

Abstract
Following acute myocardial infarction, the recovery of blood flow leads to myocardial ischemia-reperfusion (MI/R) injury, which is primarily characterized by the activation of inflammatory signals, microvascular obstruction, increased oxidative stress and excessive Ca2+ overload. It has also been demonstrated that platelets can exacerbate MI/R injury by releasing reactive oxygen species, inflammatory factors and chemokines, while also obstructing microvessels through thrombus formation. As a bioactive molecule with proinflammatory and chemotactic properties, lipocalin 2 (LCN2) exhibits a positive correlation with obesity, hyperglycemia, hypertriglyceridemia and insulin resistance index, which are all significant risk factors for ischemic cardiomyopathy. Notably, the potential role of LCN2 in promoting atherosclerosis may be related to its influence on the function of macrophages, smooth muscle cells and endothelial cells, but its effect on platelet function has not yet been reported. In the present study, the effect of a high-fat diet (HFD) on LCN2 expression was determined by detecting LCN2 expression levels in the liver and serum samples of mice through reverse transcription-quantitative PCR and enzyme linked immunosorbent assay, respectively. The effect of LCN2 on platelet function was evaluated by examining whether LCN2 affected platelet activation, aggregation, adhesion, clot retraction and P-selectin expression. To determine whether LCN2 aggravated MI/R injury in HFD-fed mice by affecting platelet and inflammatory cell recruitment, wild-type and LCN2 knockout mice fed a HFD were subjected to MI/R injury, then hearts were collected for hematoxylin and eosin staining and 2,3,5-triphenyltetrazolium chloride staining, and immunohistochemistry was employed to detect the expression of CD42b, Ly6G, CD3 and B220. Based on observing the upregulation of LCN2 expression in mice fed a HFD, the present study further confirmed that LCN2 could accelerate platelet activation, aggregation and adhesion. Moreover, in vivo studies validated that knockout of LCN2 not only mitigated MI/R injury, but also inhibited the recruitment of platelets and inflammatory cells in myocardial tissue following ischemia-reperfusion. In conclusion, the current findings suggested that the effect of HFD-induced LCN2 on aggravating MI/R injury may totally or partially dependent on its promotion of platelet function.
Keywords: high-fat diet, lipocalin 2, myocardial ischemia-reperfusion injury, platelet activation
Introduction
Acute myocardial infarction (AMI), also known as a heart attack, is a major global cause of mortality, which is frequently linked to conditions such as high cholesterol levels, high blood pressure, smoking, diabetes, obesity, lack of physical activity, metabolic syndrome and depression (1). In the case of AMI, it is crucial to restore blood flow promptly to prevent loss of heart function, decreasing the size of the damaged area and improving overall clinical prognosis. However, reperfusion, which occurs when the blood flow is restored, can actually worsen the condition and cause additional damage to the heart; this phenomenon is known as myocardial ischemia-reperfusion (MI/R) injury (2). Common techniques for reconstructing coronary arteries in AMI cases include percutaneous coronary intervention and surgical coronary artery bypass grafting. However, these methods do not offer significant benefits in terms of reducing reperfusion injury. Currently, standard medications, such as β-blockers, angiotensin-converting enzyme inhibitors and statins, are administered post-reperfusion (3). Nevertheless, these drugs do not effectively mitigate myocardial injury by addressing the persistent risk factors that induce myocardial ischemia and the activation of inflammatory signals after ischemia. Researchers have recently focused on investigating drugs that can alleviate the inflammatory cascade in MI/R injury (4–7). However, these studies often use young animals as subjects, which does not accurately reflect the situation of MI/R injury in older individuals with comorbidities such as diabetes, hypertension and metabolic syndrome (8). Therefore, the identification of more effective strategies to control MI/R injury remains a challenging and urgent task.
Platelets serve a crucial role in the entire process of ischemic heart disease, starting from the occurrence of ischemia to reperfusion (9). Physiological platelet activators (thrombin and thromboxin-A2) and external reactive oxygen species (ROS) promote the activation of platelets through extracellular signal-regulated kinase 5 after cardiac ischemia (10). Additionally, platelets release chemokines that stimulate the migration of leukocytes to the injury site within blood vessels, thereby exacerbating inflammatory damage to the myocardium. Presently, although the use of antiplatelet medications has become a vital approach to prevent and manage coronary artery disease (CAD), the mortality rate remains high (11,12), which may be due to the fact that antiplatelet drugs cannot directly eliminate the induction factors affecting platelet hyperreactivity. Therefore, it is necessary to further investigate the regulatory mechanisms that affect the enhancement of platelet reactivity, which may result in the development of more efficient antiplatelet drugs capable of achieving a balance between the antithrombotic effect and other complications, including excessive bleeding.
Lipocalin 2 (LCN2), originally considered to be a 25-kDa glycoprotein isolated from human neutrophilic granulocytes, is expressed in various tissues, including the liver, lung and kidney (13,14). LCN2 has also been detected in human atherosclerotic tissues, and it has the ability to upregulate the production of interleukin (IL)-6, IL-8 and MCP-1 in macrophages, endothelial cells and smooth muscle cells (15). In addition, LCN2 may have a significant impact on the development of metabolic syndrome (16). Notably, obese patients have been shown to exhibit increased mRNA and protein expression levels of LCN2 in their visceral adipose tissue compared with in lean subjects (17). Moreover, serum levels of LCN2 have been reported to be positively associated with obesity, hyperglycemia, hypertriglyceridemia and insulin resistance index (18–20), all of which are crucial risk factors for ischemic cardiomyopathy. Numerous studies have confirmed the crucial involvement of LCN2 in cardiovascular diseases. For example, the knockout of LCN2 in mice with myocardial ischemia has been shown to lead to a reduction in cardiac dysfunction (20). Nonetheless, it remains uncertain whether LCN2 directly affects platelet activation to regulate the occurrence of MI/R injury.
