

Abstract
Background
Achieving a definitive genetic diagnosis of unexplained multiple congenital anomalies (MCAs) in neonatal intensive care units (NICUs) infants is challenging because of the limited diagnostic capabilities of conventional genetic tests. Although the implementation of whole genome sequencing (WGS) has commenced for diagnosing MCAs, due to constraints in resources and faculty, many NICUs continue to utilize chromosomal microarray (CMA) and/or karyotyping as the initial diagnostic approach. We aimed to evaluate the diagnostic efficacy of WGS in infants with MCAs who have received negative results from karyotyping and/or CMA.
Methods
In this prospective study, we enrolled 80 infants with MCAs who were admitted to a NICU at a single center and had received negative results from CMA and/or karyotyping. The phenotypic characteristics were classified according to the International Classification of Diseases and the Human Phenotype Ontology. We assessed the diagnostic yield of trio-WGS in infants with normal chromosomal result and explored the process of diagnosing by analyzing both phenotype and genotype. Also, we compared the phenotype and clinical outcomes between the groups diagnosed with WGS and the undiagnosed group.
Results
The diagnostic yield of WGS was 26% (21/80), of which 76% were novel variants. There was a higher diagnostic yield in cases of craniofacial abnormalities, including those of the eye and ear, and a lower diagnostic yield in cases of gastrointestinal and genitourinary abnormalities. In addition, higher rates of rehabilitation therapy and gastrostomy were observed in WGS-diagnosed infants than in undiagnosed infants.
Conclusion
This prospective cohort study assessed the usefulness of trio-WGS following chromosomal analysis for diagnosing MCAs in the NICU and revealed improvements in the diagnostic yield and clinical utility of WGS.
Keywords: Congenital Abnormalities; Whole Genome Sequencing; Microarray Analysis; Karyotyping; Infants; Intensive Care Units, Neonatal
Graphical Abstract
INTRODUCTION
Multiple congenital anomalies (MCAs) consist of two or more birth defects, and are found in 20–30% of infants with birth defects, which are the fourth leading cause of neonatal mortality worldwide.1,2 Chromosomal study including chromosomal microarray (CMA), although recommended as a first-tier test for unexplained MCA, has a diagnostic yield of only 10–20% and often necessitates further genetic testing due to its limitations.3,4,5 Recently, whole genome sequencing (WGS) has shown promising results as a first-line diagnostic utility of MCA in neonatal intensive care units (NICUs), with diagnostic yield of 33%.6 Furthermore, rapid sequencing pipeline enhances both precision diagnosis and changes in clinical management, leading to an increase in research aiming to implement this system within NICUs.6,7,8 Nevertheless, transition of these methods to clinic or hospital settings requires high-throughput sequencers, complex and costly infrastructure, and bioinformatics expertise; most of the NICUs still lack the necessary conditions and resources to adopt such systems.9 Therefore, some neonatologists still prefer chromosomal testing as the first-tier diagnostic tool for MCA due to practical reasons.10 Under these conditions, there are few studies that evaluated the pure diagnostic utility of WGS for the genetic diagnosis after karyotyping or CMA in patients with MCAs.
Therefore, the present study investigated the genetic diagnostic utility of trio-WGS prospectively performed on patients with MCAs who were not diagnosed through karyotyping and/or CMA as a first-tier test, in the NICU or in those with prior NICU admission.
METHODS
Study design and participants
This prospective study was conducted using standardized protocols with specifically designed methods.11 From December 2019 to December 2022, we recruited infants in NICU or NICU discharger at Samsung Medical Center within the first year of life who presented with two or more major anomalies corresponding to code of congenital malformations and deformations (Q00-Q99) in International Classification of Diseases, 10th revision, Clinical Modification (ICD-10-CM) diagnosis codes. The inclusion criteria were infants with negative results from karyotyping and/or CMA (Supplementary Method). The exclusion criteria were as follows: patients who had identification of disorders through a neonatal screening program; congenital viral studies; and chromosomal studies before enrollment.
WGS tests were directly ordered by attending neonatologists and approved by the laboratory-based physician. Pre-test counseling was performed by physicians, and appropriate informed consent was obtained from both parents of the patients. The criteria for genetic testing were approved by the ethics committee of the Institutional Review Board of Samsung Medical Center (2019-10-138, 2021-04-189, and 2022-04-054). Patient and follow-up information were extracted from questionnaires and medical records. We documented the Human Phenotype Ontology (HPO) term used in Phenomizer. We formed a panel of multidisciplinary experts, comprising neonatologists, geneticists, and genetic laboratory staff, to discuss the trio WGS results. Geneticists curated patient-specific candidate gene lists based on the phenotype. Subsequently, we assessed the necessity for further genetic investigations, such as Sanger sequencing, RNA analysis, or functional studies. We established an official reporting system to communicate genetic diagnoses. Genetic counseling sessions involved discussions on the genetic testing results, the progression and prognosis of the genetic disorder, and the risk of recurrence in subsequent pregnancies. This information was shared with the neonatologists, who utilized it to evaluate prognosis and establish treatment strategies.
WGS
Genomic DNA from peripheral blood was sequenced using NovaSeq6000 platform (Illumina, San Diego, CA, USA) at a mean depth of 30×, with more than 97% showing coverage of ≥ 10×. Burrows–Wheeler alignment was used for alignment to a human reference genome (hg19). Subsequent variant calling and structural variants (SVs)/copy number variations (CNVs) calling were performed using Genome Analysis Toolkit, version 4.1.2 and Parliament2, respectively. Variants were annotated using ANNOVAR and AnnotSV. For prioritization of variants, both genotype-driven and phenotype-driven approaches were performed.12 In phenotype-driven approach, patient-specific candidate gene lists were made using panelAPP database (https://panelapp.genomicsengland.co.uk/panels) and HPO database (https://hpo.jax.org/app) (Supplementary Table 1). In cases where appropriate gene sets were not found in these databases, gene lists were made from resources such as gene reviews (https://www.ncbi.nlm.nih.gov/books/NBK1116/) and gene test registry (https://www.ncbi.nlm.nih.gov/gtr/genes/). Variants from candidate gene lists were classified according to the 2015 American College of Medical Genetics (ACMG)/Association for Molecular Pathology variant interpretation guidelines and the ClinGen Sequence Variant Interpretation Recommendations.13,14 Identified CNVs less than 1.5 Mb were prioritized variants containing coding regions with Online Mendelian Inheritance in Man morbid genes and were classified according to the ACMG/ClinGen guideline for CMA interpretation.15
Confirmation of copy number variants
The breakpoints were checked from CNV calling data and gap-polymerase chain reaction (PCR) with Sanger sequencing was performed for confirmation of large deletions. The gap-PCR method uses specific primers that we designed to amplify the DNA region flanking a deletion. In this method, the normal allele is too long to be amplified; therefore, PCR products can be obtained only when the allele with a large deletion is present (Supplementary Table 2).
