

Significance
Synechococcus elongatus is a model for exploring circadian biology and photosynthetic processes. Here, we present a quantitative model of 57 independently modulated gene sets, or iModulons, that form the basis of the transcriptional regulatory network in the model strain PCC 7942. These iModulons not only validate known regulatory pathways but also illuminate complex metabolic networks crucial for environmental adaptation. By detailing components such as the photosystems, carbon concentrating mechanisms, and Calvin cycle, the research offers a nuanced view of photosynthesis regulation. Additionally, it proposes regulon structures and functional roles for lesser-understood genes, enhancing our understanding of cellular responses to dynamic light changes. This comprehensive mapping provides a foundational tool for future biological inquiries into transcriptional regulation in phototrophic organisms.
Keywords: machine learning, transcriptional regulatory network, cyanobacteria, carbon fixation, circadian rhythm
Abstract
Synechococcus elongatus is an important cyanobacterium that serves as a versatile and robust model for studying circadian biology and photosynthetic metabolism. Its transcriptional regulatory network (TRN) is of fundamental interest, as it orchestrates the cell’s adaptation to the environment, including its response to sunlight. Despite the previous characterization of constituent parts of the S. elongatus TRN, a comprehensive layout of its topology remains to be established. Here, we decomposed a compendium of 300 high-quality RNA sequencing datasets of the model strain PCC 7942 using independent component analysis. We obtained 57 independently modulated gene sets, or iModulons, that explain 67% of the variance in the transcriptional response and 1) accurately reflect the activity of known transcriptional regulations, 2) capture functional components of photosynthesis, 3) provide hypotheses for regulon structures and functional annotations of poorly characterized genes, and 4) describe the transcriptional shifts under dynamic light conditions. This transcriptome-wide analysis of S. elongatus provides a quantitative reconstruction of the TRN and presents a knowledge base that can guide future investigations. Our systems-level analysis also provides a global TRN structure for S. elongatus PCC 7942.
Synechococcus elongatus PCC 7942 is a prominent model cyanobacterium valuable for scientific research and industrial biotechnology. Its ability to sequester carbon dioxide through photosynthesis holds significant implications for addressing environmental concerns (1–3), and its ability to harness sunlight as a power source greatly lowers economic and environmental expenses for bioproduction (4). Furthermore, the genetic tractability of S. elongatus makes it an attractive candidate for genetic engineering and biotechnological applications (5–7). Knowledge about genetic regulation, metabolic pathways, and cellular responses is essential for harnessing the potential of S. elongatus for biotechnological applications. Consequently, gaining a holistic understanding of the transcriptional regulatory network (TRN) holds great significance as the TRN dictates the cell’s response to environmental variations such as light and darkness, CO2 limitation, nitrogen starvation, and oxidative stress. Despite the efforts toward characterizing individual transcription factors and their regulatory patterns, a global TRN structure of S. elongatus has yet to be established (8–10).
The development of high-throughput RNA sequencing (RNA-seq) technologies has facilitated the assembly of large transcriptomic datasets that can be used to understand microbial TRNs. A method based on independent component analysis (ICA), a technique that has been used for a long time in global gene expression profile analysis starting with microarray data (11), was developed to reverse engineer the TRN by capturing independently modulated sets of genes, also known as “iModulons.” This approach has demonstrated strong capabilities in reconstructing and elucidating microbial TRNs at a systems level (12). Using ICA, the gene expression matrix (X) can be decomposed into two matrices, the iModulon matrix M, and the activity matrix A. iModulons can be extracted from the M matrix, which contains the robust independent components, and the activity of each iModulon can be found in the A matrix, which reflects the level of regulatory activity in different conditions. An iModulon comprises genes with coordinated expression patterns, and iModulon activity represents the collective behavior of these genes across different conditions. Changes in iModulon activity reflect coordinated shifts in gene expression patterns, which often correspond to specific physiological responses, providing insights into the underlying biological processes.
Different from regulons, which describe sets of coregulated genes based on bottom–up biomolecular experiments, iModulons are identified via statistical methods, enabling a top–down analysis of the regulatory changes in the transcriptome. We can perceive iModulons as data-driven parallels of regulons that can offer additional insights into condition-specific activities. iModulons often correspond to known regulators or specific regulatory mechanisms, revealing biologically relevant relationships in gene expression data. This intrinsic biological significance ensures their robustness across diverse datasets and has enabled accurate reconstruction of complex microbial regulatory networks in dozens of organisms, even facilitating the transfer of cellular functions between species (13–18).
In this study, we employ iModulon analysis to explore the transcriptional regulatory landscape of S. elongatus PCC 7942 and construct a global TRN structure for this organism. By applying ICA to 300 high-quality, publicly available RNA-seq expression datasets for S. elongatus, we extracted 57 robust iModulons that explain 67% of the variance in the expression data. Through further examination of the iModulons, we highlight the biological insights they reveal by presenting 1) regulatory iModulons that represent the effects of defined regulons, 2) four iModulons that capture functional units of photosynthesis, 3) functional iModulons that expand our current understanding of biological processes or offer promising hypotheses, and 4) iModulon activity fluctuations under dynamic light conditions. All the iModulons from this study are available on https://imodulondb.org/ for further inspection, and the results can be recreated with the files and instructions provided in https://github.com/AnnieYuan21/S.elongatus-iModulons.
Results
ICA Decomposes the S. elongatus Transcriptome into 57 iModulons.
We gathered 424 RNA-seq expression profiles for S. elongatus, comprising 31 samples generated in-house and 393 publicly available samples from NCBI’s Sequence Read Archive (SRA) as of March 2023 (Fig. 1A) (19). The expression data were processed using a standardized pipeline (Methods). After quality control, the final compendium consists of 300 high-quality expression profiles spanning 158 unique experimental conditions (Fig. 1B). Applying ICA to this compendium resulted in the identification of 57 iModulons containing a total of 959 genes (Fig. 1C). These iModulons can be classified into five main categories: Regulatory, Genomic, Functional, Single Gene, and Uncharacterized (Fig. 1D). We can calculate the explained variance of each iModulon, which quantifies how much each iModulon contributes to the expression variation in our dataset and provides a measure of how well the expression of genes within an iModulon captures the overall transcriptional changes. The top 15 iModulons with the highest explained variance include those related to global regulators like RpaA, as well as iModulons associated with key biological processes such as photosynthesis and nitrogen metabolism (Fig. 1E). There are also two uncharacterized iModulons. Unchar_3 contains 42 genes and shows decreased activity in sigma factor knockout samples, suggesting a possible compensatory response to these perturbations. Unchar_11 comprises eight genes including a proposed transcription factor. Its activity is affected by relA perturbations and darkness conditions, but its function remains unknown. The explained variance and a detailed biological category of each iModulon can be found in Fig. 1F.
Fig. 1.