The present study aimed to investigate the potential exacerbating effect of high-fat diet (HFD)-induced LCN2 on MI/R injury through its impact on platelet function. To verify the hypothesis, a series of experiments were carried out. Firstly, the expression patterns of LCN2 in the liver and serum of mice subjected to a HFD were assessed by comparing them to mice not exposed to a HFD. Secondly, the effects of LCN2 on various aspects of platelet activity, including activation, aggregation, adhesion and plaque contraction, were examined. Finally, a MI/R injury model were established in both wild-type (WT) and LCN2 knockout (LCN2−/−) mice after HFD feeding, in order to assess the extent of MI and evaluate the recruitment of platelets, neutrophils, T cells and B cells in the heart using immunohistochemistry to detect CD42b, Ly6G, CD3 and B220 expression.
Materials and methods
Establishing a mouse model of obesity
The animal experimental center at Guizhou Medical University (Guiyang, China) was responsible for raising mice under controlled conditions, namely a constant temperature of 22–24°C, a relative humidity of 55–60% and a 12-h light/dark cycle. The experimental procedures were approved by the animal experiment ethics committee of Guizhou Medical University (approval no. 2100328). To determine the effect of a HFD on LCN2 expression, male C57BL/6J WT mice (Cyagen Biosciences Inc.; weight, 18–20 g; age, 6 weeks) were fed either a HFD (60% kcal from fat; D12492 formula; Research Diets, Inc.) or normal chow diet for 24 weeks, and then blood and liver samples were collected to detect LCN2 expression. These mice (n=5/group) were given free access to food/water. To investigate the effect of LCN2 gene knockout on MI/R injury in HFD-fed mice, male WT and LCN2−/− mice (Cyagen Biosciences Inc.; weight, 18–20 g; age, 6 weeks) were fed a HFD for 24 weeks, and then underwent MI/R injury or sham surgery. The mice were divided into the following four groups: WT + HFD + sham, WT + HFD + I/R, LCN2−/− + HFD + sham and LCN2−/− + HFD + I/R. The hearts (n=5/group) collected from the four groups underwent hematoxylin and eosin (H&E) staining. The degree of MI in the hearts of the four groups was assessed through TTC staining (n=5/group). Immunohistochemistry was performed to assess the expression of CD42b in the hearts collected from the four groups (n=5/group). In addition, immunohistochemistry was employed to detect the recruitment of Ly6G+, CD3+ and B220+ cells in the myocardial tissues collected from the mice in the WT + HFD + I/R and LCN2−/− +HFD + I/R groups (n=5/group).
Identification of mouse genes
Cyagen Biosciences, Inc. provided C57BL/6J mice with a background that lacked the LCN2 gene (LCN2−/−). Mouse tail genomic DNA was extracted using lysis buffer (cat. no. GD01-02; Bioland Scientific LLC) from mouse tail tissue, which was mixed with DNA stabilization buffer. PCR was then conducted using the 2X Es Taq Master Mix (cat. no. CW0690M; Jiangsu CoWin Biotech Co., Ltd.), as follows: 35 cycles of 94°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec. The following PCR primers were utilized: LCN2, forward (F) 5′-CAACTCAGAACTTGATCCCTGCC-3′ and reverse (R) 5′-TTTCCCTAAGTCCCGTTCAATCC-3′. Agarose gel electrophoresis (2%) was used to determine the size of the PCR products and the gels were stained with nucleic acid dye SuperRed/GelRed (cat. no. BS354A; Biosharp Life Sciences). For WT mice, a PCR product of 1,287 bp was generated. By contrast, a PCR product of 586 bp was generated for LCN2−/− mice.
Establishment of a MI/R injury model in mice
Male mice were anesthetized by intraperitoneal injection of Avertin (1.25%, 250 mg/kg) (cat. no. BR4108423; Bioleaper). After anesthesia, the animals naturally laid down; their head, limbs, tail and whiskers did not respond to touch, their heartbeat and breathing were uniform in a supine position, and their muscles were relaxed. These signs indicated full anesthesia and that the subsequent experimental operations could be carried out. The tongue was pulled out with tweezers and endotracheal intubation was performed through the glottis. A 1.5-cm incision was made at the intersection of the skin between the left midclavicular line and the fourth intercostal space in mice, and hemostatic forceps were used to stretch the fourth intercostal space to expose the heart. The left anterior descending (LAD) coronary artery was identified, and a 1.5-mm suture was made at the lower margin of the left auricle through the LAD coronary artery and tied into a knot. In addition, the sham operation group underwent the same procedure with the suture passing under the LAD branch, but without binding.
H&E and TTC staining
Mice were intraperitoneally injected with 1% pentobarbital sodium (150 mg/kg) and were sacrificed by cervical dislocation. Death was verified by the absence of a heartbeat and dilated pupils. Liver and heart samples were then obtained and fixed with 4% paraformaldehyde (cat. no. P0099; 100 ml; Beyotime Institute of Biotechnology) for 24 h at room temperature. The liver and heart tissues were then dehydrated, embedded in paraffin and sliced into 5-µm sections. These tissue sections were placed onto slides and subjected to dewaxing before staining with the H&E staining kit (cat. no. C0105M; Beyotime Institute of Biotechnology). The staining procedure was carried out according to the manufacturer's instructions at room temperature, with hematoxylin staining performed for 6 min and eosin staining for 1 min. The degree of MI was assessed through TTC staining (cat. no. T8170; Beijing Solarbio Science & Technology Co., Ltd.) according to the manufacturer's instructions. Images of the tissue sections were captured using an optical microscope (80i; Nikon Corporation).
