

Abstract
Seven-day-old ICR mice were infected by intracerebral inoculation with recombinant rabies virus vaccine strain SAD-L16. Infected mice developed severe and fatal encephalitis with rabies virus-infected neurons in widespread regions of the brain. There was extensive neuronal death with predominant features of apoptosis, as assessed by light and electron microscopy, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining, and immunohistochemical staining for activated caspase-3. Although SAD-L16 is a neuroattenuated rabies virus, it is fully capable of spreading efficiently and inducing widespread neuronal apoptosis in the immature mouse brain.
Résumé
Des souris ICR âgées de 7 jours ont été infectées par inoculation intracérébrale avec la souche recombinante du virus de la rage SAD-L16. Les souris infectées développèrent une encéphalite sévère et fatale avec infection des neurones par le virus de la rage dans plusieurs régions du cerveau. Il y avait mort neuronale extensive avec des caractéristiques évidentes d’apoptose, tel que démontré par microscopie photonique et électronique, par coloration par marquage de l’extrémité de la coupure simple brin à la dUTP-biotine médiée par la déoxynucléotidyl transférase, et par coloration immunohistochimique pour la caspase-3 activée. Bien que la souche SAD-L16 soit un virus rabique neuro-atténué, elle est en mesure de se répandre efficacement et d’induire une apoptose neuronale généralisée dans le cerveau de souris immatures.
(Traduit par Docteur Serge Messier)
Introduction
Rabies is a highly neurotropic virus that produces fatal encephalomyelitis in humans and animals (1,2). Attenuated rabies virus strains have been traditionally generated by serial passages through animal brains, cell culture, or both. The attenuated SAD-B19 vaccine strain is a derivative of the Street Alabama Dufferin strain isolated from a rabid dog in Alabama in 1935, attenuated by multiple passages in baby hamster kidney (BHK) cells, and selected for vaccine production on the basis of its thermostability (3). SAD-B19 has been widely used in Europe for oral vaccination of foxes (3,4). Recently, SAD-B19 was demonstrated to be avirulent in young foxes, dogs, and other carnivores following different routes of inoculation, including the intracerebral route, while pathogenicity was noted in orally-infected wild rodents, including mice (5). Recombinant rabies virus strains are powerful tools in rabies pathogenesis research because they allow targeted exchange of genes and regulatory elements, while allowing the construction of modified rabies viruses that are highly attenuated and have potential as live vaccines (6). A recombinant clone of SAD-B19, SAD-L16 (L16), was generated from a full-length cDNA clone (7), and L16 was observed to be highly neurovirulent after intracerebral inoculation of adult and suckling mice (8). This study examines the neuropathologic changes in the encephalitis produced by intracerebral inoculation of L16 in suckling mice and notes the differences in the infection produced by the more virulent challenge virus standard (CVS) — 11 strain (9). L16 was found to cause severe encephalitis associated with widespread neuronal apoptosis.
Materials and methods
Virus
The SAD-L16 strain of fixed rabies virus was obtained from Teshome Mebatsion (Intervet International, Boxmeer, The Netherlands). The generation of recombinant rabies virus L16 has been previously described; L16 contains the authentic sequence of the SAD-B19 vaccine strain, which was derived from a full-length cDNA clone, and BSR-T7/5 cells expressing phage T7 RNA polymerase were used to recover infectious virus from cDNA (7,8).
Animals and inoculations
The experimental animal protocol followed the Canadian Council on Animal Care Guidelines on Animal Use and was approved by the Queen’s University Animal Care Committee. Timed pregnant female ICR mice (pathogen-free) were purchased (Charles River Canada, St. Constant, Quebec) and their 7-day-old offspring of either sex were used. Mice were inoculated intracerebrally with 20 μL containing 1000 focus-forming units of L16 diluted in phosphate buffered saline solution (PBSS) with 4% fetal bovine serum (10,11). Uninfected control mice of the same age were inoculated with only the diluent (mock-infected). Three to 4 infected mice and 1 to 2 mock-infected (uninfected) mice were euthanized at daily intervals for 6 d after inoculation. The mice were anesthetized with methoxyflurane and perfused with buffered 4% paraformaldehyde.
Preparation of tissue sections
Brains were removed and immersion-fixed in the same fixative for 24 h at 4°C. Coronal brain tissue sections (6 μm) were prepared after dehydration and embedding in paraffin. Tissues were stained with cresyl violet for light microscopic histological examination.
