

Abstract
Interest in the use of proteolysis-targeting chimeras (PROTACs) in cancer therapy has increased in recent years. Targeting bromodomain and extra terminal domain (BET) proteins, especially bromodomain-containing protein 4 (BRD4), has shown inhibitory effects on basal-like breast cancer (BLBC). However, the bioavailability of BRD4 PROTACs is restricted by their non-selective biodegradability and low tumor-targeting ability. We demonstrated that 6b (BRD4 PROTAC) suppresses BLBC cell growth by targeting BRD4, but not BRD2 and BRD3, for cereblon (CRBN)-mediated ubiquitination and proteasomal degradation. Compound 6b also inhibited expression of Krüppel-like factor 5 (KLF5) transcription factor, a key oncoprotein in BLBC, controlled by BRD4-mediated super-enhancers. Moreover, 6b inhibited HCC1806 tumor growth in a xenograft mouse model. The combination of 6b and KLF5 inhibitors showed additive effects on BLBC. These results suggest that BRD4-specific PROTAC can effectively inhibit BLBC by downregulating KLF5, and that 6b has potential as a novel therapeutic drug for BLBC.
Subject terms: Breast cancer, Diagnostic markers
Introduction
In 2020, breast cancer became the most frequently diagnosed cancer due to its rapid tumor growth. Patients often develop resistance to the following therapy. Basal-like breast cancer (BLBC) is associated with an aggressive clinical course, shorter survival, recurrence, and distant metastasis. It is a common malignancy, affecting women worldwide. Therefore, new methods for treating BLBC are urgently needed [1].
Bromodomain-containing protein 4 (BRD4), a member of the bromodomain and extra terminal domain (BET) family, has been implicated in the pathogenesis of breast cancer and is a novel target for cancer therapy [2]. To date, BRD4-targeted BET inhibitors (BETis) such as JQ1 and OTX015 are undergoing clinical trials. c-MYC is a well-known oncogenic transcription factor and a major driver for a wide variety of cancers. No effective drugs that directly target c-MYC are currently available. JQ1 was initially developed as a c-MYC inhibitor because c-MYC is regulated by BRD4 [3, 4]. However, drug resistance and toxic side effects of monotherapy hamper the development of BETis [5]. Future studies on the function and specific regulatory mechanism of BRD4 may reveal new mechanisms for BRD4-targeted therapy [5, 6]. BRD4 is usually upregulated in BLBC and modulates the malignancy of related genes, including the Twist/BRD4/Wnt5A axis [7], PP2A [8], Jagged1/Notch1 signaling [9], and Snail [10]. These studies indicate that targeted inhibition of BRD4 suppresses BLBC migration, invasion, and epithelial-mesenchymal transition (EMT) by downregulating these genes.
In recent years, proteolysis-targeting chimeras (PROTACs) have provided an alternative approach to modulating protein homeostasis for cancer therapy, which might help in selecting sensitive drugs to overcome small-molecule inhibitor insufficiency and reduce side effects. Numerous BET PROTACs have been reported that depend on different E3 ligases, including VHL [11], CRBN [12–14], IAP [15], MDM2 [16], AHR [17], DCAF16 [18], RNF114 [19], and RNF4 [20]. PROTAC was first proposed as a therapy in 2001, but the development of proprietary drugs has been hampered by the lack of available E3 ligand. Cereblon CRBN-based PROTACs have a lower molecular weight than VHL, and can readily be formulated as orally bioavailable PROTACs as novel anticancer agents. CRBN is the substrate receptor of Cullin4 complex, which promotes substrate protein ubiquitination and degradation. ARV-825 is the first CRBN-BRD4 PROTAC, degrading all BET protein [21].
PROTACs that target oncoproteins such as ERα, CDK4/6, PI3K, BETs, PARP1, and eEF2K, have shown great promise for breast cancer treatment [22]. The first ERα-targeting PROTAC, ARV-471, has been approved for use in phase II clinical trials to treat patients with locally advanced or metastatic ERα-positive/HER2-negative breast cancer [23]. Bai et al [12]., synthesized the first CRBN-dependent BET degrader, BETd-246, which efficiently decreased BET protein levels in BLBC cell lines in both a PDX model and MDA-MB-231/453/468 xenografts.
Most BRD4 PROTACs simultaneously degrade all three BRD proteins, thereby increasing drug toxicity [24, 25]. MZ1 and ARV-825 efficiently downregulate BET protein expression, inducing MDA-MB-231 and BT549 apoptosis, and promoting MCL1 and H2AX activation; however, no synergistic activity is observed when combined it is combined with docetaxel/olaparib/cisplatin [26]. MZ1 has strong cytotoxic effects in the naturally drug-resistant cell, HCC1954, and MZ1 is widely distributed in tissues, causing toxic side effects [26, 27]. In the BLBC xenograft model, MZ1 and ARV-825 improved tumor-targeting ability, leading to enhanced BET degradation, antitumor potency, and decreased toxicity [27]. MS83, by linking KEAP1 ligand, reduces the levels of BRD4 and BRD3, but not BRD2, and selectively degrades the BRD4-S isoform in both MDA-MB-468 and MDA-MB-231 cell lines [28]. Similarly, PROTAC molecule 24 selectively degrades BRD4-L and BRD3, but not BRD2 or BRD4-S in MCF7 [29].
Having a few representative BRD4-selective PROTAC may be preferable to using pan-BET inhibitors for cancer therapy [30, 31]. The first BRD4 BD1 (DC50/5 h = 5 nM)-specific PROTAC (ZXH-3-26) designed by Ciulli et al [32]., shows activity exclusively on BRD4 and spares degradation of BRD2 or BRD3 in cellular degradation assays. Off-target degradation can be tuned by the linker composition, but no cellular anti-proliferative efficacy against cancer cells was reported [32]. This might be because ZXH-3-26 is derived from dBET23 and dBET6 which have shown toxicity both in vivo and in vitro. dBET57, a potent and selective CRBN-based degrader of BRD4BD1, is one of the marketed drugs for solid tumors, triggers a rapid, durable, and selective degradation of BRD4 over BRD2/3, with a DC50/5 h of 500 nM for BRD4BD1 in neuroblastoma [32, 33], promoting BRD4 ternary complex for protein degradation [34]. However, dBET57 at a 300 nM concentration dramatically induces BRD2/3 degradation in neuroblastoma cell lines, including SK-N-BE (2), IMR32, and SH-SY5Y [33]. At a concentration of 1200 nM, dBET57 is significantly reduced or almost completely erased. These studies highlight the need to identify more specific and safer BRD4 degraders.
Krüppel-like factor 5 (KLF5) transcription factor is a potential therapeutic target for BLBC [35, 36]. KLF5 promotes cancer cell proliferation and the cell cycle by inducing transcription of FGF-BP1 [37] and Cyclin D1 [38], and inhibiting transcription of p27 [39] and p21 [40]. We have previously reported that BRD4 maintains high expression levels of KLF5 in HCC1806 and HCC1937 breast cancer cell lines, and that JQ-1 and compound 870 strongly inhibit the transcriptional expression of KLF5 and BLBC cell growth [41].
The therapeutic potential of selective BRD4 degraders and their mechanism of action in BLBC have not previously been reported. Based on our optimization, we developed compound 6b as a highly potent BRD4-specific degrader and investigated its therapeutic potential and mechanisms of action in BLBC in vitro and in vivo. Our study aims to confirm a new PROTAC as a selective degrader targeting BRD4 and represents a promising therapeutic approach for BLBC.
Results
Discovery of 6b as a specific degrader of BRD4 protein
We chose ABBV-075, developed by AbbVie, as a ligand for the BET protein. It has demonstrated strong efficacy in inhibiting BET protein both in vivo and in vitro, with excellent safety profiles. This compound holds great promise as a clinical candidate for oral treatment of a wide range of cancers.
