

Abstract
Rationale
Chronic obstructive pulmonary disease (COPD) is variable in its development. Lung microbiota and metabolites collectively may impact COPD pathophysiology, but relationships to clinical outcomes in milder disease are unclear.
Objectives
Identify components of the lung microbiome and metabolome collectively associated with clinical markers in milder stage COPD.
Methods
We analyzed paired microbiome and metabolomic data previously characterized from bronchoalveolar lavage fluid in 137 participants in the SPIROMICS (Subpopulations and Intermediate Outcome Measures in COPD Study), or (GOLD [Global Initiative for Chronic Obstructive Lung Disease Stage 0–2). Datasets used included 1) bacterial 16S rRNA gene sequencing; 2) untargeted metabolomics of the hydrophobic fraction, largely comprising lipids; and 3) targeted metabolomics for a panel of hydrophilic compounds previously implicated in mucoinflammation. We applied an integrative approach to select features and model 14 individual clinical variables representative of known associations with COPD trajectory (lung function, symptoms, and exacerbations).
Measurements and Main Results
The majority of clinical measures associated with the lung microbiome and metabolome collectively in overall models (classification accuracies, >50%, P < 0.05 vs. chance). Lower lung function, COPD diagnosis, and greater symptoms associated positively with Streptococcus, Neisseria, and Veillonella, together with compounds from several classes (glycosphingolipids, glycerophospholipids, polyamines and xanthine, an adenosine metabolite). In contrast, several Prevotella members, together with adenosine, 5′-methylthioadenosine, sialic acid, tyrosine, and glutathione, associated with better lung function, absence of COPD, or less symptoms. Significant correlations were observed between specific metabolites and bacteria (Padj < 0.05).
Conclusions
Components of the lung microbiome and metabolome in combination relate to outcome measures in milder COPD, highlighting their potential collaborative roles in disease pathogenesis.
Keywords: bronchoscopy, chronic obstructive pulmonary disease, metabolomics, lung function
At a Glance Commentary
Scientific Knowledge on the Subject
Chronic airway infection with specific bacteria has been linked to more advanced chronic obstructive pulmonary disease (COPD), but recent studies have implicated a broader range of microbiota. It has been unclear, however, whether airway microbiota influences COPD pathogenesis or outcomes in milder disease. Recent independent analyses from the SPIROMICS (Subpopulations and Intermediate Outcome Measures in COPD Study) found altered lung bacterial composition to be associated with measures of lung function and symptom burden. Separate analyses of the airway/lung metabolome identified specific metabolites related to these and other outcomes. Rarely have the lung microbiome and metabolome, or their potential coassociations with clinical markers in milder COPD, been examined together.
What This Study Adds to the Field
This study analyzed paired lung microbiome and metabolomic data from SPIROMICS ever-smokers with or without mild-to-moderate COPD, using an integrative analysis approach to model several clinical measures. Specific bacteria and metabolites collectively were associated, positively or negatively, with COPD status, lung function, symptom measures, and/or exacerbation history or events. Significantly correlated lung bacteria and metabolites also were identified. These findings suggest that components from the lung microbiome and metabolome may together impact pathophysiologic mechanisms in milder stage COPD.
Chronic obstructive pulmonary disease (COPD) is a smoking-associated lung disease that is heterogeneous in its development and phenotypes (1). Not all smokers develop COPD, yet some without COPD, as defined by spirometry, experience chronic respiratory symptoms and exacerbation-like events, as previously described in the SubPopulations and InteRmediate Outcome Measures in COPD Study (SPIROMICS) (2).
Airway infection has been hypothesized to contribute to COPD pathogenesis, but mechanisms remain unclear (3), including potential relationships between bacteria and the lung metabolic milieu. Existing evidence on bacterial associations with COPD outcomes derive mostly from cohorts with more advanced disease or frequent exacerbations (4–7). Also, many studies have analyzed sputum which, although easier to obtain, unavoidably represents an admixture of upper and lower airway secretions. Far fewer studies have examined the lung microbiome in milder COPD or using bronchoscopy (8–10), which allows for direct lung sampling. In recent independent studies from SPIROMICS, we reported significant relationships between the lung metabolome (untargeted metabolomics) and lung function (11), and between the lung microbiota, lung function, and symptom burden (12). These reports, however, were based on analyses of single-‘omic data, which did not allow for examination of the collective contribution of lung microbiota and metabolites to these outcomes. Because both may shape the lung environment, integrative analysis of microbiome and metabolomic data may yield new insights into the role of lung cell and microbial metabolism in COPD pathophysiology.
To address this gap, we utilized lung microbiota and metabolomic data previously generated from bronchoalveolar lavage fluid (BAL) of SPIROMICS subjects who were ever-smokers with or without mild-to-moderate COPD. For this study, we used metabolomics datasets from two independent approaches: untargeted metabolomics of a lipid-rich hydrophobic fraction and targeted metabolomics of several hydrophilic compounds implicated in airway mucoinflammation (nucleosides and/or nucleotides, amino acids, and others) (13–16). We performed paired dataset analyses (microbiome-untargeted metabolomics, microbiome-targeted metabolomics), using an integrative approach to determine features collectively associated with clinical endpoints representative of known associations with COPD trajectory. Some of these results have been previously reported in the form of an abstract (17).
Methods
Please see the online supplement for additional details.
Participants
Microbiome and metabolomic data were generated from BAL collected in the SPIROMICS bronchoscopy substudy (NCT01969344; clinicaltrials.gov) (10, 18). Institutional review boards at all participating sites approved the study, and all subjects provided written informed consent. A total of 215 subjects underwent bronchoscopy as described elsewhere (10). “Ever-smoker” was defined at SPIROMICS enrollment by smoking history ⩾ 20 pack-years (18). To identify subjects with available paired data, we first excluded 46 healthy never-smokers, given known effects of smoking on the metabolome (11), and seven with severe COPD, given our focus on milder COPD. Of the remaining subjects (Global Initiative for Chronic Obstructive Lung Disease [GOLD] 0–2), 137 had BAL microbiome data and at least one of the metabolomics datasets. Of these, 87 had paired microbiome-untargeted metabolomic data, and 126 had paired microbiome-targeted metabolomics data; Table 1). Selection of variables for modeling was informed by clinical relevance, known associations with COPD outcomes or progression (2, 3, 19), and prior reported associations with either the lung microbiome (12) or lung metabolome (11). These included lung function measures, bronchodilator response (BDR) (19), and symptom burden (COPD Assessment Test [CAT]; chronic bronchitis defined by St. Georges Respiratory Questionnaire items) (20), assessed at the annual visit most proximal to bronchoscopy. We also modeled the number of exacerbations in the year before SPIROMICS enrollment, exacerbations during SPIROMICS Year 1, and exacerbations postbronchoscopy after 1 year of follow-up.
Table 1.
Clinical Characteristics of Subjects Analyzed
| Variable | All (137 subjects) | Microbiome and Untargeted Metabolomics (87 subjects) | Microbiome and Targeted Metabolomics (126 subjects) | P Value |
|---|---|---|---|---|
| Age, yrs | 61.2 (8.8) | 61.3 (8.5) | 61 (8.9) | 0.85 |
| Sex, % male | 56.2 | 50.6 | 57.1 | 0.40 |
| Caucasian, % | 69.3 | 78.2 | 68.3 | 0.12 |
| Current smoker, % | 43.2 | 34.1 | 45.5 | 0.12 |
| Pack-years* | 38.2 (20) | 37.8 (15.6) | 38.1 (30.2) | 0.57 |
| Mild to moderate COPD, % | 43.8 | 41.4 | 40.5 | 1.00 |
| FEV1/FVC post-BD | 0.69 (0.12) | 0.70 (0.11) | 0.69 (0.12) | 0.48 |
| FEV1, % predicted | 89.5 (18.5) | 92.7 (17.5) | 89.8 (18.6) | 0.26 |
| FVC, % predicted | 100 (13.7) | 102 (14) | 100 (13.7) | 0.49 |
| FEF25–75, L/s | 2.00 (1.2) | 2.1 (1.2) | 2.0 (1.2) | 0.62 |
| FVC BDR, % change* | 1.9 (7.4) | 1.8 (6.9) | 1.7 (7.4) | 0.93 |
| FEV1 BDR, % change | 8.4 (7.8) | 8.49 (7.3) | 8.18 (7.9) | 0.78 |
| CAT score* | 9 (12) | 10 (12) | 9 (12) | 0.82 |
| BAL neutrophil, %*† | 1.2 (2.1) | 1.1 (1.9) | 1.2 (1.9) | 0.82 |
Definition of abbreviations: BD = bronchoscopy; BDR = bronchodilator response; CAT = COPD Assessment Test; COPD = chronic obstructive pulmonary disease; FEF25–75 = maximum midexpiratory flow.
For each row, data are either percentages with P values from Fisher’s exact tests between the two groups/frameworks, means (and SD) with P values from t tests, or median (and interquartile range) with P values from Wilcoxon rank-sum tests (*).
Data available for N = 86, 59, and 77, respectively.
