellular toxicity. Upon phage infection, the system is activated through a mechanism yet to be elucidated. It is possible that an activated SIR2-HerA complex can reduce the level of ATP in cells to fully activate the complex for depleting NAD+ and cell death.
Fifth, the nuclease activity of SIR2 is a completely unexpected discovery from our study. Currently, we do not have a good answer for the physiological function of this activity. However, our study opens many avenues to explore the relationships between the SIR2-HerA and phage infection. It is possible that SIR2 may also play a role in anti-plasmid surveillance.
Collectively, our study revealed that the assembly of SIR2 and HerA and the cooperation of multiple enzymatic activities in the SIR2-HerA system provide a unique and fascinating mechanism for anti-phage defense.
Limitations of the Study
Our study provides mechanistic insights into the assembly and function of the SIR2-HerA. However, a few questions remain to be addressed. First, we did not identify the catalytic center of the SIR2 nuclease. A structure of SIR2 in complex with nucleic acid may help to reveal key residues critical for cleaving nucleic acids. Second, we did not elucidate the mechanism of nucleic acid recognition by SIR2. Third, we also did not reveal the mechanism of enzymatic conversion upon complex formation. Fourth, how HerA-SIR2 is activated by phage factors remains unclear. As such, more efforts are required to gain a full understanding of this fascinating anti-phage immune system.
STAR☆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Tian-Min Fu (fu.978@osu.edu).
Materials availability
Plasmids generated in this study are available upon request from the Lead Contact with a completed Material Transfer Agreement.
Data and code availability
Accession numbers for SIR2_HerA complex 1 (12:6), SIR2_HerA complex 2 (12:5), SIR2_HerA complex 3 (12:4), SIR2 dodecamer, SIR2_HerA complex 1 (12:6) bound with ATPγS, SIR2_HerA complex 1 (12:6) bound with NAD+ are as follows: (coordinates of atomic models: 8SU9, 8SUB, 8SUW, 8SXX, 8UAE and 8UAF were deposited to Protein Data Bank), and (density map: EMD-40762, EMD-40763, EMD-40778, EMD-40860, EMD-42061 and EMD-42062 deposited to Electron Microscopy Data Bank). Original data for biochemical assays and uncropped gels were deposited to Mendeley Data (http://dx.doi.org/DOI:10.17632/xf34wcwxyf.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
The Escherichia coli (E. coli) DH5α strain was used to generate mutations and amplify plasmids. The plasmids were transformed into the E. coli BL21 (DE3) strain cultured in Luria broth (LB) medium to recombinantly express proteins including E.coli HerA (WP_021577682.1), E.coli SIR2 (WP_021577683.1), and E.coli SIR2-HerA full complex.
METHODS DETAILS
Molecular cloning, protein expression and purification
E. coli HerA (WP_021577682.1) was inserted into the pET28a vector with an N-terminal His×6 tag. E. coli SIR2 (WP_021577683.1) was inserted into the pDB-His-MBP (maltose binding protein) expression vector with an N-terminal His×6 and MBP tag followed a tobacco etch virus (TEV) protease recognition site. To express the SIR2-HerA complex, E. coli HerA and SIR2 with a C-terminal His×6-tag were cloned into the pET28a vector as a poly-cistron. All the mutations were made through site direction mutagenesis.
The recombinant plasmids were transformed into E. coli BL21 (DE3) cells (ThermoFisher Scientific) cultured in LB medium containing 50 μg/ml kanamycin at 37°C. At OD600 of 0.6–0.8, protein expression was induced at 18°C by 0.3 mM IPTG. Cells were harvested after overnight induction (~16h) and resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10 mM imidazole). After sonication, the cell debris was removed through centrifugation at 30,000 × g, 4 °C for 50 min and the supernatant was loaded onto a pre-equilibrated Ni2+-NTA agarose column, and then the column was washed with 30 column volumes (CV) of Ni2+-NTA wash buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 10 mM imidazole). The protein was eluted by Ni2+-NTA elution buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 250 mM imidazole, 0.4 mM TECP). Then, fractions containing protein of interest were pooled and incubated with TEV protease overnight at 4°.
