

Abstract
In vivo expansion of genetically modified T cells in cancer patients following adoptive transfer has been linked to both anti-tumor activity and T cell-mediated toxicities. The development of digital PCR has improved the accuracy in quantifying the in vivo status of adoptively infused T cells compared to qPCR or flow cytometry. Here, we developed and evaluated the feasibility and performance of nanoplate-based digital PCR (ndPCR) to quantify adoptively infused T cells engineered with a T cell receptor (TCR) that recognizes a human endogenous retrovirus type E (HERV-E) antigen. Analysis of blood samples collected from patients with metastatic kidney cancer following the infusion of HERV-E TCR-transduced T cells established the limit of detection of ndPCR to be 0.3 transgene copies/μL of reaction. The lower limit of quantification for ndPCR was one engineered T cell per 10,000 PBMCs, which outperformed both qPCR and flow cytometry by 1 log. High inter-test and test-retest reliability was confirmed by analyzing blood samples collected from multiple patients. In conclusion, we demonstrated the feasibility of ndPCR for detecting and monitoring the fate of TCR-engineered T cells in adoptive cell therapy.
Keywords: digital PCR, vector copy number, TCR-transduced T cells, adoptive transfer, cell therapy
Graphical abstract
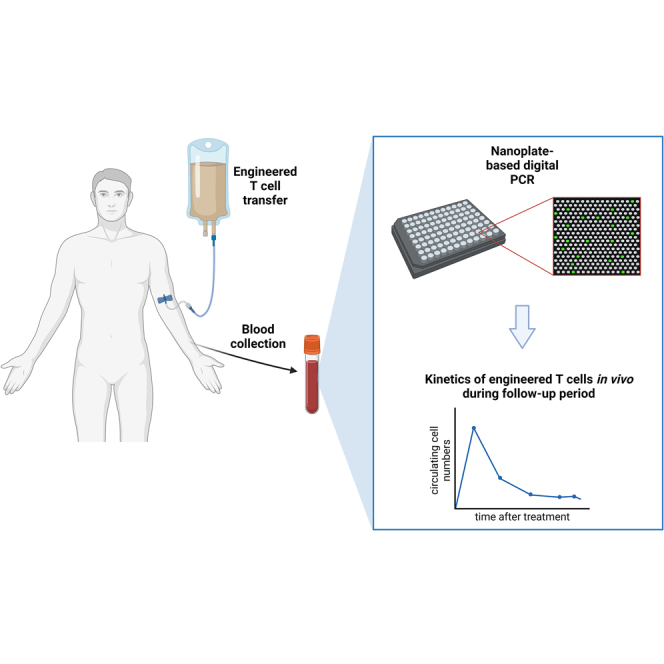
Tracking the kinetics of adoptively transferred cells in vivo has significant clinical implications. Barisic and colleagues demonstrate the feasibility of nanoplate-based digital PCR for detecting and monitoring the fate of TCR-engineered T cells in adoptive cell therapy.
Introduction
Adoptive cell therapy using genetically modified autologous or allogeneic immune cells is an emerging mode of treatment to enhance anti-tumor immunity against malignant diseases. The cells most commonly used for adoptive transfer are chimeric antigen receptor (CAR) T cells and T cell receptor (TCR)-engineered T cells.1 Their engraftment and in vivo expansion after infusion have been associated with response to therapy and the occurrence of T cell-mediated toxicities.2,3,4 Therefore, monitoring the kinetics of infused cells in vivo may have significant clinical implications.
Many investigators have shown that, following the phases of multi-log expansion and rapid contraction, CAR or TCR-engineered T cells enter a phase of gradual decline. Following their infusion, many patients will maintain low levels of these cells in the long term, at times even below the limit of accurate quantification.5,6,7,8 Given their wide availability, flow cytometry and qPCR are well-established modalities utilized for the longitudinal monitoring of engineered T cells.6 Both methods, however, have limitations in detecting and accurately quantifying low numbers of cells that are present in the circulation following adoptive transfer.
Digital PCR (dPCR) has the potential to overcome some of these limitations, as it measures absolute copy numbers instead of relative quantification, resulting in improved sensitivity compared to qPCR.9,10,11 Specifically, dPCR methods quantify target sequences in volumetrically defined partitions and apply Poisson correction to determine the absolute quantification. Partitioning the sample for dPCR can be achieved by droplet dPCR (ddPCR), which generates individual droplets, or nanoplate dPCR (ndPCR), in which samples are distributed across a chamber.12 Even though partitioning strategies may differ, the general principle upon which all dPCR systems are based is the same.