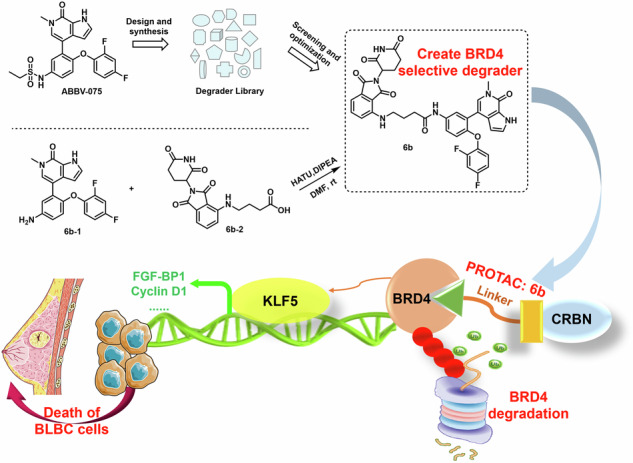
Based on our previous knowledge, we constructed a degrader library derived from ABBV-075. After subsequent screening and optimization, we identified 6b as a selective degrader of BRD4, but not BRD3 and BRD2 (Fig. 1), making it a valuable tool for BRD4 protein knockdown.
Fig. 1. Design and development of 6b.

We constructed a degrader library based on ABBV-075, then screened and optimized, identified 6b as a BET protein degrader based upon the potent and selective BRD4 inhibitor.
6b potently and selectively depletes BRD4 protein in BLBC cells
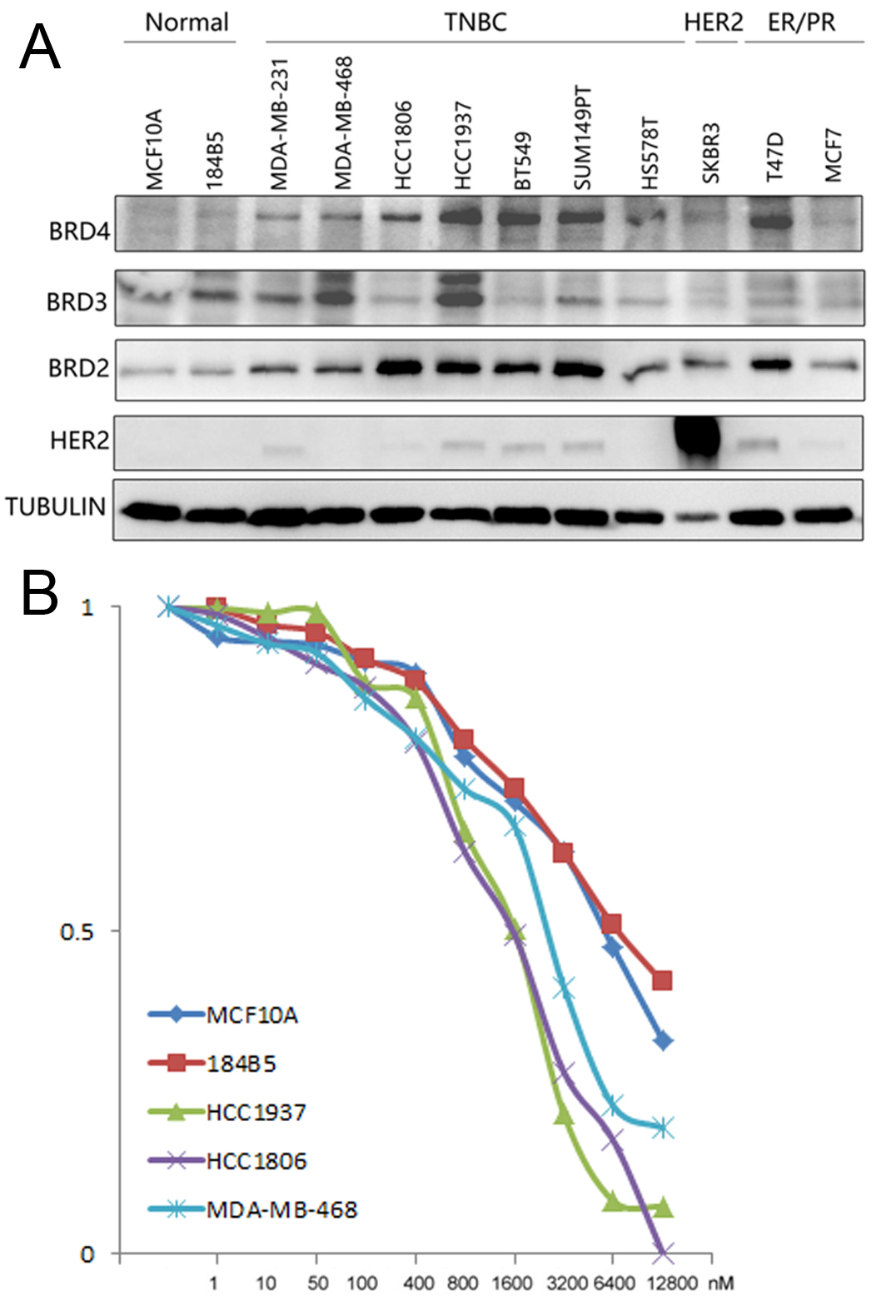
BRD2, BRD3, and BRD4 are expressed in breast tumors, whereas BRDT is rarely expressed [42]. We selected HCC1937 and HCC1806 BLBC cell lines for further study because of their high expression levels of BRD4 (Fig. S1A). We evaluated the ability of 6b and its analogs 6d, 6e, 6 f, and 6 g, to degrade BET proteins in HCC1806 and HCC1937 BLBC cell lines. Only 6b caused specific degradation of BRD4 protein (Fig. 2A, B). Compared with other cell lines, HCC1806 and HCC1937 cell lines were more sensitive to 6b (Fig. S1B). We then detected dose-dependent BRD4 degradation by 6b in BLBC cells. A near complete depletion of BRD4 proteins was observed 48 hours after the addition of 0.01 μM 6b to HCC1806 cells, and 0.1 μM 6b to HCC1937 cells (Fig. 2C). Compound 6b (0.1 μM) induced time-dependent BRD4 degradation and degraded most BRD4 protein within 12 hours (Fig. 2D).
Fig. 2. 6b is a potent and highly selective degrader of BRD4 protein.

A, B HCC1806 or HCC1937 cells were treated with 6b, 6d, 6e, 6f, and 6g at the indicated doses for 48 hours, protein expression was analyzed by WB and GAPDH was included as a loading control. C HCC1806 or HCC1937 cells were treated with 6b at the indicated doses for 48 hours for WB. D HCC1806 or HCC1937 cells were treated with 0.1 μM 6b for the indicated time for WB. E HCC1937 and HCC1806 cells were treated with 6b or DMSO (0.1 μM) for 12 h, then treated with cycloheximide (CHX, 50 μg/mL) for 1, 2, and 4 h. The cell lysates were collected for WB. The quantitative results are plotted below. GAPDH was used as the loading control. F HCC1937 and HCC1806 cells were treated with JQ1 (3 μM) or 6b (0-0.5-1 μM) for 48 h. The protein expression was measured by WB.
Next, we measured BRD4 protein stability using a cycloheximide (CHX) chase experiment to verify whether 6b promotes BRD4 protein degradation. The half-life of BRD4 protein was markedly decreased in the presence of 6b (Fig. 2E). The results confirmed that 6b selectively accelerated BRD4 protein degradation.
JQ1, a small-molecule inhibitor for BRD4, is known to effectively inhibit multiple BLBC cell lines. We used JQ1 as a positive control to test whether 6b affected the expression of BRD4 downstream target genes. Treatment of 6b for 48 hours led to a marked decline in KLF5, c-Myc, SKP2, Bcl-2, and Bcl-XL levels, and increased levels of Bim, p21, and p27 (Fig. 2F). Besides KLF5, these genes are BRD4 downstream target genes.
6b causes growth inhibition and cell cycle arrest in BLBC cell lines
When we treated HCC1806 and HCC1937 cells with different concentrations of 6b and observed cell growth over the course of 4 days using the sulforhodamine B (SRB) assay, we found that 6b significantly inhibited cell growth in a dose-dependent manner (Fig. 3A). Besides, 6b also effectively inhibited growth of other three BLBC cell lines, including Hs578T, MDA-MB-231, and SUM149PT (Fig. S2A). Interestingly, 6b does not cause significant apoptosis according to flow cytometry analysis (Fig. S2B) although 6b decreased the Bcl2 and Bcl-XL protein levels (Fig. 2F).