Confirmation of splicing aberrations
RNA was extracted from peripheral blood using the TRIzol method. One microgram of RNA was reverse transcribed into cDNA using Omniscript Reverse Transcriptase (QIAGEN, Hilden, Germany) and amplified using Platinum II Taq Hot-Start DNA Polymerase (Invitrogen, Carlsbad, CA, USA) with custom-designed primers (Supplementary Table 3). Abnormal band was extracted from the gel and Sanger sequencing was performed with purified templates.
Statistical analysis
Continuous variables were expressed as median and interquartile ranges or mean and standard deviation and compared using the Mann–Whitney U test or independent t-test. Categorical variables were expressed as percentages and frequencies and compared using the χ2 or Fisher’s exact tests. SPSS version 26.0 (IBM Corp., Armonk, NY, USA) was used for all statistical analyses, and P < 0.05 was considered statistically significant.
Ethics statement
Informed consent for the present study was obtained from both parents of the patients. The criteria for genetic testing and the study protocol were reviewed and approved by the ethics committee of the Institutional Review Board of Samsung Medical Center (2019-10-138, 2021-04-189, and 2022-04-054).
RESULTS
Demographics of clinical characteristics
A total of 82 infants with two or more malformations were enrolled in this study (Fig. 1). Two patients were excluded because their diagnoses were identified by additional CMA findings after a normal karyotype. One of the excluded infants had a cleft palate, double outlet right ventricle with ventricular septal defect, and a hypoplastic kidney, and was diagnosed with 22q13.1 duplication syndrome through CMA. The other infant with small for gestational age had a single umbilical artery and congenital ichthyosis, and was diagnosed with Xp22.3 microdeletion. We conducted WGS on 80 infants with an analysis of their families. Seventy-two probands provided trio samples, six contributed quartet samples, one from a single-parent family provided a biological specimen, and another one submitted a quintet sample.
Fig. 1. Study population.

NICU = neonatal intensive care unit, CMA = chromosomal microarray, WGS = whole genome sequencing.
Approximately 43% of infants were premature births, and 48% of infants were born with low birth weight (Table 1). All infants had records of prenatal ultrasound monitoring, and 63% presented with abnormal prenatal ultrasound findings, including fetal malformation and intrauterine growth restriction, and excluding any transient soft markers. Among the 80 infants who underwent previous genetic workups, 99% had karyotyping performed, and 68% had CMA performed. There were no significant differences in the demographic factors between the WGS-diagnosed and-undiagnosed groups.
Table 1. Clinical characteristics and genetic test of the participants in this study cohort.
| Characteristics | Total (N = 80) | WGS | P value | ||
|---|---|---|---|---|---|
| Diagnosed (n = 21) | Undiagnosed (n = 59) | ||||
| Gestational age, wk+days | 36+1 ± 3+3 | 36+4 ± 2+6 | 36+0 ± 3+4 | 0.461 | |
| Birth weight, g | 2,412 ± 832 | 2,475 ± 798 | 2,390 ± 849 | 0.690 | |
| Male sex | 43 (53.8) | 8 (38.1) | 35 (59.3) | 0.127 | |
| Small for gestational agea | 18 (22.5) | 3 (14.3) | 15 (25.4) | 0.373 | |
| Congenital microcephalya | 16 (20.0) | 3 (14.3) | 13 (22.0) | 0.709 | |
| Concurrent anomaly in the family | 7 (8.8) | 3 (14.3) | 4 (6.8) | 0.371 | |
| Preterm infant (< 37 wk) | 34 (42.5) | 8 (38.1) | 26 (44.1) | 0.798 | |
| Low birth weight (< 2,500 g) | 38 (47.5) | 9 (42.9) | 29 (49.2) | 0.800 | |
| Advanced maternal age | 44/79 (55.7) | 9/21 (42.9) | 35/58 (60.3) | 0.204 | |
| Abnormal prenatal ultrasound findings | 50 (62.5) | 13 (61.9) | 37 (62.7) | 1.000 | |
| Need for initial resuscitation | 42 (52.5) | 13 (61.9) | 29 (49.2) | 0.446 | |
| Maternal diabetes mellitus | 8 (10.0) | 1 (4.8) | 7 (11.9) | 0.674 | |
| Non-Korean parent | 3 (3.8) | 1 (4.8) | 2 (3.4) | 1.000 | |
| Previous genetic work-up | 80 (100.0) | 21 (100.0) | 59 (100.0) | 1.000 | |
| Karyotype | 79 (98.8) | 21 (100.0) | 58 (98.3) | 1.000 | |
| Chromosomal microarray | 54 (67.5) | 17 (81.0) | 37 (62.7) | 0.177 | |
| Postnatal age of WGS, days | 45 (23–92) | 56 (22–110) | 44 (24–91) | 0.930 | |
| Turnaround time of WGS, mon | 5.7 (3.3–9.1) | 5.0 (2.7–7.1) | 5.9 (3.4–9.4) | 0.219 | |
Values are presented as mean ± standard deviation, number (%), or median (interquartile rage).
WGS = whole genome sequencing.
aLess than third percentile for gestational age.
Genetic diagnosis
Among the 80 infants enrolled in this study, 21 were diagnosed using WGS, with a diagnostic rate of 26%. Of the 21 diagnosed cases, 20 had confirmed diagnoses that could explain the cause of MCAs, and one is currently undergoing functional study as a novel candidate gene (Table 2). Among the 20 confirmed diagnoses, two had CNVs in the form of deletions with an autosomal dominant (AD) disease. One was de novo and the other was inherited from his father (case 9), who had undergone surgery for branchial cleft cysts on bilateral neck. Of the 18 cases detected with pathogenic sequence variations, 14 were diagnosed as AD disorders. Of these, 13 genes had de novo variants, and one gene variant was inherited from the affected parent (case 6).16 The proband and her affected sibling had compound heterozygous variants in FLT4 gene NM_182925.4:c.[2534T>C];[4006T>C]. The mother and father, each carrying one mutation, were asymptomatic but had a family history on the maternal side. A family study revealed that the affected sister of the maternal grandfather was confirmed to have a c.2534T>C variant. Based on familial segregation and additional evidence, the c.2534T>C variant was classified as likely pathogenic with reduced penetrance. Four patients were diagnosed with autosomal recessive diseases from the carrier parents, with two homozygous and two compound heterozygous status. There was no consanguineous family history for homozygous patients and the WGS data did not show large stretches of absence of heterozygosity. Sixteen of the 23 variants from the 21 diagnosed patients were novel. One (case 17) was confirmed to be pathogenic by cDNA analysis. The infant’s phenotype corresponded to the classical Cornelia de Lange Syndrome, scoring 13 points on the clinical diagnostic criteria (Supplementary Table 4).17 Although WGS identified a de novo variant, the variant was presumed not to change the amino acid. However, the variant locates 3' of exon 31, and in-silico analysis using SpliceAI predicted that the variant is likely to affect the splicing donor site (delta score of 0.86). Targeted Sanger sequencing of cDNA confirmed the splicing effect of this variant (Fig. 2). We also detected one novel CNV in another patient (case 12) and breakpoints were delineated by gap-PCR and direct sequencing (Fig. 3).