Immunoperoxidase staining for rabies virus antigen
Immunoperoxidase staining was performed on all mouse brains for rabies virus antigen. Sections were stained for rabies virus antigen by the avidin-biotin-peroxidase complex method using a mouse monoclonal antibody against rabies virus nucleocapsid protein immunoglobulin (Ig)G 5DF12 (obtained from Alexander I. Wandeler, Centre of Expertise for Rabies, Canadian Food Inspection Agency, Nepean, Ontario) as the primary antibody, as previously described (12). Tissues from uninfected mice were used as controls. In brief, tissue sections were deparaffinized and hydrated. Sections were successively treated with 5% normal rabbit serum, primary antibody diluted 1:160 in 2% normal rabbit serum, biotinylated rabbit ant-imouse IgG (Vector Laboratories, Burlingame, California, USA) diluted 1:100 in 2% normal rabbit serum, 1% hydrogen peroxide in methanol, avidin-biotinylated horseradish peroxidase complex (Vector Laboratories), 3,3-diaminobenzidine tetrachloride (Polysciences, Warrington, Pennsylvania, USA) with 0.01% hydrogen peroxide, and 0.5% cupric sulfate in 0.15 M sodium chloride. The slides were counterstained with hematoxylin.
Immunoperoxidase staining for activated caspase-3
Immunoperoxidase staining was performed on all mouse brains for activated caspase-3. Tissue sections were deparaffinized and hydrated, and then heated in a microwave for 1 min at high power followed by 9 min at medium power in sodium citrate buffer (pH 6.0) for antigen unmasking. Sections were successively treated with 5% normal goat serum, rabbit polyclonal antibody directed against cleaved (activated) caspase-3 (Asp175) (Cell Signaling Technology, Beverley, Massachusetts, USA) diluted 1:400 in 2% normal goat serum, biotinylated goat anti-rabbit IgG (Vector Laboratories) diluted 1:100 in 2% normal goat serum, 1% hydrogen peroxide in methanol, avidin-biotinylated horseradish peroxidase complex (Vector Laboratories), 3,3-diaminobenzidine tetrachloride (Polysciences) with 0.01% hydrogen peroxide, and 0.5% cupric sulfate in 0.15 M sodium chloride. The slides were lightly counterstained with hematoxylin.
Electron microscopy
Mice were perfused with a mixture of 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M cacodylate buffer. Brain tissues were removed and immersion-fixed in the same fixative for several days at 4°C and then immersed in 1% osmium tetroxide, dehydrated in a graded series of ethanols, cleared with propylene oxide, and infiltrated with resin (Jembed resin; J.B. EM Services, Dorval, Quebec). Sections (1 μm thick) taken from 8 mice were stained with toluidine blue and examined using light microscopy. Ultrathin sections from 4 mice were stained with uranyl acetate and lead citrate and examined with an electron microscope (Hitachi H7000; Hitachi, Schaumburg, Illinois, USA) at 75 kV.
Transferase-mediated dUTP-biotin nick end labelling (TUNEL) staining
Oligonucleosomal DNA fragmentation was assessed in situ in sections using the terminal deoxynucleotidyl TUNEL method and a TdT-FragEL DNA fragmentation detection kit (Catalogue no. QIA33; Oncogene Research Products, San Diego, California, USA) with the manufacturer’s protocol for paraffin-embedded tissue sections. The TUNEL staining was performed on brains from 11 mice.
Results
Clinical observations
The L16-infected mice developed signs of limb weakness, ataxia (present in 9 out of 19), and growth retardation on day 3 post-inoculation (pi). Over the next 2 d there was progression to quadriparesis and all surviving mice became moribund by day 5 or 6 pi.
Rabies virus antigen distribution
Rabies virus antigen was first detected at 2 d pi in neurons in the brainstem, diencephalon, cerebellum (Purkinje cells), hippocampus (pyramidal layer), and cerebral cortex and also in a few ependymal cells lining the lateral ventricles. At this time the involvement of hippocampal pyramidal neurons was marked. By day 3 pi the number of infected neurons in the brainstem and cerebral cortex had markedly increased, and viral antigen was noted in deep cerebellar nuclei. There was also mild involvement of the neostriatum (Figure 1A). On day 3 pi there was increased staining in Purkinje cells (Figure 1B) and in both hippocampal pyramidal neurons and neurons in the dentate gyrus of the hippocampus (Figure 1C), while there was low background staining in the brains of mock-infected mice (Figure 1D). There was very mild multifocal involvement of the periventricular germinal matrix, but the vast majority of these immature cells were uninfected. On day 4 pi there was marked infection in the neostriatum and infection was observed in the internal granular layer of the cerebellum. Reduced staining was observed in hippocampal pyramidal neurons at late time points associated with marked neuronal loss in this region, while staining increased in the dentate gyrus. Rabies virus antigen was not observed in neurons in the external granular layer of the cerebellum.