Other investigators have already reported that absolute quantification of vector copy number (VCN) in engineered T cells by ddPCR is more sensitive and reliable than qPCR13 and can be used to monitor and quantitate CAR T cells in vivo.11,14,15,16,17 However, at present, no preclinical or clinical data exist that report on the use and precision of ndPCR to monitor and quantitate in vivo circulating TCR-engineered T cells.
Our group has recently completed a phase I trial evaluating TCR-transduced T cells targeting an antigen derived from a human endogenous retrovirus type E (HERV-E) that has tumor-restricted expression in clear cell renal cell carcinoma (ClinicalTrials.gov: NCT03354390).18 Here, we describe the development, optimization, and clinical application of a highly sensitive ndPCR method to detect and precisely quantitate HERV-E TCR-transduced T cells (HERV-E T cells) in the circulation.
Results
Custom assays for the detection of the HERV-E TCR construct confirmed specificity by ndPCR
Specific detection of the HERV-E TCR construct was accomplished by designing four different primers/probe sets, each spanning the junction regions unique to the HERV-E TCR retroviral vector. Assay TCR1 was designed to detect the junction region between the P2A site and TCRβ and assays TCRs 2–4 to amplify the junction between the T2A site and truncated CD34 (CD34t) (Figure 1; Table 1). To identify the optimal primers/probes to use in the ndPCR assay, we measured the copy numbers of HERV-E TCR and reference genes using highly purified TCR-transduced T cells from a male donor as a positive control and PBMCs from a healthy female donor as a negative control. The data are presented in Figure 2A. We found that the four primer/probe sets for detecting HERV-E TCR yielded comparable results and could reliably detect the HERV-E TCR insert, with a positive signal observed only in the sample containing pure HERV-E T cells and no signal detected in the sample containing non-manipulated PBMCs. To test intra-run conformity, all dPCRs were performed in triplicates, with all four assays demonstrating excellent reproducibility. The feasibility of using the RPPH, TERT, and USP9Y reference genes was also confirmed. RPPH and TERT showed comparable copy numbers, as both are present in two copies per genome. In contrast, USP9Y, which has one copy per male genome (Y chromosome locus), had copy numbers approximately one-half of the RPPH or TERT copies.
Figure 1.

HERV-E TCR vector map with aligned primer/probe sets used in this study
Table 1.
Primer and probe sequences used in this study
| Genes of interest | Primers and probes (5′-3′) |
|---|---|
| Ref_RPPH | F: AGTGAGTTCAATGGCTGAGG R: GGCGGAGGAGAGTAGTCT P:/5HEX/TTGGGTTAT/ZEN/GAGGTCCCCTGCG/31ABkFQ/ |
| Ref_TERT | F: GGGTTGGCTGTGTTCCG R: GACATAAAAGAAAGACCTGAGCA P for duplex assays:/5HEX/AAGTTCCTG/ZEN/CACTGGCTGATGGT/31ABkFQ/ P for quadruplex assays:/5Cy5/AAGTTCCTG/ZEN/CACTGGCTGATGGT/31ABkFQ/ |
| Ref_USP9Y | F: CAAGTATGACAGCCATCACTCA R: GTTGGAAGAGATGCTGAGACT P for duplex assays:/5HEX/CAGTAGGAG/ZEN/GGAACGACAGCCAG/3IABkFQ/ P for quadruplex assays:/5ROX/CAGTAGGAG/ZEN/GGAACGACAGCCAG/3IABkFQ/ |
| TCR set 1 | F: CTGGGTAACTTCAGTGTCTATG R: CCACGAACTTCTCTCTGTTAAA P:/56-FAM/CAAGCAGGA/ZEN/GACGTGGAGGAGAA/31ABkFQ/ |
| TCR set 2 | F: GCAAACTCAGCAAGCAAAG R: TGATGGCAATGGTCAAGAG P:/56-FAM/TGCTAACAT/ZEN/GCGGTGATGTCGAAG/31ABkFQ/ |
| TCR set 3 | F: GCAAACTCAGCAAGCAAAG R: GGATTTCGAATTCGGCTCA P:/56-FAM/TGCTAACAT/ZEN/GCGGTGATGTCGAAG/31ABkFQ/ |
| TCR set 4 | F: CCGTTGTTGTCAAGACTCATG R: TTCGAATTCGGCTCAGGC P:/56-FAM/CTGCTAACA/ZEN/TGCGGTGATGTCGAAG/31ABkFQ/ |
Figure 2.

