

Abstract
Under steady state, hematopoietic stem cells (HSCs) remain largely quiescent and are believed to be predominantly reliant on glycolysis to meet their energetic needs. However, under stress conditions such as infection or blood loss, HSCs become proliferative and rapidly produce downstream progenitor cells, which in turn further differentiate, ultimately producing mature blood cells. During this transition and differentiation process, HSCs exit from quiescence and rapidly undergo a metabolic switch from glycolysis to oxidative phosphorylation (OxPHOS). Various stress conditions, such as aging, cancer, diabetes, and obesity, can negatively impact mitochondrial function and thus can alter the metabolic reprogramming and differentiation of HSCs and progenitors during hematopoiesis. Valuable insights into glycolytic and mitochondrial functions of HSCs and progenitors under normal and stress conditions can be gained through the assessment of their extracellular acidification rate (ECAR) and oxygen consumption rate (OCR), which are indicators of cellular glycolysis and mitochondrial respiration, respectively.
Here, a detailed protocol is provided to measure ECAR and OCR in mouse bone marrow-derived lineage-negative cell populations, which include both hematopoietic stem and primitive progenitor cells (HSPCs), using the extracellular flux analyzer. This protocol describes approaches to isolate lineage-negative cells from mouse bone marrow, explains optimization of cell seeding density and concentrations of 2-deoxy-D-glucose (2-DG, a glucose analog that inhibits glycolysis) and various OxPHOS-targeted drugs (oligomycin, FCCP, rotenone, and antimycin A) used in these assays, and describes drug treatment strategies. Key parameters of glycolytic flux, such as glycolysis, glycolytic capacity, and glycolytic reserve, and OxPHOS parameters, such as basal respiration, maximal respiration, proton leak, ATP production, spare respiratory capacity, and coupling efficiency, can be measured in these assays. This protocol allows ECAR and OCR measurements on non-adherent HSPCs and can be generalized to optimize analysis conditions for any type of suspension cells.
Keywords: hematopoietic stem cells, hematopoietic progenitor cells, glycolysis, mitochondrial respiration, oxidative phosphorylation, extracellular acidification rate, oxygen consumption rate, extracellular flux, non-adherent cells
SUMMARY:
The method presented here summarizes optimized protocols for assessing cellular bioenergetics in non-adherent mouse hematopoietic stem and primitive progenitor cells (HSPCs) using the extracellular flux analyzer to measure the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) of HSPCs in real time.
INTRODUCTION:
Hematopoiesis is the process by which various types of mature blood cells with highly specialized functions are formed from HSCs1. HSCs are capable of self-renewal and differentiation into various multipotent and lineage-specific progenitor populations. These progenitors ultimately produce cells of lymphoid, myeloid, erythroid, and megakaryocyte lineages. To maintain their self-renewal capacity, HSCs remain largely quiescent and, like other tissue stem cells, are believed to rely on glycolysis rather than mitochondrial OxPHOS for ATP production2,3. Entry into the cell cycle leads to enhanced respiration and OxPHOS, resulting in elevated levels of reactive oxygen species (ROS), which are detrimental to HSC maintenance and function3. Repeated cell division thus may lead to reduced self-renewal capacity of HSCs and ultimately to their exhaustion.
In adult hematopoiesis, HSCs primarily undergo asymmetric cell division, during which one of the daughter cells retains HSC potential and continues to rely on glycolytic metabolism. The other daughter cell becomes a primitive progenitor cell that loses self-renewal capacity but proliferates and eventually gives rise to differentiated functional hematopoietic cells4. When HSCs differentiate to produce downstream progenitors, a switch from glycolysis to mitochondrial metabolism is thought to occur to supply the energy and building blocks needed to support this rapid transition5, as suggested by the observations that HSCs possess inactive mitochondrial mass6–9. In contrast, mitochondrial activity (indicated by linked ROS levels) is much higher in lineage-committed progenitors than in HSCs9–11. Metabolic changes that occur during the earliest step of hematopoiesis thus suggest a direct and crucial role of mitochondria in regulating HSC fate.
Dysfunctional mitochondria present under various stress conditions, such as aging, cancer, diabetes, and obesity12, can interfere with HSC self-renewal capacity, inducing an imbalance in HSC/progenitor differentiation by producing excessive amounts of ROS, impairing ATP production, and/or by altering other metabolic processes9,12,13. Perturbations in metabolic homeostasis in HSC/progenitor differentiation could significantly impact hematopoiesis, potentially contributing to the development of hematologic abnormalities13. Given the critical influences of glycolysis and mitochondrial OxPHOS on HSC stemness and differentiation, it is of interest to investigate both metabolic parameters under normal and stress conditions. Valuable insights into the glycolytic and mitochondrial function of HSCs and progenitor cells can be gained by assessing their ECAR and OCR, which are indicators of cellular glycolysis and mitochondrial respiration, respectively.
The Seahorse extracellular flux analyzer is a powerful apparatus equipped with two probes per well to simultaneously measure ECAR and OCR in live cells and thus can be used to assess cellular bioenergetics in real time, in response to various substrates or inhibitors. The assay cartridge used with the analyzer contains injection ports to hold up to four drugs for automated injection during the assay. A scheme of a typical glycolysis stress test is shown in Figure 1A. The assay starts with the measurement of ECAR of cells, incubated in glycolysis stress test medium containing glutamine but not glucose or pyruvate. This represents acidification occurring due to non-glycolytic activities of the cells and is reported as non-glycolytic acidification. This is followed by the injection of glucose at a saturating concentration. Via glycolysis, glucose in the cell is converted into pyruvate, which is further metabolized in the cytoplasm to produce lactate, or in mitochondria to produce CO2 and water.
Figure 1: Schematic representation of assessment of glycolytic function and mitochondrial respiration using the extracellular flux analyzer.