BAL Samples
BAL fluid was processed and stored at −80°C as previously described (10). For bacterial profiling, total DNA was extracted from cell pellets, and 16S rRNA gene sequencing was performed on an Illumina MiSeq targeting the V4 region (12). As described previously, background and negative control samples (e.g., reagents, extraction controls) were sequenced and the data processed together with the BAL-derived data (12). To identify potential contaminant sequences for removal, we used decontam (12, 21). Separately stored, unthawed aliquots of BAL supernatants were processed for earlier metabolomics studies in SPIROMICS. These included untargeted metabolites profiled by liquid chromatography-mass spectrometry (LC–MS; University of Colorado) (11), which yielded data from both the hydrophilic and hydrophobic fractions. For this study, only the hydrophobic fraction data (C18 reversed-phase chromatography MS) were analyzed with the microbiome data. We also used an unpublished targeted metabolomics dataset (LC–tandem MS [LC-MS/MS]; University of North Carolina, Chapel Hill) that measured several hydrophilic compounds (Table E1) implicated in airway inflammation, hydration, and/or with reported associations with COPD outcomes (13–16).
Data Analysis
Participants were analyzed in two groups: Framework 1, those with both BAL microbiome and untargeted metabolome data (87 subjects; 1 E1A); and Framework 2, those with both BAL microbiome and targeted metabolome data (126 subjects; (1 E1B). Participant demographics and clinical parameters did not differ between the frameworks (Table 1). We did not analyze all the datasets together in a single framework, as even fewer subjects had all three available.
Statistical analyses were performed using R (version 3.5.1) and the packages mixOmics, caret, and vegan (22). The initial set of operational taxonomic units (OTUs; ⩾97% similarity) (12) were filtered to those with approximately nonzero variance, resulting in 72 OTUs. OTU relative abundances (RAs) were transformed using centered log-ratio transformation. Metabolomics data were log-transformed. For the untargeted data, results were corrected for dilution effects based on total useful MS signal using the external scalar algorithm (4), before log-transformation.
For feature selection, for each outcome, a training set consisting of 70% of samples with nonmissing data was randomly selected, and the remaining samples (30%) were placed in a testing set (1 E1). In Framework 1, t tests and Wilcoxon rank-sum tests were used first to filter the initial set of metabolites (7,689 compounds) and OTUs to only those associated with a given outcome within the training set (Benjamini–Hochberg padj < 0.05; Table E3). This greatly reduced the number of untargeted metabolites (average number, 652) passed into the next step for feature selection. Given the fewer metabolites in Framework 2 (31 compounds), all were retained. For each outcome, we then utilized DIABLO (Data Integration Analysis for Biomarker Discovery Using Latent Variable Approaches for ‘Omics Studies) to identify initial predictive features (22). DIABLO is a supervised machine learning method to model data from multiple ‘omics datasets, using projection to latent structures and generalized canonical correlation analysis to select highly correlated features within and across datasets. To fit its requirements (categorical outcomes), clinical measures were dichotomized by clinically accepted thresholds or median values (Table E2).
DIABLO feature selection was followed by tuning with 10-fold cross-validation to determine which model produced the highest model balanced accuracy rate (BAR) out-of-sample. Selected features were then inputted into multivariable elastic net logistic regression models on the training data, plus the following covariates: age, sex, race, current smoking status, inhaled corticosteroid use, and antibiotic use within 3 months before bronchoscopy. Models were performed with 10-fold cross-validation and automated tuning. The resulting elastic net model with the highest BAR on the training data was used to predict class labels on the testing data. To ensure convergence of out-of-sample accuracy and prevent overfitting, the aforementioned steps (feature selection, elastic net regression) were repeated with new training and testing sets that were resampled each iteration, creating 100 models for each outcome and calculation of mean model statistics.
To determine microbial–metabolite correlation networks, for each framework, the most predictive microbial and metabolite features were pooled across all outcomes. Pearson correlations were calculated between the features, and networks were generated using a Euclidean distance matrix of the Pearson correlations followed by clustering of vertices using the fast-greedy algorithm (23). Piphillin was used for inferred metagenome analysis to identify predicted bacterial genes and their functional pathways (24). Pathway gene abundance count data were used to model clinical measures using DESeq2 (25).
Results
Combined Features from the Lung Microbiome and Metabolome Associate with Clinical Endpoints in SPIROMICS Subjects
For each framework, we modeled the clinical outcomes to determine 1) whether the lung microbiome and metabolome collectively are predictive of the measures and, if so, 2) which specific bacterial OTUs and metabolites, as analyzed in the context of each other, are associated with these outcomes. The main feature–outcome associations observed, collated from both frameworks, are highlighted in Table 2 and are further detailed for each framework in the following text.
Table 2.
Summary of the Main Clinical Associations Observed for Specific Lung Bacteria and Metabolites and Metabolite Classes
| Bacteria or Metabolites | Taxonomy: Phylum (Class) | Clinical Measure |
|---|---|---|
| Bacteria | ||
| Neisseria.Otu0018 | Proteobacteria (Betaproteobacteria) | ↓ FEV1, ↓ FEF25–75, + COPD, ↓ CAT, – CB |
| Streptococcus - Otu0005 or Otu0082 | Firmicutes (Bacilli) | ↓ FEV1:FVC, ↓ FEV1, + COPD, ↑ CAT, + CB |
| Veillonella.Otu0002 | Firmicutes (Negativicutes) | ↓ FEV1, ↓ BDR, ↑ CAT, + CB |
| Actinomyces.Otu0086 | Actinobacteria | ↓ FEV1, ↑ BDR, + Exac past 12 mo at baseline, + Exac in study Yr 1 |
| Fusobacterium - Otu0023 or Otu0034 | Fusobacteria | +COPD, ↓ BDR, + CB |
| Capnocytophaga.Otu0054 | Bacteroidetes (Flavobacteriia) | ↑ FEV1, ↓ BDR, – CB, – Exac past 12 mo at baseline |
| Leptotrichia.Otu0038 | Fusobacteria | ↑ FEV1, ↓ BDR, – Exac past 12 mo at baseline |