Specificity of ndPCR assays for HERV-E TCR and linearity across a range of DNA inputs
(A) ndPCR performance of four different primer/probe sets to detect HERV-E TCR (TCR 1, 2, 3, and 4) using a purified population of HERV-E T cells from a male donor (93.1% transduction efficiency; VCN per transduced cell, 2.94) and PBMCs from a female donor as a negative control. All assays were tested in technical triplicates. Error bars represent mean ± SD. (B) Serial (half-log) dilutions of gDNA derived from purified HERV-E T cells were used to test the LOD of all four assays detecting the HERV-E TCR construct. All four assays exhibited linear and reproducible copy number quantifications of the vector insert and the RPPH reference gene across a range of DNA concentrations with high concordance. All assays were tested in technical triplicates.
ndPCR produced linear and reproducible results across a range of DNA inputs
To assess the sensitivity and limits of detection of the four HERV-E TCR assays, we used serial (half-log) dilutions of gDNA derived from a HERV-E T cell product (93.1% transduction efficiency; VCN per transduced cell, 2.94) and performed duplex ndPCR using the reference RPPH gene. We diluted a starting gDNA input of 80 ng to a maximum dilution of 0.25 pg. All four TCR assay reactions were performed in technical triplicates. All four TCR assays showed a strong correlation between the two measurements (TCR and RPPH) (Figure 2B). Further, all four assays consistently showed a positive signal in the dilution containing 8 pg of gDNA in all triplicate reactions. This amount of gDNA would be expected to contain a mean of 2.4 copies of a diploid RPPH gene and a mean of 3.5 copies of a transgene used in this experiment (with VCN 2.94). According to the Poisson distribution, a mean of 3.5 copies of the transgene per tested volume unit would result in detecting at least one copy 97.15% of the time. This translates to a limit of detection (LOD) of approximately 0.3 copies/μL of reaction volume. Furthermore, when averaging triplicates of the same assay into one value, the lowest dilution where all four assays showed a positive signal was the one containing 2.5 pg of gDNA.
These data also established the fidelity of the ndPCR system, as we observed that a half-log (3.16-fold) reduction of the amount of DNA from HERV-E T cells in a sample led to a half-log decrease of the observed average copy numbers of the vector construct and the reference sequence. Even when the input vector copies were low, our data established the ndPCR assays to be highly specific and sensitive in detecting HERV-E TCR vector constructs. Because they had the best dilutional linearity, we selected primer/probe sets 1 and 4 for subsequent experiments to evaluate the dynamic range of dPCR.
ndPCR outperforms qPCR and flow cytometry in detecting and quantifying transduced T cells
We next assessed the dynamic range of ndPCR creating in vitro conditions that would mimic those expected to be observed in clinical samples collected from patients receiving TCR-transduced T cells. HERV-E T cells with 93.1% transduction efficiency and VCN per transduced cell of 2.94 were serially diluted (10-fold) with PBMCs to a maximum dilution of 1:100,000. HERV-E T cells were quantified by ndPCR using TCR assays 1 and 4 (Figure 3A) with 100 ng of gDNA input. These data established the lower limit of quantification (LLOQ) to be at least one HERV-E T cell in a background of 10,000 PBMCs. 10-fold dilutions of HERV-E T cells in PBMCs showed that VCNs were also reduced by 10-fold, corroborating that assay linearity remained stable, even in the presence of additional gDNA derived from PBMCs that lacked the vector insert.
Figure 3.

Analysis of the ndPCR dynamic range compared with qPCR and flow cytometry
(A) HERV-E T cells were serially diluted 10-fold with PBMCs, and the fractional proportion of HERV-E T cells was determined by ndPCR using TCR1 and TCR4 assays, each in duplex reactions with three reference genes. The data points shown are average of technical triplicates. (B) Aliquots of the same diluted samples were used to quantify HERV-E T cells by flow cytometry. The representative plots were gated on viable CD3+ cells; HERV-E T cells are defined as CD8+ CD34+ cells. (C) Comparison of the dynamic range of three different methods for detecting and quantifying TCR-engineered T cells in blood samples collected from patients who had received an infusion of HERV-E T cells: flow cytometry, qPCR, and ndPCR. The data points shown are averages of technical triplicates.
The same sample dilutions from the prior experiments were then used to evaluate the performance of ndPCR in quantifying HERV-E T cells compared to qPCR and flow cytometry (Figure 3B). The TCR1 assay for qPCR testing was used, as it showed the best linearity in ndPCR. We found that ndPCR was superior in detecting and quantifying HERV-E T cells in paucicellular conditions. All three duplex assays (TCR with each of the three reference genes) in dPCR were consistently positive and on a linear slope in the sample containing 1:10,000 HERV-E T cells in PBMCs. In contrast, the same three assays performed in qPCR showed that only the TCR+TERT assay performed at the same level as dPCR, being within the linear range at the 1:10,000 dilution, while the other two assays detected the HERV-E construct only up to the 1:1,000 dilution. Considering the performance within linear range, the LLOQ of the ndPCR assay outperformed both the qPCR assay and flow cytometry by one log (Figure 3C).
