

Abstract
Structure–activity relationship (SAR) studies are fundamental to drug and agrochemical development, yet only a few synthetic strategies apply to the nitrogen heteroaromatics frequently encountered in small molecule candidates1–3. Here we present an alternative approach in which we convert pyrimidine-containing compounds into various other nitrogen heteroaromatics. Transforming pyrimidines into their corresponding N-arylpyrimidinium salts enables cleavage into a three-carbon iminoenamine building block, used for various heterocycle-forming reactions. This deconstruction–reconstruction sequence diversifies the initial pyrimidine core and enables access to various heterocycles, such as azoles4. In effect, this approach allows heterocycle formation on complex molecules, resulting in analogues that would be challenging to obtain by other methods. We anticipate that this deconstruction–reconstruction strategy will extend to other heterocycle classes.
Modifying the structure of candidate compounds in structure–activity relationship (SAR) studies enables the optimization of their physiochemical properties in drug and agrochemical development5. Practitioners commonly select portions of the structure of a candidate to diversify during these studies and altering the periphery of azines, such as pyrimidines, is frequently used because they are pervasive and often form key binding interactions with the biological target6–9. Rapid access to analogue compounds is paramount; therefore, chemical reactions that make structural modifications in one or two steps are valuable, particularly during later stages of development involving complex structures1,2. Although many individual reactions can transform azines, they typically fall into only two effective strategies, transforming embedded functional groups and C–H functionalization reactions, which enable rapid diversification for SAR studies3. Molecular editing is emerging to produce analogue compounds for SAR studies but is typically associated with transforming one candidate compound to a single derivative4. We reasoned that deconstructing complex pyrimidines to a parent synthetic intermediate, with a large set of associated chemical reactions, could expand this strategy. In this way, reconstructing substituted versions of the original pyrimidine and accessing other heterocycles becomes viable through simple cyclization reactions. This approach both diversifies the structure of a candidate and can generate chemical libraries, making it a potentially useful tactic in SAR studies10.
De novo heterocycle synthesis involves cyclizing smaller chemical fragments into substituted rings, with a classic example being reactions involving 1,3-dicarbonyl compounds to form substituted azines and azoles5,11. These processes are commonly used to form building-block pyrimidines, pyrazoles and 1,2-oxazoles. Fragment coupling processes, such as that exploited to create the aminopyrimidine motif in gleevec, also use de novo synthesis; large-member libraries are created this way because of the commercial availability of partners such as amidines and hydrazines (Fig. 1a)12,13. Although valuable at earlier points in drug discovery, this strategy is far less suitable for SAR studies on more complex candidate structures. Preserving heterocycle precursors, such as 1,3-dicarbonyls or their derivatives, through several synthetic steps and diversifying at later stages is generally not viable as they tend to react with many classes of organic molecules and reagents. Therefore, developers process building-block heterocycles through rounds of multistep sequences to obtain analogue compounds. Furthermore, key heterocycles are typically extant in advanced candidates, so de novo approaches are ostensibly unsuitable for SAR studies. However, we have devised a strategy that transforms complex pyrimidine-containing structures into iminoenamines, surrogates of 1,3-dicarbonyl compounds, and then leverages de novo heterocycle synthesis to obtain substituted pyrimidine and 1,2-azole analogues. This way, practitioners can envisage a masked, diversifiable three-carbon unit in pyrimidines and strategize for late-stage SAR applications.
Fig. 1 |. Traditional de novo heterocycle synthesis and a deconstruction– reconstruction approach for heterocycle diversification.
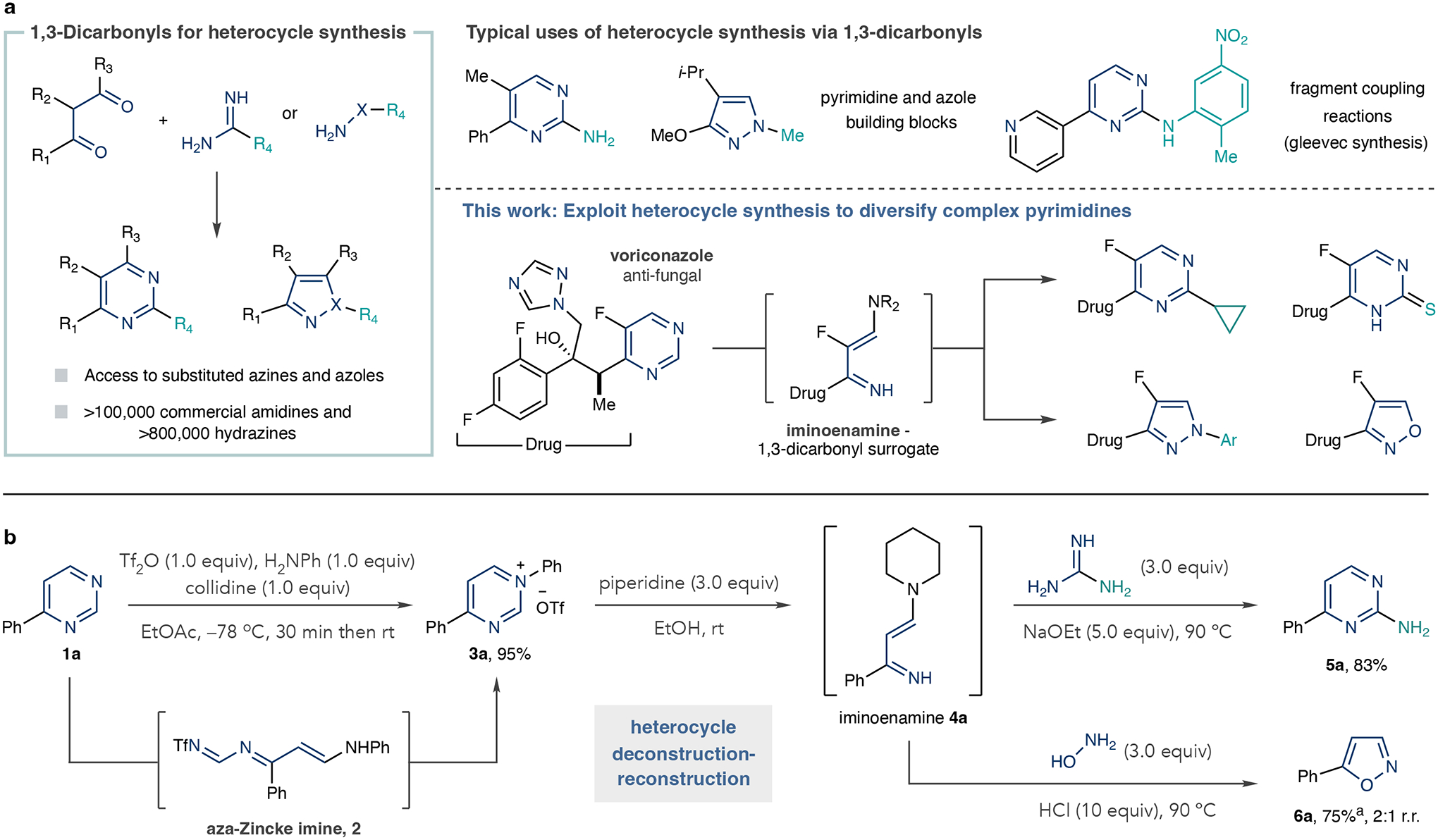
a, Pyrimidine and 1,2-azole synthesis by means of 1,3-dicarbonyls and iminoenamines as potential synthetic intermediates. b, Pyrimidine diversification model study showing access to 2-substituted analogues and azoles. Isolated yields are shown. aThe 1H NMR yield reported due to volatility of 6a. R denotes a general organic group; X, nitrogen or oxygen atom; Me, methyl; Ph, phenyl; i-Pr, iso-propyl; Ar, general aryl group; Tf, trifluoromethylsulfonyl; collidine, 2,4,6-trimethylpyridine; Et, ethyl; Ac, acetate; r.t., room temperature; r.r., regioisomeric ratio.
