

Dear Editor,
Defects in single genes involved in histone methylation have been associated with various neurodevelopmental disorders. The histone lysine methyltransferases 2 family (KMTs) plays a key role in the methylation of histone H3 lysine 4 (H3K4). One such genetic defect is responsible for Wiedemann–Steiner syndrome (WDSTS; MIM # 605130), Kabuki syndrome (MIM # 147920), Kleefstra syndrome (MIM # 617768), and Cornelia de Lange syndrome (MIM # 122470).[1] The pathogenic mechanism of WDSTS is attributed to haploinsufficiency of the KMT2A gene. Common clinical phenotypes of the disease include hypertrichosis cubiti, short palpebral fissure, short stature, and intellectual disability (ID), with an incidence of approximately 1 in 25,000-40,000.[1] The Human Gene Mutation Professional Database has recorded 491 variants as of April 2023.04. (https://www.hgmd.cf.ac.uk/). Past research suggests that WDSTS is predominantly associated with de novo variants, while inheritance from parents is uncommon. In the existing literature, only four cases of familial WDSTS have been reported with familial, with two cases reported in France and two in China.[2,3] Unfortunately, detailed clinical information for these cases is currently unavailable. This study presents a case of confirmed WDSTS within a family, which had been identified through trio whole exome sequencing (Trio-WES).
The proband, a 7-year-old female, was referred to our hospital for global developmental delay. She is the second child in the family, with the mother being 24 years old at conception and the father being 37 years old; they are nonconsanguineous. In addition, she has a healthy 10-year-old brother. The proband was delivered vaginally at 39 weeks gestation, with a birth weight of 2.8 kg (<25th percentile), length of 47 cm (<10th percentile), and head circumference of 32 cm (<25th percentile). The proband experienced postnatal failure to thrive and feeding difficulties in infancy. She achieved the ability to walk independently at 3 years of age and started speaking short words at 4 years of age. Laboratory analysis revealed blood routine, thyroid hormone, myocardial enzyme, and lactic dehydrogenase blood biochemical levels were all within normal ranges. Brain computed tomography showed no abnormalities. The Chinese Wechsler Intelligence Scale for Children indicated mild ID. Physical examination revealed that the proband had a height of 111.5 cm (<3rd percentile), weight of 17 kg (<3rd percentile), and head circumference of 47 cm (<3rd percentile). The proband exhibited characteristic features such as thick eyebrows, long eyelashes, short palpebral fissure, downslanted palpebral fissures, eyelid ptosis, flat face, and high palate [Figure 1]. There was no history of sleep disorders, recurrent infections, or abnormal emotion in the child. The proband’s mother, who also has a history of ID, is able to care for herself and shares a similar appearance with the proband. The mother underwent assessment with the Chinese revised version of the Wechsler Adult Intelligence Scale (WAIS-RC) and was diagnosed with mild ID (intelligence quotient of 65). The mother’s height is 152cm, which is significantly below the average height of 158cm for adult females in China, and has a Gallway score of 12 points indicating hirsutism. The proband’s maternal family was surveyed, and no other relatives had a similar history. Based on the family history, a genetic cause of ID syndrome was suspected. Initial chromosome karyotype and copy number variation sequencing examinations did not identify any pathogenic abnormalities in the patient. Due to the potential rarity, the patient and her family were referred to a specialized hospital for medical genetic counseling. After a multidisciplinary consultation, Trio-WES analysis was conducted, which revealed the variant presence of KMT2A (NM_001197104.2; exon27): c.10745C>A (p. Ser3582Ter). This variant was inherited from the mother and is not present in the brother, father, or maternal grandmother. According to the American College of Medical Genetics and Genomics assessment, the variant was classified as a suspected likely pathogenic variant (Very strong evidence of pathogenicity + Moderate evidence of pathogenicity 2_Supporting + evidence of pathogenicity 3) [Figure 2].[4] Based on the phenotype of the child and her mother and WES analysis, the diagnosis of Williams–Beuren Syndrome was made. Genetic counseling was provided to the family. WDSTS is an inherited autosomal dominant disorder, with de novo variants being common, and only a few families have been reported to have this condition.[5,6] Sheppard et al.[7] conducted a large-sample study of 104 cases of WDSTS from five continents, identifying a total of six family heritages, but did not provide specific clinical data. Previous research has shown that mutations in the KMT2A gene are associated with myeloid/lymphoid or mixed leukemia, but there have been no reported cases of co-occurrence of leukemia in WDSTS. Currently, there is a lack of disease-specific treatment plans for individuals with ID like WDSTS, leading to increased care, health, and social services costs for families and society. These disorders can often be recognized and diagnosed in infancy and childhood due to specific facial phenotypes. The incidence and diagnosis rate of ID are influenced by regional economic status, education levels, and the quality of medical care. There is a lack of research on ID in low- and middle-income regions, potentially leading. Diagnosis and treatment become even more challenging when rare diseases have overlapping phenotypes and genotypes. This family’s report is expected to enhance clinicians’ understanding of rare diseases and promote multidisciplinary cooperation in selecting examination methods for patients within a hierarchical medical system. This collaboration can help improve the etiological diagnosis of rare diseases, provide a basis for prenatal diagnosis, and enhance the quality of the population.