Application of ndPCR to quantify circulating TCR-engineered T cells in patient samples
The findings presented above suggest that ndPCR could potentially be used as a highly sensitivity method to accurately track the kinetics of TCR-modified cells following adoptive infusion in patients. We next studied the reproducibility of the TCR1 assay to detect and quantify circulating HERV-E T cells in PBMCs from patients who received escalating doses of HERV-E T cells in a phase I clinical trial. The transduction efficiency and VCNs of the products tested are shown in Table S1. As shown in Figure 4A, this assay reproducibly quantified, at multiple different time points, TCR-engineered T cells detected in the circulation of 3 patients who received HERV-E T cells with minimal inter-assay variability (interclass coefficient [ICC] = 0.964, 95% confidence interval [CI] [0.923–0.985]). Further, the quantification precision of the assay increased when the ndPCR analysis was changed to quadruplex format, in which the TCR construct and three reference genes were assessed simultaneously in the same reaction well (ICC = 0.999, 95% CI [0.997–1]) (Figure 4B). Last, the test-retest reliability between three independent replicates yielded an ICC = 0.999, 95% CI (0.997–0.999), indicating high test-retest precision. In summary, these data show that ndPCR can be used to accurately quantitate and longitudinally monitor TCR-engineered T cells in patient samples.
Figure 4.

The kinetics of infused HERV-E T cells in patient samples
The graphs show the fractional abundance of HERV-E T cells (% of PBMCs) at multiple time points post infusion. (A) The data show high concordance of the 3 assays using three different reference genes (interclass coefficient [ICC] = 0.964, 95% CI [0.923–0.985]). Patient A11-HERVE-10 did not reach the 30 day post-treatment time point. The data points shown are averages of technical triplicates. (B) The quantification precision of the assay increased between calculations when the ndPCR analysis was changed to the quadruplex format, where the TCR construct and three reference genes were assessed simultaneously in the same reaction well (ICC = 0.999, 95% CI [0.997–1]). The data points shown are averages of technical triplicates.
Discussion
For decades, flow cytometry and quantitative PCR have been used as the gold standard for detecting and quantifying engineered T cells in patient samples after adoptive transfer.19,20 Flow cytometry assays can also be used to assess the phenotypic and functional properties of adoptively transferred T cells. However, flow cytometry-based analysis in this context requires the existence of a suitable marker on engineered T cells and the availability of a larger sample input than PCR. Further, high background noise and variabilities in standardization between laboratories hinder flow-based assays’ quantitative sensitivity and ability to detect relatively small changes in cell frequencies.19 The main downside of qPCR is its susceptibility to inhibitors, requiring laborious assay development, as well as its limited resolution for fine quantitative discrimination and limited sensitivity for detecting rare targets. Furthermore, qPCR relies on reference samples to generate standard curves for indirect quantification of targets.
dPCR has improved accuracy and precision compared to qPCR. The sample partitioning and independent assaying of each partition reduce the impact of pipetting errors, minimize the susceptibility to inhibitors, and enable absolute and direct target quantification.21 This translates into the ability to detect rare targets in complex samples with a wider dynamic range. Other groups have exploited higher sensitivity of dPCR in trials evaluating micro-chimerism,22 minimal-residual disease detection in hematologic malignancies,23 and liquid biopsy.24 In vivo monitoring of engineered T cells by dPCR is an active area of research.8,13
The data presented in this analysis establish ndPCR as a novel dPCR-based approach to quantitate the status of TCR-transduced T cells both in preclinical models as well as in patients. For clinical use, this approach appears to be particularly useful in patients undergoing longitudinal monitoring, when the circulating numbers of gene-modified cells are low.
Using 4 custom designed primer/probe assays, we found that the LLOQ of this assay was extremely sensitive, with an LOD of 8 pg of gDNA or 0.3 copies/μL of ndPCR reaction, which translated into the ability to detect one in 10,000 TCR-transduced HERV-E T cells in a background of PBMCs. Importantly, our data establish that this ndPCR-based approach had a 1-log wider linear range for quantifying the vector construct in sequential samples collected from patients who received HERV-E T cells compared to both a flow cytometry- and qPCR-based analysis. It is important to acknowledge, however, that PCR and flow cytometry results may be discordant because not all copies detectable by PCR will necessarily result in equimolar surface TCR expression, which likely further limits the linear dynamic range of flow cytometry compared to PCR. Taken altogether, these data establish the feasibility, ease of use, and quantitative precision of ndPCR for the in vivo monitoring of adoptively infused TCR-modified T cells.