Immunohistochemistry
The heart tissues of mice collected from different treatment groups were fixed with 4% paraformaldehyde solution for 24 h and dehydrated in an ascending series of alcohol at room temperature. After permeabilization with xylene for 30 min, heart tissues were then embedded in paraffin and sliced into 5-µm sections. These tissue sections were attached to slides and dewaxed with xylene. For antigen retrieval, they were placed in dyeing tanks containing citrate buffer, and were boiled in a pressure cooker for 15 min. To inhibit the activity of endogenous peroxidase, a 3% solution of hydrogen peroxide was applied for 10 min at room temperature. The heart tissue sections were then blocked with goat serum (cat. no. SAP-9100; Beijing Zhongshan Jinqiao Biotechnology Co., Ltd.) at 24°C for 30 min, and were subsequently incubated with primary antibodies [anti-CD42b antibody (1:200; cat. no. ab183345; Abcam), anti-CD3 antibody (1:200; cat. no. ab16669; Abcam), anti-B220 antibody (1:200; cat. no. ab10558; Abcam) and anti-Ly6G antibody (1:200; cat. no. ab238132; Abcam)] at 4°C for 12 h, followed by incubation with a horseradish peroxidase (HRP)-IgG secondary antibody (1:50; cat. no. A0208; Beyotime Institute of Biotechnology) at 24°C for 60 min. The expression of specific proteins in the heart tissues was visualized using DAB staining. Images of the tissue sections were captured using an optical microscope (80i; Nikon Corporation). Three visual field images of the infarct area were obtained from each histochemical section. The DAB-positive cell number in each image was manually counted by ImageJ (version 1.8.0; National Institutes of Health), and the mean value was defined as the positive cell number in the section.
Platelet separation
All blood samples used in the present study were obtained from healthy volunteers (age, 18–26 years) who provided written informed consent. Blood samples were collected from 18 volunteers, including 10 men and 8 women, with an mean age of 24 years, between March 2021 and March 2023. The Human Ethics Committee of Guizhou Medical University approved the experimental procedures [(approval no. 2021 (35)]. The collected blood was stored in a VACUETTE® vessel and mixed with 0.109 M sodium citrate at a ratio of 9:1. Subsequently, centrifugation at 120 × g for 20 min at 25°C was conducted to obtain the washed platelets for further experiments.
ELISA
After mice were intraperitoneally injected with 1% pentobarbital sodium (150 mg/kg), the blood serum was obtained by centrifuging blood samples (150 µl) collected from the tail veins of mice at 3,000 × g for 15 min at room temperature. The serum LCN2 levels were determined using a mouse ELISA kit (cat. no. ab199083; Abcam) according to the manufacturer's instructions.
Detection of platelet aggregation, activation and adhesion
Platelets were exposed to LCN2 (2 µg/ml; cat. no. 1757-LC-050; R&D Systems, Inc.) for 5 min at 37°C, and were then stimulated with thrombin (0.04 U/ml; cat. no. T6884; Sigma-Aldrich; Merck KGaA), collagen (1 µg/ml; cat. no. P/N 385; Chrono-Log Corporation) or adenosine diphosphate (ADP; 2.5 µM; cat. no. A2754; Sigma-Aldrich; Merck KGaA) for 5 min at 37°C. An equivalent volume of normal saline was used as a vehicle. Aggregation of platelets was then assessed using an aggregator (Model 700; Chrono-Log Corporation), while constant stirring was maintained at a rate of 1,200 rpm for 5 min. The expression levels of P-selectin were analyzed by performing flow cytometric analysis. Platelets were stimulated with or without LCN2 (2 µg/ml; cat. no. 1757-LC-050; R&D Systems, Inc.) for 5 min at 37°C, and were then exposed to thrombin for 5 min at 37°C; untreated platelets were used as a control. After incubating with FITC-conjugated P-selectin antibodies (0.2 µg/ml; cat. no. ab33279; Abcam) for 20 min at 37°C, the expression of P-selectin on the platelet surface was detected by flow cytometry (CytoFLEX S; Beckman Coulter, Inc.). Flow cytometry data were analyzed using CytExpert software (version 2.3.1.22; Beckman Coulter, Inc.).
To investigate the adhesion of platelets, cellular coverslips were categorized into three distinct groups: The first group was coated with 1% bovine serum albumin (cat. no. A8020; Beijing Solarbio Science & Technology Co., Ltd.) for 1 h at 20°C and subsequently incubated with non-treated platelets (2×107). The remaining two groups were coated with 5 µg/ml collagen for 14 h at 4°C, and incubated with 2 µg/ml LCN2-stimulated platelets (2×107) and unstimulated platelets (2×107) at 37°C. After a 45-min incubation, the platelets were stained with iFluor™ 680-phalloidin (1 µg/ml; cat. no. 40788ES75; Shanghai Yeasen Biotechnology Co., Ltd.) for 25 min at 37°C, and the adhesion of platelets was observed using a fluorescence microscope.
Assessment of clot retraction
Platelets were incubated with or without LCN2 (2 µg/ml) for 5 min at 37°C, and subsequently stimulated with thrombin (0.4 U/ml) at 37°C. The process of clot retraction was monitored and recorded at intervals of 0, 20, 40 and 60 min after thrombin stimulation by capturing images of platelet clots.
