

Abstract
Immunotherapy for the treatment of metastatic melanoma remains a major clinical challenge. The melanoma microenvironment may lead to local T cell tolerance in part through downregulation of costimulatory molecules, such as B7.1 (CD80). We report the results from the first clinical trial, to our knowledge, using a recombinant vaccinia virus expressing B7.1 (rV-B7.1) for monthly intralesional vaccination of accessible melanoma lesions. A standard 2-dose–escalation phase I clinical trial was conducted with 12 patients. The approach was well tolerated with only low-grade fever, myalgias, and fatigue reported and 2 patients experiencing vitiligo. An objective partial response was observed in 1 patient and disease stabilization in 2 patients, 1 of whom is alive without disease 59 months following vaccination. All patients demonstrated an increase in postvaccination antibody and T cell responses against vaccinia virus. Systemic immunity was tested in HLA-A*0201 patients who demonstrated an increased frequency of gp100 and T cells specific to melanoma antigen recognized by T cells 1 (MART-1), also known as Melan-A, by ELISPOT assay following local rV-B7.1 vaccination. Local immunity was evaluated by quantitative real-time RT-PCR, which suggested that tumor regression was associated with increased expression of CD8 and IFN-γ. The local delivery of vaccinia virus expressing B7.1 was well tolerated and represents an innovative strategy for altering the local tumor microenvironment in patients with melanoma.
Introduction
The identification of tumor-associated antigens (TAAs) has led to a variety of vaccine strategies for the treatment of cancer. Despite reports that vaccines can generate systemic antigen-specific T cell responses, there have been few objective clinical responses (1). In order for T cells to effectively eradicate cancer, they must migrate into established tumors in large numbers and maintain effective cytotoxic functions. There is increasing evidence that the tumor microenvironment is a critical factor in the balance between tumor growth and immune control (2, 3). A variety of mechanisms appears to regulate this balance, including defects in tumor-antigen expression and presentation, local release of inhibitory cytokines, downregulation of DC and T cell effector function, and systemic or local increases in regulatory T cell populations (4–9). Recently, murine studies demonstrated that adoptively transferred gp100-specific pmel-1 T cells trafficked equally well to gp100-expressing melanoma tumors and antigen-negative tumors but mediated regression following vaccination and IL-2 treatment only in gp100-expressing lesions (10). This suggests that local activation of tumor-reactive T cells is a critical event in eliciting tumor regression. One mechanism that may prevent activation of antigen-specific T cells within the tumor microenvironment and result in local tolerance is the lack of T cell costimulation due to decreased expression of costimulatory molecules by tumor cells (11, 12). Thus, effective vaccination against established tumors likely requires both induction of systemic tumor-reactive T cells that can traffic to sites of tumor growth and the local activation of these T cells.
Strategies for manipulating the tumor microenvironment have been proposed, and recent reports highlight the importance of providing appropriate local danger signals (13). The introduction of proinflammatory chemokines, such as DC-LIGHT, into poorly immunogenic murine B16 melanoma cells resulted in significant T cell–mediated rejection of the established tumors (14). Direct injections of TLR agonists have also been used to stimulate tumor immunity in mice through activation of local DC populations (15). Molecular profiling of melanoma lesions following systemic IL-2 administration suggested a correlation between clinical responses and local inflammatory chemokine expression (16). The ability of IL-2 to reverse tolerance of immunized T cells in vitro also suggests that local tolerance may be reversible in vivo with appropriate stimulation (17, 18). Collectively, these studies provide the rationale for directly expressing strong immunomodulatory signals with tumor vaccines to promote local tumor immunity.
Costimulatory signals promote T cell proliferation and cytokine release and in general lower the threshold for T cell activation. This is especially important when T cells recognize weak antigens, such as TAAs (19). B7.1 (CD80) is among the best-characterized costimulatory molecules and is expressed on professional antigen-presenting cells where it acts as a ligand for CD28 and CTL-associated antigen 4 (CTLA-4) (20). Whereas CTLA-4 is induced following T cell activation, CD28 is constitutively expressed on naive T cells. Once a T cell receptor recognizes its cognate antigen, CD28-B7.1 ligation provides a potent activation signal characterized by an increased production of IL-2 and related cytokines, enhanced expression of CD25, and inhibition of activation-induced cell death in the T cell. The potential role of B7.1-mediated signaling in tumor immunotherapy has been demonstrated in murine studies in which B7.1-transduced tumor cells were rejected more readily than nontransduced cells (21). Preliminary in vitro studies demonstrated that a recombinant vaccinia virus engineered to express B7.1 (rV-B7.1) significantly enhanced T cell proliferation and activation, while in vivo studies demonstrated rejection of experimental murine tumors injected with the recombinant virus (22–24).
Recombinant vaccinia vectors have been used to express defined TAAs in cancer patients (25). To date, these vectors have largely been confined to systemic administration with an acceptable safety profile and evidence of T cell induction against expressed antigens in selected patients (26, 27). Vaccinia virus triggers the release of chemokines and proinflammatory cytokines, resulting in the recruitment and activation of macrophages, DCs, and T cells at the site of infection (28). Local replication of vaccinia virus also promotes continued influx of T cells. Once recruited, vaccinia virus may also induce local DC maturation, resulting in activation of both CD8+ and CD4+ T cells (29).
Malignant melanoma is increasing in incidence faster than almost any other cancer in the United States, with over 53,000 new cases diagnosed annually (30). The median survival for patients with stage IV disease is typically fewer than 12 months, and treatment options are limited. Melanoma lesions are typically accessible for direct injection and often contain T cell infiltrates capable of mediating at least partial regression of established lesions. However, there is evidence that infiltrating T cells may lose effector function after exposure to the tumor microenvironment (12). Thus, we evaluated the safety and immune effects of local delivery of vaccinia virus expressing B7.1 in a dose-escalation phase I clinical trial of patients with metastatic melanoma. The generation of local immunity was monitored by objective measurement and collection of fine-needle aspirates (FNAs) for quantitative real-time PCR (qRT-PCR) analysis. In addition, systemic immunity was documented by determining the frequency of melanoma-specific T cell responses in peripheral blood samples. Our results suggest that local vaccination with vaccinia virus expressing B7.1 is well tolerated and results in objective tumor responses in selected patients. Furthermore, vaccinated patients appeared to develop systemic antitumor immunity as evidenced by an increase in gp100- and MART-1–specific T cells following local immunization and development of autoimmune vitiligo. Thus, local delivery of B7.1 by vaccinia virus is a feasible strategy for initiating local and systemic immunity against established tumors.
Results
Local delivery of vaccinia virus expressing B7.1 is well tolerated and feasible in patients with metastatic melanoma.
Twelve patients with metastatic melanoma who each had at least 1 accessible cutaneous, subcutaneous, or lymph node lesion that measured 1 cm or greater in diameter were enrolled in a phase I clinical trial. All subjects provided written, informed consent. The first 6 patients received monthly intralesional injections of rV-B7.1 at a dose of 4.26 × 107 PFU. The next 6 patients received a dose of 4.26 × 108 PFU using a similar treatment schedule. All patients completed pretreatment radiologic imaging, which was repeated after the third vaccination. Monthly booster immunizations were received by patients in cases where there was no evidence of disease progression upon restaging. The site of the injected (index) lesion used for vaccination and location of known metastatic disease for each patient is listed in Table 1.
Table 1.