| Pasteurellaceae_unclassified.Otu0087 | Proteobacteria (Gammaproteobacteria) | ↑ FEV1:FVC, ↑ FEV1, ↑ FEF25–75, ↓ CAT |
| Prevotella.Otu003 | Bacteroidetes (Bacteroidia) | ↑ FEV1, ↑ FEF25–75, – COPD, ↓ BDR, + CAT, + CB, – Exac postbronch-Yr 1 |
| Prevotella.Otu004 | Bacteroidetes (Bacteroidia) | ↑ FEV1, – Exac past 12 mo at baseline |
| Prevotella.Otu0045 | Bacteroidetes (Bacteroidia) | ↑ FEV1, ↑ FEF25–75, – COPD, ↓ CAT |
| Pseudomonas.Otu0029 | Proteobacteria (Gammaproteobacteria) | ↑ FEV1, – CB |
| Metabolite | Metabolite Class | Clinical Measure |
|---|---|---|
| Untargeted panel, hydrophobic fraction | ||
| DG(42:3) | Diglyceride (diacylglycerol) | ↑ BDR, ↑ CAT, + CB |
| Galα1-3(Fucα1-2)Galβ1-4(Fucα1-3) GlcNAcβ1-3Galβ1-4Glcβ-Cer(d18:1/16:0) (“Class-Glycosphingolipid-1” in Figure 1B) | Glycosphingolipid | ↓ FEV1, + CB |
| NeuAcα2-3Galβ1-4GlcNAcβ1-3Galβ1-4(Fucα1-3) GlcNAcβ1-3Galβ1-4GlcNAcβ1-3Galβ1-4 GlcNAcβ1-3Galβ1-4Glcβ-Cer(d18:1/24:0) (“Class-Glycosphingolipid-2” in Figure 1B) | Glycosphingolipid | ↓ FEV1 |
| GalNAcα1-3GalNAcβ1-3Galα1-3Galβ1- 4Glcβ-Cer(d18:1/22:0) (“Class-Glycosphingolipid-3” in Figure 1B) | Glycosphingolipid | + Exac postbronch-Yr 1 |
| LysoPC(17:0) | Lysophospholipid | ↓ FEV1,↑ BDR |
| LysoPC(22:0) | Lysophospholipid | + Exac postbronch-Yr 1 |
| LysoPE(22:0) | Lysophospholipid | + Exac postbronch-Yr 1 |
| LysoPE(22:6) | Lysophospholipid | ↓ FEV1 |
| PA(21:4) | Phosphatidic Acid | ↓ FEV1:FVC, + COPD, ↑ BDR |
| PC(O-18:0_O-3:1) | Phosphatidylcholine | ↓ FEV1, ↓ FEF25-75 , + Exac study Yr 1, + Exac postbronch-Yr 1 |
| PC(18:2_18:2) | Phosphatidylcholine | ↑ FEF25–75 , ↓ CAT |
| PE(P-38:3) | Phosphatidylethanolamine | ↓ FEV1, ↓ FEF25–75 , + COPD, ↑ BDR |
| PS(42:7) | Phosphatidylserine | ↓ FEV1, ↓ FEV25–75, ↑ BDR |
| PS(29:0) or PS(29:0)_isomer | Phosphatidylserine | ↑ FEV1, – Exac postbronch-Yr 1 |
| PS(O-36:0) | Phosphatidylserine | ↑ FEV1:FVC, ↑ FEV1 , ↑ FEF25–75 |
| Betaine aldehyde | Quaternary ammonium salts | ↑ FEV1, ↑ FEF25–75, – Exac postbronch-Yr 1 |
| 2-amino-14,16-dimethyloctadecan-3-ol | Sphingolipid | ↑ FEV1:FVC, ↑ FEF25–75, ↓ BDR |
| 3,11-dioxopregna-4,17(20)-dien-21-oic acid methyl ester (“Class-Steroid Ester-1” in Figure 1B) | Steroid Ester | ↓ FEV1, + COPD, + CB |
| 392.7607@6.112 | NA | ↓ FEV1:FVC, + COPD, ↑ BDR, + Exac postbronch-Yr 1 |
| Targeted panel | ||
| Adenine | Purine | ↓ FEV1, ↑ BDR, ↑ CAT, + Exac past 12 mo at baseline |
| Adenosine | Purine metabolism | ↑ FEV1:FVC, ↑ FEV1, – COPD, ↓ CAT, – CB, |
| AMP | Purine metabolism | ↑ FEV1:FVC, ↑ FEV1, – COPD, ↑ BDR, ↑ CAT, + CB |
| Glutathione | Glutathione metabolism | ↑ FEV1, – COPD, ↓ BDR, ↓ CAT |
| Glutathione disulfide | Glutathione metabolism | ↑ FEV1, + COPD, ↑ BDR, ↑ CAT, + Exac past 12 mo at baseline |
| Lactate | Organic acid | ↑ FEV1:FVC, ↑ FEV1, – COPD, ↓ BDR, ↑ CAT, + Exac in study Yr 1 |
| Leu-Pro | Dipeptide | ↓ FEV1, + COPD, ↑ BDR, + CB |
| Methionine | Amino acid | ↑ FEV1, – COPD, ↓ CAT, – Exac past 12 mo at baseline, – Exac study Yr 1 |
| 5′-methylthioadenosine | S-methyl derivative of adenosine | ↑ FEV1:FVC, ↑ FEV1, ↓ CAT, – Exac study Yr 1 |
| Putrescine | Polyamine | ↓ FEV1:FVC, ↓ FEV1, +COPD, ↑ BDR, ↑ CAT, + CB, + Exac past 12 mo at baseline, + Exac study Yr 1 |
| Spermidine or spermine | Polyamine | ↓ FEV1, + COPD, ↑ BDR, ↑ CAT, + CB, + Exac past 12 mo at baseline |
| Sialic acid | Acidic monosaccharide | ↑ FEV1:FVC, ↑ FEV1, – COPD, ↓ BDR, ↓ CAT |
| Tyrosine | Amino acid | ↑ FEV1:FVC, ↑ FEV1, – COPD, , ↓ CAT, – CB |
| Xanthine | Purine metabolism | ↓ FEV1:FVC, ↓ FEV1, + COPD, ↑ BDR, ↑ CAT, +CB, + Exac past 12 mo at baseline, + Exac study Yr 1 |
Definition of abbreviations: BDR = bronchodilator response; CAT = COPD Assessment Test score; CB = chronic bronchitis; COPD = chronic obstructive pulmonary disease; Exac = exacerbations; FEF25–75 = maximum midexpiratory flow; NA = not applicable; Postbronch-Yr 1 = postbronchoscopy, Year 1.
This table highlights features with consistent association patterns across outcomes or categories from either framework.
Framework 1: Lung Microbiome–Untargeted Metabolome (Hydrophobic Fraction)
In Framework 1, for which 87 subjects had available paired data, the mean out-of-sample classification accuracy across the iterative models exceeded 50% (P < 0.05) for nearly all clinical measures, indicative of better than chance performance (Figure 1A). These overall model results indicated that, in this cohort, features from the lung microbiome and untargeted metabolome collectively associated with lung function, COPD status, symptom burden, and exacerbations reported during SPIROMICS Year 1 or postbronchoscopy after 1 year). The covariates in these models (age, sex, race, smoking status, recent antibiotic use, and inhaled steroid use) were selected in an average of 68%, 73%, 62%, 61%, 77%, and 79% of final elastic net models, respectively, across all outcomes modeled, highlighting their importance as predictors.
Figure 1.
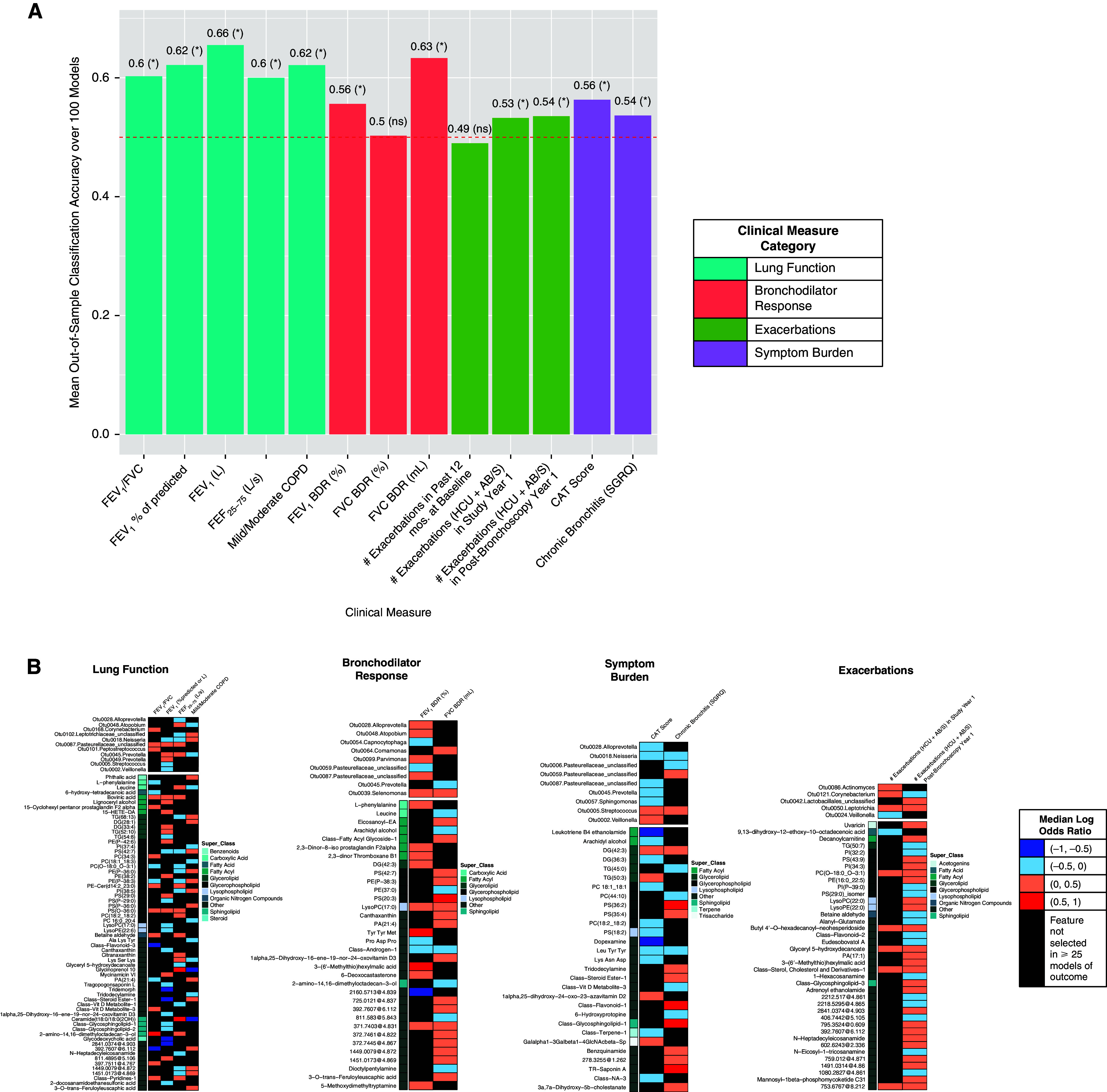
Classification accuracies and the lung bacteria and untargeted metabolomics features (hydrophobic fraction) most strongly associated with each clinical outcome, as modeled in Framework 1. Results from DIABLO feature selection followed by elastic net models adjusted for age, sex, race, current smoking, inhaled corticosteroid use, and recent antibiotic use. (A) Mean out-of-sample classification accuracies. Red dashed line = 50% accuracy (random chance). Asterisks indicate mean model performance > random chance (one-sided t test). (B) Most predictive microbial and untargeted metabolomic features from adjusted elastic net models for outcomes whose classification accuracy exceeded random chance. Bacterial OTUs are displayed alphabetically, and metabolites are displayed by class membership with superclasses of interest indicated. See Table E5 for full IDs and class information. Metabolite names with >50 characters were relabeled as “Class-Name-#”. Superclasses are intended to highlight metabolite groups of interest; in particular, lipids. “Other” refers to compounds for which the superclass was unknown or the metabolite name, as displayed, provides indication of metabolite class. BDR = bronchodilator response; CAT = COPD Assessment Test; COPD = chronic obstructive pulmonary disease; FEF25–75 = maximum midexpiratory flow; HCU+AB/S = exacerbation requiring healthcare utilization and antibiotics/steroid treatment; ns = not significant; OTU = operational taxonomic unit; SGRQ = St. George’s Respiratory Questionnaire.
