

Abstract
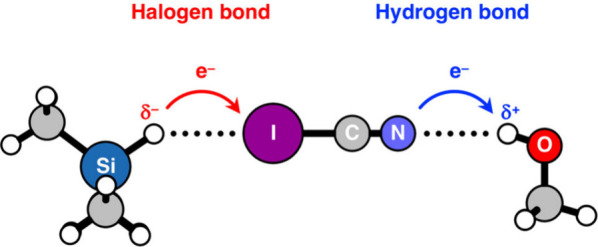
Herein, we dismiss a recent proposal by Civiš, Hobza, and co-workers to modify the IUPAC definition of hydrogen bonds in order to expand the scope from protonic Y–Hδ+ to hydridic Y–Hδ− hydrogen-bond donor fragments [J. Am. Chem. Soc.2023, 145, 8550]. Based on accurate Kohn–Sham molecular orbital (KS-MO) analyses, we falsify the conclusion that interactions involving protonic and hydridic hydrogens are both hydrogen bonds; they are not. Instead, our quantitative KS-MO, energy decomposition, and Voronoi deformation density analyses reveal two fundamentally different bonding mechanisms for protonic Y–Hδ+ and hydridic Y–Hδ− fragments which go with charge transfer in opposite directions. On one hand, we confirm the IUPAC definition for regular hydrogen bonds in the case of protonic Y–Hδ+ fragments. On the other hand, complexes involving Y–Hδ− fragments are, in fact, acceptors in other well-known families of Lewis-acid/base interactions, such as halogen bonds, chalcogen bonds, and pnictogen bonds. These mechanisms lead to the same spectroscopic phenomenon in both the Y–Hδ+ and Y–Hδ− fragments, that is, the redshift in the Y–H stretching frequency, which is, thus, not an exclusive indicator for hydrogen bonding.
Introduction
The hydrogen bond is a key chemical interaction in biological, supramolecular, and organic chemistry.1 IUPAC defines the hydrogen bond (HB) as an attractive interaction between a hydrogen atom from a molecule or a molecular fragment Y–H, in which Y is more electronegative than H, and an atom or group of atoms (Z), in which there is evidence of bond formation (i.e., a Y–H···Z hydrogen-bonded complex).2 According to molecular orbital (MO) theory, the reason behind the stability of the HB is twofold: (i) the electrostatic attraction between the protonic hydrogen in the Y–Hδ+ fragment and Z; and (ii) the covalent donor–acceptor interaction stemming from the charge transfer from the HOMO of Z into the empty σ* Y–H antibonding orbital.3 The covalency of HB is manifested in the characteristic Y–H bond elongation and a decrease, i.e., redshift, in the vibrational frequency associated with the Y–H bond stretching mode.3b,4
Hydridic hydrogens are also known to engage in intramolecular interactions that do not fit the IUPAC definition of the HB. For example, in the case of the charge inverted hydrogen bonds (CIHB),5 a designation coined by Jabłoński to cover systems of the type Y–Hδ−···Z, in which Y is less electronegative than H and Z is an electron-deficient fragment. In this point of view, hydrogen bonding refers to all interactions involving a hydrogen atom that can either be protonic, forming a regular HB, or hydridic, forming a CIHB. Recently, Civiš, Hobza, and co-workers proposed a generalization of the IUPAC definition of HB in order to cover both the protonic and hydridic forms within the same definition.6 According to the authors, CIHBs still hold many important features of the HB, such as the donation of charge into the σ* Y–Hδ− antibonding orbital and the elongation, i.e., redshift, of the Y–Hδ− bond. This implies that, in CIHB, hydridic hydrogens behave as Lewis acids and the electron-deficient Z fragment behaves as a Lewis base; thus, the bonding mechanism of HB and CIHB would be the same.
In this work, we challenge the idea that hydridic hydrogen bonds involve only an inversely polarized H-bond donor but otherwise are, in essence, electronically similar to protonic hydrogen bonds. To this end, we analyze the bonding mechanism of a series of MemYH···NH3, MemYH···NCI, and MemYH···ICN complexes (Y = C, Si, Ge, N, P, As, O, S, Se; m = 3, 2, 1), using Kohn–Sham molecular orbital (KS-MO) theory (Scheme 1). Our model systems feature Y–H groups with protonic and hydridic hydrogens and allow for a systematic and quantitative comparison of how bonding mechanisms change in nature along the series. We show that true hydrogen bonds, i.e., in which the H-bond acts as a Lewis acid accepting charge in its σ* Y–H LUMO, can only be formed with protonic hydrogens. On the contrary, complexes involving hydridic hydrogens lead to a completely different bonding mechanism in which the molecular fragment containing the hydrogen atom acts as a Lewis base, not as a Lewis acid.7 Thus, CIHB is not a correct designation for interactions involving hydridic hydrogens, and hence, there is no need to change the IUPAC definition of the hydrogen bond.
Scheme 1. Schematic MemYH···NH3, MemYH···NCI, and MemYH···NCI Model Complexes (Y = C, Si, Ge, N, P, As, O, S, Se; m = 3, 2, 1).

Results and Discussion
The MemYH Fragments
We start by analyzing the protonic or hydridic character of the H atom of the studied model MemYH fragments, in which Y is an element belonging to groups 14, 15, and 16 and periods 2, 3, and 4 of the periodic table (Y = C, Si, Ge, N, P, As, O, S, Se, and m = 3, 2, and 1). For this purpose, we analyze the Voronoi deformation density (VDD) charge on the H atom (QH) directly bound to atom Y (see Theoretical Methods). In general, we find that Y atoms with an electronegativity higher than H (χH = 2.20)8 cause a charge depletion on H, i.e., a positive VDD charge on H, whereas Y atoms with an electronegativity lower than H result in an accumulation of charge on H, i.e., a negative VDD charge on H. For example, QH is +154 milli-electrons for MeOH and −92 milli-electrons for Me3SiH, where the electronegativity of O and Si are χO = 3.44 and χSi = 1.90, respectively (Figure 1). Thus, the nature of atom Y greatly influences the charge of the H atom in the MemYH monomer, making them either protonic or hydridic.