Patient characteristics

The median age of the patients was 54 years (range, 34–74) and all had an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1 (Table 1). All patients had received and failed prior therapy, which consisted of IFN-α (n = 7), chemotherapy (n = 5), and/or IL-2 (n = 4). Toxicity was assessed using the National Cancer Institute Common Toxicity Criteria. The 3 most frequently encountered adverse events were grade 1 fever (n = 9), grade 1 fatigue (n = 7), and grade 1 myalgias (n = 5). There were 2 grade 1 skin rashes, 1 at each dose of vaccine (Table 2). Of the 12 subjects, there was 1 incident of new onset grade 2 vitiligo in a patient enrolled at the lower vaccine dose and 1 grade 1 vitiligo exacerbation in a patient who had vitiligo prior to study enrollment. Overall, there was no correlation between adverse events and the dose of vaccine administered. Thus, patients were able to tolerate both doses of rV-B7.1 vaccine without significant adverse events.
Table 2.
Incidence of adverse events listed by vaccine dose

Local delivery of vaccinia virus expressing B7.1 induced objective tumor regression in selected patients.
One patient withdrew from the study before the second dose and could not be fully evaluated. Responses were classified according to standard response evaluation criteria in solid tumors (Table 3), and 2 of the 11 (18.2%) evaluable patients, both at the 4.26 × 107 PFU vaccine dose, had an objective partial response of the index lesion (31). Another 4 patients (36.3%) had stable lesions 3 months after starting vaccination. Two patients had stable disease at distant sites of documented metastases, and 1 patient had a complete response of metastatic tumors. Patient 3 presented with mediastinal and left inguinal melanoma 4 years after excision of a left foot primary. After the third vaccination, she experienced complete regression of all mediastinal lesions, while the left inguinal mass remained stable by objective measurements. An FNA of this inguinal lesion revealed necrotic tumor 4 weeks after completion of the third vaccine, and the mass was surgically excised. Interestingly, following the third vaccination, the patient also exhibited profound vitiligo around her lower right lip and at a skin graft donor site in the right lateral thigh (Figure 1). Now, more than 59 months after completing treatment, she shows no evidence of recurrent melanoma and has had no other therapy. Patient 4 was a 47-year-old man with melanoma metastatic to the right parotid gland, spleen, lymph nodes, lung, and liver. After completion of the first cycle of rV-B7.1 vaccine, follow-up CT scans showed his nontarget disease to be stable while he had a greater than 50% regression of the index parotid mass. He elected to continue treatment with rV-B7.1, and his disease remained stable through 2 additional cycles of vaccination (9 total vaccinations). Fourteen months after entering the trial, the patient died of splenic rupture related to progressive melanoma. Patient 11 was a 42-year-old woman with a primary anal melanoma and metastases to her lungs, lymph nodes, and left adrenal gland. She completed 1 cycle of rV-B7.1, and postvaccination CT scans demonstrated stable disease for 22 months, after which time her disease progressed. The remaining patients all experienced disease progression after the first cycle of vaccine therapy.
Table 3.
Clinical responses of patients treated with intralesional rV-B7.1 vaccine

Figure 1.
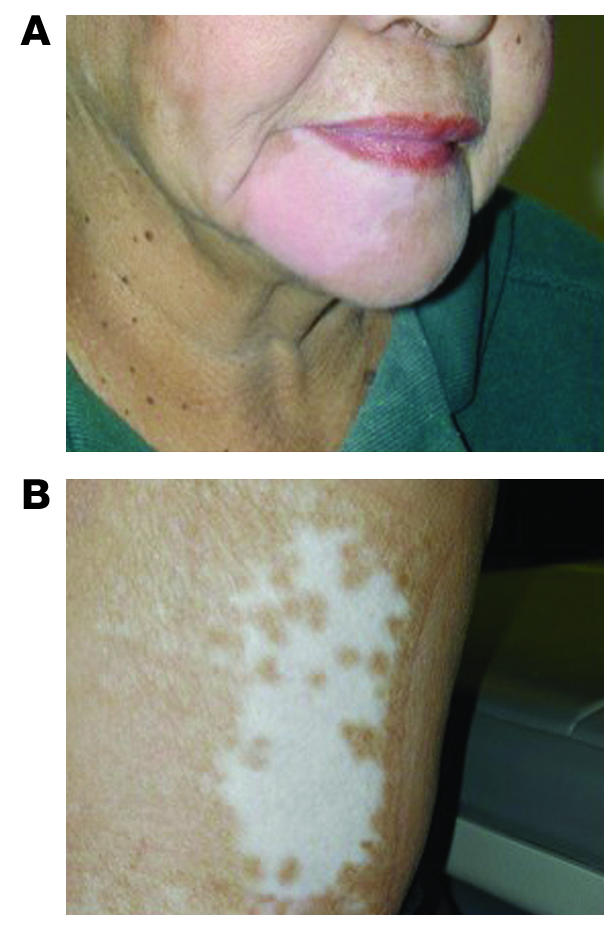
Patient 3 received 3 intratumoral injections of rV-B7.1 (4.26 × 107 PFU) in a left inguinal subcutaneous melanoma lesion. Following the last vaccine she developed new autoimmune vitiligo involving the right lower chin (A) and a skin graft donor site on the right lateral thigh (B). She had a complete response of metastatic disease, and excision of the left inguinal melanoma revealed necrotic tumor. She has had no evidence of recurrent disease for more than 59 months.
Melanoma patients injected with rV-B7.1 develop antivaccinia virus–antibody responses.
Pre- and postvaccination antivaccinia virus antibodies were evaluated by ELISA. Patients were required to have had smallpox vaccination in the past for eligibility in the clinical trial, and prevaccination titers (greater than 400) were positive in 4 out of 12 (33%) patients. Following intratumoral administration of rV-B7.1, all patients exhibited an increase in postvaccination antivaccinia titers (Table 4). In the low-dose (4.26 × 107 PFU) group, patients showed an average fold titer increase of 41.3 ± 26.0 (range 8–64); and in the high-dose (4.26 × 108 PFU) group, patients experienced an average fold titer increase of 56 ± 40.2 (range 16–128).
Table 4.
Antivaccinia antibody titers

Melanoma patients injected with rV-B7.1 develop antivaccinia virus T cell responses.
All patients tested showed an average fold increase of 29.6 ± 50.7 (range 1–144.4) in antivaccinia T cell precursor frequencies against vaccinia lysate in an IFN-γ ELISPOT assay (Table 5). The fold increase in antivaccinia T cell precursor frequency was compared to the fold increase in antivaccinia antibody titers and showed a correlation of –0.45 with 95% confidence interval: –0.93 to –0.28 (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI24624DS1).
Table 5.
ELISPOT results for HLA-A2 positive vaccine patients

Melanoma patients injected with intralesional rV-B7.1 developed systemic T cell responses against defined melanoma antigens.
The expression of 2 defined melanoma antigens, gp100 and melanoma antigen recognized by T cells 1 (MART-1), also known as Melan-A, was confirmed by immunohistochemical staining of FNAs taken from index lesions prior to vaccination (Supplemental Figure 2). We observed some minor variability in antigen expression in individual lesions throughout the trial, but, as previously reported in other studies, gp100 and MART-1 were expressed in all HLA-A*0201 patients (n = 6) who were selected for T cell analysis (32, 33). Pre- and postimmunization PBMCs were subjected to in vitro stimulation (IVS) using a single HLA-A2–restricted MART-1 peptide and a mixture of 3 HLA-A2–restricted gp100 peptides. The tumor antigen–specific immune response was monitored in PBMCs using IFN-γ ELISPOT assay (Table 5). The postvaccination frequency of gp100-specific T cells increased in 5 of 6 patients (fold increases ranged 1.1–35.9), while MART-1–specific T cells increased in 4 of 6 patients (fold increases ranged 1.21–23.5). The patient who experienced long-term survival following vaccination (patient 3) initially had little change in antigen-specific T cell responses but developed a significant increase in both gp100- and MART-1–specific T cells 6 months after starting vaccination (Figure 2).