Proteins were further purified by gel filtration using a column of Superose™ 6 increase 10/300 GL (Cytiva) in a buffer consisting of 50 mM Tris-HCl pH 8.0, 300 mM NaCl, and 0.4 mM TECP.
Cryo-EM data collection
To prepare grids, 3 μL sample at 1.8 mg/ml was applied to a glow discharged Quantifoil R1.2/1.3 400 mesh gold grid (Electron Microscopy Sciences) followed by 4 s blotting with 100% humidity at 4 °C using an FEI Vitrobot Mark IV (Thermo Fisher). All grids were screened using a ThermoFisher Glacios microscope (OSU Center for Electron Microscopy and Analysis).
For the SIR2-HerA complex, 7,541 micrographs were collected using a 300 kV Titan Krios microscope equipped with a K3 direct electron detector (Thermo Fisher) in counting mode with a nominal magnification of 81,000×, and a physical pixel size of 1.11 Å with defocus values ranging from −1.0 to −2.0 μm. Each micrograph stack contained 40 frames with a total electron dose of 50 e− /Å2. Using the same parameters, we collected 2,314 micrographs, 3,102 micrographs, and 2,320 micrographs for SIR2-HerA-ATPγS, SIR2-HerA-NAD+, and HerA, respectively.
For SIR2, we collected 4,788 micrographs using a 300 kV Titan Krios microscope equipped with a K3 direct electron detector (Thermo Fisher) in super-resolution mode with a nominal magnification of 81,000×, and a physical pixel size of 0.899 Å with defocus values ranging from −1.0 to −2.0 μm. Each micrograph stack contained 40 frames with a total electron dose of 50 e− /Å2.
Cryo-EM data processing
As illustrated in Figures S1 & S2, all the datasets were imported into cryoSPARC (v4.1.1) for implementation of patch motion correction, and patch contrast transfer function (CTF) estimation 24. Initial particles were picked by blob picking from 200 micrographs for generating 2D classes. Representative 2D classes were selected as templates for template-guided particle picking from all micrographs.
For the SIR2-HerA complex, 5,877,984 particles were picked and extracted. After two rounds of 2D classification, 2,333,530 particles were selected and merged for ab-initio reconstruction to generate three initial models for subsequent homogeneous refinement. 215,970 particles were subjected to non-uniform refinement with a C1 symmetry, resulting in a 2.83 Å map for the SIR2-HerA complex with a stoichiometry of 12:6. 200,494 particles were used to generate a 2.89 Å map for the SIR2-HerA complex with a stoichiometry of 12:5. 62,864 particles were processed with C1 symmetry to generate a 3.15 Å map for the SIR2-HerA complex with a stoichiometry of 12:4.
For the SIR2-HerA complex with ATPγS, we picked 1,728,175 particles for 2D classification. After two rounds of 2D classification, 582,471 particles were selected and merged for further ab-initio reconstruction to generate three initial 3D models. Eventually, 232,183 particles were selected for further non-uniform refinement with C1 symmetry, resulting in a 3.25 Å map.
For the SIR2-HerA complex with NAD+, 1,582,857 particles were picked for two rounds of 2D classification, and 700,842 particles were selected and merged for further ab-initio reconstruction to generate three initial models. A class of 306,385 particles were selected for further non-uniform refinement with a C1 symmetry, resulting in a 3.18 Å map.
For HerA, 2,105,123 particles were picked for 2D classification to generate diverse oligomeric HerA as illustrated in Figure 1.
For SIR2, 2,610,601 particles were picked for two rounds of 2D classification. 322,818 particles were selected to generate two initial 3D models for further heterogeneous and homogeneous refinement. The best model with 272,075 particles was selected for non-uniform refinement, resulting in a 3.60 Å map.
All reported resolutions were estimated based on the gold-standard Fourier shell correlation (FSC) = 0.143 criterion 32.