We next examined the specific features identified by the models for their direction and strength of association with each outcome (Figure 1B). Median adjusted log odds ratios (ORs) for each feature–outcome pair ranged between −1 and 1 (0.36 < median adjusted OR < 2.72). Bacteria associated with COPD status, lower forced expiratory volume in 1 second (FEV1), or a CAT score ⩾10 included Neisseria-Otu0018 (mean RA: 1.6% vs. 1.1% for COPD vs. no COPD), Streptococcus-Otu0005 (mean RA: 11.3% vs. 5.9% for FEV1 <80% vs. ⩾80%; 10.6% vs. 3.8% for CAT score ⩾10 vs. <10), and Veillonella-Otu0002 (mean RA: 16% vs. 9% for CAT ⩾10 vs. <10). We had previously culture-isolated streptococci from BAL of subjects with higher relative abundance of Streptococcus OTUs (see Supplemental Methods in the online supplement) (12). On the basis of earlier generated sequence data from these isolates (rnpB locus plus full-length 16S rRNA gene), we identified Otu0005 as S. pneumoniae. In contrast, several bacteria were associated with higher lung function, non-COPD status, or fewer symptoms; most notably, Prevotella-Otu0045, which associated with all three.
As Framework 1 utilized data from the hydrophobic fraction of BAL, compounds from several lipid classes were found to associate with the clinical measures. This included several glycosphingolipids, lysophospholipids, and glycerophospholipids that associated with lower lung function (e.g., FEV1, FEF25–75), COPD status, or occurrence of at least one exacerbation in the year after bronchoscopy (Table 2). Several of these also associated with positive BDR, which has been linked to indicators of worse COPD (19). Additional metabolites positively associated with BDR included the diglyceride DG(42:3), 2,3-dinor thromboxane B1, and 2,3-dinor-8-iso prostaglandin F2alpha (Figure 1B).
These data provided a unique opportunity to explore connections between microbes and metabolites in the same compartment, about which little is known in the lungs. Significant correlations (padj < 0.01) were observed between specific bacteria and metabolites (Figure 2). In particular, we noted the following. Prevotella-Otu0049, which associated with higher FEV1, strongly correlated with a ceramide—Ceramide(t18:0/18:0(2OH)—that itself was associated with higher FEF25–75 and no COPD status (Figure 1B). Leptotrichia-Otu0050 strongly correlated with the lysophospholipid LysoPC (22:0), and both were positively associated with exacerbations postbronchoscopy after 1 year. Actinomyces-Otu0086, associated with exacerbations during SPIROMICS Year 1, also displayed multiple positive correlations with metabolites, including the diglyceride DG(42:3) and the phosphatidylserines PS(P–29:0) and PS(35:4). These metabolites, respectively, also associated with positive BDR, lower FEV1, and chronic bronchitis (Figure 1B). In contrast, Corynebacterium-Otu0121 correlated negatively with many metabolites, including 2,3-dinor thromboxane B1 and several unknown ones. More bacteria–metabolite correlations were evident when a higher significance threshold was applied (padj < 0.05; Figure E2), including additional bacteria and compounds from the aforementioned classes. Network-based cluster analysis identified four clusters of connected bacteria and metabolites, with the most connections observed for the Actinomyces and Corynebacterium OTUs (Figure E3A).
Figure 2.
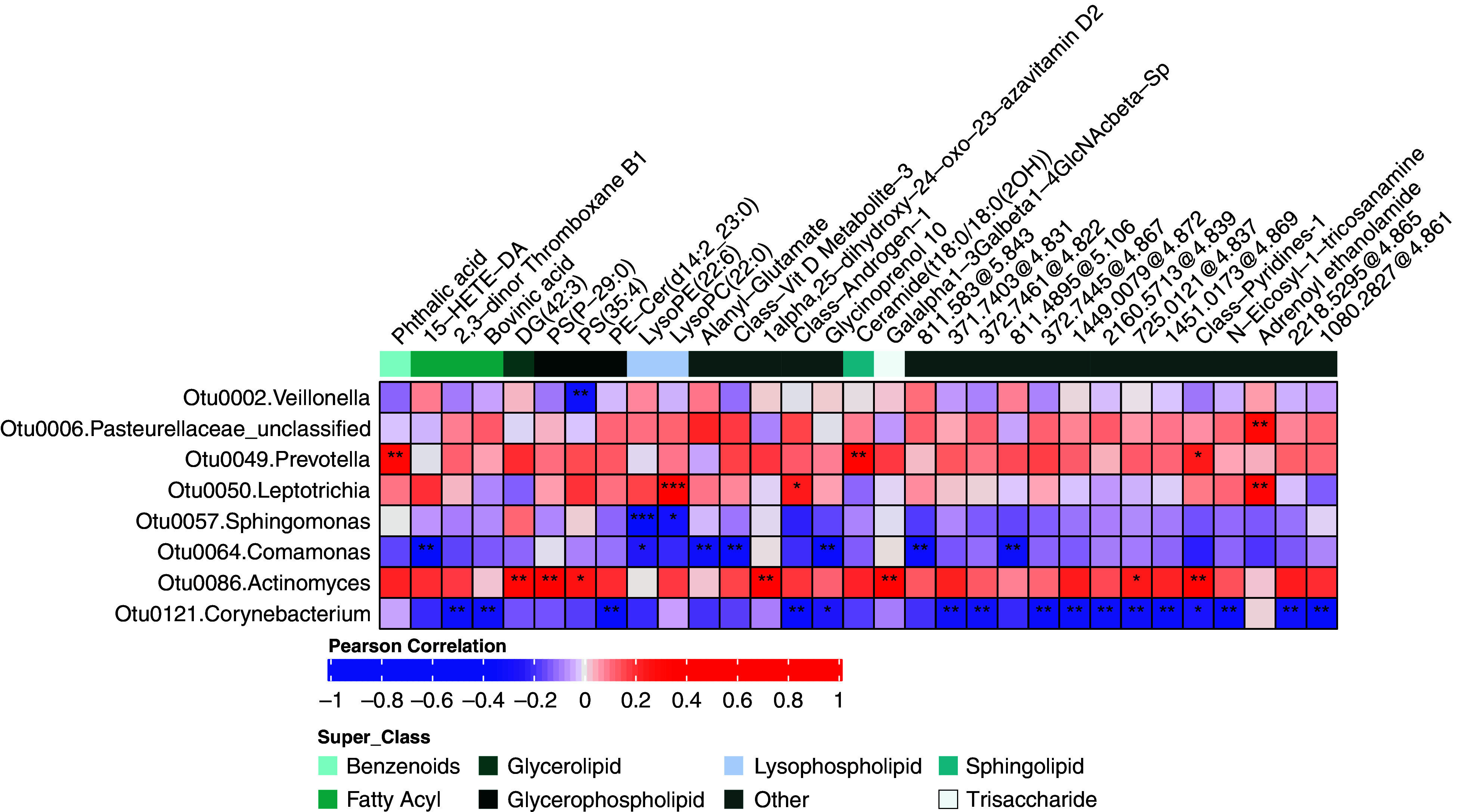
Correlation heatmap between lung microbiota and untargeted metabolomics features that were most strongly associated with the clinical measures. Only OTUs and metabolites having at least one significant correlation based on Padj < 0.01 (Benjamini–Hochberg corrected) are shown. *0.01 ⩽ Padj < 0.05, **0.001 ⩽ Padj < 0.01, and ***Padj < 0.001. OTU = operational taxonomic unit.
Framework 2: Lung Microbiota–Targeted Metabolome (LC-MS/MS)
In Framework 2, for which 126 subjects had available paired data, the mean out-of-sample classification accuracy across 100 iterative models significantly exceeded 50% (P < 0.05) for all the clinical measures except exacerbations in the year postbronchoscopy (Figure 3A). Across all outcomes modeled, the included covariates (age, sex, race, smoking status, recent antibiotic use, and inhaled steroid use) were, on average, selected in 76%, 62%, 44%, 46%, 56%, and 76% of the final elastic net models, respectively.
Figure 3.