Figure 2.

Local delivery of rV-B7.1–induced gp100- and MART-1–specific T cell response in peripheral blood lymphocytes 6 months after vaccination. T cell response was measured by IFN-γ ELISPOT assay after 7 days of IVS with an HLA-A*0201–restricted MART-1 peptide or a mixture of 3 gp100 peptides. In patient 3, a dramatic increase in both gp100 and MART-1 T cell responses was seen by IFN-γ ELISPOT assay 3 months after completion of the vaccine trial (month 6), and she is alive without disease recurrence more than 59 months later without other therapy. Pre-Tx, pretreatment.
Differential gene expression of CD8, IFN-γ, and IL-10 in regressing and stable lesions compared with that in nonregressing lesions following intralesional rV-B7.1 vaccination.
Total RNA was successfully extracted from 21 FNAs from 6 patients. Samples were tested for RNA integrity by RNA nanochip electropherogram technology and then selected for 2 consecutive rounds of amplification, as described in Methods. We were able to amplify RNA (aRNA) approximately 102- to 103-fold from samples in 5 patients, which were available for qRT-PCR analysis. The pre- versus postvaccination cycle threshold (CT) values of gene expression for the endogenous control phosphoglycerate kinase 1 (PGK1) were compared to rule out differences in aRNA assessment during processing prior to qRT-PCR analysis. There were no differences found among all 5 prevaccination (20.08 ± 0.98) and 16 postvaccination (20.1 ± 1.2) PGK1 CT values from patients 3, 4, 5, 7, and 9. Among the prevaccination samples, we measured the difference in CT values for IFN-γ (7.6 ± 4.54), IL-10 (11.1 ± 3.57), and CD8 (4.6 ± 2.5) without finding notable differences among patients (Supplemental Table 1).
The gene expression profile of the index lesion in stable disease (SD) was not markedly altered, exhibiting less than a 10-fold difference (greater than 0.1) in CD8, IFN-γ, or IL-10 (Figure 3A). The 2 lesions exhibiting a partial response (PR) demonstrated a general increase in CD8 (patient 4) and IFN-γ and IL-10 (patients 4 and 5). Patient 4 experienced a less than 800-fold increase in IFN-γ transcription levels, correlating with a less than 200-fold increase in CD8 transcription levels in postvaccination samples compared with preimmunization specimens (Figure 3A). In the 2 regressing lesions, the IFN-γ levels were higher than the IL-10 expression. In contrast, the 2 nonresponding index lesions in patients with progressive disease (PD) were characterized by a decrease in all 3 markers, most notably IFN-γ, which decreased more than 100-fold (0.02 ± 0.02), and to a lesser degree, CD8, with a decrease of 7-fold (0.3 ± 0.04).
Figure 3.

The gene expression levels of CD8, IFN-γ, and IL-10 in the tumor microenvironment were altered by local rV-B7.1 vaccination. RNA was amplified from FNAs obtained before and after vaccination. (A) Logarithmic changes of target genes corrected by PKG-1 endogenous control values are shown for 5 index lesions. Postvaccination gene expression of CD8, IFN-γ, and IL-10 in index lesions from patients 3, 4, 5, 7 and 9 classified as SD, PR, or PD are shown. ND, not determined. (B) Postvaccination gene expression levels of CD8, IFN-γ, and IL-10 are shown for patient 4, classified as PR, throughout the administration of 3 vaccine cycles (9 injections of rV-B7.1). The specific injection number within each cycle is indicated. Post, postvaccination; pre, prevaccination. (C) Measurements of the index lesion area (cm2) of patient 4 corresponding to the gene transcript levels in B are shown.
Patient 4 underwent 3 cycles of vaccination for a total of 9 separate injections and experienced a partial response in the index lesion with stable disease elsewhere for 14 months. During the first vaccination cycle, there was an increase in CD8 and IFN-γ transcription levels, whereas following the second cycle, IL-10 gene expression appeared to be increased above the IFN-γ and CD8 genes (Figure 3B). This trend was reversed again during the third cycle of vaccination, which was characterized by a rebound in CD8 and IFN-γ transcription levels and an accompanying decrease in IL-10 gene expression. These changes were associated with an initial regression in the size of the index lesion during the first treatment cycle, a rapid increase in tumor size associated with IL-10 gene expression, and stabilization during the third cycle of vaccination (Figure 3C).
Discussion
This is the first report, to our knowledge, evaluating the safety and feasibility of direct intratumoral injection of recombinant vaccinia virus expressing a costimulatory molecule (rV-B7.1) in patients with melanoma. The maximum dose administered was 4.26 × 108 PFU virus, which was the highest concentration of virus that could be administered with available technology. The adverse events related to vaccination were not dose dependent and consisted of low-grade fever (75%), fatigue (58%), and myalgias (42%). These reactions compare favorably with other immunotherapy strategies that employ systemic cytokines, such as IL-2, which induce a significantly greater toxicity profile. The clinical trial was not designed to determine clinical responses, yet we noted that 2 (18%) injected lesions underwent an objective partial response and another 3 lesions (27.3%) were stable during the course of the clinical trial. The disease stability in these 3 patients correlated with prolonged survival (mean, 31.6 months). The most interesting response was in 1 patient who also developed vitiligo (Figure 1) and remains alive without disease recurrence 59 months after vaccination without other therapy. Overall, these results support the feasibility of this approach and justify further investigation of this strategy for patients with metastatic melanoma (34, 35).
Vaccinia virus is a protypical poxvirus composed of a double-stranded DNA core and is capable of encoding large eukaryotic genes engineered for expression by vaccinia-specific promoters (36, 37). Infection of tumor cells by recombinant vaccinia viruses results in a lytic process 7 hours after exposure, and the virus is generally cleared from patients within 5–7 days, especially following repeated exposure. The rV-B7.1 vaccine was constructed using a synthetic early/late vaccinia promoter leading to short term cell surface B7.1 expression for several hours prior to lysis of infected melanoma cells (data not shown). In addition to B7.1 expression, the presence of vaccinia virus itself in the local microenvironment led to release of proinflammatory cytokines and chemokines, which may serve as a potent adjuvant to amplify local immunity. Despite these advantages, the use of repetitive boosting with vaccinia constructs may be inhibited by the rapid appearance of neutralizing antivaccinia antibody and subsequent clearance of virus before local antitumor immunity can be firmly established (38). This may have been even more profound in the current trial since all patients were required to have prior smallpox vaccination. In fact, some patients did exhibit low levels of preexisting antivaccinia antibody titers (Table 4), but all patients responded with a significant increase in both antivaccinia antibody titers and T cell responses after vaccination. We observed an inverse correlation between antibody and T cell responses against vaccinia, which suggests that other host factors may play a role in the type of immune response that occurs following exposure to vaccinia virus. Patients able to mount a Th1-type response are more likely to develop cell-mediated immunity, and an association between antigen-specific Th1 responses and tumor immunity following immunotherapy has been previously reported (39).
