

Abstract
Tumour-infiltrating T cells offer a promising avenue for cancer treatment, yet their states remain to be fully characterized. Here we present a single-cell atlas of T cells from 308,048 transcriptomes across 16 cancer types, uncovering previously undescribed T cell states and heterogeneous subpopulations of follicular helper, regulatory, and proliferative T cells. We identified a unique stress response state, TSTR, characterized by heat shock gene expression. TSTR cells are detectable in situ in the tumour microenvironment across various cancer types, mostly within lymphocyte aggregates or potential tertiary lymphoid structures in tumor beds or surrounding tumor edges. T cell states/compositions correlated with genomic, pathological, and clinical features in 375 patients from 23 cohorts, including 171 patients who received immune checkpoint blockade (ICB) therapy. We also found significantly upregulated heat shock gene expression in intratumoural CD4/CD8 cells following ICB treatment, particularly in non-responsive tumors, suggesting a potential role of TSTR cells in immunotherapy resistance. Our well-annotated T-cell reference maps, web portal, and automatic alignment/annotation tool could provide valuable resources for T-cell therapy optimization and biomarker discovery.
Introduction
Tumour-infiltrating T cells (TILs) are a crucial component of the TIME and have demonstrated anticancer efficacy in various settings, such as chimeric antigen receptor (CAR) T-cell therapy, TIL therapy, and immune checkpoint blockade (ICB) therapy. However, TILs are phenotypically and functionally diverse, and their characteristics determine the effectiveness and potential side effects of anticancer therapies1–7. As T-cell-directed or combinational therapies rapidly expand to treat different cancer types with varying responses, a comprehensive understanding of TIL biology is essential for effectively stratifying patients and advancing future therapies. The application of single-cell RNA sequencing (scRNA-seq) has revolutionized our understanding of cell states and heterogeneity within the TIME8–18. A recent pan-cancer study characterized various TIL states and paths to exhaustion19. However, further analysis of TILs is necessary due to their remarkable heterogeneity. Additional scRNA-seq datasets from complementary diseases, tissue types, and conditions are critical to fully capture all possible TIL states in the TIME and better characterize heterogeneous subsets. Moreover, investigating the biological relevance and clinical significance of TIL subsets in larger patient cohorts, particularly in the context of immunotherapy, is imperative. Furthermore, cross-study comparisons remain challenging due to inconsistencies in the markers and gene signatures used to define TIL states. Although automatic annotation tools20,21 are available, they lack the desired level of granularity, as they were not specifically designed for TILs.
In this study, we analyzed T cells from 27 datasets across 16 cancer types13,15,16,18,22–34. More than 65% of the analyzed T cells were not present in the previous pan-cancer study19. We characterized 32 distinct T cell states, further dissected the heterogeneous regulatory, follicular helper, and proliferative subsets, and highlighted the stress response state by integrating single-cell and spatial profiling data. We investigated the genomic, pathological, and clinical correlates of these T cell states in large patient cohorts, and in the context of ICB therapy. We also created well-annotated T-cell reference maps, an interactive web portal, and an automatic alignment/annotation tool to support efficient single-cell profiling of T cells.
Results
Pan-cancer analysis of T cells
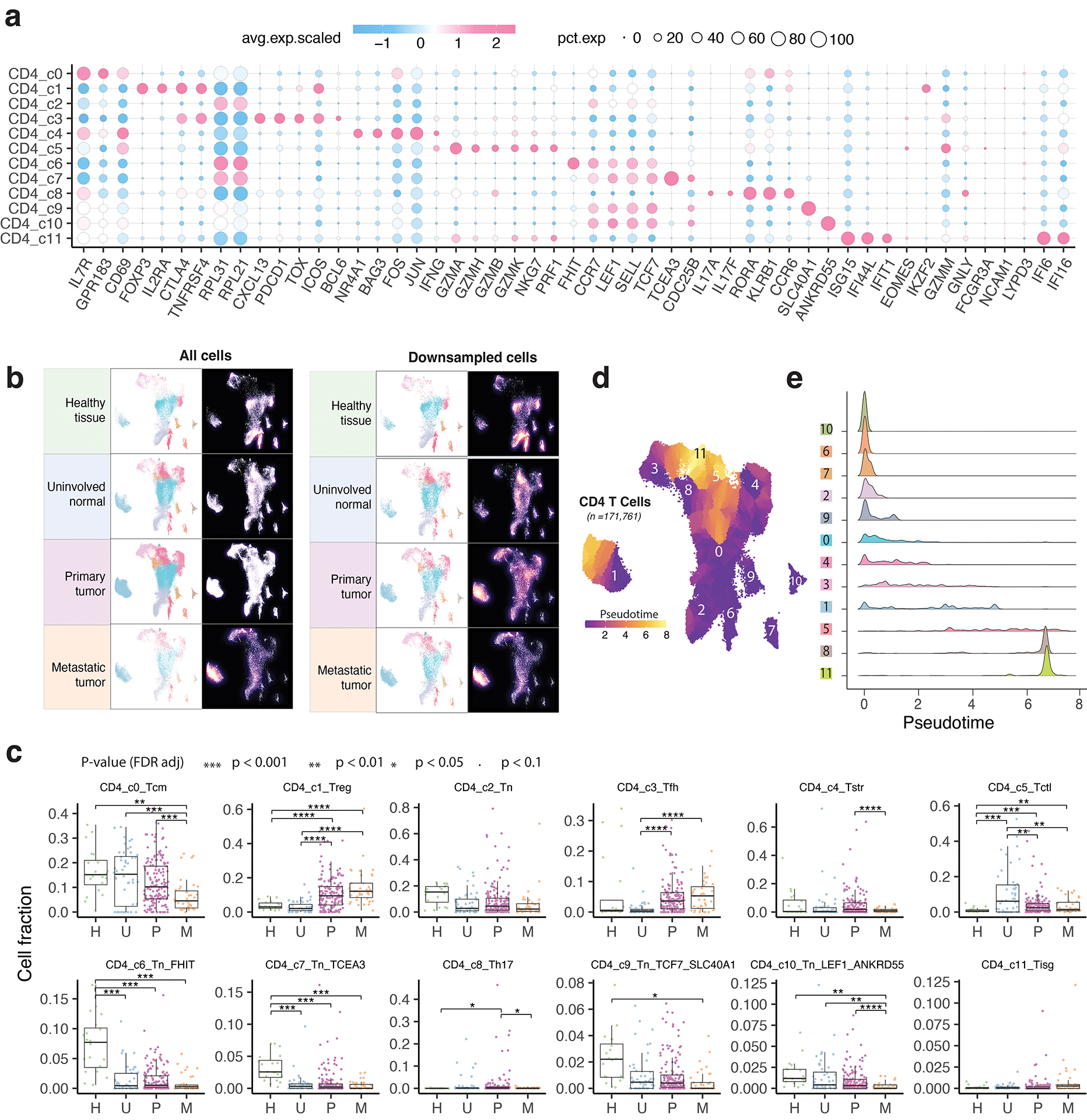
We collected scRNA-seq data from T cells across 16 cancer types and 9 non-neoplastic/normal tissue types, comprising 486 samples from 324 individuals (Fig. 1a–b; Supplementary Tables 1–2). 16 out of 27 datasets13,15,16,18,22–34 were not present in the previous pan-cancer study22 (Supplementary Tables 1). 308,048 high-quality T cells were identified following rigorous quality control (Supplementary Fig. 1; Methods). We identified 6 major types of T cells: CD4, CD8, gamma delta T cells (Tgd), natural killer T cells (NKT), mucosal-associated invariant T cells (MAIT), and proliferative T cells (Extended Data Fig. 1). CD4 T cells were the most abundant subset (Fig. 1c) and their cellular fractions varied substantially across tissues of different locations/conditions (Fig. 1d).
Figure 1. Pan-cancer analysis of T cells - data collection and major T cell types.
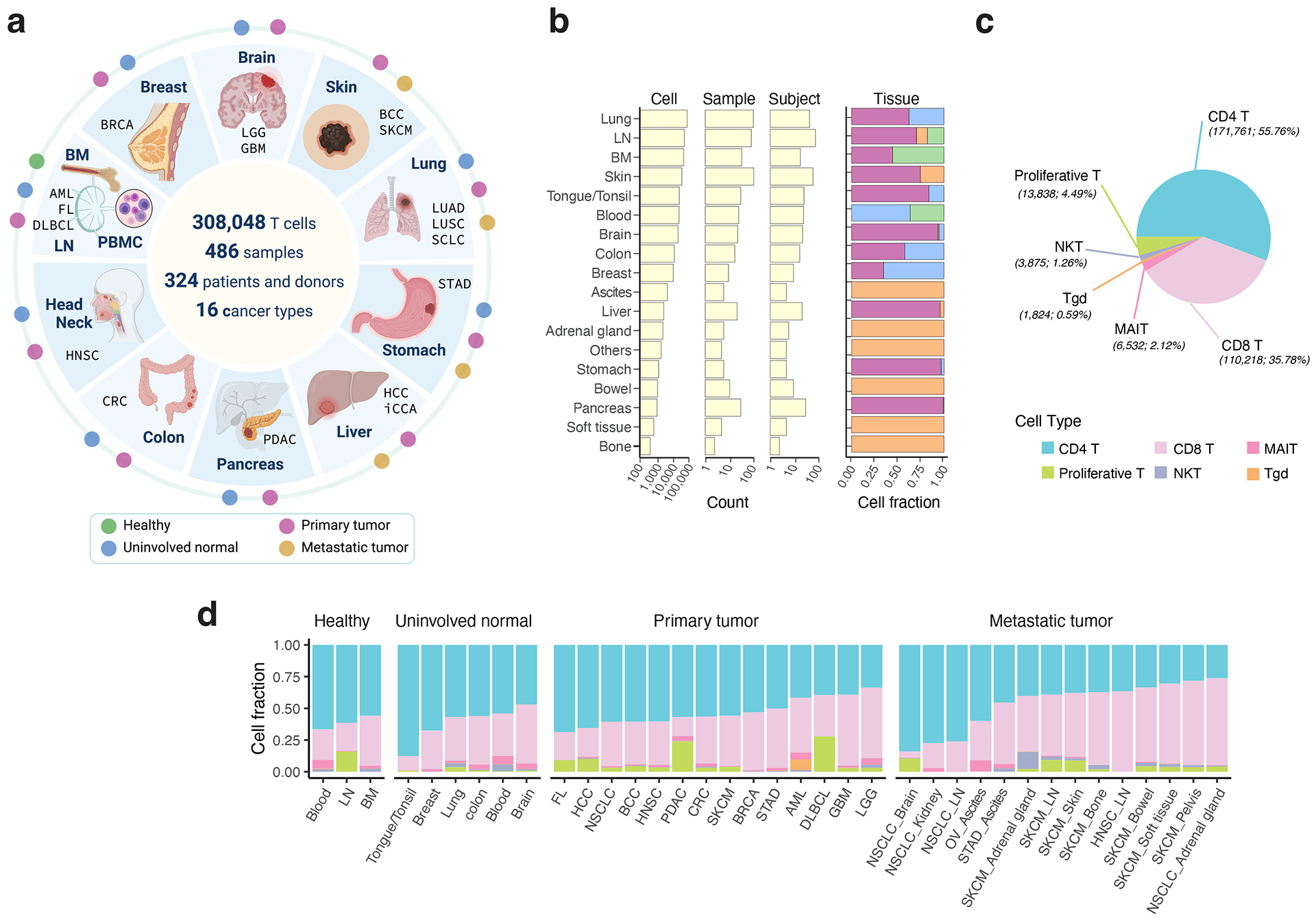
a) Schematic depicting the study design (created with BioRender.com). We used 17 published and 10 in-house datasets. Detailed information on cohorts and samples is provided in Supplementary Tables 1 and 2. b) Bar graphs showing summary statistics for the number of cells, samples, and subjects collected by organ (left) and their tissue compositions (right). Tissue color codes are consistent with panel a. c) Pie chart depicting the cellular frequencies of the 6 major T cell types in all analyzed samples. d) Bar graphs displaying relative cellular fractions of the 6 major T cell types across various cohorts of the four main tissue groups. In our study, the analyzed metastatic tumors were biopsies taken from metastases. BM, bone marrow; LN, lymph node; PBMC, peripheral blood mononuclear cell. For uninvolved normal tissues and metastatic tumors, their corresponding organs/sites of sample collection are labeled. Cancer types are labeled using the TCGA study abbreviations.
Transcriptional diversity of CD8 T cells
Unsupervised clustering analysis identified 14 clusters of CD8 T cells (Fig. 2a), each observed in multiple datasets (Supplementary Fig. 2a–b). Based on differentially expressed genes (DEGs) (Supplementary Table 3), canonical immune markers (Fig. 2b–d; Extended Data Fig. 2a), and curated gene signatures (Fig. 2e; Supplementary Table 4), we defined 14 transcriptional states: naïve-like (TN, c3, c13), transitional effector (t-TEFF, c0), effector (TEFF, c2, c8, c10, c11), central memory (TCM, c6), resident memory (TRM, c12), stress response (TSTR, c4), interferon response (TISG, c5), senescent-like (TSEN, c9), precursor exhausted (p-TEX, c7), and exhausted (TEX, c1) CD8 T cells (Fig. 2a).
Figure 2. Transcriptional diversity of CD8 T cells.
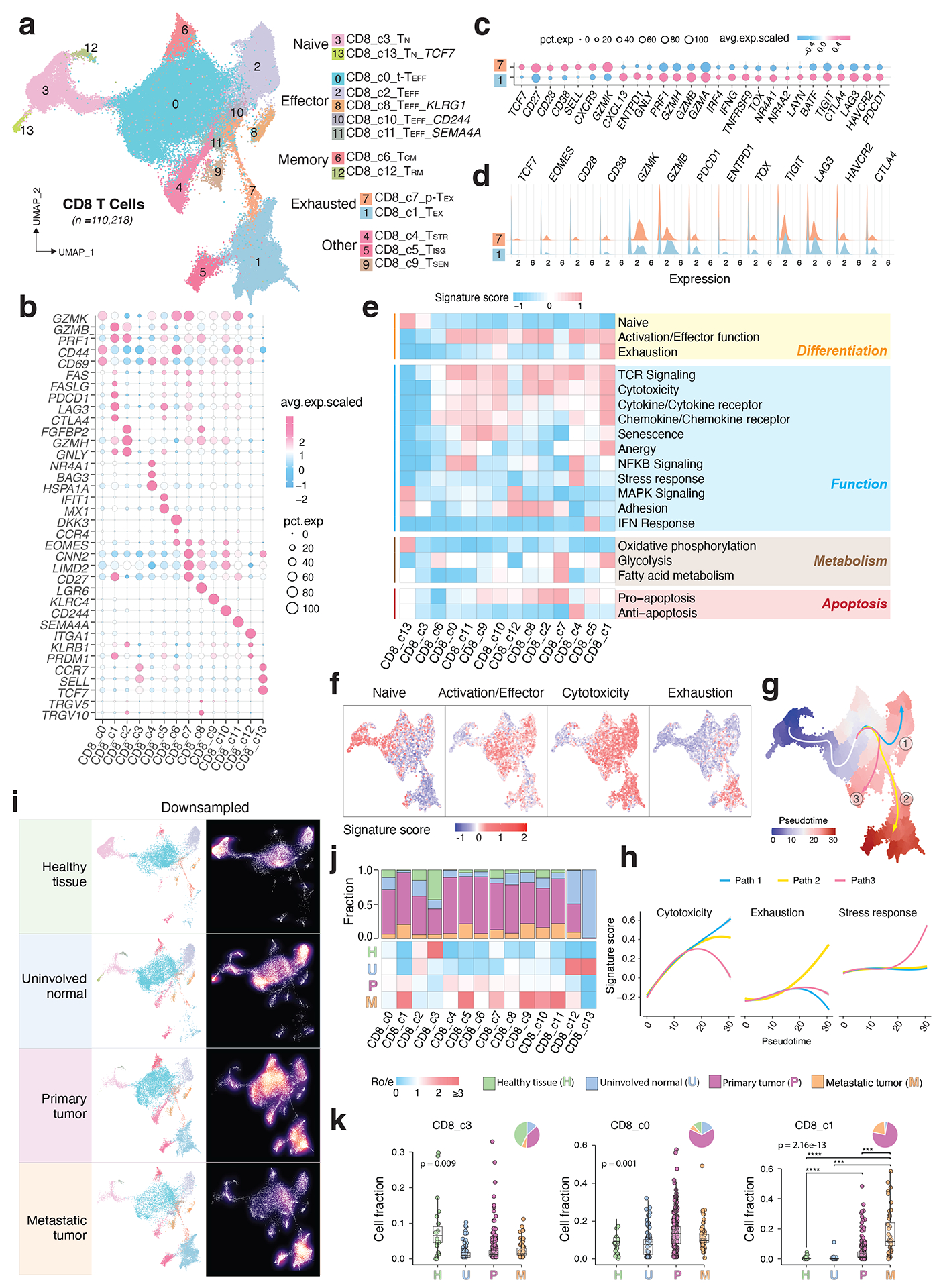
a) The UMAP view of 14 CD8 T cell clusters. b) Marker gene expression across defined T cell clusters. Bubble size is proportional to the percentage of cells expressing a gene and color intensity is proportional to average scaled gene expression. c) Bubble plot and d) ridge plot showing key marker gene expression between the two CD8 T cell clusters. e) Heatmap illustrating expression of 19 curated gene signatures across CD8 T cell clusters. Heatmap was generated based on the scaled gene signature scores. f) Expression of 4 representative gene signatures selected from e). g) Monocle 3 trajectory analysis of CD8 T cell differentiation revealing three main divergent trajectories. Cells are color coded for their corresponding pseudotime. h) Two-dimensional plots showing expression scores for 3 representative gene signatures in cells of paths 1 (blue), path 2 (yellow), and path 3 (pink), respectively, along the inferred pseudotime. i) UMAP view of CD8 T cell states (left) and cell density (right) displaying CD8 T cell distribution across 4 main tissue groups. Downsampling was applied, and 11,592 cells were included for each group. High relative cell density is shown as bright magma. j) Distribution of CD8 T cell states across tissue groups. Top bar plot showing the relative proportion of cells from 4 different tissue types for each CD8 T cell subset. Heatmap showing tissue prevalence estimated by Ro/e. k) Box plots showing cellular fractions of three CD8 T cell subsets across tissue groups. Each dot represents a sample. Pie charts displaying tissue composition. H, normal tissues from healthy donors; U, tumour adjacent uninvolved tissues; P, primary tumour tissues; and M, metastatic tumour tissues. The one-sided Games-Howell test was applied to calculate the p values between tissue types (sample number n = 20, 51, 156, 39), followed by FDR (false discovery rate) correction. FDR adjusted p-value: *≤0.05; **≤0.01; ***≤0.001, ****≤0.0001. For CD8_c1, p = 7.27e-10 (H vs. P), p = 4.21e-6 (H vs. M), p = 2.84e-4 (U vs. M), p = 8.38e-4 (P vs. M). Boxes, median ± interquartile range; whiskers, 1.5× interquartile range.