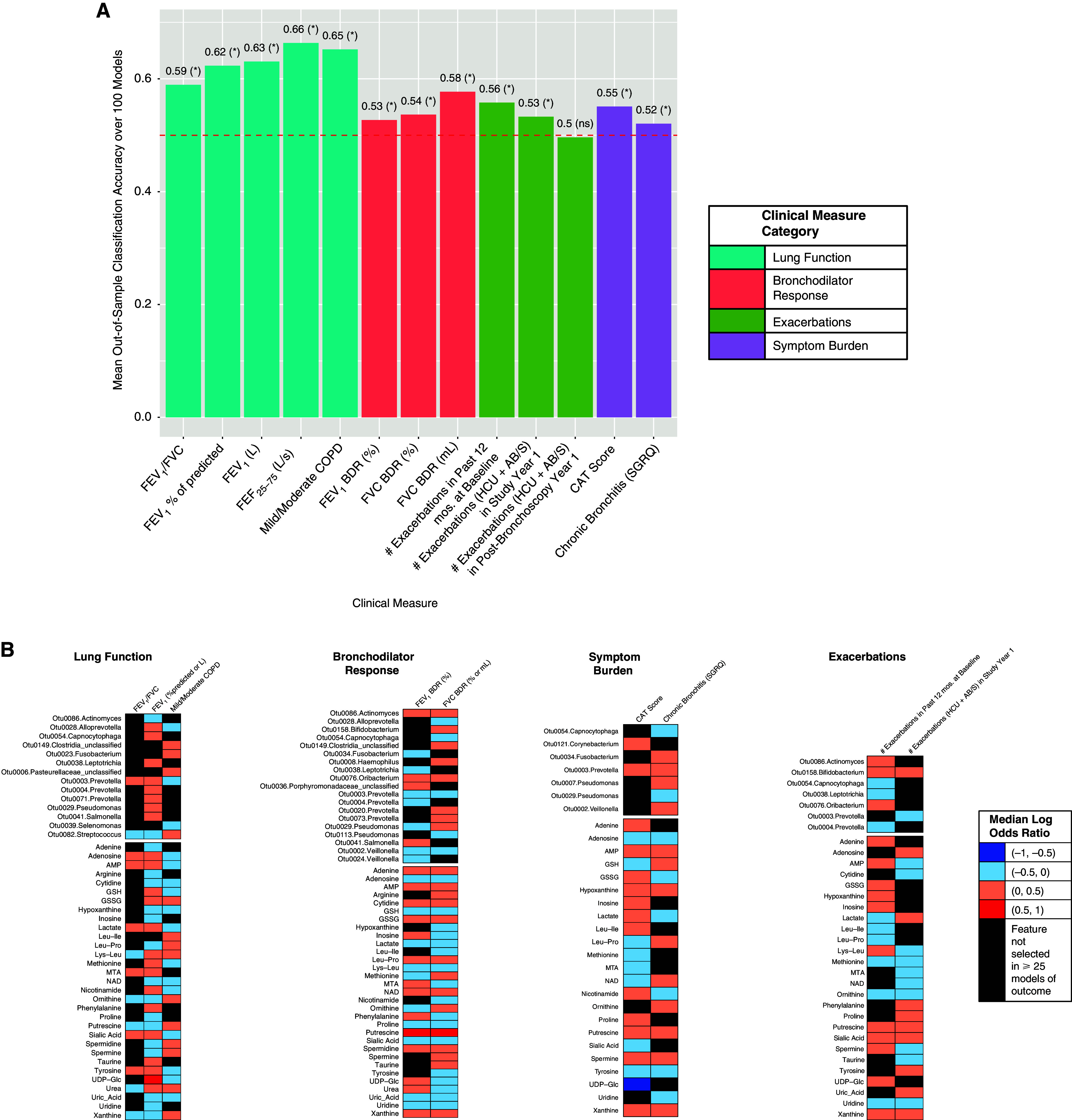

Classification accuracies and the lung bacterial and metabolites from targeted metabolomics most strongly associated with each clinical outcome, as modeled in Framework 2. (A) Mean out-of-sample classification accuracies for each outcome. Asterisks indicate mean model performance > random chance (one-sided t test). (B) The most predictive features from adjusted elastic net models for outcomes whose classification accuracy exceeded random chance. (C) Correlation heatmap between lung microbiota and metabolites that were most strongly associated with the clinical measures. Only OTUs and metabolites having at least one significant correlation based on Padj < 0.05 (Benjamini–Hochberg correction) are shown. *0.01 ⩽ Padj < 0.05, **0.001 ⩽ Padj < 0.01, and ***Padj < 0.001. For definition of abbreviations, see Figure 1.
We noted the following feature–outcome associations, displayed in Figure 3B and highlighted in Table 2. Lung bacteria associated with COPD status and/or lower FEV1 included Streptococcus-Otu0082, two Fusobacterium members, and Actinomyces-Otu0086. In contrast to these low prevalence bacteria, Prevotella-Otu0003 associated with non-COPD status and higher FEV1 (mean RA: 9.5% vs. 7.5% for no COPD vs. COPD; mean RA: 10.0% vs. 6.0% for FEV1 ⩾80% vs. <80%). However, this Prevotella OTU also associated positively with both CAT score and chronic bronchitis, suggestive of a possible role in symptomatic ever-smokers with preserved lung function.
Among the targeted metabolites, COPD status associated with glutathione disulfide, polyamines (putrescine, spermidine or spermine), leucine-proline, and xanthine. These also associated with greater symptoms (CAT score ⩾10, chronic bronchitis, or both). Putrescine, a polyamine breakdown product of amino acid metabolism, was very strongly predictive of BDR by both FEV1 and forced vital capacity (selected in 91 of 100 iterative models). In contrast, targeted metabolites negatively associated with COPD or symptoms, and positively related to FEV1, included adenosine, 5′-methylthioadenosine (MTA), glutathione, methionine, sialic acid, and tyrosine. Notably, from the correlation analyses (Figure 3C), Prevotella-Otu0003 displayed significant relationships with adenosine, AMP, and MTA, congruent with their individual associations with better lung function and non-COPD status, and these features constituted one main cluster in the network analysis (Figure E3B).
Functional Potential of Lung Bacteria by Inferred Metagenome Analysis
Metabolites derive from metabolism by the host, microbes, or both (co-metabolism). We therefore explored whether the identified bacterial community might possess predicted genes in pathways related to the metabolites identified. We used Piphillin (24) to infer bacterial metagenomes and explored whether predicted functional orthologues from the Kyoto Encyclopedia of Genes and Genomes Orthology Database (ko) related with the clinical measures (Table ES4). Negative relationships existed between FEV1 and several predicted bacterial pathways, including for purine metabolism (ko00230), amino acids (ko00400, ko01230, and ko00290), and pyruvate metabolism (ko00620). Positive BDR was associated with the phosphoenolpyruvate-dependent phosphotransferase system (log2 fold difference = 0.50, padj = 0.017), a major system by which bacteria uptake sugars for energy. CAT score and predicted bacterial genes in the sphingolipid signaling pathway (ko04071) were negatively related. Altogether, these results support the possibility that lung bacteria harbor the capacity to shape the metabolic environment and potentially impact pathophysiologic mechanisms in COPD.
Lung Microbiota and Metabolome Features Are Predictive of BAL Neutrophil Percentage
Neutrophilic inflammation, often observed in COPD, has been associated with airway metabolite biomarkers (13, 15). We explored whether the profiled bacteria and metabolites may together predict BAL neutrophil percentages ascertained previously by flow cytometry (26). Neutrophil percentages were overall low, but modeling this per Framework 1 (above or below median of 1.1%; 59 subjects) yielded a mean out-of-sample accuracy of 59.2% (P < 0.05 vs. chance). BAL neutrophil percentage was positively associated with Streptococcus-Otu0005, Leptotrichia-Otu0050, and several glycerophospholipids and sphingolipids—most notably, ganglioside GM3 (d18:0/20:0) (Figure 4A). In contrast, BAL neutrophils negatively associated with two Prevotella members (Otus 0003 and 0045), noted earlier to relate to better lung function. Prevotella-Otu0003 also displayed negative correlations with the phosphatidylinositol PI(22:0_20:3) and the lysophospholipid LysoPE(22:4) (Figure 4B), both metabolites that were positively related to BAL neutrophils (Figure 4A).
Figure 4.