T cell costimulation is a significant factor in the induction of T cell responses and may also be critical for antitumor immunity (20). B7.1 is the ligand for at least 2 T cell surface receptors, CD28 and CTLA-4. Ligation of CD28 delivers a positive stimulatory signal to T cells that have engaged the T cell receptor, which results in T cell proliferation, release of IL-2, and inhibition of apoptosis through increased expression of Bcl-XL (20, 40, 41). In contrast, CTLA-4 is induced following T cell activation and exerts a negative regulatory role following initial T cell activation. The importance of B7.1 in generating autoreactive T cells has been demonstrated in experiments on NOD mice, in which diabetes can be accelerated by the introduction of B7.1 into murine islet cells (42). The role of B7.1 in tumor immunity has also been suggested by the observation that tumor cells escape detection by the lack of costimulatory molecule expression on the cell surface (43). Showing results similar to those of the NOD mouse experiments, murine tumors transduced with B7.1 are rejected more efficiently than B7.1-negative tumor cells (44). Furthermore, enhanced tumor rejection was observed after vaccination with recombinant viral vectors expressing a model tumor antigen and B7.1 (45, 46). In other murine studies, recombinant vaccinia viruses expressing B7.1 or B7.2 were used to directly infect transplantable colon carcinoma cells. Although infection of murine carcinoma cells with wild-type vaccinia virus resulted in progressive tumor growth, tumor cells infected with either B7.1- or B7.2-expressing viruses completely prevented tumor growth after transplantation into immunocompetent mice (22). These results suggest that the inclusion of B7.1 is important for rejection of local tumors, although we were unable to directly test this in the current phase I clinical trial.
Established melanomas often contain tumor-infiltrating lymphocyte populations, which can mediate limited regression in selected cases (47, 48). However, a recent report has documented that both Melan-A– and cytomegalovirus-specific T cells were functionally impaired in lymph nodes containing metastatic melanoma (12). These potentially reactive T cells were found to lack perforin, suggesting that the local tumor microenvironment induces local tolerance through direct effects on antigen-specific T cells. Thus, the introduction of a viral vector encoding immunomodulatory molecules provides a strategy for inducing strong local inflammatory responses that may be capable of reversing local T cell tolerance. In a murine–transitional cell carcinoma model, vaccination with vaccinia virus expressing the male-specific transplantation antigen H-Y and GM-CSF was able to induce systemic H-Y–specific CTL responses only with injection into the tumor site, not after injection in the contralateral normal flank (49). Our data is consistent with this observation, as we documented an increase in melanoma-specific T cells in PBMCs following local vaccination with rV-B7.1. In addition, local immunity was evident, as documented by an increase in CD8 and IFN-γ transcripts in rV-B7.1–injected melanoma lesions undergoing a partial clinical response (Figure 3A).
While the inclusion of B7.1 or other immunomodulatory molecules is likely important for the induction of host immunity, it is also possible that vaccinia-induced lysis of local melanoma cells initiated an immune response through cross-presentation of melanoma antigens following engulfment by professional antigen-presenting cells. The contribution of nonspecific vaccinia effects, however, seems less likely since mice immunized with a murine MC38 colon carcinoma cell vaccine infected with vaccinia virus expressing B7.1 experienced complete protection against tumor challenge with native MC38, whereas wild-type vaccinia-infected MC38 vaccination had no effect (50). A similar effect was observed in a lung metastasis treatment model in which only tumor cells infected with vaccinia-B7.1, not vaccinia virus, were effective. The influence of local costimulatory molecules delivered by poxvirus vectors was also evaluated using a nonreplicating fowlpox virus (51). In this model, intratumoral vaccination with fowlpox virus expressing carcinoembryonic antigen (CEA) and 3 costimulatory molecules (B7.1, ICAM-2, and LFA-3) induced regression of implanted MC38-CEA colon carcinomas, whereas intratumoral wild-type fowlpox virus had no effect on tumor growth. Infection of human DCs with avipoxviruses has also demonstrated increased T cell activation when costimulatory molecules are expressed by the virus compared with infection of DCs with wild-type control poxviruses (52, 53). Thus, while we could not ethically justify a direct comparison of the rV-B7.1 to wild-type vaccinia virus in this phase I clinical trial, there is compelling evidence of the superiority of recombinant poxviruses expressing costimulatory molecules in activating T cell responses and mediating therapeutic responses in murine tumor models.
Local delivery of rV-B7.1 avoids the need for antigen identification prior to vaccination and offers the opportunity of inducing a wide range of antigen-specific immune responses. This general strategy has also been successful using local recombinant IL-2 administration, which induced regression in 85% of injected melanoma lesions in one study (54). Recently, the induction of melanoma regression associated with a dense inflammatory response has also been seen following local application of the TLR agonist, imiquimod (55). This potential for using poxviruses to deliver immunomodulatory molecules to the tumor microenvironment has also been evaluated in a small clinical trial that used an IL-12 expressing canarypox virus administered as an intratumoral vaccination to 9 patients with metastatic melanoma (56). One patient exhibited a complete clinical response, and vaccinated lesions were characterized by an enhanced T cell infiltrate.
Although T cell activation at the site of tumor growth has been implicated as an essential component of successful tumor rejection by CD8+ T cells, protective antitumor T cell priming occurs in secondary lymphoid tissue (10, 11). This has led to interest in targeting vaccines to local lymph node basins to improve the likelihood of activating an effective T cell response (57–60). The benefit of this approach was demonstrated in a murine model using a lymphocytic choriomeningitis virus peptide vaccine, which induced 106 more potent CTL responses when administered by intrasplenic injection compared to subcutaneous vaccination (61). Thus, it is intriguing that the 3 patients in our trial who achieved a significant survival advantage all had index lesions localized to lymph node sites. Despite the superiority of lymph nodes for priming T cell responses, such cells may be rendered anergic when they encounter peripheral tumors. Consistent with our data, others have shown that such T cell ignorance can be overcome by expression of B7.1 at peripheral tumor sites (11).
The success of these local treatment approaches introduces some difficulty in monitoring immune responses, an important aspect of modern tumor vaccine investigation. In this report, we utilized a qRT-PCR method that used a relatively small amount of material collected through serial FNAs of injected lesions. We previously validated this approach, and identification of differential gene expression profiles may provide a tool for predicting response to treatment (62). In a melanoma trial employing a peptide vaccine and IL-2, 8 of 27 metastatic lesions completely regressed, and 1 lesion demonstrated an objective partial response (63). Similar to our results with local poxvirus vaccination, regression was associated with significantly increased INF-γ transcript levels in regressing compared to nonregressing lesions following peptide vaccination and IL-2 administration. Thus, the evaluation of local gene profiles through validated qRT-PCR analysis may represent a method for sampling the tumor microenvironment and predicting response to local interventions.
The data presented in this report documents the safety profile and feasibility of rV-B7.1 as a vaccine for local delivery into established melanoma lesions. Vaccination resulted in systemic antivaccinia antibody and T cell responses in all patients and was associated with mild low-grade constitutional symptoms and, in 3 patients, autoimmune vitiligo. The vaccine did not encode melanoma antigens, yet evidence of systemic immunity was provided by an increase in gp100- and MART-1–specific T cell responses in HLA-A*0201–bearing patients. We also demonstrated the feasibility of using a qRT-PCR assay for profiling the tumor microenvironment in serial FNAs of injected lesions. An increase in CD8 and IFN-γ gene transcripts was associated with clinical regression in our trial, consistent with previous reports. Local delivery of poxviruses expressing other immunomodulatory genes represents a method for altering the local tumor microenvironment and provides a mechanism for inducing systemic antitumor immunity.