Model building and refinement
The initial models of HerA and SIR2 were predicted by AlphaFold 25, and fitted into cryo-EM maps using Chimera 26. Then, manual adjustments were done in Coot to generate the final model 27. Real-space refinement was performed in Phenix 28. All the models were validated using Molprobity 29. All the structural figures were generated using PyMOL31 and ChimeraX30.
NADase assay
To test the NADase activities of the SIR2-HerA complex and SIR2 alone, ε-NAD+ assays were performed using a modified method based on a published protocol 7. In detail, a reaction mixture containing 0.5 μM protein, with concentrations in the range from 50 uM ε-NAD+ to 50 nM (and buffer as a control), 20 mM Tris pH 8.0, 75 mM KCl, and 2 mM MgCl2 was prepared on ice in a 96-well plate (Thermo Fisher Scientific). The plate was then transferred to a preheated Spark Multimode Microplate reader (TECAN) immediately. All measurements took place at 33 °C with an excitation wavelength of 360 nm and emission wavelength of 465 nm. The excitation bandwidth and emission bandwidth are both 35 nm.
ATPase assay
The ATPase activity was measured by an ATPase/GTPase Assay Kit (Sigma-Aldrich, #113CB04A30). Briefly, we prepared a 4 mM solution of ATP phosphate standard and generated a standard curve following the kit instruction. Protein was incubated with ATP in the buffer of 20 mM Tris pH 8.0, 75 mM KCl, and 2 mM MgCl2 at 37 °C. The reaction products were monitored by Biotek synergy ht microplate reader at 600 nm. ATPase activities were calculated based on the standard curve.
Nuclease and helicase assay
For multiple turnover nuclease assay, 400 nM protein and 800 nM 5’ Cys-labeled nucleic acids substrate (5’ overhang dsDNA, 3’ overhang dsDNA, blunt dsDNA, dual forked dsDNA, ssDNA, ssRNA, dsRNA) were incubated in reaction buffer (25 mM Tris-HCl pH 8.0, 75 mM KCl, and 5 mM MgCl2) at 30 °C. Samples were separated on 12% PAGE gel in 1 x TBE buffer.
For single turnover nuclease assay, the concentrations of protein and substrate (5’ Cys-labeled ssDNA or ssRNA) were changed to 1,000 nM and 100 nM, separately. After incubation, samples were separated on 12% PAGE gel (DNA samples) or 15% urea gel (RNA samples).
For helicase assay, 2 mM ATP was added in the same reaction system (25 mM Tris-HCl pH 8.0, 75 mM KCl, and 5 mM MgCl2) as the cleavage assay (substrates including 5’ overhang dsDNA, 3’ overhang dsDNA, blunt dsDNA, and dual forked dsDNA), and incubated at 30 °C. Samples were evaluated on 12% PAGE gel in 1 x TBE buffer.
All the gel results were visualized by Sapphire biomolecular imager (Azure biosystems). The intensity of each band on the gel was quantified by image J 33.
Bacterial growth assay
Phages were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen DSMZ): T5 (DSM 16353). Phages were propagated by inoculating into E. coli BL21 (DE3) with an OD600 (optical density) of 0.3 in LB medium. Phages were harvested by centrifugation and the supernatant was filtered through a 0.22-μm filter to get rid of remaining bacteria and bacterial debris. Phage titer was determined by a small drop plaque assay. E. coli with/without the SIR2-HerA system was cultured to OD600 of 0.3 at 37 °C for the growth assay. 180 μl cultured bacterial cells were transferred into wells of a 96-well plate followed by adding 20μl of phage solution at different MOIs. Infected cells were incubated at 37°C with continuous shaking in SpectraMax Absorbance Plus Microplate Readers (Molecular Devices) and OD600 was taken every five minutes for 6 hours for generating a growth curve.
Cellular NAD+ levels determined by UPLC-MS/MS
Bacterial cells at OD600 of 0.3 were infected by T5 phage with an MOI of 3 at 37°C with 220 rpm shaking for the duration of the experiment. Samples were collected at time points of 15, 30, 50 min by centrifugation at 4 °C for 5 min at 4000 × g. The supernatant was discarded, and 1 mL 80 % methanol was used to resuspend the precipitates. After sonication and centrifugation at 13,800 × g for 10 min, the supernatant was pooled and dried in a SpeedVac concentrator (HEREXI) and redissolved with 100 μL 10 % acetonitrile for further UPLC-MS/MS analysis.