RT-qPCR
Liver tissues were collected for extraction of total RNA using a DNA/RNA co-extraction kit (cat. no. DP422; Tiangen Biotech Co., Ltd.). Subsequently, cDNA synthesis was carried out using the FastKing gDNA Dispelling RT SuperMix (cat. no. KR118; Tiangen Biotech Co., Ltd.) according to the manufacturer's instruction. qPCR was performed according to the protocol specified by the manufacturer of FastFire qPCR PreMix (SYBR Green) (cat. no. FP207; Tiangen Biotech Co., Ltd.). The qPCR program consisted of an initial phase of pre-denaturation at 95°C for 1 min, followed by 40 cycles at 95°C for 5 sec, 55°C for 10 sec and 72°C for 15 sec. For the expression analysis of the target genes, GAPDH was employed as the internal reference for normalization. The primer sequences were as follows, LCN2, F 5′-AAGGCAGCTTTACGATGT-3′, R 5′-TGGTTGTAGTCCGTGGTG-3′; GAPDH, F 5′-GCACAGTCAAGGCCGAGAAT-3′ and R 5′-GCCTTCTCCATGGTGGTGAA-3′. Data were normalized to GAPDH levels as a reference. Subsequently, the gene expression levels were calculated using the 2−ΔΔCq method (21).
Statistical analysis
The data were analyzed using GraphPad Prism v5.01 (Dotmatics), and the results are presented as the mean ± SEM derived from ≥3 independent experiments. Differences between two groups were assessed using unpaired Student's t-test, while differences among multiple groups were determined by one-way ANOVA and Tukey's post-hoc test. P<0.05 was considered to indicate a statistically significant difference.
Results
LCN2 expression is increased in the liver and serum of mice fed a HFD
Notably, LCN2 has been shown to be highly expressed in adipose tissue, and to serve a role in both obesity and obesity-related diseases (22,23). Considering that obesity is an important risk factor for CAD and MI/R injury, a mouse model of obesity was established in the present study through the use of a HFD. The results revealed a significant increase in the weight of the mice (Fig. 1A), as well as the accumulation of large lipid droplets in their liver tissues (Fig. 1B) following administration of a HFD. Additionally, a significant upregulation of LCN2 levels was observed in both the liver (Fig. 1C) and serum (Fig. 1D) of mice fed a HFD.
Figure 1.
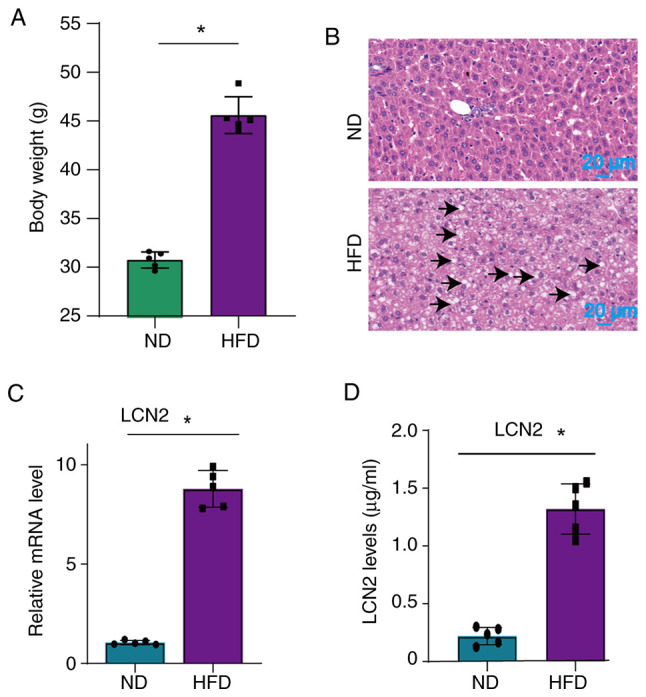
Expression of LCN2 in the liver and serum of HFD-fed mice. (A) Body weight was measured in mice that were fed a ND or HFD. (B) Liver tissue was analyzed using hematoxylin and eosin staining to observe pathological changes caused by a HFD. Lipid droplets were indicated by black arrows. (C) Reverse transcription-quantitative PCR was performed to assess the expression levels of LCN2 in the livers of mice who were or were not fed a HFD. (D) Levels of LCN2 in the serum of mice who were or were not fed a HFD were measured by ELISA. Data are presented as the mean ± SEM (n=5 mice/group). *P<0.05. HFD, high-fat diet; LCN2, lipocalin 2; ND, normal diet.
LCN2 potentiates platelet function
In cardiovascular disorders, the formation of atherosclerotic plaques and microthrombi are the consequences of platelet aggregation, adhesion and activation (24). These processes also contribute to the worsening of MI/R injury. Previous research has indicated that endogenous proteins possessing pro-inflammatory properties can enhance platelet hyperresponsiveness, and exacerbate atherosclerosis and MI/R injury by promoting platelet aggregation (25,26). The present study investigated the effects of LCN2 on platelet function. When platelet agonists (thrombin, collagen and ADP) were applied, incubation with LCN2 promoted platelet aggregation (Fig. 2A). Additionally, compared with the untreated platelets, a higher number of platelets adhered to collagen-coated coverslips following treatment with LCN2 (Fig. 2B). Furthermore, it was observed that LCN2-treated platelets had more pronounced clot retraction (Fig. 2C) and higher expression of P-selectin (Fig. 2D) upon stimulation with thrombin.
Figure 2.