The two TN clusters displayed a naïve-like phenotype with high expression of naïve gene signature (Fig. 2b, e). The t-TEFF cluster showed high expression of GZMK, CXCR4, and early activation markers CD44 and CD69 (Fig. 2b; Extended Data Fig. 2a). The four TEFF clusters highly expressed effector molecules (e.g., FGFBP2, CX3CR1, FCGR3A, and KLRG142,43), cytolytic activity-related genes, and consistently, high cytotoxicity gene signature and TCR signaling (Extended Data Fig. 2a; Fig. 2e). The TCM cluster exhibited high expression of GZMK, CD44, EOMES, CD28, CCR7, exclusively expressed DKK337–38, and downregulated activation markers (e.g., NKG7, PRDM1, ID2, HOPX, FGFBP2), consistent with its TCM phenotype. The TRM cluster displayed high expression of IL7R, PRDM1, TGFBR2, and upregulated ITGA1, along with downregulated SIPR1, CCR7, SELL, consistent with their tissue retention property. The TSTR cluster was characterized by unique expression of stress-related heat shock genes (e.g., HSPA1A, HSPA1B)39–41 and a stress response gene signature (Fig. 2e), along with the highest expression of NF-κB signaling, a primary regulator of cellular stress response42. The TISG cluster displayed high expression of interferon-stimulated genes (ISGs) and the interferon (IFN) response signature (Supplementary Table 3; Fig. 2e). The TSEN cluster demonstrated the highest expression of T cell senescence signature43, low CD27 and cytotoxicity. The TEX cluster was characterized by the highest expression of exhaustion-related markers, HAVCR2 (TIM-3), LAG3, TIGIT, PDCD1 (PD-1), CTLA4, LAYN, and transcription factors (TFs) such as TOX, with low expression of TCF7 (TCF1) (Fig. 2c–e). In accordance with previous work19–44, TEX cells also highly expressed cytotoxicity markers and CXCL13, ENTPD1 (CD39), and TNFRSF9 (4–1BB), indicating that they were likely antigen-experienced. The p-TEX cluster (c7) bridges TEX and other TEFF clusters (Fig. 2a). Relative to TEX, p-TEX cells had lower expression of inhibitory checkpoint receptors, exhaustion-related TFs, cytotoxicity genes, and terminally differentiated T cell markers, but higher expression of TCF7, CD27, CD28, and EOMES (Fig. 2c–e).
To understand the differentiation trajectories of CD8 T cells, we first projected the expression of naïve, activation/effector, cytotoxicity, and exhaustion gene signatures onto the UMAP, which is following its expected dynamics (Fig. 2f). Monocle 345–47 analysis revealed three main paths (Fig. 2g; Extended Data Fig. 2b): all started with TN, followed by t-TEFF, with path 1 ending in a terminally differentiated TEFF state, path 2 connected with p-TEX and ending in the TEX state, and path 3 ending in the TSTR state. These paths imply divergent cell fates, which are also supported by expression kinetics of related gene signatures along the inferred pseudotime axis (Fig. 2h). We observed substantial changes in the CD8 T cell landscape. Tumor tissues exhibited decreased fractions of TN cells and increased fractions of TEFF (c10, c11), TCM, TSTR, TISG, TSEN, p-TEX, and TEX cells. TEX fractions were low or undetectable in healthy/uninvolved normal tissues but were elevated in various primary tumours and highly enriched in metastases [one-sided Games-Howell test, FDR adjusted p-value: 2.8e-4 (U vs. M), and 8.4e-4 (P vs. M)] (Fig. 2i–k; Extended Data Fig. 2c–d; Supplementary Fig. 2c).
CD4 T cell states and heterogeneous TREG and TFH populations
We identified 12 different CD4 T cell states (Fig. 3a): TN (c2, c6, c7, c9, c10), T follicular helper (TFH, c3), T helper 17 (Th17, c8), TCM (c0), T regulatory (TREG, c1), cytotoxic (CTL, c5), stress response (TSTR, c4), and IFN response (TISG, c11) states (Supplementary Table 5). Each state was observed across multiple datasets (Supplementary Fig. 3a–b). TN clusters expressed high levels of naive markers (Fig. 3b–c; Extended Data Fig. 3a; Supplementary Table 5). TCM cluster highly expressed CD69, GPR183, IL7R, KLF2, TOB1, and the anti-apoptosis gene signature (Supplementary Table 6). The CTL cluster markedly expressed cytolytic activity-related genes and chemokine/chemokine receptors (Fig. 3b–c). The TREG cluster exhibited a classical TREG gene signature (IL2RA, FOXP3, CTLA4, and TNFRSF4). The TFH cluster was characterized by high expression of ICOS, TNFRSF4, BCL6, TOX, CXCL13, PDCD1, and CTLA4. The Th17 cluster showed high expression of RORA and IL17A/F. The CD4 TSTR cluster highly expressed heat shock genes and stress response signature and the CD4 TISG cluster was marked by IFN response. Our annotation of these cell states was also supported by published gene signatures19,23,48 (Supplementary Fig. 4). CD4 T cell states and compositions varied significantly between normal and tumor tissues (Fig. 3d–f; Extended Data Fig. 3b–c; Supplementary Fig. 3c). TN c6 and c7 were highly abundant in healthy tissues but depleted in uninvolved normal and tumor tissues. TCM subset was abundant in healthy/uninvolved normal tissues but decreased in primary tumours and further reduced in metastases. Conversely, TREG and TFH subsets were low in healthy/uninvolved normal tissues but highly enriched in tumour tissues [one-sided Games-Howell test, FDR adjusted p-values: for TREG, 4.1e-13 (U vs. P) and 4.0e-16 (U vs. M); for TFH, 0.023 (U vs. P) and 9.0e-8 (U vs. M)].
Figure 3. The landscape of CD4 T cells.
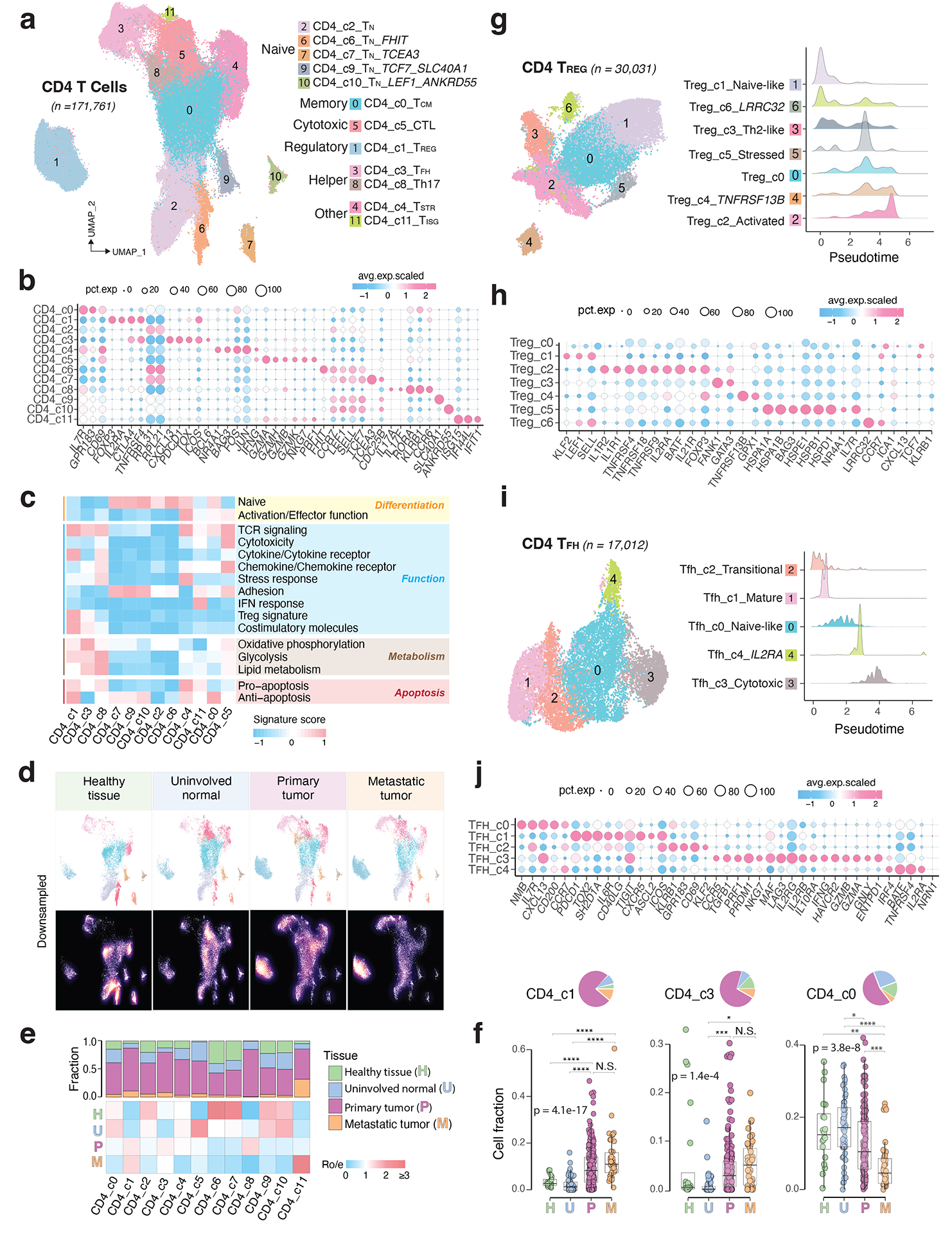
a) UMAP view of 12 CD4 T cell clusters. b) Bubble plot showing marker gene expression across defined clusters. More marker genes are shown in Extended Data Fig. 3a and a list of the top 50 most significant DEGs are provided in Supplementary Table 5. c) Heatmap displaying expression of 16 curated gene signatures (as listed in Supplementary Table 6) across CD4 T cell clusters. d) UMAP view of CD4 T cell states (top) and cell density (bottom) demonstrating CD4 T cell distribution across 4 tissue groups. Downsampling was applied, and 10,703 cells were included for each group. High relative cell density is shown as bright magma. e) Distribution of CD4 T cell states across different tissues. (Top) bar plot showing the relative proportion of cells from 4 tissue types and (bottom) heatmap showing tissue prevalence estimated by Ro/e. f) Box plots comparing cellular fractions of three CD4 T cell subsets across tissue types. Each dot represents a sample. Pie charts displaying tissue composition. The one-sided Games-Howell test was applied to calculate the p values between those 4 tissue types (sample number n = 20, 53, 158, 39), followed by FDR correction. FDR adjusted p-value: * ≤0.05; **≤0.01; ***≤0.001, ****≤0.0001. For CD4_c1, pHvsP = 4.08e-13, pHvsM = 4.03e-6, pUvsP = 4.08e-13, pUvsM = 4.03e-16. For CD4_c3, pUvsP = 4.05e-4, pUvsM = 0.021. For CD4_c0, pHvsM = 0.002, pUvsP = 0.023, pUvsM = 9e-8, pPvsM = 8.06e-5. Boxes, median ± interquartile range; whiskers, 1.5× interquartile range. g) UMAP plot of seven CD4 Treg subclusters (left) and (right) ridge plots displaying the distribution of inferred pseudotime across Treg subclusters. h) Marker gene expression across Treg subclusters. The complete list of significant DEGs is provided in Supplementary Table 7. i) UMAP plot of five CD4 Tfh subclusters (left) and (right) ridge plots illustrating the distribution of inferred pseudotime across Tfh subclusters. j) Marker gene expression across Tfh subclusters. A list of significant DEGs is provided in Supplementary Table 8.
CD4 TREG and TFH clusters displayed relatively lower purity (Supplementary Fig. 3a) and greater variability in inferred pseudotime (Extended Data Fig. 3d–e). Further subclustering analysis identified 7 TREG subclusters (Fig. 3g–h; Supplementary Table 7; Supplementary Fig. 5). TREG c1 highly expressed naïve markers, while c2 displayed high expression of co-stimulatory molecules (e.g., TNFRSF4, TNFRSF18, TNFRSF9), and cytokine receptors (e.g., IL1R1, IL1R2, IL21R), suggesting their highly activated phenotype (Fig. 3h). TREG c0 bridged the naïve and activated states, while c3 had a Th2-like profile with high expression of GATA3. TREG c4 expressed high levels of TNFRSF13B56. TREG c5 marks a stress response state39–41. TREG c6 expressed LRRC32, a TREG-specific receptor for TGF-β50. Among these subsets, TREG c2 fractions were significantly increased in tumours (Supplementary Fig. 5d). Utilizing the same approach, we identified 5 TFH subclusters (Fig. 3i–j; Supplementary Table 8). TFH c1 expressed high levels of PDCD1, TIGIT, TOX2, CXCR5, CD40LG, ICOS, and ASCL2, suggesting a mature Tfh state. TFH c3 appears to be at a late developmental stage (Fig. 3i, right). TFH c0 expressed high IL7R, CCR7, SELL, and CXCL13. TFH c2 expressed KLRB1, ICOS, CD69, GPR183, and KLF2, indicating a transitional state. TFH c4 expressed IRF4 and BATF, two TFs for TFH differentiation. In summary, we characterized 12 TREG and TFH subsets, highlighting their heterogeneous nature.
States of unconventional and proliferative T cells
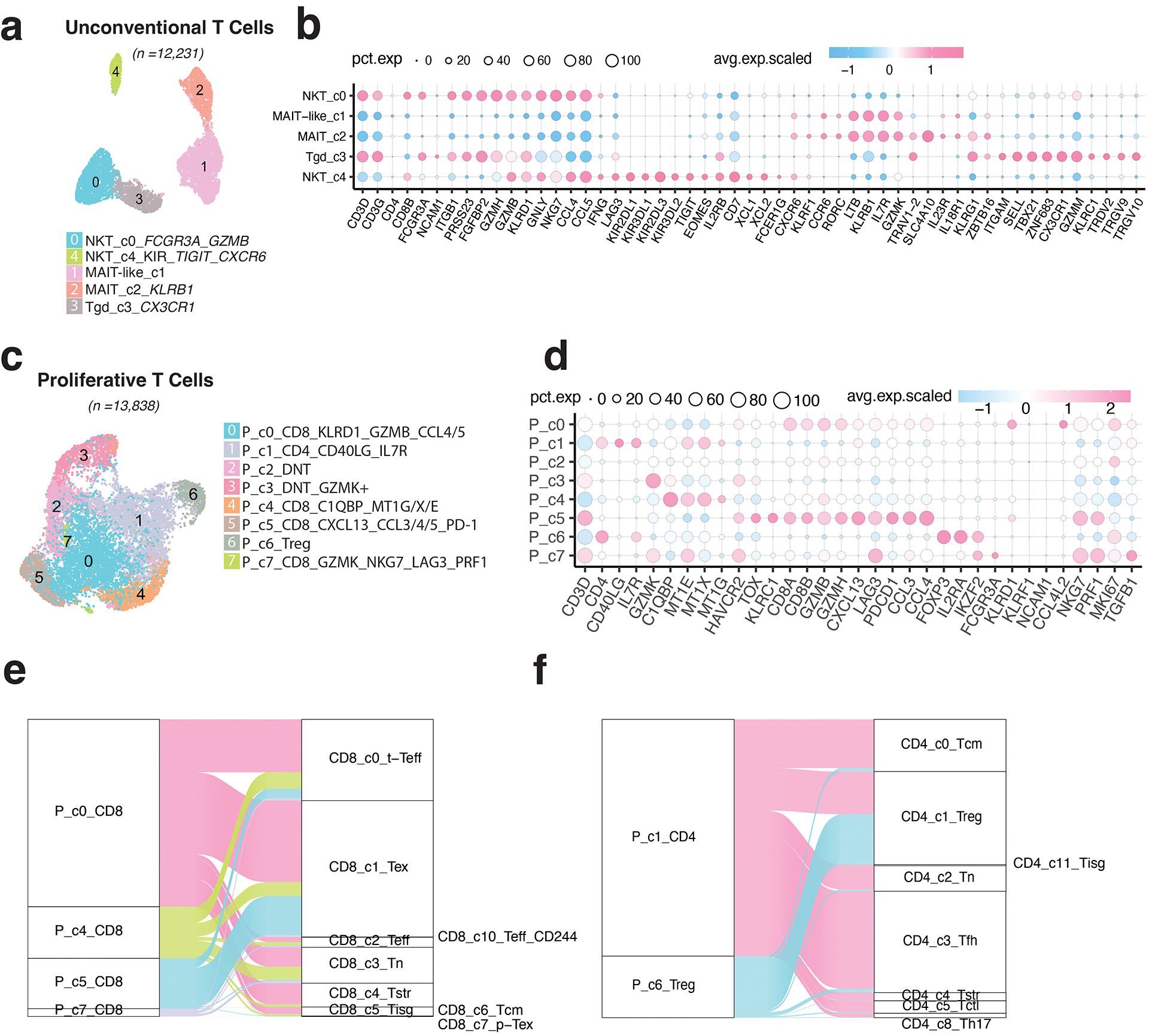
Five unconventional T cell subsets were identified: NKT (c0, c4), MAIT (c2), MAIT-like (c1), and Tgd (c3) clusters (Extended Data Fig. 4a–b; Supplementary Table 9; Supplementary Fig. 6a). NKT c0 was CD8+FCGR3A+, with high expression of cytotoxicity genes, activation markers, and chemokines. NKT c4 cells were EOMES+, expressing XCL1/2, CXCR6, TIGIT, LAG3, and inhibitory killer cell immunoglobulin-like receptors (KIRs), suggestive of tissue-resident NKT cells. MAIT c2 expressed TRAV1–2, SLC4A10, and high levels of GZMK and KLRB1, similar to previously reported CD161+ MAIT cells51. MAIT-like c1 displayed a similar profile but had low TRAV1–2 and no SLC4A10. Tgd c3 showed a unique expression of Tgd cell-related markers (Extended Data Fig. 4b; Supplementary Fig. 6b). Among them, NKT c0 fractions were decreased in primary tumours (Supplementary Fig. 6c).