Fig. 3. 6b displays strong growth inhibition and cell cycle arrest in BLBC cell lines.

A HCC1806 or HCC1937 cells were treated with 6b (0-0.2-0.4-0.8-1.6-3.2 μM) for 4 days for cell viability assay. B HCC1937 and HCC1806 cells were treated with 6b (0-0.05-0.1-0.2 μM) for 48 hours for Annexin V-PI apoptosis analysis. The quantitative results are plotted below. C HCC1937 and HCC1806 cells were treated with 6b (0-0.05-0.1-0.2 μM) for 48 hours and cell lysates were collected for WB. D 6b (0-0.05-0.1-0.2 μM) was used to treat HCC1937 and HCC1806 cells for 48 h, DNA synthesis was measured by the EdU assay, nuclei were stained with Hoechst 33342 (blue channel) and green channel represents EdU. The total numbers of cells and EdU-incorporated cells in each sample were counted under a fluorescent microscope, percentages of EdU-positive proliferating cells vs. total cells are shown. E Colony formatting unit assays of BLBC cells were performed on different formulations and incubated with the freshly prepared medium for another 9 day (n = 3). *p < 0.05, **p < 0.01. *** p < 0.001. t-test.
Because 6b inhibits cell growth, it is likely that it causes cell cycle arrest. To test this, we treated the HCC1806 and HCC1937 cells with 6b at concentrations of 0, 0.05, 0.1 and 0.2 μM for 48 hours and measured cell cycle progression using flow cytometry. The proportion of cells in the G1 phase increased in a dose-dependent manner after the administration of 6b (Fig. 3B). We then examined the protein levels of cell cycle regulators. Treatment with 6b decreased cyclinD1 expression and increased the levels of p21 and p27 but not CDK4/6 (Fig. 3C). These results demonstrate that 6b induces cell cycle arrest in the G1 phase in BLBC cells.
To further investigate whether 6b inhibits cell proliferation, we chose HCC1806 and HCC1937 cell lines to measure DNA synthesis using the EdU incorporation assay. As shown in Fig. 3D, 6b gradually inhibited DNA synthesis in both cancer cell lines in a dose-dependent manner. Furthermore, treating the cells with 0.05 μM 6b almost eliminated colony formation, as determined by colony formation assays (Fig. 3E). These results indicate that 6b has a strong anti-proliferative effect on BLBC cells.
6b promotes BRD4 ubiquitination and degradation dependent on CRBN
As a PROTAC molecule, 6b recruits CUL4CRBN to ubiquitinate BRD4 and induces its proteasomal degradation. We treated HCC1806 and HCC1937 cells with the proteasome inhibitor, MG132. As expected, the 6b-induced decrease in BRD4 expression was blocked (Fig. 4A). Furthermore, we demonstrated that 6b increased the ubiquitination of exogenous and endogenous BRD4 (Fig. 4B, C).
Fig. 4. 6b promotes BRD4 ubiquitination and degradation dependent on CRBN.

A HCC1937 and HCC1806 cells were treated with indicated 6b and at the same time MG132 (3 μM) were added for 8 h. Cell lysates were collected for WB. B HEK293 cells were co-transfected with plasmids expressing 3×Flag-BRD4 and HA-Ub for 2 days. Flag-BRD4 was immunoprecipitated with anti-Flag M2 beads under denaturing conditions. Ubiquitinated BRD4 proteins were detected by anti-BRD4 antibody. C HCC1937 and HCC1806 cells were treated with 6b (0-0.01-0.1 μM) for 48 h and then MG132 (20 μM) were added for the next 8 h. Cell lysates were collected for WB. D CRBN was knocked down by two different siRNAs in HCC1937 and HCC1806 cells, as analyzed by RT-qPCR. E. CRBN was knocked down by two different siRNAs in HCC1937 and HCC1806 cells for 48 h. 48 h later, cells were treated with 6b for indicated concentration, and cell lysates were collected for WB. F. CRBN knockdown by two different siRNAs abrogated the inhibitory effects of 6b in HCC1937 and HCC1806 cells. Cell viability was measured by the SRB assay. *p < 0.05, **p < 0.01. ***p < 0.001. t-test.
To elucidate the role of CRBN in 6b-induced BRD4 degradation, we knocked down CRBN in HCC1806 and HCC1937 cells (Fig. 4D). CRBN knockdown blocked 6b-induced BRD4 degradation (Fig. 4E) and significantly decreased the cytotoxicity of 6b in BLBC cells (Fig. 4F). These results suggest that the 6b-mediated BRD4 degradation and BLBC growth inhibition depends on CRBN.
Overexpression of BRD4 rescues 6b-induced BLBC cell growth inhibition
To confirm the role of 6b in promoting BRD4 degradation, we tested whether ectopic BRD4 expression could rescue the 6b-induced changes in BRD4 downstream target genes and growth inhibition using HCC1806 and HCC1937 cells stably expressing BRD4 (Fig. 5A). Consistent with the increased expression levels of BRD4, the downregulation of KLF5, c-MYC, and KLF5 target genes, including FGF-BP1, and Cyclin D1 proteins, by 6b was partially restored. Consistently, the cell growth inhibition caused by 6b was significantly rescued by stable overexpression of BRD4 in both BLBC cell lines (Fig. 5B). Overexpression of BRD4 in BLBC cells also rescued them from 6b-induced cell cycle arrest, as determined by flow cytometry (Fig. 5C, D). BRD4 overexpression reduced G1 arrest and p21 protein accumulation. Collectively, these results demonstrated that 6b induced cell cycle arrest and that BLBC cell growth inhibition mainly relied on targeting BRD4 for degradation.
Fig. 5. BRD4 overexpression rescue 6b induced BLBC growth.

A HCC1937 and HCC1806 cells were infected with expressing an empty vector pCDH or BRD4 were analyzed for expression of the transduced proteins by WB using antibodies against BRD4. GAPDH was used to show equal protein loading. Relative growth is shown below. B HCC1937 and HCC1806 wild-type or stable overexpression BRD4 cells were treated with 6b 0-0.8-1.6 μM for 72 h and cell viability was measured by SRB assay. C HCC1937 and HCC1806 cells stably expressing vector control or exogenous BRD4 were treated with 0.2 cc of 6b for 48 h and stained with propidium iodide for cell-cycle analysis. The data represent one of three independent experiments. Statistic analyze were shown below. D BRD4 overexpression suppressed the expression increase of cell cycle protein p21 by 6b. *p < 0.05, **p < 0.01. ***p < 0.001. t-test.
6b suppresses BLBC tumor growth in mice
To investigate the functional role of 6b in suppressing BLBC tumor growth, we inoculated HCC1806 cells into nude mice and treated them with 5 or 10 mg/kg 6b every two days. A 5 mg/kg dose of 6b inhibited tumor growth (Fig. 6A). The tumor growth curves are shown in Fig. 6B and C. No apparent changes were observed in the mouse body weight for any of the treatments (Fig. 6C). We detected KLF5, BRD4, and Ki-67 protein levels in mouse xenograft tissues by immunohistochemistry and found that 6b dramatically decreased the expression of these proteins (Fig. 6D). Finally, we detected BRD4, Ki67, and KLF5 expression levels in tumors treated with control or 6b (Fig. 6E). The BRD4 and Ki67 levels in tumors treated with 10 mg/kg were lower than those in control group.
Fig. 6. 6b significantly suppressed HCC1806 xenograft and UM1 patient-derived xenograft tumor growth in nude mice.