Figure 1.

Equilibrium geometries, Voronoi deformation density charges (in milli-electrons) on the indicated hydrogen atom, and the electronegativity of Y on the Pauling scale7 (χY; in gray) of the MemYH fragments (Y = C, Si, Ge, N, P, As, O, S, Se; m = 3, 2, 1). The electronegativity of H is χH = 2.20.8 Computed at ZORA-BLYP-D3(BJ)/TZ2P.
We recall that, according to the IUPAC definition,2 a hydrogen bond is only formed when the Y atom is more electronegative than H. In other words, the Me3SiH, Me3GeH, Me2PH, and Me2AsH fragments with hydridic H should be unable to engage in a hydrogen bond with an electron-rich fragment, i.e., a Lewis base (Figure 1). In the following section, we quantify this statement and explain why hydrogen bonds involving a hydric hydrogen can indeed not be formed.
On the Nonexistence of Hydridic Hydrogen Bonds
To evaluate the ability of protonic and hydridic hydrogens to engage in hydrogen bonding, we study the interaction between the MemYH fragments and NH3, a well-known Lewis base and hydrogen-bond acceptor, to form complexes of the type MemYH···NH3 (see Scheme 1a). The equilibrium geometries, electronic bond energies ΔE, and the charge depletion on the NH3 fragment are reported in Figure 2 (see Table S1 for additional data). We find that MemYH···NH3 complexes exist only for the MemYH fragments with protonic or close to neutral hydrogens, namely, for Y = C, N, O, S, and Se, in which the most stable and shortest is MeOH···NH3, with ΔE = −7.2 kcal mol–1 and rH···N = 1.93 Å, and the least stable and longest is Me3CH···NH3, with ΔE = −1.0 kcal mol–1 and rH···N = 2.68 Å (Figure 2). The fragments with hydridic hydrogens (Y = Si, Ge, P, As), but also the slightly protonic Me3CH, form structurally completely different bonding motifs of the type Me3YH···HNH2, which are less stable, ranging from Me2AsH···HNH2, with ΔE = −2.6 kcal mol–1, to Me3CH···HNH2, with ΔE = −1.4 kcal mol–1 (Figure 2).
Figure 2.

Equilibrium geometries (in Å), electronic bond energies (in kcal mol–1; in brackets), and the charge depletion on the NH3 fragment (ΔQNH3; in milli-electrons; in blue) of the MemYH···NH3 hydrogen-bonded complexes (Y = C, N, O, S, Se) and the MemYH···HNH2 complexes (Y = C, Si, Ge, P, As). Computed at ZORA-BLYP-D3(BJ)/TZ2P.
The different bonding motifs in protonic MemYH···NH3 versus those in hydridic MemYH···HNH2 are accompanied by different bonding mechanisms. In MemYH···NH3, we find a depletion of charge on NH3 upon binding, i.e., positive ΔQNH3, that is, there is charge transfer from NH3 into the MemYH fragment (see Figure 2). The charge transfer mechanism from NH3 to MemYH is the characteristic covalent component of the hydrogen bond, where the lone pair orbital of NH3 (LPN) donates charge into the empty σ* Y–H antibonding orbital of MemYH (σ*Y–H; see Scheme 2).3 As the σ*Y–H becomes more populated, the Y–H bond elongates (see Table S1), resulting in the characteristic redshift in the vibrational frequency associated with the Y–H bond stretching mode. In contrast, there is no charge transfer in the MemYH···HNH2 bonding motif involving a hydridic hydrogen, that is, there is no charge depletion or accumulation on NH3 and, hence, no hydrogen bond in these complexes (see Figure 2). Next, we explain why hydridic fragments cannot form MemYH···NH3 hydrogen-bonded complexes.
Scheme 2. Schematic Molecular Orbital Diagram for the Attractive (Green) and Repulsive (Red) Orbital Interactions in MemYH···NH3 (Y = C, N, O, S, Se).

Stable hydrogen-bonded complexes are formed because the electrostatic attraction between Y–Hδ+ and the electron-rich bond acceptor, together with the covalent, charge transfer interaction, overcomes the destabilizing Pauli repulsion between the occupied σ Y–H bonding orbital (σY–H) and the occupied orbitals of the bond acceptor (see Scheme 2).3 Protonic hydrogens are depleted of electron density, which exposes their positively charged nucleus to the electron density of an incoming electron-rich fragment, resulting in a stronger electrostatic attraction. On the other hand, hydridic hydrogens have an excess of electron density that: (i) screens their positively charged nucleus, causing a weaker electrostatic attraction between Y–Hδ− and the hydrogen-bond acceptor; and (ii) increases the size of σY–H on the H atom, resulting in a more destabilizing Pauli repulsion between σY–H and the occupied orbitals of the bond acceptor. As a result, hydrogen-bonded complexes involving a hydridic hydrogen are not formed because the attractive electrostatic and charge transfer interactions are not strong enough to overcome the repulsive wall arising from the Pauli repulsion. These findings emerge from our energy decomposition and quantitative KS-MO analyses that will be explained in detail in the next section.