We identified 8 proliferative T cell subclusters: CD8+ (c0, c4, c5, c7), CD4+ (c1, c6), and double negative (DNT, c2, c3) T cells (Extended Data Fig. 4c–d; Supplementary Fig. 7a–b; Supplementary Table 10). Subcluster c0 displayed characteristics of activated CD8 T cells, while c4 exhibited lower cytotoxicity and higher expression of C1QB and MT1G/E/X52,53. Subcluster c5, mapped to CD8 TEX cluster after regressing out proliferative cell markers, displayed the highest levels of cytotoxicity and expression of inhibitory checkpoint receptors (Extended Data Fig. 4d–e). Subcluster c7 was characterized by high expression of PRF1, NKG7, GZMK, LAG3, TIM-3, and TGF-b1. Subcluster c1 was activated CD4+ T cells and c6 was proliferative TREG (Extended Data Fig. 4f). The DNT c3 had the highest GZMK expression and c2 showed the lowest cytotoxicity levels. Among these subsets, c0 and c6 showed relatively higher fractions in tumours (Supplementary Fig. 7c). Our findings highlight the transcriptional heterogeneity among proliferative T cells, often overlooked in single-cell studies.
Transcriptional similarity and co-occurrence patterns
We analyzed the phenotypic relationships of these T cell states. Unsupervised hierarchical clustering analysis revealed 4 main branches (Fig. 4a). TN subsets clustered tightly in the 2nd branch and TEFF subsets formed the 3rd branch. CD4 and CD8 TSTR were grouped together in the 4th branch, as were the CD4 and CD8 TISG subsets. Tgd subset formed a separate branch due to distinctive profiles. We also investigated T cell state co-occurrence through spearman correlation analysis of cluster frequencies (Fig. 4b; Supplementary Table 11). CD4 and CD8 TSTR, and CD4 and CD8 TISG subsets showed strong positive correlations in primary and metastatic tumours, indicating their co-occurrence in the TIME. In primary tumors, CD8 TEX, CD4 TREG, and TFH exhibited positive co-occurrence, which, together with CD4/CD8 TSTR exhibited negative correlations with CD4/CD8 TN, CD8 TEFF, and CD4 CTL subsets (Fig. 4b, left). In metastatic tumors, CD8 TEX was negatively correlated with CD4/CD8 TN and TEFF subsets (Fig. 4b, right), indicating mutual exclusivity.
Figure 4. Transcriptional similarity and co-occurrence patterns of T cell subsets and correlations with genomic, molecular, and pathological features.
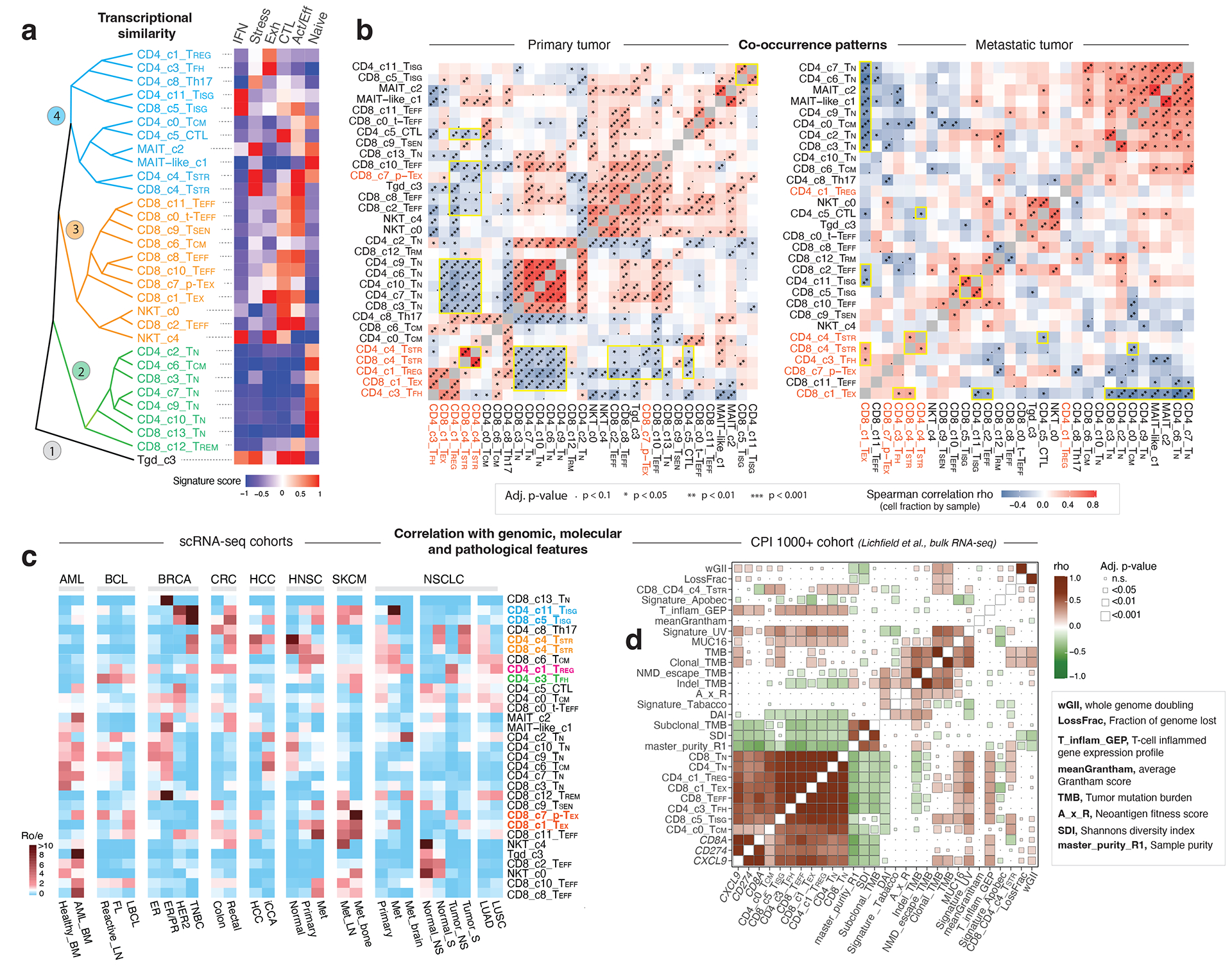
a) The dendrogram on the left displays transcriptional similarity among 31 T cell subsets. The computed Euclidean distance matrix was used for unsupervised hierarchical clustering analysis, which revealed 4 major “branches” that are colored (from bottom to top) in black, green, orange, and cyan, respectively. The heatmap on the right shows the expression of 6 curated gene signatures across T-cell clusters. The heatmap was generated based on the scaled gene signature scores. IFN, IFN response; stress, stress response; Exh, exhaustion; CTL, cytotoxicity; Act/Eff, activation/effector function. b) T cell state co-occurrence in primary tumours (left) and metastatic tumours (right). Sample-level Spearman correlation analysis was performed based on cluster frequencies of 31 non-proliferative T cell subsets. Positive co-occurrence patterns are in ‘warm’ color, and negative co-occurrence patterns are in ‘cold’ color. Color intensity is proportional to the Spearman correlation coefficient. Asterisks indicate the statistical significance based on FDR-adjusted two-sided p-values. c) Correlation with genomic, molecular, and pathological features in 16 scRNA-seq cohorts across 8 cancer types with corresponding information available, and d) the CPI1000+ cohorts. The heatmap in c) displays the distribution of T cell states across different cancer types and subtypes, as estimated by Ro/e. FL, follicular lymphoma; LBCL, large B cell lymphoma; iCCA, intrahepatic cholangiocarcinoma; NS, never smoker; S, smoker. Cancer types are labeled using the TCGA study abbreviations. The heatmap in d) illustrates correlations with TMB and additional mutation quality characteristics as well as known biomarkers of ICB therapy response (Litchfield et al.). The size of the square is proportional to statistical significance (FDR-adjusted two-sided p-value) and the color intensity is proportional to the Spearman correlation coefficient (rho). An annotation of the abbreviations is listed on the right.
Correlations with genomic, molecular, pathological features
Next, we examined their correlations with major clinical, histopathological, and molecular features in scRNA-seq datasets (Supplementary Table 2). Among the 8 cancer types examined, CD8 TEX and CD4/CD8 TISG states were observed more frequently than expected in metastases derived from head & neck squamous cell carcinoma (HNSC), non-small cell lung cancer (NSCLC), and melanoma (Fig. 4c). The CD4/CD8 TSTR states were associated with aggressive phenotypes across cancer types including triple-negative breast cancer (TNBC), rectal adenocarcinoma, hepatocellular carcinoma (HCC), HNSC, and NSCLC in smokers.
We then analyzed bulk RNA-seq data from TCGA cohorts using gene signatures derived from our scRNA-seq data (Supplementary Table 12). To ensure specificity, we merged similar subsets (e.g., CD4/CD8 TSTR, CD4 TN, CD8 TEFF) and focused on cell states with unique gene signatures. Tumours with low T-cell infiltration levels were excluded (Supplementary Table 13; Supplementary Fig. 8a). We found a significant positive correlation between CD8 TEX signature scores and TMB in LUAD and MSI-driven uterine corpus endometrial carcinoma (UCEC) (Extended Data Fig. 5a), consistent with previous studies61. We observed variations in TMB correlations across genotypic/molecular subtypes with individual cancer types. For instance, positive correlations were found between TMB and TREG, TFH, TEX, and CD8 TEFF states in EVB+ stomach adenocarcinoma (STAD), while negative correlations were seen in genomically stable STAD. In KRAS-mutant LUADs, there was a strong positive correlation between TMB and CD8 TEX, while no such correlation was found in EGFR-mutant LUADs. Similar variation was observed in bladder urothelial carcinoma with and without 9p21 loss. These findings suggest the diversity in TIL states and landscapes associated with cancer genotypic/molecular subtypes.
We further assessed the association of T cell subsets with overall survival (OS) in TCGA cohorts (Extended Data Fig. 5b). Higher CD4 TCM was linked to increased OS in sarcoma. In cancers related to oncogenic viruses, such as HPV+ HNSC, cervical squamous cell carcinoma, and endocervical adenocarcinoma (predominantly HPV), significant associations were observed between T cell subset abundance and improved OS. Conversely, no such association was found in HPV-NHSC, and in LGG, T cell subsets were linked to reduced OS, reflecting the immunosuppressive nature of glioma TIME. Unlike LGG, multiple T cell subsets were associated with improved OS in melanoma, consistent with a high burden of ultraviolet light mutations known for their enriched immunogenic potential62.
Correlations with TMB and ICB response in CPI1000+ cohorts
We also analyzed the CPI1000+ cohorts56. Of the 1,008 patients, 562 with available genomic, expression, and clinical response data were included. UV signature mutations were positively correlated with the abundance of CD8 TN, TEFF, TISG, and TEX, while a negative association was observed between the APOBEC mutation signature and CD8 TEX (Fig. 4d). Clonal TMB was positively correlated with CD8 TEX, CD4/CD8 TSTR, and CD8 TISG. Additionally, PD-L1 expression in immune and tumour cells was strongly associated with the levels of CD8 TEX, CD8 TISG, and TREG in urothelial carcinoma64 (Supplementary Fig. 8b).
These patients received single-agent checkpoint inhibitors without prior ICB treatment56 and predominantly had baseline pretreatment specimens. We found no significant difference in the levels of individual T cell states between responders and non-responders. We then tested all possible combinations of T cell states in the 3 largest cohorts: renal cell carcinoma58, melanoma56, and urothelial cancer57. In the renal and melanoma cohorts, we observed low response rates in patients with high levels of CD4/CD8 TSTR and low CD4 TFH or low CD8 TEX. Conversely, high response rates were observed in patients with low CD4/CD8 TSTR and high CD4 TFH or high CD4/CD8 TN (Extended Data Fig. 6). These results suggest that the combination of high CD4/CD8 TSTR and low CD4 TFH in pre-treatment tumors is associated with an unfavorable response to ICB therapy. However, these findings were not observed in the urothelial cancer cohort64, possibly due to the dominant immunosuppressive mechanism in this cancer type being TGF-β signaling from fibroblasts64. While the correlation between low CD4 TFH and poor response to ICB therapy is expected, it’s worth noting that the TSTR state has been previously underappreciated.
TSTR cells are detectable in situ across cancer types
To account for the potential stress-induced expression of heat shock genes in T cells during tissue dissociation59, we aimed to validate TSTR cells within intact cells through in situ hybridization and target RNA detection. Specifically, we performed RNAscope on an LN metastasis from a melanoma patient (Fig. 5a, i) and examined the expression of HSPA1B, a top DEG in CD4/CD8 TSTR clusters, in distinct regions of melanoma cells (Fig. 5a, ii) and peritumoral lymphocytes (Fig. 5a, iv) from the same tissue section. We found HSPA1B expression in both melanoma cells (Fig. 5a, iii) and peritumoral lymphocytes (Fig. 5a, v).
Figure 5. Detection of TSTR cells in situ using multiple different spatial profiling approaches.
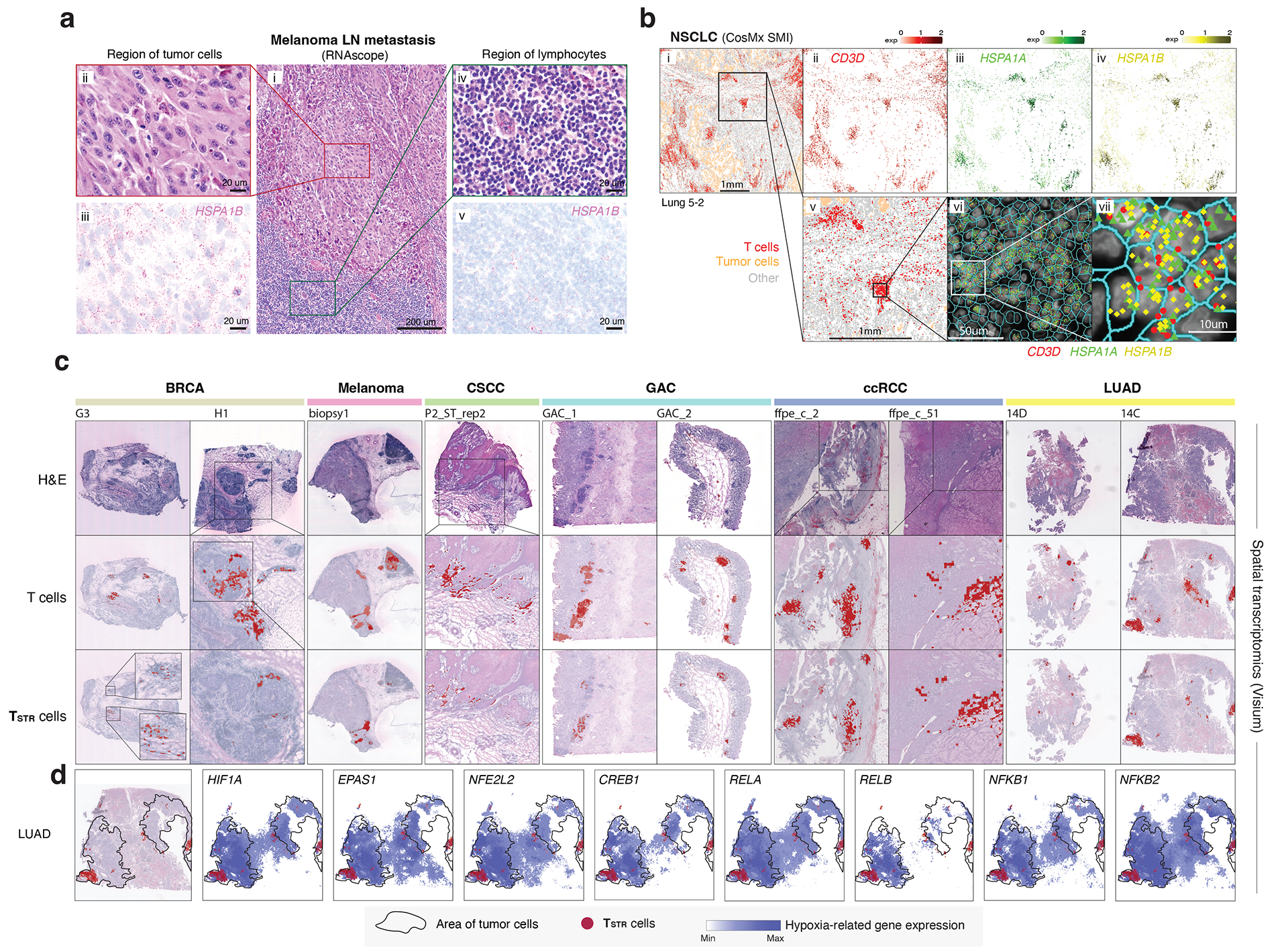
a) Detection of HSPA1B expression in peritumoral lymphocytes in a melanoma lymph-node (LN) metastasis by RNAscope. (i) H&E of the sample at low magnification (40x, scale bar 200 μm), with high-magnification (400x, scale bar 20 μm) areas showing H&E and RNAscope on melanoma cells (ii & iii) and peritumoral lymphocytes (iv & v) demonstrating that both tumor cells and peritumoral lymphocytes express HSPA1B RNA. No tissue section replicate was available for this sample. b) Detection of TSTR cells in an NSCLC sample by CosMx. A representative tissue section (Lung 5–2) is shown. Two consecutive tissue sections of Lung 5–2 are presented in Extended Data Fig. 7. (i) Cells in physical locations (x, y coordinates). Color denotes cell type. Spatial mapping of CD3D (ii), HSPA1A (iii), and HSPA1B (iv) expression in T cells (the same area as i). (v) A zoom-in view of a representative area of (i) showing two lymphocyte aggregates. (vi) a zoom-in view of (v) showing subcellular localization of CD3D, HSPA1A, and HSPA1B transcripts. (vii) a zoom-in view of (vi) showing co-localization of CD3D, HSPA1A, and HSPA1B transcripts. c) Pan-cancer detection of TSTR cells by spatial transcriptomics. Representative tissue sections of 6 cancer types are shown. (top row) H&E stained tissue image. (middle row) Mapping of T cells and (bottom row) the TSTR cells on the same histology image (Melanoma, GAC, LUAD) or a high-magnification image (BRCA, CSCC, ccRCC). BRCA, breast cancer; CSCC, cutaneous squamous cell carcinoma; GAC, gastric adenocarcinoma; ccRCC, clear cell renal cell carcinoma; LUAD, lung adenocarcinoma. d) Co-mapping of TSTR cells and hypoxia-related gene expression by spatial transcriptomics in a LUAD sample (section 14C) as shown in c). (first on the left) Mapping of TSTR cells (in red) on the same image as shown in c). The black curve outlines the two tumor areas. (the remaining images on the right) Spatial co-mapping of TSTR cells (in red) and hypoxia-related gene expression (in blue, the darker the color, the higher the level of gene expression) on the same capture area.