Table 2. Molecular summary of infants diagnosed through whole genome sequencing.
| No. | Disease | Gene | Inheritance pattern | Ref. seq. | DNA change | AA change | Type of variant | Zygosity | Inheritance | Classification | Evidence | Known or novel | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identified variants in genetically confirmed cases | |||||||||||||
| 1 | CHARGE syndrome | CHD7 | AD | NM_017780.3 | c.2429_2430del | p.(Ser810Cysfs*14) | Frameshift | Het | De novo | PV | PVS1, PS2, PM2 | Novel | |
| 2 | CHARGE syndrome | CHD7 | AD | NM_017780.3 | c.2958-19C>G | p.(?) | Splicing | Het | De novo | LPV | PS2, PM2, PP3 | Novel | |
| 3 | Intellectual developmental disorder, autosomal recessive 18, with or without epilepsy | MED23 | AR | NM_004830.3 | c.2996-1G>A | p.(?) | Splicing | Het | Paternal | LPV | PVS1_M, PM2, PM3 | Novel | |
| c.431_433del | p.(Lys144del) | In-frame | Het | Maternal | VUS | PM2, PM4 | Novel | ||||||
| 4 | Ehlers-Danslos syndrome | COL5A2 | AD | NM_000393.3 | c.3039+2T>C | p.(?) | Splicing | Het | De novo | PV | PVS1_S, PS2, PM2 | Novel | |
| 5 | Kabuki syndrome | KMT2D | AD | NM_003482.3 | c.3103C>T | p.(Gln1035*) | Nonsense | Het | De novo | PV | PVS1, PS2, PM2 | Novel | |
| 6 | Lymphatic malformation 1 | FLT4 | AD | NM_182925.4 | c.2534T>C | p.(Leu845Pro) | Missense | Het | Maternal | LPV | PM1, PM2, PP1, PP3 | Novel | |
| 7 | CHARGE syndrome | CHD7 | AD | NM_017780.3 | c.8745dup | p.(Leu2916Ilefs*25) | Frameshift | Het | De novo | PV | PVS1, PS2, PM2 | Novel | |
| 8 | Epidermolysis bullosa simplex 1A, generalized severe; Epidermolysis bullosa simplex 1B, generalized intermediate; Epidermolysis bullosa simplex 1C, localized | KRT14 | AD | NM_000526.4 | c.377T>A | p.(Leu126Gln) | Missense | Het | De novo | LPV | PS2, PM2, PM5, PP3 | Novel | |
| 9 | Branchio-oto-renal syndrome | EYA1 | AD | NC_000008.10(NM_000503.6) | c.203-2100_419-1133delinsG (exon 5-6 deletion) | p.(?) | CNV | Het | Paternal | LPV | PM2, PM4, PP1, PP4 | Known42 | |
| 10 | Godenhar syndrome; Rubinstein-Taybi syndrome | CREBBP | AD | NM_004380.2 | c.14dup | p.(Leu5Phefs*22) | Frameshift | Het | De novo | PV | PVS1, PS2, PM2 | Novel | |
| 11 | Neurodevelopmental disorder with neonatal respiratory insufficiency, hypotonia, and feeding difficulties | PURA | AD | NM_005859.4 | c.583del | p.(Leu195Serfs*30) | Frameshift | Het | De novo | PV | PVS1, PS2, PM2 | Novel | |
| 12 | Anterior segment dysgenesis 3, multiple subtypes; Axenfeld-Rieger syndrome, type 3 | FOXC1 | AD | NC_000006.11 | g.1596640_1655705delinsGAG (whole gene deletion) | p.(?) | CNV | Het | De novo | PV | PVS1, PS2, PM2 | Novel | |
| 13 | Polycystic kidney disease 4, with or without hepatic disease | PKHD1 | AR | NM_138694.3 | c.6840G>A | p.(Trp2280*) | Nonsense | Het | Paternal | PV | PVS1, PM2, PM3 | Known43 | |
| c.8408G>A | p.(Cys2803Tyr) | Missense | Het | Maternal | LPV | PM2, PM3, PM5 | Novel | ||||||
| 14 | Noonan syndrome 1 | PTPN11 | AD | NM_002834.3 | c.228G>C | p.(Glu76Asp) | Missense | Het | De novo | PV | PS2, PS3_M, PS4_M, PM2 | Known44 | |
| 15 | Neurodevelopmental disorder with dysmorphic facies and skeletal and brain abnormalities | HNRNPR | AD | NM_001102398.1 | c.1609dup | p.(Ala537Glyfs*10) | Frameshift | Het | De novo | PV | PVS1, PS2, PM2 | Known45 | |
| 16 | Thrombophilia 7 due to antithrombin III deficiency | SERPINC1 | AR | NM_000488.3 | c.235C>T | p.(Arg79Cys) | Missense | Hom | Paternal/Maternal | PV | PS3, PS4_M, PM2, PM5, PP4 | Known46 | |
| 17 | Cornelia de Lange syndrome 1 | NIPBL | AD | NM_133433.3 | c.5808G>A | r.5710_5808del | Splicing | Het | De novo | LPV | PS2, PM2, PM4 | Novel | |
| p.(Lys1904_Val1936del) | |||||||||||||
| 18 | Ciliary dyskinesia, primary, 40 | DNAH9 | AR | NM_001372.3 | c.12844-1G>C | p.(?) | Splicing | Hom | Paternal/Maternal | LPV | PS3, PVS1_M, PM3 | Known47 | |
| 19 | CHARGE syndrome | CHD7 | AD | NM_017780.3 | c.5968C>T | p.(Gln1990*) | Nonsense | Het | De novo | PV | PVS1, PS2, PM3 | Known48 | |
| 20 | Diets-Jongmans syndrome | KDM3B | AD | NM_016604.3 | c.1987G>C | p.(Ala663Pro) | Missense | Het | De novo | LPV | PS2, PM2 | Novel | |
| Novel candidate gene | |||||||||||||
| 21 | No known Mendelian disease | OLIG3 | NA | NM_175747.2 | c.536_539dup | p.(Val181Profs*162) | Frameshift | Hom | Maternal isoUPD | VUS | NA | Novel | |
AA = amino acid, AD = autosomal dominant, Het = heterozygous, PV = pathogenic variant, LPV = likely pathogenic variant, AR = autosomal recessive, VUS = variant of uncertain significance, CNV = copy number variant, Hom = homozygous, NA = not applicable, isoUPD = isouniparental disomy.