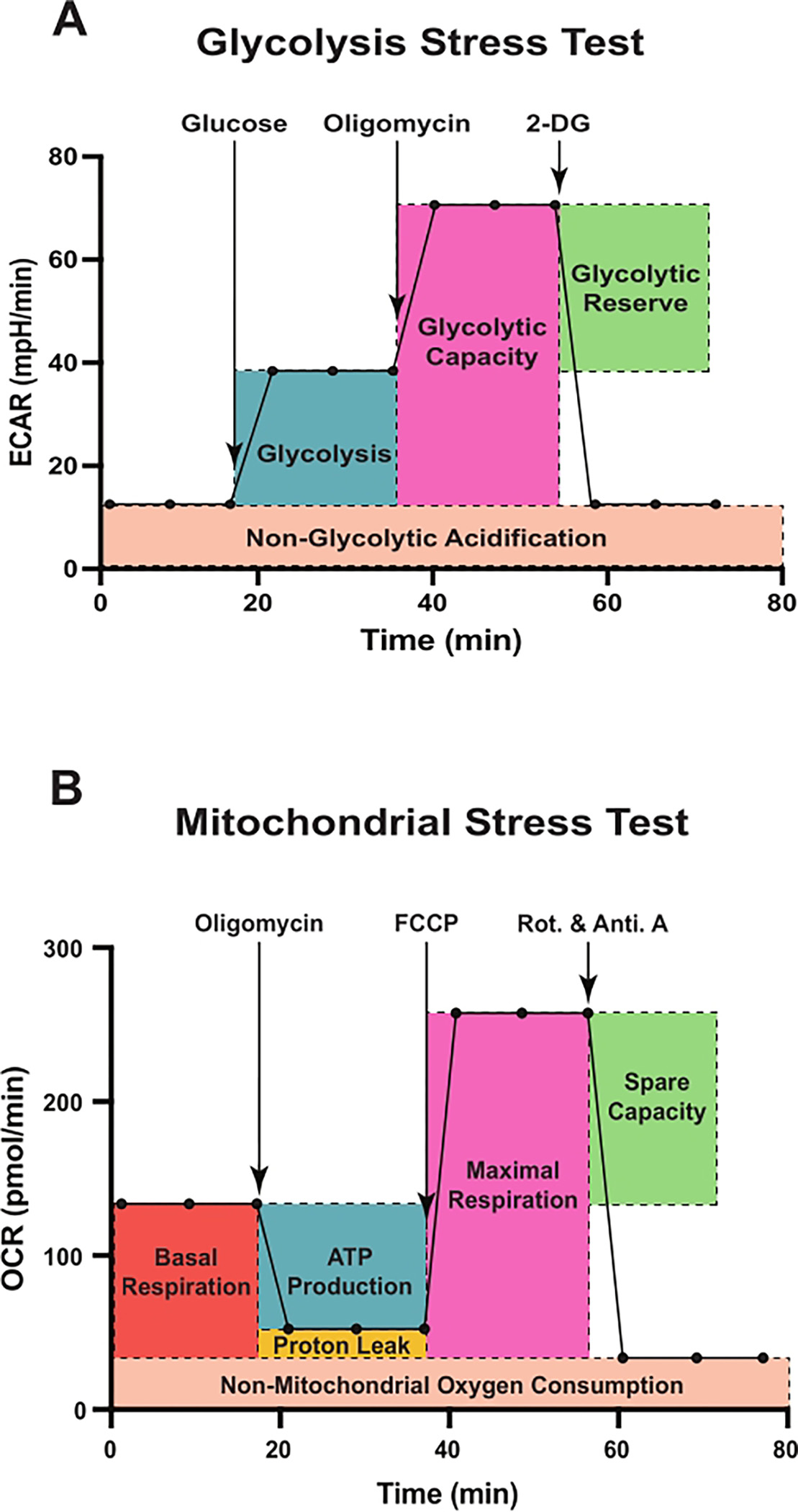
Sequences and injection timings of various effector compounds used in the glycolytic stress test (A) and the mitochondrial stress test (B) are shown. (A) Glycolytic parameters, glycolysis, glycolytic capacity, glycolytic reserve, and non-glycolytic acidification, and (B) mitochondrial function parameters, basal respiration, ATP-linked respiration, maximal respiration, non-mitochondrial oxygen consumption, proton leak, and spare respiratory capacity are outlined. Abbreviations: ECAR = extracellular acidification rate; 2-DG = 2-deoxy-D-glucose; OCR = oxygen consumption rate; FCCP = carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone; Rot. = rotenone; Anti. A = antimycin A.
Conversion of glucose to lactate causes net production and subsequent release of protons into the extracellular medium, resulting in a rapid increase in the ECAR14–16. This glucose-stimulated change in ECAR is reported as glycolysis under basal conditions. The second injection consists of oligomycin (an inhibitor of ATP synthase, a.k.a. complex V17), which inhibits mitochondrial ATP production. During oligomycin-mediated OxPHOS inhibition, cells maximally upregulate glycolysis to meet their energetic demands. This causes a further increase in ECAR, revealing the maximum glycolytic capacity of the cells. The difference between the maximum glycolytic capacity and basal glycolysis is referred to as glycolytic reserve. Finally, 2-DG is injected, which causes a significant drop in ECAR, usually close to non-glycolytic acidification levels. 2-DG is a glucose analog that competitively binds to hexokinase, resulting in inhibition of glycolysis18. Thus, the 2-DG-induced decrease in ECAR further confirms that glycolysis is indeed the source of ECAR observed after glucose and oligomycin injections.
Figure 1B displays the schematic for a typical mitochondrial stress test. The assay starts with baseline OCR measurement of the cells, incubated in mitochondrial stress test medium containing glucose, glutamine, and pyruvate. Following basal OCR measurements, oligomycin is injected in this assay, which inhibits complex V, thereby reducing electron flow through the electron transport chain (ETC)17. Consequently, OCR is reduced in response to oligomycin injection, and this decrease in OCR is linked to mitochondrial ATP production. The second injection consists of carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP), a protonophore and an uncoupler of mitochondrial OxPHOS17. FCCP collapses the mitochondrial proton gradient by allowing the flow of protons across the mitochondrial inner membrane. Because of the FCCP injection, electron flow through the ETC is derepressed, and complex IV consumes oxygen at the maximal level. The difference between maximal OCR and basal OCR is referred to as the spare respiratory capacity, which is a measure of the cell’s ability to respond to increased energy demand under stress conditions. Finally, a mixture of two ETC inhibitors (rotenone, a complex I inhibitor, and antimycin A, a complex III inhibitor17) is injected, which completely shuts down electron flow, and OCR decreases to a low level. OCR measured after rotenone and antimycin A injection corresponds to non-mitochondrial OCR driven by other processes inside the cells. Non-mitochondrial OCR enables the calculation of basal respiration, proton leak, and maximal respiration.
Basal respiration represents the difference between baseline OCR and non-mitochondrial OCR. Proton leak refers to the difference between OCR after oligomycin injection and non-mitochondrial OCR. Maximal respiration represents the difference between OCR after FCCP injection and non-mitochondrial OCR. Coupling efficiency is calculated as the percentage of ATP production rate to basal respiration rate. This method paper provides a detailed protocol to measure ECAR and OCR in lineage-negative HSPCs using the Seahorse XFe96 extracellular flux analyzer. This protocol describes approaches to isolate mouse lineage-negative HSPCs, explains the optimization of cell-seeding density and concentrations of various drugs used in extracellular flux assays, and describes drug treatment strategies.
PROTOCOL:
All vertebrate animal experiments were approved by and performed in accordance with the regulations of the University of Michigan Committee on Use and Care of Animals.