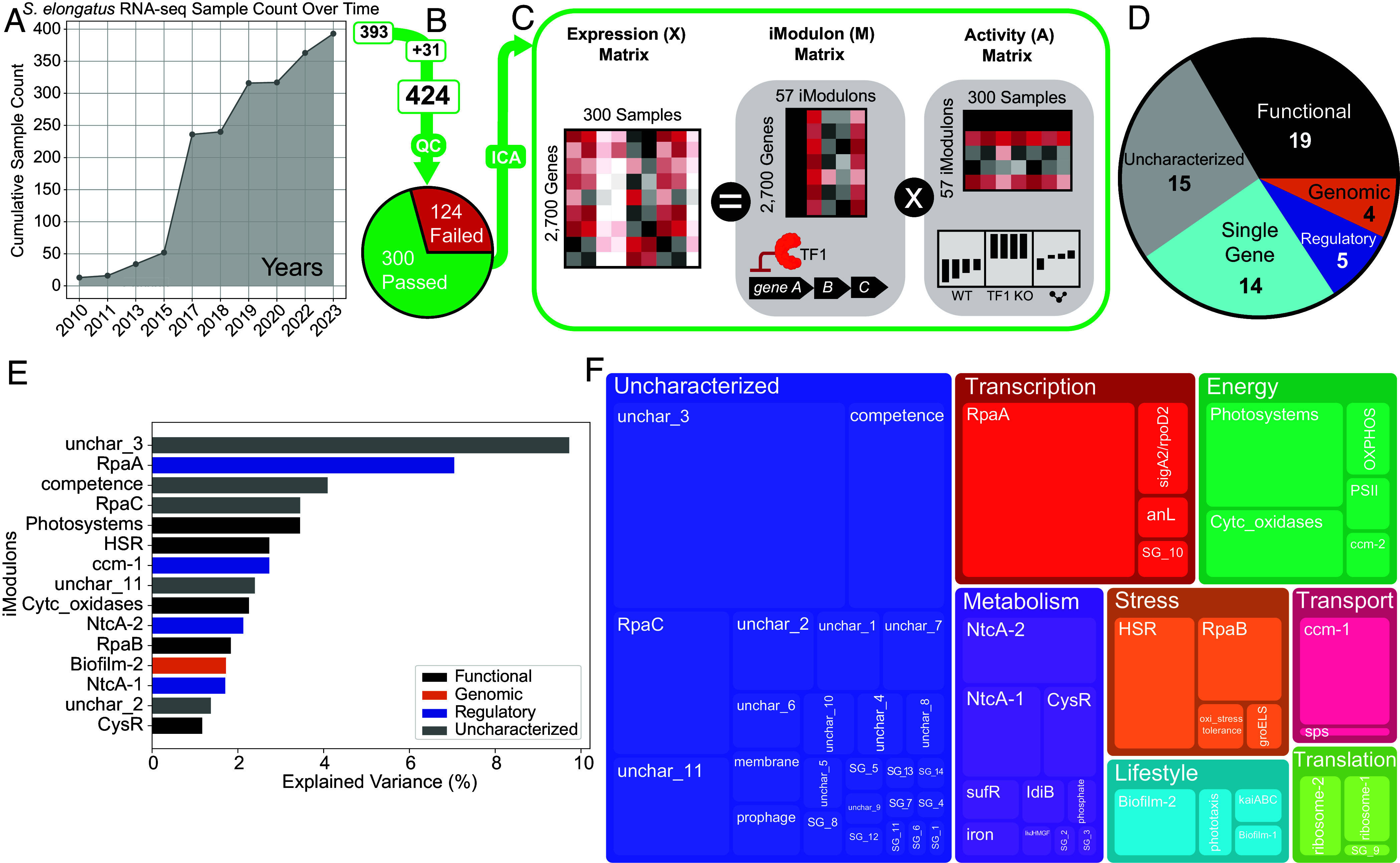
ICA decomposes the transcriptome of S. elongatus PCC 7942. (A) Line graph depicting the increase in publicly available high-throughput RNA-seq for S. elongatus as seen in NCBI’s SRA database over time. (B) The 393 public samples were combined with 31 new in-house samples and processed using our RNA-seq and quality control pipeline. The final dataset contains 300 high-quality samples. (C) ICA takes in the expression data matrix X (2,700 genes × 300 samples) and produces the iModulon matrix M (2,700 genes × 57 iModulons) and Activity matrix A (57 iModulons × 300 samples). The condition-independent iModulon matrix demonstrates the relationship between iModulon genes and an underlying biological signal, while the condition-dependent activity matrix corresponds to the activity level of the iModulon across different experimental conditions in the compendium. (D) Main categories of the 57 iModulons. Regulatory iModulons have significant overlaps with a known regulon, while Genomic iModulons result from genomic changes such as knockouts. Functional iModulons are iModulons that are related to a particular biological function but are not linked to a specific regulator. Single Gene iModulons contain one gene that has a significantly higher weighting than all other genes, and Uncharacterized iModulons contain genes whose functions are yet to be defined. (E) Top 15 iModulons with the highest explained variance and their categories. (F) Treemap showing biological and functional categories of all 57 iModulons. The sizes of the boxes represent the explained variance of the corresponding iModulon. The explained variance of each iModulon is knowledge-based and biologically meaningful, as the iModulons are annotated with specific functional roles.
iModulons Capture the Activity of Known Regulators.
We first describe four regulatory iModulons whose structures significantly overlap with well-characterized regulons from the literature: RpaA, NtcA-1, NtcA-2, and ccm-1. We show that the regulatory iModulons affirm the utility of iModulons in general, recovering meaningful biological signals while providing additional insights into the genes associated with the regulator and related biological processes.
The RpaA iModulon captures the activity of the master transcription factor RpaA, which is a key regulator of the circadian clock system in S. elongatus. RpaA binds to 170 downstream gene targets (8), 61 of which were captured by the RpaA iModulon, including rpaA itself (Fig. 2A). The iModulon is most active in the moments leading to and during dusk, which is consistent with the behavior of RpaA. Compared to the regulon, the iModulon captures more dusk genes than dawn genes (20). Interestingly, all 16 dawn genes in this iModulon are negatively weighted, suggesting the opposite regulatory role of RpaA for dusk and dawn genes (Dataset S1) (Fig. 2B).
Fig. 2.

iModulons show high consistency with known regulons. (A) Venn diagram between the RpaA regulon and RpaA iModulon. (B) Activity of the RpaA iModulon for samples during Clear Day and constant Low Light conditions. The Clear Day condition is defined by a maximal light intensity of 600 μmol photons m−2 s−1, peaking at 6 h after dawn. The Low Light condition has a constant light intensity of 50 μmol photons m−2 s−1. (C) Venn diagram between the NtcA, NtcB regulons, and the two NtcA iModulons. (D) Diagram of the nitrogen metabolic pathways with genes from the NtcA-1 and NtcA-2 iModulons mapped on. (E) Integrated map depicting the interplay between carbon and nitrogen metabolisms with the carbon and nitrogen associated iModulons.
The incomplete overlap between the RpaA iModulon and regulon may be explained by the iModulon’s ability to capture the genes that are most responsive to fluctuations in light intensity. It has been suggested that light changes affect the expression of dusk genes more significantly than that of dawn genes (21). We hypothesize that the RpaA iModulon preferentially identifies the RpaA-regulated genes that exhibit the most pronounced transcriptional changes in response to dynamic light conditions, which are well represented in the data compendium [PRJNA412032 (21)]. Additionally, overlapping regulation by other transcription factors and unique expression patterns of certain genes may contribute to this incomplete overlap (12, 22).
Nitrogen metabolism in cyanobacteria is tightly regulated by complex molecular networks involving proteins such as NtcA, PII, PipX, and other regulators (23). The regulatory targets for these proteins are not fully characterized, but the NtcA and NtcB regulons have been defined (9, 24). We identified two iModulons, NtcA-1 and NtcA-2, that overlap with the regulons of NtcA and NtcB (Fig. 2C).
NtcA-1 encapsulates genes essential for nitrogen transport and conversion to ammonium including the ABC-type nitrite and nitrate transporters (nrtABCD), nitrogen reductases (nirA and narB), cyanate transporters, and its catabolic enzyme cyanase (cynABDS). NtcA-2 also contains nirA, nrtA, and cynA, along with ntcA, ntcB, glutamine synthetases glnN, and glnT (SI Appendix, Fig. S1), all known to play key roles in nitrogen metabolism. Mapping the nitrogen-associated genes from both iModulons onto metabolic pathways reveals their alignment with key steps in nitrogen assimilation and the GS/GOGAT cycle (Fig. 2D). Notably, several uncharacterized genes in the NtcA-2 iModulon are predicted to be part of the NtcA-PipX regulon (10). Synpcc7942_0839 is a proposed nitrilase, and Synpcc7942_1745 is suggested to be a nitrite transporter (25). Further investigations revealed that several genes in this iModulon, including Synpcc7942_0840, Synpcc7942_2529, and Synpcc7942_1707, have been shown to significantly impact the organism’s fitness when using L-glutamine as a nitrogen source and under nitrate stress conditions (26). Among these, Synpcc7942_2529 (gifB) encodes a glutamine synthetase inactivating factor that directly binds to and down-regulates glutamine synthetase activity. In Synechocystis PCC 6803, gifB plays a crucial role in fine-tuning glutamine synthetase activity during transitions between different nitrogen sources (27, 28). These results provide compelling evidence supporting the functional annotations of the remaining uncharacterized genes in these iModulons as being linked to nitrogen metabolism. They may participate in the GS/GOGAT cycle, downstream amino acid metabolism, and other related metabolic processes.