We conceived of this alternative approach to exploit de novo heterocycle synthesis based on the unexpected reaction in Fig. 1b. Our laboratory has previously disclosed that pyridines react with Tf2O and dibenzylamine to form ring-opened Zincke imines, which can be halogenated and recyclized to form 3-halopyridines14. When we tested an alternative procedure involving 4-phenylpyrimidine 1a and aniline as a nucleophile15, we did not observe ring-opened aza-Zincke imine 2, but instead obtained N-phenylpyrimidinium salt 3a in high yield16. It was then trivial to excise the C2 carbon atom using ethanolic solutions of piperidine and obtained a high yield of iminoenamine 4a along with minor amounts of a vinamidinium salt as judged by the crude 1H nuclear magnetic resonance (NMR) spectra with an internal standard (Supplementary Fig. 1). Iminoenamines are derivatives of 1,3-dicarbonyl compounds and represent key building blocks for de novo heterocycle synthesis. Figure 1b shows that in situ recyclization using guanidine under basic conditions forms 2-aminopyrimidine 5a in good yield. Alternatively, adding hydroxylamine and hydrochloric acid to the crude mixture containing 4a forms 1,2-oxazole 6a as a mixture of regioisomers. These ‘scaffold hopping’ processes have recently gained attention in the burgeoning area of molecular editing17–23. Sarpong reported that hydrazines react with NTf-pyrimidinium salts and form pyrazoles by means of a ring-opening, ring-closing process24. Here, rather than a one-to-one mapping of structures, iminoenamines enable access to several heterocyclic scaffolds25. This pilot study successfully demonstrates the principle of using de novo synthesis on existing heterocycles; in the next phase, we aimed to show its generality on various pyrimidine starting materials and cyclization partners.
Figure 2 shows representative examples of pyrimidinium salts formed by means of the ring-opening, ring-closing process, as well as the scope of 2-substituted pyrimidines and 1,2-azoles obtainable after the deconstruction–reconstruction steps. A wide variety of pyrimidines undergo the ring-opening, ring-closing protocol with aniline to form the corresponding pyrimidinium salts and Fig. 2 shows six representative examples (see Supplementary Fig. 4 for 14 more substrates). In general, 4-monosubstituted pyrimidines work well and can include (hetero)aryl groups, alkyl, alkoxy, amido and carbonyl substituents (3b–3d; Supplementary Fig. 4). In the case of C4 amide-substituted salts, we observed significant aniline triflylation and low yields of the pyrimidinium salt. However, we found that using 2,4,6-trimethylaniline mitigates this unwanted side reaction, resulting in a reasonable yield of salt 3d (vide infra). The 5-monosubstituted pyrimidines form mixtures of pyrimidinium salts, aza-Zincke imines and aniline-derived iminoenamines (Supplementary Fig. 3). This mixture is less suitable for isolation and characterization at this stage but can be directly taken into the subsequent heterocycle-forming steps (vide infra). On the other hand, 4,5-disubstituted pyrimidines form salts well (3e and 3f), with the chemoselectivity between the pyridine and pyrimidine being notable. We also showed that fused heterocycles, such as NTs-protected deazapurine, are viable substrates (3g). However, quinazolines were not successful in the salt-forming process. Other limitations include 4-halopyrimidines, 4-methylpyridimidines and 2-substituted pyrimidines. These cases result in low yields or competitive side reactions, although we are currently investigating how different anilines can alleviate these issues. We provide further information on the limitations of the ring-opening and ring-closing process in Supplementary Fig. 2.
Fig. 2 |. Scope of pyrimidinium salts, 2-substituted pyrimidines and 1,2-azoles.