A Gross morphology of HCC1806 xenografts tumor after intraperitoneal injection with 6b (5 mg or 10 mg kg−1). DMSO is a negative control for 24 days (n = 6). B 6b significantly inhibited tumor volumes. C 6b significantly decreased tumor weights. D 6b did not significantly change mouse body weights. E. The KLF5, BRD4, and Ki-67 levels were quantified by Image J after staining with specific antibodies in paraffin-embedded tissues, scale bar equals 50 μM. F Gross morphology of UM1 xenografts tumor after mammary fat pad implantation with 6b (5 mg or 10 mg kg−1). DMSO serves as a negative control (n = 6). G 6b significantly inhibited UM1 tumor growth. H 6b significantly decreased tumor weights I. 6b did not affect the mouse bodyweight. J The KLF5, BRD4, and Ki-67 levels were quantified by Image J after staining with specific antibodies in paraffin-embedded tissues, scale bar equals 50 μM. The data are represented as means ± SD. n ≥ 6 for mice in each group. (*p < 0.05, **p < 0.01. ***p < 0.001. t-test).
We determined whether 6b suppresses growth of a BLBC patient-derived xenograft (PDX). We established UM1 PDX xenografts and treated the mice with 5 or 10 mg/kg 6b every two days. As expected, 6b significantly inhibited UM1 tumor growth (Fig. 6F–I) in nude mice without affecting the body weight of the mice (Fig. 6J). We detected KLF5, BRD4, and Ki-67 protein levels in mouse xenograft tissues by immunohistochemistry. 6b showed a good inhibitory effect on UM1 PDX model in terms of both tumor growth and degradation of target protein (Fig. 6F, H, and J).
6b suppressed BLBC cell growth through KLF5 in part
KLF5 is an important transcription factor that maintains BLBC growth and is a BRD4 downstream target gene [41]. KLF5 regulates the transcription of downstream target genes, such as FGF-BP1, Cyclin D1, and p21. We studied whether ectopic expression of KLF5 could rescue 6b induced growth inhibition and whether stable knockdown of KLF5 could sensitize BLBC cells to 6b. We generated HCC1806 and HCC1937 cells stably overexpressing KLF5 or stably depleting KLF5. Consistent with previous results in Fig. 2D, we use 0.01 and 0.1 μM 6b to treat cells and detect protein expression by Western blotting. However, these concentrations were not ideal for detecting cell viability (HCC1937 IC50 = 1.7 μM; HCC1806 IC50 = 1.3 μM) and higher concentrations (0.8-1.6 μM) of 6b were more appropriate for this assay. As expected, KLF5 overexpression dramatically increased the BLBC cell viability in the presence of 6b (Fig. 7A, E). Stable knockdown of KLF5 sensitized BLBC cells to 6b (Fig. 7B, E). Consistent with previous results, KLF5 overexpression and depletion positively changed the expression of FGF-BP1. We found that 6b regulated the expression of KLF5 downstream target genes, including FGF-BP1, p27, and p21 (Fig. 7C).
Fig. 7. 6b inhibits BLBC growth through KLF5 in part.

A HCC1937 and HCC1806 cells were infected with expressing empty vector pCDH or KLF5 and were analyzed for expression of the transduced proteins by WB using antibodies against KLF5. GAPDH was used to show equal protein loading. Relative growth is shown below. B HCC1937 and HCC1806 cells were infected with shRNA expressing an empty vector shControl or shKLF5-3’UTR/#5/#7 were analyzed for expression. GAPDH was used to show equal protein loading. Relative growth is shown below. C HCC1937 and HCC1806 cells were treated with 0-0.25-0.5-1 μM of 6b for 48 h and protein expression was analyzed by WB using indicated antibodies. D HCC1937 and HCC1806 cells were treated with 0.1-1 μM of 6b with FZU004, EPI, or PTX for 48 h and protein expression was analyzed by WB using indicated antibodies. Cell viability shown below is measured by SRB assays, the quantitative data are compared to negative control. E Cells stably over-expressing or knockdown KLF5 relative growth was detected by SRB assay. F HCC1806 cells were treated with 6b plus FZU004 or EPI for 48 h, stained with Annexin V/7AAD, and analyzed by flow cytometry, the quantitative data and percentages of Annexin V-positive cells are shown. *p < 0.05, **p < 0.01. ***p < 0.001. t-test.
The combination of 6b with KLF5 inhibitor shows better antitumor effects in BLBC
We previously reported that FZU-00,04 [43] and the chemotherapeutic drugs EPI and PTX [44] inhibit KLF5 expression through different mechanisms. We wondered whether 6b, in combination with these compounds, would better inhibit BLBC. To test this hypothesis, the HCC1806 and HCC1937 cells were treated with these combinations. As shown in Fig. 7D, the combination of 6b with the three drugs additively inhibited the expression of KLF5 and its target genes, FGF-BP1 and CyclinD1. The combination of 6b with the three drugs consistently decreased the viability of BLBC cells. Based on the above, the addition of these compounds could enhance 6b anticancer property, use flow cytometry determine HCC1806 apoptosis rate (Fig. 7F) made clear results that KLF5 has a major role in the BLBC growth, 6b can be used as a potential degrader to inhibit BLBC.
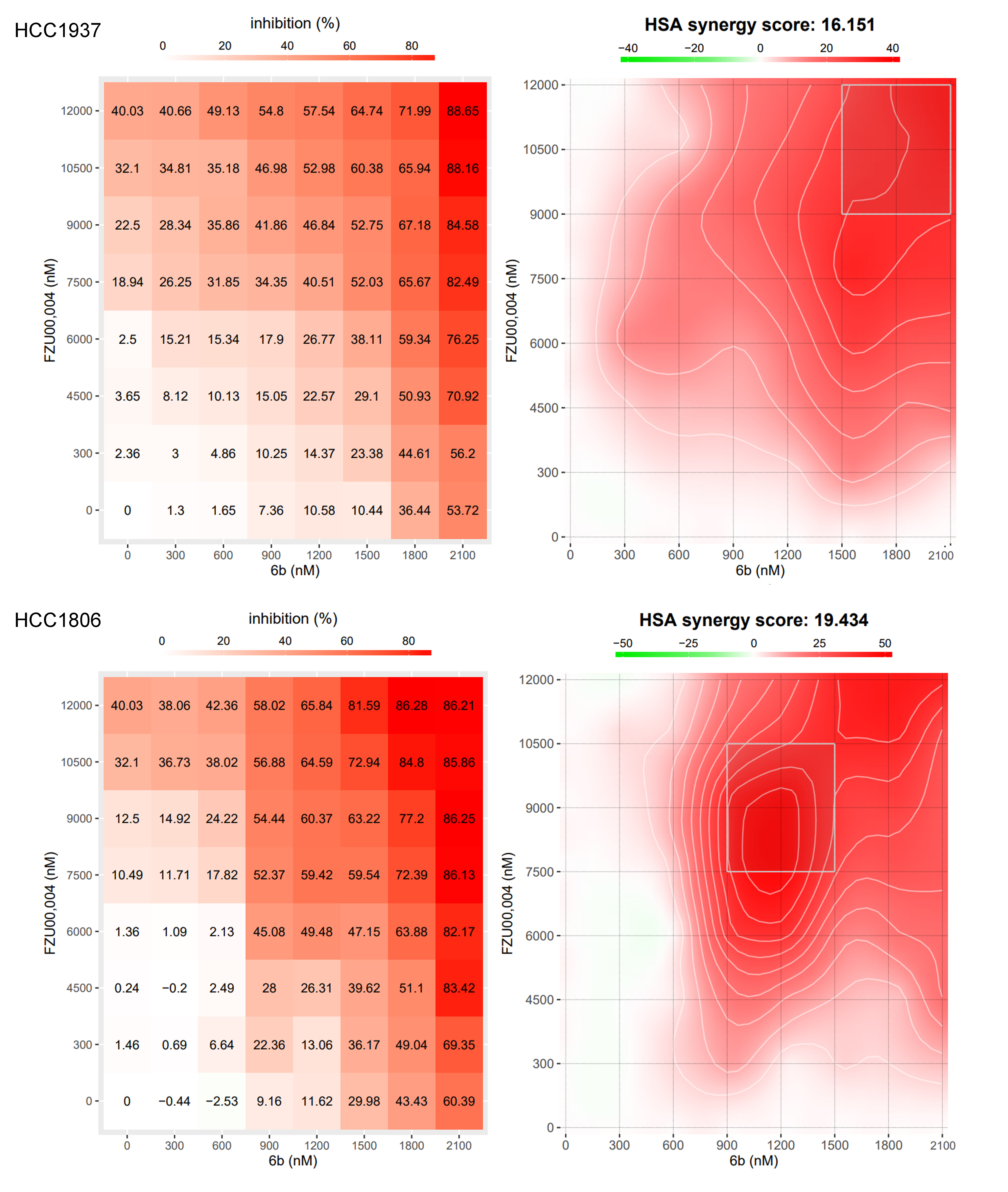
To test whether 6b in combination with KLF5 inhibitors, will better inhibit BLBC in vivo, we selected implanted HCC1806 cells into the breast fat pad of nude mice and treated them with 5 mg/kg 6b, 50 mg/kg FZU-00,004, or in combination every day. Monotherapy with 6b or FZU-00,004 could moderately reduce tumor growth, compared with monotherapy, combination slightly decreased tumor volume but was not statistically significant when measured in vivo (Fig. 8B). After final treatment, we then sacrificed the mice and weighed the wet weight of tumor, the combination therapy could modestly reduce tumor growth (Fig. 8D). No apparent changes were observed in the mouse body weight for any of the treatments (Fig. 8C and E). We detected KLF5, BRD4, and Ki-67 protein levels in xenograft tissues by immunohistochemistry and found that both monotherapy and combination therapy could decrease the expression of these proteins (Fig. 8F, G). We further test whether 6b and FZU-00,004 exhibited synergistic effects in BLBC. The SRB assay was employed to measure the cell viability inhibition when treated with 6b PROTAC and FZU-00,004. The analysis of combination effects was performed using the SynergyFinder web application 2.0 (https://synergyfinder.fimm.fi/). As anticipated, synergistic effects were identified for 6b PROTAC in combination with FZU-00,004 in HCC1937 and HCC1806 cell lines, with synergy scores of 16.2 and 19.4, respectively (Fig S3). The heatmap illustrated that the least synergistic concentration of 6b and FZU00,004 was 1.5 μM and 9 μM in HCC1937, and 0.9 μM and 7.5 μM in HCC1806. These findings suggest that KLF5 blockade can partially potentiate the efficacy of 6b in antineoplastic therapy.