Next, we sought to confirm the existence of TSTR cells in situ within the TIME using an orthogonal technology, we analyzed the public CosMx dataset from NSCLC patients60. We found high expression of HSPA1A and HSPA1B in T cells, primarily, within lymphocyte aggregates (LAs) near the tumor bed or myeloid-enriched stroma (Fig. 5b, i–iv). We confirmed the co-expression of CD3D, HSPA1A, and HSPA1B in T cells at subcellular resolution in all 3 tissue sections from tumor lung-5 (Fig. 5b, v-vii; Extended Data Fig. 7, left) and in tissue sections from other lung cancers (Supplementary Fig. 9), as well as in HCC (Extended Data Fig. 7, right).
We then conducted a pan-cancer analysis to map TSTR cells and examine their spatial relationships. We analyzed Visium data (10x Genomics) across 6 cancer types: melanoma, LUAD, basal cell carcinoma (BCC), cutaneous squamous cell carcinoma (CSCC), clear cell RCC (ccRCC), and gastric adenocarcinoma (GAC) (Supplementary Table 14). Regions highly expressing T cell markers were examined at high magnification to confirm the presence of lymphocytes (Supplementary Figs. 10a–c and 11a–b), and TSTR cells were mapped spatially based on their expression of HSPA1A/HSPA1B within T-cell-enriched spots (Supplementary Figs. 10d and 11c). We successfully mapped TSTR cells in 33 tissue sections across all 6 cancer types that contained T-cell-enriched spots (Fig. 5c; Extended Data Fig. 8–9; Supplementary Fig. 12). TSTR cells were primarily localized within LAs, particularly those within tumor beds or surrounding tumor edges.
Finally, we investigated the relationship between TSTR cells and tumor hypoxia in the TIME. Previous studies have shown that hypoxic conditions can activate TFs including HIF-1α (HIF1A), HIF-2α (EPAS2), RELA, RELB, NFKB1, NFKB2, CREB (CREB1), and Nrf2 (NFE2L2), which are essential for hypoxic adaptation61. Using Visium data, we measured the expression levels of these TFs and plotted them alongside the spatial distribution of TSTR cells. TSTR cells were predominantly located near or within hypoxic cancer cell domains (Fig. 5d; Extended Data Fig. 8–9; Supplementary Fig. 12). Additionally, we combined the Visium data to assess whether T-cell enriched spots with high levels of stress signals were more likely to be hypoxic. Our analysis showed significant correlations between the expression levels of HSPA1A/HSPA1B and hypoxia-related TFs only in a subset of cancer types examined (Supplementary Fig. 13). We did not observe any significant correlation between the expression levels of these TFs in cancer cells and TSTR cell fractions in CosMx and scRNA-seq cohorts. Further functional studies are necessary to determine the underlying mechanisms of stress response in T cells.
Enrichment of CD4/CD8 TSTR cells in non-responsive tumors
Since certain TSTR signature genes can be expressed by cancer cells and other cell lineages, deconvolution of bulk RNA-seq data may not provide an accurate estimate of the actual TSTR abundance in a given sample. We, therefore, attempted to assess the clinical relevance of TSTR cells using publicly available scRNA-seq datasets. We obtained scRNA-seq data from 6 cohorts18,44,48,62–64 of patients who underwent anti-PD-1/PD-L1 therapy, which included a total of 247 samples from 133 patients18,44,48,62–64 (Supplementary Table 15). We applied TCellMap to align T cells uniformly from these public scRNA-seq datasets with the T cell maps built in this study (Extended Data Fig. 10; Methods).
In the BCC cohort 18 (Fig. 6a), we observed enriched CD4 TFH cells in the responsive (R) tumors before and after ICB therapy (Fig. 6b). Notably, we also observed highly enriched CD4 and CD8 TSTR cells in non-responsive (NR) tumors before and especially after ICB therapy (Fig. 6b). Further supporting these results, we observed a significant upregulation of HSPA1A and HSPA1B expression in both CD4 and CD8 T cells in NR (vs. R) tumors following ICB treatment and in the post- (vs. pre-) ICB timepoint (Fig. 6c–d; two-sided Welch’s t-test, FDR adjusted p-value < 2.2e-16 for all comparisons for HSPA1A). In the NSCLC cohort51 (Fig. 6e), we observed enriched CD8 TSTR cells in LN metastases compared to primary tumors at the pre-ICB timepoint. During ICB treatment, there was a greater enrichment of CD4 and CD8 TSTR cells in the NR tumors (Fig. 6f). We consistently observed a significant upregulation of HSPA1A and HSPA1B expression in both CD4 and CD8 T cells in LN-Met (vs. primary tumors) before ICB treatment, and in NR (vs. R) tumors during ICB treatment (Fig. 6g–h; two-sided Welch’s t-test, FDR adjusted p-value < 2.2e-16 for all comparisons for HSPA1A).
Figure 6. Significant enrichment of CD4/CD8 TSTR cells following ICB therapy across cancer types, primarily, in non-responsive tumors.
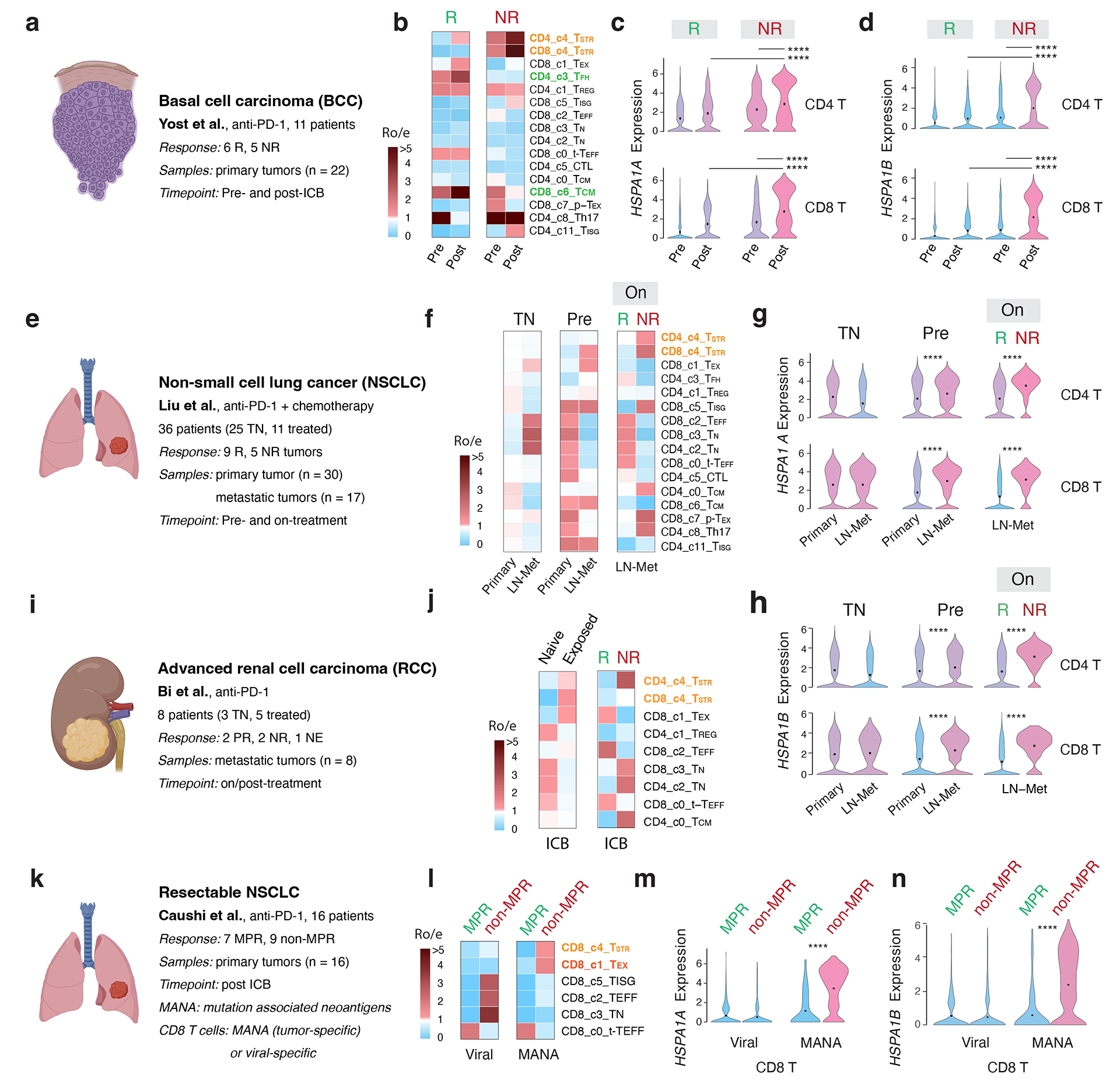
a, e, i, k) Description of the cohort, patients, and samples (created with BioRender.com). a-d) The BCC cohort. b) Enriched CD4/CD8 TSTR cells in non-responsive (NR) tumors. c) Significantly higher expression of HSPA1A (all p values < 2.2e-16) and d) HSPA1B in CD4 and CD8 T cells from NR vs. responsive (R) tumors, and at post (vs. pre)-ICB timepoint. Pre, pre-ICB; Post, post-ICB treatment (For CD4 T, pNR_Pre-vs-Post = 1.16e-11, pPost_R-vs-NR = 2.16e-8. For CD8 T, pNR_Pre-vs-Post < 2.2e-16, pPost_R-vs-NR = 1.33e-10). e-h) The NSCLC cohort. f) Enriched CD4/CD8 TSTR cells in NR tumors during ICB treatment. g) Significantly higher expression of HSPA1A and h) HSPA1B in CD4/CD8 T cells from NR vs. R tumors on ICB treatment, and in LN-met vs. primary tumors at pre-ICB timepoint. TN, treatment naïve; On, on-ICB treatment; LN-Met, lymph node metastasis (For CD4 T, pPre_Primary-vs-LN-Met < 2.2e-16, pLN-Met_R-vs-NR = 6.89e-12. For CD8 T, pPre_Primary-vs-LN-Met = 3.07e-10, pPost_R-vs-NR = 8.29e-12). i-j) The advanced RCC cohort from Bi et al. j) Enriched CD4/CD8 TSTR cells from tumors exposed to ICB treatment, and enriched CD4 TSTR cells in NR tumors post-ICB treatment. k-n) The resectable NSCLC cohort. l) Enriched CD8 TSTR cells in tumors from patients with no major pathological response (non-MPR) post-ICB treatment among MANA-specific CD8 T cells. (m) Significantly higher expression of HSPA1A (all p values < 2.2e-16) and n) HSPA1B in CD8 T cells in tumors from non-MPR patients compared to those from MPR patients post-ICB treatment, among MANA-specific CD8 T cells (all p values < 2.2e-16). MPR, defined as < 10% viable tumor at the time of surgery; MANA, mutation-associated neoantigens. MANA-specific CD8 T cells were identified using the MANA functional expansion of specific T cells (MANAFEST) assay. Viral (EBV and influenza)-specific T cells were identified using the viral functional expansion of specific T cells (ViralFEST) assay, as described in the original study. For c), d), g), h), m), and n), two-sided Welch’s t-test was applied to calculate p values: (****p≤0.0001), followed by FDR correction.
In the RCC cohort62 (Fig. 6i), we observed an enrichment of CD8 TSTR and CD4 TSTR cells in tumors exposed to ICB and highly enriched CD4 TSTR cells in NR tumors (Fig. 6j; Supplementary Fig. 14a). In both cohorts of resectable breast cancer48, the CD4/CD8 TSTR cell fractions increased 2–3 times in on-treatment tumors from patients with limited/no T cell clonal expansion compared to those with clonal expansion (p = 0.015 and p = 0.026, respectively). However, the proportion of TSTR among T cells was generally low and not presented in the data. In the advanced TNBC cohort 64, we observed increased expression of HSPA1A and HSPA1B in CD4 and CD8 T cells from NR (vs. R) tumors before and after treatment in patients who were treated with paclitaxel (Supplementary Fig. 14b), but not in those received paclitaxel plus anti-PD-L1.
Finally, we investigated whether CD8 TSTR cells are specific for mutation-associated neoantigens (MANA). Among several studies that have successfully detected MANA-specific T cells, the study by Caushi et al.63 made their scRNA-seq and scTCR-seq data on CD8 T cells available (Fig. 6k). MANA- and viral-specific (EBV, influenza) CD8 T cells were identified using the MANA and viral functional expansion of specific T cells assay, respectively63. We found enriched CD8 TSTR cells in the MANA (but not viral)-specific CD8 T cells in tumours from patients with no major pathological response (non-MPR) (Fig. 6l). Consistently, we observed significantly higher expression levels of HSPA1A and HSPA1B in MANA-specific CD8 T cells in tumours from non-MPR patients compared to those from MPR patients (Fig. 6m–n; two-sided Welch’s t-test, FDR adjusted p-value < 2.2e-16 for all comparisons).
Discussion
In this study, we provide a high-resolution T-cell reference catalogue with well-defined cell states and gene signatures for the research community. The unprecedented scale of the datasets has enabled us to elucidate 32 T cell states, including previously undescribed and overlooked states22. We were able to further dissect TREG, TFH, and proliferative T cell subsets and discover the TSTR state, leading to an improved understanding of the transcriptional heterogeneity of T cells. Importantly, all data have been harmonized together in a common analytical framework, providing a robust T-cell reference map for the community. To facilitate the use of this resource, we have built a user-friendly, interactive Single-Cell Research Portal (SCRP, https://singlecell.mdanderson.org/TCM/) for visualizing and querying the T cell maps built in this study (Supplementary Fig. 15). Additionally, we provide TCellMap (https://github.com/Coolgenome/TCM), an R script that automatically aligns and annotates T cells from a query scRNA-seq dataset with our reference maps (Extended Data Fig. 10).
The identification of the TSTR state is intriguing. In previous scRNA-seq studies, the expression of stress-related genes in T cells was thought to be a potential artifact related to tissue dissociation, and TSTR cells have been largely overlooked. In a recent study, the expression of HSPA1A, HSPH1, and HSPA6 in glioma-infiltrating CD3E+ T cells was confirmed by RNA in situ hybridization72. By integrating data from multiple independent single-cell and spatial profiling platforms, this study presents the first, most comprehensive pan-cancer characterization of TSTR cells at cellular, subcellular resolution, and within the tissue context. We demonstrate that TSTR cells are detectable in situ within the TIME across 6 cancer types examined. Notably, TSTR cells were mostly mapped to LAs or likely tertiary lymphoid structures (TLSs) within the tumour beds or surrounding tumour edges, indicating that they may play a role in the TIME. Given the acknowledged role of TLSs in cancer73, it would be of great interest to understand the crosstalk between CD4/CD8 TSTR cells, other T, B/plasma, and dendritic cell subsets that coexist with TSTR cells in TLSs, as well as its impact on the function of TLSs and anti-tumor immunity.