Fig. 2. Genetic diagnosis using RT-PCR and cDNA analysis. (A) RT-PCR amplifications of the mRNA of case 17. The lower band (386 bp) represents the aberrant NIPBL gene transcript. (B) Diagram showing regions of the NIPBL where the splicing variant described was identified as NM_133433.3(NIPBL):c.5808G>A. The cDNA sequence shows skipping of exon 31.

RT-PCR = reverse transcription polymerase chain reaction.
Fig. 3. Genetic diagnosis using gap PCR and direct sequencing. (A) Electrophoresis of gap-PCR amplifications of Case 12. (B) Diagram showing a 59 kb deletion on chromosome 6p25.3 including the entire FOXC1 gene and part of the adjacent GMDS gene. The band was sequenced, and the breakpoint was delineated as NC_000006.11:g.1596640_1655705delinsGAG.

PCR = polymerase chain reaction.
Suggestion of novel candidate gene for pontocerebellar hypoplasia
One patient (case 21) exhibited symptoms of pontocerebellar hypoplasia (Supplementary Table 4). CMA revealed a loss of heterozygosity across chromosome 6, suggesting isouniparental disomy (isoUPD) (Supplementary Fig. 1). WGS showed that all variants found in homozygous pattern on chromosome 6 were maternally originated, confirming maternal UPD(6). A frameshift variant, NM_175747.2:c.536_539dup, on OLIG3 was highly suspected to be a candidate gene, as recent reports have indicated its role in early cerebellar development by determining the differentiation of neurons.18,19,20 OLIG3 knock-out mice had reduced or inhibited development of precerebellar neurons originating from the caudal rhombic lip, which differentiate into various brainstem and cerebellar cell types, and exhibited dysregulation of respiratory system development and cyanosis.21 This case is currently undergoing functional testing to elucidate the precise etiology.
Comparative analysis of organ system involvement between WGS-diagnosed and undiagnosed groups
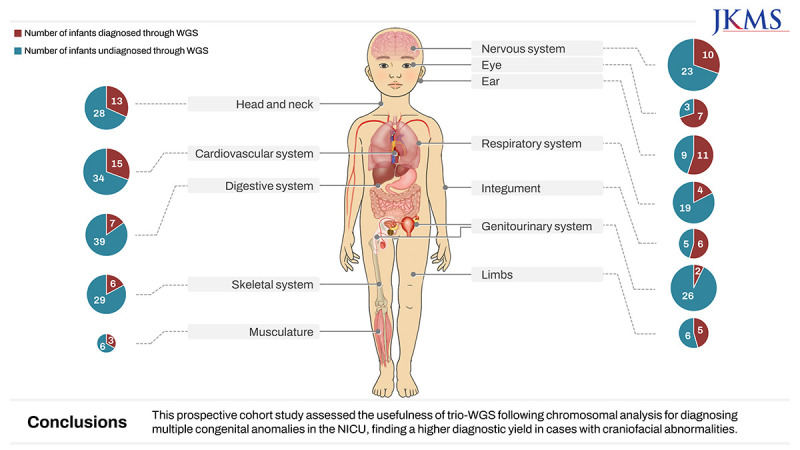
In the total patient group, the top three organ systems identified by Q-codes were the cardiovascular system (63%), followed by the nervous and digestive systems (45%) (Supplementary Fig. 2, Supplementary Table 5). The incidence of eye anomalies was significantly higher in the group diagnosed using WGS than in the undiagnosed group (29% vs. 5%), although the rate of digestive system involvement was significantly lower in the WGS-diagnosed group than in the undiagnosed group (14% vs. 55%). According to the HPO terms, the incidence rates of eye anomalies and defects in the ear and integument were significantly higher in the WGS-diagnosed group than in the undiagnosed group (Fig. 4, Table 3). The involvement of the digestive and genitourinary system significantly decreased the rate of diagnosis by WGS.
Fig. 4. Distribution of infants by organ system based on HPO term.

HPO = Human Phenotype Ontology, WGS = whole genome sequencing.
Table 3. Counts for top-level organ system HPO terms, based on the up-propagation of terms used in Phenomizer according to the ontology.
| HPO category | Total (N = 80) | Whole genome sequencing | P value | ||
|---|---|---|---|---|---|
| Diagnosed (n = 21) | Undiagnosed (n = 59) | ||||
| Abnormality of the cardiovascular system | 49 (61.3) | 15 (71.4) | 34 (57.6) | 0.307 | |
| Abnormality of the abdomen | 46 (57.5) | 7 (33.3) | 39 (66.1) | 0.011* | |
| Structural anomaly | 38 (47.5) | 3 (14.3) | 35 (59.3) | < 0.001* | |
| Functional anomaly | 18 (22.5) | 5 (23.8) | 13 (22.0) | 1.000 | |
| Abnormality of head and neck | 41 (51.3) | 13 (61.9) | 28 (47.5) | 0.314 | |
| Abnormality of prenatal development or birth | 39 (48.8) | 10 (47.6) | 29 (49.2) | 1.000 | |
| Abnormality of the skeletal system | 35 (43.8) | 6 (28.6) | 29 (49.2) | 0.128 | |
| Growth abnormality | 35 (43.8) | 6 (28.6) | 29 (49.2) | 0.128 | |
| Abnormality of the nervous system | 33 (41.3) | 10 (47.6) | 23 (39.0) | 0.607 | |
| Abnormality of the genitourinary system | 28 (35.0) | 2 (9.5) | 26 (44.1) | 0.007* | |
| Abnormality of the respiratory system | 23 (28.8) | 4 (19.0) | 19 (32.2) | 0.400 | |
| Abnormality of the ear | 20 (25.0) | 11 (52.4) | 9 (15.3) | 0.002* | |
| Structural anomaly | 11 (13.8) | 6 (28.6) | 5 (8.5) | 0.032* | |
| Functional anomaly | 14 (17.5) | 8 (38.1) | 6 (10.2) | 0.007* | |
| Abnormality of the integument | 11 (13.8) | 6 (28.6) | 5 (8.5) | 0.032* | |
| Abnormality of the limbs | 11 (13.8) | 5 (23.8) | 6 (10.2) | 0.146 | |
| Abnormality of the eye | 10 (12.5) | 7 (33.3) | 3 (5.1) | 0.006* | |
| Abnormality of the musculature | 9 (11.3) | 3 (14.3) | 6 (10.2) | 0.691 | |
| Abnormal muscle tone (hypotonia) | 7 (8.8) | 2 (9.5) | 5 (8.5) | 1.000 | |
| Abnormality of connective tissue | 8 (10.0) | 1 (4.8) | 7 (11.9) | 0.674 | |
| Neoplasm | 6 (7.5) | 0 (0.0) | 6 (10.2) | 0.332 | |
| Abnormality of the endocrine system | 5 (6.3) | 2 (9.5) | 3 (5.1) | 0.602 | |
| Abnormality of metabolism/homeostasis | 4 (5.0) | 1 (4.8) | 3 (5.1) | 1.000 | |
| Abnormality of the immune system | 3 (3.8) | 1 (4.8) | 2 (3.4) | 1.000 | |
| Abnormality of blood and blood-forming tissues | 2 (2.5) | 2 (9.5) | 0 (0.0) | 0.066 | |
Values are presented as number (%).