Fig. 8. The combination of 6b with KLF5 inhibitor shows a better antitumor effect in the HCC1806 xenograft model.

A Gross morphology of HCC1806 xenograft tumors after mammary fat pad implantation with 6b (5 mg kg−1) or FZU-00,004 (50 mg kg−1) or combination (6b: 5 mg kg−1, FZU-00,004: 50 mg kg−1). DMSO serves as a negative control (n = 6). B 6b, FZU-00,004 or 6b+FZU inhibited tumor volumes, growth curves (mean ± SEM). C 6b, FZU-00,004 or 6b+FZU did not significantly change mouse body weights. D 6b, FZU-00,004, or 6b+FZU significantly decreased tumor weights. E. The mice were weighed at the end of the experiment. F, G The KLF5, BRD4 and Ki-67 levels were quantified by Image J after staining with specific antibodies in paraffin-embedded tissues, the scale bar equals 50 μM. The data are represented as means SD. n ≥ 6 for mice in each group. (*p < 0.05, **p < 0.01. ***p < 0.001. t-test).
Discussion
In this study, we investigated the selective, efficient, and therapeutic potential of the small-molecule BRD4 degrader 6b in BLBC, which exhibited excellent growth-inhibitory activity in the majority of BLBC cell lines evaluated. We demonstrated that 6b specifically recruits BRD4 to CUL4CRBN E3 ligase for ubiquitination and degradation in BLBC. Importantly, 6b inhibited BLBC growth via BRD4 and its downstream target KLF5. Compound 6b exhibited anti-BLBC activity in vivo without significant toxicity. The combination of KLF5 inhibitors and 6b showed additive effects on BLBC. Therefore, 6b is a leading compound with potential for use as an anticancer drug.
Therefore, BRD4 is considered a therapeutic target for BLBC. Previous studies have shown that BRD4 inhibitors, such as JQ-1, and BRD4 degraders, such as MZ1 and ARV-825, largely induce G2/M arrest and have profound effects on caspase-dependent apoptosis [26]. Our data show that a key difference in the actions between the BET protein degrader and 6b in BLBC cells is the robust induction of apoptosis by the BET degrader and the minimal to moderate induction of apoptosis by 6b. 6b induced G1 phase cell cycle arrest (Fig. 3B) with minimal apoptosis (data not shown). Compared with another BRD4 selective degrader, ZHX3-26, 6b showed more potent growth inhibition in xenograft mouse tumor models, effectively suppressing BLBC tumor growth at well-tolerated dosing schedules. Compared with dBET57, 6b was more selective in BRD4 degradation. This study demonstrates that BRD4-selective degrader 6b can effectively and safely inhibit BLBC growth both in vitro and in vivo.
KLF5, an important oncogenic transcription factor in BLBC, was downregulated by 6b. We have previously reported that KLF5 is controlled by a super-enhancer and BRD4 in BLBC. BRD4 inhibitors such as JQ-1 and compound 870 strongly downregulate KLF5 expression in BLBC cells. 6b consistently inhibited the expression of KLF5 and its downstream target gene FGF-BP1 in BLBC cells. More importantly, KLF5 overexpression partially rescued the inhibitory effect of 6b, and KLF5 knockdown by shRNA enhanced the cytotoxicity of 6b on BLBC cells (Fig. 7). These data suggest that KLF5 is a key mediator of BET degrader-induced growth arrest in BLBC cells. Therefore, it would be worth designing a polymerized PROTAC to target both KLF5 and BRD4 in BLBC.
ARV-471, the first oral PROTAC targeting ERα to enter clinical trials, has inspired a greater enthusiasm for breast cancer treatment. All efforts are now directed toward its bioavailability, systemic exposure, and stability in clinical testing. In the future, 6b, which has prolonged efficacy, increased potency, and a higher selectivity profile, needs to be tested in clinical trials to bring clinical benefit to patients. We plan to design a peptide based on the BRD4 protein structure with high binding affinity for accurate delivery of PROTAC to the tumor site and effective degradation of BRD4, using (AI)-aided peptide drug design using a hotspot-centric Rosetta computational approach.
Unlike small molecule drugs, PROTACs could overcome drug resistance by not directly binding target protein for a long time and by high strength. For example, targeted degradation of CDK4/6 overcomes CDK4/6 inhibitors resistance [45], and targeted BTK C481S mutated protein degradation could overcome the resistance of Ibrutinib caused by the mutation [46]. Recently, Guan et al [47]. developed a promising nucleic acid-based PROTAC that blocks transcription and targets the degradation of endogenous c-MYC/MAX. This PROTAC not only restrains BLBC cell proliferation but also suppresses BLBC tumor growth when administered in combination with palbociclib [47]. This new approach guides our future research direction. We may design a PROTAC that degrades both KLF5 and BRD4. In addition, the combination of PROTAC with either chemotherapy or antibody drugs may represent a promising alternative strategy for cancer therapy.
In summary, our study provides compelling preclinical data that selectively targeting BRD4 protein for degradation is a promising therapeutic strategy for BLBC. Compound 6b specifically recruits BRD4 for CUL4CRBN E3 ligase for ubiquitination and degradation in BLBC cells. It shows excellent anti-BLBC activity in vitro and in vivo which is dependent on the degradation of BRD4 and downregulation of KLF5. The clinical significance of 6b warrants further investigation.