Figure 1.
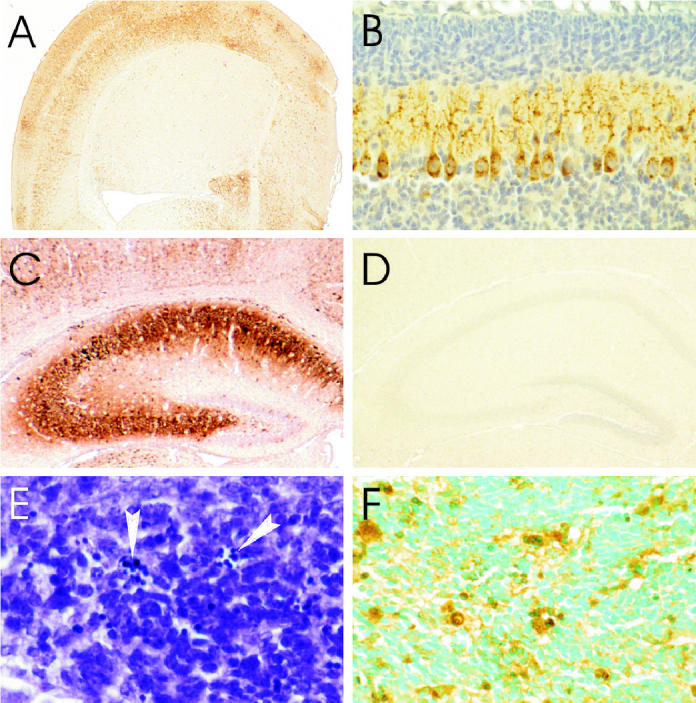
Immunoperoxidase staining for rabies virus antigen in a cerebral hemisphere 3 d postinoculation (pi) showing heavy involvement of the cerebral cortex with relative sparing of the neostriatum (A) in cerebellar Purkinje cells with involvement of their processes in the molecular layer (B), in pyramidal neurons of the hippocampus, and in a few neurons in the dentate gyrus (C), along with low background staining in the hippocampus of a mock-infected mouse (D). Histopathology of periventricular germinal matrix (E) 5 d pi showing a few apoptotic cells with nuclear chromatin condensations (arrowheads), and transferase-mediated dUTP-biotin nick end labeling (TUNEL) and staining is present in cells in periventricular germinal matrix (F) 4 d pi, immunoperoxidase — hematoxylin (A to D), cresyl violet (E), TUNEL staining — methyl green (F). Magnification: A × 10; B × 100; C, D × 25; E × 385; F × 230.
Histopathologic changes
Mock-infected mice showed only occasional apoptotic neurons in the regional areas of the brain, likely due to naturally occurring neuronal death during postnatal development. The earliest pathologic changes in infected mice were observed at 2 d pi in scattered pyramidal neurons of the hippocampus and cerebellum, including neurons in deep cerebellar nuclei and Purkinje cells (Table I), along with cytological features of apoptosis, including karyorrhectic condensation of nuclear chromatin and cytoplasmic shrinkage. By day 3 pi, there was extensive apoptotic cell death of hippocampal pyramidal neurons, most prominently in the CA2 field (Figures 2A and B). Apoptotic changes were also present at this time in neurons in the brainstem, diencephalon, dentate gyrus of the hippocampus (particularly involving the inner layers), and cerebral cortex. By days 3 to 4 pi mononuclear inflammatory changes were present in the leptomeninges, perivascular regions, and brain parenchyma, and there was neuronal apoptosis in all of the regional areas of the brain. Ongoing loss of hippocampal pyramidal neurons on days 4 and 5 pi resulted in a near-complete loss of neurons in all CA fields (Figures 2C and 3B). Neuronal apoptosis also became prominent in the brainstem, diencephalon, cerebellum (deep cerebellar nuclei, internal granular layer, and Purkinje cells) (Figures 3C and D), dentate gyrus of hippocampus (Figure 2D), and cerebral cortex (Figures 2E to H) at late time points. A small number of cells also showed apoptotic changes in the periventricular germinal matrix (Figure 1E). Apoptosis of neurons in the external granular cell layer of the cerebellum was not noted to be more frequent than in mock-infected animals.