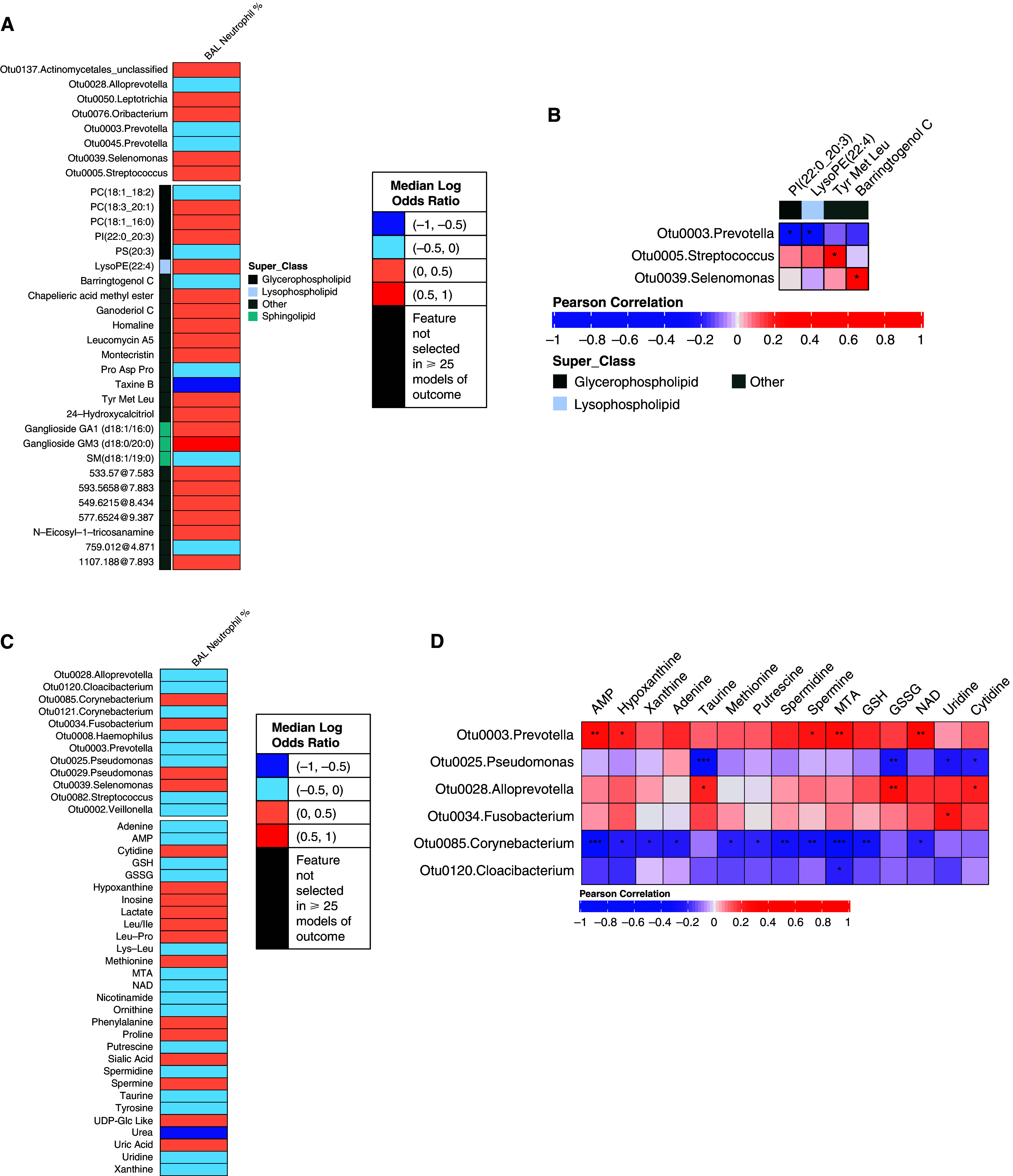
Bacterial and metabolite features associated with BAL neutrophil percentages and correlations between these features per Framework 1 (A and B) or Framework 2 (C and D). *0.01 ⩽ Padj < 0.05, **0.001 ⩽ Padj < 0.01, and ***Padj < 0.001.
Modeling BAL neutrophil percentage per Framework 2 (above or below median of 1.2%; 77 subjects) yielded a mean out-of-sample accuracy of 52.5% (P < 0.05). Targeted metabolites positively associated with neutrophils included hypoxanthine, inosine, lactate, leucine-proline, phenylalanine, and uric acid, together with Fusobacterium-Otu0034 and other bacteria (Figure 4C). Features negatively associated with lung neutrophils included ones mentioned earlier (Prevotella-Otu0003, glutathione, MTA, and tyrosine) that associated with no COPD and better lung function. Correlations between specific bacterial and metabolites in this subset of subjects are shown in Figure 4D.
Discussion
In this study from SPIROMICS, we provide new evidence that components of both the lung microbiome and lung metabolome together are associated with clinical endpoints in smokers with or without mild-to-moderate COPD. These findings reflect consideration of the lung microbiome and metabolome from an ecological perspective and emphasize their potential collaborative roles in disease-shaping pathways. Our study moves beyond single -‘omics analytical focus of such data, which predominate in the literature, to shed light on potential microbial-metabolic contributions to COPD pathogenesis. In addition to the new insights related to clinical impairment in milder disease, the findings motivate new hypotheses about the likely bidirectional relationships between metabolites and microbes and their role in lung inflammation.
Few studies of the lung microbiome and metabolome in COPD exist, and to our knowledge, this is the first to pursue an integrative approach to their analysis, which included untargeted and targeted metabolomics data from BAL. There are several novel observations. Specific members of the Prevotella genus (e.g., Otu0003 or Otu0045) associated with better lung function and lower likelihood of COPD. A recent analysis of bronchial brush data (9) from former smokers also noted an overall abundance of Prevotella associated negatively with indicators of COPD severity and positively with expression of genes involved in tight junction promotion. We extend those observations by including current smoking as a covariate and identifying lung metabolites possibly involved. Collectively, our two studies suggest a potential protective or homeostatic role for specific Prevotella. As mentioned, Prevotella-Otu0003 positively correlated with AMP and adenosine, which play key roles in regulating airway surface liquid volume (27). Prevotella-Otu0003 also correlated with MTA and spermine and negatively associated with BAL neutrophils. Although the species identity of Prevotella-Otu0003 is currently unknown, pathways for purine metabolism, methionine salvage, and polyamines have been described in bacteria, including Prevotella (28, 29). It is also possible that metabolites create or reflect a lung environment that promotes Prevotella and other microbes. Interestingly, compounds targeting methionine salvage and polyamines have been investigated in preclinical and clinical studies (30, 31).
In contrast, several lung bacteria and correlated metabolites associated with lower FEV1, COPD status, or higher CAT score and/or chronic bronchitis. This included two Streptococcus members—notably, Otu0005, which we identified as S. pneumoniae from cultured BAL. A recent murine study found that aspiration of oral commensal bacteria activated Th17 responses, and pretreatment with these commensals reduced susceptibility to S. pneumoniae respiratory challenge (32). We extend those findings with human evidence that additional bacteria, together with specific metabolites from different classes, may further shape the inflammatory and metabolic milieu in milder lung disease. This includes other anaerobes or fastidious bacteria (Veillonella, Neisseria, Actinomyces, and Fusobacterium), lipids from several superclasses, polyamines, and other metabolites. Members of these bacterial genera or metabolite classes have been previously implicated in COPD, but in studies that focused on only the microbial or metabolite components alone (11, 12) or assayed from other specimen types (15, 16, 33–35). We note that several of the BAL lipid classes represented in the clinical associations included metabolite pathways that have been associated with COPD phenotype in prior plasma-based metabolomic studies (e.g., glycosphingolipids, lysophospholipids) (33–35), supporting the potential importance of these pathways in the COPD lung environment (33).
The bacteria–metabolite correlations observed could reflect direct or indirect interactions. Bacteria have well-documented mechanisms for sphingolipid production or manipulation and metabolism of amino acids and nucleotides (24, 34–38). Specific bacteria could also trigger lung inflammatory responses that, by altering the metabolic milieu, could, in turn, be reflected in the correlations. We noted with interest that specific members of the Actinobacteria phylum displayed many strongly significant lung metabolite correlations (Actinomycetes and Corynebacterium OTUs). These are representatives of a biologically diverse phylum, inclusive of nonpathogenic species known for their capacity to produce many secondary metabolites (39, 40). Additional studies will be needed to understand potential causal relationships and functional mechanisms by which the identified bacteria modulate the lung metabolic milieu.
Strengths of this study include the multicenter nature of the well-characterized SPIROMICS cohort, the focus on ever-smokers with or without milder stage disease, and the complementary tools used to examine relationships between the paired-omics data and clinical measures. We applied a rigorous analytical workflow to extract microbial and metabolite features in the context of each other, coupled with elastic net regression models that allowed for covariate adjustment. We believe that this approach, together with resampling, greatly increased the probability of identifying only those molecular features most likely related to a given outcome. Mean overall model performances did not differ significantly between the two frameworks, suggesting that the metabolomic features, when examined together with the microbiome were similarly predictive of the outcomes in those groups. We also explored potential interactions between bacteria and metabolites, including the potential for the lung bacterial community to participate in functional gene pathways intersecting with some of the identified metabolites.
This study also has limitations. We did not have full subject overlap across the three datasets—a common challenge. However, our approach allowed for inclusion in each framework of the greatest number of subjects whose clinical characteristics did not differ significantly. Although we split samples into training and testing sets for feature selection and modeling (repeated 100 times for each outcome), there was no opportunity for external validation. Validation studies in independent cohorts would be ideal, although identifying such with matching characteristics and data types are known challenges. The scope of a future study could be to pursue such in a new cohort and consider an integrated analysis using the three types of -omics data simultaneously. We observed consistent classification accuracies between the frameworks for each outcome, but the specific features predictive for some measures differed between frameworks. In some cases, different OTUs from the same bacterial genus associated with the same outcome in either framework. Similar patterns were observed within metabolite classes. Because DIABLO evaluates features in the context of the other paired dataset, different OTUs might be selected because of the difference in compound class coverage between metabolomics datasets. There were class imbalances for some outcomes because of the dichotomization need for DIABLO. Although this was based on accepted clinical thresholds for most outcomes, this may have killed information analytically. Because of the study’s cross-sectional nature, our findings, although statistically predictive within this cohort, remain associative. Last, speciation of some features was not possible because of known limitations of 16S rRNA gene sequences or metabolite databases. Confirmation of specific untargeted metabolites (e.g., those marked “MSI 3” in Table E5) by MS/MS was attempted, but because of sample degradation in the years since the untargeted data were generated, we were unable to assign more confident annotation. Nonetheless, we did affirm several feature–outcome associations observed in our prior analyses of either the lung microbiome or metabolomic data alone that used alternate statistical methods (11, 12).