Materials and Methods
Chemistry
The synthesis of 6b-1 and 6b-2 was referred to reported synthetic routes, Fig. S4 [48, 49]. To a solution of 6b-2 (430 mg, 1.2 mmol, 1.2 eq.) in N, N-dimethylformamide (5 mL), N, N-Diisopropylethylamine (0.49 mL, 3 mmol, 3.0 eq.) and HATU (456 mg, 1.2 mmol, 1.2 eq.) were added and the reaction mixture was stirred at room temperature for 1 h. Then, 6b-1 (367 mg, 1 mmol, 1.0 eq.) was added, and the reaction and stirred at room temperature for 4 h. The reaction mixture was poured into water (100 mL) and was extracted with ethyl acetate (3 ×100 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified via flash chromatography (PE/EA 1:1 - > 1:0) to yield 396 mg (56%) of the desired product 6b. 1H-NMR (CDCl3, 400 MHz): δ 10.36 (s, 1H), 9.09 (s, 1H), 8.21 (s, 1H), 7.55-7.52 (m, 2H), 7.41 (t, J = 7.48 Hz, 1H), 7.14-7.11 (m, 2H), 7.02-7.00 (m, 1H), 6.8-6.79 (m, 4H), 6.69 (t, J = 7.6 Hz, 1H), 6.36 (s, 1H), 6.29 (t, J = 5.36 Hz, 1H), 4.90-4.87 (m, 1H), 3.63 (s, 3H), 3.37-3.35 (m, 2H), 2.84-2.68 (m, 3H), 2.48 (t, J = 6.84 Hz, 2H), 2.07-2.00 (m, 3H) ppm; 13C-NMR (CDCl3, 100 MHz): δ 171.69, 169.77, 169.25, 167.63, 154.95, 150.64, 146.91, 136.37, 134.01, 132.49, 130.56 129.28, 127.72, 126.81, 123.70, 123.27, 120.51, 118.05, 116.89, 112.04, 111.84, 111.21, 111.17, 110.14, 105.16, 103.68, 53.55, 49.04, 41.83, 36.52, 34.14, 31.57, 31.52, 29.81, 29.47, 24.79, 22.87; LC-MS: calculated for C37H30F2N6O7 [M + H] + , 709.22; found 709.65.
Chemicals, reagents, and antibodies
MG132, and cycloheximide (CHX) were purchased from Sigma (St. Louis, MO, USA). FZU-00,004 was synthesized by Haijun Chen (College of Chemistry, Fuzhou University, China). Epirubicin, EPI purchased from Sigma-Aldrich (USA). Paclitaxel, PTX purchased from MedChemExpress (China).
Antibodies for BRD2 (D89B4) (#5848; 1:1000), BRD4 (E2A7X) (#13440; 1:1000), c-Myc (E5Q6W) (#18583; 1:1000), CyclinD1 (#55506; 1:1000) p21Waf1/Cip1 (12D1) (#2947; 1:2000), p27 (#2552; 1:1000), CDK4(D9G3E) (#12790; 1:1000), CDK6 (D4S8S) (#13331; 1:1000), SKP2(#4358; 1:1000), Bcl-2 (D55G8) (#4223; 1:1000), BCLxL(#2762; 1:1000), Bim(#2819; 1:1000), β-actin (13E5) (#4970; 1:5000), and GAPDH (14C10) (#2118; 1:5000) were purchased from Cell Signaling Technology. Antibody for BRD3 (#ab264294; 1:2000) were purchased from abcam. Antibodies for KLF5 (#AF3758, 1:1000) and FGF-BP1 (MAB1593; 1:500) were purchased from R&D Systems.
Cell culture
MCF10A, 184B5, HEK293T, HCC1937, HCC1806, MDA-MB-468, MDA-MB-231, SUM149PT, Hs578T cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) Cell lines were reauthenticated with short tandem repeat analysis and tested for mycoplasma contamination every 6 months after thawing in our experiments. HEK293T, Hs578T, MDA-MB-231, and MDA-MB-468 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS); HCC1937 and HCC1806 cells were cultured in Roswell Park Memorial Institute (RMPI)-1640 medium supplemented with 10% FBS; MCF10A and 184B5 cell lines were maintained in DMEM/F12 medium supplemented with EGF (10 ng/mL), insulin (10 μg/mL), cholera toxin (100 ng/mL), hydrocortisone (0.5 μg/mL) with 10% horse serum. SUM149PT cell lines were maintained in Ham’s F-12 medium supplemented with insulin (5 μg/mL), hydrocortisone (1 μg/mL), 10 mM HEPES with 10% FBS. All the cells were cultured at 37 °C in a humidified atmosphere with 5% CO2.
Lentivirus preparation and transfection
All transfections for plasmids and siRNAs were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were cultured at 5 × 105 cells/well in 6-well plates. After incubation for 24 h, the cells were transfected with CRBN siRNA at 25 nM final concentration, The siRNA target sequences for the human CRBN gene are siCRBN#1 5’- GCAGGACTTTGCACGATGA-3’, siCRBN#2 5’- GTAGCTGCTTGTCTTCCTA-3’. The sequence chosen for preparing the KLF5 shRNA construct in the pSIH1-H1-Puro shRNA vector were: shKLF5#5 5’- CGAUUACCCUGGUUGCACA-3’, shKLF5#7 5’- GAUGUGAAAUGGAGAAGUA-3’, shKLF5 3’UTR 5’- GCTGTAATGTATATGGCTTTA-3’. Lentiviruses prepared from HEK293FT cells. Stable knockdown cells were selected with puromycin (1 μg/mL; Sigma) in cell culture media for 48 h after transfection. Cell lysates were then collected, and protein expression was detected by Western blotting (WB). Full-length wild-type human KLF5 or BRD4 were cloned into pCDH vectors. Lentiviruses expressing KLF5 or BRD4 were packaged and applied to infect target cells.
Ubiquitination assays
Plasmids were transfected into HEK293T cells for 48 h. Cells were harvested in lysis buffer (50 mM Tris-Cl and 1.5% SDS; pH 6.8) using a six-well plate. Each well contained 150 μL lysis buffer. The cell lysate was boiled for 15 min to denature proteins. BSA buffer (50 mM Tris-Cl, 180 mM NaCl, 0.5% CA630, and 0.5% BSA; pH 6.8; 1.2 mL cell−1) was added to dilute the samples. Flag-M2 beads (30 μL per sample; prewashed with BSA buffer 3×) were added to immunoprecipitate Flag-BRD4 overnight with rotation in a cold room (4 °C). Beads were washed 5× with 1 mL ice-cold BSA buffer, resuspended in 30 μL of 1× SDS-PAGE sample buffer, boiled for 7 min, and centrifuged for 2 min at 12,000 g. The supernatant was subjected to a Western blot.
Endogenous ubiquitination, performed by HCC1937 or HCC1806 cells was treated with 6b (0-0.01-0.1 μM). After 2 days, the cells were treated with 20 μM MG132 for 8 h to enrich polyubiquitinated BRD4 proteins. The cells were harvested using the same method above. Protein A/G beads (30 μL per sample; prewashed with BSA buffer three times) were added to cell lysates were immunoprecipitated with the anti-BRD4 antibody with rotation in a cold room (4 °C) and analyzed by immunoblotting with anti-BRD4 to detect ubiquitination.
Colony formation assay
HCC1937 or HCC1806 cells were seeded into 6-well plates (2000 cells/well), allowed to attach overnight, changed with fresh medium containing with indicated 6b concentration, compared with DMSO (vehicle), and incubated for 9 days (change fresh medium every three days). Then the colonies were fixed with methanol for 30 min and stained with 0.1% crystal violet for 15 min. After that, the cells were washed with PBS and the colonies (>50 cells) were photographed.
EdU Assay
HCC1937 or HCC1806 cells incubated with different concentrations of 6b were inoculated into 6-well plates, and the prepared EdU (Invitrogen, Carlsbad, CA) working solution was added to label the cells and then incubated for 2 h. Then cells were fixed with 4% paraformaldehyde in a humidified incubator, and the prepared solution was added after removing the fixative. Cells were incubated with the permeabilizer for 10 min, then add click reaction solution and Hoechst 33342 to cover the surface of cells and incubated against light.
Apoptosis and cell cycle
HCC1937 or HCC1806 cells treated with different concentrations of 6b. Apoptosis was detected by using FITC Annexin V Apoptosis Detection Kit I (BD Biosciences, San Jose, CA, USA). Cells were collected, washed with PBS, and then re-suspend in 1× binding buffer with 5 μL of PE Annexin V for 5 min and 5 μl of 7-AAD for 15 min in the dark at 37 °C in the dark, 400 μL of 1× binding buffer was added to each sample. The samples were analyzed via flow cytometry.