Carbon and nitrogen metabolism are closely related in cyanobacteria, as they need to be highly integrated and precisely regulated to ensure a balanced energy production and nutrient assimilation. We present the ccm-1 iModulon, a regulatory iModulon of CmpR, which regulates the acquisition of inorganic carbon in response to cellular CO2 levels (24) (SI Appendix, Fig. S2). The iModulon contains the bicarbonate transport systems cmpABCD and sbtA, the NDH-I3 complex (ndhF3-ndhD3-cupA-cupS) containing the high-affinity CO2-uptake system genes, and parts of the NDH-core complex (29, 30). It also includes the mnh genes that are proposed to be involved in Na+/H+ antiport and to improve the efficiency of HCO3−-dependent photosynthesis (31). Linking the ccm-1 iModulon with the NtcA iModulons, as well as the ccm-2 iModulon discussed later, provides a comprehensive map that delineates the connections between carbon and nitrogen metabolism and the regulatory networks that govern these processes (Fig. 2E).
Four iModulons Extract Functional Units for Photosynthesis.
While regulatory iModulons uncover genes related to a specific regulator, functional iModulons are associated with a biological function, but not directly tied to a particular regulator. They are valuable for unraveling intricate cellular processes into interconnected functional units, simplifying the structure of the complex system under investigation. We describe four iModulons that extract major functional units of photosynthesis, deconstructing the complexity of this essential process (Fig. 3).
Fig. 3.
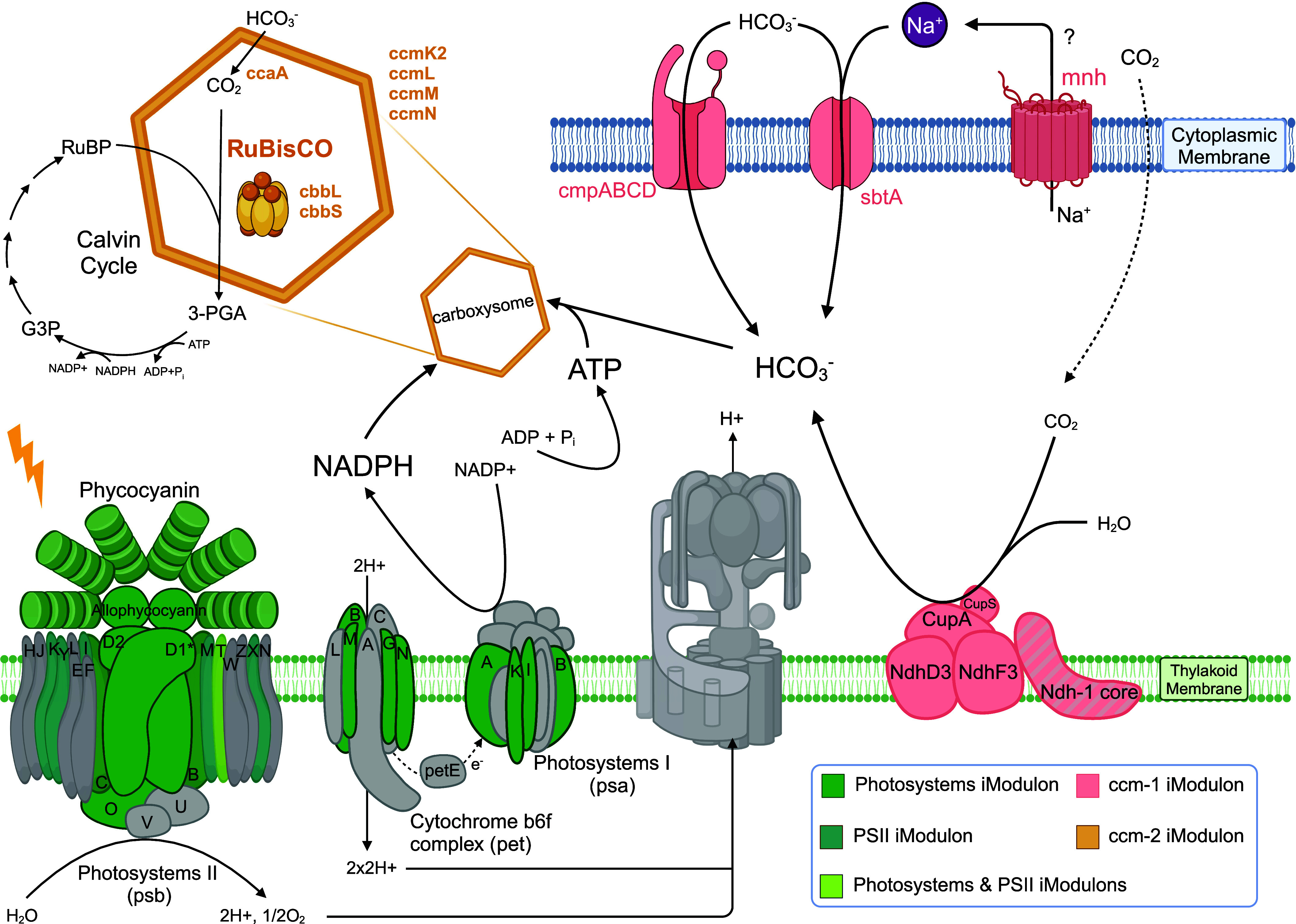
iModulons capture functional units of photosynthesis. (Created with BioRender.com). Four iModulons map to different functional units of photosynthetic metabolism. The proposed structures and reactions are created by incorporating existing models and integrating organism-specific features identified through a comprehensive literature review. The colors show which iModulon the gene coding for the corresponding protein is in. Gray means the gene is not in the listed four iModulons. Colored components with gray stripes indicate some, but not all, genes of the protein complex are captured by iModulons. The ccm-1 and ccm-2 iModulons reveal the light-independent reactions on the Top and on the Right, whereas the PSII and Photosystems iModulons capture genes in the photosystems involved in the light-dependent reactions.
Photosynthesis comprises two stages: the light-dependent reactions, which take place in the thylakoid membranes and harness sunlight, and the light-independent reactions in the stroma. The Photosystems and PSII iModulon genes are highly enriched in the light-dependent reactions, namely, the photosystems, light-harvesting complex, and cytochrome complex (Fig. 3). Within the two iModulons, there are 16 hypothetical proteins with unknown functions (Datasets S2 and S3). Given the coherence observed between the iModulons and the photosystems, we propose that these uncharacterized genes are associated with photosynthesis. In fact, Synpcc7942_1090 clusters with photosystem proteins by the predictions of the STRING database and Synpcc7942_0551 with photosystem proteins, RNA polymerase, and sigma factors (32). This association strongly suggests that the remaining uncharacterized genes likely contribute to the light-dependent reactions, offering a basis for further investigations to uncover their functions.
The Photosystems and PSII iModulons capture genes crucial for absorbing sunlight and initiating photosynthesis, including those involved in water splitting, the photosynthetic electron transport chain, and the production of adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide phosphate (NADPH). The activities of these iModulons are correlated with each other, with both exhibiting low activity in darkness. The Photosystems iModulon, which contains the light-harvesting complex, displays great sensitivity to light changes (SI Appendix, Fig. S3).