Table I.
Morphological changes of apoptosis were evaluated in neurons in the midbrain, cerebellum (deep cerebellar nuclei and Purkinje cells), thalamus, hippocampus (CA1 and CA3 regions), and cerebral cortex (40× objective). A semiquantitative evaluation of the severity of apoptotic changes was performed on days 2 to 6 postinoculation (pi) with the following grading scheme: 0, no significant changes; 1, mild changes; 2, moderate changes; 3, severe changes; and 4, very severe changes or neuronal loss. The identity of all slides was masked during scoring in order to prevent bias in the evaluation. Rating scale scores are expressed as the mean score, standard error: 0, no significant changes; 1, mild changes; 2, moderate changes; 3, severe changes; and 4, very severe changes or neuronal loss
| Day | Midbrain | Cerebellum deep cerebellar nuclei | Cerebellum Purkinje cells | Thalamus | Hippocampus CA1 | Hippocampus CA3 | Cerebral cortex |
|---|---|---|---|---|---|---|---|
| 2 | 2.2, 0.4 | 2.3, 0.3 | 2.3, 0.3 | 1.5, 0.2 | 1.5, 0.2 | 1.5, 0.2 | 1.5, 0.2 |
| 3 | 3.0, 0.6 | 3.5, 0.5 | 2.8, 0.5 | 2.5, 0.3 | 3.3, 0.5 | 4.0, 0 | 3.8, 0.3 |
| 4 | 3.8, 0.2 | 4.0, 0 | 3.5, 0.2 | 4.0, 0 | 4.0, 0 | 4.0, 0 | 4.0, 0 |
| 5 | 4.0, 0 | 3.7, 0.3 | 3.7, 0.2 | 3.7, 0.2 | 4.0, 0 | 3.8, 0.2 | 4.0, 0 |
| 6 | 4.0, 0 | 4.0, 0 | 4.0, 0 | 4.0, 0 | 4.0, 0 | 4.0, 0 | 4.0, 0 |
Figure 2.
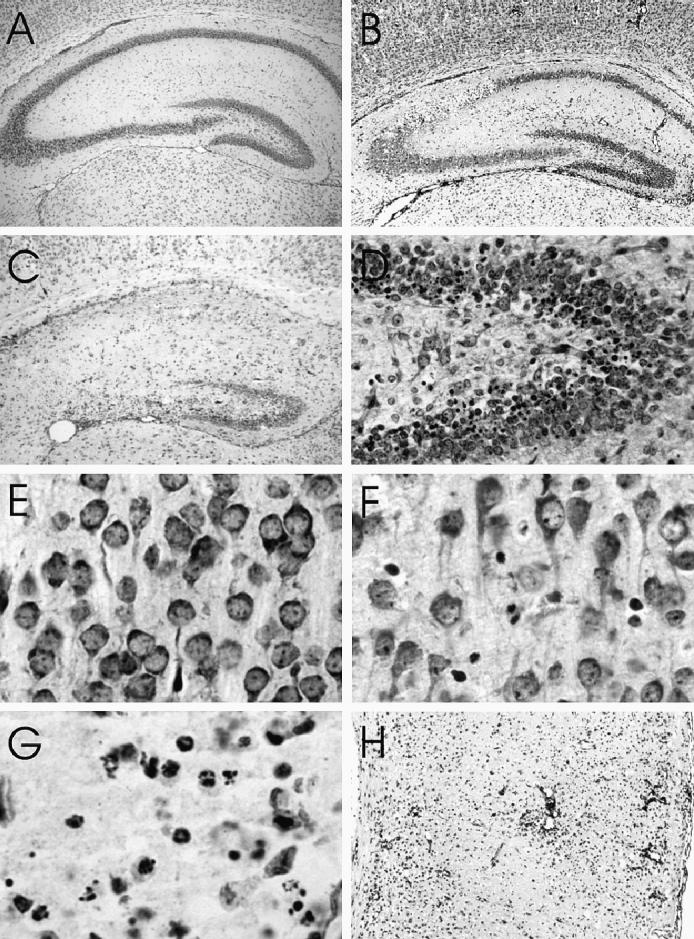
Histopathologic examination showing hippocampi of a (A) mock-infected and (B) L16-infected mouse 3 d postinoculation (pi) showing selective severe neuronal loss in the CA2 region and (C) L16-infected mouse 5 d pi showing almost complete loss of pyramidal neurons. (D) Dentate gyrus of the hippocampus 5 d pi showing condensation of nuclear chromatin of many neurons in the inner layer. Cerebral cortex of a mock-infected mouse (E) and L16-infected mice 4 d pi showing condensation of nuclear chromatin of a few neurons (F) and 5 d pi showing condensation of nuclear chromatin of many neurons (G) and severe neuronal loss and perivascular inflammatory inflitrates (H). Cresyl violet. Magnification: A, B × 20; C × 30; D × 100; E, F × 320; G × 390; H × 40.