To ensure reliability in quantification, we utilized a panel of three different reference genes (RPPH, TERT, and USP9Y) that exhibited comparable results in quantifying HERV-E transgene copies with high inter-test reliability. The inter-test reliability was further increased by using the multichannel capabilities of ndPCR instruments and by designing a quadruplex assay that measured all three reference genes and the HERV-E TCR in the same reaction. This combined approach created a highly sensitive and precise method to rapidly quantify gDNA that required less sample input and reduced the need for reagents, reducing costs.
The role of different forms of dPCR for monitoring engineered T cells in vivo is an active area of research. Very recent published data reported that the performance of ddPCR for measuring VCN in CAR T cell products is comparable to ndPCR,25 with ddPCR being reported to have higher test-retest reliability than ndPCR, while ndPCR showed better linearity across a range of DNA inputs. Therefore, the decision to use ddPCR or ndPCR for adoptive cell therapy monitoring will ultimately depend on individual validation results and test performance in clinical trials as well as multiple other real-world factors, including availability of in-house instrumentation, setup, and running costs and preferences of researchers.
It is important to consider that the sensitivity to detect engineered T cells in patient samples by PCR is a function of the mean VCN per transduced cell and the amount of the input gDNA. If the VCN per infused cell is low, then the sensitivity to detect cellular transgenes by PCR will decrease or will require larger gDNA sample input to maintain the same level of sensitivity. Although using nanoplates with more partitions could potentially increase the sensitivity of ndPCR, this approach would require a larger sample input. It is also important to consider that, despite its improved linearity, ndPCR-based analysis can only be used to quantitate the in vivo kinetics of genetically modified and adoptively infused T cells and cannot provide in vivo phenotypic and functional data on cell populations observed using a flow cytometry-based analysis.
Another limitation of our method is the assumption that the mean VCN per transduced cell does not change in vivo over time. Other groups have reported that cell engraftment, expansion fitness, and in vivo persistence may not be the same for all infused cells. This likely explains the apparent lack of clear correlation between the infused cell dose and in vivo expansion kinetics post infusion. There are limited data showing whether VCN remains stable over time in vivo. Efforts to generate T cell products with a more defined and standardized composition will prove beneficial to tracking the engraftment results.26
Last, our method is limited to tracking engineered cells in blood. Tracking their biodistribution in other organs necessitates alternative approaches, such as whole-body imaging methods.27
In conclusion, we have demonstrated that ndPCR can be used as a sensitive method to precisely quantify HERV-E TCR-engineered T cells in patient samples. The linear range of detecting HERV-E T cells was 1 log lower with ndPCR than with qPCR and flow cytometry. This sets a new benchmark for absolute quantification of transgenes and supports the use of ndPCR as a feasible analytical approach for the quantification of TCR-engineered T cells, particularly when the number of circulating engineered T cells is small. Taken together, our findings justify the application of ndPCR for the precise monitoring of engineered T cells in other clinical trials.
Materials and methods
Samples
For assay establishment and evaluation, TCR-transduced T cells targeting HERV-E antigen were produced. PBMCs were isolated from de-identified buffy coats of healthy donors procured at the NIH Department of Transfusion Medicine by Ficoll-Paque density gradient centrifugation. T cells were isolated using the Pan T Cell Isolation Kit (Miltenyi Biotec), cultured in T cell medium (TexMacs medium, [Miltenyi] supplemented with 3% heat-inactivated human AB serum [Sigma-Aldrich], 1% penicillin-streptomycin-glutamine [Gibco], rhIL-7 [10 ng/mL, PeproTech], and rhIL-15 [5 ng/mL, PeproTech]) and activated using ImmunoCult Human CD3/CD28/CD2 T Cell Activator (STEMCELL Technologies). Three days post activation, CD4+ T cells were depleted using CD4 MicroBeads (Miltenyi). The next day (4 days post activation), retroviral spinoculation was performed with a retrovirus encoding the HERV-E TCR and CD34t as a selection marker (Figure 1) at an MOI of 4, as described elsewhere.28 On day 7 post transduction, TCR-transduced T cells were enriched using CD34 MicroBeads (Miltenyi). Enriched cells were expanded until desired cell numbers were reached, aliquoted, and frozen. PBMCs from healthy donors were used as a negative control.
Patients and patient material
Peripheral blood samples were collected at baseline and at pre-planned sequential time points post cell infusion from three patients treated with TCR-transduced HERV-E T cells (ClinicalTrial.gov: NCT03354390). These samples were then processed using Ficoll-Paque density centrifugation to obtain the PBMC fraction. The PBMCs were then frozen and used for DNA isolation as described below.
The clinical trial protocol was approved by the institutional review board at each study site and was conducted in accordance with the International Conference on Harmonization Good Clinical Practice, the Declaration of Helsinki, and local regulations on the conduct of clinical research. All patients provided written informed consent before participating in the study.