The ATP and NADPH generated during the light-dependent reactions are utilized in the light-independent reactions, which consist of the carbon concentrating mechanism (ccm) and the Calvin cycle (Fig. 3). During this stage of photosynthesis, carbon from atmospheric carbon dioxide is concentrated and fixed through the Calvin cycle and incorporated into organic molecules essential for growth and metabolism. The ccm-1 iModulon contains genes responsible for promoting efficient carbon fixation by concentrating CO2 around the carboxylating enzyme, ribulose bisphosphate carboxylase-oxygenase (RuBisCO), in the carboxysome. Genes in the ccm-2 iModulon are mainly involved in the carboxysome. These genes encode both the structural proteins, CcmK2, CcmN, CcmM, and CcmL, and proteins involved in the RuBisCO complex, CcaA, CbbL, and CbbS (33, 34). RuBisCO catalyzes the first step of carbon fixation in the Calvin cycle, where inorganic CO2 is converted to organic molecules using ATP and NADPH. The reduced activity of both the ccm-1 and ccm-2 iModulons in darkness aligns with the observed decrease in CO2 fixation rates, likely due to restricted RuBisCO activities and the dormancy of Ci transporters (31, 35).
Photosynthesis involves complex biochemical reactions in various cellular structures. iModulons simplify this complexity by capturing major functional units, enabling focused investigation into each component’s role, interactions, and regulation. This systems-level perspective not only deepens our understanding of photosynthesis elements but also reveals how they interact with other cellular processes within the broader TRN, facilitating a comprehensive grasp of this fundamental process and its integration with overall cellular function.
iModulons Generate Hypotheses for Regulon Structures and Expand upon Existing Knowledge.
iModulon analysis can propose structure for unexplored regulons and expand our current understanding of regulatory frameworks. This utility can be demonstrated by three notable results.
First, the CysR iModulon provides a promising framework for outlining the structure of the CysR regulon. CysR regulates the expression of genes encoding the sulfate permease complexes, therefore regulating the transport of sulfur, and utilization of sulfur-containing compounds (36). Although it has been shown that CysR influences the expression of multiple genes, the structure of its regulon remains undefined. The CysR iModulon contains 29 genes (Fig. 4A), including the cysPTUVWA genes essential for sulfur uptake, sulfur binding proteins sbpA and sbpB, and the CysR-regulated plasmid gene srpA (37). Additionally, this iModulon captures numerous other genes located within the proposed sulfur-regulated region of the pANL plasmid (38). Given this comprehensive gene composition related to sulfur metabolism and CysR regulation, the CysR iModulon likely serves as a solid foundational structure for the CysR regulon.
Fig. 4.
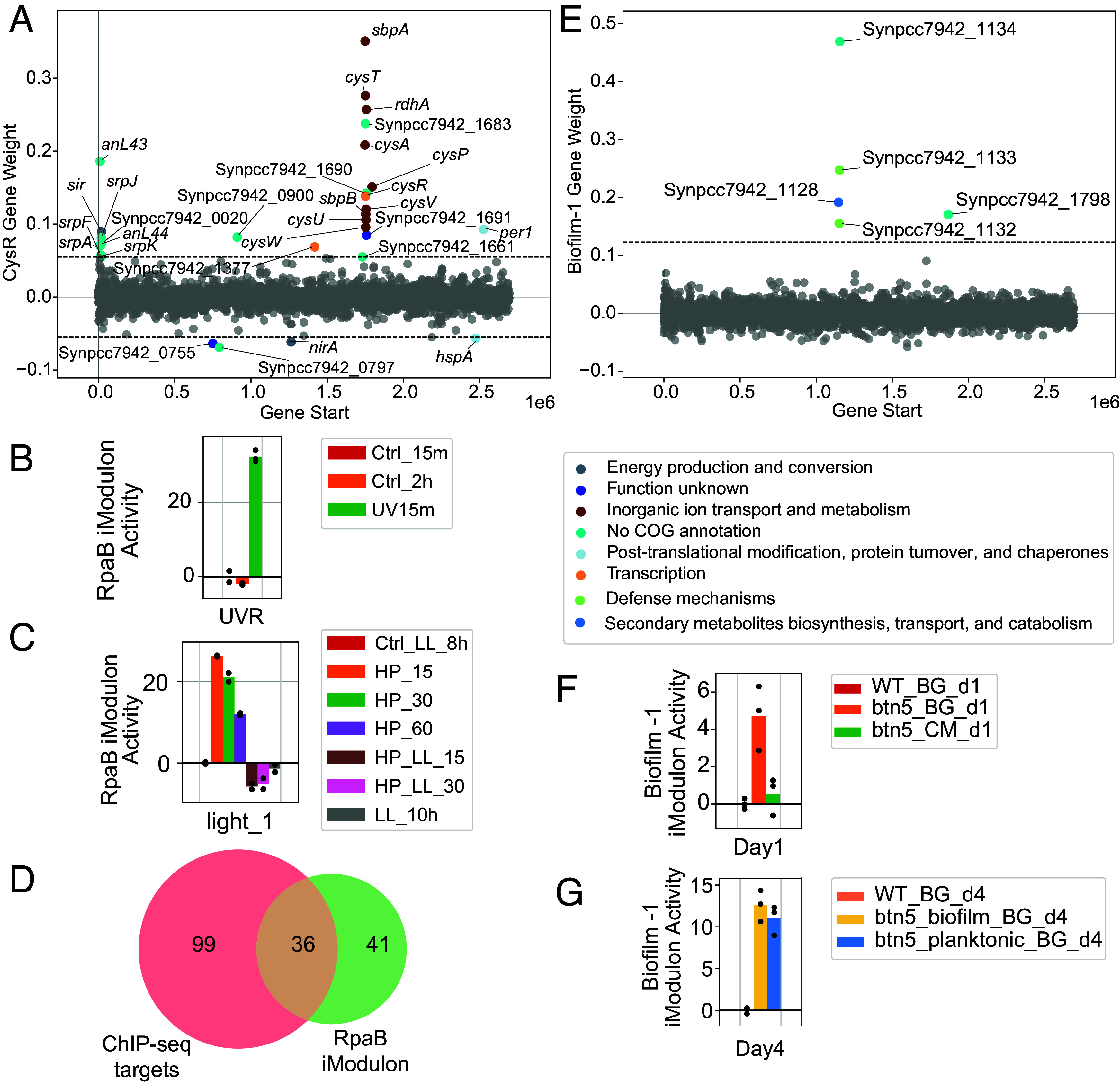
iModulons generate knowledge and iModulon activities exhibit fluctuations during dynamic light changes. (A) Gene weight plot for the CysR iModulon. (B) The activity of the RpaB iModulon for a UV radiation study acquired from public data (PRJNA854269). (C) The activity of the RpaB iModulon for a high light exposure study acquired from public data (PRJNA412032). Starting from low light condition at 8 h (Ctrl_LL_8h), the samples were exposed to high light pulse for 15, 30, and 60 min (HP_15, HP_30, HP_60) before recovering to low light for 15 and 30 min (HP_LL_15, HP_LL_30), ending at low light 10 h (LL_10h). (D) Venn diagram showing the overlap between genes in the RpaB iModulons and gene targets of RpaB identified with ChIP-seq (21). (E) Gene weight plot for the Biofilm-1 iModulon. (F) The activity of the Biofilm-1 iModulon for the biofilm project on day 1. The conditions are wildtype samples in fresh BG-11 (WT_BG_d1), pilB::Tn5 Mutant in fresh BG-11 media (btn5_BG_d1), and pilB::Tn5 mutant in conditioned medium (btn5_CM_d1). (G) The activity of the Biofilm-1 iModulon for the biofilm project on day4. The conditions are wildtype samples in fresh BG-11 (WT_BG_d4), pilB::Tn5 mutant biofilm fraction in fresh BG-11 media (btn5_biofilm_BG_d4), and pilB::Tn5 mutant planktonic fraction in fresh BG-11 media (btn5_planktonic_BG_d4) (39).
Second, the RpaB iModulon captures the activity of the high light response regulator RpaB. RpaB binds to the High Light Regulatory 1 (HLR1) sequences and represses the expression of the high light-inducible genes under nonstressed conditions. Under high light, RpaB~P is dephosphorylated, dissociating from HLR1 and derepressing these genes (40). While a tentative structure of the RpaB regulon in Synechocystis PCC 6803 has been proposed (41), the full regulon structure of RpaB for S. elongatus PCC 7942 is underdefined.