Methods
Human subjects and vaccination protocol.
A standard dose-escalation phase I clinical trial received ethical approval from the Committee on Clinical Investigation at the Albert Einstein College of Medicine and the Institutional Review Board of Columbia University, and all subjects provided written informed consent. Patients with metastatic melanoma were eligible contingent upon the following: (a) age of 18 years or more; (b) histologically proven stage IV metastatic melanoma without brain metastasis and with lesions greater than or equal to 1 cm in size and accessible to percutaneous injection; (c) history of prior vaccinia vaccination; (d) ECOG performance status of 0–1; (e) negative β–human chorionic gonadotrophin in women of childbearing potential; (f) at least 4 weeks from completing surgery or prior chemotherapy and complete recovery from same; (g) at least 2 weeks from completing prior radiation therapy without evidence of bone marrow toxicity and complete recovery from such therapy; (h) at least 8 weeks from completing prior immunotherapy and complete recovery from same; (i) life expectancy greater than 3 months; (j) no history of eczema or other illness requiring systemic corticosteroids or contraindicating vaccinia administration; (k) adequate organ function as defined by a wbc count greater than or equal to 3,000/mm3, platelet count greater than or equal to 100,000/mm3, hemoglobin level greater than or equal to 10 g/100 ml, serum creatinine less than or equal to 2.0 mg/dl, serum direct bilirubin levels less than or equal to 1.5 mg/dl, serum transaminases and alkaline phosphatase levels less than 2 times the upper normal limits, less than 2-fold elevation of prothrombin and partial thromboplastin time in patients not receiving anticoagulation medications, and no evidence of congestive heart failure, serious cardiac arrhythmias, recent prior myocardial infarction, clinical coronary artery disease, or cirrhosis; (l) no significant allergy or hypersensitivity to eggs; (m) no history of another malignancy in the preceding 2 years other than stage I cervical carcinoma or basal cell carcinoma of the skin; (n) ability to avoid contact with high-risk individuals (immunosuppressed individuals, pregnant women, children less than 3 years of age, and patients with eczema or other open skin conditions) for a period of 7–10 days after immunization.
Subjects received monthly intralesional injections of rV-B7.1 provided by the Cancer Therapy Evaluation Program (National Cancer Institute). The rV-B7.1 study design has been previously reported (64), and each cycle consisted of 3 vaccine doses. Two vaccine concentrations, 4.26 × 107 and 4.26 × 108 PFU, were used. Prior to each injection, a history was taken and physical examination performed, and blood was collected for complete blood count, antinuclear antibody titer, serum electrolyte levels, and coagulation studies. An FNA of the treatment lesion was conducted to collect a baseline sample for immunologic assay. Two weeks after vaccination, serum and T cells were obtained for immunologic studies. Four weeks after the third injection, patients underwent radiologic imaging to assess disease status; those patients without objective disease progression and who still met eligibility criteria were offered booster vaccinations following the same schema.
Immunohistochemistry.
Cytospin preparations of sequentially obtained FNA material were fixed in acetone and stained with anti–MART-1 murine IgG2b (M2-7C10) (16, 19) and anti-Pmel17/gp100 mAb HMB45 (BioGenex). For secondary staining, biotinylated goat anti-mouse IgG (KPL) was used, followed by avidin-biotin-peroxidase (VECTASTAIN Elite Kit; Vector Laboratories).
HLA-typing.
HLA typing was determined by standard PCR assay as previously described (65).
Antibody titers.
Patient sera was collected before and 2 weeks after each vaccination and stored at –20°C. Ninety-six–well ELISA plates were coated with vaccinia virus (Wyeth strain) lysate overnight at 4°C. On day 2, plates were washed and then blocked with 5% milk solution. Appropriate dilutions of control and sample sera were made using the same milk solution and incubated for 1 hour at 37°C. For antivaccinia antibody detection, HRP-goat anti-human IgG was added, followed by the HRP substrate (TMB Microwell Peroxidase system; KPL), and OD was read at 450 nm. The dilution factor of patient sera (1:2) was corrected and titers were determined based on the value of the naive sera times 3. A positive postvaccination response was indicated by a 2-fold increase in titer compared with preimmunization titers. Positive prevaccination responses were defined as titers greater than 400 OD.
Systemic T cell responses.
PBMCs were isolated by density gradient centrifugation using Ficoll-Hypaque (Pharmacia) before and 2 weeks after each vaccination. PBMCs were stored at –80°C until analysis. The precursor frequency of melanoma antigen-specific T lymphocytes was determined by IFN-γ ELISPOT assay as previously described (66). Since, with the use of a nonantigen encoding viral vaccine, low precursor frequencies were expected, IVS was performed as follows: pre- and postvaccination PBMCs were thawed and incubated with 10 μg/ml MART-126-35(EAAGIGILTV) or a gp100 1:1:1 peptide mix (gp100154–162[KTWGQYWQV], gp100209[ITDQVPFSV], gp100619–627[RLMKQDFSV]). On day 2, rIL-2 (300 IU/ml) and rIL-7 (10 ng/ml; PeproTech) were added to the cultures for 7 days. Following IVS, ELISPOT assays were set up on 96-well nitrocellulose plates (Millititer HA; Millipore) coated overnight with 10 μg/ ml murine anti-human IFN-γ monoclonal antibody (BD Biosciences — Pharmingen). After washing, wells were blocked for 2 hours at 37°C with 200 μl of complete media containing AimV (GIBCO; Invitrogen Corp.), 5% human AB serum (Bioreclamation), 1% glutamine and penicillin-streptomycin. Pre- and postvaccination PBMCs were collected, washed, and plated onto anti–IFN-γ mAB–coated wells at 1 or 2 × 105 cells per well with 10 μg/ml of the corresponding HLA-A2–restricted peptides (gp100209 and gp100209-2M, MART-1A26–35 and MART-1A26–35/2L). After a 24-hour incubation at 37°C and 5% CO2, plates were washed several times with PBS containing Tween 20 (0.05%) and incubated with 2 μg/ml of biotinylated mouse anti-human IFN-γ monoclonal antibody (BD Biosciences — Pharmingen) at 4°C overnight. Subsequently, wells were washed with PBS containing Tween 20 (0.05%) and incubated with a 1:4,000 dilution of avidin alkaline phosphatase (GIBCO-BRL; Invitrogen Corp.) for 2 hours, followed by washing with PBS. After adding 100 μl of 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium (BCIP/NBT) phosphatase substrate (KPL) for 1 hour, plates were washed and allowed to air dry. Concanavalin A (5 μg/ml) was added as a positive control and PBS alone served as a negative control. T cell responses against vaccinia virus were also tested as a positive control and in order to verify successful vaccination. For antivaccinia assay, viral lysates were produced using an overnight culture of vaccinia-infected (TBC-Wy; Therion Biologics) BSC-40 cells (MOI 10). The following day, cells were harvested and lysed in a hypotonic lysis buffer (1 mM Tris), and lysates (2.5 μl/ml) were added to pre- and postvaccination PBMCs.