Figure 3.
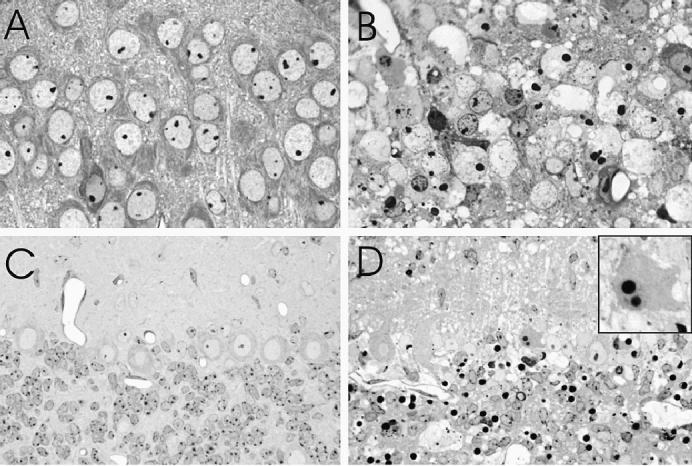
Histopathology in pyramidal neurons of the hippocampus (CA3 field) (A and B) and in the cerebellum (C and D) after mock-infection (A and C) and 4 to 5 d postinnoculation (pi) with L16 (B and D). The L16-infected pyramidal neurons show condensation of nuclear chromatin and cytoplasmic shrinkage and associated neuropil vacuolation (B). In the cerebellum there are nuclear condensations in many neurons in the internal granular layer and in scattered Purkinje cells (inset). There is also prominent vacuolation of the neuropil, especially in the molecular layer (D). Toluidine blue stained resin embedded sections. Magnification: A, B × 370; C, D × 230, inset × 1200.
Electron microscopy
Ultrastructural examination of both the cerebellum and hippocampus showed a predominant pattern of apoptotic cell death, characterized by dense nuclear chromatin condensations (Figure 4A), cell shrinkage with intact cytoplasmic membranes and relative preservation of organelle integrity. Some dying cells also contained numerous autophagic-type vacuoles (Figure 4A). In addition, some shrunken cerebellar Purkinje cells (Figure 4A) and hippocampal pyramidal neurons (Figure 4B) lacked prominent nuclear chromatin condensations. These cells exhibited dilation of the nuclear envelope; endoplasmic reticulum; Golgi apparatus; and, to a lesser extent, of mitochondria, in combination with preservation of cytoplasmic membrane integrity, a pattern characteristic of type 3B (“cytoplasmic”) cell death described by Clarke and colleagues (13,14). Swollen astrocyte processes were frequently seen adjacent to dying neurons (Figure 4A) and accounted for much of the neuropil vacuolation seen by light microscopy (Figures 3B and D).
Figure 4.