The activity of the RpaB iModulon closely matches the documented behavior of RpaB. The iModulon includes the rpaB and rpaC genes that are negatively weighted along with many other genes related to PSI and PSII. The three genes known to be controlled by RpaB (hliA, nblA, and rpoD3) are positively weighted in this iModulon, suggesting a repressive role of RpaB on their expression. The iModulon exhibits elevated activity under UV and high light, and reduced activity under shade, consistent with the proposed role of RpaB in light responses (Fig. 4 B and C). Additionally, the RpaB iModulon’s activity is negatively correlated with the Photosystems iModulon, indicating a potential positive regulatory role of RpaB for the photosynthetic apparatus (SI Appendix, Fig. S4). The iModulon also contains many genes related to light acclimation and state transitions, potentially under RpaB control. The genes in the RpaB iModulon overlap with the gene targets of RpaB identified with ChIP-seq (21) (Fig. 4D). The iModulon’s structure also shows a similarity to the proposed RpaB regulon in Synechocystis PCC 6803, with several homologous genes present (41). Collectively, these findings indicate that the RpaB iModulon provides a robust basis for defining the RpaB regulon in S. elongatus PCC 7942.
Third, two iModulons related to biofilm formation demonstrate the iModulon’s ability to facilitate more efficient knowledge mining. Wildtype S. elongatus does not form biofilms under standard growth conditions, but an inactivation of the pilB gene restores this ability. Our dataset includes a study that provided biofilm-relevant conditions, such as samples from planktonic supernatants and the biofilm, as well as mutants of the pilB gene (39). We observe two iModulons related to biofilm formation.
The Biofilm-1 iModulon contains five genes (Fig. 4E) and shows increased activity for the pilB mutants in fresh BG-11 media, both in the planktonic and biofilm fraction (Fig. 4 F and G). It also shows decreased activity in conditioned media, aligning with the impact of conditioned media on the transcript levels of Synpcc7942_1133 and Synpcc7942_1134 (42) (Fig. 4F). While all five genes were identified as significantly differentially expressed in any comparisons in the original study, iModulon analysis provided additional insight by grouping them together, revealing a unique functional relationship between them. This iModulon appears to have isolated genes required for the secretion of the EbfG proteins, a set of small secreted proteins known to be essential for biofilm formation. Synpcc7942_1133 (pteB) is crucial for the secretion of the peptides EbfG1-4, and both loci are required, but not sufficient, for biofilm formation (42, 43). Synpcc7942_1132, a component of the Type I secretion system, may be involved in the secretion of EbfG (43, 44). While there is limited literature on the other two genes, Synpcc7942_1128 and Synpcc7942_1798, their appearance alongside the other three genes suggests their potential involvements in the secretion of EbfG proteins (39), making them compelling subjects for further investigations.
The Biofilm-2 iModulon shows significantly low activity across all pilB mutants, and many of the genes in this iModulon can also be found in the genes that are differentially expressed between the mutants and the wildtype samples (such as pilT and pilC) (39). This result suggests that Biofilm-2 is a genomic iModulon stemming from the pilB mutation, with additional genes related to biofilm formation and pilus assembly. The inactivation of the pil genes is known to have pleiotropic effects, impacting behaviors including natural competence and phototaxis (39). The Biofilm-2 iModulon activity correlates with the phototaxis and proposed competence iModulons, suggesting the interconnected nature of these diverse processes (SI Appendix, Fig. S5). Interestingly, all the genes in the Biofilm-1 iModulon, except for Synpcc7942_1128, are also present in Biofilm-2, but they are negatively weighted. This indicates that the Biofilm-1 genes are oppositely regulated compared to most of the positively weighted genes in Biofilm-2, which is likely to be the result of the pilB inactivation. This result also supports the finding that the pilB mutation leads to enhanced expression of genes that facilitate the secretion of EbfG proteins.
iModulon Activity Patterns Reveal the Cell’s Response to Dynamic Light Conditions.
The composition of iModulons reveals the connections among genes, while the activity levels of iModulons offer valuable insights into the expression and regulation of these genes across diverse conditions. We now delve into the activity patterns of selected iModulons under light-related conditions. We present how iModulon activity patterns can reveal the activity of different biological processes under various light conditions.
We observe iModulons whose activities show the most distinct patterns under different light conditions in Fig. 5. Under the “Clear Day” condition that resembles natural daylight, the Photosystems iModulon shows high activity at dawn, peaking around 6 h after dawn when light intensity is highest, and then gradually decreasing. A similar trend is observed for the ribosome iModulons. This result aligns with the observation that many genes encoding photosystem components as well as some genes involved in ribosomal proteins are dawn genes (20, 21). RpaA and RpaB collaboratively orchestrate the circadian and clock-related processes (21). The RpaA and the RpaB iModulons display monotonic activity change throughout the day. RpaA activity increases from dawn to dusk, while RpaB activity drops, suggesting increased activity for both regulators (Fig. 5A). The competence and RpaC iModulons transition from negative to positive activity around 9 h, with the activity of the competence iModulon continuing to increase in the dark, peaking in the middle of the night before decreasing (Fig. 5B). In the absence of light, the Photosystems iModulon is less active, and the ccm-1 iModulon shows decreased activity, as Ci acquisition becomes redundant and energetically futile (45, 46).
Fig. 5.

iModulon activities respond to changing light conditions. (A) The activity of selected iModulons under a clear day condition. On a clear day, light intensity varies in a parabolic manner, with maximum photon intensity of 600 μmol photons m−2 s−1. (B) The activity of selected iModulons in the dark. (C) iModulons whose activities display significant changes during shade pulse. The condition is defined by dividing the light intensity value of the clear day condition by 10-fold between 8 and 9 h after dawn. Samples were taken at clear day 8 h (CD_8h), 15, 30, and 60 min during shade pulse exposure (SP_15m - SP_60m), 15 and 30 min of recovery after switching back to clear day (SP_CD_15m, SP_CD_30m) and cleary day 10 h (CD_10h), time after exposure, and recovery. (D) iModulons whose activities display significant changes during high light pulse, where the samples in constant low light condition (50 μmol photons m−2 s−1) were exposed to the light intensity of clear day condition between 8 and 9 h after dawn (21). Samples were taken at low light 8 h (LL_8h), 15, 30, and 60 min during high light pulse exposure (HP_15m - HP_60m), 15 and 30 min of recovery after switching back to low light (HP_LL_15m, HP_LL_30m) and low light 10 h (LL_10h), time after exposure and recovery.
While there are also other iModulons that exhibit activity changes under natural light/dark conditions, some appear most sensitive to abrupt shifts in light levels (Fig. 5 C and D). One of them is the competence iModulon, which contains 41 genes, 7 of which are associated with the competence machinery in S. elongatus (47). This iModulon exhibits increased activity in the dark, corresponding to the enhanced natural competence of the organism in response to darkness. The 4% explained variance indicates that this iModulon plays an important role in the cell, and it is evident that it is under the control of the circadian clock. However, with 19 of its 41 genes uncharacterized, this iModulon remains poorly understood and worthy of further investigation.
Another promising target for further investigation is the RpaC iModulon, which has an explained variance of 3.5% and appears to be regulated by the circadian clock during the day. This iModulon contains 31 genes, including rpaC. RpaC is known to be essential for state transitions in Synechocystis, as a lack of this protein inhibits state transition (48). However, the same effect is not observed for rpaC mutants in S. elongatus PCC 7942 (49). Complete segregation of an rpaC mutation renders the cells nonviable, suggesting the importance of rpaC in S. elongatus (49). Further investigation of the RpaC iModulon may provide valuable insights into the function of this gene and its regulatory mechanisms in S. elongatus.