For all ELISPOT assays, IFN-γ spot-forming units were counted by CTL ImmunoSpot Analyzer (BD Biosciences). T cell precursor frequency was calculated as the number of spot-forming units from PBMC + APC + peptide subtracted by the background (PBMC + APC) per number of PBMC seeded. IFN-γ ELISPOT assay was validated to measure T cell responses against vaccinia virus using PBMC from 2 healthy donors vaccinated with smallpox vaccine (Dryvax; Wyeth Laboratories). During the qualification phase, assays (n = 2 or 3) were conducted on separate days using standard conditions. We evaluated the use of fresh and cryopreserved/thawed PBMCs from the same volunteer and the average number of spots/well were comparable (data not shown). Fresh PBMC, 5 × 105 cells/well were incubated with various concentrations of vaccinia lysate (5, 2.5, and 1.5 μl /well) and 2.5 μl /well was chosen for future assays. The interassay reproducibility was determined in 2 consecutive assays resulting in comparable relative standard deviations.
Statistical analysis.
The fold change in T cell response was estimated using empirical Bayes method after background adjustments. The average background reactivity for each patient at each time point was calculated based on 2 or 3 wells from the same plate. The ELISPOT readings were then adjusted by subtracting the background; if a reading was below the background, the adjusted spot count would be 0. To avoid numerical problems in calculating the fold changes, half spot (per 105 cells) was added to each count. The fold change for an individual patient was estimated by the following: [(Y + ν/mpost)/(X + ν/mpre)] × [(a + a0/mpre)/(a + (a0 – 1)/mpost)], where X and Y are the respective average numbers of spots before and after vaccination and mpre and mpost are the respective numbers of replicates (wells) over which averages were taken. The parameters ν, a, and a0 were well-specific positive numbers and were estimated by maximizing a marginal likelihood. The parameter ν served as an attenuation factor to avoid false positives when prevaccination ELISPOT readings were low.
The antivaccinia antibody responses were measured by fold change of titers as determined by ELISA assay. Correlation between antivaccinia T cell and antibody responses was summarized using Pearson’s correlation coefficient, with a 95% confidence interval (via bootstrap).
RNA isolation and amplification.
Total RNA was isolated from FNA samples using RNeasy mini-kits (QIAGEN) and amplified as previously described (67). Two rounds of amplification, each consisting of a combination of in vitro transcription (IVT) and template-switching PCR, were performed. The quality of aRNA after each round of IVT was monitored by electrophoresis through denaturaing gels. RNA products were separated on an RNA 6000 chip using Agilent 2100 bioanalyzer (Agilent Technologies). Quality analysis using electropherograms of aRNA were obtained using the Agilent bioanalyzer. The size of aRNA after the first IVT ranged from 500 to 6,000 nucleotides and was from 500 to 4,000 nucleotides after the second IVT. For qRT-PCR, 5–10 μg of aRNA was reverse transcribed into cDNA using the High-Capacity cDNA Archive Kit (Applied Biosystems), and cDNA was stored at –20°C until use.
qRT-PCR.
The levels of gene expression for CD8, IFN-γ, IL-10, and a reference housekeeping gene, PGK1, were determined in FNA-derived cDNA samples by qRT-PCR using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). Primers and probes were obtained using TaqMan Assays-on-Demand gene expression products (Applied Biosystems), which are built on 5′-nuclease chemistry and consist of 2 unlabeled PCR primers (900 nM) and a FAM dye-labeled TaqMan MGB probe (250 nM) as a reporter as well as a 3′ end with the quencher dye TAMRA. For each sample, we used 200-400 ng of cDNA per well for the reference and target genes. An ABI PRISM 7900HT Sequence Detection System was programmed for the initial step of 2 minutes at 50°C and 10 minutes at 95°C, followed by 40 thermal cycles of 15 seconds at 95°C and 1 minute at 60°C. Each measurement was set up in triplicate. To validate the amount and quantity of source RNA, transcript levels of the reference gene, PGK1, were measured. For each sample, target values were corrected by PGK1 copies to obtain normalized values independent of variation in starting material. Each primer and probe pair was validated using the comparative CT method for relative quantification (ΔΔCT) where the amount of target gene copies are normalized to a reference gene (PGK1) using mitogen-stimulated PBMCs from healthy donors as source RNA. The correlation between the CT and the fold difference in RNA concentration for each probe and primer set was measured by creating a 10-fold serial dilution curve over the entire detection scale. For each probe and primer pair combination used in this study, the correlation coefficient of detection was at least 0.99. When we performed the relative efficiency plot of target versus reference gene, the absolute value of the slope of log input (ng cDNA) versus ΔCT was less than 0.1. The fold difference between pre- and postvaccination samples was calculated assuming 100% efficient PCR where each CT is normalized to PGK1 using the comparative 2–ΔΔCT method described previously (68): fold difference = 2||CT1(target) – CT1(PGK1)| – |CT2(target) – CT2(PGK1)||; fold difference = 2|| – CT1| – | – CT2||, where the CT1(target) and CT2(target) are the CT values for the post- and prevaccination samples with the target gene and CT1(PGK1) and CT2(PGK1) are the CT values for post- and prevaccination samples with the internal control PGK1. A 2-fold difference in gene expression was defined to be within the discrimination ability of the assay.
Supplementary Material
Acknowledgments
This work was supported by grants RO1 93696 from the NIH and T98052 from the Doris Duke Charitable Foundation. Additional support was provided by NIH/National Center for Research Resources grant RR00645. We thank M. Bereta for critical review of the manuscript.
Footnotes
Heidi Hörig and Francesco M. Marincola are co-senior authors.
Nonstandard abbreviations used: aRNA, amplified RNA; CEA, carcinoembryonic antigen; CT, cycle threshold; CTLA-4, CTL-associated antigen 4; ECOG, Eastern Cooperative Oncology Group; FNA, fine-needle aspirate; IVS, in vitro stimulation; IVT, in vitro transcription; MART-1, melanoma antigen recognized by T cells 1; PD, progressive disease; PGK1, phosphoglycerate kinase 1; PR, partial response; qRT-PCR, quantitative real-time PCR; rV-B7.1, recombinant vaccinia virus expressing B7.1; SD, stable disease; TAA, tumor-associated antigen.
Conflict of interest: J. Donovan, K. Dolan, K. Manson, and D. Panicali are employees and stockholders of Therion Biologics Corp.