Cell cycle analysis cells were collected, centrifuged at 1000 rpm for 5 min, washed, and fixed overnight at 4 °C in 75% ethanol. Washed with cold PBS and stained with the 0.5 mL PI/RNase Buffer (BD Biosciences, San Jose, CA, USA) at room temperature for 15 min, dark area. The percentage of cells in the G1, S, and G2/M phases of the cell cycle was analyzed by using a flow cytometer.
Xenograft nude mouse model
All experiments involving animals were handled according to the protocol (SMKX-20160305-08) approved by the Animal Ethics Committee of the Kunming Institute of Zoology, CAS. All the mice were maintained in a temperature-controlled and pathogen-free environment with 12 h light/dark cycles and access to food and water ad libitum. All the animal experiments were performed in accordance with relevant guidelines and local regulations. Each group contains ≥ 6 animals, this study does not involve the extent of blinding.
HCC1806 cells were resuspended in matrigel (Becton Dickinson, Franklin Lakes, NJ) with 1640 medium (1:1 volumetric mixture) and injected subcutaneously into the fat pads (Fig. 6A) or mammary fat pad implantation (Fig. 6F and Fig. 8: 0.1 mL or 1 × 106 cells per injection site) of female BALB/c nude mice (4–6 weeks). Tumor sizes were calculated using the formula (0.5 × L × W). When the tumors grew to ∼20 mm3, the mice were randomly distributed into three groups and administered either 6b (5 or 10 mg kg−1) or DMSO control (5 mg kg−1) by intraperitoneal injection once every 2 days. we add in order 10% DMSO (containing 6b), 40% PEG300, 5% Tween-80, 45% Saline, the solubility was good. Tumor volumes were measured with calipers once every 2 days. After 25 days, the mice were sacrificed and their tumor xenografts were immediately dissected.
The ERα-PR-HER2- invasive ductal carcinoma UM1 patient-derived xenograft tissues, which were maintained in fat-pads of BALB/c nude mice without culture in vitro, were collected and dissociated as described previously [50]. 1 × 106 UM1 cell implanted into mammary fat pads of the BALB/c nude mice (4–6 weeks). Tumor sizes were calculated using the formula (0.5 × L × W). When the tumors grew to ∼20 mm3, the mice were randomly distributed into three groups and administered either 6b (5 or 10 mg kg−1) or DMSO control (5 mg kg−1) by intraperitoneal injection once every 2 days. Tumor volumes were measured with calipers once every 2 days. After 15 days, the mice were sacrificed and their tumor xenografts were immediately dissected.
Immunohistochemistry (IHC) assay
For KLF5, BRD4, and Ki67 staining, the slides were deparaffinized, rehydrated, and the pressure cooker heated for 2.5 min in EDTA for antigen retrieval. Endogenous peroxidase activity was inactivated by adding an endogenous peroxidase blocker (Ori-Gene, Beijing, China) for 15 min at room temperature. Slides were incubated overnight at 4 °C with anti-KLF5 (1:1000) or anti-BRD4 (1:1000). After 12 h, the slides were washed three times with PBS and incubated with secondary antibodies (hypersensitive enzyme-labeled goat anti-mouse/rabbit IgG polymer (Ori-Gene, Beijing, China) at room temperature for 20 min, DAB concentrate chromogenic solution (1:200 dilution of concentrated DAB chromogenic solution), counterstained with 0.5% hematoxylin, dehydrated with graded concentrations of ethanol for 3 min each (70–80%–90–100%), and finally stained with dimethyl benzene immune stained slides were evaluated by light microscopy. The IHC signal was scored using the ‘Allred Score’ method.
Statistics
All experiments were repeated three times independently, and GraphPad Prism 8.0 was used for data analysis. Students’ t-tests were employed when the variance between two groups was similar, while Welch’s t-tests were utilized if the variance was not the same. The subcutaneous tumor growth curves were compared using two-way ANOVA. statistical significance was indicated in the figures as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Author contributions
Ceshi Chen: Supervision, Project administration, Funding acquisition. Yu Rao: Supervision, Project administration, Funding acquisition. Xia Liu: Supervision. Yanjie Kong: Writing original draft, Investigation, Funding acquisition. TianlongLan: Investigation. Luzhen Wang: Investigation. Chen Gong: Investigation. Wenxin Lv: Investigation. Hailin Zhang: Investigation. Chengang Zhou: Investigation. Xiuyun Sun: Investigation. Wenjing Liu: Investigation. Haihui Huang: Investigation. Xin Weng: Investigation. Chang Cai: Investigation. Wenfeng Peng: Investigation. Meng Zhang: Investigation. Dewei Jiang: Investigation. Chuanyu Yang: Investigation.
Funding
This study was supported in part by grants National Key Research and Development Program of China (2020YFA0112300 to Chen, C. 2020YFE0202200, 2021YFA1300200 to Rao, Y.), The National Nature Science Foundation of China (U2102203 to Chen, C., 82273216 to Kong, Y., 82125034 to Rao, Y.). Shenzhen Science and Technology program (RCYX20221008092911040 to Kong, Y.), Shenzhen Municipal Government of China (JCYJ20210324103603011 to Kong, Y.), Shenzhen Planned Projects for Post-doctors Stand out Research Funds to Kong, Y., Guangdong Basic and Applied Basic Research Foundation (2022A1515220051 to Kong, Y.). Biomedical Projects of Yunnan Key Science and Technology Program (202302AA310046), Yunnan Fundamental Research Projects (202101AS070050), and Yunnan Revitalization Talent Support Program (Yunling Shcolar Project to CC). Yunnan (Kunming) Academician Expert Workstation (grant No. YSZJGZZ-2020025 to CC).
Data availability
Data will be made available on request.
Competing interests
The author declares no competing interests.
Ethics statement
All methods were performed in accordance with the relevant guidelines and regulations (reference number: SMKX-20160305-08) approved by the Animal Ethics Committee of the Kunming Institute of Zoology, CAS and informed consent was obtained from all participants. This work does not involve human research.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xia Liu, Email: mdcg2014bl@163.com.
Yu Rao, Email: yrao@tsinghua.edu.cn.
Ceshi Chen, Email: chenc@mail.kiz.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41388-024-03121-1.