When exposed to shade pulse on a clear day, the Photosystems, ribosome-1, and ribosome-2 iModulons experience a significant drop in activity, likely due to the reduced light availability and the cells’ reprogramming of transcription and translation. They recover their activities when light is restored (Fig. 5C). As expected, the RpaB iModulon shows decreased activity in the shade and recovers in the light. In contrast, the RpaA, RpaC, and Competence iModulons exhibit increased activity during the shade pulse. Under high light pulse, most iModulons display the opposite activity trends compared to the shade pulse (Fig. 5D). The exception is the Photosystems iModulon, whose activity also drops when exposed to high light pulse conditions. This result is likely due to photoinhibition, a phenomenon caused by an imbalance between the rate of photodamage to PSII and the rate of PSII complex repair, as excess light energy can be toxic, damaging the photosynthetic machinery and other cellular components (50).
The analysis of iModulon activities allows us to identify iModulons under the control of the circadian clock, as well as iModulons sensitive to changes in light levels. This decomposition of the complex light response into individual iModulons facilitates targeted investigations and provides insights into the underlying regulatory mechanisms. Furthermore, iModulons can be clustered together based on their similar activity patterns, suggesting they may respond to the same or related stimuli, which gives a close approximation to the concept of “stimulons” (SI Appendix, Fig. S6). Combined with the activity profiles of individual iModulons, this approach offers a powerful means to understand the expression of diverse gene groups with various biological functions under different environmental conditions. The activity pattern of all the iModulons under light-related conditions can be found in SI Appendix, Figs. S7–S10.
Discussion
In this study, we used ICA to deconvolute a compendium of 300 curated RNA-seq profiles of S. elongatus into 57 robust iModulons. This computed iModulon structure is knowledge-enriched, allowing us to explain 67% of the variance in the transcriptome across 158 unique experimental conditions. By examining these iModulons in detail, we revealed the complex metabolic and regulatory networks that enable S. elongatus to thrive in diverse environments. Through iModulon analysis, we uncover regulatory iModulons that accurately reflect the activity of known transcriptional regulators and their associated regulons. Our results complement and validate existing knowledge of gene regulatory mechanisms associated with RpaA, and with nitrogen and carbon metabolism.
A key strength of the iModulon framework is its ability to deconstruct complex cellular processes, like photosynthesis, by capturing its major functional units. By isolating and analyzing iModulons corresponding to components like the photosystems, carbon concentrating mechanism, and Calvin cycle, iModulons allow us to dissect and reconnect the underlying TRNs that orchestrate these fundamental cellular processes. This systems-level perspective provides insights that would be difficult to obtain through the study of individual genes or pathways in isolation.
Moreover, iModulon analysis can generate data-driven hypotheses to characterize regulons and expand our understanding of transcriptional regulation. The CysR and RpaB iModulons offer a framework for defining the gene targets and functional scope of these regulators, and the biofilm iModulons highlight genes essential for EbfG secretion. Additionally, iModulons identify poorly annotated genes that covary with known pathway components, suggesting potential functional associations worthy of further investigation. iModulons thus can generate testable hypotheses about regulatory mechanisms beyond well-studied systems.
Furthermore, iModulons describe dynamic transcriptional shifts that occur under changing light conditions, a critical environmental factor for phototrophic organisms like S. elongatus. Their activities reveal the temporal patterns of gene expression for photosynthesis, carbon/nitrogen metabolism, competence, and circadian regulation, providing insights into the integrated response of the cell to diurnal light cues. This enhances our understanding of the sophisticated mechanisms by which S. elongatus, and potentially other phototrophs, sense and respond to fluctuating light availability in their natural environments.
In summary, iModulons not only expand our knowledge about S. elongatus, but also provide a framework for formulating testable hypotheses to guide future investigations. By offering a systems-level perspective on the transcriptome, iModulon analysis presents a quantitative reconstruction of the TRN of S. elongatus. This TRN structure provides genetic and regulatory mechanisms underlying key cellular functions and physiological states. The comprehensiveness of this iModulon-based TRN is defined by the diversity of the conditions represented in our dataset. Theoretically, if we could capture all possible transcriptional interactions through an exhaustive set of conditions, we could establish a comprehensive quantitative TRN model. This study represents a global TRN structure has been established for this S. elongatus, laying the groundwork for future refinements and expansions of this model as we expand and diversify the experimental conditions in our data collection.
Methods
The methods in this study for public RNA-seq data acquisition and processing, performing ICA, and iModulon characterization were adapted from the iModulonMiner workflow (51).
Public Data Acquisition and RNA-seq Processing.
The 393 public RNA-seq datasets were collected from NCBI Sequence Read Archive (SRA) repository using step 1 of the iModulonMiner workflow (https://github.com/SBRG/iModulonMiner/tree/main/1_download_metadata) and was combined with the in-house collection. During the analysis, it was determined that 10 of the 31 in-house samples generated in 2021 had been published and were already in the public domain at the time we collected the public datasets (52). Therefore, these 10 samples were also included in the 393 public samples. As a result, the final 424 samples analyzed consisted of 414 unique samples, with 393 public samples and 21 novel in-house samples. All samples were processed with a standardized RNA-seq processing pipeline using NextFlow v22.10.0 (https://github.com/SBRG/iModulonMiner/tree/main/2_process_data). Briefly, the raw FASTQ files were downloaded from NCBI using fastq-dump (https://github.com/ncbi/sra-tools/wiki/HowTo:-fasterq-dump). Next, Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) was applied with default options for read trimming, followed by FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reads were aligned to the genome using Bowtie (53). RSEQC was used to infer read direction before generating read counts using featureCounts (54, 55). The quality control metrics are compiled using MultiQC (56), and the final expression dataset is reported in units of log-transformed Transcripts per Million (log-TPM).
Culture Conditions and Sample Preparation.
Ten samples from the in-house UV study were published and the growth condition and sample preparation are described in the published study (52). The original experimental design called for 12 samples, with 4 conditions in triplicate. However, 1 post UV control sample was lost during the experiment. For the publicly released dataset, an additional sample from the experimental condition associated with this control condition was deliberately omitted, so the comparisons were done for this pair of control/experimental samples in duplicate (rather than triplicate). In our iModulon analysis, we included all 11 samples from the original dataset.
Twenty in-house samples for transcription factors (Synpcc7942_0110, Synpcc7942_0616, Synpcc7942_1684) deletion and control experiment cultures were grown as follows: Cultures in flasks were diluted to density of OD750 = 0.05 and were grown under constant light. When culture density reached to OD750 ~ 0.2, all samples were synchronized with 12 h dark incubation. Before harvesting, “light samples” were kept under constant light for an additional 28 h and the “dark samples” were kept under light for an additional 24 h and then incubated under dark for 4 h, a total of 28 h growth. Afterward, 10 mL of OD750 = 0.4 cells were harvested using Qiagen RNAprotect Bacteria Reagent according to the manufacturers’ protocols. Following centrifugation, cell pellets were immediately frozen at −80 °C.
RNA Extraction and Library Preparation.
Total RNA was isolated and purified using a Zymo Research Quick-RNA Fungal/Bacterial Microprep Kit from frozen cell pellets previously harvested using Qiagen RNAprotect Bacteria Reagent according to the manufacturers’ protocols. Ribosomal RNA was removed from 1 μg Total RNA with the use of a QIAseq FastSelect - 5S/16S/23S kit (Qiagen). The resulting rRNA- subtracted RNA was made into libraries with a KAPA RNA HyperPrep kit incorporating short Y-adapters and barcoded PCR primers. The libraries were quantified with a fluorescent assay (dsDNA AccuGreen quantitation kit, Biotium) and checked for proper size distribution and average size with a TapeStation (D1000 Tape, Agilent). Library pools were then assembled and a 1X SPRI bead cleanup performed to remove traces of carryover PCR primers. The final library pool was quantified and run on an Illumina instrument (NextSeq, Novaseq).
Quality Control and Metadata Curation.