DNA isolation
Genomic DNA was isolated from samples using the PureLink Mini DNA Isolation Kit (Thermo Fisher Scientific, USA) according to the manufacturer-recommended protocol, aliquoted, and stored at −20°C until use. The concentration of DNA was determined spectrophotometrically using NanoDrop 1000 (Thermo Fisher Scientific).
Design of PCR primers and probes
Custom primers and probes specific for the junction regions between the P2A site and TCRβ or T2A site and CD34t within the HERV-E TCR retroviral vector were designed using NCBI Primer-BLAST and synthesized by Integrated DNA Technologies (IDT) (Figure 1). Reference copy number assays (RPPH, TERT, and USP9Y) were purchased from IDT. The sequences of all primers and probes used in this study are listed in Table 1.
ndPCR
The Qiacuity Probe PCR kit (QIAGEN) was used to set up the reactions per the manufacturer’s protocol. The sample input for ndPCR reactions was 100 ng of gDNA. The restriction enzyme used to fragment gDNA before partitioning was XBaI (Anza 12 XBaI, Thermo Fisher Scientific) per the manufacturer’s recommendations. dPCR was performed using the Qiacuity One instrument (QIAGEN) and Qiacuity 96-well nanoplates with 8.5k partitions. DNA amplification was carried out using the following cycling parameters: initial heat activation at 95°C for 2 min, followed by 40 cycles of denaturation (95°C, 15 s) and combined annealing/extension (60°C, 30 s). We used default imaging parameters for each dye channel. Data analysis was done using the Qiacuity Software Suite (v.2.1).
The reference genes RPPH and TERT are both present in 2 copies per diploid genome, and USP9Y is present in one copy per male diploid genome (located on the Y chromosome). Assays were designed as either duplex PCRs (TCR + one of the reference genes) or quadruplex assays (TCR + all 3 reference genes). All samples were assessed in triplicates.
qPCR
The DNA concentrations, samples, master mix, primers and probes, and cycling parameters used in qPCR were identical to those used in ndPCR. The 10-fold serial dilutions of gDNA extracted from HERV-E T cells were used to generate a standard curve to estimate PCR efficiency and to quantify the signal intensities. All samples were assessed in triplicates. The qPCR was done using the LightCycler96 real-time qPCR system (Roche Diagnostics).
Flow cytometry
The cells were analyzed using the following antibodies: anti-CD3 (clone UCHT1, BUV395, BD Biosciences), anti-CD4 (clone OKT4, Pacific Blue, BioLegend), anti-CD8 (clone SK1, fluorescein isothiocyanate, BioLegend), anti-CD34 (clone 581, APC, BioLegend). The viability dye used was Live/Dead Yellow (Thermo Fisher Scientific). Samples were acquired using the LSR Fortessa (BD Biosciences) instrument, and data were analyzed using the BD Biosciences FACSDiva and FlowJo (v.10.8.1) packages.
Statistical analysis
To measure the inter-assay and test-retest reliabilities of the developed assays, we used the ICC. For inter-assay reliability, we tested the agreement between the independent measurements yielded by three different reference genes for each clinical sample. For test-retest reliability, we evaluated the consistency of independent triplicate measurements for each clinical sample time point. An absolute-agreement two-way model was used to calculate the ICC and 95% CI.
Algorithm for quantifying circulating transduced T cells in clinical samples
To calculate the fraction of circulating transduced cells in patient samples, we used previously published algorithms8,14 with certain modifications.
The mean VCN of the bulk infusion product was calculated per the following formula:
The correction of the mean VCN of the bulk infusion product for its purity (transduction efficiency) to yield the mean VCN per transduced cell was calculated per the following formula:
The purity of the product was determined by flow cytometry using the CD34t selection marker (i.e., % CD34+ cells).
The same reference and TCR assays were used in patient samples after infusion to determine the absolute copy numbers of the TCR and reference genes. Reference gene copy number indicates the total number of genome equivalents (i.e., cells) in the sample:
The number of transduced cells in the sample is then calculated using the following formula:
The percentage of transduced cells within PBMCs was calculated in the following manner:
Data and code availability
Data are available on reasonable request. All data relevant to the study are included in the article.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, the Dean R. O’Neill Renal Cell Cancer Research Fund, the Commissioned Corps of the United States Public Health Service, and T-Cure BioScience. The graphical abstract and Figure 1 were created with BioRender.
Author contributions
S.B. conceived the study, designed and conducted the experiments, analyzed the data, created figures, and wrote the paper. E.C. conceived the study, conducted the experiments, and wrote the paper. R.N., L.C., A.P., and R.N.R. gave advice and wrote the paper. X.T. performed the statistical analysis. G.M.S. and M.I.N. conducted the experiments, gave advice, and reviewed the paper. R.W.C. initiated the hypothesis, provided financial support, designed the experiments, gave advice, and wrote the paper.