1. Day before the assay (Total time: ~10 min)
1.1. Hydration of the sensor cartridge (Step time: ~10 min)
1.1.1. Open the extracellular flux assay kit and remove the sensor cartridge and utility plate assembly. Save loading guide flats for use the next day.
1.1.2. Manually separate the sensor cartridge (the top green part with lid) from the utility plate (lower 96-well microplate) and place it upside-down next to the utility plate.
1.1.3. Using a multichannel pipette, fill each well of the utility plate with 200 μL of calibrant, included with flux assay kit.
1.1.4. Place the sensor cartridge back onto the utility plate, making sure to completely submerge the sensors in the calibrant.
1.1.5. Place the assembled sensor cartridge and utility plate with calibrant in a non-CO2 37 °C incubator overnight. To prevent evaporation of the calibrant, ensure that the incubator is properly humidified. If using a regular oven incubator for non-CO2 37 °C incubations, place an open beaker containing water inside the incubator to humidify it. If available, use an XF prep station for all non-CO2 37 °C incubations.
NOTE: Alternatively, the sensor cartridge can be hydrated overnight with 200 μL per well of sterile ultrapure water in a non-CO2 37 °C incubator. Water should be replaced with 200 μL per well of 37 °C prewarmed calibrant, at least 45–60 min before loading the injection ports on the day of assay.
2. Day of the assay (Total time: ~9 h 30 min)
NOTE: The total time indicated above includes cumulative durations of steps 2.1–2.4 in addition to either step 2.5 or step 2.6.
2.1. Preparation of cell-adhesive-coated microplates (Step time: ~1 h 30 min)
NOTE: Perform all steps in a biosafety cabinet.
2.1.1. Prepare 2.5 mL of the cell adhesive solution (22.4 μg/mL, see the Table of Materials) per 96-well cell culture microplate by dissolving 56 μg of the cell adhesive in an appropriate volume of filter-sterilized 0.1 M sodium bicarbonate (pH 8.0) and immediately adding 1 N sodium hydroxide at half the volume of cell adhesive stock used. Vortex or pipette up and down to mix.
2.1.2. Open the 96-well cell culture microplate (included with the flux assay kit) and dispense 25 μL of the prepared cell adhesive solution at the bottom of each well. Cover the microplate with the lid and incubate for 30 min at room temperature inside the hood.
2.1.3. After incubation, remove and discard excess cell adhesive solution using a multichannel pipette or aspirator, and wash each well twice with 200 μL of sterile ultrapure water. Air-dry the plate, with the lid removed, inside the hood for 30–45 min.
NOTE: Cell-adhesive-coated cell culture microplates may be stored for up to one week at 4 °C. Precoated microplates stored at 4 °C must be allowed to warm to room temperature inside the hood before seeding cells.
2.2. Preparation of assay media (Step time: ~30 min)
- 2.2.1. Preparation of glycolysis stress test assay medium
-
2.2.1.1. Supplement 100 mL of base medium (see the Table of Materials) with 1 mL of 200 mM L-glutamine.NOTE: The final L-glutamine concentration in the assay medium is 2 mM.
- 2.2.1.2. Warm the L-glutamine-supplemented medium to 37 °C in a water bath.
- 2.2.1.3. Adjust the pH of the warm medium to 7.4 with 1 N NaOH.
- 2.2.1.4. Filter-sterilize the medium with a 0.2 μm filter and keep it at 37 °C until ready to use.
-
- 2.2.2. Preparation of mitochondrial stress test assay medium
-
2.2.2.1. Supplement 100 mL of the base medium with 0.45 g of glucose, 1 mL of 200 mM L-glutamine, and 1 mL of 100 mM sodium pyruvate.NOTE: The final assay medium contains 25 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate.
- 2.2.2.2. Warm glucose, L-glutamine, and the sodium pyruvate-supplemented medium to 37 °C in a water bath.
- 2.2.2.3. Adjust the pH of the warm medium to 7.4 with 1 N NaOH.
- 2.2.2.4. Filter-sterilize medium with a 0.2 μm filter and keep at 37 °C until ready to use.
-
2.3. Harvest mouse lineage-negative HSPCs (Step time: ~3 h)
2.3.1. Prepare the assay buffer by supplementing Hank’s balanced salt solution with 4% fetal bovine serum. Prepare 1x enrichment buffer by diluting the 5x stock with sterile distilled water. Keep both buffers on ice until use.
2.3.2. Humanly euthanize mice using CO2 followed by cervical dislocation and remove the hindlimbs with scissors. Carefully remove all tissues from the bones, and cut the ends from both sides of the femur and tibia using scissors. Flush out bone marrow cells from the femur and tibia using the assay buffer. Pass the flushed cells through a sterile 70 μm filter, centrifuge the filtrate at 200 × g for 5 min at 4 °C, and discard the supernatant.
2.3.3. Lyse the red blood cells by resuspending the cell pellet in 500 μL of ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.2). Following lysis for 1 min on ice, wash the samples by adding 1 mL of assay buffer and centrifuge at 200 × g for 5 min at 4 °C. Discard the supernatant.
2.3.4. After washing, incubate the samples with a cocktail of biotinylated antibodies directed against CD2 (1:200 dilution), CD3 (1:200 dilution), CD5 (1:200 dilution), CD8 (1:200 dilution), Ter-119 (1:200 dilution), B220 (1:200 dilution), and Gr-1 (1:800 dilution) for 45 min on ice in a total volume of 500 μL of assay buffer.
2.3.5. Wash the samples with 1 mL of assay buffer with centrifugation as above.
2.3.6. Wash the samples with 1 mL of 1x enrichment buffer with centrifugation as above.
2.3.7. Incubate the samples with 20 μL of streptavidin nanobeads and 100 μL of 1x enrichment buffer on ice for 15 min.
2.3.8. Wash the samples with 1 mL of 1x enrichment buffer with centrifugation as above.
2.3.9. Resuspend the samples in 2.5 mL of 1x enrichment buffer. Place them on a separation magnet for 5 min. Collect the supernatants separately in 15 mL conical tubes.
-
2.3.10. Resuspend magnet-separated samples in an additional 2.5 mL of 1x enrichment buffer, and place them again on the separation magnet for 5 min. Collect and combine supernatants to the prior collections in 15 mL conical tubes from step 2.3.9.
NOTE: The supernatants contain the lineage-negative cell fractions.
2.3.11. Centrifuge the 15 mL conical tubes containing the negatively selected cells for 5 min at 200 × g at 4 °C.
2.3.12. Resuspend the cell pellets in 1 mL of assay buffer and count the cell number using trypan blue manually or via an automated cell counter such as a Countess 3.
NOTE: Following the above-described approach, ~6 × 106 lineage-negative HSPCs should be readily purified from the hindlimbs of a single mouse. If reagents, such as assay buffer, 1x enrichment buffer, and lineage-specific antibody cocktail, are prepared on the day prior to the experiment, it takes approximately 2.5–3 h to enrich HSPCs from 4 mice. It would take an additional 10 min for each mouse beyond this number.