The log-transformed dataset was further processed using our quality control (QC) pipeline to remove poor-quality expression profiles not suitable for subsequent analyses (https://github.com/SBRG/iModulonMiner/tree/main/3_quality_control). Samples were filtered based on per base sequence quality, per sequence quality scores, per base n content, and adapter content. Any sample that failed a global correlation with the other samples was also discarded. Furthermore, the metadata of all the samples were manually curated from literature to include information such as strain description, base media, nutrient sources, experimental treatments (such as light conditions), and growth stages if disclosed. Samples with a low correlation with its biological replicates (R2 < 0.91) or without biological replicates were filtered out. Several samples from the projects “ppGpp,” “rpaA,” “clock,” and “in_house” were manually added back to the collection despite a lack of biological replicates as they represent unique and interesting conditions. A reference condition was selected for each project, and the final log-TPM data were normalized to reference conditions specific to each project.
Running ICA to Identify Robust Components.
ICA was performed using the FastICA algorithm from scikit-learn (https://github.com/SBRG/iModulonMiner/tree/main/4_optICA) (57–59). FastICA was applied to the log-TPM data with 100 iterations with random seeds and a convergence tolerance of 10−7. The resulting independent components (ICs) were clustered using DBSCAN (60). To identify robust ICs using a maximum distance threshold of 0.1 and a minimum cluster seed size of 50. To account for identical with opposite signs, the following distance metric was used for computing the distance matrix:
where ρx, y is the Pearson correlation between components a and b. The final robust ICs were defined as the centroids of the computed cluster. To determine the optimal dimensionality for the ICA, the procedure was applied to the S. elongatus compendium multiple times, varying the number of dimensions from 10 to 270 in increments of 10. The optimal dimensionality was identified by comparing the number of independent components (ICs) with single genes to the number of ICs correlated (Pearson R > 0.7) with the ICs in the largest dimension (final components). The selected dimensionality was the one where the number of nonsingle gene ICs matched the number of final components. The optimal dimension for this study was 130 (SI Appendix, Fig. S11).
Compiling Annotations and Computing iModulon Enrichments.
Regulon information for S. elongatus was manually curated from Biocyc (61), RegPrecise, and from the literature that reported potential and ChIP-seq TF-DNA-binding events and were reported in the TRN data. Gene annotations were pulled for the reference genome from NCBI. We also included information from Cluster of Orthologous Groups (COG) and KEGG using EggNOG mapper (62, 63), Gene Ontology (GO) annotations using AmiGO2 (64), Uniprot IDs using the Uniprot ID mapper (65), and operon clusters from Biocyc and Cyanocyc (61, 66). The gene annotation pipeline can be found at (https://github.com/SBRG/iModulonMiner/blob/main/5_characterize_iModulons/1_create_the_gene_table.ipynb). To calculate regulator enrichment against known regulons and GO and KEGG enrichments, Fisher’s exact test with Benjamini–Hochberg false discovery rate (FDR) correction was used with a FDR of 10−5 and 0.01, respectively.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (CSV)
Dataset S02 (CSV)
Dataset S03 (CSV)
Acknowledgments
We would like to thank Dr. Kevin Rychel, Dr. Saugat Poudel, Reo Yoo, Siddharth Chauhan, and Dr. Cameron Lamoureux for informative discussions, and Dr. Elliot Weiss for providing early access to ultraviolet (UV)-treated samples, which have since been published and are cited in the manuscript. We would also like to thank Dr. Rakefet Schwarz for helpful suggestions for the manuscript. Portions of this paper were developed from T.A.B’s Master’s thesis. Research reported in this publication was supported by the Y.C. Fung Endowed Chair in Bioengineering to B.O.P. and National Institute of General Medical Sciences of the NIH under Award Number R35GM118290 to S.S.G.
Author contributions
A.V.S. and B.O.P. designed research; Y.Y. performed research; Y.Y. and A.V.S. contributed new analytic tools; C.S. and R.S. collected experimental data; Y.Y. analyzed data; and Y.Y., T.A.B., A.V.S., C.S., R.S., S.S.G., and B.O.P. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Susan S. Golden, Email: sgolden@ucsd.edu.
Bernhard O. Palsson, Email: bpalsson@ucsd.edu.
Data, Materials, and Software Availability
All code and data used to generate the results in this paper can be found on GitHub (https://github.com/AnnieYuan21/S.elongatus-iModulons) (67). The results of this study are also presented as interactive dashboards on https://imodulondb.org (68). The general iModulon analysis pipeline can be found at https://github.com/SBRG/iModulonMiner (69).
Supporting Information
References
- 1.Kajiwara S., Yamada H., Ohkuni N., Ohtaguchi K., Design of the bioreactor for carbon dioxide fixation by Synechococcus PCC7942. Energy Convers. Manag. 38, S529–S532 (1997). [Google Scholar]
- 2.Samiotis G., Stamatakis K., Amanatidou E., Assessment of Synechococcus elongatus PCC 7942 as an option for sustainable wastewater treatment. Water Sci. Technol. 84, 1438–1451 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Chen P.-H., et al. , Enhancing CO2 bio-mitigation by genetic engineering of cyanobacteria. Energy Environ. Sci. 5, 8318–8327 (2012). [Google Scholar]
- 4.Machado I. M. P., Atsumi S., Cyanobacterial biofuel production. J. Biotechnol. 162, 50–56 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Santos-Merino M., Garcillán-Barcia M. P., de la Cruz F., Engineering the fatty acid synthesis pathway in Synechococcus elongatus PCC 7942 improves omega-3 fatty acid production. Biotechnol. Biofuels 11, 239 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi S. Y., et al. , Improvement of squalene production from CO2 in Synechococcus elongatus PCC 7942 by metabolic engineering and scalable production in a photobioreactor. ACS Synth. Biol. 6, 1289–1295 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Lan E. I., et al. , Metabolic engineering of cyanobacteria for photosynthetic 3-hydroxypropionic acid production from CO2 using Synechococcus elongatus PCC 7942. Metab. Eng. 31, 163–170 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Markson J. S., Piechura J. R., Puszynska A. M., O’Shea E. K., Circadian control of global gene expression by the cyanobacterial master regulator RpaA. Cell 155, 1396–1408 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopatovskaya K. V., Seliverstov A. V., Lyubetsky V. A., NtcA and NtcB regulons in cyanobacteria and rhodophyta chloroplasts. Mol. Biol. 45, 522–526 (2011). [PubMed] [Google Scholar]
- 10.Espinosa J., et al. , PipX, the coactivator of NtcA, is a global regulator in cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 111, E2423–E2430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong W., Vanderburg C. R., Gunshin H., Rogers J. T., Huang X., A review of independent component analysis application to microarray gene expression data. Biotechniques 45, 501–520 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sastry A. V., et al. , The Escherichia coli transcriptome mostly consists of independently regulated modules. Nat. Commun. 10, 5536 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rychel K., Sastry A. V., Palsson B. O., Machine learning uncovers independently regulated modules in the Bacillus subtilis transcriptome. Nat. Commun. 11, 6338 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauhan S. M., et al. , Machine learning uncovers a data-driven transcriptional regulatory network for the crenarchaeal thermoacidophile Sulfolobus acidocaldarius. Front. Microbiol. 12, 753521 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoo R., et al. , Machine learning of all Mycobacterium tuberculosis H37Rv RNA-seq data reveals a structured interplay between metabolism, stress response, and infection. mSphere 7, e0003322 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan Y., et al. , Pan-genome analysis of transcriptional regulation in six Salmonella enterica serovar Typhimurium strains reveals rheir different regulatory structures. mSystems 7, e00467-22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bajpe H., Rychel K., Lamoureux C. R., Sastry A. V., Palsson B. O., Machine learning uncovers the Pseudomonas syringae transcriptome in microbial communities and during infection. mSystems 8, e0043723 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choe D., et al. , Advancing the scale of synthetic biology via cross-species transfer of cellular functions enabled by iModulon engraftment. Nat. Commun. 15, 2356 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leinonen R., Sugawara H., Shumway M., The sequence read archive. Nucleic Acids Res. 39, D19–D21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijayan V., Zuzow R., O’Shea E. K., Oscillations in supercoiling drive circadian gene expression in cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 106, 22564–22568 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piechura J. R., Amarnath K., O’Shea E. K., Natural changes in light interact with circadian regulation at promoters to control gene expression in cyanobacteria. Elife 6, e32032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamoureux C. R., et al. , A multi-scale expression and regulation knowledge base for Escherichia coli. Nucleic Acids Res. 51, 10176–10193 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esteves-Ferreira A. A., Inaba M., Fort A., Araújo W. L., Sulpice R., Nitrogen metabolism in cyanobacteria: Metabolic and molecular control, growth consequences and biotechnological applications. Crit. Rev. Microbiol. 