In summary, the results of this integrative analysis of the lung microbiome and metabolome highlight the ecological context of microbial–metabolic interactions in the lung and their potential collaborative roles in COPD development or progression. Further studies are needed to understand how specific interactions may be targeted for therapeutic or even preventative goals and how clinical interventions potentially affect these systems.
Acknowledgments
Acknowledgments
The authors thank the SPIROMICS participants and participating physicians, investigators, and staff for making this research possible. More information about the study and how to access SPIROMICS data is available at www.spiromics.org. The authors would like to acknowledge the University of North Carolina at Chapel Hill BioSpecimen Processing Facility for sample processing, storage, and sample disbursements (https://bsp.web.unc.edu/).
Members of the SPIROMICS Research Group: The authors would like to acknowledge the following current and former investigators of the SPIROMICS sites and reading centers: Neil E. Alexis, M.D.; Wayne H. Anderson, Ph.D.; Mehrdad Arjomandi, M.D.; Igor Barjaktarevic, M.D., Ph.D.; R. Graham Barr, M.D., Dr.P.H.; Lori A. Bateman, M.Sc.; Surya P. Bhatt, M.D.; Eugene R. Bleecker, M.D.; Richard C. Boucher, M.D.; Russell P. Bowler, M.D., Ph.D.; Stephanie A. Christenson, M.D.; Alejandro P. Comellas, M.D.; Christopher B. Cooper, M.D., Ph.D.; David J. Couper, Ph.D.; Gerard J. Criner, M.D.; Ronald G. Crystal, M.D.; Jeffrey L. Curtis, M.D.; Claire M. Doerschuk, M.D.; Mark T. Dransfield, M.D.; Brad Drummond, M.D.; Christine M. Freeman, Ph.D.; Craig Galban, Ph.D.; MeiLan K. Han, M.D., M.S.; Nadia N. Hansel, M.D., M.P.H.; Annette T. Hastie, Ph.D.; Eric A. Hoffman, Ph.D.; Yvonne J. Huang, M.D.; Robert J. Kaner, M.D.; Richard E. Kanner, M.D.; Eric C. Kleerup, M.D.; Jerry A. Krishnan, M.D., Ph.D.; Lisa M. LaVange, Ph.D.; Stephen C. Lazarus, M.D.; Fernando J. Martinez, M.D., M.S.; Deborah A. Meyers, Ph.D.; Wendy C. Moore, M.D.; John D. Newell, Jr., M.D.; Robert Paine III, M.D.; Laura Paulin, M.D., M.H.S.; Stephen P. Peters, M.D., Ph.D.; Cheryl Pirozzi, M.D.; Nirupama Putcha, M.D., M.H.S.; Elizabeth C. Oelsner, M.D., M.P.H.; Wanda K. O’Neal, Ph.D.; Victor E. Ortega, M.D., Ph.D.; Sanjeev Raman, M.B.B.S., M.D.; Stephen I. Rennard, M.D.; Donald P. Tashkin, M.D.; J. Michael Wells, M.D.; Robert A. Wise, M.D.; and Prescott G. Woodruff, M.D., M.P.H. The project officers from the Lung Division of the NHLBI were Lisa Postow, Ph.D., and Lisa Viviano, B.S.N.
Footnotes
A complete list of SPIROMICS Research Group members may be found before the beginning of the References.
Supported by: contracts from the NHLBI (HHSN268200900013C, HHSN268200900014C, HHSN268200900015C, HHSN268200900016C, HHSN268200900017C, HHSN268200900018C, HHSN268200900019C, and HHSN268200900020C), as well as grants from the NHLBI (U01 HL137880 and U24 HL141762) to SPIROMICS, and supplemented by contributions made through the Foundation for the National Institutes of Health and the COPD Foundation from AstraZeneca/MedImmune; Bayer; Bellerophon Therapeutics; Boehringer-Ingelheim Pharmaceuticals, Inc.; Chiesi Farmaceutici S.p.A.; Forest Research Institute, Inc.; GlaxoSmithKline; Grifols Therapeutics, Inc.; Ikaria, Inc.; Novartis Pharmaceuticals Corporation; Nycomed GmbH; ProterixBio; Regeneron Pharmaceuticals, Inc.; Sanofi; Sunovion; Takeda Pharmaceutical Company; and Theravance Biopharma and Mylan. This work was also supported by NIH grants R01HL121774-S1 (Y.J.H. and G.B.H.), R01AI129958 (Y.J.H.), and R01HL121774 (to G.B.H and P.W.)
Author Contributions: Conception and design: N.R., R.P.B., C.R.E., and Y.J.H. Acquisition of data: C.C.-Q., L.A.B., I.B., R.G.B., A.P.C., C.B.C., D.J.C., C.M.F., M.K.H., R.J.K., W.L., F.J.M., V.E.O., S.P.P., R.P., P.W., J.L.C., G.B.H., R.P.B., C.R.E., N.R., and Y.J.H. Data curation and analysis: S.S.M., C.C.-Q., K.O., J.R.E.-D., L.A.B., G.L., C.B.C., D.J.C., C.M.F., C.R.E., N.R., and Y.J.H. Writing – first draft: S.M.M. and Y.J.H. Review and editing: all authors.
Data Sharing: All data used in this study and accompanying data dictionary can be obtained from the SPIROMICS Genomics and Informatics Coordinating Center (https://www2.cscc.unc.edu/spiromics/contact-gic). The 16S rRNA gene sequence data used in this study were previously submitted to the National Center for Biotechnology Information Sequence Read Archive under PRJNA67315.
This version of the article was corrected on September 15, 2024 (see https://www.atsjournals.org/doi/10.1164/rccm.v210erratum4)
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202110-2241OC on May 10, 2022
Author disclosures are available with the text of this article at www.atsjournals.org.
Contributor Information
for the SPIROMICS Research Group:
Neil E. Alexis, Wayne H. Anderson, Mehrdad Arjomandi, Lori A. Bateman, Surya P. Bhatt, Eugene R. Bleecker, Richard C. Boucher, Stephanie A. Christenson, Gerard J. Criner, Ronald G. Crystal, Claire M. Doerschuk, Mark T. Dransfield, Brad Drummond, Craig Galban, Nadia N. Hansel, Annette T. Hastie, Eric A. Hoffman, Richard E. Kanner, Eric C. Kleerup, Jerry A. Krishnan, Lisa M. LaVange, Stephen C. Lazarus, Deborah A. Meyers, Wendy C. Moore, John D. Newell, Jr., Laura Paulin, Cheryl Pirozzi, Nirupama Putcha, Elizabeth C. Oelsner, Wanda K. O’Neal, Sanjeev Raman, Stephen I. Rennard, Donald P. Tashkin, J. Michael Wells, and Robert A. Wise
References
- 1.Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. GOLD; 2021. [accessed June 10, 2021]. Available from: http://www.goldcopd.org [Google Scholar]
- 2. Woodruff PG, Barr RG, Bleecker E, Christenson SA, Couper D, Curtis JL, et al. SPIROMICS Research Group. Clinical significance of symptoms in smokers with preserved pulmonary function. N Engl J Med . 2016;374:1811–1821. doi: 10.1056/NEJMoa1505971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Martinez FJ, Han MK, Allinson JP, Barr RG, Boucher RC, Calverley PMA, et al. At the root: defining and halting progression of early chronic obstructive pulmonary disease. Am J Respir Crit Care Med . 2018;197:1540–1551. doi: 10.1164/rccm.201710-2028PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pragman AA, Kim HB, Reilly CS, Wendt C, Isaacson RE. The lung microbiome in moderate and severe chronic obstructive pulmonary disease. PLoS One . 2012;7:e47305. doi: 10.1371/journal.pone.0047305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garcia-Nuñez M, Millares L, Pomares X, Ferrari R, Pérez-Brocal V, Gallego M, et al. Severity-related changes of bronchial microbiome in chronic obstructive pulmonary disease. J Clin Microbiol . 2014;52:4217–4223. doi: 10.1128/JCM.01967-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mayhew D, Devos N, Lambert C, Brown JR, Clarke SC, Kim VL, et al. AERIS Study Group. Longitudinal profiling of the lung microbiome in the AERIS study demonstrates repeatability of bacterial and eosinophilic COPD exacerbations. Thorax . 2018;73:422–430. doi: 10.1136/thoraxjnl-2017-210408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Z, Locantore N, Haldar K, Ramsheh MY, Beech AS, Ma W, et al. Inflammatory endotype-associated airway microbiome in COPD clinical stability and exacerbations: a multicohort longitudinal analysis. Am J Respir Crit Care Med . 2020;203:1488–1502. doi: 10.1164/rccm.202009-3448OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grønseth R, Haaland I, Wiker HG, Martinsen EMH, Leiten EO, Husebø G, et al. The Bergen COPD microbiome study (MicroCOPD): rationale, design, and initial experiences Eur Clin Respir J 2014126196. DOI: 10.3402/ecrj.v1.26196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramsheh MY, Haldar K, Esteve-Codina A, Purser LF, Richardson M, Müller-Quernheim J, et al. Lung microbiome composition and bronchial epithelial gene expression in patients with COPD versus healthy individuals: a bacterial 16S rRNA gene sequencing and host transcriptomic analysis Lancet Microbe 20212e300–e310.. (Internet). [DOI] [PubMed] [Google Scholar]