Effect of LCN2 on platelet function. (A) After platelets were incubated with or without LCN2, they were further stimulated with ADP, thrombin or collagen to detect platelet aggregation (5 platelet samples were collected from 5 volunteers). (B) After platelets from different treatment groups were incubated on cellular coverslips for 45 min, they were stained with iFluor™ 680-phalloidin, and the adhesion of platelets was observed under a fluorescence microscope (5 platelet samples were collected from 5 volunteers). (C) After incubation with or without LCN2, the platelets were stimulated with thrombin. Clot retraction was observed after stimulation with thrombin for 0, 20, 40 and 60 min (5 platelet samples were collected from 5 volunteers). (D) Upon stimulation with or without LCN2, the platelets were incubated with thrombin, and untreated platelets were used as a control. The expression of P-selectin on the platelet surface was detected by flow cytometry (3 platelet samples were collected from 3 volunteers). Data are presented as the mean ± SEM. *P<0.05. ADP, adenosine diphosphate; BSA, bovine serum albumin; LCN2, lipocalin 2.
Effect of LCN2 knockout on MI/R injury in HFD-treated mice
Obesity serves an important role in the development of cardiovascular diseases. One of the main contributors to MI/R injury in obese individuals is the increase in endogenous bioactive molecules, which stimulate platelet activation (25). The gene phenotypes of WT mice and LCN2−/− mice were determined by PCR (Fig. 3A). For WT mice, a PCR product of 1,287 bp was generated, whereas a PCR product of 586 bp was generated for LCN2−/− mice. The PCR product of 586 bp in LCN2−/− mice indicated that LCN2 was knocked out, whereas the PCR product of 1,287 bp in WT mice indicated that LCN2 was not knocked out. These mice were then subjected to MI/R injury or sham operation after being fed a HFD. Subsequently, cardiac pathology was observed by H&E staining (Fig. 3B). After sham operation, the myocardia of WT mice and LCN2−/− mice were closely arranged, with normal structure and no obvious inflammatory cell infiltration. WT mice and LCN2−/− mice with MI/R injury exhibited notable histological features of myocardial tissue injury and inflammatory cell infiltration. In the case of MI/R injury, there was less structural damage to the myocardium and relatively less inflammatory cell infiltration in LCN2−/− mice compared with that in the WT mice (Fig. 3B). TTC staining showed no obvious ischemic features in the myocardia after sham operation in WT mice and LCN2−/− mice. After MI/R injury, the myocardial infarct size of LCN2−/− mice was smaller than that of WT mice (Fig. 3C).
Figure 3.

Effect of LCN2 knockout on MI/R injury in HFD-fed mice. (A) Mouse tails were utilized for DNA extraction, followed by PCR amplification and electrophoresis. For WT mice, a PCR product of 1,287 bp was obtained; conversely, LCN2−/− mice had a product of 586 bp. Subsequently, both WT and LCN2−/− mice underwent MI/R injury and sham surgery after being fed a HFD. (B) Mouse hearts were procured for hematoxylin and eosin staining. (C) Degree of infarction was assessed through TTC staining. Data are presented as the mean ± SEM (n=5). *P<0.05. HFD, high-fat diet; LCN2, lipocalin 2; MI/R, myocardial ischemia-reperfusion; TTC, 2,3,5-triphenyltetrazolium chloride; WT, wild-type.
Effect of LCN2 knockout on the recruitment of platelets and inflammatory cells in the myocardial tissue of HFD-fed mice subjected to MI/R injury
During MI/R injury, platelets tend to form microthrombi within the microvessels of the heart and worsen myocardial damage by releasing certain bioactive molecules (24). Thus, the current study aimed to investigate whether the absence of LCN2 affects the accumulation of platelets induced by I/R injury in myocardial tissue. As shown in Fig. 4A, the number of CD42b+ platelets in myocardial tissues was lower in LCN2−/− mice compared with that in WT mice following reperfusion post-ischemia. These findings suggested that LCN2 induced by HFD may enhance platelet accumulation in myocardial tissue during MI/R injury. Notably, activated platelets form complexes with leukocytes and adhere to microvessels within the heart, thereby exacerbating MI/R injury. Suppression of platelet activation can reduce the recruitment of inflammatory cells in myocardial tissue, ultimately mitigating MI/R injury (24,25). In the present study, it was observed that LCN2 gene knockout decreased the accumulation of Ly6G+ cells within the region of myocardial injury induced by I/R, while having no significant impact on the recruitment of CD3+ or B220+ cells (Fig. 4B). These results indicated that LCN2 knockout might alleviate MI/R injury by inhibiting platelet and Ly6G+ cell recruitment.
Figure 4.

Effect of LCN2 knockout on the recruitment of platelets and inflammatory cells in myocardial tissue after I/R injury in mice induced by a HFD. (A) WT and LCN2−/− mice were administered a HFD, and were then subjected to MI/R injury. Immunohistochemistry was performed to assess the expression of CD42b in the collected hearts. (B) WT and LCN2−/− mice were fed a HFD and were subjected to MI/R injury. Immunohistochemistry was then employed to detect the recruitment of Ly6G+, CD3+ and B220+ cells in myocardial tissues. Data are presented as the mean ± SEM (n=5). *P<0.05. HFD, high-fat diet; LCN2, lipocalin 2; MI/R, myocardial ischemia-reperfusion; WT, wild-type.
Discussion
In diseases related to obesity, an increase in platelet volume is observed. Additionally, there is an increase in the expression of thrombolane B2, prostaglandin F2, soluble P-selectin and soluble CD40L. These changes occur due to continuous stimulation from free fatty acids, oxidized low-density lipoprotein, inflammatory cytokines, enhanced oxidative stress, elevated stress proteins and other factors. Additionally, platelets release a higher number of particles, indicating a hyperreactive state (27–29). This hyperreactivity of platelets leads to their adherence to damaged endothelial cells, which serves a role in the development of atherosclerosis. Furthermore, activated platelets secrete certain specific bioactive molecules, such as myeloperoxidase and ROS, which participate in MI/R injury (30–34). Therefore, understanding how obesity impacts platelet function is crucial in identifying its role in exacerbating MI/R injury.