2.4. Seeding cells in cell-adhesive-coated microplates (Step time: ~1 h)
2.4.1. Centrifuge cells from step 2.3.12 at 200 × g for 5 min at room temperature.
2.4.2. Remove the supernatant and resuspend the cell pellet in the appropriate assay medium (glycolysis stress test medium or mitochondrial stress test medium). Centrifuge at 200 × g for 5 min at room temperature.
2.4.3. Repeat step 2.4.2. Remove the supernatant and resuspend the cells in the appropriate warmed assay medium to the concentration of 2.5 × 105 cells per 50 μL or 5 × 106 per mL.
2.4.4. Pipette 50 μL of the cell suspension along the side of each well of the room-temperature cell-adhesive-coated 96-well cell culture microplate. Pipette 50 μL of the assay medium into the corner background measurement wells. Use a multichannel pipette for cell plating to ensure consistency.
2.4.5. Create a centrifuge balance plate by adding 50 μL of water per well of a non-coated 96-well cell culture microplate.
2.4.6. Centrifuge the cells in the plate at 200 × g for 1 min at room temperature. Do not apply brake. Transfer the plate to a non-CO2 37 °C incubator for 25–30 min to ensure the cells have completely attached. Visually confirm under a microscope that the cells are stably adhered to the microplate surface.
NOTE: As ~6 × 106 lineage-negative HSPCs can be harvested from the hindlimbs of a single mouse, HSPCs isolated from 4 mice (~2.4 × 107 cells) are needed to fill an entire 96-well plate if the aim is to screen multiple interventions ex vivo in parallel. However, if the aim is to investigate the impact of a genetic alteration or an intervention on the metabolic parameters of HSPCs in vivo, then HSPCs isolated from multiple pairs of control and test mice can be analyzed in multiple technical replicates in parallel using a single plate.
2.5. Performing glycolysis stress test using the extracellular flux analyzer (Step time: ~3 h 30 min)
- 2.5.1. Loading sensor cartridge with injecting compounds
-
2.5.1.1. Prepare 100 mM glucose, 20 μM oligomycin, and 500 mM 2-DG solutions in prewarmed glycolysis stress test assay medium (pH 7.4).NOTE: All injecting compound solutions are made at 10x concentration. The final well concentrations are 10 mM for glucose, 2 μM for oligomycin, and 50 mM for 2-DG (see Table 1).
- 2.5.1.2. Remove the hydrated sensor cartridge from the non-CO2 37 °C incubator from step 1.1.5. Remove air bubbles from the calibrant in the utility plate by lifting the sensor cartridge out of the calibrant and replacing it on to the same utility plate with the calibrant.
- 2.5.1.3. Place the A/D loading guide flat (included in the extracellular flux assay kit) on the top of the sensor cartridge, orienting it so that the letter ‘A’ is located on the upper left-hand corner to ensure that only ports A are available for loading. Using a multichannel pipette, dispense 20 μL of 100 mM glucose solution in ports A.
- 2.5.1.4. Replace the A/D loading guide flat with the B/C loading guide flat, orienting so that the letter ‘B’ is located on the upper left-hand corner to ensure that only ports B are available for loading. Using a multichannel pipette, dispense 22 μL of 20 μM oligomycin solution in ports B.
- 2.5.1.5. Re-orient the B/C loading guide flat to locate the letter ‘C’ on the upper left-hand corner for loading ports C. Using a multichannel pipette, dispense 25 μL of 500 mM 2-DG solution in ports C.
-
2.5.1.6. Remove and discard the loading guide flats.NOTE: It is important that each port series of all 96 wells, including those of the background wells, be loaded completely with the same volume of the injecting compound.
-
- 2.5.2. Template creation, calibration, and measurements
- 2.5.2.1. Create or load the template for the glycolysis stress test on the controller. Enter details regarding injection strategies, treatment conditions, and cell types, and press generate groups. Go to the plate map and assign wells to each group to be analyzed. Assign the 4 corner wells for background measurements.
- 2.5.2.2. In protocol, make sure calibration and equilibration are checked in the initialization step.
- 2.5.2.3. For baseline measurement and measurements after each injection (glucose, oligomycin, and 2-DG), set the number of measurement cycles to 3 and Mix – Wait – Measure times to 3 min – 0 min – 3 min (see Table 2).
-
2.5.2.4. Remove the lid from the compounds loaded and hydrated sensor cartridge, submerged in calibrant in the utility plate. Place it on the work tray of the extracellular flux analyzer and begin the run.NOTE: The first step is the calibration, which usually takes ~20 min.
-
2.5.2.5. Take out the microplate containing the cells from the non-CO2 37 °C incubator from step 2.4.6 after 25–30 min of incubation is over. Without disturbing the cells, slowly add 130 μL of the prewarmed glycolysis stress test assay medium (pH 7.4) per well to make up the medium volume in each well to 180 μL and return the plate to the non-CO2 37 °C incubator for additional 15–20 min.NOTE: A total incubation time of 45–60 min after centrifugation is preferred.
- 2.5.2.6. After the calibration is over, replace the utility plate with the assay microplate (without lid) containing the cells. Press continue to start the measurements, which will take ~1.5 h for completion of the assay.
- 2.5.2.7. After the measurements are over, collect the assay microplate containing the cells and remove the assay medium without disturbing the cells. Wash once gently with 250 μL of 1x phosphate-buffered saline without dislodging the cells.
- 2.5.2.8. Add 10 μL of RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 1% NP-40, 0.5 % Na deoxycholate, 0.1% sodium dodecylsulfate, 150 mM NaCl, 2 mM EDTA, 50 mM NaF), supplemented with 1x protease inhibitor cocktail, to each well and agitate the plate on a shaker for 10 min. Freeze the whole plate at −80 °C.
- 2.5.2.9. Thaw the plate, and perform a protein measurement assay for data normalization following the manufacturer’s instructions.
- 2.5.2.10. Retrieve and analyze the data. Open the data file using Wave Desktop Software. Click on Normalize and paste the normalization values for each well. Click on apply to normalize the data. Click on Export and select glycolysis stress test report generator to export the analyzed data to a report generator. Alternatively, export the data to an external application.
Table 1:
Injection strategy for the glycolysis stress test.
| Injection Port | Port volume | Injecting compound (10x concentrated) | Final compound concentration in the well |
|---|---|---|---|
| A | 20 μL | Glucose (100 mM) | 10 mM |
| B | 22 μL | Oligomycin (20 μM) | 2 μM |
| C | 25 μL | 2-DG (500 mM) | 50 mM |
Abbreviation: 2-DG = 2-deoxy-D-glucose.