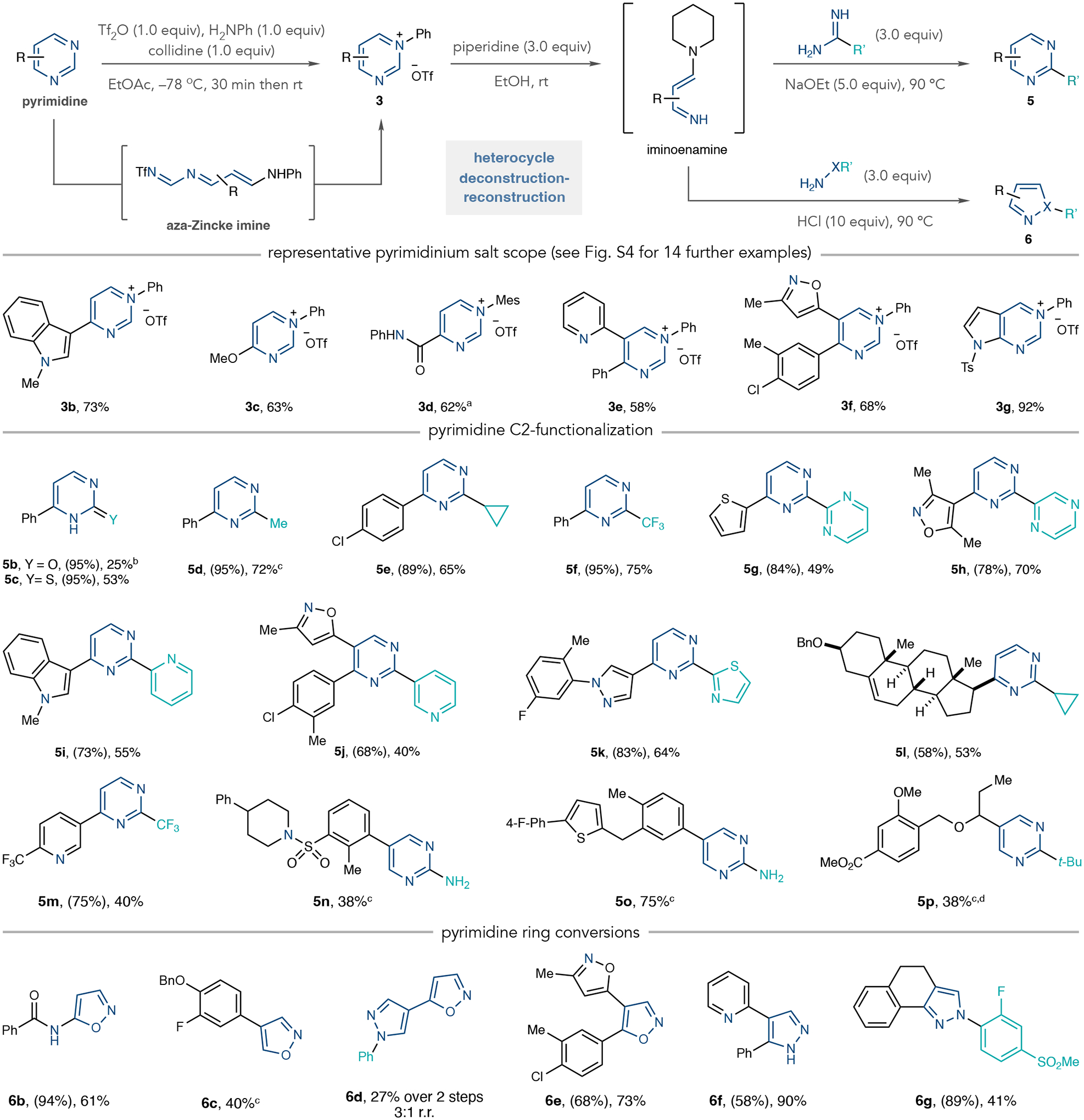
Isolated yields are shown. Yields in parentheses are for pyrimidinium salts. aInstead of aniline, 2,4,6-trimethylaniline was used. bInstead of piperidine, 6.0 equivalents of pyrrolidine was used. cIsolated as a 17:1 ratio with 1a. cOne-pot protocol: 4-nitroaniline used instead of aniline, then the solvent was exchanged for ethanol (EtOH). In the ring-closing step, 10–20 equivalents of amidine or hydroxylamine were used. dSodium methoxide (NaOMe) and methanol (MeOH) used instead of sodium ethoxide (NaOEt) and EtOH. Mes, 2,4,6-trimethylphenyl; Ts, tosyl; Bn, benzyl; t-Bu, tert-butyl.
With this robust method for pyrimidinium salt formation in hand, we next examined the scope of the deconstruction–reconstruction process. Cyclizations with urea and thiourea result in pyrimidinone 5b and thiopyrimidine 5c, respectively. Constructing 2-substituted pyrimidines with alkyl groups, such as methyl and cyclopropyl, is effective using the corresponding amidines in the cyclization step (5d and 5e). Similarly, using trifluoroacetamidine forms CF3-substituted pyrimidine 5f in good yield. The process can also form biheteroaryls, such as 5g–5k, in moderate to good yields. It is important to note that forming the C–heteroatom and C–C bonds in 5b–5k would typically necessitate a (pseudo)halide precursor and require nucleophilic aromatic substitution (SNAr) or metal-catalysed cross-coupling reactions that could be challenging to implement on complex azines26,27. Furthermore, there are only a few pyrimidine C–H functionalization reactions that function on complex substrates28. Others reported a 2-selective C–H amination reaction by means of pyrimidine N-oxides29. As the intermediate is distinct from the NTf-pyridinium salts in this approach, the output scope of these processes is likely to be complementary29. Minisci-type processes are ways to construct C–C bonds but they tend to give unselective mixtures of regioisomers or are biased towards the C4 position. Directed metalation reactions are viable for pyrimidine 2-halogenation in simple substrates30.
We continued to test this deconstruction–reconstruction approach to effect net C–H functionalization reactions. To that end, we transformed a pyrimidine-containing analogue of pregnenolone into 2-cyclopropylated structure 5l. Polyazines are compatible and we did not observe any reactions on the pyridine moiety in 5m, demonstrating chemoselectivity relevant to drug and agrochemical-type molecules31. We next examined a series of 5-monosubstituted pyrimidines which resemble drug-like fragments. As described above, these structures form intermediate mixtures at the pyrimidinium salt stage, so we developed a one-pot procedure by exchanging the solvent for ethanol and recyclizing to 2-substituted pyrimidines 5n–5p in reasonable yields. The method also works well to convert pyrimidines into azoles; we formed 1,2-oxazole 6b from an amido-substituted pyrimidinium salt. The 4-arylated 1,2-oxazole 6c is another example of deconstructing and reconstructing a 5-substituted pyrimidine. This chemistry also functions on molecules possessing several pyrimidines. Using 4,5-bipyrimidine, we performed a round of pyrimidinium salt formation, cleavage to the iminoenamine and recyclization with N-phenylhydrazine to install a pyrazole. Then, a second round culminating instead with hydroxylamine resulted in pyrazole-1,2-oxazole heteroaryl 6d. The process also functions on 4,5-disubstituted pyrimidines and provides access to disubstituted azoles 6e–6g. See Supplementary Fig. 5 for 20 more examples of 2-substituted pyrimidines and azoles formed through this approach. At present, isoureas and N-substituted guanidines are unsuccessful in the recyclization step (Supplementary Fig. 2). We also found that N-acyl- and N-sulfonylhydrazines result in N–H pyrazoles, indicating cleavage of those groups during recyclization.
At this stage, we turned this deconstruction–reconstruction approach towards modifying biologically active molecules into diverse sets of derivatives in line with strategies used in drug and agrochemical development. First, we converted the fungicide fenarimol (1b) into amino, cyclopropyl, and phenyl derivatives 5q–5s in one-pot processes (Fig. 3a). Second, to test a more challenging pyrimidine-containing structure, we selected dabrafenib, a small molecule treatment for melanoma, as a case study32. In Fig. 3a, 1c is a precursor to dabrafenib containing a pyrimidine with a 2-position C–H bond. In a one-pot process that formed the corresponding N-arylpyrimidinium salt in situ, we sequentially added piperidine in ethanol to form iminoenamine 4b followed by guanidine under basic conditions to install the C2–NH2 group, resulting in the active drug dabrafenib (5t). Using the same one-pot protocol, we also successfully obtained cyclopropylated and trifluoromethylated analogues 5u and 5v using amidines in the pyrimidine reconstruction stage. We found that scaffold hopping to other heterocycles was viable in this endeavour. In these cases, we obtained higher overall yields from 1c when we isolated its N-arylpyrimidinium salt. Cleavage to iminoenamine 4b followed by recyclization with hydrazines formed pyrazoles 6h and, notably, 6i as a single regioisomer. The same protocol, using hydroxylamine, constructs 1,2-oxazole 6j as a 16:1 regioisomeric mixture. These six derivatives of 1c highlight the capacity of the three-carbon iminoenamine intermediate 4b to serve as a versatile moiety for complex pyrimidine diversification.