References
- 1.Rosenberg S, Yang J, Restifo N. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Kempen L, Ruiter D, van Muijen G, Coussens L. The tumor microenvironment: a critical determinant of neoplastic evolution. Eur. J. Cell Biol. 2003;82:539–548. doi: 10.1078/0171-9335-00346. [DOI] [PubMed] [Google Scholar]
- 3.Henning T, Kraus M, Brischwein M, Otto A, Wolf B. Relevance of tumor microenvironment for progression, therapy, and drug development. Anticancer Drugs. 2004;15:7–14. doi: 10.1097/00001813-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Mizoguchi H, O’Shea J, Longo D. Alterations in signal transduction molecules in T lymphocytes from tumor-bearing mice. Science. 1992;258:1732–1733. doi: 10.1126/science.1465616. [DOI] [PubMed] [Google Scholar]
- 5.Zou W, et al. Stromal-derived factor-1 in human tumors recruits and alters the function of plasmacytoid precursor dendritic cells. Nat. Med. 2001;7:1339–1346. doi: 10.1038/nm1201-1339. [DOI] [PubMed] [Google Scholar]
- 6.Dong R, et al. Dendritic cells from CML patients have altered actin organization, reduced antigen processing, and impaired migration. Blood. 2002;101:3560–3567. doi: 10.1182/blood-2002-06-1841. [DOI] [PubMed] [Google Scholar]
- 7.Liyannage U, Moore T, Joo H. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- 8.Lu S, et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res. 2004;64:4405–4410. doi: 10.1158/0008-5472.CAN-04-1032. [DOI] [PubMed] [Google Scholar]
- 9.Viguier M, et al. Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J. Immunol. 2004;173:1444–1453. doi: 10.4049/jimmunol.173.2.1444. [DOI] [PubMed] [Google Scholar]
- 10.Palmer D, et al. Vaccine-stimulated, adoptively transferred CD8+ T cells traffic indiscriminately and ubiquitously while mediating specific tumor destruction. J. Immunol. 2004;173:7209–7216. doi: 10.4049/jimmunol.173.12.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ochsenbein A, et al. Roles of tumour localization, second signals, and cross priming in cytotoxic T-cell induction. Nature. 2001;411:1058–1064. doi: 10.1038/35082583. [DOI] [PubMed] [Google Scholar]
- 12.Zippelius A, et al. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64:2865–2873. doi: 10.1158/0008-5472.can-03-3066. [DOI] [PubMed] [Google Scholar]
- 13.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 14.Yu P, Lee Y, Liu W. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat. Immunol. 2004;5:141–149. doi: 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- 15.Doxsee C, et al. The immune response modifier and Toll-like receptor 7 agonist S-27609 selectively induces IL-12 and TNF-alpha production in CD11c+CD11b+CD8- dendritic cells. J. Immunol. 2003;171:1156–1163. doi: 10.4049/jimmunol.171.3.1156. [DOI] [PubMed] [Google Scholar]
- 16.Panelli M, et al. Gene-expression profiling of the response of peripheral blood mononuclear cells and melanoma metastases to systemic IL-2 administration. Genome Biol. 2002;3:research0035.1–research0035.17. doi: 10.1186/gb-2002-3-7-research0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schols D, Vandekerckhove B, Jones D, Roncarolo M. IL-2 reverses human T cell unresponsiveness induced by thymic epithelium in SCID-hu mice. J. Immunol. 1994;152:2198–2206. [PubMed] [Google Scholar]
- 18.Monsurro V, et al. Quiescent phenotype of tumor-specific CD8+ T cells following immunization. Blood. 2004;104:1970–1978. doi: 10.1182/blood-2004-02-0525. [DOI] [PubMed] [Google Scholar]
- 19.van der Merwe P, Davis S. Molecular interactions mediating T cell antigen recognition. Annu. Rev. Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 20.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat. Rev. Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 21.Gajewski T, Fallarino F, Uyttenhove C, Boon T. Tumor rejection requires a CTLA4 ligand provided by the host or expressed on the tumor: superiority of B7-1 over B7-2 for active tumor immunization. J. Immunol. 1996;156:2909–2917. [PubMed] [Google Scholar]
- 22.Hodge J, Abrams S, Schlom J, Kantor J. Induction of antitumor immunity by recombinant vaccinia viruses expressing B7-1 or B7-2 costimulatory molecules. Cancer Res. 1994;54:5552–5555. [PubMed] [Google Scholar]
- 23.Kalus R, et al. The use of combination vaccinia vaccines and dual-gene vaccinia vaccines to enhance antigen-specific T-cell immunity via T-cell costimulation. Vaccine. 1999;17:893–903. doi: 10.1016/s0264-410x(98)00275-8. [DOI] [PubMed] [Google Scholar]
- 24.Hodge J, Grosenbach D, Aarts W, Poole D, Schlom J. Vaccine therapy of established tumors in the absence of autoimmunity. Clin. Cancer Res. 2003;9:1837–1849. [PubMed] [Google Scholar]
- 25.Essajee S, Kaufman H. Poxvirus vaccines for cancer and HIV therapy. Expert Opin. Biol. Ther. 2004;4:575–588. doi: 10.1517/14712598.4.4.575. [DOI] [PubMed] [Google Scholar]
- 26.Baldwin P, et al. Vaccinia-expressed human papillomavirus 16 and 18 e6 and e7 as a therapeutic vaccination for vulval and vaginal intraepithelial neoplasia. Clin. Cancer Res. 2003;9:5205–5213. [PubMed] [Google Scholar]
- 27.Kaufman H, et al. Phase II randomized study of vaccine treatment of advanced prostate cancer (E7897): a trial of the Eastern Cooperative Oncology Group. J. Clin. Oncol. 2004;22:2122–2132. doi: 10.1200/JCO.2004.08.083. [DOI] [PubMed] [Google Scholar]
- 28.Smith S, Kotwal G. Immune response to poxvirus infections in various animals. Crit. Rev. Microbiol. 2002;28:149–185. doi: 10.1080/1040-840291046722. [DOI] [PubMed] [Google Scholar]
- 29.Matsuoka T, et al. Characteristics of immunity induced by viral antigen or conferred by antibody via different administration routes. Clin. Exp. Immunol. 2002;130:386–392. doi: 10.1046/j.1365-2249.2002.02003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamberg L. “Epidemic” of malignant melanoma: true increase or better detection? [news] JAMA. 2002;287:2201. [PubMed] [Google Scholar]
- 31.Therasse P, Arbuck S, Eisenhauer E. New guidelines to evaluate the response to treatment in solid tumours. J. Natl. Cancer. Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 32.Hofbauer G, et al. High-frequency of melanoma-associated antigen or HLA class I loss does not correlate with survival in primary melanoma. J. Immunother. 2004;27:73–78. doi: 10.1097/00002371-200401000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Khong H, Wang Q, Rosenberg S. Identification of multiple antigens recognized by tumor-infiltrating lymphocytes from a single patient: tumor escape by antigen loss and loss of MHC expression. J. Immunother. 2004;27:184–190. doi: 10.1097/00002371-200405000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eigentler T, Caroli U, Radny P, Garbe C. Palliative therapy of disseminated malignant melanoma: a systematic review of 41 randomised clinical trials. Lancet Oncol. 2003;4:748–759. doi: 10.1016/s1470-2045(03)01280-4. [DOI] [PubMed] [Google Scholar]
- 35.Komenaka I, Hoerig H, Kaufman H. Immunotherapy for melanoma. Clin. Dermatol. 2004;22:251–265. doi: 10.1016/j.clindermatol.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Moss B. Vaccinia virus: a tool for research and vaccine development. Science. 1991;252:1662–1667. doi: 10.1126/science.2047875. [DOI] [PubMed] [Google Scholar]