UPLC-MS/MS analysis was carried out on a Nexera LC-30 UPLC system coupled with AB Sciex 6500+ triple quadrupole mass spectrometer (AB SCIEX). The UPLC was performed using a Waters HSS T3 column (2.1*100 mm, 1.7 um, Waters) with the mobile phase A of 10 mM ammonium formate in ultra-pure water with 0.2 % acetic acid and the mobile phase B of Acetonitrile. The flow rate was 300 μL/min with the listed gradients: 3 % B (0–2 min), increased to 90 % B (2–4 min), 90 % B (4–5 min), decreased to 3 % B (5–5.5 min), 3 % B (5.5–7 min). The column temperature was 35 ℃, and the injection volume was 10 μL with electrospray as the ionization source. Data analysis was performed in both positive and negative ionization modes. Mass spectrometers for positive ionization mode are elaborated as below: IonSpray Voltage of 4500 V, desolvation temperature of 500 °C, Curtain Gas 35 psi, Collision Gas Medium, and Ion source Gas 1 = 50 psi, Ion source Gas 2 = 40 psi. Mass spectrometers for negative ionization mode are as follows: IonSpray Voltage of −4500 V, desolvation temperature of 500 °C, Curtain Gas 35 psi, Collision Gas Medium, and Ion source Gas 1 = 50 psi, Ion source Gas 2 = 40 psi. Declustering potential (DP) and collision energy (CE) of MRM transitions of NAD+ are 662.2/540.0 (negative ionization mode, DP of −30 V and CE of −21 V). Data were acquired using Analyst 1.7.2™ software and analyzed using SCIEX OS 2.0 software (AB Sciex).
QUANTIFICATION AND STATISTICAL ANALYSIS
For cryo-EM structure determination, the numbers of particles used for each of the 3D reconstructions are listed (Figure S1 & S2), and fourier shell correlation (FSC) analyses for resolution determination were performed in cryoSPARC (v4.1.1) (Punjani et al., 2017). For in vitro NADase assay (Figure3D–F), the intensities were monitored by a preheated Spark Multimode Microplate reader (TECAN), and automatically generated a corresponding Excel data sheet. Each experiment was replicated three times, and the error bars indicate standard deviations. For ATPase assay (Figure 4J–K), the intensities of reaction products were monitored by Biotek synergy ht microplate reader at 600 nm, and automatically generated a corresponding data sheet. Each experiment was replicated three times, and the error bars indicate standard deviations. For single turnover nuclease assay (Figure 6G–I), measurements of fluorescent intensity were performed from each gel. Experiments were replicated three times, and the error bars indicate standard deviations. For Bacterial grow assay (Figure 7A–D), OD600 of cells was taken by plate reader. Experiments were replicated three times, and the error bars indicate standard deviations. For cellular NAD+ levels evaluated by UPLC-MS/MS (Figure S7F), data were acquired using Analyst 1.7.2™ software and analyzed using SCIEX OS 2.0 software (AB Sciex). Each sample was replicated twice. No statistical analysis was used in this study.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli DH5α Chemically Competent Cell | NEB | C2987P |
| E. coli BL21 (DE3) Chemically Competent Cell | NEB | C2527H |
| Escherichia phage T5 | DSMZ | DSM 16353 |
| Deposited Data | ||
| SIR2_HerA complex 1 (12:6) map | This study | EMDB: EMD-40762 |
| SIR2_HerA complex 1 (12:6) model | This study | PDB: 8SU9 |
| SIR2_HerA complex 2 (12:5) map | This study | EMDB: EMD-40763 |
| SIR2_HerA complex 2 (12:5) model | This study | PDB: 8SUB |
| SIR2_HerA complex 3 (12:4) map | This study | EMDB: EMD-40778 |
| SIR2_HerA complex 3 (12:4) model | This study | PDB: 8SUW |
| SIR2 Dodecamer map | This study | EMDB: EMD-40860 |