Fig. 3 |. Applications of the pyrimidine diversification strategy to biologically active molecules and further reaction development.
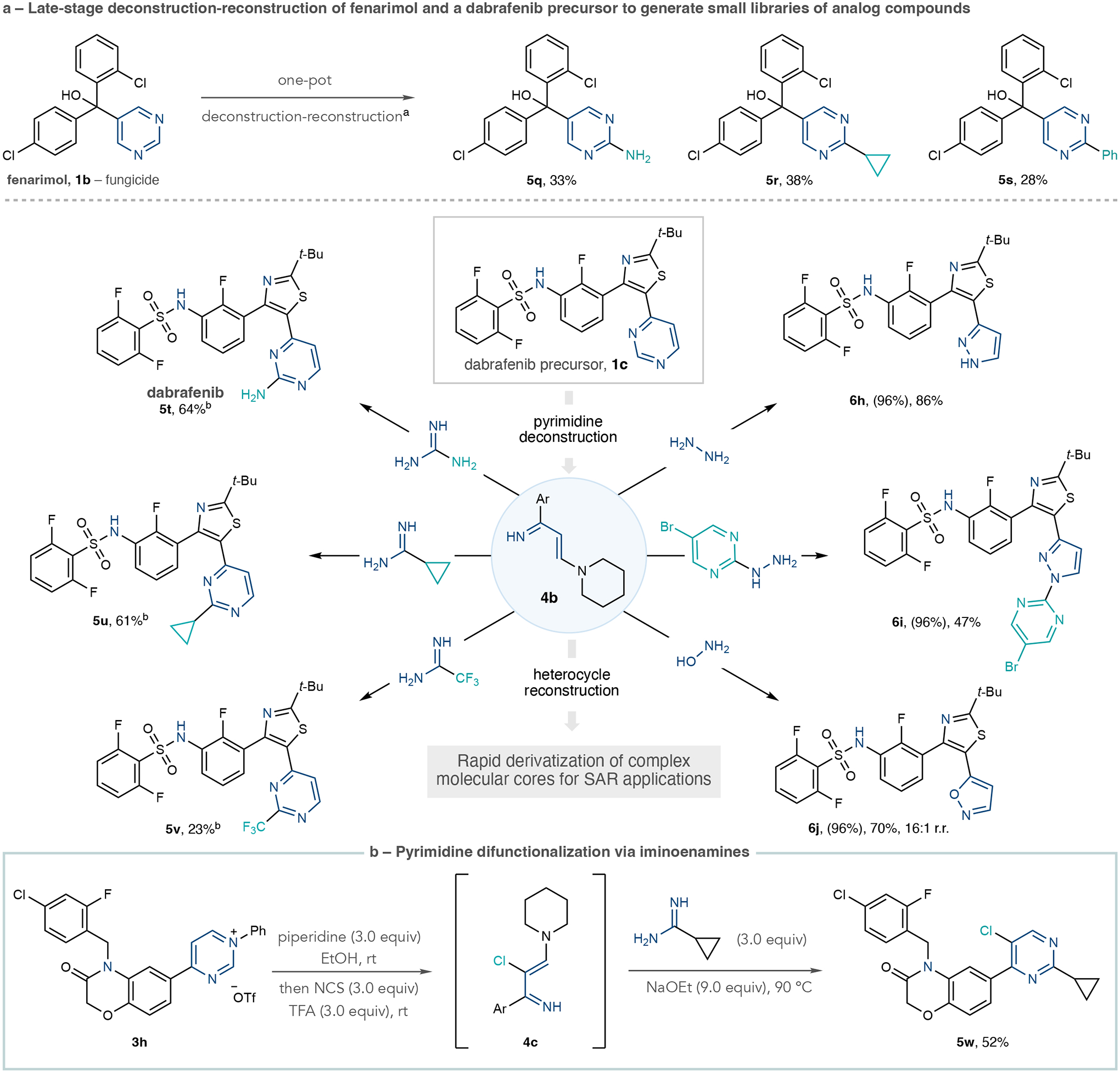
a, Transformations of fenarimol and a dabrafenib precursor. b, Pyrimidine halogenation and difunctionalization. Isolated yields are shown. Yields in parentheses are for pyrimidinium salts. aOne-pot protocol A: Tf2O (1.0 equiv.), 4-nitroaniline (1.0 equiv.), collidine (1.0 equiv.), EtOAc, −78 °C to room temperature. Solvent exchange to EtOH, then piperidine (3.0 equiv.) at room temperature, then amidine (10–20 equiv.), NaOEt (5.0 equiv.). bOne-pot protocol B: same as protocol A except using aniline (1.0 equiv.), amidine (3.0 equiv.) at 70 °C and pyrimidinium salt formation conducted in CH2Cl2. NCS, N-chlorosuccinimide; TFA, trifluoroacetic acid.
Figure 3b shows that it is possible to functionalize the pyrimidine C2 and C5 positions using iminoenamine intermediates. We cleaved pyrimidinium salt 3h using the standard protocol, then added N-chlorosuccinimide and trifluoroacetic acid directly to the reaction mixture to form chloroiminoenamine 4c. Subjecting the crude mixture to the pyrimidine reconstruction process with cyclopropaneamidine resulted in 2,5-disubstituted product 5w. The C–Cl bond can be inherently valuable in biologically active molecules and can be used as a functional group for further derivatization at a distinct position of the heterocycle (see Extended Data Fig. 1a for an extra example of pyrimidine C5-chlorination)33. Furthermore, this net C–H difunctionalization process is distinct from typical periphery modification reactions which functionalize one position and provides two points to diversify pyrimidines during SAR studies. Further investigations on pyrimidine C5-functionalization using this approach are underway.