- 37.Moss B. Genetically engineered poxviruses for recombinant gene expression, vaccination, and safety. Proc. Natl. Acad. Sci. U. S. A. 1996;93:11341–11348. doi: 10.1073/pnas.93.21.11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scholl S, et al. Recombinant vaccinia virus encoding human MUC1 and IL2 as immunotherapy in patients with breast cancer. J. Immunother. 2000;23:570–580. doi: 10.1097/00002371-200009000-00007. [DOI] [PubMed] [Google Scholar]
- 39.Ikeda H, et al. The critical role of type-1 innate and acquired immunity in tumor immunotherapy. Cancer Sci. 2004;95:697–703. doi: 10.1111/j.1349-7006.2004.tb03248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraser J, Irving B, Crabtree G, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251:313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]
- 41.Boise L, Noel P, Thompson C. CD28 and apoptosis. Curr. Opin. Immunol. 1995;7:620–625. doi: 10.1016/0952-7915(95)80067-0. [DOI] [PubMed] [Google Scholar]
- 42.Wong F, et al. The role of lymphocyte subsets in accelerated diabetes in nonobese diabetic-rat insulin promoter-B7-1 (NOD-RIP-B7-1) mice. J. Exp. Med. 1998;187:1985–1993. doi: 10.1084/jem.187.12.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schwartz R. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 44.Yu X, Abe R, Hodes R. The role of B7-CD28 costimulation in tumor rejection. Int. Immunol. 1998;10:791–797. doi: 10.1093/intimm/10.6.791. [DOI] [PubMed] [Google Scholar]
- 45.Hodge J, et al. Admixture of a recombinant vaccinia virus containing the gene for the costimulatory molecule B7 and a recombinant vaccinia virus containing a tumor-associated antigen gene results in enhanced specific T-cell responses and antitumor immunity. Cancer Res. 1995;55:3598–3603. [PubMed] [Google Scholar]
- 46.Akagi J, et al. Therapeutic antitumor response after immunization with an admixture of recombinant vaccinia virus expressing a modified MUC-1 gene and the murine T-cell costimulatory molecule B7. J. Immunother. 1997;20:38–47. doi: 10.1097/00002371-199701000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Pastorfide G, et al. Image analysis of stage I melanoma (1.00 - 2.50 mm): lymphocytic infiltrates related to metastasis and survival. J. Cutan. Pathol. 1992;19:390–397. doi: 10.1111/j.1600-0560.1992.tb00611.x. [DOI] [PubMed] [Google Scholar]
- 48.Halliday G, Patel A, Hunt M, Tefany F, Barnetson R. Spontaneous regression of human melanoma/nonmelanoma skin cancer: association with infiltrating CD4+ T cells. World J. Surg. 1995;19:352–358. doi: 10.1007/BF00299157. [DOI] [PubMed] [Google Scholar]
- 49.Yang A, Monken C, Lattime E. Intratumoral vaccination with vaccinia-expressed tumor antigen and granulocyte macrophage colony-stimulating factor overcomes immunological ignorance to tumor antigen. Cancer Res. 2003;63:6956–6961. [PubMed] [Google Scholar]
- 50.Hodge J, Schlom J. Comparative studies of a retrovirus versus a poxvirus vector in whole tumor-cell vaccines. Cancer Res. 1999;59:5106–5111. [PubMed] [Google Scholar]
- 51.Kudo-Saito C, Schlom J, Hodge J. Intratumoral vaccination and diversified subcutaneous/ intratumoral vaccination with recombinant poxviruses encoding a tumor antigen and multiple costimulatory molecules. Clin. Cancer Res. 2004;10:1090–1099. doi: 10.1158/1078-0432.ccr-03-0145. [DOI] [PubMed] [Google Scholar]
- 52.Tsang K, et al. The infection of human dendritic cells with recombinant avipox vectors expressing a costimulatory molecule transgene (CD80) to enhance the activation of antigen-specific cytolytic T cells. Cancer Res. 2001;61:7568–7576. [PubMed] [Google Scholar]
- 53.Zhu M, et al. Enhanced activation of human T cells via avipox vector-mediated hyperexpression of a triad of costimulatory molecules in human dendritic cells. Cancer Res. 2001;61:3725–3734. [PubMed] [Google Scholar]
- 54.Radny P, et al. Phase II trial of intralesional therapy with interleukin-2 in soft-tissue melanoma metastases. Br. J. Cancer. 2003;89:1620–1626. doi: 10.1038/sj.bjc.6601320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michalopoulos P, Yawalkar N, Bronnimann M, Kappeler A, Braathen L. Characterization of the cellular infiltrate during successful topical treatment of lentigo maligna with imiquimod. Br. J. Dermatol. 2004;151:903–906. doi: 10.1111/j.1365-2133.2004.06176.x. [DOI] [PubMed] [Google Scholar]
- 56.Triozzi P, et al. Intratumoral administration of a recombinant canarypox virus expressing interleukin 12 in patients with metastatic melanoma. Hum. Gene Ther. 2005;16:91–100. doi: 10.1089/hum.2005.16.91. [DOI] [PubMed] [Google Scholar]
- 57.Bedrosian I, et al. Intranodal administration of peptide-pulsed mature dendritic cell vaccines results in superior CD8+ T-cell function in melanoma patients. J. Clin. Oncol. 2003;21:3826–3835. doi: 10.1200/JCO.2003.04.042. [DOI] [PubMed] [Google Scholar]
- 58.Maier T, et al. Vaccination of patients with cutaneous T-cell lymphoma using intranodal injection of autologous tumor-lysate-pulsed dendritic cells. Blood. 2003;102:2338–2344. doi: 10.1182/blood-2002-08-2455. [DOI] [PubMed] [Google Scholar]
- 59.Brown K, et al. Adenovirus-transduced dendritic cells injected into skin or lymph node prime potent simian immunodeficiency virus-specific T cell immunity in monkeys. J. Immunol. 2003;171:6875–6882. doi: 10.4049/jimmunol.171.12.6875. [DOI] [PubMed] [Google Scholar]
- 60.Chang G, et al. A pilot clinical trial of vaccination with dendritic cells pulsed with autologous tumor cells derived from malignant pleural effusions in patients with late-stage lung carcinoma. Cancer. 2005;103:763–771. doi: 10.1002/cncr.20843. [DOI] [PubMed] [Google Scholar]
- 61.Johansen P, et al. Direct intralymphatic injection of peptide vaccines enhances immunogenicity. Eur. J. Immunol. 2005;35:568–574. doi: 10.1002/eji.200425599. [DOI] [PubMed] [Google Scholar]
- 62.Mocellin S, Rossi C, Pilati P, Nitti D, Marincola F. Quantitative real-time PCR: a powerful ally in cancer research. Trends Mol. Med. 2003;9:189–195. doi: 10.1016/s1471-4914(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 63.Mocellin S, Ohnmacht G, Wang E, Marincola F. Kinetics of cytokine expression in melanoma metastases classifies immune responsiveness. Int. J. Cancer. 2001;93:236–242. doi: 10.1002/ijc.1328. [DOI] [PubMed] [Google Scholar]
- 64.Kaufman H, et al. A phase I trial of intra lesional RV-B7.1 vaccine in the treatment of malignant melanoma. Hum. Gene Ther. 2000;11:1065–1082. doi: 10.1089/10430340050015374. [DOI] [PubMed] [Google Scholar]
- 65.Bunce M, et al. Phototyping: comprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 & DQB1 by PCR with 144 primer mixes utilizing sequence-specific primers (PCR-SSP) Tissue Antigens. 1995;46:355–367. doi: 10.1111/j.1399-0039.1995.tb03127.x. [DOI] [PubMed] [Google Scholar]
- 66.Horig H, et al. Phase I clinical trial of a recombinant canarypoxvirus (ALVAC) vaccine expressing human carcinoembryonic antigen and the B7.1 costimulatory molecule. Cancer Immunol. Immunother. 2000;49:504–514. doi: 10.1007/s002620000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang E, Miller L, Ohnmacht G, Liu E, Marincola F. High-fidelity mRNA amplification for gene profiling. Nat. Biotechnol. 2000;18:457–459. doi: 10.1038/74546. [DOI] [PubMed] [Google Scholar]
- 68.Freeman W, Walker S, Vrana K. Quantitative RT-PCR: pitfalls and potential. Biotechniques. 1999;26:112–125. doi: 10.2144/99261rv01. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.