Table 2:
Extracellular flux analyzer program for the glycolysis stress test.
| 1 | Calibration | ||||
| 2 | Equilibration | ||||
| 3 | Injections and measurements | ||||
| Cycles | Mix | Wait | Measure | ||
| Baseline (Non-glycolytic acidification) | 3 times | 3 min | 0 min | 3 min | |
| Inject Port A: Glucose | 3 times | 3 min | 0 min | 3 min | |
| Inject Port B: Oligomycin | 3 times | 3 min | 0 min | 3 min | |
| Inject Port C: 2-DG | 3 times | 3 min | 0 min | 3 min | |
Abbreviation: 2-DG = 2-deoxy-D-glucose.
NOTE: Alternatively, the total nuclear DNA content in each well can be used to normalize the data. A fluorescent dye, such as CyQuant that binds to nucleic acid, can be used to assess the total nuclear DNA content per well.
2.6. Performing a mitochondrial stress test using the extracellular flux analyzer (Step time: ~3 h 30 min)
- 2.6.1. Loading sensor cartridge with injecting compounds
-
2.6.1.1. Prepare 20 μM oligomycin, 20 μM FCCP, and 5 μM rotenone + 5 μM antimycin A solutions in prewarmed mitochondrial stress test assay medium (pH 7.4).NOTE: All injecting compound solutions are made at 10x concentration. The final well concentrations are 2 μM for oligomycin, 2 μM for FCCP, and 0.5 μM each for rotenone and antimycin A (see Table 3).
- 2.6.1.2. Remove the hydrated sensor cartridge from the 37 °C incubator from step 1.1.5 and remove air bubbles as described previously in step 2.5.1.2.
-
2.6.1.3. By following steps 2.5.1.3 to 2.5.1.6, dispense 20 μL of 20 μM oligomycin in ports A, 22 μL of 20 μM FCCP in ports B, and 25 μL of a mixture of 5 μM rotenone and 5 μM antimycin A in ports C.NOTE: It is important that each port series of all 96 wells, including those of the background wells, be loaded completely with the same volume of injecting compound.
-
- 2.6.2. Template creation, calibration, and measurements
- 2.6.2.1. Create or load the template for the mitochondrial stress test on the controller. Enter details regarding injection strategies, treatment conditions, and cell types, and press generate groups. Go to the plate map and assign wells to each group to be analyzed. Assign the 4 corner wells for background measurements.
- 2.6.2.2. In protocol, make sure calibration and equilibration are checked in the initialization step.
- 2.6.2.3. For baseline measurement and measurements after each injection (oligomycin, FCCP, and rotenone + antimycin A), set the number of measurement cycles to 3 and Mix – Wait – Measure times to 3 min – 0 min – 3 min (see Table 4).
- 2.6.2.4. Start the run to perform the calibration as in step 2.5.2.4.
- 2.6.2.5. Add 130 μL of the prewarmed mitochondrial stress test assay medium (pH 7.4) per well as in step 2.5.2.5.
- 2.6.2.6. Start the measurements; retrieve and analyze the data as in steps 2.5.2.6 to 2.5.2.10. Export the analyzed data to the mitochondrial stress test report generator or an external application.
Table 3:
Injection strategy for the mitochondrial stress test.
| Injection Port | Port volume | Injecting compound (10x concentrated) | Final compound concentration in the well |
|---|---|---|---|
| A | 20 μL | Oligomycin (20 μM) | 2 μM |
| B | 22 μL | FCCP (20 μM) | 2 μM |
| C | 25 μL | Rotenone (5 μM) + antimycin A (5 μM) | 0.5 μM each |
Abbreviation: FCCP = carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone.
Table 4:
Extracellular flux analyzer program for the mitochondrial stress test.
| 1 | Calibration | ||||
| 2 | Equilibration | ||||
| 3 | Injections and measurements | ||||
| Cycles | Mix | Wait | Measure | ||
| Baseline measurements | 3 times | 3 min | 0 min | 3 min | |
| Inject Port A: Oligomycin | 3 times | 3 min | 0 min | 3 min | |
| Inject Port B: FCCP | 3 times | 3 min | 0 min | 3 min | |
| Inject Port C: Rotenone & Antimycin A | 3 times | 3 min | 0 min | 3 min | |
Abbreviation: FCCP = carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone.
REPRESENTATIVE RESULTS:
Using this protocol, the cell number and the concentrations of various OxPHOS-targeting drugs (used in the extracellular flux assays) were optimized to measure ECAR and OCR of HSPCs isolated from 24-week-old female C57BL/6 mice. First, the glycolysis stress test was performed to optimize cell number and oligomycin concentration. A varying number of HSPCs per well ranging from 5 × 104 to 2.5 × 105 were used in this assay. As shown in Figure 2A and Figure 2C, the non-glycolytic acidification rate rises with increased cell number from 5 × 104 to 2.5 × 105 but increases only minimally between 2 × 105 to 2.5 × 105 cells. As expected, injection of 10 mM glucose stimulates glycolysis at all cell numbers, with a maximum increase in ECAR observed at 2.5 × 105 cells per well. However, injection of 2 μM of oligomycin does not further increase ECAR, as otherwise expected. The use of 1 μM oligomycin yielded similar results (not shown). These results could be interpreted to indicate that these oligomycin concentrations were not optimal in these assays.
Figure 2: Assessment of the glycolytic function in mouse HSPCs.