44, 541–560 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Novichkov P. S., et al. , RegPrecise 3.0 – A resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genomics 14, 745 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maeda S., Konishi M., Yanagisawa S., Omata T., Nitrite transport activity of a novel HPP family protein conserved in cyanobacteria and chloroplasts. Plant Cell Physiol. 55, 1311–1324 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Wetmore K. M., et al. , Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons. mBio 6, e00306-15 (2015), 10.1128/mbio.00306-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Domínguez M., Reyes J. C., Florencio F. J., Glutamine synthetase inactivation by protein–protein interaction. Proc. Natl. Acad. Sci. U.S.A. 96, 7161–7166 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muro-Pastor M. I., Reyes J. C., Florencio F. J., Cyanobacteria perceive nitrogen status by sensing intracellular 2-oxoglutarate levels. J. Biol. Chem. 276, 38320–38328 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Burnap R. L., Hagemann M., Kaplan A., Regulation of CO2 concentrating mechanism in cyanobacteria. Life 5, 348–371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodger F. J., Bryant D. A., Price G. D., Transcriptional regulation of the CO2-concentrating mechanism in a euryhaline, coastal marine cyanobacterium, Synechococcus sp. Strain PCC 7002: Role of NdhR/CcmR. J. Bacteriol. 189, 3335–3347 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price G. D., Badger M. R., Woodger F. J., Long B. M., Advances in understanding the cyanobacterial CO2-concentrating-mechanism (CCM): Functional components, Ci transporters, diversity, genetic regulation and prospects for engineering into plants. J. Exp. Bot. 59, 1441–1461 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Szklarczyk D., et al. , The STRING database in 2023: protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rae B. D., Long B. M., Badger M. R., Price G. D., Structural determinants of the outer shell of β-carboxysomes in Synechococcus elongatus PCC 7942: Roles for Ccm K2, K3–K4, CcmO, and CcmL. PLoS One 7, e43871 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Niederhuber M. J., Lambert T. J., Yapp C., Silver P. A., Polka J. K., Superresolution microscopy of the β-carboxysome reveals a homogeneous matrix. Mol. Biol. Cell 28, 2734–2745 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Y., Huang F., Dykes G. F., Liu L.-N., Diurnal regulation of in vivo localization and CO2-fixing activity of carboxysomes in Synechococcus elongatus PCC 7942. Life 10, 169 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laudenbach D. E., Grossman A. R., Characterization and mutagenesis of sulfur-regulated genes in a cyanobacterium: Evidence for function in sulfate transport. J. Bacteriol. 173, 2739–2750 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholson M. L., Laudenbach D. E., Genes encoded on a cyanobacterial plasmid are transcriptionally regulated by sulfur availability and CysR. J. Bacteriol. 177, 2143–2150 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Y., Holtman C. K., Magnuson R. D., Youderian P. A., Golden S. S., The complete sequence and functional analysis of pANL, the large plasmid of the unicellular freshwater cyanobacterium Synechococcus elongatus PCC 7942. Plasmid 59, 176–192 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simkovsky R., et al. , Transcriptomic and phenomic investigations reveal elements in biofilm repression and formation in the cyanobacterium Synechococcus elongatus PCC 7942. Front. Microbiol. 13, 899150 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moronta-Barrios F., Espinosa J., Contreras A., In vivo features of signal transduction by the essential response regulator RpaB from Synechococcus elongatus PCC 7942. Microbiology 158, 1229–1237 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Riediger M., et al. , Biocomputational analyses and experimental validation identify the regulon controlled by the redox-responsive transcription factor RpaB. iScience 15, 316–331 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schatz D., et al. , Self-suppression of biofilm formation in the cyanobacterium Synechococcus elongatus. Environ. Microbiol. 15, 1786–1794 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Parnasa R., et al. , Small secreted proteins enable biofilm development in the cyanobacterium Synechococcus elongatus. Sci. Rep. 6, 32209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suban S., et al. , A cyanobacterial sigma factor F controls biofilm-promoting genes through intra- and intercellular pathways. Biofilm 8, 100217 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaplan A., Zenvirth D., Marcus Y., Omata T., Ogawa T., Energization and activation of inorganic carbon uptake by light in cyanobacteria 1. Plant Physiol. 84, 210–213 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tan X., et al. , The primary transcriptome of the fast-growing cyanobacterium Synechococcus elongatus UTEX 2973. Biotechnol. Biofuels 11, 218 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taton A., et al. , The circadian clock and darkness control natural competence in cyanobacteria. Nat. Commun. 11, 1688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emlyn-Jones D., Ashby M. K., Mullineaux C. W., A gene required for the regulation of photosynthetic light harvesting in the cyanobacterium Synechocystis 6803. Mol. Microbiol. 33, 1050–1058 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Joshua S., Mullineaux C. W., The rpaC gene product regulates phycobilisome–photosystem II interaction in cyanobacteria. Biochim. Biophys. Acta 1709, 58–68 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Bailey S., Grossman A., Photoprotection in cyanobacteria: Regulation of light harvesting. Photochem. Photobiol. 84, 1410–1420 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Sastry A. V., et al. , Mining all publicly available expression data to compute dynamic microbial transcriptional regulatory networks. bioRxiv [Preprint] (2021). 10.1101/2021.07.01.450581 (Accessed 25 May 2024). [DOI]
- 52.Weiss E. L., et al. , An unexpected role for leucyl aminopeptidase in UV tolerance revealed by a genome-wide fitness assessment in a model cyanobacterium. Proc. Natl. Acad. Sci. U.S.A. 119, e2211789119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L., Wang S., Li W., RSeQC: Quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Ewels P., Magnusson M., Lundin S., Käller M., MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hyvarinen A., Fast and robust fixed-point algorithms for independent component analysis. IEEE Trans. Neural Netw. 10, 626–634 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Pedregosa F., et al. , Scikit-learn: Machine learning in Python. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 59.McConn J. L., Lamoureux C. R., Poudel S., Palsson B. O., Sastry A. V., Optimal dimensionality selection for independent component analysis of transcriptomic data. BMC Bioinformatics 22, 584 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ester M., Kriegel H.-P., Sander J., Xu X., “A density-based algorithm for discovering clusters in large spatial databases with noise” in Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD’96) (AAAI Press, 1996), pp. 226–231.
- 61.Karp P. D., et al. , The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform. 20, 1085–1093 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanehisa M., Furumichi M., Sato Y., Ishiguro-Watanabe M., Tanabe M., KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 49, D545–D551 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huerta-Cepas J., et al. , Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol. Biol. Evol. 34, 2115–2122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.The Gene Ontology Consortium, The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.The UniProt Consortium, UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moore L. R., et al. , CyanoCyc cyanobacterial web portal. Front. Microbiol. 15, 1340413 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yuan Y., Data from “S. elongatus-iModulons”. GitHub. https://github.com/AnnieYuan21/S.elongatus-iModulons. Deposited 3 June 2024.
- 68.Independent component analysis of prokaryotic transcriptomes. iModulonDB. https://imodulondb.org/. Accessed 18 July 2024.
- 69.SBRG, iModulonMiner. Github. https://github.com/SBRG/iModulonMiner. Deposited 4 April 2024.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (CSV)
Dataset S02 (CSV)
Dataset S03 (CSV)
Data Availability Statement
All code and data used to generate the results in this paper can be found on GitHub (https://github.com/AnnieYuan21/S.elongatus-iModulons) (67). The results of this study are also presented as interactive dashboards on https://imodulondb.org (68). The general iModulon analysis pipeline can be found at https://github.com/SBRG/iModulonMiner (69).