| SIR2 Dodecamer model | This study | PDB: 8SXX |
| SIR2_HerA complex 1 (12:6) bound with ATPγS map | This study | EMDB: EMD-42061 |
| SIR2_HerA complex 1 (12:6) bound with ATPγS model | This study | PDB: 8UAE |
| SIR2_HerA complex 1 (12:6) bound with NAD+ map | This study | EMDB: EMD-42062 |
| SIR2_HerA complex 1 (12:6) bound with NAD+ model | This study | PDB: 8UAF |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ε-NAD+ sodium salt | Sigma-Aldrich | N2630-5MG |
| beta-NAD+ sodium salt | Sigma-Aldrich | N0632-1G |
| NTP (ATP, UTP, CTP, GTP) | NEB | N0450S |
| dNTP (dATP, dTTP, dCTP, dGTP) | NEB | N0447S |
| ATPγS | Roche | 11162306001 |
| HerA recombinant protein | This paper | N/A |
| SIR2 recombinant protein | This paper | N/A |
| SIR2-HerA complex recombinant protein | This paper | N/A |
| Critical commercial assays | ||
| ATPase/GTPase Activity Assay Kit | Sigma-Aldrich | MAK113 |
| Oligonucleotides | ||
| 5’Cy3-ssRNA1 /cy3/rArCrArCrArCrUrArUrArCrArArCrCrUrArCrUrArCrCrUrCrArArCrC | This paper | Figure 6F, 6H, 6I |
| Complementary_ssRNA2 rUrGrArGrGrUrArGrUrArGrGrUrUrGrUrArUrArGrU | This paper | Figure 6F |
| 5’Cy3-ssDNA4 /cy3/TACTTTACATAAATTGTAAAATTTATTTAATTAACAAC | This paper | Figure 6A, 6B, 6D, 6E, 6G, 6I, 6J, S7A, S7C, S7D, S7E |
| Complementary_blunt_ssDNA5 GTTGTTAATTAAATAAATTTTACAATTTATGTAAAGTA | This paper | Figure 6A, 6B, 6E, S7A, S7C |
| Complementary_5’_Overhang_ssDNA6 TTTTTTGAACTTTTTTTGTCAAGTTGTTAATTAAATAAATTTTACAATTTATGTAAAGTA | This paper | Figure 6A, 6B, 6E, S7A, S7E |
| Complementary_3’ Overhang_ssDNA7 GTTGTTAATTAAATAAATTTTACAATTTATGTAAAGTAAATTGAGATCTCTTTTTTTGTCA | This paper | Figure 6A, 6B, 6E, S7A |
| Complementary_dual_forked_ssDNA8 TTTTTTTTTTTTTTTTTTTTTTAATTTTACAATTTATGTAAAGTA | This paper | Figure 6A, 6B, S7A, S7D |
| Complementary_dual_forked_3’overhand_ssDNA9 TTTTTTTTTTTTTTTTTTTTTTAATTTTACAATTTATGTAAAGTATTTTTTTTTTTTTTT | This paper | Figure S7D |
| Recombinant DNA | ||
| pET28a-His×6-HerA | This paper | N/A |
| pDB-His×6-MBP-TEV-SIR2 | This paper | N/A |
| pDB-His×6-MBP-TEV-SIR2H227A | This paper | N/A |
| pDB-His×6-MBP-TEV-SIR2D311A | This paper | N/A |
| pDB-His×6-MBP-TEV-SIR2H313A | This paper | N/A |
| pET28a-SIR2-HerA-His×6 | This paper | N/A |
| pET28a-SIR2-HerA H227A-His×6 | This paper | N/A |
| pET28a-SIR2-HerA D311A-His×6 | This paper | N/A |
| pET28a-SIR2-HerA H313A-His×6 | This paper | N/A |
| Software and Algorithms | ||
| cryoSPARC (v4.1.1) | Punjani et al. 24 | https://cryosparc.com |
| AlphaFold v2 | John Jumper et al.25 | https://github.com/deepmind/alphafold |
| UCSF Chimera | Pettersen et al. 26 | https://www.cgl.ucsf.edu/chimera/ |
| Coot | Emsley and Cowtan 27 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PHENIX | Adams et al.28 | https://phenix-online.org |
| Molprobity | Williams et al. 29 | http://molprobity.manchester.ac.uk |
| ChimeraX v1.5 | Pettersen et al. 30 | https://www.rbvi.ucsf.edu/chimerax/ |
| PyMOL 2.3.0 | Schrödinger 31 | https://pymol.org/2/ |
| Analyst_1.7.2 | AB Sciex | https://sciex.com/support/software-support/software-downloads |
| SCIEX OS 2.0 | AB Sciex | https://sciex.com/products/software/sciex-os-software |
| Other | ||
| Superose™ 6 Increase 10/300 GL | Cytiva | 29-0915-96 |
| HiTrap Heparin HP affinity column | Cytiva | GE17-0407-01 |
| Black 96-well plates | Thermo Fisher Scientific | 237105 |
| HSS T3 column | Waters | Cat#186003539 |
| Quantifoil R 1.2/1.3, 400 mesh, Gold | Electron Microscopy Sciences | Cat#Q4100AR1.3 |
ACKNOWLEDGMENTS