- 10. Wells JM, Arenberg DA, Barjaktarevic I, Bhatt SP, Bowler RP, Christenson SA, et al. Safety and tolerability of comprehensive research bronchoscopy in chronic obstructive pulmonary disease. Results from the SPIROMICS bronchoscopy substudy. Ann Am Thorac Soc . 2019;16:439–446. doi: 10.1513/AnnalsATS.201807-441OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Halper-Stromberg E, Gillenwater L, Cruickshank-Quinn C, O’Neal WK, Reisdorph N, Petrache I, et al. Bronchoalveolar lavage fluid from COPD patients reveals more compounds associated with disease than matched plasma. Metabolites . 2019;9:157. doi: 10.3390/metabo9080157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Opron K, Begley LA, Erb-Downward JR, Freeman C, Madapoosi S, Alexis NE, et al. Lung microbiota associations with clinical features of COPD in the SPIROMICS cohort. NPJ Biofilms Microbiomes . 2021;7:14. doi: 10.1038/s41522-021-00185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Esther CR, Jr, Coakley RD, Henderson AG, Zhou Y-H, Wright FA, Boucher RC. Metabolomic evaluation of neutrophilic airway inflammation in cystic fibrosis. Chest . 2015;148:507–515. doi: 10.1378/chest.14-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Esther CR, Jr, Hill DB, Button B, Shi S, Jania C, Duncan EA, et al. Sialic acid-to-urea ratio as a measure of airway surface hydration. Am J Physiol Lung Cell Mol Physiol . 2017;312:L398–L404. doi: 10.1152/ajplung.00398.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Esther CR, Jr, Lazaar AL, Bordonali E, Qaqish B, Boucher RC. Elevated airway purines in COPD. Chest . 2011;140:954–960. doi: 10.1378/chest.10-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Esther CR, Jr, O’Neal WK, Anderson WH, Kesimer M, Ceppe A, Doerschuk CM, et al. Identification of sputum biomarkers predictive of pulmonary exacerbations in chronic obstructive pulmonary disease. Chest . 2022;161:1239–1249. doi: 10.1016/j.chest.2021.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang YJ, Madapoosi S, Cruickshank-Quinn C, Opron K, Erb-Downward J, Begley L, et al. Integrated analysis of the lung microbiome and metabolome reveals bacterial and metabolite features co-associated with clinical outcomes in COPD. Am J Respir Crit Care Med . 2021;203:A1393. [Google Scholar]
- 18. Couper D, LaVange LM, Han M, Barr RG, Bleecker E, Hoffman EA, et al. SPIROMICS Research Group. Design of the Subpopulations and Intermediate Outcomes in COPD Study (SPIROMICS) Thorax . 2014;69:491–494. doi: 10.1136/thoraxjnl-2013-203897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fortis S, Comellas A, Make BJ, Hersh CP, Bodduluri S, Georgopoulos D, et al. COPDGene Investigators–Core Units; COPDGene Investigators–Clinical Centers. Combined forced expiratory volume in 1 second and forced vital capacity bronchodilator response, exacerbations, and mortality in chronic obstructive pulmonary disease. Ann Am Thorac Soc . 2019;16:826–835. doi: 10.1513/AnnalsATS.201809-601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim V, Zhao H, Regan E, Han MK, Make BJ, Crapo JD, et al. COPDGene Investigators. The St. George’s Respiratory Questionnaire definition of chronic bronchitis may be a better predictor of COPD exacerbations compared with the classic definition. Chest . 2019;156:685–695. doi: 10.1016/j.chest.2019.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davis NM, Proctor DM, Holmes SP, Relman DA, Callahan BJ. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome . 2018;6:226. doi: 10.1186/s40168-018-0605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singh A, Shannon CP, Gautier B, Rohart F, Vacher M, Tebbutt SJ, et al. DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics . 2019;35:3055–3062. doi: 10.1093/bioinformatics/bty1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Clauset A, Newman MEJ, Moore C. Finding community structure in very large networks. Phys Rev E Stat Nonlin Soft Matter Phys . 2004;70:066111. doi: 10.1103/PhysRevE.70.066111. [DOI] [PubMed] [Google Scholar]
- 24. Iwai S, Weinmaier T, Schmidt BL, Albertson DG, Poloso NJ, Dabbagh K, et al. Piphillin: improved prediction of metagenomic content by direct inference from human microbiomes. PLoS One . 2016;11:e0166104. doi: 10.1371/journal.pone.0166104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol . 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Freeman CM, Crudgington S, Stolberg VR, Brown JP, Sonstein J, Alexis NE, et al. Design of a multi-center immunophenotyping analysis of peripheral blood, sputum and bronchoalveolar lavage fluid in the Subpopulations and Intermediate Outcome Measures in COPD Study (SPIROMICS) J Transl Med . 2015;13:19. doi: 10.1186/s12967-014-0374-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lazarowski ER, Tarran R, Grubb BR, van Heusden CA, Okada S, Boucher RC. Nucleotide release provides a mechanism for airway surface liquid homeostasis. J Biol Chem . 2004;279:36855–36864. doi: 10.1074/jbc.M405367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banerji R, Kanojiya P, Patil A, Saroj SD. Polyamines in the virulence of bacterial pathogens of respiratory tract. Mol Oral Microbiol . 2021;36:1–11. doi: 10.1111/omi.12315. [DOI] [PubMed] [Google Scholar]
- 29. North JA, Miller AR, Wildenthal JA, Young SJ, Tabita FR. Microbial pathway for anaerobic 5′-methylthioadenosine metabolism coupled to ethylene formation. Proc Natl Acad Sci USA . 2017;114:E10455–E10464. doi: 10.1073/pnas.1711625114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, Dong X, et al. Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of 5′-methylthioadenosine phosphorylase. J Biol Chem . 2011;286:4902–4911. doi: 10.1074/jbc.M110.198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. LoGiudice N, Le L, Abuan I, Leizorek Y, Roberts SC. Alpha-difluoromethylornithine, an irreversible inhibitor of polyamine biosynthesis, as a therapeutic strategy against hyperproliferative and infectious diseases. Med Sci (Basel) . 2018;6:E12. doi: 10.3390/medsci6010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wu BG, Sulaiman I, Tsay JJ, Perez L, Franca B, Li Y, et al. Episodic aspiration with oral commensals induces a MyD88-dependent, pulmonary T-helper cell type 17 response that mitigates susceptibility to Streptococcus pneumoniae. Am J Respir Crit Care Med . 2021;203:1099–1111. doi: 10.1164/rccm.202005-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ran N, Pang Z, Gu Y, Pan H, Zuo X, Guan X, et al. An updated overview of metabolomic profile changes in chronic obstructive pulmonary disease. Metabolites . 2019;9:E111. doi: 10.3390/metabo9060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowler RP, Jacobson S, Cruickshank C, Hughes GJ, Siska C, Ory DS, et al. Plasma sphingolipids associated with chronic obstructive pulmonary disease phenotypes. Am J Respir Crit Care Med . 2015;191:275–284. doi: 10.1164/rccm.201410-1771OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cruickshank-Quinn CI, Jacobson S, Hughes G, Powell RL, Petrache I, Kechris K, et al. Metabolomics and transcriptomics pathway approach reveals outcome-specific perturbations in COPD. Sci Rep . 2018;8:17132. doi: 10.1038/s41598-018-35372-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heaver SL, Johnson EL, Ley RE. Sphingolipids in host-microbial interactions. Curr Opin Microbiol . 2018;43:92–99. doi: 10.1016/j.mib.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 37. Rolando M, Buchrieser C. A comprehensive review on the manipulation of the sphingolipid pathway by pathogenic bacteria. Front Cell Dev Biol . 2019;7:168. doi: 10.3389/fcell.2019.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kilstrup M, Hammer K, Ruhdal Jensen P, Martinussen J. Nucleotide metabolism and its control in lactic acid bacteria. FEMS Microbiol Rev . 2005;29:555–590. doi: 10.1016/j.femsre.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 39. Al-Shaibani MM, Radin Mohamed RMS, Sidik NM, Enshasy HAE, Al-Gheethi A, Noman E, et al. Biodiversity of secondary metabolites compounds isolated from phylum actinobacteria and its therapeutic applications. Molecules . 2021;26:4504. doi: 10.3390/molecules26154504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oliveira A, Oliveira LC, Aburjaile F, Benevides L, Tiwari S, Jamal SB, et al. Insight of genus Corynebacterium: ascertaining the role of pathogenic and non-pathogenic species. Front Microbiol . 2017;8:1937. doi: 10.3389/fmicb.2017.01937. [DOI] [PMC free article] [PubMed] [Google Scholar]