First, we analyze the bonding mechanism of the representative MeOH···NH3, Me3CH···NH3, and Me3SiH···NH3 hydrogen-bonded complexes using our canonical energy decomposition analysis (EDA)9 as a function of the H···N bond distance (rH···N), while keeping the geometries of the fragments frozen to the equilibrium geometries of the complexes (Figure 3; see Theoretical Methods). The Me3SiH···NH3 complex is unbound; therefore, in this case, we have the Me3SiH and NH3 fragments approach each other along the rH···N in their respective equilibrium geometries.
Figure 3.

Energy decomposition analysis (in kcal mol–1) and the repulsive occupied–occupied overlap (S) as a function of the H···N bond distance for the representative Me3SiH···NH3, Me3CH···NH3, and MeOH···NH3 complexes. Computed at ZORA-BLYP-D3(BJ)/TZ2P.
The interaction energy curve, ΔEint(rH···N), goes from stabilizing, i.e., binding, for MeOH···NH3 to weakly stabilizing for Me3CH···NH3 to destabilizing, i.e, unbound, for Me3SiH···NH3, reflecting the trends in hydrogen-bond stability of the complexes (Figure 3a). This trend is a direct result of the Pauli repulsion, ΔEPauli(rH···N), which becomes significantly more destabilizing from MeOH···NH3 to Me3CH···NH3 to Me3SiH···NH3 (Figure 3c). Along this series, Y becomes less electronegative than H, which significantly affects the size of the occupied σY–H orbital on the H atom. The orbital amplitude on the H atom substantially increases along MeOH to Me3CH to Me3SiH (see Figure 4), causing the repulsive orbital overlap between the σY–H orbital of MemYH and the LPN orbital of NH3 to increase along MeOH···NH3, Me3CH···NH3, and Me3SiH···NH3 and, thus, leading to more build-up of Pauli repulsion upon bond formation (see Figure 3f). Note that the reduced Pauli repulsion is also the reason why MeOH···NH3 can form a shorter hydrogen bond compared to the other complexes (Figure 3). In addition, the electrostatic interactions curve, ΔVelstat(rH···N), also becomes less stabilizing along MeOH···NH3, Me3CH···NH3, and Me3SiH···NH3, especially around the equilibrium hydrogen-bond bond length (Figure 3b). This is a direct consequence of the decreasing electronegativity of Y along O, C, and Si, which makes the donating H atom less positively (or more negatively) charged along the same series, leading to a weaker electrostatic attraction between MemYH and the electron-rich NH3 fragment. In summary, a hydridic hydrogen atom causes the MemYH···NH3 complexes to experience a substantial build-up in steric repulsion and a weakening of electrostatic attraction, resulting in the nonexistence of hydridic hydrogen bonds in MemYH···NH3.
Figure 4.

Electron density, occupied σ Y–H bonding orbital, and empty σ* Y–H antibonding orbital (contour plots from 0.9 to 0.03 au) for the representative Me3SiH, Me3CH, and MeOH fragments. Computed at ZORA-BLYP-D3(BJ)/TZ2P.
Hydridic Hydrogens as Bond Acceptors
We have shown that hydridic hydrogens are unable to engage in hydrogen bonds with electron-rich molecules like NH3. In this section, we address the question: can hydrogen bonding be the most dominant interaction when a hydridic hydrogen engages in an intermolecular interaction with electron-poor halogen-bond donors as proposed by Civiš, Hobza, and co-workers?6 To answer this question, we study the bonding mechanism for the interaction between MemYH and ICN, where ICN can form a hydrogen bond with the nitrogen side and a halogen bond with the iodine side,10 according to the IUPAC definitions of hydrogen bonds2 and halogen bonds10a (see Scheme 1). The equilibrium geometries, electronic bond energies ΔE, and the charge rearrangement in the ICN fragment are reported in Figure 5.
Figure 5.

Equilibrium geometries (in Å), electronic bond energies (in kcal mol–1; in brackets), and the charge accumulation (red) and depletion (blue) on the ICN fragment (in milli-electrons) of the MemYH···NCI complexes (Y = C, N, O, S, Se) and MemYH···ICN (Y = C, Si, Ge, P, As). Computed at ZORA-BLYP-D3(BJ)/TZ2P.
Similar to MemYH···NH3, the MemYH fragments involving protonic and close to neutral hydrogens (Y = C, N, O, S, Se) form regular Y–Hδ+···Nδ− hydrogen bonds, in which charge flows into the hydrogen-bond donor MemY–H, i.e., positive ΔQICN (Figure 5). This bond has a strength of up to 4.6 kcal mol–1 for MeOH···NCI (Figure 5). On the other hand, the MemYH fragments involving a hydridic hydrogen (Y = Si, Ge, P, As) form regular Hδ−···Iδ+–C halogen bonds, in which charge flows out of the halogen-bond acceptor MemY–H into the halogen-bond donor I–CN, i.e., negative ΔQICN (Figure 5). There is one intermediate situation, namely, the Me3CH fragment, which shows both Me3CH···NCI and Me3CH···ICN bonding modes. As will become clear, this change in bonding mode occurs because we go from protonic MemYH···NCI hydrogen bonds to MemYH···ICN halogen bonds, not hydridic MemYH···ICN hydrogen bonds, in line with the current IUPAC definitions of hydrogen bonds2 and halogen bonds.10a
The bonding mechanism in MemYH···NCI involves a regular Y–Hδ+···Nδ− hydrogen bond and hence is similar to that of the MemYH···NH3 complexes (vide supra). The ICN fragment acts as a hydrogen-bond acceptor on the N side through which the LPN orbital of the ICN fragment donates charge into the empty σ*Y–H orbital of MemYH fragments involving protonic hydrogens (Scheme 3, right side). However, as explained in the previous section, this donor–acceptor interaction is not strong enough to overcome the buildup of steric repulsion when involving MemYH fragments with hydridic hydrogens and, thus, MemYH···NCI complexes cannot be found for Y = Si, Ge, P, and As.