The roles of TSTR cells in tumor immunobiology and immunotherapy response remain largely unknown. Our analysis of 16 scRNA-seq studies and the CPI1000+ cohorts63 collectively suggests that the presence of TSTR cells within TIME is biologically relevant and has potentially significant clinical implications. Our results demonstrate that CD4/CD8 TSTR cells are enriched in aggressive cancer subtypes or metastases and are associated with an unfavorable response to ICB therapy, more consistently and strongly than other known T cell subsets (TEX, TREG, or TFH) in the examined cohorts38,51,55,70,71. Interestingly, we found that the expression of stress response signature was massively upregulated following anti-PD-1/PD-L1 therapy, primarily in NR tumours, and TSTR cells were predominantly upregulated in tumour-specific CD8 T cells. These findings suggest that TSTR cells represent a distinct resistance mechanism to ICB therapy that warrants further validation in larger, longitudinal cohorts. Moreover, our trajectory analysis of CD8 T cells suggests that TSTR cells are likely from a diverged differentiation path, which is an intriguing area for further investigation. It is essential to track the differentiation trajectories of CD8 T cells, their cells of origin, phenotypic transition, and antigen specificity during disease progression and the course of ICB therapy. We also need to investigate the causes of stress responses in TILs mechanistically. Their inconsistent and puzzling relationships with hypoxia68 across cancer types may be indicative of other properties of the tumor, which requires additional investigation to determine the causality.
This study has several limitations. First, the analysis of T-cell receptor (TCR) repertoire was limited by the unavailability of paired scTCR-seq data for most of the datasets collected. Second, the lack of paired primary-metastatic tumors prevented the inference of changes in TIL states with tumor progression. Third, the Visium platform does not provide single-cell resolution, potentially leading to the omission of diffuse T-cell infiltrates in the TIME. Therefore, further spatial profiling of TILs at cellular, subcellular resolution in larger cohorts could result in a comprehensive understanding of their spatial neighborhoods, multicellular modules, and signaling hubs74.
In summary, our work addresses several challenges faced by the research community, such as the need of a reliable TIL reference map and the need for an automatic tool to align/annotate T cells to a desired level of granularity, in order to facilitate T-cell therapy optimization, biomarker discovery, and clinical applications. Notably, our study sheds light on the potential involvement of TSTR cells in immunotherapy resistance, and this finding may guide future efforts in the development of biomarkers and therapeutic targets. Moreover, investigating stress response in T cells in the context of CAR T-cell therapy and TIL therapy could be an interesting avenue for future research. Finally, our findings may stimulate further research on stress response in other TIME cell types, which have promising translational potential.
METHODS
scRNA-Seq data collection
Transcriptome data for T cells in 486 samples from 324 individuals across 27 scRNA-seq datasets (Fig. 1a–b; Supplementary Tables 1–2) were obtained, including 10 generated internally in the Advanced Technology Genomics Core (ATGC) facility at the University of Texas MD Anderson Cancer Center (MDACC), and the rest from public studies13,15,16,28,22–34. The data accession numbers and references for public datasets13,15,16,18,22–34 including those generated by us are provided in Supplementary Table 1. Detailed clinical information on patients and samples is provided in Supplementary Table 2. For the collection of public datasets, we included all accessible datasets that were released on or before June 2020. Samples that passed quality control and datasets with at least 500 cells were selected. The normal bone marrow dataset from healthy donors was downsampled to 30,000 cells (about 20% of the original size). Additional filters included quality filtering and doublets removal (as described in the following sections). In addition, six scRNA-seq datasets18,44,48,62–64 from patients who received ICB therapy were included for validation purposes. The data accession numbers and references for these datasets18,44,48,62–64 and detailed clinical information are provided in Supplementary Table 15. Furthermore, as a demo of TCellMap, four additional scRNA-seq datasets were collected and processed. The data accession numbers and references for these datasets are provided in Supplementary Table 16.
scRNA-Seq data generation
We included in-house scRNA-seq data from unique patient cohorts, such as early-stage lung cancers with tumour and matched normal lung tissues, a large lymphoma cohort including follicular lymphoma (FL) and large B-cell lymphoma (LBCL), low-grade glioma (LGG) and aggressive glioblastoma (GBM) together with non-neoplastic brain tissues, HPV+ head and neck cancer (HNSC), paired primary-metastatic stomach adenocarcinomas (STAD) together with matched normal stomach tissues and PBMC samples, and a cohort of acute myeloid leukemia (AML) with longitudinal samples collected during ICB therapy. In addition, we generated scRNA-seq data on normal bone marrow (BM), PBMC samples, and reactive lymph nodes (LN) from healthy donors.
For these in-house cohorts, all datasets (except the STAD) were generated in the ATGC facility at MDACC. All experiments were compliant with the review board of MDACC, and the studies were conducted in accordance with the Declaration of Helsinki. For the LUAD (LC_1) study, as we previously described75, all samples were obtained under the waiver of consent from banked or residual tissues approved by MDACC internal review board (IRB) protocols (PA14–0077 and LAB90–020). For the rest of the cohorts, written informed consent was provided by all patients. Tumor specimens were collected with informed consent in accordance with the MDACC IRB-approved protocols (LN_1, LN_2, and BRCA_2: PA19–0420; GBM: 2012–0441; AML: PA12–0305; HNSC_2: 2019–1059, LAB02–039, and PA18–0782; LUAD LC_5: PA14–0276; OV: 2017–0264). For the STAD dataset, the study was approved by the Ethics Committee of Zhejiang Cancer Hospital (# IRB-2020–109) and all patients provided written informed consent to participate. All patients were at stage IV and treatment-naïve prior to sample collection. Fresh tumors or biopsies were placed in 10% FBS RPMI 1640 media after collection and transferred to the laboratory for immediate processing. The tissues were minced and enzymatically digested37. Following red blood cell removal, cells were filtered, counted, and stained with SYTOX Blue viability dye (S34857, Life Technologies), followed by fluorescence-activated single cell sorting (FACS) to collect viable singlet cells. The methods for sample collection, processing, library preparation, and sequencing for our in-house cohorts were described in our previous studies14,22,23,31,68. We selected samples that passed quality control and datasets that had at least 500 cells. Additional filters included quality filtering and doublets removal (as described in the following sections).
scRNA-seq data integration, quality control, and data filtering
Raw scRNA-seq datasets generated in-house were pre-processed (demultiplex cellular barcodes, read alignment, and generation of gene count matrix) using Cell Ranger Single Cell Software Suite (v3.1.0, 10x Genomics). Quality control metrics were generated and evaluated. For previously published scRNA-Seq datasets, cell annotation tables (including quality control metrics, cell types, etc.) were obtained from the original publications. T cell clusters were selected based on either available cell type annotation or identification using Seurat (version 3.1.0)76 with default parameters and based on the unique expression of T-cell marker genes (e.g., CD3D, CD3G). Normalization was performed using Seurat, dividing the UMI counts of genes by the total UMI count of each cell, and scaling by 1e4 for computational efficiency. Count data, when unavailable, were replaced with CPM/TPM data. All normalized data was log2-transformed.
We integrated all datasets using the Seurat (version 4.0) rPCA approach. First, the T cell expression matrix of each dataset was normalized by NormalizeData function with default parameters. Next, we applied the FindVariableFeatures function with default parameters to detect HVGs for each normalized matrix. The SelectIntegrationFeatures function was then applied with nfeatures = 1000 to choose genes for integrating multiple datasets. We then removed cell cycle-related genes from the gene set to reduce the cell-cycle effect on data integration. The ScaleData and RunPCA functions were applied sequentially with parameter features set to these genes. After that, the matrix for each dataset was scaled and principal component analysis (PCA) was performed. We used the FindIntegrationAnchors function with reduction=”rpca” to find a set of anchors between all matrices, which were used to integrate the matrices with the IntegrateData function and parameter dims=1:50. Finally, we applied the ScaleData function to scale the integrated matrix with default parameters.
The data matrices were annotated with the sample, patient, and project IDs, and then filtered to remove likely cell debris and doublets, using similar approaches as described in our previous studies14,2,,23,31,68. Briefly, cells with low complexity libraries (in which detected transcripts are aligned to < 200 genes), likely dying or apoptotic cells (where >15% of transcripts are derived from the mitochondria), and cells with high-complexity libraries (in which detected transcripts are aligned to > 6,500 genes) were removed. UMAP (uniform manifold approximation and projection)77 plots were generated, and the expression of canonical marker genes was reviewed to further identify and clean doublets. T cells co-expressing discrepant markers of other cell lineages (e.g., cells in a T-cell cluster showed expression of canonical marker genes of epithelial or B, myeloid, and stromal cell lineages) were further cleaned. For studies with paired single-cell V(D)J sequencing data generated on the same libraries, cells with both productive T cell receptors and B cell receptors or had ≥2 productive T cell receptors were further removed. The filtering process was repeated to ensure high-quality cells, resulting in 308,048 cells for further analysis.
Batch effect evaluation and correction
We evaluated the significance of batch effects and the performance of two commonly used batch correction methods, harmony71 and reciprocal principal component analysis (rPCA)72, using the silhouette score80. The score measures how similar an object is to its own cluster (batch) compared to other clusters (batches) and ranges from −1 to +1, where a high value indicates that the object is well-matched to its own cluster (batch), and poorly matched to neighboring clusters (batches). We computed the silhouette score for each cell using the following formula. For cell (cell in batch ), let
be the mean distance between and all other data points in the same cluster, where is the number of points belonging to batch , and is the distance between cell and in the batch . Let
be the smallest mean distance of to all points in any other cluster, of which is not a member. The cluster with this smallest mean dissimilarity is said to be the “neighboring cluster” of because it is the next best-fit cluster for point . Then the silhouette score of cell is
HVGs were identified using the FindVariableFeatures function of Seurat69, and PCA was performed using the top 2,000 HVGs. Harmony and rPCA were applied to remove batch effects in the PCA space when clustering major cell lineages, and each cell’s silhouette score was computed as described previously74. The average score of all data points of a cluster was used to quantitatively assess overall batch mixing. For major cell types, CD4+ and CD8+ T cells, the silhouette scores were computed using the 20% downsampled data, and this process was repeated 20 times. The silhouette scores obtained from the downsampling analysis were then aggregated and averaged. Consistent with the results of a recent benchmark study of batch-effect correction methods for scRNA-seq data74, the Seurat rPCA approach72 showed better performance (lower silhouette score) than harmony (Supplementary Fig. 1a), displaying a good ability to mix batches while preserving cell type purity.
Unsupervised cell clustering and subclustering analysis
HVGs were further filtered to remove mitochondrial genes, ribosomal genes, and T cell receptor genes that could potentially influence cell clustering results. The Shared Nearest Neighbor (SNN) graph was constructed using the FindNeighbors function, and unsupervised clustering was performed with the FindClusters function. Clustering analysis was conducted separately on cycling and non-cycling cells due to their distinct expression of cell proliferation markers25. Multiple rounds of clustering and subclustering analysis were performed to identify major cell types (e.g., CD4 T cells, CD8 T cells, NKT cells, MAIT cells, proliferative T cells) and distinct cell transcriptomic states within each major cell type. UMAP70 was performed with the Seurat function RunUMAP for dimensionality reduction and 2-D visualization of the single cell clusters. The number of significant principal components (PCs) was determined based on the elbow plot generated with the ElbowPlot function of Seurat (Supplementary Fig. 1c). ROGUE75, an entropy-based statistic, was applied to quantify the purity of identified cell clusters. Various resolution and PC parameters for unsupervised clustering were evaluated, and the resulting UMAP plots and cluster marker genes were reviewed to determine the optimal number of clusters and guide the proper clustering of our scRNA-seq datasets (Supplementary Fig. 2a and 3a). For CD4 and CD8 T cells, the first 50 PCs, calculated using 1,978 HVGs identified by Seurat were used for unsupervised clustering with the resolution set to 0.3, yielding a total of 14 and 12 cell clusters, respectively (Figs. 2a and 3a). For unconventional T cells, the first 5 PCs, and 1,792 HVGs identified by Seurat were used for unsupervised clustering with a resolution set to 0.3, yielding a total of 5 cell clusters (Extended Data Fig. 4a). For proliferative T cells, the first 15 PCs, and 1,748 HVGs identified by Seurat were used for unsupervised clustering with a resolution set to 0.3, yielding a total of 8 cell clusters (Extended Data Fig. 4c).
Determination of major T cell types and cell states
To define major cell types and cell states, we integrated information from multiple steps. First, we identified differentially expressed genes (DEGs) using the FindAllMarkers function in Seurat R package69 for major cell types. DEG lists were filtered based on the following criteria: expressed in at least 20% of cluster cells; expression fold change >1.2, and the FDR q-value <0.05. Second, we generated feature and bubble plots for top 50 DEGs and immune cell markers (Supplementary Tables 3, 5, 7–10). Third, gene signature scores were calculated for curated gene sets related to T cell functional states (Supplementary Tables 4 and 6) for each cell cluster of CD4/CD8 T cells using Seurat’s AddModuleScore function. Finally, we integrated multiple layers of information, including cluster distribution, cluster-specific genes (particularly the top 50 DEGs), expression of cluster marker genes and canonical immune cell markers, as well as functional gene signatures and carefully reviewed by our multidisciplinary team, along with by an extensive literature search to carefully annotate cell transcriptomic states.
Single-cell trajectory inference
We applied Monocle3 (version 0.2.0)47 to reconstruct the cellular differentiation trajectory of CD8 and CD4 T cell subsets. Specifically, the clusters were divided into large, well-separated partitions using the function cluster_cells and fitted a principal graph within each partition using the function learn_graph. The principal graph, displayed on the UMAP as “skeleton lines”, indicating the differentiation trajectories. Based on prior knowledge, we selected the naïve T cell cluster CD8_c3 and CD4_c6 as the root for the trajectory for CD8 and CD4 T cells, respectively. We ran the learn_graph function with Euclidean distance ratio set to 0.1 and 0.2, minimal branch length set to 15 and 30, and geodesic distance ratio set to 0.8 and 0.3 to build the CD8 and CD4 T cell trajectories, respectively.
Quantification of tissue enrichment of T cell subsets
We utilized the Ro/e approach, as previously described8 to assess the enrichment or depletion of each T cell subset in specific tissue types (or cancer types/subtypes). Briefly, we calculated the ratio of observed cell number to random expectation using a chi-square test in each cluster across different tissue groups (or cancer types/subtypes). An Ro/e value > 1 indicates enrichment, while an Ro/e value < 1 indicates depletion of cells in a specific tissue or cancer types/subtype.
Quantifying T cell subset transcriptome similarity and inferring co-occurrence patterns
To evaluate the phenotypic relationships and transcriptome similarity among 31 non-proliferative T cell subsets identified in this study, we employed unsupervised hierarchical cluster analysis based on the Euclidean distance matrix to generate a dendrogram. Additionally, we conducted sample-level Spearman correlation analysis of cell cluster frequencies across distinct tissue groups (healthy, uninvolved normal, primary tumor, and metastatic tumor tissues) to investigate the co-occurrence patterns of various T cell states in both normal and tumor tissues, taking into account both positive and negative associations.
T cell deconvolution analysis and correlation of T cell subsets with clinical and histopathological variables in scRNA-seq, TCGA, and CPI1000+ cohorts
To examine the correlation between T-cell states and clinical variables, we first collected corresponding information for our scRNA-seq, TCGA, and CPI1000+ cohorts56. Clinical and histopathological data for scRNA-seq cohorts were downloaded from original studies (Supplementary Table 2). Bulk mRNA-seq expression data (normalized) for TCGA cohorts were downloaded from the NCI Cancer Genomic Data Commons (NCI-GDC: https://gdc.cancer.gov). Clinical annotation, survival data and the tumour mutational burden (TMB) was obtained from previous TCGA Pan-cancer studies76,77. Clinical data for the CPI1000+ cohorts were obtained from the original study56, and mutation quality characteristics and bulk expression data were kindly shared by Drs. Litchfield and Swanton. A total of 1, 008 patients, 562 patients with genomic data, expression data, and clinical response data, were included in the analysis. To analyze TCGA cohorts, samples with low T cell frequencies were identified using MCP-counter78 based on MCP-counter-inferred T cell gene signature scores, and the bottom 25 % samples were excluded from subsequent T cell deconvolution analysis (Supplementary Table 13).
T cell deconvolution was performed on normalized bulk expression data from tumour samples of TCGA cancer types and their genotypic/molecular subtypes (n = 52) as well as those of samples of the CPI1000+ cohorts using unique gene signatures from 9 T cell states in this study (Supplementary Table 12). The gene signatures were extracted from the top 30 most significant DEGs followed by additional filtering to ensure the uniqueness of the gene signatures. For T-cell deconvolution analyses in both cohorts, we included only patients with available bulk expression data. Spearman’s correlation analysis was applied to quantify correlations between levels of signature gene expression and tumour mutation burden (TMB) across cohorts of main cancer types as well as their genotypic/molecular subtypes, followed by FDR (false discovery rate) correction of resulting p-values for multiple hypothesis testing. For survival analysis, the Cox proportional hazards model was fit using patient groups dichotomized by the median level of signature expression (high or low). Similarly, as described above, the p-values were adjusted from multiple testing.
T cell reference mapping using the R script TCellMap
We developed an R script (TCellMap.R, https://github.com/Coolgenome/TCM) based on the Seurat R package (v4) to map T cells from a query dataset to our T cell maps. The bioinformatics flow involves extracting T cells (as described in the section “scRNA-seq data integration, quality control and data filtering”), normalizing and scaling query, identifying HVGs, removing cell cycle-related genes from DEGs (Supplementary Tables 3 and 5) and HVGs, performing PCA, and finding transfer anchors by FindTransferAnchors function with reference.reduction=”rpca” on intersection of DEGs and HVGs, mapping query data to reference data by MapQuery function with default parameters, and automatically annotating cells, as shown in Extended Data Fig. 10. The results could be exported as a plain text or visualized in UMAP plots. To evaluate the method’s effectiveness, we performed leave-one-out cross-validation on annotated single-cell datasets with ≥ 5,000 T cells. The prediction accuracy was computed by comparing the automatically assigned T cell states with their original cell labels (Extended Data Fig. 10b). Additionally, we demonstrated the T cell reference mapping by processing 4 additional query datasets (as listed in Supplementary Table 16) using the methods described above (Extended Data Fig. 10c).