Currently, the focus of research on the impact of platelets on MI/R injury lies primarily in the analysis of the active molecules that are secreted during platelet activation, the surface molecules of platelets and the increased expression of endogenous bioactive molecules affecting platelet function. A previous study confirmed a significant positive correlation between plasma galectin-3 and platelet aggregation in patients with CAD. Moreover, galectin-3 was found to enhance platelet activation and thrombosis in animal models (25). Thus, the present findings supplement the understanding of the involvement of LCN2 in cardiovascular disease and contribute valuable insights to clinical science. Currently, known antiplatelet drugs do not aim to reduce the expression of endogenous molecules that activate platelets (24). This particular phenomenon may help to explain the uncertainty surrounding the efficacy of antiplatelet drugs in MI/R treatment.
The current research strategy was similar to that of Chen et al. In this study, the endogenous molecule galectin-3 was found to enhance platelet function to aggravate MI/R injury (25). Since a HFD, which is a risk factor of cardiovascular disease, could induce an increase in LCN2 expression, the present study further confirmed that LCN2 could promote platelet aggregation, adhesion and plaque retraction through in vitro experiments. These findings suggested that, similar to the function of galectin-3, LCN2 may have a role in enhancing platelet function. Subsequently, the current study further explored whether LCN2 could enhance the local accumulation of platelets in injured myocardium during MI/R in HFD-treated mice, as platelet accumulation has been considered to be an important factor in exacerbating I/R-induced myocardial injury. Under a HFD, the pathological damage of myocardial tissues was significantly lower in LCN2−/− mice than in WT mice when suffering from MI/R injury. Furthermore, fewer platelets accumulated in the injured myocardial tissues of LCN2−/− mice, which indicated that LCN2 was an important inducer of MI/R injury in HFD mice. Combined with the in vitro evidence that LCN2 promoted platelet function, it was hypothesized that HFD-induced LCN2 may aggravate MI/R injury, which could be fully or partially dependent on the enhancement of platelet activation and aggregation. In addition to the aforementioned results, it was observed that fewer Ly6G+ cells, rather than CD3+ and B220+ cells, accumulated in the injured myocardium of LCN2−/− mice when MI/R injury occurred. A previous study reported that LCN2 can aggravate psoriasis by inducing neutrophil chemotaxis and activation (35), and the present results also suggested that the effect of LCN2 on neutrophil function may serve a role in promoting MI/R injury. However, additional research is needed to fully confirm these possible mechanisms.
LCN2 has previously been reported to be involved in heart disease, as demonstrated by Jang et al (36). In this previous study, a mouse model of cardiomyopathy was established using adriamycin. Notably, it was observed that LCN2 knockout mice experienced milder myocardial injury after adriamycin induction, and this effect of LCN2 was related to its function of inhibiting autophagic flow. While the present study supports the potential pro-atherosclerotic effects of LCN2, specifically its regulatory function on platelets, it also raises important scientific questions for further exploration. Firstly, it is known that MI/R injury is a consequence of functional changes in multiple cell types. Beyond cardiomyocytes and cardiac microvascular endothelial cells, T helper (Th)17 cell differentiation has been implicated in the MI/R process (37). Notably, a previous study demonstrated that LCN2 exacerbated psoriasis-like skin inflammation by enhancing the Th17 response (38), which suggests that LCN2 may potentially worsen MI/R injury by enhancing the Th17 reaction. Furthermore, LCN2 is capable of activating various signaling pathways, including JAK/STAT and NF-κB, which have important roles in MI/R injury (39,40). Therefore, it is worth investigating whether LCN2 aggravates MI/R injury by influencing the activation of these signaling pathways. Moreover, potential receptors for LCN2, such as the glycoproteins GP330 and SLC22A17, have been identified (40). However, it remains unclear whether these receptors are expressed in platelets and whether LCN2 exerts its function through these receptors. Finally, exploring the development of drugs that effectively inhibit LCN2 function could lead to clinical applications for improving MI/R injury. These scientific questions will be the focus of further exploration in subsequent research.
In conclusion, the present study demonstrated that HFD-induced LCN2 exacerbated MI/R injury by promoting platelet activation, aggregation and adhesion.
Acknowledgements
Not applicable.
Funding Statement
The present study was supported by the Guizhou Provincial Health Commission (grant nos. gzwkj2022-031, gzwkj2022-254 and gzwkj2022-174), the Guizhou Administration of Traditional Chinese Medicine (grant no. QZYY-2021-145), and the Guiyang Science and Technology Bureau [grant no. (2019)9-12-3].
Availability of data and materials
The data generated in the present study may be requested from the corresponding author.
Authors' contributions
XL and QW conceived and designed the experiments. PL performed the experiments. PL, JC and MW analyzed the data and prepared the figures. XL and PL confirm the authenticity of all the raw data. All authors contributed to the article, and read and approved the final version of the manuscript.