We thank Drs. Michael Kearse and Wen Tang at the Ohio State University (OSU) for the NADase assay and providing the RNA substrates, respectively. We thank Dr. Mark Parthun at OSU for helpful discussion. Grids screening were performed at OSU CEMAS with the assistance of Drs. Giovanna Grandinetti and Yoshie Narui. Cryo-EM data were collected with the assistance of Dr Patrick Mitchell at the Stanford-SLAC Cryo-Electron Microscopy Center supported by grants from the NIH National Institute of Health Common Fund Transformative High Resolution Cryo-Electron Microscopy program, Dr. Omar Davulcu at PNCC, and Drs. Giovanna Grandinetti and Yoshie Narui at OSU CEMAS, respectively.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. We support inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
All authors declare they have no competing interests.
Reference
- 1.Bernheim A & Sorek R The pan-immune system of bacteria: antiviral defence as a community resource. Nat Rev Microbiol 18, 113–119, doi: 10.1038/s41579-019-0278-2 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Millman A, Melamed S, Amitai G & Sorek R Diversity and classification of cyclic-oligonucleotide-based anti-phage signalling systems. Nat Microbiol 5, 1608–1615, doi: 10.1038/s41564-020-0777-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wein T & Sorek R Bacterial origins of human cell-autonomous innate immune mechanisms. Nat Rev Immunol 22, 629–638, doi: 10.1038/s41577-022-00705-4 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Tal N & Sorek R SnapShot: Bacterial immunity. Cell 185, 578–578 e571, doi: 10.1016/j.cell.2021.12.029 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Doron S et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, doi: 10.1126/science.aar4120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao L et al. Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077–1084, doi: 10.1126/science.aba0372 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koopal B et al. Short prokaryotic Argonaute systems trigger cell death upon detection of invading DNA. Cell 185, 1471–1486 e1419, doi: 10.1016/j.cell.2022.03.012 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millman A et al. An expanded arsenal of immune systems that protect bacteria from phages. Cell Host Microbe 30, 1556–1569 e1555, doi: 10.1016/j.chom.2022.09.017 (2022). [DOI] [PubMed] [Google Scholar]
- 9.Duncan-Lowey B & Kranzusch PJ CBASS phage defense and evolution of antiviral nucleotide signaling. Curr Opin Immunol 74, 156–163, doi: 10.1016/j.coi.2022.01.002 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Gao LA et al. Prokaryotic innate immunity through pattern recognition of conserved viral proteins. Science 377, eabm4096, doi: 10.1126/science.abm4096 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Y et al. Molecular basis of RADAR anti-phage supramolecular assemblies. Cell 186, 999–1012 e1020, doi: 10.1016/j.cell.2023.01.026 (2023). [DOI] [PubMed] [Google Scholar]
- 12.Duncan-Lowey B et al. Cryo-EM structure of the RADAR supramolecular anti-phage defense complex. Cell 186, 987–998 e915, doi: 10.1016/j.cell.2023.01.012 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garb J et al. Multiple phage resistance systems inhibit infection via SIR2-dependent NAD(+) depletion. Nat Microbiol 7, 1849–1856, doi: 10.1038/s41564-022-01207-8 (2022). [DOI] [PubMed] [Google Scholar]
- 14.Xu Y et al. Mechanisms of helicase activated DNA end resection in bacteria. Structure 30, 1298–1306 e1293, doi: 10.1016/j.str.2022.06.005 (2022). [DOI] [PubMed] [Google Scholar]