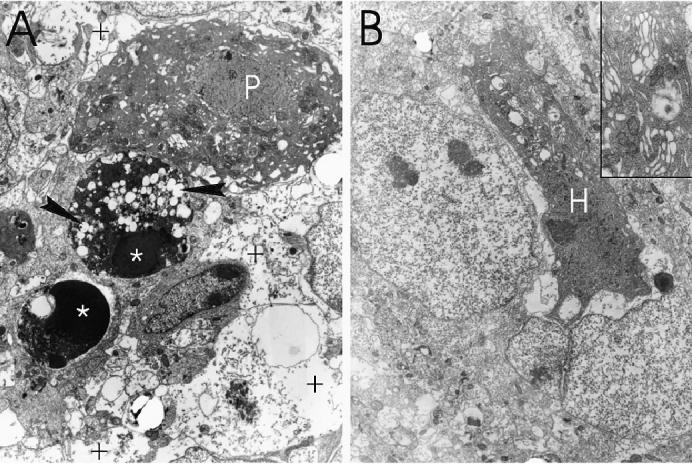
Ultrastructure of cerebellar neurons (A) and hippocampal neurons (CA3 field) (B) 4 to 5 d postinoculation (pi) with L16. A shrunken Purkinje cell (P) and a hippocampal pyramidal neuron (H) show characteristic features of type 3B (“cytoplasmic”) cell death, with expansion of the perinuclear space and prominent dilation of the endoplasmic reticulum and Golgi apparatus (B, inset). Dense nuclear chromatin condensations are seen in 2 internal granule cell neurons (*), one of which contains numerous autophagic-type vacuoles (arrowheads). Swollen astrocyte processes are present (+). Magnification: A × 3000; B × 2300, inset × 14 000.
Transferase-mediated dUTP-biotin nick end labelling staining
At day 2 pi, positive TUNEL staining was observed in scattered hippocampal pyramidal neurons; in neurons in the internal granular layer of the cerebellum; and, rarely, in neurons in the cerebral cortex and diencephalon. By day 3, and increasing on days 4 and 5 pi, TUNEL staining involved many neurons in the brainstem, diencephalon, cerebellum (deep cerebellar nuclei, internal granular layer, and Purkinje cells) (Figure 5A), cerebral cortex (Figure 5B), and hippocampus (including dentate gyrus) (Figure 5C), while there was low background staining in the brains of mock-infected mice (Figure 5D). The TUNEL staining was also noted in a few neurons in the periventricular germinal matrix (Figure 1F) and in the external granular layer of the cerebellum (Figure 5A). Morphological features of apoptotic cell death above the background level in mock-infected animals were observed in all of these regions except the external granular layer of the cerebellum.
Figure 5.
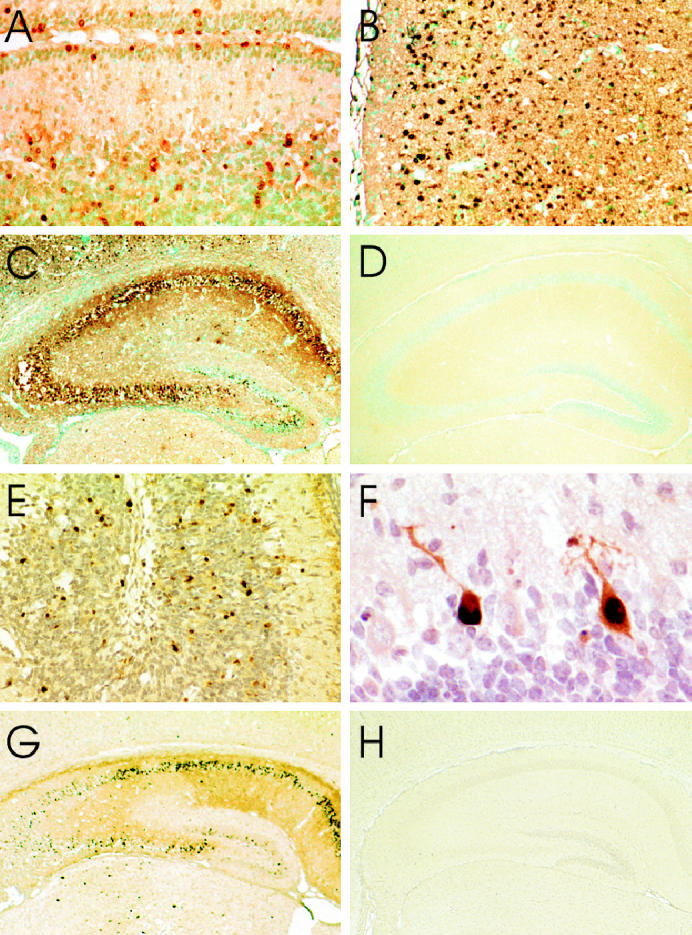
Transferase-mediated dUTP-biotin nick end labelling (TUNEL) and staining (A to D) and immunoperoxidase staining for activated caspase-3 (E to H) in L16-infected mouse brains. The TUNEL staining is present in neurons in the cerebellar external and internal granular layers (A) 6 d postinoculation (pi), in many neurons in the cerebral cortex (B) 4 d pi, and in hippocampal pyramidal neurons and neurons in the dentate gyrus of the hippocampus (C) 5 d pi, while there is low background staining in the hippocampus of a mock-infected mouse (D). Activated caspase-3 staining is present in many neurons in the internal granular layer of the cerebellum (E) 5 d pi, in some Purkinje cells (F) 4 d pi, in both hippocampal pyramidal neurons and in neurons in the dentate gyrus of the hippocampus (G) 4 d pi, and there is low background staining in the hippocampus of a mock-infected mouse (H). A to D, TUNEL staining — methyl green; E to H, Immunoperoxidase — hematoxylin. Magnification: A × 150; B × 95; C, D × 25; E × 130; F × 390; G, H × 20.