Ethics approval and consent to participate
The experimental procedures carried out in the present study were approved by the animal experiment ethics committee of Guizhou Medical University (approval no. 2100328) and the Human Ethics Committee of Guizhou Medical University [approval no. 2021 (35)]. All blood samples used in the current study were obtained from healthy volunteers who provided written informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Harrington DH, Stueben F, Lenahan CM. ST-elevation myocardial infarction and non-ST-elevation myocardial infarction: Medical and surgical interventions. Crit Care Nurs Clin North Am. 2019;31:49–64. doi: 10.1016/j.cnc.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, Li CY, Li CJ. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46:1650–1667. doi: 10.1159/000489241. [DOI] [PubMed] [Google Scholar]
- 3.Visseren FLJ, Mach F, Smulders YM, Carballo D, Koskinas KC, Bäck M, Benetos A, Biffi A, Boavida JM, Capodanno D, et al. 2021 ESC guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. 2021;42:3227–3337. doi: 10.1093/eurheartj/ehab484. [DOI] [PubMed] [Google Scholar]
- 4.Algoet M, Janssens S, Himmelreich U, Gsell W, Pusovnik M, Van den Eynde J, Oosterlinck W. Myocardial ischemia-reperfusion injury and the influence of inflammation. Trends Cardiovasc Med. 2023;33:357–366. doi: 10.1016/j.tcm.2022.02.005. [DOI] [PubMed] [Google Scholar]
- 5.Wei Y, Xing J, Su X, Li X, Yan X, Zhao J, Tao H. IL-38 attenuates myocardial ischemia-reperfusion injury by inhibiting macrophage inflammation. Immun Inflamm Dis. 2023;11:e898. doi: 10.1002/iid3.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang J, Hu S, Gao Y, Wei X, Qu Y, Gao R, Lv Y, Wang J, Wang Y, Yang J, et al. Galangin alleviated myocardial ischemia-reperfusion injury by enhancing autophagic flux and inhibiting inflammation. Eur J Pharmacol. 2023;945:175621. doi: 10.1016/j.ejphar.2023.175621. [DOI] [PubMed] [Google Scholar]
- 7.Chen LQ, Wang WS, Li SQ, Liu JH. Minocycline relieves myocardial ischemia-reperfusion injury in rats by inhibiting inflammation, oxidative stress and apoptosis. Eur Rev Med Pharmacol Sci. 2022;26:3001–3009. doi: 10.26355/eurrev_202204_28631. [DOI] [PubMed] [Google Scholar]
- 8.Seeger JP, Benda NM, Riksen NP, van Dijk AP, Bellersen L, Hopman MT, Cable NT, Thijssen DH. Heart failure is associated with exaggerated endothelial ischaemia-reperfusion injury and attenuated effect of ischaemic preconditioning. Eur J Prev Cardiol. 2016;23:33–40. doi: 10.1177/2047487314558377. [DOI] [PubMed] [Google Scholar]
- 9.Wereski R, Kimenai DM, Bularga A, Taggart C, Lowe DJ, Mills NL, Chapman AR. Risk factors for type 1 and type 2 myocardial infarction. Eur Heart J. 2022;43:127–135. doi: 10.1093/eurheartj/ehab581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cameron SJ, Ture SK, Mickelsen D, Chakrabarti E, Modjeski KL, McNitt S, Seaberry M, Field DJ, Le NT, Abe J, Morrell CN. Platelet extracellular regulated protein kinase 5 is a redox switch and triggers maladaptive platelet responses and myocardial infarct expansion. Circulation. 2015;132:47–58. doi: 10.1161/CIRCULATIONAHA.115.015656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schanze N, Bode C, Duerschmied D. Platelet contributions to myocardial ischemia/reperfusion injury. Front Immunol. 2019;10:1260. doi: 10.3389/fimmu.2019.01260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Meijden PEJ, Heemskerk JWM. Platelet biology and functions: New concepts and clinical perspectives. Nat Rev Cardiol y. 2019;16:166–179. doi: 10.1038/s41569-018-0110-0. [DOI] [PubMed] [Google Scholar]
- 13.Jaberi SA, Cohen A, D'Souza C, Abdulrazzaq YM, Ojha S, Bastaki S, Adeghate EA. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed Pharmacother. 2021;142:112002. doi: 10.1016/j.biopha.2021.112002. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh S, Stepicheva N, Yazdankhah M, Shang P, Watson AM, Hose S, Liu H, Weiss J, Zigler JS, Jr, Valapala M, et al. The role of lipocalin-2 in age-related macular degeneration (AMD) Cell Mol Life Sci. 2020;77:835–851. doi: 10.1007/s00018-019-03423-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eilenberg W, Stojkovic S, Piechota-Polanczyk A, Kaun C, Rauscher S, Gröger M, Klinger M, Wojta J, Neumayer C, Huk I, Demyanets S. Neutrophil gelatinase-associated lipocalin (NGAL) is associated with symptomatic carotid atherosclerosis and drives pro-inflammatory state in vitro. Eur J Vasc Endovasc Surg. 2016;51:623–631. doi: 10.1016/j.ejvs.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 16.Mosialou I, Shikhel S, Luo N, Petropoulou PI, Panitsas K, Bisikirska B, Rothman NJ, Tenta R, Cariou B, Wargny M, et al. Lipocalin-2 counteracts metabolic dysregulation in obesity and diabetes. J Exp Med. 2020;217:e20191261. doi: 10.1084/jem.20191261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Silva C, Rotellar F, Gil MJ, Cienfuegos JA, Salvador J, Frühbeck G. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J Mol Med (Berl) 2009;87:803–813. doi: 10.1007/s00109-009-0486-8. [DOI] [PubMed] [Google Scholar]
- 18.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, Chow WS, Wat NM, Xu JY, Hoo RL, Xu A. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34–41. doi: 10.1373/clinchem.2006.075614. [DOI] [PubMed] [Google Scholar]