Scheme 3. Schematic Molecular Orbital Diagram (Contour Plots from 0.9 to 0.03 a.u.) for the Attractive (Green) and Repulsive (Red) Orbital Interactions in the Halogen-Bonded MemYH···ICN and Hydrogen-Bonded MemYH···NCI Complexes (Computed at ZORA-BLYP-D3(BJ)/TZ2P).

The hydridic MemYH···ICN complexes are constructed via a different bonding mechanism, namely, a halogen-bond,3b,11 which is characterized by the covalent, donor–acceptor interaction between the occupied σY–H orbital of MemYH and the empty σ* I–C antibonding orbital of ICN (σ*I–C) (Scheme 3, left side). Consequently, upon binding, the charge flows from MemYH into the ICN fragment, as indicated by the negative ΔQICN (see Figure 5). Note that the hydrogen-bonding and halogen bonding mechanisms go with charge transfer in opposite directions; that is, ΔQICN is positive for the former and negative for the latter. Therefore, the negative ΔQICN is clear evidence that there is no hydrogen-bonding mechanism that is able to overcome the halogen bond in the MemYH···ICN complexes. This is because ICN is a poor hydrogen-bond acceptor, i.e., weak Lewis base, on the I side due to its very low-lying I lone-pair (LPI) orbital (see Scheme 3).
The magnitude of the various pairwise donor–acceptor orbital interactions is approximately proportional to their orbital overlap squared (S2) divided by the orbital energy gap (Δε). For Me3SiH···ICN, the S and Δε for the σY–H → σ*I–C halogen-bond interaction are 0.19 and 4.0 eV, respectively, and its associated S2/Δε is 9.4 × 103 (see Figure 6 and Table S5 for all the MemYH···ICN complexes). The LPI orbital of ICN has a large amplitude on the I side and overlaps with the empty σ*Y–H of Me3SiH, in which S is 0.15 (Figure 6). However, this σ*Y–H ← LPI hydrogen-bond interaction is significantly weaker due to a large Δε of 13.3 eV, leading to an S2/Δε of only 1.7 × 103, which is about five times weaker than that of the σY–H → σ*I–C halogen-bond interaction (Figure 6). Therefore, the magnitude of the hydrogen-bonding mechanism is negligible and dominated by the halogen bond in Me3SiH···ICN. For this reason, Me3SiH···ICN and the other MemYH···ICN complexes involving hydridic hydrogens should be seen as halogen-bonded, not hydridic hydrogen-bonded, complexes.
Figure 6.
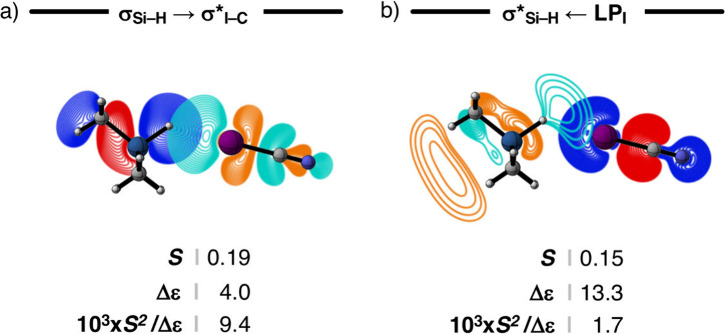
Donor–acceptor orbital overlap (S), energy gap (Δε, in eV), and the orbital stabilization for a) the σSi–H → σ*I–C halogen bond and b) the σ*Si–H ← LPI hydrogen bond in the representative Me3SiH···ICN complex (contour plots from 0.9 to 0.03 au). Computed at ZORA-BLYP-D3(BJ)/TZ2P.
The Redshift
The analyzed MemYH···NH3 hydrogen-bonded systems feature the characteristic Y–H bond elongation and, thus, the redshift in the Y–H bond stretching frequency. For example, in the series of MeOH···NH3, MeSH···NH3, and MeSeH···NH3, the Y–H bond elongates by 0.016 Å, 0.010 Å, and 0.012 Å, and the symmetric Y–H bond stretching frequency in the complexes decreases by −316 cm–1, −125 cm–1, and −134 cm–1, respectively, in respect to the isolated MemYH monomers (Table 1). The Y–H bond elongation in hydrogen-bond donors is the manifestation of the covalent nature of hydrogen bonds, that is, it is caused by the donation of charge into the empty σ*Y–H orbital via donor–acceptor interactions.3b,4 In the series of MeOH···NH3, MeSH···NH3, and MeSeH···NH3, there is a depletion of charge in the NH3 fragment, i.e., positive ΔQNH3, while the population in empty σ*Y–H orbital increases to 0.04 0.03, and 0.03 electrons along the same series.
Table 1. Y–H Bond Elongation (in Å), Change in the Y–H Stretching Frequency (in cm–1), Charge in the Voronoi Deformation Density Charge on the NH3 or ICN Fragments (in Milli-electrons), and Populations in the Filled σY–H and Empty σ*Y–H Orbitals for Representative MemYH···NH3 (Y = O, S, Se) and MemYH···ICN (Y = C, Si, Ge) Complexes (Computed at ZORA-BLYP-D3(BJ)/TZ2Pa).
| Complex | ΔrY–H | ΔυY–H | ΔQNH3/ICN | Pop.σY–H | Pop.σ*Y–H |
|---|---|---|---|---|---|
| MeOH···NH3 | 0.016 | –316 | +41 | 2.00 | 0.04 |
| MeSH···NH3 | 0.010 | –125 | +28 | 2.00 | 0.03 |
| MeSeH···NH3 | 0.012 | –134 | +34 | 2.00 | 0.03 |
| Me3CH···ICN | 0.003 | –47 | –11 | 1.99 | 0.00 |
| Me3SiH···ICN | 0.014 | –87 | –35 | 1.94 | 0.00 |
| Me3GeH···ICN | 0.019 | –98 | –39 | 1.93 | 0.01 |
See complete data in Tables S1–S4 in the Supporting Information.