Activated caspase-3 staining
Immunohistochemical staining for activated caspase-3 was first observed on day 3 pi in neurons in the diencephalon, hippocampus (pyramidal neurons and dentate gyrus), and cerebral cortex. Increased staining was present on days 4 to 6 pi and also involved the cerebellum, including Purkinje cells and the internal granular layer (Figures 5E to G) with low background staining in mock-infected mouse brains (Figure 5H). Staining was not noted in neurons in deep cerebellar nuclei and staining was only present in the brainstem neurons of 2 mice.
Discussion
L16 produced severe and fatal encephalitis after intracerebral inoculation of young mice. There was rapid viral spread within the brain and infection of numerous neurons in multiple brain regions developed by 3 d pi. The neuronal infection was associated with neuronal death with features of apoptosis and marked neuronal loss. Typical morphologic features of apoptotic cell death were observed using both light and electron microscopy. In addition, ultrastructural elements of autophagic cell death and also of type 3B (“cytoplasmic”) cell death were seen in some cells, consistent with the morphological diversity of cell death that can be seen in vivo (13,14). The encephalitis resulted in severe neuronal loss in regional areas, including the hippocampus and cerebellum, which was much more severe than previously observed in CVS-infected suckling mice (9). Positive TUNEL staining, indicating oligonucleosomal fragmentation of DNA, and immunohistochemical staining for activated caspase-3, a downstream executioner of the apoptotic program (15), provide strong supporting biochemical evidence of a predominant apoptotic mechanism of neuronal death in this model.
The attenuated L16 strain produced neuronal apoptosis in the mouse brain in the present model, and the more neurovirulent CVS-11 strain also produced widespread neuronal apoptosis in the brains of 6-day-old mice when inoculated by the intracerebral route (9). The CVS-infected mice survived for 1 d less (4 d), but, unlike L16-infected mice, they did not demonstrate marked loss in hippocampal pyramidal neurons or death of Purkinje cells. In addition, the apoptotic neuronal changes were less severe in the internal granular layer of the cerebellum in the CVS-infected mice. However, CVS-infected mice demonstrated apoptosis in neurons in the external granular layer of the cerebellum. This likely occurred by indirect mechanisms because these neurons were uninfected. In summary, L16 and CVS demonstrated some differences in their neuronal targets and in the severity of neuronal injury that they induced in specific regional areas of the brain.
The L16 vaccine strain is considered to be highly attenuated in vitro and in vivo under certain conditions. It is highly attenuated in carnivores (5), which makes it an effective vaccine strain. L16 remains highly neurovirulent in both young and adult mice after intracerebral inoculation, as well as in young mice after peripheral inoculation (8,16). The present study indicates that neuronal apoptosis is extensive in the fatal infection of young mice. The destruction of virus infected cells by apoptosis has been proposed as an innate host cellular response that acts to limit viral propagation during infection (17–19), and it is likely that this is an important mechanism by which neuroinvasion is limited in the adult mouse. However, once L16 gains access to the mouse brain, by either a peripheral route or by intracerebral inoculation, it spreads efficiently and induces widespread neuronal apoptosis. This study suggests that neuroattenuation of L16 predominantly occurs by a restriction in susceptibility of carnivore hosts and also by restriction of neuroinvasiveness, particularly in adult hosts, because the virus remains highly neurovirulent and capable of inducing severe cytopathology by an apoptotic mechanism in vivo. Hopefully, a better understanding of how rabies virus injures and kills neurons will put us one step closer to conquering this ancient disease.