HPO = Human Phenotype Ontology.
*P < 0.05.
Clinical implementation
Although the rate of surgery during the NICU stay was lower in the WGS-diagnosed group compared to the undiagnosed group (48% vs. 70%), for infants diagnosed with WGS, it significantly increased during the follow-up period (Table 4). The rates of gastrostomy (33% vs. 9%), rehabilitation therapy (86% vs. 58%), and genetic counseling from clinical geneticists (79% vs. 7%) in the WGS-diagnosed group compared to the undiagnosed group.
Table 4. Clinical implementation of whole genome sequencing.
| Clinical implementation | Total (N = 80) | Whole genome sequencing | P value | ||
|---|---|---|---|---|---|
| Diagnosed (n = 21) | Undiagnosed (n = 59) | ||||
| Duration of NICU admission, days | 46 (22–97) | 47 (17–102) | 41 (23–90) | 0.933 | |
| Death | 9 (11.3) | 3 (14.3) | 6 (10.2) | 0.691 | |
| Start of target medication | 1 (1.3) | 1 (4.8) | 0 (0.0) | 0.262 | |
| Surgery | 63 (78.8) | 15 (71.4) | 48 (81.4) | 0.363 | |
| Before NICU discharge | 51 (63.8) | 10 (47.6) | 41 (69.5) | 0.107 | |
| Rehabilitation | 52 (65.0) | 18 (85.7) | 34 (57.6) | 0.032* | |
| Tracheostomy | 4 (5.0) | 1 (4.8) | 3 (5.1) | 1.000 | |
| Gastrostomy | 12 (15.0) | 7 (33.3) | 5 (8.5) | 0.011* | |
| Genetic counselling | 19 (23.8) | 15 (71.4) | 4 (6.8) | < 0.001* | |
| Long term follow-up | 65/75 (86.7) | 17/19 (89.5) | 48/56 (85.7) | 1.000 | |
Values are presented as number (%) or median (interquartile ranges).
NICU = neonatal intensive care unit.
*P < 0.05.
In one infant, the initiation of targeted medication was facilitated through a WGS diagnosis, altering the family’s clinical management approach (case 16). The neonate underwent surgery for an intracardiac thrombus and was diagnosed with antithrombin III deficiency, and the patient’s parents were identified as heterozygous carriers for this variant through trio-WGS. She was initiated on warfarin treatment to prevent further thrombosis. Following family planning, the proband’s younger sister was born, and she received prophylactic antithrombin treatment until the genetic test results were proven to be normal. In another case diagnosed with Axenfeld-Rieger Syndrome Type 3, the early detection of increased intraocular pressure through regular ophthalmological examinations and parental education allowed for the delay of glaucoma, a complication of the disease (case 12).
DISCUSSION
The diagnostic yield in this study was 26%, which aligns with the range observed in other studies reported a wide range of 20–50% for MCA through WGS.1,5,8,22,23,24 This result represents a 12% increase in the diagnostic rate compared with the period when traditional diagnostic methods were utilized in our NICU (Supplementary Fig. 3). These findings are consistent with previous studies showing that WGS can improve diagnostic rates by approximately 10–20% compared to conventional diagnostic testing including whole exome sequencing (WES) or clinical exome sequencing (ES).25,26 Contrary to many studies suggesting that CNVs are a major cause of congenital anomalies, in our study, they accounted for only 10%.4,27,28 In this study, not all patients performed CMA, and those who found abnormalities in CMA were excluded. Although CNV calling was performed in WGS, the accuracy is limited, and the fact that only CNVs of 1.5 Mb or less may be included in this study may affect the CNV detection rate.
Although many recent studies have demonstrated the advantages of genome sequencing (GS)/ES in the initial tier of diagnosis for congenital anomalies, owing to a higher diagnostic yield,29 CMA is still widely recommended as the first-tier test in many countries.4,23 Although WES/WGS takes an advantage of detecting CNVs, it may not be as effective as CMA in detecting large genomic alterations or certain CNVs.30 Even if the CNVs are called in ES/GS, it is not easy to distinguish true CNVs among hundreds or thousands of CNVs. In the present study, we utilized WGS as a secondary test for MCA since South Korean government reimburses the cost for CMA, but not for ES/GS. Although targeted panel sequencing is reimbursed, it may have limited utility considering genetic heterogeneity of congenital anomalies.
The reason we adopted WGS instead of WES in the present study is that WGS gives a chance to identify causative variants in deep intronic or regulatory elements. Even in coding region analysis, it can be more useful by providing more uniform coverage, especially in GC-rich regions, with better variant detection performance. In addition, WGS is more suitable for CNV detection than WES due to its genome-wide coverage by providing information on sequences involving breakpoints located in non-coding regions. A previous study showed that WGS increased the diagnostic rate by 5.6% compared to WES due to the better coverage and detection of SVs and non-coding variants of WGS.31 In our investigation, although we could not find deep intronic or regulatory mutations, we could detect two CNV cases. Case 9 revealed an approximately 5.5 kb deletion in EYA1 through WGS despite the previous negative CMA result. Case 12, where only karyotyping was performed without CMA, exhibited an approximately 60 kb deletion in FOXC1. Although the CNVs smaller than 400 kb may be detectable with CMA depending on the number of markers utilized, WGS could provide breakpoints at the base pair level, which is not possible with CMA. This information may offer valuable insights, especially in this case, as the size of the deletion and the specific genes involved can significantly influence the patient’s phenotype.32,33
Through genotype-phenotype analysis, we ascertained the high and low diagnostic yields of WGS in the phenotypes of specific organ systems. Consistent with previous studies, we observed a higher diagnostic yield in cases with craniofacial abnormalities, including the eye, ear, and integument. Conversely, a lower diagnostic yield was observed in cases with abdominal abnormalities.22,34,35 Based on these results, particularly in cases of abnormalities in the eyes, ears, and integument, it is suggested to consider genetic testing such as WGS is recommended.