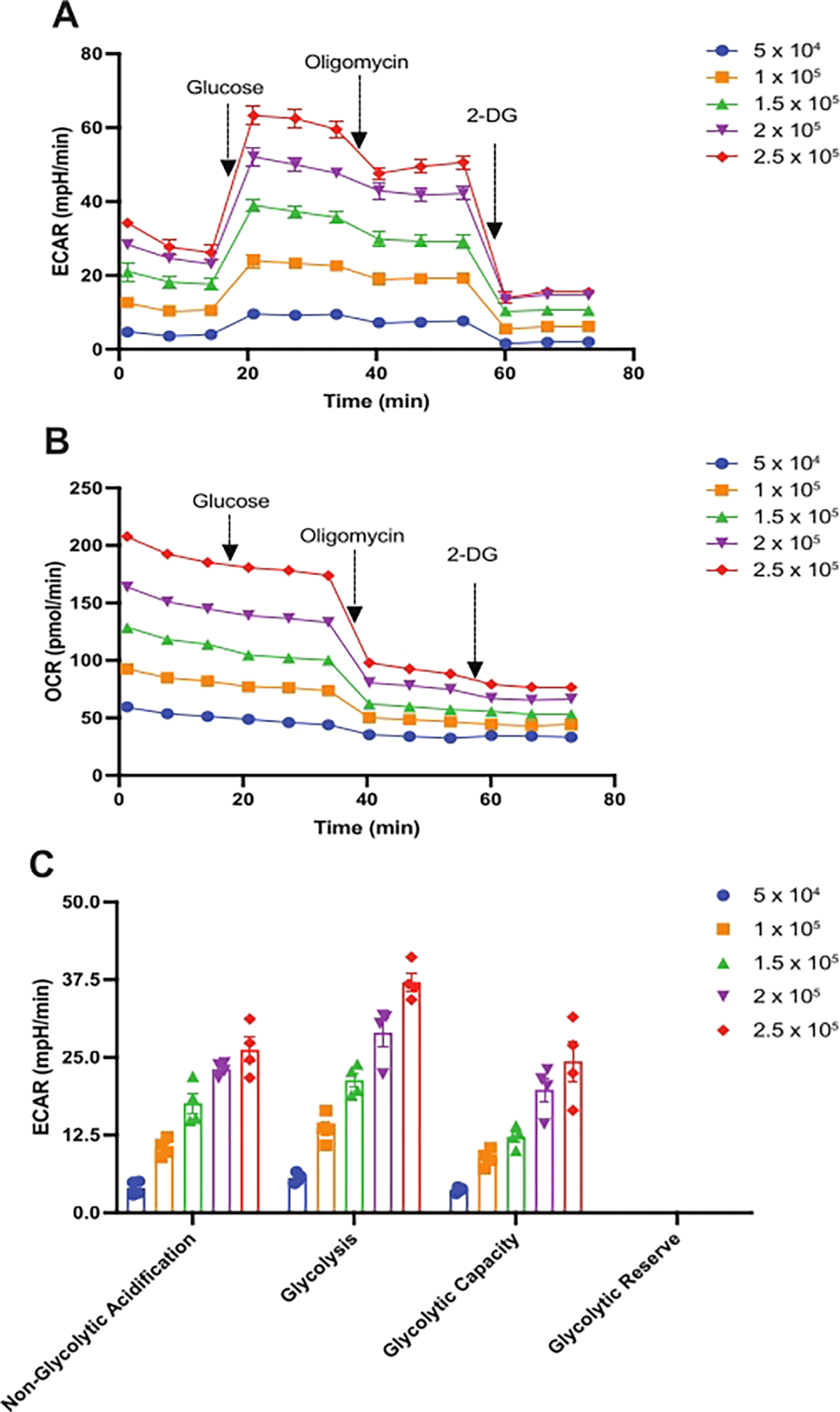
The glycolysis stress test was performed to measure (A) the extracellular acidification rates (ECAR, mpH/min) and (B) the oxygen consumption rates (OCR, pmol/min) of HSPCs isolated from 24-week-old female C57BL/6 mice. At times indicated, glucose (10 mM), oligomycin (2 μM), and 2-DG (50 mM) were injected. (C) ECAR is calculated as non-glycolytic acidification, glycolysis, glycolytic capacity, and glycolytic reserve per 5 × 104, 1 × 105, 1.5 × 105, 2 × 105, and 2.5 × 105 HSPCs/well. Data are presented as mean ± SEM, n = 4. Abbreviations: HSPCs = hematopoietic stem and primitive progenitor cells; ECAR = extracellular acidification rate; 2-DG = 2-deoxy-D-glucose; OCR = oxygen consumption rate.
However, OCR data obtained in the same set of glycolysis stress tests show that both tested oligomycin concentrations (1 μM and 2 μM) were indeed effective, as suggested by the significant drop in OCR following oligomycin injection due to complex V inhibition (see Figure 2B for 2 μM oligomycin; not shown for 1 μM oligomycin). At both tested oligomycin concentrations, maximal decreases in OCR were observed at 2.5 × 105 cells per well. Finally, injection of 50 mM 2-DG resulted in a significant reduction of ECAR, implying that glycolysis is the source of ECAR observed in these experiments. Taken together, it may be concluded that HSPCs achieve maximum glycolysis following glucose injection in these assays, and they possess little-to-no glycolytic reserve. This indicates that, like purified HSCs, lineage-negative HSPCs, which include the HSC fraction used in this assay, also rely predominantly on glycolysis for their ATP production. In cells with a high glycolytic rate, there may not be a significant increase in ATP demand upon oligomycin-mediated inhibition of mitochondrial ATP production. Cells could easily manage the loss of mitochondrial ATP without further upregulating glycolysis19. Glycolysis stress test parameters—non-glycolytic acidification, glycolysis, glycolytic capacity, and glycolytic reserve as shown in Figure 2C–were calculated as described previously in the introduction and protocol sections. Based on these data, 2.5 × 105 cells per well and 2 μM of oligomycin were selected for further studies.
The mitochondrial stress test was used for the optimization of FCCP concentration. In this assay, 2.5 × 105 HSPCs were used per well, as optimized previously via the glycolysis stress test. As shown in Figure 3A, the assay begins with the measurement of baseline OCR followed by 2 μM oligomycin injection, causing a significant reduction in OCR via the inhibition of complex V. Following post-oligomycin injection OCR measurements, FCCP was injected at varying concentrations: 0.5 μM, 1 μM, 1.5 μM, and 2 μM. As indicated earlier, FCCP reverses the oligomycin-induced repression of electron flow through ETC by uncoupling proton transport from OxPHOS and forces complex IV to consume oxygen maximally. As shown in Figure 3A, FCCP stimulates OCR in HSPCs in a dose-dependent manner with a maximal increase in OCR observed at 2 μM of FCCP. Finally, a mixture of 0.5 μM rotenone and 0.5 μM antimycin A was injected, which completely shuts down electron flow through the ETC, and OCR decreases to its minimal level. OCR measured after rotenone and antimycin A injection corresponds to non-mitochondrial oxygen consumption. Other mitochondrial stress test parameters—basal respiration, maximal respiration, proton leak, ATP production, spare respiratory capacity, and coupling efficiency as shown in Figure 3B—were calculated as described previously in the introduction and protocol sections. Finally, 2 μM of FCCP was selected for further studies.
Figure 3: Assessment of the mitochondrial respiration in mouse HSPCs.