Acknowledgments
The authors are grateful for recombinant rabies virus vaccine strain SAD-L16 from Teshome Mebatsion (Intervet International, B.V., Boxmeer, The Netherlands) and for monoclonal antibody 5DF12 from Alexander I. Wandeler (Centre for Rabies Expertise, Canadian Food Inspection Agency, Nepean, Ontario). This work was supported by a research contract with Intervet International, B.V. (Boxmeer, The Netherlands), Canadian Institutes of Health Research grant MOP — 64376, and the Queen’s University Violet E. Powell Research Fund (all to A.C. Jackson).
Footnotes
Dr. Jackson’s current address is the Kingston General Hospital, Connell 725, 76 Stuart Street, Kingston, Ontario K7L 2V7.
References
- 1.Jackson AC. Human disease. In: Jackson AC, Wunner WH, eds. Rabies. San Diego: Academic Press, 2002:219–244.
- 2.Niezgoda M, Hanlon CA, Rupprecht CE. Animal rabies. In: Jackson AC, Wunner WH, eds. Rabies. San Diego: Academic Press, 2002:163–218.
- 3.Blancou J, Meslin F-X. Modified live-virus rabies vaccines for oral immunization of carnivores. In: Meslin F-X, Kaplan MM, Koprowski H, eds. Laboratory Techniques in Rabies. 4th ed. Geneva: World Health Organization, 1996:324–337.
- 4.Schneider LG. Rabies virus vaccines. Dev Biol Stand. 1995;84:49–54. [PubMed] [Google Scholar]
- 5.Vos A, Neubert A, Aylan O, et al. An update on safety studies of SAD B19 rabies virus vaccine in target and non-target species. Epidemiol Infect. 1999;123:165–175. doi: 10.1017/s0950268899002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schnell MJ, Tan GS, Dietzschold B. The application of reverse genetics technology in the study of rabies virus (RV) pathogenesis and for the development of novel RV vaccines. J Neurovirol. 2005;11:76–81. doi: 10.1080/13550280590900436. [DOI] [PubMed] [Google Scholar]
- 7.Schnell MJ, Mebatsion T, Conzelmann KK. Infectious rabies viruses from cloned cDNA. EMBO J. 1994;13:4195–4203. doi: 10.1002/j.1460-2075.1994.tb06739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mebatsion T. Extensive attenuation of rabies virus by simultaneously modifying the dynein light chain binding site in the P protein and replacing Arg333 in the G protein. J Virol. 2001;75:11496–11502. doi: 10.1128/JVI.75.23.11496-11502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson AC, Park H. Apoptotic cell death in experimental rabies in suckling mice. Acta Neuropathol. 1998;95:159–164. doi: 10.1007/s004010050781. [DOI] [PubMed] [Google Scholar]
- 10.King AA. Cell culture of rabies virus. In: Meslin F-X, Kaplan MM, Koprowski H, eds. Laboratory Techniques in Rabies. 4th ed. Geneva: World Health Organization, 1996:114–130.
- 11.Mims CA. Intracerebral injections and the growth of viruses in the mouse brain. Br J Exp Pathol. 1960;41:52–59. [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson AC, Ye H, Phelan CC, et al. Extraneural organ involvement in human rabies. Lab Invest. 1999;79:945–951. [PubMed] [Google Scholar]
- 13.Borsello T, Mottier V, Castagne V, Clarke PG. Ultrastructure of retinal ganglion cell death after axotomy in chick embryos. J Comp Neurol. 2002;453:361–371. doi: 10.1002/cne.10411. [DOI] [PubMed] [Google Scholar]
- 14.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. Anat Embryol (Berl) 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 15.Friedlander RM. Apoptosis and caspases in neurodegenerative diseases. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 16.Rasalingam P, Rossiter JP, Mebatsion T, Jackson AC. Comparative pathogenesis of the SAD-L16 strain of rabies virus and a mutant modifying the dynein light chain binding site of the rabies virus phosphoprotein in young mice. Virus Res 2005 (In press). [DOI] [PubMed]
- 17.Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Trends Microbiol. 2000;8:410–418. doi: 10.1016/S0966-842X(00)01830-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allsopp TE, Fazakerley JK. Altruistic cell suicide and the specialized case of the virus-infected nervous system. Trends Neurosci. 2000;23:284–290. doi: 10.1016/s0166-2236(00)01591-5. [DOI] [PubMed] [Google Scholar]
- 19.Fazakerley JK, Allsopp TE. Programmed cell death in virus infections of the nervous system. Curr Top Microbiol Immunol. 2001;253:95–119. doi: 10.1007/978-3-662-10356-2_5. [DOI] [PubMed] [Google Scholar]