Detection of TSTR cells in situ by RNAscope
RNA in situ hybridization is a method to detect target RNA within intact cells. RNAscope 2.5 LS Reagent Kit with red chromogen (ACDBio, cat. no. 322150) was used on a Leica Bond RXm automated stainer. The procedure recommended in the user manual was followed including a 15-minute 95°C antigen retrieval in Tris-EDTA buffer, a 15-minute protease digestion, and subsequent one-minute probe hybridization. The Hs-HSPA1B target probe (ACDBio, cat. no. 1101828-C1) was run alongside positive Hs-PPIB (ACDBio, cat. no. 313908) and negative dapB (ACDBio, cat. no. 312038) control probes. Stained slides were dried at 60°C for 30 minutes and mounted using VectaMount Permanent Mounting Medium (Vector Laboratories, cat. no. H-5000).
Detection of TSTR cells in situ using the CosMx SMI datasets
To verify the detection of TSTR cells in situ, we analyzed the public CosMx Spatial Molecular Imager (SMI, NanoString) datasets, 60 including five non-small-cell lung cancer (NSCLC) samples (eight tissue sections) and one hepatocellular carcinoma (HCC) sample (one tissue section) (https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/). The methods for data processing, cell segmentation, and cell type identification were described in the original study60. Cellular spatial contexts (x, y coordinates), along with transcriptome data and cell type annotations were extracted, then co-expression patterns of T cell markers and heat shock genes in the same tissue section were examined. Spatial locations of these transcripts were mapped to the corresponding cells (based on the x, y coordinates) to confirm the co-expression of CD3D, HSPA1A, and HSPA1B in the same cells at cellular and subcellular resolutions across all tissue sections.
Collection and generation of spatially resolved transcriptomics (SRT) data
We downloaded four published SRT datasets, including breast cancer (BC)79, Melanoma80, cutaneous squamous cell carcinoma (CSCC) 82, and clear cell renal cell carcinoma (ccRCC) 91. The BC and melanoma datasets were generated using the spatial transcriptomics technology, and the CSCC and ccRCC datasets were generated using the Visium spatial platform (10X Genomics). We also generated SRT data on lung adenocarcinoma (LUAD) and gastric adenocarcinoma (GAC) samples using the Visium spatial platform as described in our previous study69. Briefly, the FFPE tissue blocks with DV200 >50% were selected for sectioning. Appropriate-sized sections were placed within the frames of capture areas on the Visium Spatial Gene Expression Slide (PN-1000188, 10X Genomics) with one section in each capture area (6.5 × 6.5 mm). Tissues were deparaffinized, stained, and decross-linked, followed by probe hybridization, ligation, release, and extension. Visium spatial gene expression FFPE libraries were constructed with Visium Human Transcriptome Probe Kit (PN-1000363) and Visium FFPE Reagent Kit (PN-1000361) following the manufacturer’s guidance, and sequenced on the Illumina NovaSeq 6000 platforms to achieve a depth of at least 75,000 mean read pairs and 2,000 median genes per spot.
SRT data processing and analysis, mapping the spatial locations of T cells and TSTR cells
We obtained gene expression count matrices and histology images for the four public SRT datasets from their original studies79–82 and processed our in-house data using the Space Ranger pipeline (v-1.3.0, 10x Genomics) with default parameters. The processed data were then analyzed using the TESLA python package v1.2.2 (https://github.com/jianhuupenn/TESLA). We applied TESLA to map the spatial locations of T cells directly on histology images and to detect the tissue border by the “cv2_detect_contour” function with parameter “apertureSize=5, L2gradient=True”, then enhance gene expression at the super-pixel level within the tissue border by the “imputation” function with parameter “s=1, k=2, num_nbs=10”. Next, we used the “annotation” function to annotate cell types. T cells were identified based on their expression of T cell markers (e.g., CD3D, CD3G). The “visualize_annotation” function was then applied to project the spatial locations of T cells directly on histology images. For each tissue section, regions with detectable T cell signals were pathologically reviewed and analyzed at high magnification to confirm lymphocyte presence (Extended Data Figs. 7 and 8). Next, we examined the heat shock gene (e.g., HSPA1A and HSPA1B) expression in spots with T cell signals and further mapped the spatial locations of TSTR cells. In addition, to examine whether the presence of TSTR cells in the TIME was associated with tumor hypoxia, we curated a list of transcription factors (TFs) that are reported to be activated under hypoxic conditions61 including HIF1A, EPAS2, RELA, RELB, NFKB1, NFKB2, CREB1, and NFE2L2. In the same manner, as described above, we projected hypoxia-related gene activity on histology image and then overlaid the hypoxia signal and the spatial locations of TSTR cells on the same images using a custom script (https://github.com/Coolgenome/TCM/blob/main/res_largerT.py#L230).
To investigate the relationship between heat shock gene expression and hypoxia within the TIME, we leveraged the Seurat (v4)72 rPCA workflow to correct the potential batch effect in each Visium dataset. This was conducted in the manner described in the scRNA-seq data integration, quality control, and data filtering section. We then applied Seurat’s dimensionality reduction, clustering, and visualization workflow with default parameters, as detailed in the Determination of major T cell types and cell states section. Through this workflow, we identified clusters of spots enriched with T cells based on their marker gene expression, and performed Spearman correlation analysis to assess the association between the expression levels of heat shock genes and 8 hypoxia-related TFs within T cell-enriched spots. To ensure statistical significance, we performed Holm adjustment on the resulting p-values.
Collection of scRNA-seq data from patients received ICB therapy, data processing and analysis
We collected additional scRNA-seq data from 247 samples from 133 patients across 6 cohorts and 4 cancer types to evaluate the clinical relevance of T cell subsets including TSTR cells, in the context of ICB therapy. The data accession numbers and references for these datasets and detailed clinical information are provided in Supplementary Table 15. The scRNA-seq cohort from Yost et al.18 was included in the original data collection used to build the T-cell atlas. For the scRNA-seq datasets from Liu et al. 84 (GSE179994) and Bi et al.62 (SCP1288), CD4 and CD8 T cells were extracted based on cell type annotation provided by the original studies. For scRNA-seq datasets from Caushi et al.63 (GSE173351) and Zhang et al. 64 (GSE169246) without a cell type annotation, read count matrices were merged using Seurat, followed by quality filtering and batch effect correction as described in the section “scRNA-seq data processing, quality control and data filtering”, and unsupervised cell clustering analysis to identify and extract CD4 T and CD8 T cells. For the scRNA-seq dataset from Caushi et al.63 (GSE173351), the original study defined tumor-specific and viral-specific CD8+ TCR clonotypes by the MANAFEST and viraFEST assay, respectively. We integrated their TCR clonotype data and scRNA-seq data and identified CD8 T cells that were defined as tumor-specific or viral-specific.
TCellMap (https://github.com/Coolgenome/TCM) was then applied to uniformly align T cells extracted from each scRNA-seq dataset to the CD4 and CD8 T cell maps built in this study in the same manner as described in the section “T cell reference mapping using the R script TCellMap” (Extended Data Fig. 10). Annotation was added, and the abundance of each T cell subset was then quantified. For each T cell subset, we calculated Ro/e values to quantify their tissue prevalence between groups. For group-level analysis based on Ro/e values, only T cell subsets with ≥ 100 total cells were included (≥ 30 tumor- or viral-specific CD8 T cells for the scRNA-seq dataset from Caushi et al.63). In addition to Ro/e values, we also measured the expression levels of heat shock genes (e.g., HSPA1A and HSPA1B) in all CD4 and CD8 T cells and compared their expression levels between groups. The clinical response information for each dataset (Supplementary Table 15) was defined by the original studies. Comparison analysis was performed at multiple levels, such as between responsive (R) and non-responsive (NR) tumors, between different time points (e.g., pre- vs. post-ICB), and/or tissue types (e.g., primary vs. metastatic; ICB-naïve vs. ICB-exposed) for cohorts with available metadata.
Additional statistical analyses
In addition to the bioinformatics approaches and statistical analyses described above, all other statistical analyses were performed using statistical software R v3.6.0. To compare the fractions of different T cell clusters and subclusters across tissue groups in our single-cell studies (box plots in main and supplementary figures), and to compare expression levels of gene signatures across patient groups defined by PD-L1 expression (Supplementary Fig. 8b), the Games-Howell pairwise test was applied to calculate the p-values, followed by FDR correction for multiple hypothesis testing. For cell fraction comparisons across tissue or patient groups, samples that had < 200 T cells were excluded. Sex was not considered in the study design because there was insufficient statistical power to perform sex-specific analyses for most of the scRNA-seq datasets included in this study. All statistical significance testing in this study was two-sided unless specified, and results were considered statistically significant at P-values or FDR q-values < 0.05. When a p-value reported by R (v3.6.0) was smaller than 2.2e-16, it was reported as “P < 2.2 × 10−16”.
Extended Data
Extended Data Fig. 1. Major T cell types.
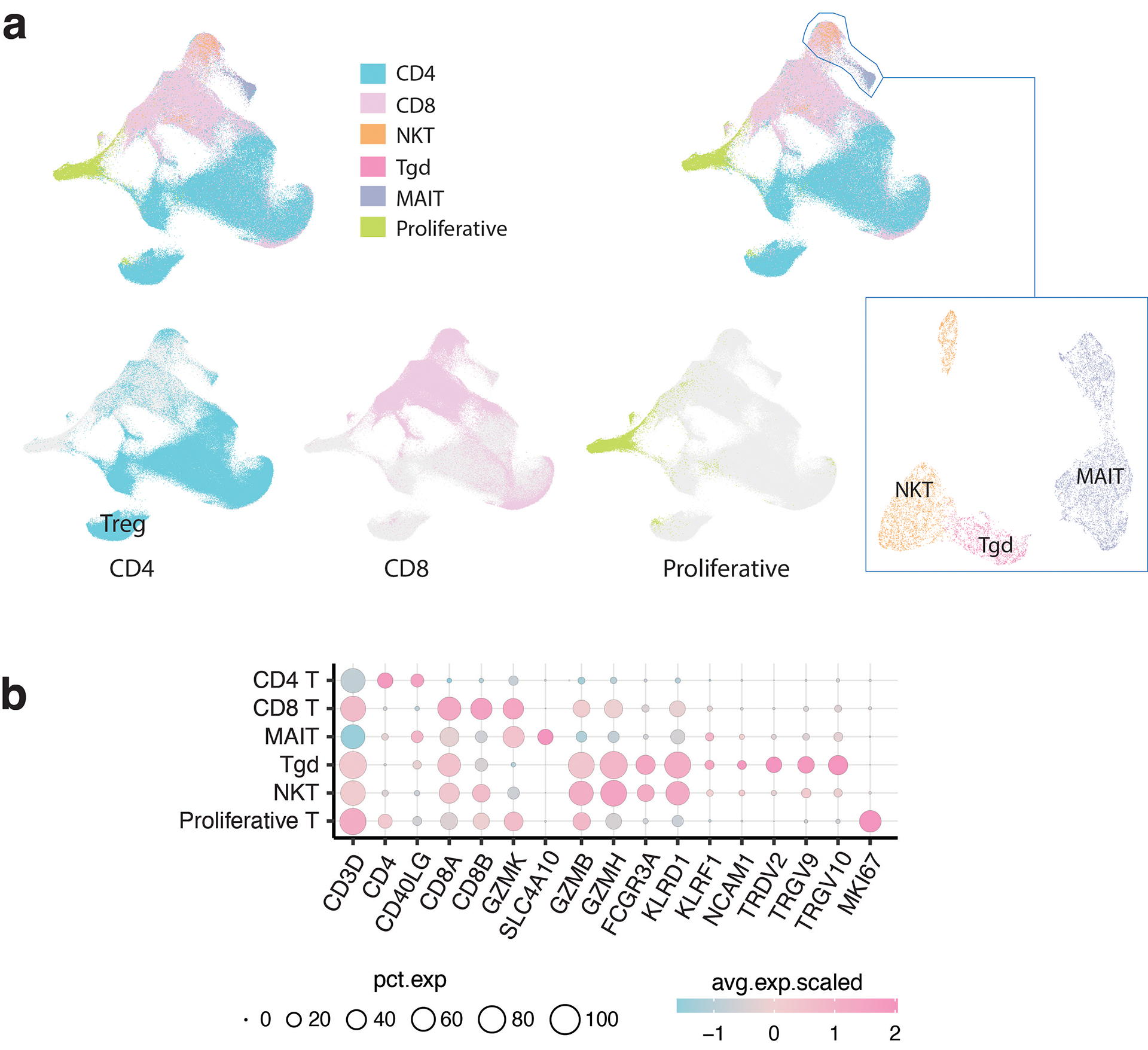
a) Global UMAP of all T cells and major T cell types. The subpopulations of CD4, CD8, proliferative, and unconventional T cells were further separated and defined by subsequent clustering analysis. b) Bubble plot showing the average expression levels and cellular fractions of representative marker genes across six major T cell types.
Extended Data Fig. 2. Characterization of CD8 T cell clusters.
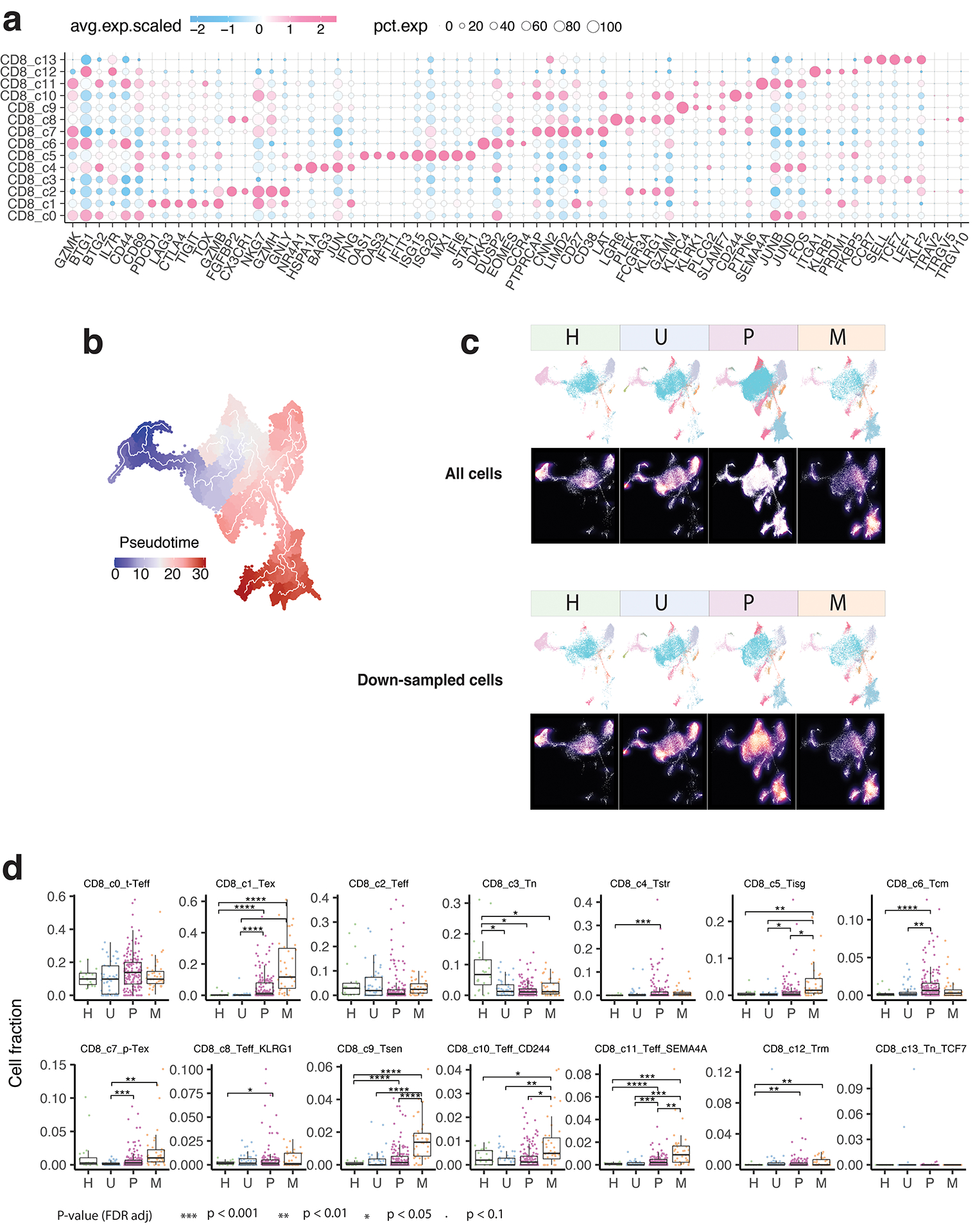
a) Bubble plot showing the average expression levels and cellular fractions of selected marker genes in 14 defined CD8 T cell clusters. The complete list of the top 50 most significant differentially expressed genes (DEGs) is provided in Supplementary Table 3. b) Monocle 3 trajectory analysis of CD8 T cell differentiation demonstrating multiple possible routes. c) The UMAP and density plots before and after downsampling analysis. UMAP (top) and density plots (bottom) of CD8 T cells demonstrating T cell distribution across four main tissue groups. High relative cell density is shown as bright magma. For CD8 T cells, the downsampled cell number is 11,592 cells for each tissue group. d) Box plot showing cell fractions of CD8 T cell subsets across four tissue groups. Each dot represents a sample. H, normal tissues from healthy donors; U, tumor-adjacent uninvolved tissues; P, primary tumor tissues; M, metastatic tumor tissues. The one-sided Games-Howell test was applied to calculate the p values between those four tissue groups (n = 20, 51, 156, 39), followed by FDR (false discovery rate) correction. FDR-adjusted p value: *≤0.05; **≤0.01; ***≤0.001; ****≤0.0001. Boxes, median ± the interquartile range; whiskers, 1.5× interquartile range.