- 15.Rzechorzek NJ et al. Structure of the hexameric HerA ATPase reveals a mechanism of translocation-coupled DNA-end processing in archaea. Nat Commun 5, 5506, doi: 10.1038/ncomms6506 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahdash Z et al. Mechanistic insight into the assembly of the HerA-NurA helicase-nuclease DNA end resection complex. Nucleic Acids Res 45, 12025–12038, doi: 10.1093/nar/gkx890 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Min J, Landry J, Sternglanz R & Xu RM Crystal structure of a SIR2 homolog-NAD complex. Cell 105, 269–279, doi: 10.1016/s0092-8674(01)00317-8 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Avalos JL, Boeke JD & Wolberger C Structural basis for the mechanism and regulation of Sir2 enzymes. Mol Cell 13, 639–648, doi: 10.1016/s1097-2765(04)00082-6 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Sanders BD, Jackson B & Marmorstein R Structural basis for sirtuin function: what we know and what we don’t. Biochim Biophys Acta 1804, 1604–1616, doi: 10.1016/j.bbapap.2009.09.009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finnin MS, Donigian JR & Pavletich NP Structure of the histone deacetylase SIRT2. Nat Struct Biol 8, 621–625, doi: 10.1038/89668 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Zhao K, Chai X & Marmorstein R Structure of the yeast Hst2 protein deacetylase in ternary complex with 2’-O-acetyl ADP ribose and histone peptide. Structure 11, 1403–1411, doi: 10.1016/j.str.2003.09.016 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Zhao K, Chai X & Marmorstein R Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli. J Mol Biol 337, 731–741, doi: 10.1016/j.jmb.2004.01.060 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Cheng R et al. A nucleotide-sensing endonuclease from the Gabija bacterial defense system. Nucleic Acids Res 49, 5216–5229, doi: 10.1093/nar/gkab277 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296, doi: 10.1038/nmeth.4169 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature, doi: 10.1038/s41586-021-03819-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pettersen EF et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612, doi: 10.1002/jcc.20084 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams CJ et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci 27, 293–315, doi: 10.1002/pro.3330 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pettersen EF et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci 30, 70–82, doi: 10.1002/pro.3943 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schrödinger L. a. D., W.,. The PyMOL Molecular Graphics System. Version 2.5 Schrödinger, LLC (2022). [Google Scholar]
- 32.Rosenthal PB & Henderson R Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333, 721–745, doi: 10.1016/j.jmb.2003.07.013 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Schneider CA, Rasband WS & Eliceiri KW NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675, doi: 10.1038/nmeth.2089 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession numbers for SIR2_HerA complex 1 (12:6), SIR2_HerA complex 2 (12:5), SIR2_HerA complex 3 (12:4), SIR2 dodecamer, SIR2_HerA complex 1 (12:6) bound with ATPγS, SIR2_HerA complex 1 (12:6) bound with NAD+ are as follows: (coordinates of atomic models: 8SU9, 8SUB, 8SUW, 8SXX, 8UAE and 8UAF were deposited to Protein Data Bank), and (density map: EMD-40762, EMD-40763, EMD-40778, EMD-40860, EMD-42061 and EMD-42062 deposited to Electron Microscopy Data Bank). Original data for biochemical assays and uncropped gels were deposited to Mendeley Data (http://dx.doi.org/DOI:10.17632/xf34wcwxyf.1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.