By confirming the variance in genetic diagnostic yields across different organ systems through the application of HPO terms and Q codes, the incorporation of these terminologies into clinical protocols has been validated.36 Q codes are a classification within the ICD system for categorizing congenital anomalies, deformations, and chromosomal abnormalities. The ICD system designations play a crucial role in the storage, search, and management of patient medical information within electronic medical records globally. Analyzing such databases enables easier communication and provides necessary support in handling insurance claims and compensation processes for genetic testing and treatments.37
Unlike other studies in which a high incidence of surgical intervention following a diagnosis through ES/GS has been reported,24 this study found no difference in the rates of surgeries between infants diagnosed with WGS and those not diagnosed. This outcome can be attributed to the fact that most surgeries in this cohort were related to cardiac or gastrointestinal anomalies, such as tracheoesophageal fistula and imperforate anus, with no genetic diseases identified in this group. Additional analysis is necessary to identify these specific gastrointestinal diseases separately and to search for variations. The increase in specific medical interventions such as rehabilitation therapy and gastrostomy in infants diagnosed with definitive diseases may be an indirect result of the ability to predict the prognosis and course of the disease. In the WGS-undiagnosed group, genetic counseling by geneticists was less frequent than in the diagnosed group, likely due to the limited availability of genetic counselors. Nonetheless, all patients received thorough explanations about their results from pediatricians, including the limitations of tests and risk of genetic diseases, even without a definitive genetic diagnosis.
WGS can facilitate early diagnosis during the neonatal period by identifying genetic variants where timely intervention can enhance long-term prognosis.38 For early diagnosed MCA infants, supportive treatments such as rehabilitation and gastrostomy can be implemented more proactively. For genetically undiagnosed infants, it may be helpful to reanalyze genomic data periodically and/or when new symptoms appear. Meanwhile, it is also important to shorten the time required for genetic diagnosis, but in this study, rapid diagnosis was not possible due to a lack of various resources. Nonetheless, applying advanced technologies such as rapid WGS integrated with the latest pipelines and artificial intelligence for analysis and interpretation can shorten the time to diagnosis, enabling timely intervention before irreversible outcomes occur.39,40
We utilized trio-based WGS analysis, an efficient approach given that most pathogenic variants in our study were de novo. This approach can shorten the time for selecting gene sets based on phenotypes and aid in classifying novel missense variants as pathogenic that would otherwise be mostly classified as variants of uncertain significance (VUS). However, the cost of WGS is a significant consideration for clinical implementation. Given that a substantial portion of diagnosed patients resulted from previously reported or high-impact variants, such as nonsense, frameshift, and splicing variants, which could be identified solely through proband analysis, singleton WGS could be an alternative approach. However, to make this feasible, a robust interpretation management system must be in place. This system would require continuous updating and curation of gene lists for relevant disease categories to ensure accurate and efficient diagnosis.
Since this study excluded infants diagnosed with CNV-related disease through CMA, it is impossible to compare the clinical information between MCA infants with CNV-related diseases and those with non-CNV disease. Previous studies have indicated that infants diagnosed with non-CNV diseases have a greater number of involved organs than those diagnosed with CNV-related diseases, despite no differences in specific phenotype or clinical information between the two groups.22 Therefore, further study is needed to analyze the clinical characteristics that would determine whether WGS, rather than CMA, should be chosen as the first-tier test in MCA infants.
The strengths of this study are as follows. WGS was performed on at least three members from each family to improve the diagnostic yield. Utilizing trio sequencing in WGS in our study has led to success in identifying the genetic cause in many cases, providing critical information for accurate diagnosis. Furthermore, by adopting a multidisciplinary approach, we were able to perform both genotype-driven and phenotype-driven analyses, thereby enhancing the diagnostic yield of genetic diseases that might have been overlooked with conventional genetic testing. This integrative approach also allowed us to conduct subsequent functional studies, leading to the identification of novel genetic diseases. Continuous collaboration between multiple specialists may be necessary for elucidating the causes of rare diseases.
Our study had several limitations. Most of variants in non-coding regions, especially regulatory regions, found in this study were classified as variants of uncertain significance. Although extensive functional studies have demonstrated that variants in many non-coding regions are associated with genetic diseases,12 interpretation of variants found in deep intronic and regulatory regions was limited due to difficulties in determining their splicing effects and impact on gene expression. Further research will be needed to elucidate the functions of non-coding regions. In addition, we reviewed only 1.5 Mb or less of the CNVs called in WGS. There are yet no well-established bioinformatics pipeline, protocol, and quality-control standards for CNV analysis through WGS. Mappability issues of repeat regions, GC-content bias, sequence read quality, and difficulty in identifying duplications make accurate CNV analysis difficult in WGS.41
In conclusion, this is the first prospective cohort study to investigate the utility of trio-WGS after chromosomal analysis for diagnosing MCA in an NICU setting. This study presents evidence that early adoption of trio-WGS following karyotyping/CMA significantly increases the genetic diagnostic yield for MCAs and aids in identifying novel genetic etiologies of rare diseases.
ACKNOWLEDGMENTS
The authors would like to thank Da Hyeun Lee and Chan Mi Moon, an audiovisual engineer at Samsung Medical Information & Media Services, for providing the medical illustrations.
Footnotes
Funding: This work was supported by a Research Program funded by the Korea Disease Control and Prevention Agency (2021-ER0706-00 and 2022-ER0503-00) and an intramural grant from the Korea National Institute of Health (2019-NI-093-00, 2022-NI-060-00, and 2022-NI-060-01).
Disclosure: The authors have no potential conflicts of interest to disclose.
- Conceptualization: Jang JH, Chang YS.
- Data curation: Ahn SY, Sung SI, Kim JM, Cho HW.
- Formal analysis: Kim JA, Park JH, Park MH.
- Investigation: Sung SI, Park WS.
- Methodology: Yang M, Jo HS, Park HY.
- Validation: Ahn SY, Park WS.
- Writing - original draft: Yang M, Kim JA, Jo HS.
- Writing - review & editing: Jang JH, Chang YS.
SUPPLEMENTARY MATERIALS
Karyotyping and chromosomal microarray
Genes targeted in the phenotype-driven analysis
Primer sequences used for the gap-PCR for confirmation of copy number variant in this study
Primer sequences used for amplification of target region of cDNA for confirmation of splicing aberration in this study
Clinical phenotype of infants diagnosed through whole genome sequencing.