Declaration of interests
The authors (E.C., M.I.N., and R.W.C.) declare a filed patent, WO2018006054A1, licensed by T-Cure BioScience.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2024.101324.
Supplemental information
References
- 1.Barisic S., Childs R.W. Graft-Versus-Solid-Tumor Effect: From Hematopoietic Stem Cell Transplantation to Adoptive Cell Therapies. Stem Cell. 2022;40:556–563. doi: 10.1093/stmcls/sxac021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y., et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/nejmoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster S.J., Bishop M.R., Tam C.S., Waller E.K., Borchmann P., McGuirk J.P., Jäger U., Jaglowski S., Andreadis C., Westin J.R., et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019;380:45–56. doi: 10.1056/nejmoa1804980. [DOI] [PubMed] [Google Scholar]
- 4.Scholler J., Brady T.L., Binder-Scholl G., Hwang W.T., Plesa G., Hege K.M., Vogel A.N., Kalos M., Riley J.L., Deeks S.G., et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore T., Wagner C.R., Scurti G.M., Hutchens K.A., Godellas C., Clark A.L., Kolawole E.M., Hellman L.M., Singh N.K., Huyke F.A., et al. Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunol. Immunother. 2018;67:311–325. doi: 10.1007/s00262-017-2073-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qi C., Gong J., Li J., Liu D., Qin Y., Ge S., Zhang M., Peng Z., Zhou J., Cao Y., et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat. Med. 2022;28:1189–1198. doi: 10.1038/s41591-022-01800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melenhorst J.J., Chen G.M., Wang M., Porter D.L., Chen C., Collins M.A., Gao P., Bandyopadhyay S., Sun H., Zhao Z., et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature. 2022;602:503–509. doi: 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mika T., Maghnouj A., Klein-Scory S., Ladigan-Badura S., Baraniskin A., Thomson J., Hasenkamp J., Hahn S.A., Wulf G., Schroers R. Digital-Droplet PCR for Quantification of CD19-Directed CAR T-Cells. Front. Mol. Biosci. 2020;7:84. doi: 10.3389/fmolb.2020.00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hindson C.M., Chevillet J.R., Briggs H.A., Gallichotte E.N., Ruf I.K., Hindson B.J., Vessella R.L., Tewari M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods. 2013;10:1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lou Y., Chen C., Long X., Gu J., Xiao M., Wang D., Zhou X., Li T., Hong Z., Li C., et al. Detection and Quantification of Chimeric Antigen Receptor Transgene Copy Number by Droplet Digital PCR versus Real-Time PCR. J. Mol. Diagn. 2020;22:699–707. doi: 10.1016/j.jmoldx.2020.02.007. [DOI] [PubMed] [Google Scholar]
- 11.de la Iglesia-San Sebastián I., Carbonell D., Bastos-Oreiro M., Pérez-Corral A., Bailén R., Chicano M., Muñiz P., Monsalvo S., Escudero-Fernández A., Oarbeascoa G., et al. Digital PCR Improves Sensitivity and Quantification in Monitoring CAR-T Cells in B Cell Lymphoma Patients. Transplant. Cell. Ther. 2024;30:306.e1–306.e12. doi: 10.1016/J.JTCT.2023.12.672. [DOI] [PubMed] [Google Scholar]
- 12.Tan L.L., Loganathan N., Agarwalla S., Yang C., Yuan W., Zeng J., Wu R., Wang W., Duraiswamy S. Current Commercial dPCR Platforms: Technology and Market Review. Crit. Rev. Biotechnol. 2023;43:433–464. doi: 10.1080/07388551.2022.2037503. [DOI] [PubMed] [Google Scholar]
- 13.Lin H.T., Okumura T., Yatsuda Y., Ito S., Nakauchi H., Otsu M. Application of Droplet Digital PCR for Estimating Vector Copy Number States in Stem Cell Gene Therapy. Hum. Gene Ther. Methods. 2016;27:197–208. doi: 10.1089/hgtb.2016.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fehse B., Badbaran A., Berger C., Sonntag T., Riecken K., Geffken M., Kröger N., Ayuk F.A. Digital PCR Assays for Precise Quantification of CD19-CAR-T Cells after Treatment with Axicabtagene Ciloleucel. Mol. Ther. Methods Clin. Dev. 2020;16:172–178. doi: 10.1016/j.omtm.2019.