Next, we developed processes that transforms pyrimidines into pyridines using a deconstruction–reconstruction approach (Fig. 4). After forming the pyridmidinium salt, a modified version of the C2 cleavage step using pyrrolidine as a nucleophile forms intermediate vinamidinium salts34. Previous reports have shown that vinamidinium salts react with ketone-derived enolates and then ammonium salts to form substituted pyridines35. We adapted a procedure from Marcoux using the lithium enolate of commercially available acetyltrimethylsilane followed by a mixture of ammonium acetate (NH4OAc) and acetic acid (AcOH) at 95 °C. As shown in Fig. 4a, we formed pyridines 7a–7d from pyrimidine precursors; we propose that a C6 C–Si bond cleaves after forming the pyridine under the reaction conditions. This modification ensures the formal replacement of a pyrimidine N-atom with a C-atom, which would be a useful manoeuvre in SAR studies4,36. Figure 4b shows that choosing the appropriate nucleophile can introduce substituents at the C5 or C2 positions during N to C replacement. For example, using acetyltrimethylsilane as the nucleophile when transforming 3j results in monosubstituted pyridine 7e as described above. However, using the dimethylhydrazone derived from butyraldehyde under basic conditions forms disubstituted pyridine 7f, with a C5 ethyl substituent. In this way, the reconstruction step uses the wealth of aldehydes and the β-substituent resides at the pyridine C5 position. When transforming pyrimidine 1f, we used methyl ketones in the recyclization step to selectively incorporate C2 substituents in pyridines 7g and 7h35. Extended Data Fig. 1b shows two more examples of converting pyrimidine 1f into 2-substituted pyridines and Supplementary Fig. 6 provides a guide for atom incorporation for the transformations in Fig. 4.
Fig. 4 |. Pyrimidine to pyridine conversions through deconstruction– reconstruction.
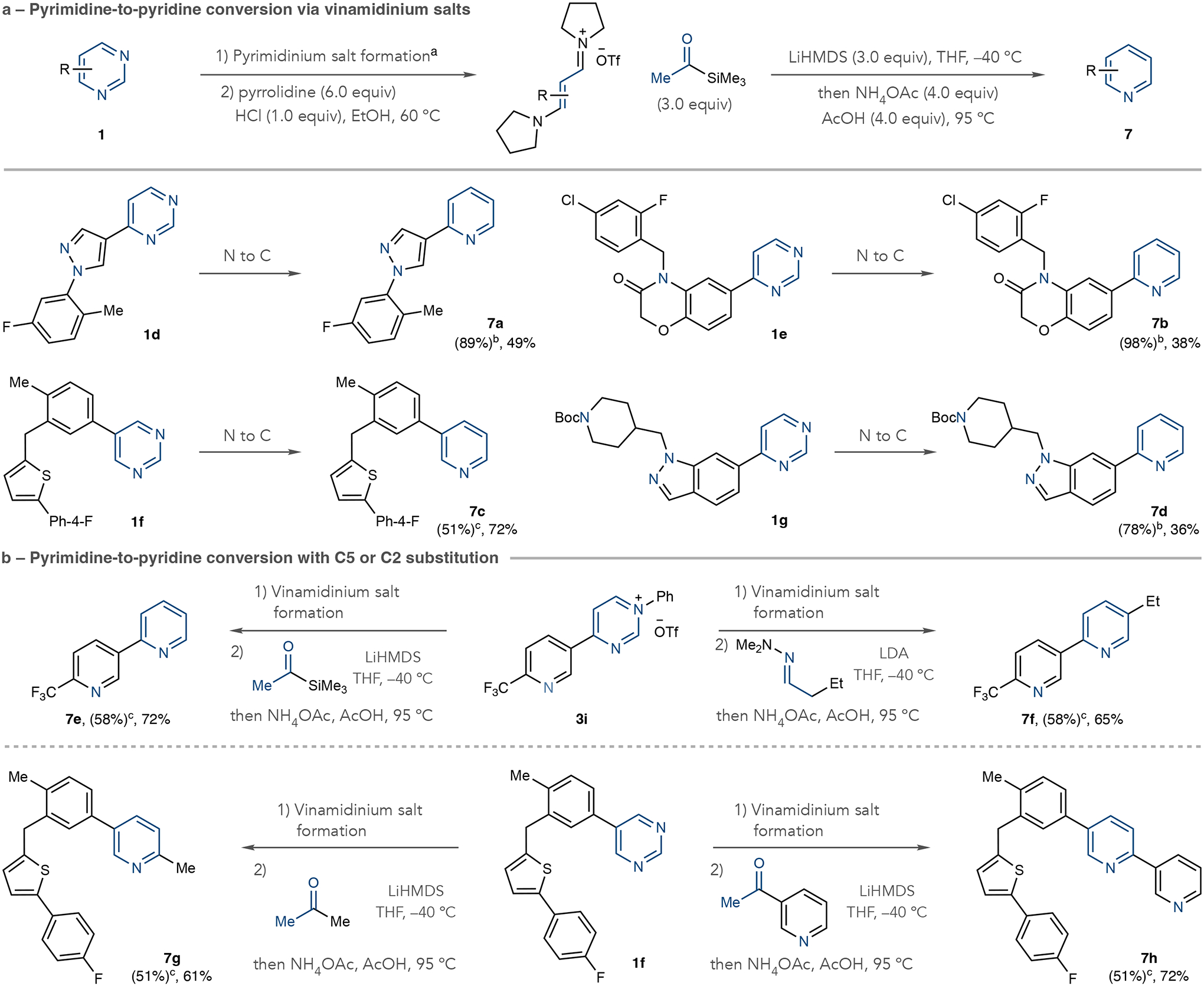
a, Transformations using acetyltrimethylsilane as a nucleophile. b, Pyridine formation using hydrazones or methyl ketones resulting in C5 or C2 substitution. Isolated yields are shown. aPyrimidinium salt formation: Tf2O (1.0 equiv.), aniline (1.0 equiv.), collidine (1.0 equiv.), EtOAc, −78 °C to room temperature. bIsolated yields of pyrimidinium salts. Crude vinamidinium salts were used in the recyclization step. cIsolated yields of vinamidinium salts either directly from pyrimidines or pyrimidinium salts. LiHMDS, lithium hexamethyldisilazide; LDA, lithium diisopropylamine; THF, tetrahydrofuran.
In the final part of this project, we studied the mechanism of N-arylpyrimidinium salt formation computationally by performing quantum chemical calculations at the ωB97X-D/def2-TZVP//ωB97X-D/6–31+G(d,p) level of theory, with a solvent model based on density (SMD) description of ethyl acetate. Figure 5a shows the computed potential energy surface for the reaction of 1a with Tf2O and aniline. The reaction between 1a and Tf2O is exergonic (−4.3 kcal mol−1) and proceeds in a single step through TS-Act (Gibbs free energy change between the reactants and the transition state, ΔG‡ = 12.6 kcal mol−1). Aniline then adds to the resulting NTf-pyrimidinium with nucleophilic attack at C6 (ΔG‡ 15.1 kcal mol−1 through TS-Add-C6) preferred in place of C2 (ΔG‡ 15.9 kcal mol−1 through TS-Add-C2), which corresponds to a kinetic selectivity of 8:1 at −78 °C. These transition structure (TS) energies reflect the electronic Fukui (ƒ +) indices that indicate greater electrophilicity at C6 versus C2 (0.15 versus 0.10). Although the C6-adduct formed in endergonic by 8.9 kcal mol−1, deprotonation of the ammonium species by collidine occurs in an energetically barrierless process to produce a stable intermediate, Int-I (Gibbs free energy change, ΔG = −20.7 kcal mol−1) and indicates that the base drives this process forward.