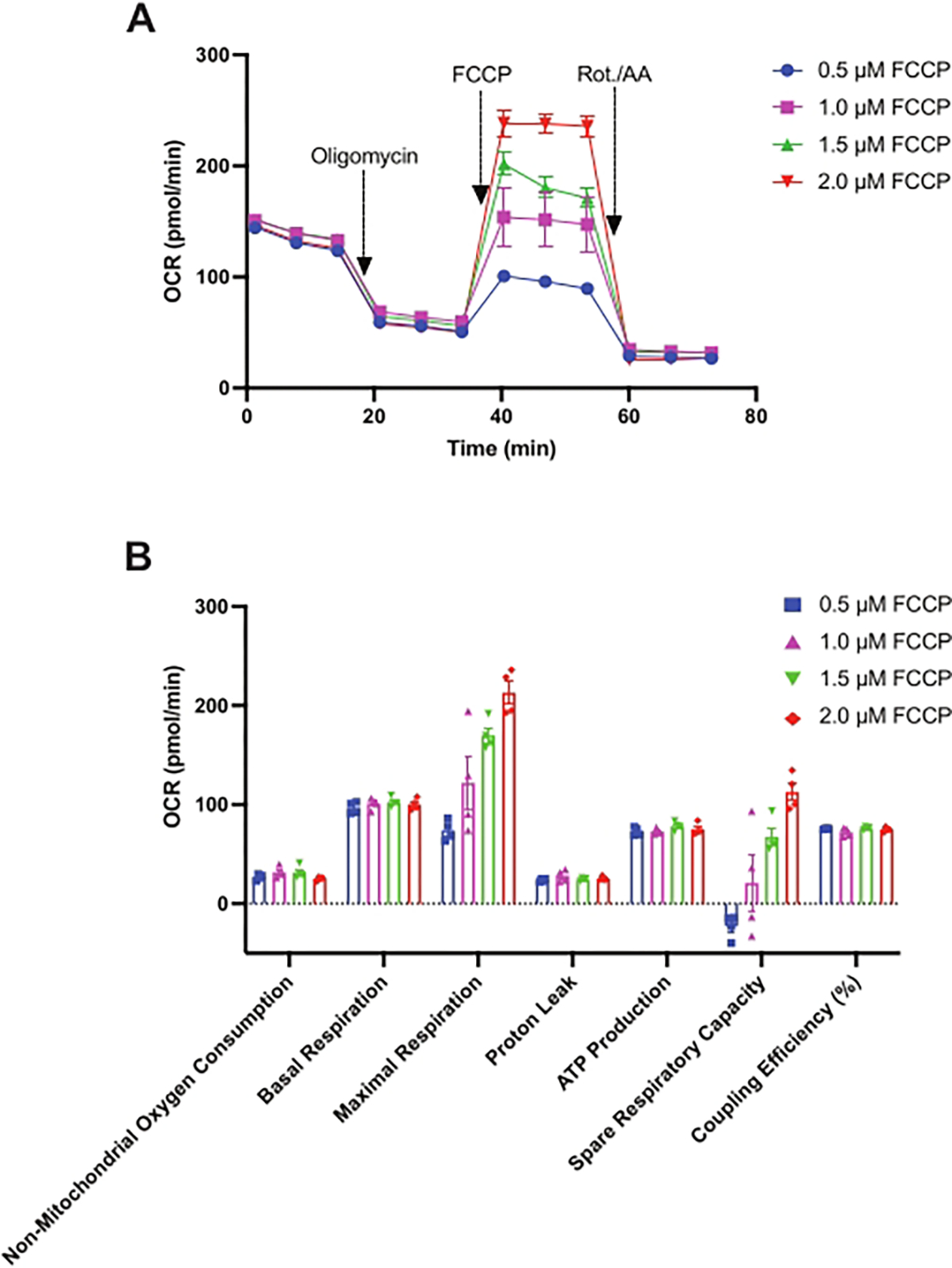
A mitochondrial stress test was performed to measure (A) the oxygen consumption rates (pmol/min) of HSPCs isolated from 24-week-old female C57BL/6 mice. At times indicated, oligomycin (2 μM), FCCP (0.5 μM, 1 μM, 1.5 μM, 2 μM), and rotenone and antimycin A (Rot./AA, 0.5 μM each) were injected. (B) OCR is calculated as non-mitochondrial oxygen consumption, basal respiration, proton leak, maximal respiration (per 0.5 μM, 1 μM, 1.5 μM, and 2 μM FCCP), ATP production, spare respiratory capacity (per 0.5 μM, 1 μM, 1.5 μM, and 2 μM FCCP), and coupling efficiency per 2.5 × 105 HSPCs/well. Data are presented as mean ± SEM, n = 4. Abbreviations: HSPCs = hematopoietic stem and primitive progenitor cells; OCR = oxygen consumption rate; FCCP = carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone; Rot. = rotenone; AA = antimycin A.
DISCUSSION:
This method paper describes an optimized protocol for the assessment of cellular bioenergetics (glycolysis and OxPHOS) in mouse HSPCs using the Seahorse extracellular flux analyzer. This device is a powerful tool that simultaneously measures the ECAR and OCR of live cells, which are the metrics of glycolysis and mitochondrial respiration, respectively. Thus, it can be used to assess the cellular bioenergetics in real time. Further, the 96-well microplate-based platform offers high-throughput quantification with high sensitivity, allowing simultaneous analysis of multiple samples using a single plate, compared to another high-resolution respirometer, the Oroboros O2k, which can only analyze 2 samples simultaneously, or the classical Clark electrode20.
The extracellular flux analyzer has primarily been used for the analysis of adherent cell types, as it requires cells to be present in a monolayer, rendering it more challenging to analyze cells grown in suspension. Moreover, following drug injection from the port, turbulence occurs during the drug-media mixing cycle in the wells prior to the measurements, which may dislodge the cells. Therefore, cells need to be firmly attached to the bottom of the well. The protocols described here have used a cell-adhesive formulation of non-immunogenic polyphenolic proteins extracted from the marine mussel, Mytilus edulis, to prepare an adherent monolayer of murine HSPCs.
Another limitation of this technology is the per-assay cost, which is very high compared to the Clark-type electrode and the Oroboros O2k. The cartridges are relatively expensive and cannot be reused. It is estimated that the cost of the extracellular flux analyzer itself is ~4 times higher than Oroboros O2k20. However, given the semi-automation and high-throughput capability of the extracellular flux analyzer, a much greater amount of data can be gathered per run than with O2k.
The results obtained here show that 2.5 × 105 HSPCs per well are required to obtain reliable data using a 96-well-based extracellular flux analyzer. This adds another challenge in performing flux assays using HSPCs, which are difficult to obtain in large quantities from a mouse and require flow cytometric sorting to obtain a pure population. Moreover, flow-sorting adds substantial time to the experiment and could alter the metabolic phenotype of the progenitor cells. To overcome the limitations associated with flow cytometry-based purification, the strategy discussed here utilized lineage-specific antibodies bound to magnetic beads to deplete lineage-committed progenitors, thus enriching the lineage-negative HSPCs. To further enrich for more primitive HSPCs, an additional cKit enrichment step can be added; however, this comes with the cost of additional ex vivo time (~1.5 h) and a reduction in cell number. The utilization of such a protocol would require optimization outside of the scope of this paper.