Counts for top-level organ system of Q-codes according to the ICD-10-CM
Plotting the variant allele frequencies for the variants across the chromosome 6. The X-axis represents the genomic position of chromosome 6 and the Y-axis represents the variant allele frequencies of the variants found in case No. 21. The arrow indicates a stretch of homozygous pattern of the maternally originated variants on chromosome 6, indicating an isouniparental disomy. This figure was created using Python 3.10.9.
Distribution of infants by organ system based on Q-codes.
Comparison of the traditional approach to diagnosis of genetic diseases in NICU infants with this study group.
References
- 1.Marouane A, Olde Keizer RACM, Frederix GWJ, Vissers LELM, de Boode WP, van Zelst-Stams WAG. Congenital anomalies and genetic disorders in neonates and infants: a single-center observational cohort study. Eur J Pediatr. 2022;181(1):359–367. doi: 10.1007/s00431-021-04213-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agopian AJ, Evans JA, Lupo PJ. Analytic methods for evaluating patterns of multiple congenital anomalies in birth defect registries. Birth Defects Res. 2018;110(1):5–11. doi: 10.1002/bdr2.1115. [DOI] [PubMed] [Google Scholar]
- 3.Jang W, Kim Y, Han E, Park J, Chae H, Kwon A, et al. Chromosomal microarray analysis as a first-tier clinical diagnostic test in patients with developmental delay/intellectual disability, autism spectrum disorders, and multiple congenital anomalies: a prospective multicenter study in Korea. Ann Lab Med. 2019;39(3):299–310. doi: 10.3343/alm.2019.39.3.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kritioti E, Theodosiou A, Parpaite T, Alexandrou A, Nicolaou N, Papaevripidou I, et al. Unravelling the genetic causes of multiple malformation syndromes: a whole exome sequencing study of the Cypriot population. PLoS One. 2021;16(7):e0253562. doi: 10.1371/journal.pone.0253562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.NICUSeq Study Group. Krantz ID, Medne L, Weatherly JM, Wild KT, Biswas S, et al. Effect of whole-genome sequencing on the clinical management of acutely ill infants with suspected genetic disease: a randomized clinical trial. JAMA Pediatr. 2021;175(12):1218–1226. doi: 10.1001/jamapediatrics.2021.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott AM, du Souich C, Lehman A, Guella I, Evans DM, Candido T, et al. RAPIDOMICS: rapid genome-wide sequencing in a neonatal intensive care unit-successes and challenges. Eur J Pediatr. 2019;178(8):1207–1218. doi: 10.1007/s00431-019-03399-4. [DOI] [PubMed] [Google Scholar]
- 8.Olde Keizer RACM, Marouane A, Kerstjens-Frederikse WS, Deden AC, Lichtenbelt KD, Jonckers T, et al. Rapid exome sequencing as a first-tier test in neonates with suspected genetic disorder: results of a prospective multicenter clinical utility study in the Netherlands. Eur J Pediatr. 2023;182(6):2683–2692. doi: 10.1007/s00431-023-04909-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daoud H, Luco SM, Li R, Bareke E, Beaulieu C, Jarinova O, et al. Next-generation sequencing for diagnosis of rare diseases in the neonatal intensive care unit. CMAJ. 2016;188(11):E254–E260. doi: 10.1503/cmaj.150823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waggoner D, Wain KE, Dubuc AM, Conlin L, Hickey SE, Lamb AN, et al. Yield of additional genetic testing after chromosomal microarray for diagnosis of neurodevelopmental disability and congenital anomalies: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG) Genet Med. 2018;20(10):1105–1113. doi: 10.1038/s41436-018-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jo HS, Yang M, Ahn SY, Sung SI, Park WS, Jang JH, et al. Optimal protocols and management of clinical and genomic data collection to assist in the early diagnosis and treatment of multiple congenital anomalies. Children (Basel) 2023;10(10):1673. doi: 10.3390/children10101673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Austin-Tse CA, Jobanputra V, Perry DL, Bick D, Taft RJ, Venner E, et al. Best practices for the interpretation and reporting of clinical whole genome sequencing. NPJ Genom Med. 2022;7(1):27. doi: 10.1038/s41525-022-00295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.ClinGen Working Group. ClinGen sequence variant interpretation recommendation - version 1.0. [Updated 2017]. [Accessed March 1, 2024]. https://clinicalgenome.org/working-groups/sequence-variant-interpretation/
- 15.Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) Genet Med. 2020;22(2):245–257. doi: 10.1038/s41436-019-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiang Q, Chen J, Xiao X, Xu B, Xie H, Wang H, et al. Case report: The compound heterozygotes variants in FLT4 causes autosomal recessive hereditary lymphedema in a Chinese family. Front Genet. 2023;14:1140406. doi: 10.3389/fgene.2023.1140406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kline AD, Moss JF, Selicorni A, Bisgaard AM, Deardorff MA, Gillett PM, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. 2018;19(10):649–666. doi: 10.1038/s41576-018-0031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Storm R, Cholewa-Waclaw J, Reuter K, Bröhl D, Sieber M, Treier M, et al. The bHLH transcription factor Olig3 marks the dorsal neuroepithelium of the hindbrain and is essential for the development of brainstem nuclei. Development. 2009;136(2):295–305. doi: 10.1242/dev.027193. [DOI] [PubMed] [Google Scholar]
- 19.Müller T, Anlag K, Wildner H, Britsch S, Treier M, Birchmeier C. The bHLH factor Olig3 coordinates the specification of dorsal neurons in the spinal cord. Genes Dev. 2005;19(6):733–743. doi: 10.1101/gad.326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowenstein ED, Rusanova A, Stelzer J, Hernaiz-Llorens M, Schroer AE, Epifanova E, et al. Olig3 regulates early cerebellar development. eLife. 2021;10:e64684. doi: 10.7554/eLife.64684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z, Li H, Hu X, Yu L, Liu H, Han R, et al. Control of precerebellar neuron development by Olig3 bHLH transcription factor. J Neurosci. 2008;28(40):10124–10133. doi: 10.1523/JNEUROSCI.3769-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang H, Xiao F, Dong X, Lu Y, Cheng G, Wang L, et al. Diagnostic and clinical utility of next-generation sequencing in children born with multiple congenital anomalies in the China neonatal genomes project. Hum Mutat. 2021;42(4):434–444. doi: 10.1002/humu.24170. [DOI] [PubMed] [Google Scholar]