Fig. 5 |. Computational studies of pyrimidine ring-opening.
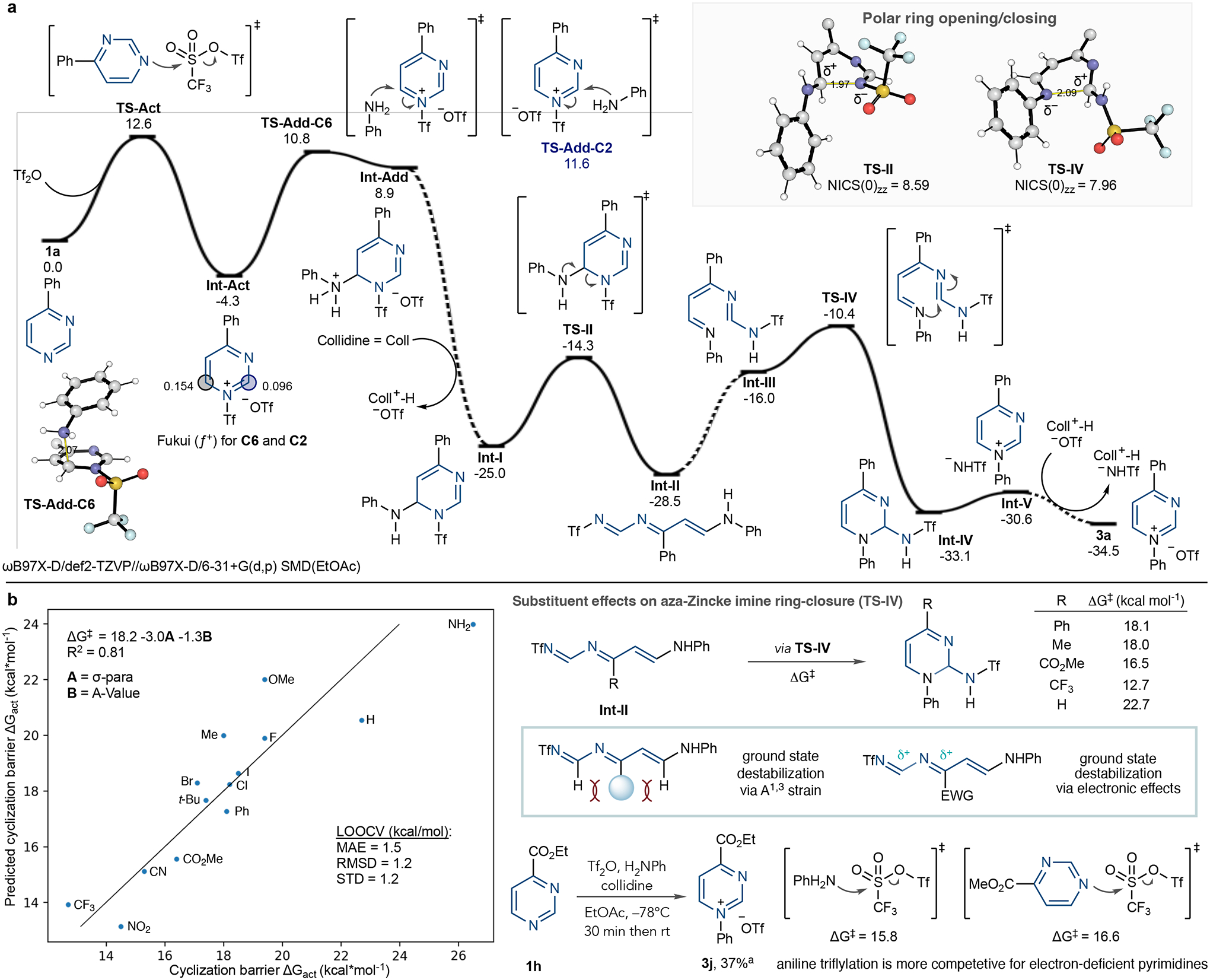
a, Quantum chemical computed reaction mechanism between pyrimidine 3a, Tf2O and aniline; relative Gibbs energies (195.15 K, 1 mol l−1) in kcal mol−1. b, Computational study of the steric and electronic effect of substituents on the cyclization of Int-II and NTf-pyrimidinium salt formation. a1H NMR yield reported. LOOCV, leave-one-out cross-validation; MAE, mean absolute error; r.m.s.d., root mean square deviation; s.d., standard deviation.
Next, Int-I undergoes facile ring-opening (ΔG‡ 10.7 kcal mol−1 through TS-II) to form aza-Zincke imine Int-II (shown in its most stable E, Z, E configuration), which is thermodynamically favoured by 3.5 kcal mol−1. Int-II then tautomerizes and isomerizes to Int-III before ring-closing through TS-IV (ΔG‡ 18.1 kcal mol−1, relative to Int-II) to form dihydropyrimidine derivative Int-IV in exergonic fashion (ΔG −4.6 kcal mol−1). Ring closure is the largest barrier along the reaction coordinate, making TS-IV rate-limiting. Ejection of a triflamide anion yields a stable ion pair (Int-V) and subsequent anion metathesis results in the pyrimidinium triflate 3a (overall ΔG‡ −34.5 kcal mol−1).
We performed nucleus independent chemical shift calculations (NICS(0)zz, B3LYP/6–311+G(d,p) level of theory) at the ring centre of each ring-opening and ring-closing TS, to investigate whether these structures exhibit aromatic shielding patterns characteristic of pericyclic transformations37. These calculations gave positive values of 8.6 and 8.0 ppm for TS-II and TS-IV, respectively. For context, the TS for the 6π-electrocyclization of 1,3,5-hexatriene has a NICS(0)zz value of −29.5 ppm and a barrier of 33.1 kcal mol−1 at the same level of theory (Supplementary Fig. 10). Thus, because no aromaticity is found in these TSs, they describe polar processes rather than electrocyclic reactions. These polar reactions have relatively low barriers, may be considered as pseudo-pericyclic and can proceed at much lower temperatures38,39. For the minor regioisomer, corresponding to aniline adding at C2, the ring-closing barrier is raised significantly by 8.5 kcal mol−1 relative to the major regioisomeric pathway shown. Notably, the ring-closing TS for this minor pathway shows a lower NICS(0)zz value (4.5 ppm), consistent with less polar (and greater pericyclic) character (Supplementary Fig. 9).