Extended Data Fig. 3. Characterization of CD4 T cell clusters.
a) Bubble plot showing marker gene expression across 12 defined CD4 T cell clusters. The complete list of the top 50 most significant DEGs is provided in the Supplementary Table 5. b) The UMAP and density plots before and after downsampling analysis. UMAP (left) and density plots (right) of CD4 T cells demonstrating T cell distribution across four main tissue groups. High relative cell density is shown as bright magma. For CD4 T cells, the downsampled cell number is 10,703 cells for each tissue group. c) Box plot showing cell fractions of 12 CD4 T cell subsets across four tissue groups. Each dot represents a sample. H, normal tissues from healthy donors; U, tumor-adjacent uninvolved tissues; P, primary tumor tissues; and M, metastatic tumor tissues. The one-sided Games-Howell test was applied to calculate the p values between those four tissue groups (n = 20, 53, 158, 39), followed by FDR (false discovery rate) correction. FDR-adjusted p value: *≤0.05; **≤0.01; ***≤0.001; ****≤0.0001. Boxes, median ± the interquartile range; whiskers, 1.5× interquartile range. d) Monocle 3 trajectory analysis of CD4 T cells. Cells are color coded for their corresponding pseudotime. e) Ridge plots show the distribution of inferred pseudotime across CD4 T cell clusters.
Extended Data Fig. 4. Characterization of unconventional T cells and proliferative T cells.
a) UMAP view of 5 innate T cell clusters. b) Bubble plot showing marker gene expression across 5 innate T cell clusters. The complete list of top 50 most significant DEGs is provided in the Supplementary Table 9. c) UMAP view of 8 proliferative T cell clusters. d) Bubble plot showing marker gene expression across 8 proliferative T cell clusters. The complete list of top 50 most significant DEGs is provided in the Supplementary Table 10. e) Sankey diagram showing the mapping of four proliferative CD8 subsets to the rest of CD8 T cell clusters after regressing out cell proliferative markers. f) Sankey diagram showing the mapping of two proliferative CD4 subsets (P_c6_Treg and P_c1) to the rest of CD4 T cell clusters after regressing out cell proliferative markers.
Extended Data Fig. 5. Correlations with tumor mutational burden (TMB) and patient survival in TCGA cohorts.
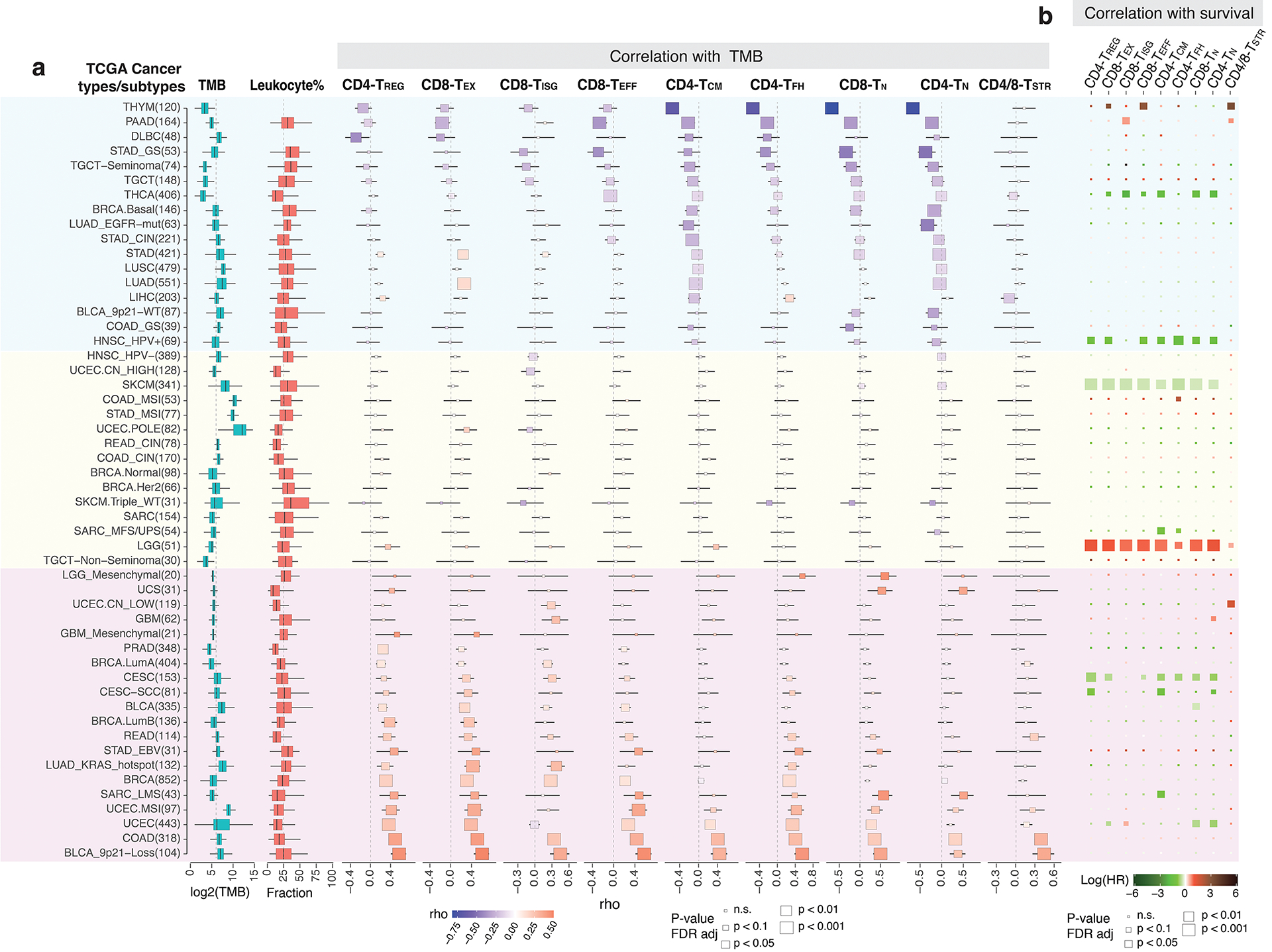
a) Correlation between the abundance of 9 T cell states (estimated via T cell deconvolution analysis using unique gene signatures in Supplementary Table 12) and TMB across 52 cancer types and their genotypic/molecular subtypes (labeled on the left with numbers indicating sample size). A total of 11,051 TCGA tumors with bulk RNA-seq data available were included and samples with low abundance of T cells (the bottom 25% of the ranked data) as estimated using MCP-counter were excluded (Supplementary Table 13). TMB and leukocyte fractions were from TCGA pan-cancer study by Thorsson et al.77. The annotation of cancer types and their genotypic/molecular subtypes was adopted from our recent study by Han et al. (Nature Communication, 12, 5606, 2021). The size of the rectangle is proportional to statistical significance (p-value, two-sided spearman correlation test, FDR-adjusted) and the color intensity is proportional to Spearman correlation coefficient (rho). Boxes, median ± interquartile range; whiskers, 1.5× interquartile range. b) Correlation with patient overall survival (OS). The size of the rectangle is proportional to statistical significance (FDR-adjusted p-value) and the color intensity is proportional to log scaled hazard ratio (HR).
Extended Data Fig. 6. Correlation with patient survival in the CPI1000+ cohorts.
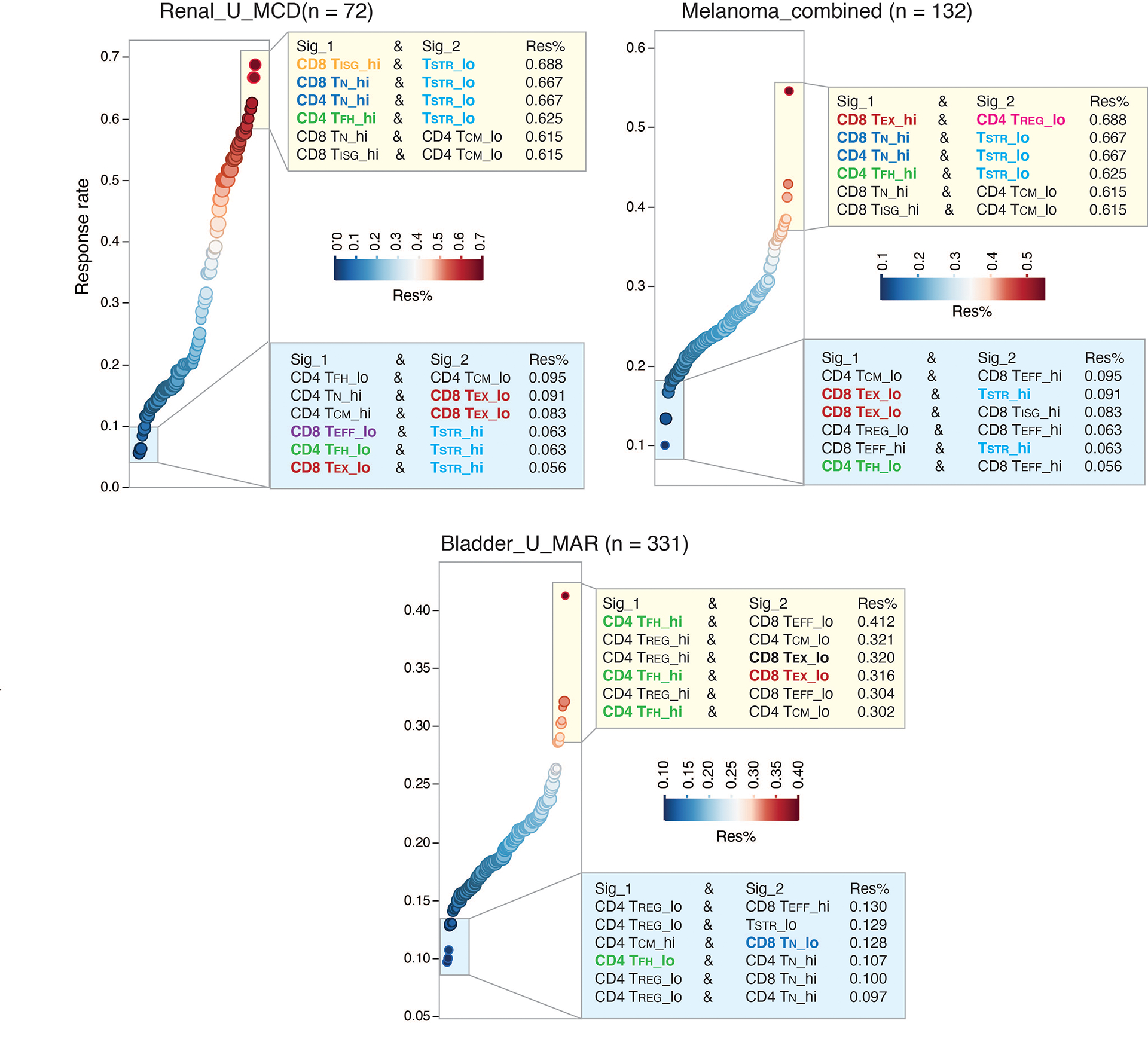
Association with ICT response in three large cohorts of cancer patients from the CPI1000+ cohort with both RNA sequencing and clinical response data available are shown. Samples predominantly represented baseline pretreatment specimens, treated with single-agent immune checkpoint inhibitor (CPI) and without prior CPI treatment. Patients of the bladder cancer cohort (Bladder_U_MAR) and renal cancer cohort (Renal_U_MCD) received single-agent anti-PD-L1 therapy and patients of the combined melanoma cohort received either single-agent anti-CTLA-4 or anti-PD-1 therapy. More details on the clinical data (for example, drug treatment and biopsy timepoint, radiological response) of these patients can be found in the Supplementary Table 1 of the original study by Litchfield et al.56. Immune deconvolution was performed on normalized gene expression data from the original study using the 9 gene signatures included in the Supplementary Table 12. For each cohort, we assessed the radiological response rates in patient groups with all the different possible combinations of T cell state gene signature expression. Patient groups showing the highest ICT radiological response rates (among top 6) or the lowest response rates (among bottom 6) are shown. Sig_1, T cell state 1; Sig_2, T cell state 2; Res%, response rate. Hi, high expression group; lo, low expression group. Hi and lo groups were split based on the group median value of gene signature expression. Recurrently presented gene signatures are highlighted in color.
Extended Data Fig. 7. Detection of in situ HSPA1A and HSPA1B expression in tumor-infiltrating T cells in NSCLC and HCC samples by CosMx.
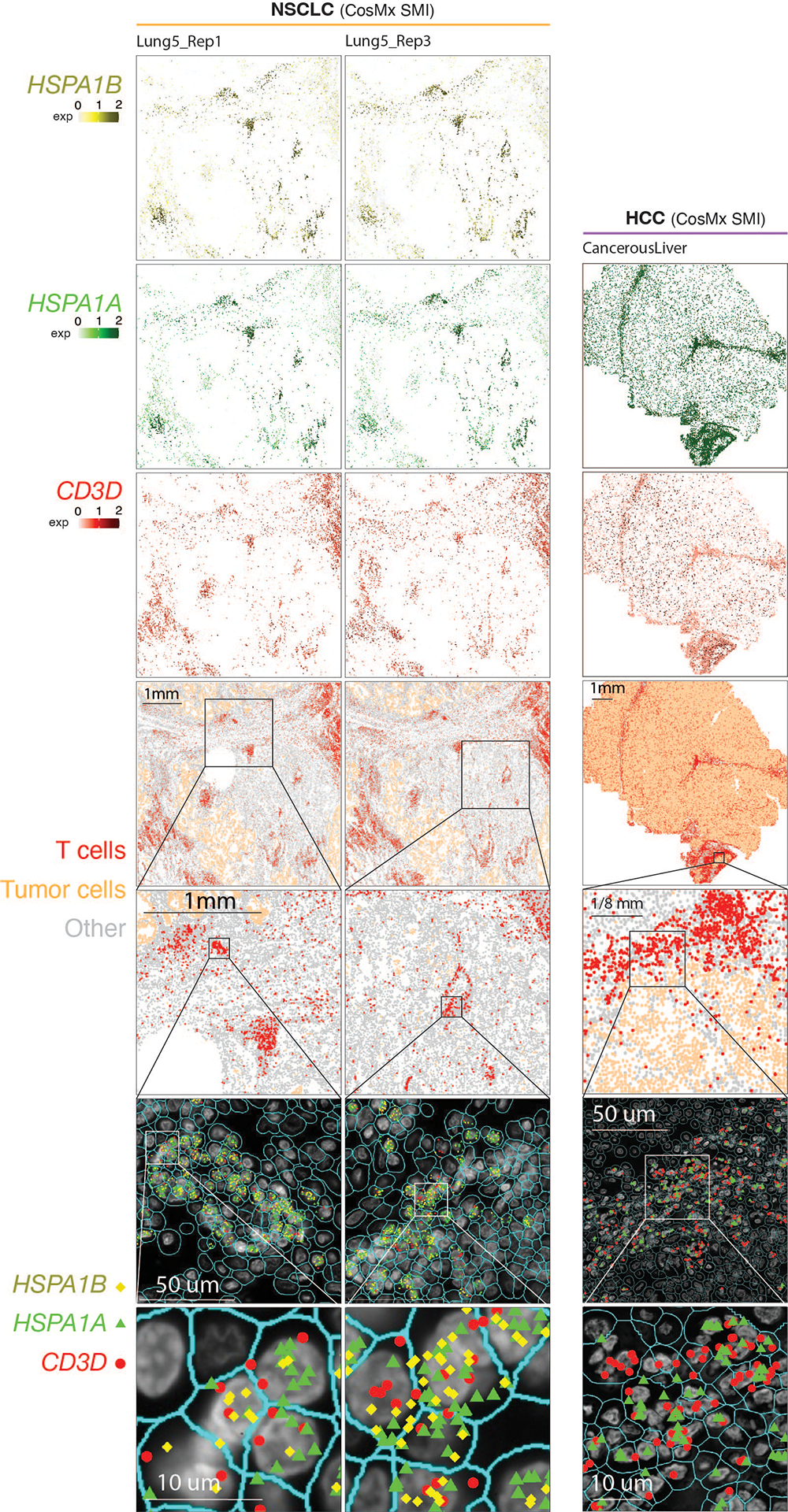
Two consecutive tissue sections from a NSCLC sample (sections ‘Lung 5_Rep1’ and ‘Lung 5-Rep3’) and one tissue section from a hepatocellular carcinoma (HCC) sample (section ‘CancerousLiver’) were profiled. (Column 1) Cells in physical locations (x, y coordinates). Color denotes cell type. Spatial mapping of CD3D (Column 2), HSPA1A (Column 4, Row 1/3/5), and HSPA1B (Column 3, Row 1/3) expression in T cells (note that, the HCC data does not include HSPA1B). (Column 2, Row 2/4/6) A zoom-in view of a representative area of their corresponding images in Column 1 showing lymphocyte aggregates enriched with T cells. (Column 3, Row 2/4/6) a zoom-in view of their corresponding images in Column 2 showing subcellular localization of CD3D, HSPA1B, and/or HSPA1A transcripts. (Column 4, Row 2/4/6) a further zoom-in view of their corresponding images in Column 3 showing co-localization of CD3D, HSPA1B, and/or HSPA1A transcripts in the same cells. Cell segmentation was done by the original study60. The outlines of cell nuclei were determined based on DAPI staining and the cell boundaries were determined based on morphology markers for membrane (for example, CD298) combined with a machine learning approach60.
Extended Data Fig. 8. Co-mapping of TSTR cells and hypoxia-related gene expression using spatial transcriptomics.