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu A., Liu H., Shi R., Cai Y., Ma J., Shao L., Rong V., Gkitsas N., Lei H., Highfill S.L., et al. Application of droplet digital PCR for the detection of vector copy number in clinical CAR/TCR T cell products. J. Transl. Med. 2020;18:191–197. doi: 10.1186/s12967-020-02358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haderbache R., Warda W., Hervouet E., da Rocha M.N., Trad R., Allain V., Nicod C., Thieblemeont C., Boissel N., Varlet P., et al. Droplet digital PCR allows vector copy number assessment and monitoring of experimental CAR T cells in murine xenograft models or approved CD19 CAR T cell-treated patients. J. Transl. Med. 2021;19:265. doi: 10.1186/s12967-021-02925-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arcila M.E., Patel U., Momeni-Boroujeni A., Yao J., Chan R., Chan J., Rijo I., Yu W., Chaves N., Patel H., et al. Validation of a High-Sensitivity Assay for Detection of Chimeric Antigen Receptor T-Cell Vectors Using Low-Partition Digital PCR Technology. J. Mol. Diagn. 2023;25:634–645. doi: 10.1016/J.JMOLDX.2023.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nadal R., Barisic S., Scurti G.M., Cherkasova E., Chen L., Wood K., Highfill S.L., Wells B., Aue G., Shalabi R., et al. Final results of a phase I trial of HERV-E TCR transduced T cells for the treatment of HLA-A∗11 patients with metastatic clear cell renal cell carcinoma (mccRCC) J. Clin. Oncol. 2024;42:435. doi: 10.1200/JCO.2024.42.4_suppl.435. [DOI] [Google Scholar]
- 19.Novosiadly R., Kalos M. High-content molecular profiling of T-cell therapy in oncology. Mol. Ther. Oncolytics. 2016;3 doi: 10.1038/mto.2016.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kunz A., Gern U., Schmitt A., Neuber B., Wang L., Hückelhoven-Krauss A., Michels B., Hofmann S., Müller-Tidow C., Dreger P., et al. Optimized Assessment of qPCR-Based Vector Copy Numbers as a Safety Parameter for GMP-Grade CAR T Cells and Monitoring of Frequency in Patients. Mol. Ther. Methods Clin. Dev. 2020;17:448–454. doi: 10.1016/j.omtm.2020.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Basu A.S. Digital Assays Part I: Partitioning Statistics and Digital PCR. SLAS Technol. 2017;22:369–386. doi: 10.1177/2472630317705680. [DOI] [PubMed] [Google Scholar]
- 22.Lombard C.A., Fabre A., Ambroise J., Ravau J., André F., Jazouli N., Najimi M., Stéphenne X., Smets F., Vaerman J.L., Sokal E.M. Detection of human microchimerism following allogeneic cell transplantation using droplet digital pcr. Stem Cell. Int. 2019;2019 doi: 10.1155/2019/8129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petiti J., Lo Iacono M., Dragani M., Pironi L., Fantino C., Rapanotti M.C., Quarantelli F., Izzo B., Divona M., Rege-Cambrin G., et al. Novel multiplex droplet digital pcr assays to monitor minimal residual disease in chronic myeloid leukemia patients showing atypical bcr-abl1 transcripts. J. Clin. Med. 2020;9:1457. doi: 10.3390/jcm9051457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm M., Andersson E., Osterlund E., Ovissi A., Soveri L.M., Anttonen A.K., Kytölä S., Aittomäki K., Osterlund P., Ristimäki A. Detection of KRAS mutations in liquid biopsies from metastatic colorectal cancer patients using droplet digital PCR, Idylla, and next generation sequencing. PLoS One. 2020;15 doi: 10.1371/journal.pone.0239819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy L.A., Marians R., Kohler M.E., Fry T.J., Winters A.C. Detection of Vector Copy Number in Bicistronic CD19xCD22 CAR T Cell Products with Digital PCR. Blood. 2021;138:4001. doi: 10.1182/blood-2021-153889. [DOI] [Google Scholar]
- 26.Turtle C.J., Hanafi L.A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kist de Ruijter L., van de Donk P.P., Hooiveld-Noeken J.S., Giesen D., Elias S.G., Lub-de Hooge M.N., Oosting S.F., Jalving M., Timens W., Brouwers A.H., et al. Whole-body CD8+ T cell visualization before and during cancer immunotherapy: a phase 1/2 trial. Nat. Med. 2022;28:2601–2610. doi: 10.1038/s41591-022-02084-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spear T.T., Callender G.G., Roszkowski J.J., Moxley K.M., Simms P.E., Foley K.C., Murray D.C., Scurti G.M., Li M., Thomas J.T., et al. TCR gene-modified T cells can efficiently treat established hepatitis C-associated hepatocellular carcinoma tumors. Cancer Immunol. Immunother. 2016;65:293–304. doi: 10.1007/s00262-016-1800-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on reasonable request. All data relevant to the study are included in the article.