In the halogen-bonded MemYH···ICN complexes, we find that the Y–H bond also elongates and, thus, the Y–H bond stretching frequency redshifts. For example, in the series of Me3CH···ICN, Me3SiH···ICN, and Me3GeH···ICN, the Y–H bond elongates by 0.003 Å, 0.014 Å, and 0.019 Å, and the symmetric Y–H bond stretching frequency in the halogen-bond acceptors decreases by −47 cm–1, −87 cm–1, and −98 cm–1, respectively (Table 1). However, since the hydrogen-bonding mechanism is negligible in the MemYH···ICN complexes (vide supra), the population in the empty σ*Y–H orbital does not significantly change and, therefore, cannot be responsible for the Y–H bond elongation in these complexes (Table 1). In fact, the redshift in the MemYH halogen-bond acceptors is a consequence of their Lewis basicity. In the previous section, we showed that the halogen-bond donor–acceptor mechanism in the MemYH···ICN complexes is the donation of charge from the filled σY–H orbital of MemYH into the empty σ*I–C orbital of ICN. For example, in the series of Me3CH···ICN, Me3SiH···ICN, and Me3GeH···ICN, the population in the filled σY–H orbital is reduced to 1.99 electrons, 1.94 electrons, and 1.94 electrons respectively, while there is charge accumulation in the ICN fragment, i.e., negative ΔQICN (Table 1). Consequently, the Y–H bond becomes longer and, thus, redshifts, as the σY–H bonding orbital loses electrons due to the halogen bond. Therefore, the redshift in the Y–H bond stretching frequency should also be expected in situations when hydridic Y–Hδ− fragments behave as a Lewis base and hence is not an exclusive feature of hydrogen-bond donors.
A similar phenomenon is found when the hydridic hydrogen of Me3SiH engages in a stabilizing intermolecular interaction with the different electron-poor molecules studied by Civiš, Hobza, and co-workers,6 that is, ICF3, BrCN, S(CN)2, P(CN)3, and K+. In all these complexes, the Si–H bond elongates and redshifts upon complex formation. However, this is not because Me3SiH behaves a hydrogen-bond donor, but as a bond acceptor. We find that there is charge transfer from Me3SiH into the electron-poor fragments and the population in the filled σ Si–H bonding orbital is reduced, which, in fact, characterizes different intermolecular interactions, e.g., halogen bonds, chalcogen bonds, and pnictogen-bonds11 (see Table S6; see addition analyses on palladium hydride, boron hydride, beryllium hydride, and lithium hydride in Tables S7 and S8). A bonding motif in which a hydridic hydrogen acts as the acceptor of a hydrogen bond exists in the particular form of dihydrogen bonds (DHB); this has been described elsewhere.7 Thus, we have presented clear evidence that the interaction between the hydridic hydrogen of Me3YH and an electron-poor molecule should not be seen as any kind of hydrogen bonding but instead as a different intermolecular interaction named after the nature of the electron-poor bond donor.
Conclusions
We have quantum chemically analyzed the bonding mechanism of a series of MemYH···NH3 and MemYH···ICN complexes, in which Y = C, Si, Ge, N, P, As, O, S, Se and m = 3, 2, 1, using quantitative Kohn–Sham molecular orbital (KS-MO) theory. We showed that MemY–Hδ+ fragments involving protonic hydrogens (i.e., Y = C, N, O, S, Se) form regular Y–Hδ+···Nδ− hydrogen bonds, whose stability is due to the Hδ+···Nδ− electrostatic attraction and the charge transfer from the hydrogen-bond acceptor NH3 into the hydrogen-bond donor MemY–Hδ+. The MemY–Hδ− fragments involving hydridic hydrogens (i.e., Y = Si, Ge, P, As) are unable to act as hydrogen-bond donors and, instead, behave as Lewis bases, donating charge into electron-poor fragments, such as ICN, forming a Hδ−···Iδ+–C halogen bond. Therefore, our findings do not support the proposal to change the IUPAC definition of hydrogen bonds to include molecules with partially negatively charged H as hydrogen-bond donors.6
Regular hydrogen bonds, as in MemY–Hδ+···NH3, involve charge transfer into the empty σ* Y–H antibonding orbital of the hydrogen-bond donor fragment. This hydrogen-bond donor–acceptor interaction is negligible in the MemY–Hδ−···ICN complexes, and the charge transfer goes in the opposite direction from the occupied σ Y–H bonding orbital into the ICN fragment. This is because ICN is a poor electron donor on the I side, which leads to a weaker hydrogen-bond donor–acceptor interaction in MemY–Hδ−···ICN. In turn, the covalent component in MemY–Hδ−···ICN is dominated by the halogen-bond donor–acceptor interaction between the occupied σ Y–H bonding orbital and the empty σ* I–C antibonding orbital of ICN. Therefore, the MemY–Hδ− fragments are halogen-bond acceptors, not hydrogen-bond donors.
The accumulation of charge in the empty σ* Y–H antibonding orbital causes the Y–H bond elongation and, thus, the typical redshift in the of the Y–H stretching frequency for regular MemY–Hδ+···NH3 hydrogen bonds. However, the elongation the Y–H bond can also be caused by other factors, e.g., the depletion of charge in the occupied σ Y–H bonding orbital in halogen-bonded MemY–Hδ−···ICN complexes. Therefore, the redshift in the Y–H stretching frequency should not be used as the unique but as one of the diagnostics to characterize hydrogen bonds.