Our computation studies indicate that the size and electronic properties of the pyrimidine substituents significantly impact the energy barriers for product formation. We described the effect of 14 different C4-substituents on the barrier for ring closure (TS-IV) using a bivariate linear regression model featuring tabulated A-values and Hammett σp constants (Fig. 5b). In the aza-Zincke imine intermediates (for example, Int-II), groups at the 4-position experience destabilizing steric interactions through allylic strain (A1,3) relative to the cyclic geometry in TS-IV. Ring-closing barriers, therefore, decrease in response to increasing ground state destabilization caused by larger 4-substituents, as exemplified by 4-phenylpyrimidine (ΔG‡ 18.1 kcal mol−1) and 4-methylpyrimidine (ΔG‡ 18.0 kcal mol−1) relative to pyrimidine (ΔG‡ 22.7 kcal mol−1). Electronic effects at the 4-position are also apparent as a result of conjugation with two imine-like carbons; electron-withdrawing groups, such as trifluoromethyl, further destabilize Int-II, lowering the cyclization barrier (ΔG‡ 12.7 kcal mol−1). Although a 4-CO2Me group has a relatively low TS-IV barrier (ΔG‡ 16.5 kcal mol−1), we observed moderate yield when transforming 1h into pyrimidinium salt 3j. We suspected that electron-deficient pyrimidines may not react efficiently with Tf2O in the first step of the salt-forming process. Indeed, we obtained similar barriers for triflyl-transfer from Tf2O to the pyrimidine 1h (ΔG‡ 16.6 kcal mol−1) as for aniline (ΔG‡ 15.8 kcal mol−1), suggesting that the unproductive formation of N-phenyltriflamide occurs competitively. Supplementary Table 3 suggests that more sterically hindered anilines may mitigate this unwanted pathway.
Extended Data
Extended Data Fig. 1 |. Additional examples of pyrimidine functionalization and pyrimidine to pyridine conversion.
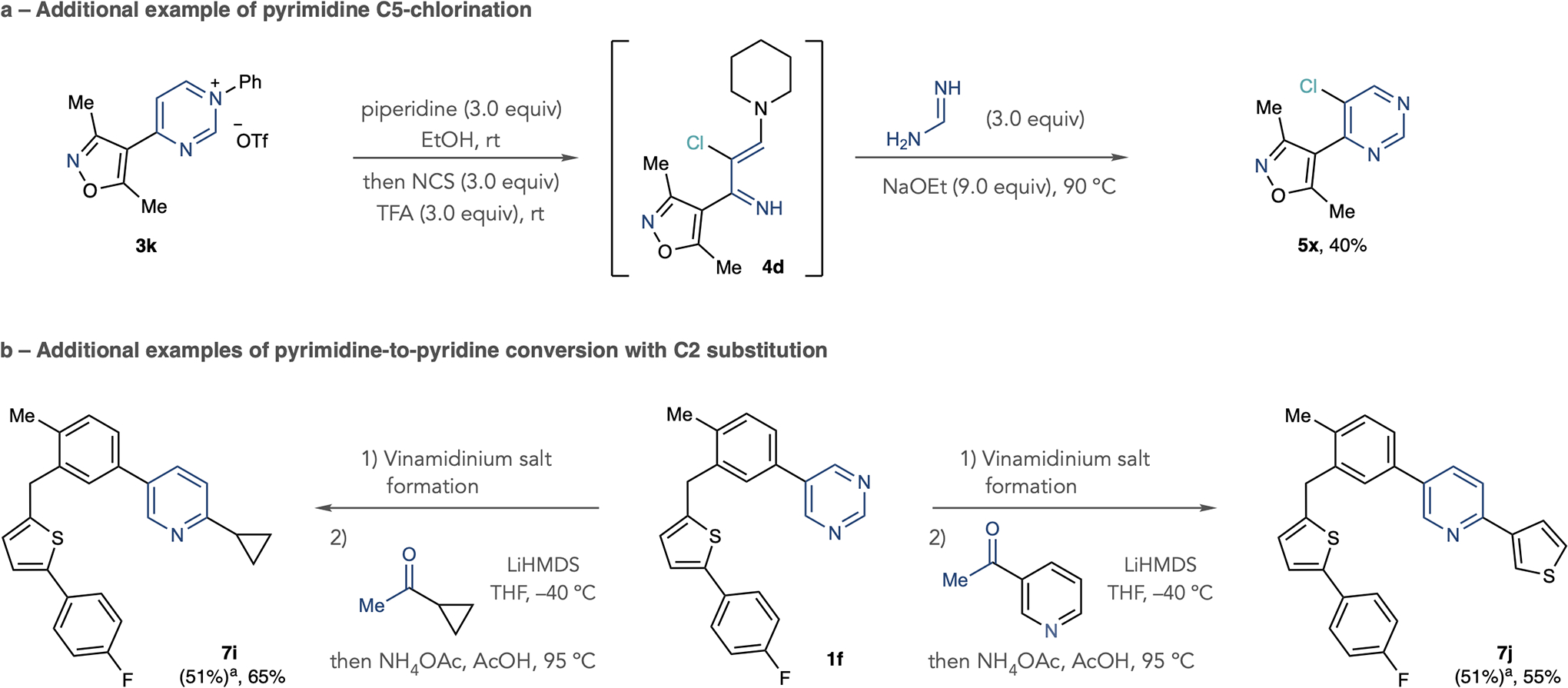
a, Additional example of pyrimidine halogenation. b, Additional examples of pyrimidine to pyridine conversion using methyl ketones. Isolated yields are shown. Vinamidinium salt formation: Tf2O (1 equiv), 4-trifluoromethylaniline (1 equiv), collidine (1 equiv), EtOAc, −78 °C to room temperature, then pyrrolidine (6 equiv), EtOH, 60 °C. aIsolated yield of vinamidinium salt from pyrimidine.
Supplementary Material
Acknowledgements
This work was supported by funds from the National Institutes of Health under award no. R01 GM144591 and from the Albert I. Meyers Foundation at Colorado State University. R.S.P. acknowledges support from the NSF (CHE-1955876) and computational resources from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) through allocation TG-CHE180056. This work also used the Alpine high performance computing resource at the University of Colorado Boulder, jointly funded by the University of Colorado Boulder, the University of Colorado Anschutz and Colorado State University.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-024-07474-1.
Competing interests A provisional patent has been filed for this work. The authors declare no other competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-024-07474-1.