- 20.Jang Y, Lee JH, Wang Y, Sweeney G. Emerging clinical and experimental evidence for the role of lipocalin-2 in metabolic syndrome. Clin Exp Pharmacol Physiol. 2012;39:194–199. doi: 10.1111/j.1440-1681.2011.05557.x. [DOI] [PubMed] [Google Scholar]
- 21.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 22.Wu G, Li H, Zhou M, Fang Q, Bao Y, Xu A, Jia W. Mechanism and clinical evidence of lipocalin-2 and adipocyte fatty acid-binding protein linking obesity and atherosclerosis. Diabetes Metab Res Rev. 2014;30:447–456. doi: 10.1002/dmrr.2493. [DOI] [PubMed] [Google Scholar]
- 23.Abella V, Scotece M, Conde J, Gómez R, Lois A, Pino J, Gómez-Reino JJ, Lago F, Mobasheri A, Gualillo O. The potential of lipocalin-2/NGAL as biomarker for inflammatory and metabolic diseases. Biomarkers. 2015;20:565–571. doi: 10.3109/1354750X.2015.1123354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schanze N, Hamad MA, Nührenberg TG, Bode C, Duerschmied D. Platelets in myocardial ischemia/reperfusion injury. Hamostaseologie. 2023;43:110–121. doi: 10.1055/a-1739-9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Fu W, Zheng Y, Yang J, Liu Y, Qi Z, Wu M, Fan Z, Yin K, Chen Y, et al. Galectin 3 enhances platelet aggregation and thrombosis via Dectin-1 activation: A translational study. Eur Heart J. 2022;43:3556–3574. doi: 10.1093/eurheartj/ehac034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Liu G, Hong Y, Han J, Yang Z, Yang Y, Li H, Wang S, Jue L, Wang Q. Regulation of atherosclerosis by toll-like receptor 4 induced by serum amyloid 1: A systematic in vitro study. Biomed Res Int. 2022;2022:4887593. doi: 10.1155/2022/4887593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anfossi G, Russo I, Trovati M. Platelet dysfunction in central obesity. Nutr Metab Cardiovasc Dis. 2009;19:440–449. doi: 10.1016/j.numecd.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Santilli F, Vazzana N, Liani R, Guagnano MT, Davì G. Platelet activation in obesity and metabolic syndrome. Obes Rev. 2012;13:27–42. doi: 10.1111/j.1467-789X.2011.00930.x. [DOI] [PubMed] [Google Scholar]
- 29.Barale C, Russo I. Influence of cardiometabolic risk factors on platelet function. Int J Mol Sci. 2020;21:623. doi: 10.3390/ijms21020623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mauler M, Herr N, Schoenichen C, Witsch T, Marchini T, Härdtner C, Koentges C, Kienle K, Ollivier V, Schell M, et al. Platelet serotonin aggravates myocardial ischemia/reperfusion injury via neutrophil degranulation. Circulation. 2019;139:918–931. doi: 10.1161/CIRCULATIONAHA.118.033942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davidson SM, Andreadou I, Barile L, Birnbaum Y, Cabrera-Fuentes HA, Cohen MV, Downey JM, Girao H, Pagliaro P, Penna C, et al. Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc Res. 2019;115:1156–1166. doi: 10.1093/cvr/cvy314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gumiężna K, Baruś P, Sygitowicz G, Wiśniewska A, Ochijewicz D, Pasierb K, Klimczak-Tomaniak D, Kuca-Warnawin E, Kochman J, Grabowski M, et al. Immature platelet fraction in cardiovascular diagnostics and antiplatelet therapy monitoring. Cardiol J. 2023;30:817–824. doi: 10.5603/CJ.a2023.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margraf A, Zarbock A. Platelets in inflammation and resolution. J Immunol. 2019;203:2357–2367. doi: 10.4049/jimmunol.1900899. [DOI] [PubMed] [Google Scholar]
- 34.Huilcaman R, Venturini W, Fuenzalida L, Cayo A, Segovia R, Valenzuela C, Brown N, Moore-Carrasco R. Platelets, a key cell in inflammation and atherosclerosis progression. Cells. 2022;11:1014. doi: 10.3390/cells11061014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shao S, Cao T, Jin L, Li B, Fang H, Zhang J, Zhang Y, Hu J, Wang G. Increased lipocalin-2 contributes to the pathogenesis of psoriasis by modulating neutrophil chemotaxis and cytokine secretion. J Invest Dermatol. 2016;136:1418–1428. doi: 10.1016/j.jid.2016.02.538. [DOI] [PubMed] [Google Scholar]
- 36.Jang HM, Lee JY, An HS, Ahn YJ, Jeong EA, Shin HJ, Kim KE, Lee J, Koh JS, Roh GS. LCN2 deficiency ameliorates doxorubicin-induced cardiomyopathy in mice. Biochem Biophys Res Commun. 2022;588:8–14. doi: 10.1016/j.bbrc.2021.12.048. [DOI] [PubMed] [Google Scholar]
- 37.Li D, Yang Z, Gao S, Zhang H, Fan G. Tanshinone IIA ameliorates myocardial ischemia/reperfusion injury in rats by regulation of NLRP3 inflammasome activation and Th17 cells differentiation. Acta Cir Bras. 2022;37:e370701. doi: 10.1590/acb370701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hau CS, Kanda N, Tada Y, Shibata S, Uozaki H, Fukusato T, Sato S, Watanabe S. Lipocalin-2 exacerbates psoriasiform skin inflammation by augmenting T-helper 17 response. J Dermatol. 2016;43:785–794. doi: 10.1111/1346-8138.13227. [DOI] [PubMed] [Google Scholar]
- 39.Chen PC, Ho CH, Fan CK, Liu SP, Cheng PC. Antimicrobial peptide LCN2 inhibited uropathogenic escherichia coli infection in bladder cells in a high-glucose environment through JAK/STAT signaling pathway. Int J Mol Sci. 2022;23:15763. doi: 10.3390/ijms232415763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim SL, Shin MW, Seo SY, Kim SW. Lipocalin 2 potentially contributes to tumorigenesis from colitis via IL-6/STAT3/NF-κB signaling pathway. Biosci Rep. 2022;42:BSR20212418. doi: 10.1042/BSR20212418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data generated in the present study may be requested from the corresponding author.