Theoretical Methods
Computational Details
All calculations were carried out using the Amsterdam Density Functional (ADF) 2023.101 program.12 All stationary points and energies were obtained using relativistic, dispersion-corrected density functional theory at ZORA-BLYP-D3(BJ)/TZ2P (see Tables S9, S10, and S11 in the Supporting Information for the Cartesian coordinates). This approach comprises the BLYP level of the generalized gradient approximation (GGA); the exchange functional developed by Becke (B), and the GGA correlation functional developed by Lee, Yang, and Parr (LYP).13 The empirical DFT-D3(BJ) correction developed by Grimme and co-workers,14 which contains the damping function proposed by Becke and Johnson,15 is used to account for nonlocal dispersion interactions. Scalar relativistic effects are accounted for using the zeroth-order regular approximation (ZORA).16 This level has been proven to accurately describe weak interactions.17 Molecular orbitals (MO) were expanded into a large, uncontracted set of Slater-type orbitals (STOs) containing diffuse functions: TZ2P.18 The basis set is of triple-ζ quality augmented with polarization functions, i.e., one 2p and one 3d set on H; one 3d and one 4f set on C, N, O, Si, P, S; one 4d and one 4f set on Ge, As, Se; one 5d and one 4f set on I. All electrons were included in the variational process; i.e., no frozen core approximation was applied. The accuracies of both the Zlm fitting scheme19a and the Becke integration grid19b were set to ‘EXCELLENT’.
Bond Analyses
Insight into the bonding mechanism is obtained by analyzing the intermolecular interaction between MemYH (Y = C, Si, Ge, N, P, As, O, S, Se) and the NH3 or ICN fragments using the activation strain model,9 which is a fragment-based approach to understanding the energy profile associated with a chemical process in terms of the original reactants. Thus, the total bond energy ΔE is decomposed into the strain energy ΔEstrain, which is associated with the geometrical deformation of the individual reactants as the process takes place, plus the actual interaction energy ΔEint between the deformed reactants (eq 4).
| 4 |
The interaction energy ΔEint between the deformed reactants is further analyzed in the conceptual framework provided by the quantitative Kohn–Sham MO model.9 To this end, it is decomposed into physically meaningful terms, using a quantitative energy decomposition analysis (EDA) as implemented in ADF (eq 5).9 The analyses are done as a function of the H···N (rH···N) or H···I (rH···I) bond distances along the range of 1.5 to 3.5 Å. Since ΔEstrain is negligible (see Tables S1 and S3), we performed the analyses starting from the equilibrium geometry of the complex while keeping all other geometrical parameters frozen. Values in the equilibrium geometries are shown in Tables S2 and S4.
| 5 |
ΔVelstat is the classical Coulomb interaction between the unperturbed charge distributions of the deformed reactants which is usually stabilizing and comprises four components: (i) the electron–electron electrostatic repulsion between the electron densities of fragments 1 and 2, ΔVelstat,ρ1ρ2; (ii) the nuclei–electron electrostatic attraction between the nuclei of fragment 1 and the electron density of fragment 2, ΔVelstat,n1ρ2; (iii) the electron–nuclei electrostatic attraction between the electron density of fragment 1 and the nuclei of fragment 2, ΔVelstat,ρ1n2; and (iv) the nuclei–nuclei electrostatic repulsion between the nuclei of fragments 1 and 2, ΔVelstat,n1n2.
The Pauli repulsion energy (ΔEPauli) comprises the destabilizing interactions between occupied orbitals on either fragment (more precisely, between same-spin occupied spinorbitals on either fragment) and arises from the antisymmetrization of the Hartree wave function due to the Pauli principle. The orbital-interaction energy (ΔEoi) accounts for charge transfer, that is, the interaction between occupied orbitals of one fragment with unoccupied orbitals of the other fragment, including the interactions of the highest occupied and lowest unoccupied MOs (HOMO–LUMO), and polarization, that is, empty–occupied orbital mixing on one fragment, due to the presence of another fragment. ΔEdisp accounts for the empirical dispersion corrections as introduced by Grimme et al.14 To facilitate the analyses, the ASM and EDA were performed using the PyFrag 2019 program.20
Voronoi Deformation Density (VDD) Charge Analysis
The Voronoi Deformation Density (VDD) charge analysis allows for the quantification of the flow of electronic charge as a consequence of chemical-bond formation.21 In our model MemYH fragments, the VDD atomic charge of atom H directly bound to Y (QH) is computed by the spatial integration of the deformation density over the Voronoi cell of atom H, which is the space defined by the bond midplanes on and perpendicular to all bond axes between atom H and its neighboring atoms (eq 1).
| 1 |
Herein, the deformation density Δρ(r) = [ρ(r) – ∑iρi(r)] is the density change going from a superposition of the original atomic densities at the positions of the molecule to the actual density of that molecule. This atomic or so-called promolecular density is defined as the sum of the (spherically averaged) ground-state atomic densities ∑iρi(r). This is the fictitious state in which the charge density has not been affected by chemical bonding and in which all atoms have zero charge. Then, QH represents the amount of charge that, due to chemical bonding, flows from a position closer to another nucleus to a position closer to the nucleus of H (QH < 0), or from a position closer to the nucleus of H to a position closer to another nucleus (QH > 0).