Data availability
All data are available in the main text or the Supplementary Information.
References
- 1.Cernak T, Dykstra KD, Tyagarajan S, Vachal P & Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 45, 546–576 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Zhang L & Ritter T A perspective on late-stage aromatic C–H bond functionalization. J. Am. Chem. Soc 144, 2399–2414 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Josephitis CM, Nguyen HMH & McNally A Late-stage C–H functionalization of azines. Chem. Rev 123, 7655–7691 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jurczyk J et al. Single-atom logic for heterocycle editing. Nat. Synth 1, 352–364 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joule JA & Mills K Heterocyclic Chemistry 5th edn (Wiley, 2009). [Google Scholar]
- 6.Baumann M & Baxendale IR An overview of the synthetic routes to the best selling drugs containing 6-membered heterocycles. Beilstein J. Org. Chem 9, 2265–2319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitaku E, Smith DT & Njardarson JT Analysis of the structural diversity, substitution patterns and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 57, 10257–10274 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Bhutani P et al. U.S. FDA approved drugs from 2015–June 2020: a perspective. J. Med. Chem 64, 2339–2381 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Nadar S & Khan T Pyrimidine: an elite heterocyclic leitmotif in drug discovery-synthesis and biological activity. Chem. Biol. Drug Des 100, 818–842 (2022). [DOI] [PubMed] [Google Scholar]
- 10.Blakemore DC et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem 10, 383–394 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Li JJ & Corey EJ Name Reactions in Heterocyclic Chemistry II (Wiley, 2011). [Google Scholar]
- 12.Liu YF et al. A facile total synthesis of imatinib base and its analogues. Org. Process Res. Dev 12, 490–495 (2008). [Google Scholar]
- 13.CAS SciFindern database search for commercial fragments: amidines 178,649, hydrazines 830,858 (CAS SCIFINDER, accessed November 2023); https://scifinder-n.cas.org. [Google Scholar]
- 14.Boyle BT, Levy JN, de Lescure L, Paton RS & McNally A Halogenation of the 3-position of pyridines through Zincke imine intermediates. Science 378, 773–779 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Selingo JD et al. A general strategy for N-(hetero)arylpiperidine synthesis using Zincke imine intermediates. J. Am. Chem. Soc 146, 936–945 (2023). [DOI] [PubMed] [Google Scholar]
- 16.Bartholomew GL et al. 14N to 15N isotopic exchange of nitrogen heteroaromatics through skeletal editing. J. Am. Chem. Soc 146, 2950–2958 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barczynski P & Vanderplas HC Ring transformations in reactions of heterocyclic-compounds with nucleophiles. Conversion of 5-nitropyrimidine into 2-substituted 5-nitropyrimidine and 2-amino-5-nitropyridines by amidines. J. Org. Chem 47, 1077–1080 (1982). [Google Scholar]
- 18.Hu Y, Stumpfe D & Bajorath J Recent advances in scaffold hopping. J. Med. Chem 60, 1238–1246 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Lamberth C Agrochemical lead optimization by scaffold hopping. Pest Manag. Sci 74, 282–292 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Woo J et al. Scaffold hopping by net photochemical carbon deletion of azaarenes. Science 376, 527–532 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dherange BD, Kelly PQ, Liles JP, Sigman MS & Levin MD Carbon atom insertion into pyrroles and indoles promoted by chlorodiazirines. J. Am. Chem. Soc 143, 11337–11344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reisenbauer JC, Green O, Franchino A, Finkelstein P & Morandi B Late-stage diversification of indole skeletons through nitrogen atom insertion. Science 377, 1104–1109 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Patel SC & Burns NZ Conversion of aryl azides to aminopyridines. J. Am. Chem. Soc 144, 17797–17802 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Bartholomew GL, Carpaneto F & Sarpong R Skeletal editing of pyrimidines to pyrazoles by formal carbon deletion. J. Am. Chem. Soc 144, 22309–22315 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishiwaki N, Ogihara T, Takami T, Tamura M & Ariga M New synthetic equivalent of nitromalonaldehyde treatable in organic media. J. Org. Chem 69, 8382–8386 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Terrier F Modern Nucleophilic Aromatic Substitution (Wiley-VCH, 2013). [Google Scholar]
- 27.Crawley ML & Trost BM Applications of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective (John Wiley & Sons, 2012). [Google Scholar]
- 28.Verbitskiy EV, Rusinov GL, Chupakhin ON & Charushin VN Recent advances in direct C–H functionalization of pyrimidines. Synthesis 50, 193–210 (2018). [Google Scholar]
- 29.Ham WS, Choi H, Zhang J, Kim D & Chang S C2-selective, functional-group-divergent amination of pyrimidines by enthalpy-controlled nucleophilic functionalization. J. Am. Chem. Soc 144, 2885–2892 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Groll K et al. Regioselective metalations of pyrimidines and pyrazines by using frustrated Lewis pairs of BF3.OEt2 and hindered magnesium- and zinc-amide bases. Angew. Chem. Int. Ed 52, 6776–6780 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Dolewski RD, Fricke PJ & McNally A Site-selective switching strategies to functionalize polyazines. J. Am. Chem. Soc 140, 8020–8026 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rheault TR et al. Discovery of dabrafenib: a selective inhibitor of Raf kinases with antitumor activity against B-Raf-driven tumors. ACS Med. Chem. Lett 4, 358–362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiodi D & Ishihara Y “Magic chloro”: profound effects of the chlorine atom in drug discovery. J. Med. Chem 66, 5305–5331 (2023). [DOI] [PubMed] [Google Scholar]
- 34.Mao Y, Tian SX, Zhang W & Xu GY Preparation and application of vinamidinium salts in organic synthesis. Chin. J. Org. Chem 36, 700–710 (2016). [Google Scholar]
- 35.Marcoux JF et al. A general preparation of pyridines and pyridones via the annulation of ketones and esters. J. Org. Chem 66, 4194–4199 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Pearson TJ et al. Aromatic nitrogen scanning by ipso-selective nitrene internalization. Science 381, 1474–1479 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Wannere CS, Corminboeuf C, Puchta R & Schleyer P Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem. Rev 105, 3842–3888 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Zhou C & Birney DM A density functional theory study clarifying the reactions of conjugated ketenes with formaldimine. A plethora of pericyclic and pseudopericyclic pathways. J. Am. Chem. Soc 124, 5231–5241 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Kukier GA, Turlik A, Xue XS & Houk KN Violations. How nature circumvents the Woodward–Hoffmann rules and promotes the forbidden conrotatory 4n + 2 electron electrocyclization of Prinzbach’s vinylogous sesquifulvalene. J. Am. Chem. Soc 143, 21694–21704 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the Supplementary Information.