References
- 1.Kim C, Gao R, Sei E, Brandt R, Hartman J, Hatschek T, et al. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173:879–893.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao J, Hou B, Zhu Q, Yang L, Jiang X, Zou Z, et al. Engineered bioorthogonal POLY-PROTAC nanoparticles for tumour-specific protein degradation and precise cancer therapy. Nat Commun. 2022;13:4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y, Xu B, Shi J, Li J, Lu X, Xu L, et al. BRD4 modulates vulnerability of triple-negative breast cancer to targeting of integrin-dependent signaling pathways. Cell Oncol. 2020;43:1049–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med. 2018;24:1599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duan W, Yu M, Chen J. BRD4: New hope in the battle against glioblastoma. Pharmacol Res. 2023;191:106767. [DOI] [PubMed] [Google Scholar]
- 6.Djamai H, Berrou J, Dupont M, Coude MM, Delord M, Clappier E, et al., Biological effects of BET inhibition by OTX015 (MK-8628) and JQ1 in NPM1-mutated (NPM1c) Acute Myeloid Leukemia (AML). Biomedicines, 2021. 9. [DOI] [PMC free article] [PubMed]
- 7.Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanz-Alvarez M, Cristobal I, Luque M, Santos A, Zazo S, Madoz-Gurpide J, et al., Expression of phosphorylated BRD4 is markedly associated with the activation status of the PP2A pathway and shows a strong prognostic value in triple negative breast cancer patients. Cancers, 2021. 13. [DOI] [PMC free article] [PubMed]
- 9.Andrieu G, Tran AH, Strissel KJ, Denis GV. BRD4 regulates breast cancer dissemination through Jagged1/Notch1 signaling. Cancer Res. 2016;76:6555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu L, Chen Z, Lin X, Tian L, Su Q, An P, et al. Inhibition of BRD4 suppresses the malignancy of breast cancer cells via regulation of Snail. Cell Death Differ. 2020;27:255–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AM, et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc Natl Acad Sci USA. 2016;113:7124–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai L, Zhou B, Yang CY, Ji J, McEachern D, Przybranowski S, et al. Targeted degradation of BET proteins in triple-negative breast cancer. Cancer Res. 2017;77:2476–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, et al. Hijacking the E3 Ubiquitin Ligase Cereblon to efficiently target BRD4. Chem Biol. 2015;22:755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohoka N, Ujikawa O, Shimokawa K, Sameshima T, Shibata N, Hattori T, et al. Different degradation mechanisms of inhibitor of Apoptosis Proteins (IAPs) by the Specific and Nongenetic IAP-Dependent Protein Eraser (SNIPER). Chem Pharm Bull (Tokyo). 2019;67:203–9. [DOI] [PubMed] [Google Scholar]
- 16.Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2-recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79:251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohoka N, Tsuji G, Shoda T, Fujisato T, Kurihara M, Demizu Y, et al. Development of small molecule chimeras that recruit AhR E3 ligase to target proteins. ACS Chem Biol. 2019;14:2822–32. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nat Chem Biol. 2019;15:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spradlin JN, Hu X, Ward CC, Brittain SM, Jones MD, Ou L, et al. Harnessing the anti-cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol. 2019;15:747–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward CC, Kleinman JI, Brittain SM, Lee PS, Chung CYS, Kim K, et al. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem Biol. 2019;14:2430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Q, Ding X, Huang T, Zhang S, Li Y, Xu L, et al. BRD4 degrader ARV-825 produces long-lasting loss of BRD4 protein and exhibits potent efficacy against cholangiocarcinoma cells. Am J Transl Res. 2019;11:5728–39. [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Xue J, Li J. Chemical degrader enhances the treatment of androgen receptor-positive triple-negative breast cancer. Arch Biochem Biophys. 2022;721:109194. [DOI] [PubMed] [Google Scholar]
- 23.Qin H, Zhang Y, Lou Y, Pan Z, Song F, Liu Y, et al. Overview of PROTACs targeting the estrogen receptor: achievements for biological and drug discovery. Curr Med Chem. 2022;29:3922–44. [DOI] [PubMed] [Google Scholar]
- 24.Wang F, Liu H, Blanton WP, Belkina A, Lebrasseur NK, Denis GV. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem J. 2009;425:71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houzelstein D, Bullock SL, Lynch DE, Grigorieva EF, Wilson VA, Beddington RS. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol Cell Biol. 2002;22:3794–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noblejas-Lopez MDM, Nieto-Jimenez C, Burgos M, Gomez-Juarez M, Montero JC, Esparis-Ogando A, et al. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. 2019;38:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He S, Gao F, Ma J, Ma H, Dong G, Sheng C. Aptamer-PROTAC Conjugates (APCs) for tumor-specific targeting in breast cancer. Angew Chem Int Ed Engl. 2021;60:23299–305. [DOI] [PubMed] [Google Scholar]
- 28.Wei J, Meng F, Park KS, Yim H, Velez J, Kumar P, et al. Harnessing the E3 Ligase KEAP1 for targeted protein degradation. J Am Chem Soc. 2021;143:15073–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan Z, Lyu X, Lin D, Wu G, Gong Y, Ren X, et al. Selective degradation of cellular BRD3 and BRD4-L promoted by PROTAC molecules in six cancer cell lines. Eur J Med Chem. 2023;254:115381. [DOI] [PubMed] [Google Scholar]
- 30.Sarnik J, Poplawski T, Tokarz P. BET Proteins as attractive targets for cancer therapeutics. Int J Mol Sci, 2021. 22. [DOI] [PMC free article] [PubMed]
- 31.Wroblewski M, Scheller-Wendorff M, Udonta F, Bauer R, Schlichting J, Zhao L, et al. BET-inhibition by JQ1 promotes proliferation and self-renewal capacity of hematopoietic stem cells. Haematologica. 2018;103:939–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol. 2018;14:706–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jia SQ, Zhuo R, Zhang ZM, Yang Y, Tao YF, Wang JW, et al. The BRD4 inhibitor dBET57 exerts anticancer effects by targeting superenhancer-related genes in neuroblastoma. J Immunol Res. 2022;2022:7945884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zengerle M, Chan KH, Ciulli A. Selective small molecule induced degradation of the BET Bromodomain Protein BRD4. ACS Chem Biol. 2015;10:1770–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo Y, Chen C. The roles and regulation of the KLF5 transcription factor in cancers. Cancer Sci. 2021;112:2097–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takagi K, Miki Y, Onodera Y, Nakamura Y, Ishida T, Watanabe M, et al. Kruppel-like factor 5 in human breast carcinoma: a potent prognostic factor induced by androgens. Endocr Relat Cancer. 2012;19:741–50. [DOI] [PubMed] [Google Scholar]
- 37.Zheng HQ, Zhou Z, Huang J, Chaudhury L, Dong JT, Chen C. Kruppel-like factor 5 promotes breast cell proliferation partially through upregulating the transcription of fibroblast growth factor binding protein 1. Oncogene. 2009;28:3702–13. [DOI] [PubMed] [Google Scholar]
- 38.Chen C, Benjamin MS, Sun X, Otto KB, Guo P, Dong XY, et al. KLF5 promotes cell proliferation and tumorigenesis through gene regulation and the TSU-Pr1 human bladder cancer cell line. Int J Cancer. 2006;118:1346–55. [DOI] [PubMed] [Google Scholar]
- 39.Wang C, Nie Z, Zhou Z, Zhang H, Liu R, Wu J, et al. The interplay between TEAD4 and KLF5 promotes breast cancer partially through inhibiting the transcription of p27Kip1. Oncotarget. 2015;6:17685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia H, Wang C, Chen W, Zhang H, Chaudhury L, Zhou Z, et al. Kruppel-like factor 5 transcription factor promotes microsomal prostaglandin E2 synthase 1 gene transcription in breast cancer. J Biol Chem. 2013;288:26731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CH, Yang N, Zhang Y, Ding J, Zhang W, Liu R, et al. Inhibition of super enhancer downregulates the expression of KLF5 in basal-like breast cancers. Int J Biol Sci. 2019;15:1733–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu Q, Liu Z, Gao Z, Luo Y, Li F, Yang C, et al. KLF5 inhibition potentiates anti-PD1 efficacy by enhancing CD8(+) T-cell-dependent antitumor immunity. Theranostics. 2023;13:1381–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui Q, Sun J, Yuan J, Li J, Yang C, Du G, et al. DNA damage chemotherapeutic drugs suppress basal-like breast cancer growth by down-regulating the transcription of the FOXO1-KLF5 axis. Genes Dis. 2024;11:91–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kargbo RB. Targeted degradation of CDK4/6: An innovative approach to overcoming cancer drug resistance. ACS Med Chem Lett. 2023;14:1162–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun Y, Zhao X, Ding N, Gao H, Wu Y, Yang Y, et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018;28:779–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X, Zhang Z, Gao F, Ma Y, Wei D, Lu Z, et al. c-Myc-targeting PROTAC based on a TNA-DNA bivalent binder for combination therapy of triple-negative breast cancer. J Am Chem Soc. 2023;145:9334–42. [DOI] [PubMed] [Google Scholar]
- 48.Liu H, Ding X, Liu L, Mi Q, Zhao Q, Shao Y, et al. Discovery of novel BCR-ABL PROTACs based on the cereblon E3 ligase design, synthesis, and biological evaluation. Eur J Med Chem. 2021;223:113645. [DOI] [PubMed] [Google Scholar]
- 49.McDaniel KF, Wang L, Soltwedel T, Fidanze SD, Hasvold LA, Liu D, et al. Discovery of N-(4-(2,4-Difluorophenoxy)-3-(6-methyl-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridin-4-yl)phenyl)ethanesulfonamide (ABBV-075/Mivebresib), a Potent and Orally Available Bromodomain and Extraterminal Domain (BET) Family Bromodomain Inhibitor. J Med Chem. 2017;60:8369–84. [DOI] [PubMed] [Google Scholar]
- 50.Liu R, Shi P, Nie Z, Liang H, Zhou Z, Chen W, et al. Mifepristone suppresses basal triple-negative breast cancer stem cells by down-regulating KLF5 Expression. Theranostics. 2016;6:533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.