Besides the above regular VDD atomic charges relative to noninteracting neutral atoms, the VDD method allows for the analysis of changes in atomic charges (ΔQ), i.e., charge-density rearrangements, caused by interactions between molecular fragments (eq 2).
| 2 |
Equation 2 defines the charge rearrangements in an atom A of a fragment i (ΔQA) in the final density of the overall complex ρcomplex(r) relative to the sum of the initial molecular fragment densities ∑fragment,iρi(r)i. This reveals how the interactions between the molecular fragments affect the electron density distribution in atom A of fragment i. The sum of the ΔQA of all atoms in a fragment i gives the amount of charge transfer into (ΔQfragment,i < 0) or out of (ΔQfragment,i > 0) the Voronoi cell of a fragment i due to the interaction between the molecular fragments (eq 3).
| 3 |
Acknowledgments
We thank The Netherlands Organization for Scientific Research (NWO). This work was carried out on the Dutch national e-infrastructure with the support of SURF Cooperative.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c07821.
Activation strain, energy decomposition, and VDD analyses; and Cartesian coordinates of all stationary points. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Berg J. M.; Tymoczko J. L.; Stryer L.. Biochemistry, 5th Edition; W. H. Freeman: New York, 2002; p 8. [Google Scholar]; b Zhang S.; Liu C.; Song Y.; Ruan J.; Quan P.; Fang L. High drug-loading and controlled-release hydroxyphenyl-polyacrylate adhesive for transdermal patch. J. Controlled Release 2023, 353, 475–489. 10.1016/j.jconrel.2022.11.058. [DOI] [PubMed] [Google Scholar]; c Desiraju G. R. Supramolecular Synthons in Crystal Engineering-A New Organic Synthesis. Angew. Chem., Int. Ed. 1995, 34, 2311–2327. 10.1002/anie.199523111. [DOI] [Google Scholar]; d Young M. C.; Holloway L. R.; Johnson A. M.; Hooley R. J. A Supramolecular Sorting Hat: Stereocontrol in Metal–Ligand Self-Assembly by Complementary Hydrogen Bonding. Angew. Chem., Int. Ed. 2014, 53, 9832–9836. 10.1002/anie.201405242. [DOI] [PubMed] [Google Scholar]; e Pupo G.; Gouverneur V. Hydrogen Bonding Phase-Transfer Catalysis with Alkali Metal Fluorides and Beyond. J. Am. Chem. Soc. 2022, 144, 5200–5213. 10.1021/jacs.2c00190. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Mayfield A. B.; Metternich J. B.; Trotta A. H.; Jacobsen E. N. Stereospecific Furanosylations Catalyzed by Bis-thiourea Hydrogen-Bond Donors. J. Am. Chem. Soc. 2020, 142, 4061–4069. 10.1021/jacs.0c00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunan E.; Desiraju G. R.; Klein R. A.; Sadlej J.; Scheiner S.; Alkorta I.; Clary D. C.; Crabtree R. H.; Dannenberg J. J.; Hobza P.; Kjaergaard H. G.; Legon A. C.; Mennucci B.; Nesbitt D. J. Defining the Hydrogen Bond: An Account (IUPAC Technical Report). Pure Appl. Chem. 2011, 83, 1619–1636. 10.1351/PAC-REP-10-01-01. [DOI] [Google Scholar]
- a van der Lubbe S. C. C.; Fonseca Guerra C. The Nature of Hydrogen Bonds: A Delineation of the Role of Different Energy Components on Hydrogen Bond Strengths and Lengths. Chem. Asian J. 2019, 14, 2760–2769. 10.1002/asia.201900717. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wolters L. P.; Bickelhaupt F. M. Halogen Bonding versus Hydrogen Bonding: A Molecular Orbital Perspective. ChemistryOpen 2012, 1, 96–105. 10.1002/open.201100015. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Fonseca Guerra C.; Bickelhaupt F. M.; Snijders J. G.; Baerends E. J. The Nature of the Hydrogen Bond in DNA Base Pairs: The Role of Charge Transfer and Resonance Assistance. Chem. Eur. J. 1999, 5, 3581–3594. . [DOI] [Google Scholar]; d Shaik S.; Danovich D.; Zare R. N. Valence Bond Theory Allows a Generalized Description of Hydrogen Bonding. J. Am. Chem. Soc. 2023, 145, 20132–20140. 10.1021/jacs.3c08196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Allerhand A.; von Rague Schleyer P. A Survey of C-H Groups as Proton Donors in Hydrogen Bonding. J. Am. Chem. Soc. 1963, 85, 1715–1723. 10.1021/ja00895a002. [DOI] [Google Scholar]; b Wang C.; Danovich D.; Shaik S.; Mo Y. A Unified Theory for the Blue- and Red-Shifting Phenomena in Hydrogen and Halogen Bonds. J. Chem. Theory Comput. 2017, 13, 1626–1637. 10.1021/acs.jctc.6b01133. [DOI] [PubMed] [Google Scholar]
- Jabłoński M. Ten years of charge-inverted hydrogen bonds. Struct. Chem. 2020, 31, 61–80. 10.1007/s11224-019-01454-2. [DOI] [Google Scholar]
- Civiš S.; Lamanec M.; Špirko V.; Kubišta J.; Špet’ko M.; Hobza P. Hydrogen Bonding with Hydridic Hydrogen–Experimental Low-Temperature IR and Computational Study: Is a Revised Definition of Hydrogen Bonding Appropriate?. J. Am. Chem. Soc. 2023, 145, 8550–8559. 10.1021/jacs.3c00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The structures of the type MemYH···HNH2 reported in Figure 2 can be seen as containing weak dihydrogen bonds. See, for example:; Hugas D.; Simon S.; Duran M.; Fonseca Guerra C.; Bickelhaupt F. M. Dihydrogen Bonding: Donor–Acceptor Bonding (AH···HX) versus the H2 Molecule (A–H2–X). Chem. Eur. J. 2009, 15, 5814–5822. 10.1002/chem.200802641. [DOI] [PubMed] [Google Scholar]