To maintain their pluripotency, HSCs must remain quiescent and preferentially reside in the hypoxic environment inside the bone marrow9,21. Quiescent HSCs are believed to rely predominantly on glycolysis to fulfill their modest energy requirements, as high levels of ROS, a byproduct of mitochondrial respiration, are detrimental to their stemness2,3. HSCs possess relatively high, but largely inactive mitochondrial mass, maintaining cellular ROS at low levels to allow maintenance of HSCs pluripotency9. However, during commitment and differentiation, mitochondria become the primary source of ATP production in HSCs5,9. Thus, mitochondria play a key function in transitioning HSCs from quiescence to the metabolically active state required for their lineage commitment and differentiation. In addition to producing ATP via OxPHOS, mitochondria play many other important roles in hematopoietic cell homeostasis, including ROS regulation, apoptosis, calcium signaling, and the synthesis of heme and many other critical metabolite intermediates9. Alterations in mitochondrial functions could significantly impact HSC/progenitor differentiation pathways and may contribute to various hematological disorders, such as hematopoietic malignancies, congenital dyserythropoiesis and sideroblastic anemias, and myelodysplastic syndromes13. In malignant hematopoietic cells, dysfunctional mitochondria play a critical role in conferring resistance to apoptosis induced by various cytotoxic drugs13.
Owing to the central role of mitochondria in maintaining HSC/progenitor homeostasis, valuable insights into the physiological status of these cells can be obtained by assessing their mitochondrial OxPHOS under normal and stress conditions. The identification of novel modulators of mitochondrial activities could identify novel therapeutic targets for treating hematologic abnormalities. The protocol described in this paper can be used to screen the effects of chemical compounds or metabolic CRISPR libraries on the OCR and ECAR of HSCs under normal and pathological conditions, e.g., HSCs harvested from genetically engineered mouse models of hematological disorders. Such screening could also be useful in elucidating metabolic pathways that sustain HSC potency in their hypoxic niche, which would be useful in devising strategies for in vitro expansion of HSCs while maintaining their pluripotency for therapeutic purposes. Given the high-throughput nature of this analysis, the protocol described here could easily be adapted to screen a large number of bioenergetic modulators in HSCs, hematopoietic progenitors, and malignant hematopoietic cells.
In summary, the methods presented here describe optimized protocols for measuring ECAR and OCR in primary mouse HSPCs using the extracellular flux analyzer. Results obtained from the glycolysis stress test have shown that 2.5 × 105 cells per well and 2 μM oligomycin are optimal for further analysis. Using 2.5 × 105 cells per well and 2 μM oligomycin, the mitochondrial stress test was performed to optimize the FCCP concentration, and 2 μM of FCCP was found to induce maximum OCR. Although the current study is mainly focused on mouse HSPCs, the protocol and this approach could easily be adapted to optimize analysis conditions for any type of suspension cells.
Supplementary Material
ACKNOWLEDGMENTS:
Work in Lombard laboratory is supported by the NIH (NIGMS R01GM101171, NIEHS R21ES032305), DoD (CA190267, CA170628, NF170044, and ME200030), and Glenn Foundation for Medical Research. Work in Li laboratory is supported by NIH (NHLBI 5R01HL150707).
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/63045.
DISCLOSURES:
The authors declare that there is no conflict of interest.
REFERENCES:
- 1.Rieger MA, Schroeder T Hematopoiesis. Cold Spring Harbor Perspectives in Biology. 4 (12), a008250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papa L, Djedaini M, Hoffman R Mitochondrial role in stemness and differentiation of hematopoietic stem cells. Stem Cells International. 2019, 4067162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snoeck HW Mitochondrial regulation of hematopoietic stem cells. Current Opinion in Cell Biology. 49, 91–98 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bujko K, Kucia M, Ratajczak J, Ratajczak MZ Hematopoietic stem and progenitor cells (HSPCs). Advances in Experimental Medicine and Biology. 1201, 49–77 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Ito K, Suda T Metabolic requirements for the maintenance of self-renewing stem cells. Nature Reviews Molecular Cell Biology. 15 (4), 243–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norddahl GL et al. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell. 8 (5), 499–510 (2011). [DOI] [PubMed] [Google Scholar]
- 7.de Almeida MJ, Luchsinger LL, Corrigan DJ, Williams LJ, Snoeck HW Dye-independent methods reveal elevated mitochondrial mass in hematopoietic stem cells. Cell Stem Cell. 21 (6), 725–729 e724 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim M, Cooper DD, Hayes SF, Spangrude GJ Rhodamine-123 staining in hematopoietic stem cells of young mice indicates mitochondrial activation rather than dye efflux. Blood. 91 (11), 4106–4117 (1998). [PubMed] [Google Scholar]
- 9.Filippi MD, Ghaffari S Mitochondria in the maintenance of hematopoietic stem cells: new perspectives and opportunities. Blood. 133 (18), 1943–1952 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue S et al. Mitochondrial respiration defects modulate differentiation but not proliferation of hematopoietic stem and progenitor cells. FEBS Letters. 584 (15), 3402–3409 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Simsek T et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 7 (3), 380–390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhatti JS, Bhatti GK, Reddy PH Mitochondrial dysfunction and oxidative stress in metabolic disorders - A step towards mitochondria based therapeutic strategies. Biochimica et Biophysica Acta - Molecular Basis of Disease. 1863 (5), 1066–1077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontenay M, Cathelin S, Amiot M, Gyan E, Solary E Mitochondria in hematopoiesis and hematological diseases. Oncogene. 25 (34), 4757–4767 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Nadanaciva S et al. Assessment of drug-induced mitochondrial dysfunction via altered cellular respiration and acidification measured in a 96-well platform. Journal of Bioenergetics and Biomembranes. 44 (4), 421–437 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Nicholls DG et al. Bioenergetic profile experiment using C2C12 myoblast cells. Journal of Visualized Experiments: JoVE. (46), 2511 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD The contributions of respiration and glycolysis to extracellular acid production. Biochimica et Biophysica Acta. 1847 (2), 171–181 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Nicholls DG, Ferguson SJ in Bioenergetics.eds Nicholls DG, Ferguson SJ (eds) Fourth Edition, Ch. 9, Academic Press, 255–302 (2013). [Google Scholar]
- 18.Aft RL, Zhang FW, Gius D Evaluation of 2-deoxy-D-glucose as a chemotherapeutic agent: mechanism of cell death. British Journal of Cancer. 87 (7), 805–812 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Traba J, Miozzo P, Akkaya B, Pierce SK, Akkaya M An Optimized protocol to analyze glycolysis and mitochondrial respiration in lymphocytes. Journal of Visualized Experiments: JoVE. (117), 54918 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horan MP, Pichaud N, Ballard JW Review: quantifying mitochondrial dysfunction in complex diseases of aging. The Journals of Gerontology, Series A: Biological Sciences and Medical Sciences. 67 (10), 1022–1035 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Morrison SJ, Scadden DT The bone marrow niche for haematopoietic stem cells. Nature. 505 (7483), 327–334 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.