Figure 1.
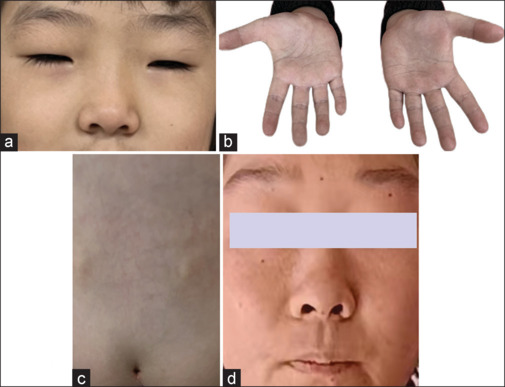
Clinical photographs showing dysmorphic features of the proband and her mother. (a) The proband exhibited characteristic features such as thick eyebrows, long eyelashes, and short palpebral fissure. (b) Clinodactyly of the fifth finger. (c) Sacral dimple. (d) The proband’s mother exhibited characteristic features such as flat face
Figure 2.
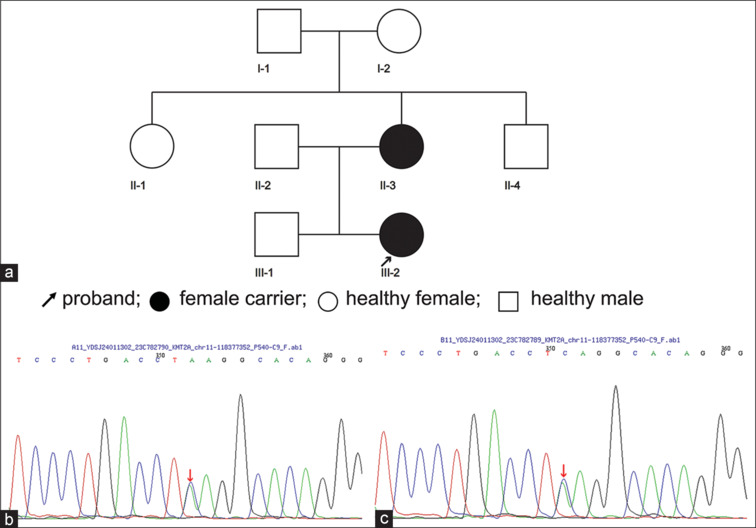
(a) Pedigrees of WDSTS. (b) Sanger sequencing chromatograms of KMT2A in the proband’s mother. (c) Sanger sequencing chromatograms of KMT2A in the proband. WDSTS = Wiedemann–Steiner syndrome
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the ClinVar repository, SUB14223807.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
This research was funded and supported by the Chengdu Bureau of Science and Technology Project (2021-YF05-01658-SN).
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
We thank the families which participated in this study. We are grateful to the family for its willing participation and cooperation.
REFERENCES
- 1.Al Ojaimi M, Banimortada BJ, Othman A, Riedhammer KM, Almannai M, El-Hattab AW. Disorders of histone methylation: Molecular basis and clinical syndromes. Clin Genet. 2022;102:169–81. doi: 10.1111/cge.14181. [DOI] [PubMed] [Google Scholar]
- 2.Bruno LP, Doddato G, Valentino F, Baldassarri M, Tita R, Fallerini C, et al. New candidates for autism/intellectual disability identified by whole-exome sequencing. Int J Mol Sci. 2021;22:13439. doi: 10.3390/ijms222413439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen K, Yang Y, Yang L, Xiao F, Wu X, Huang H, et al. Analysis of gene variation and clinical characteristics of Wiedemann-Steiner syndrome. Chin J Pediatr. 2022;60:119–23. doi: 10.3760/cma.j.cn112140-20210720-00608. [DOI] [PubMed] [Google Scholar]
- 4.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baer S, Afenjar A, Smol T, Piton A, Gérard B, Alembik Y, et al. Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: A study of 33 French cases. Clin Genet. 2018;94:141–52. doi: 10.1111/cge.13254. [DOI] [PubMed] [Google Scholar]
- 6.Bogaert DJ, Dullaers M, Kuehn HS, Leroy BP, Niemela JE, De Wilde H, et al. Early-onset primary antibody deficiency resembling common variable immunodeficiency challenges the diagnosis of Wiedeman-Steiner and Roifman syndromes. Sci Rep. 2017;7:3702. doi: 10.1038/s41598-017-02434-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheppard SE, Campbell IM, Harr MH, Gold N, Li D, Bjornsson HT, et al. Expanding the genotypic and phenotypic spectrum in a diverse cohort of 104 individuals with Wiedemann-Steiner syndrome. Am J Med Genet A. 2021;185:1649–65. doi: 10.1002/ajmg.a.62124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the ClinVar repository, SUB14223807.