- CRC Handbook of Chemistry and Physics, 84th ed.; Lide D. R., Ed.; CRC Press: Boca Raton, FL, 2003. [Google Scholar]
- a Vermeeren P.; van der Lubbe S. C. C.; Fonseca Guerra C.; Bickelhaupt F. M.; Hamlin T. A. Nat. Protoc. 2020, 15, 649–667. 10.1038/s41596-019-0265-0. [DOI] [PubMed] [Google Scholar]; b Vermeeren P.; Hamlin T. A.; Bickelhaupt F. M. Chemical Reactivity from an Activation Strain Perspective. Chem. Commun. 2021, 57, 5880–5896. 10.1039/D1CC02042K. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bickelhaupt F. M.; Baerends E. J. In Reviews in Computational Chemistry; Lipkowitz K. B., Boyd D. B., Eds.; Wiley: Hoboken, NJ, 2000; pp 1–86. [Google Scholar]; d Hamlin T. A.; Vermeeren P.; Fonseca Guerra C.; Bickelhaupt F. M. In Complementary Bonding Analysis, Grabowski S., Ed.; De Gruyter: Berlin, 2021; pp 199–212. [Google Scholar]
- a Desiraju G. R.; Ho P. S.; Kloo L.; Legon A. C.; Marquardt R.; Metrangolo P.; Politzer P.; Resnati G.; Rissanen K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. 10.1351/PAC-REC-12-05-10. [DOI] [Google Scholar]; b Terraneo G.; Resnati G.; Metrangolo P. In Iodine Chemistry and Applications; Kaiho T., Ed.; Wiley Online Books: 2014; pp 159–194. [Google Scholar]; c Horibe T.; Tsuji Y.; Ishihara K. Thiourea–I2 as Lewis Base–Lewis Acid Cooperative Catalysts for Iodochlorination of Alkene with In Situ-Generated I–Cl. ACS Catal. 2018, 8, 6362–6366. 10.1021/acscatal.8b01565. [DOI] [Google Scholar]; d Vermeeren P.; Hamlin T. A.; Fernández I.; Bickelhaupt F. M. How Lewis Acids Catalyze Diels–Alder Reactions. Angew. Chem., Int. Ed. 2020, 59, 6201–6206. 10.1002/anie.201914582. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Vermeeren P.; Wolters L. P.; Paragi G.; Fonseca Guerra C. Cooperative Self-Assembly in Linear Chains Based on Halogen Bonds. ChemPlusChem 2021, 86, 812–819. 10.1002/cplu.202100093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a de Azevedo Santos L.; Ramalho T. C.; Hamlin T. A.; Bickelhaupt F. M. Intermolecular Covalent Interactions: Nature and Directionality. Chem. Eur. J. 2023, 29, e202203791 10.1002/chem.202203791. [DOI] [PubMed] [Google Scholar]; b Wang C.; Danovich D.; Mo Y.; Shaik S. On The Nature of the Halogen Bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. 10.1021/ct500422t. [DOI] [PubMed] [Google Scholar]
- a ADF2023.1, SCM Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com. [Google Scholar]; b te Velde G.; Bickelhaupt F. M.; Baerends E. J.; Fonseca Guerra C.; van Gisbergen S. J. A.; Snijders J. G.; Ziegler T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. 10.1002/jcc.1056. [DOI] [Google Scholar]
- a Becke A. D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- a Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]; b Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Johnson E. R.; Becke A. D. Exchange-hole dipole moment and the dispersion interaction: High-order dispersion coefficients. J. Chem. Phys. 2006, 124, 024101 10.1063/1.2139668. [DOI] [PubMed] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. 10.1063/1.467943. [DOI] [Google Scholar]
- a Vermeeren P.; Wolters L. P.; Paragi G.; Fonseca Guerra C. Cooperative Self-Assembly in Linear Chains Based on Halogen Bonds. ChemPlusChem 2021, 86, 812–819. 10.1002/cplu.202100093. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- van Lenthe E.; Baerends E. J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. 10.1002/jcc.10255. [DOI] [PubMed] [Google Scholar]
- a Franchini M.; Philipsen P. H. T.; van Lenthe E.; Visscher L. Accurate Coulomb Potentials for Periodic and Molecular Systems Through Density Fitting. J. Chem. Theory Comput. 2014, 10, 1994–2004. 10.1021/ct500172n. [DOI] [PubMed] [Google Scholar]; b Franchini M.; Philipsen P. H. T.; Visscher L. The Becke Fuzzy Cells Integration Scheme in the Amsterdam Density Functional Program Suite. J. Comput. Chem. 2013, 34, 1819–1827. 10.1002/jcc.23323. [DOI] [PubMed] [Google Scholar]
- a Sun X.; Soini T. M.; Wolters L. P.; van Zeist W.-J.; Fonseca Guerra C.; Hamlin T. A.; Bickelhaupt F. M.. PyFrag 2019, Vrije Universiteit Amsterdam, The Netherlands. [Google Scholar]; b Sun X.; Soini T. M.; Poater J.; Hamlin T. A.; Bickelhaupt F. M. Pyfrag 2019 – Automating the exploration and analysis of reaction mechanisms. J. Comput. Chem. 2019, 40, 2227–2233. 10.1002/jcc.25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwland C.; Vermeeren P.; Bickelhaupt F. M.; Fonseca Guerra C. Understanding Chemistry with the Symmetry-Decomposed Voronoi Deformation Density (VDD) Charge Analysis. J. Comput. Chem. 2023, 44, 2108–2119. 10.1002/jcc.27184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.