- 23.Malinowski J, Miller DT, Demmer L, Gannon J, Pereira EM, Schroeder MC, et al. Systematic evidence-based review: outcomes from exome and genome sequencing for pediatric patients with congenital anomalies or intellectual disability. Genet Med. 2020;22(6):986–1004. doi: 10.1038/s41436-020-0771-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kingsmore SF, Cole FS. The role of genome sequencing in neonatal intensive care units. Annu Rev Genomics Hum Genet. 2022;23(1):427–448. doi: 10.1146/annurev-genom-120921-103442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20(4):435–443. doi: 10.1038/gim.2017.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halldorsson BV, Eggertsson HP, Moore KHS, Hauswedell H, Eiriksson O, Ulfarsson MO, et al. The sequences of 150,119 genomes in the UK Biobank. Nature. 2022;607(7920):732–740. doi: 10.1038/s41586-022-04965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manning M, Hudgins L Professional Practice and Guidelines Committee. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12(11):742–745. doi: 10.1097/GIM.0b013e3181f8baad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Southard AE, Edelmann LJ, Gelb BD. Role of copy number variants in structural birth defects. Pediatrics. 2012;129(4):755–763. doi: 10.1542/peds.2011-2337. [DOI] [PubMed] [Google Scholar]
- 29.Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med. 2018;3(1):16. doi: 10.1038/s41525-018-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perovic D, Damnjanovic T, Jekic B, Dusanovic-Pjevic M, Grk M, Djuranovic A, et al. Chromosomal microarray in postnatal diagnosis of congenital anomalies and neurodevelopmental disorders in Serbian patients. J Clin Lab Anal. 2022;36(6):e24441. doi: 10.1002/jcla.24441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim YG, Kwon H, Park JH, Nam SH, Ha C, Shin S, et al. Whole-genome sequencing in clinically diagnosed Charcot-Marie-Tooth disease undiagnosed by whole-exome sequencing. Brain Commun. 2023;5(3):fcad139. doi: 10.1093/braincomms/fcad139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edgar AZ, Baby S. Axenfeld-Rieger syndrome. [Updated October 24, 2022]. [Accessed March 1, 2024]. https://www.ncbi.nlm.nih.gov/books/NBK538504/
- 33.Haliburton GD, McKinsey GL, Pollard KS. Disruptions in a cluster of computationally identified enhancers near FOXC1 and GMDS may influence brain development. Neurogenetics. 2016;17(1):1–9. doi: 10.1007/s10048-015-0458-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang L, Wei Z, Chen X, Hu L, Peng X, Wang J, et al. Use of medical exome sequencing for identification of underlying genetic defects in NICU: experience in a cohort of 2303 neonates in China. Clin Genet. 2022;101(1):101–109. doi: 10.1111/cge.14075. [DOI] [PubMed] [Google Scholar]
- 35.Normand EA, Braxton A, Nassef S, Ward PA, Vetrini F, He W, et al. Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder. Genome Med. 2018;10(1):74. doi: 10.1186/s13073-018-0582-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong X, Xiao T, Chen B, Lu Y, Zhou W. Precision medicine via the integration of phenotype-genotype information in neonatal genome project. Fundam Res. 2022;2(6):873–884. doi: 10.1016/j.fmre.2022.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison JE, Weber S, Jakob R, Chute CG. ICD-11: an international classification of diseases for the twenty-first century. BMC Med Inform Decis Mak. 2021;21(Suppl 6):206. doi: 10.1186/s12911-021-01534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith LD, Willig LK, Kingsmore SF. Whole-exome sequencing and whole-genome sequencing in critically ill neonates suspected to have single-gene disorders. Cold Spring Harb Perspect Med. 2015;6(2):a023168. doi: 10.1101/cshperspect.a023168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peterson B, Hernandez EJ, Hobbs C, Malone Jenkins S, Moore B, Rosales E, et al. Automated prioritization of sick newborns for whole genome sequencing using clinical natural language processing and machine learning. Genome Med. 2023;15(1):18. doi: 10.1186/s13073-023-01166-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao D, Liu C, Wang L, Ai-Ouran R, Deisseroth C, Pasupuleti S, et al. AI-MARRVEL - a knowledge-driven AI system for diagnosing Mendelian disorders. NEJM AI. 2024;1(5):10.1056/aioa2300009. doi: 10.1056/aioa2300009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teo SM, Pawitan Y, Ku CS, Chia KS, Salim A. Statistical challenges associated with detecting copy number variations with next-generation sequencing. Bioinformatics. 2012;28(21):2711–2718. doi: 10.1093/bioinformatics/bts535. [DOI] [PubMed] [Google Scholar]
- 42.Kim DH, Yang M, Jo HS, Park J, Jang J, Shin S, et al. A preterm infant with feeding aspiration diagnosed with BOR syndrome, confirmed case by whole-genome sequencing and structural variant calling. Children (Basel) 2022;10(1):76. doi: 10.3390/children10010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hou L, Du Y, Zhang M, Su P, Zhao C, Wu Y. Novel mutations of PKHD1 and AHI1 identified in two families with cystic renal disease. Int J Clin Exp Pathol. 2018;11(5):2869–2874. [PMC free article] [PubMed] [Google Scholar]
- 44.Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet. 2006;78(2):279–290. doi: 10.1086/499925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duijkers FA, McDonald A, Janssens GE, Lezzerini M, Jongejan A, van Koningsbruggen S, et al. HNRNPR variants that impair homeobox gene expression drive developmental disorders in humans. Am J Hum Genet. 2019;104(6):1040–1059. doi: 10.1016/j.ajhg.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koide T, Odani S, Takahashi K, Ono T, Sakuragawa N. Antithrombin III Toyama: replacement of arginine-47 by cysteine in hereditary abnormal antithrombin III that lacks heparin-binding ability. Proc Natl Acad Sci U S A. 1984;81(2):289–293. doi: 10.1073/pnas.81.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen W, Zhang Y, Shen L, Zhu J, Cai K, Lu Z, et al. Biallelic DNAH9 mutations are identified in Chinese patients with defective left-right patterning and cilia-related complex congenital heart disease. Hum Genet. 2022;141(8):1339–1353. doi: 10.1007/s00439-021-02426-5. [DOI] [PubMed] [Google Scholar]
- 48.Janssen N, Bergman JE, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33(8):1149–1160. doi: 10.1002/humu.22086. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Karyotyping and chromosomal microarray
Genes targeted in the phenotype-driven analysis
Primer sequences used for the gap-PCR for confirmation of copy number variant in this study
Primer sequences used for amplification of target region of cDNA for confirmation of splicing aberration in this study
Clinical phenotype of infants diagnosed through whole genome sequencing.
Counts for top-level organ system of Q-codes according to the ICD-10-CM
Plotting the variant allele frequencies for the variants across the chromosome 6. The X-axis represents the genomic position of chromosome 6 and the Y-axis represents the variant allele frequencies of the variants found in case No. 21. The arrow indicates a stretch of homozygous pattern of the maternally originated variants on chromosome 6, indicating an isouniparental disomy. This figure was created using Python 3.10.9.
Distribution of infants by organ system based on Q-codes.
Comparison of the traditional approach to diagnosis of genetic diseases in NICU infants with this study group.