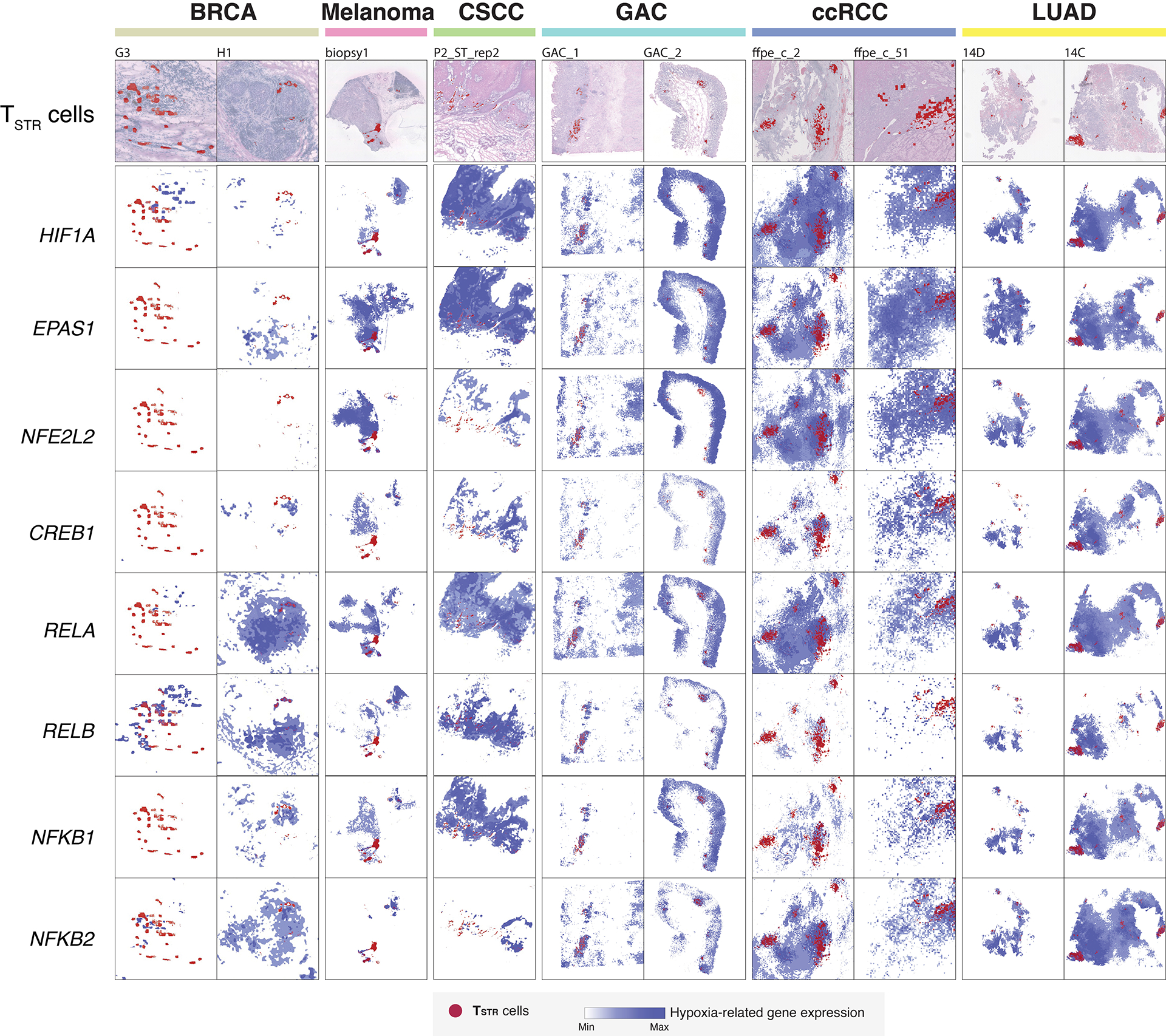
(Top row) Mapping of TSTR cells (in red) on the histology image based on corresponding spatial transcriptomics data generated from the same tissue section. (Rest of the rows) spatial co-mapping of TSTR cells (in red) and hypoxia-related gene expression (in blue, the darker the color, the higher the level of gene expression) in the same regions as shown in the top row. BRCA, breast cancer; CSCC, cutaneous squamous cell carcinoma; GAC, gastric adenocarcinoma; ccRCC, clear cell renal cell carcinoma; LUAD, lung adenocarcinoma.
Extended Data Fig. 9. Pan-cancer detection of TSTR cells and Co-mapping of TSTR cells and hypoxia-related gene expression using spatial transcriptomics.
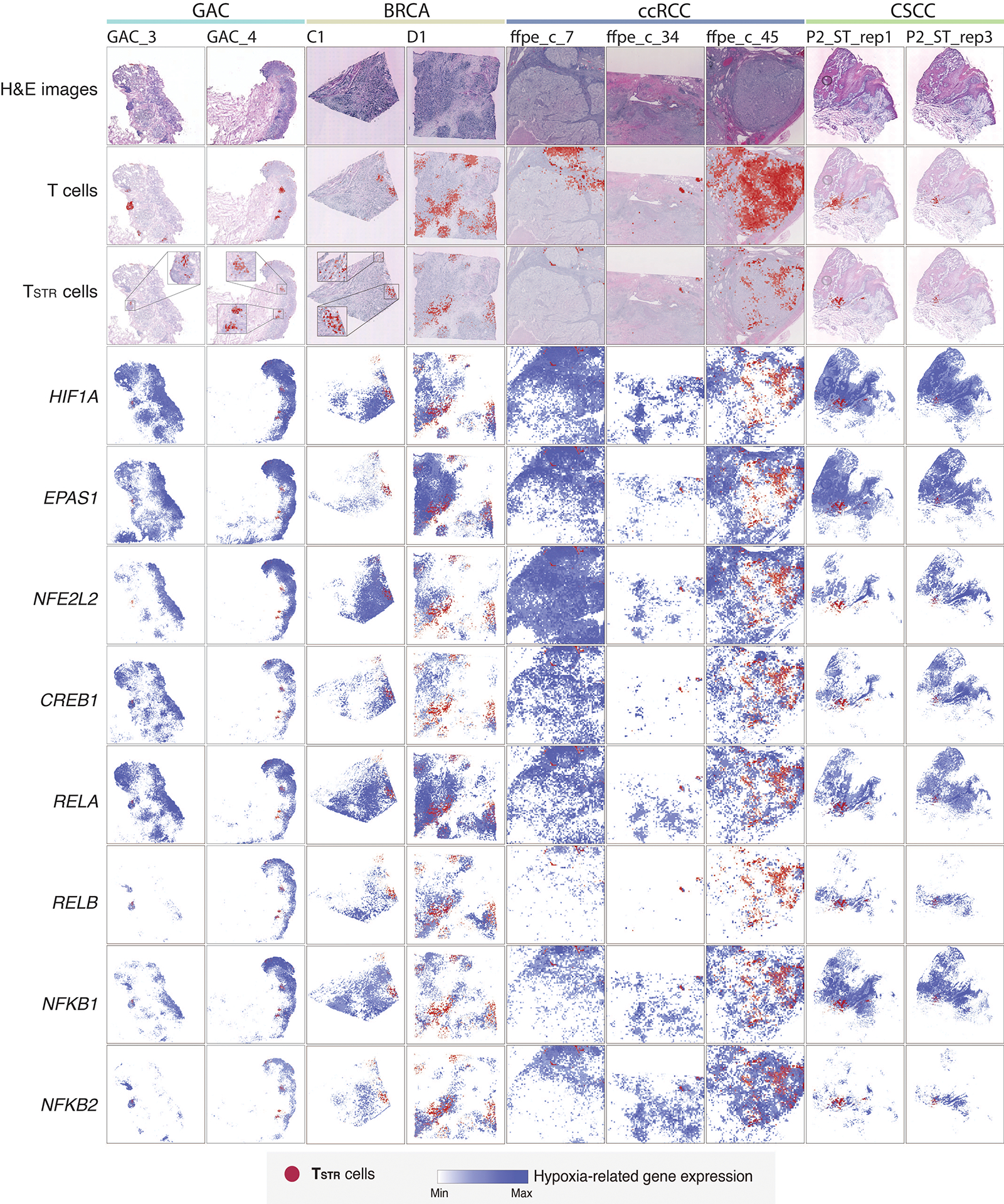
Extra representative tissue sections of 4 cancer types are shown. (Top row) H&E stained tissue images. (Second row) Mapping of T cells and (third row) the TSTR cells on the same histology images (GAC, BRCA, ccRCC, CSCC) or a high-magnification image (ccRCC). GAC, gastric adenocarcinoma; BRCA, breast cancer; ccRCC, clear cell renal cell carcinoma; CSCC, cutaneous squamous cell carcinoma. (Rest of the rows) spatial co-mapping of TSTR cells (in red) and hypoxia-related gene expression (in blue, the darker the color, the higher the level of gene expression) in the same regions as shown in the top row.
Extended Data Fig. 10. The workflow of TCellMap.
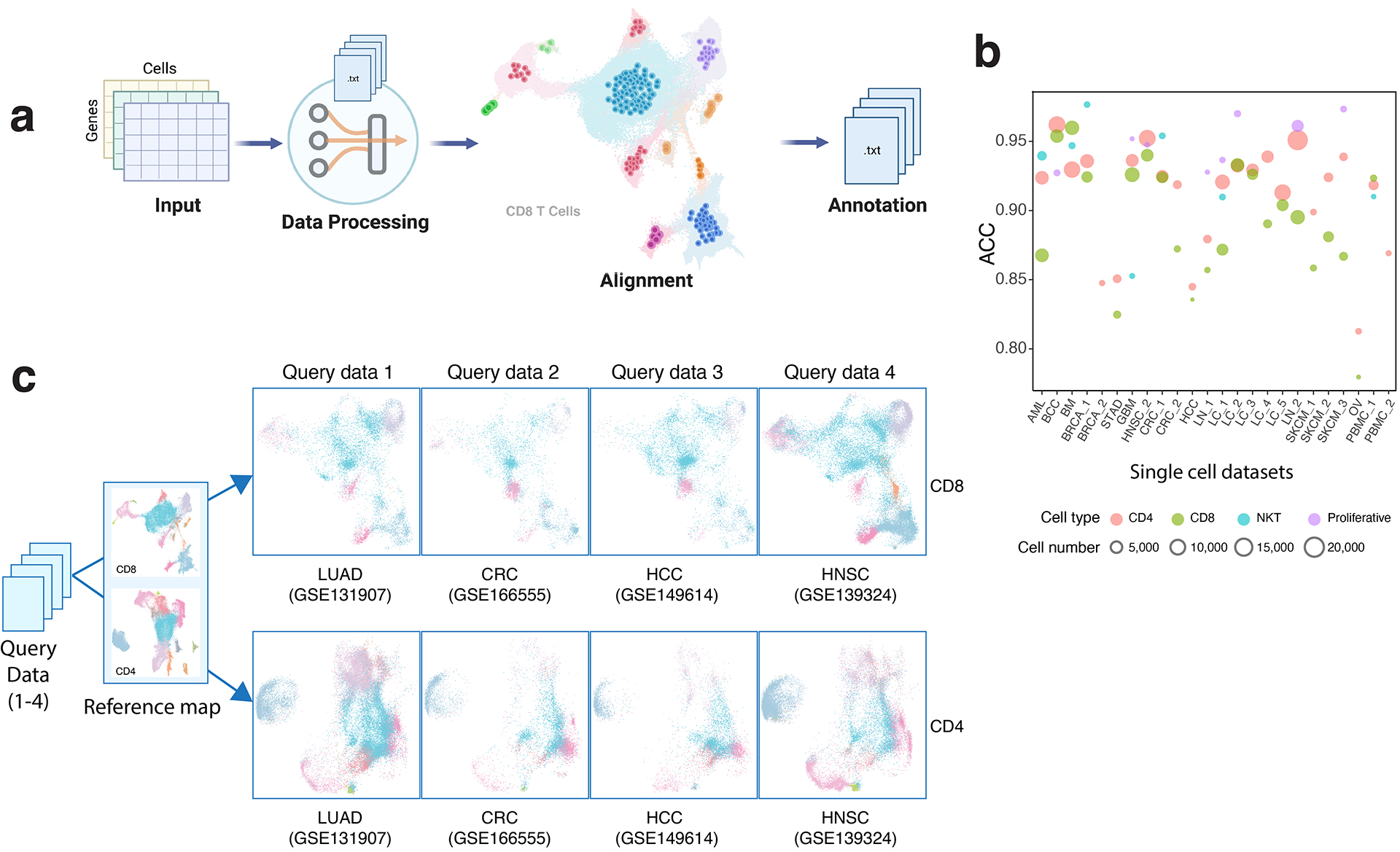
a) Schematic view of the bioinformatic flow of TCellMap, created with BioRender.com. b) Leave-one-out cross-validation of the performance of TCellMap using scRNA-seq datasets included in this study. Scatter plot showing the accuracy (ACC) of T cell state prediction. A total of 24 scRNA-seq datasets with ≥5,000T cells were selected (x axis), and the prediction accuracy was calculated by comparing T cell states automatically assigned for 32 states of the 5 major cell types using the reference maps with that manually annotated by this study. The size of the bubble corresponds to the number of T cells in each scRNA-seq dataset. c) Visualization of the output of TCellMap. Four scRNA-seq datasets that were not included in original data collection of this study were used as the query datasets. UMAP views of CD8 (top) and CD4 (bottom) T cells mapped in each query dataset. Cell clusters are color coded in the same way as in Fig. 2a (CD8 T cells map) and Fig. 3a (CD4 T cell map). LUAD, lung adenocarcinoma; CRC, colorectal carcinoma; HCC, hepatocellular cell carcinoma; HNSC, head and neck cancer. The gene expression count matrices were downloaded from the Gene Expression Omnibus (GEO) database and the accession codes (GSE#) are labeled for each dataset. Further details of each query dataset are provided in the Supplementary Table 16.
Supplementary Material
Acknowledgements
This study was supported in part by the NIH/NCI grants R01CA266280 (to L.W.), U01CA264583 (to H.K. and L.W.), the start-up research fund, and the University Cancer Foundation via the Institutional Research Grant (IRG) Program at the University of Texas MD Anderson Cancer Center (MDACC), and The Break Through Cancer Foundation (to L.W.), the Cancer Prevention and Research Institute of Texas (CPRIT) awards RP200385 (to L.W. and M.R.G.), RP220101 (to H.K. and L.W.), and RP150079 (to H. K.), research funding from Johnson and Johnson Lung Cancer Initiative (to H.K.), the DOD team grants CA160445 and CA200990 (to J.A.A.), a Leukemia and Lymphoma Society Scholar Award (to M.R.G.), and the generous philanthropic contributions to MDACC Moon Shots Program. In addition, A.M. was supported by Sheikh Khlaifa bin Zayed Foundation and the MDACC Moon Shots Program in Pancreas Cancer. L.W., M.R.G., and H.K. are Andrew Sabin Family Foundation Fellows at MDACC. M.G. is a CPRIT Scholar in Cancer Research. C.S. is supported by the Francis Crick Institute which receives its core funding from Cancer Research UK (CC2041), the UK Medical Research Council (CC2041), and the Wellcome Trust (CC2041). K.L. was supported by the UK Medical Research Council (MR/V033077/1), the Rosetrees Trust and Cotswold Trust (A2437), Melanoma Research Alliance, and Cancer Research UK (C69256/A30194). A.S. is supported by an NCI T32CA217789 MDACC postdoctoral fellowship. This study was also supported by MD Anderson Cancer Center Support Grant (CA016672) and a grant from the Emerson Collective to A.A.J. We thank Gaston Benavides, Peter Wang, Rui Jiang, Alex Liu, Kim-Anh Vu from the University of Texas MD Anderson Cancer Center for their excellent technical support in developing and testing the SCRP web portal.
Competing Interest Statement
A.A.J. has served as a consultant for Guidepoint, Gerson Lehrman Group, Nuprobe, AvengeBio, Agenus, AstraZeneca, Iovance, Bristol-Myers Squibb, Eisai, GSK/Tesaro, Macrogenics, Instill Bio, Immune-Onc Therapeutics, Obsidian, Alkermes, and Roche/Genentech, and he receives research support to his institution from AstraZeneca, Bristol-Myers Squibb, Merck, Eli Lilly, Pfizer, Aravive, and Iovance. Shareholder: AvengeBio. A.M. receives royalties from Cosmos Wisdom Biotechnology and Thrive Earlier Detection, an Exact Sciences Company. A.M. is also a consultant for Freenome and Tezcat Biotechnology. A.R. has received honoraria and serves on the scientific advisory board of Adaptive Biotechnologies. H.K. reports funding from Johnson and Johnson. M.R.G. receives research funding from Allogene, Kite/Gilead, Sanofi and Abbvie, has received honoraria from Monte Rosa Therapeutics, Daiichi Sankyo and Tessa Therapeutics, and has stock ownership interest in KDAc Therapeutics. X.L. reports consulting or advisory role for EMD Serono (Merck KGaA), AstraZeneca, Spectrum Pharmaceutics, Novartis, Eli Lilly, Boehringer Ingelheim, and Bristol-Myers Squibb, Dachii, Hengrui therapeutics, and she receives research funding from Lilly (Inst), Boehringer Ingelheim (Inst) and regeneron (Inst). Y.Y.E reports research funding from Spectrum Pharmaceuticals, AstraZeneca, Takeda, Xcovery, Lilly, Elevation Oncology, Turning Point Therapeutics and he serves as a consultant for Lilly, AstraZeneca, Turning Point Therapeutics. J.V. H. reports consulting or advisory role for AstraZeneca, Bristol Myers Squibb, Spectrum Pharmaceuticals, Guardant Health, Hengrui Pharmaceutical, GlaxoSmithKline, EMD Serono, Lilly, Takeda, Sanofi/Aventis, Genentech/Roche, Boehringer Ingelheim, Catalyst Biotech, Foundation medicine, Novartis, Mirati Therapeutics, BrightPath Biotheraputics, Janssen, Nexus Health Systems, Pneuma Respiratory, Kairos Ventures, Roche, Leads Biolabs, and he receives research funding from AstraZeneca (Inst), Spectrum Pharmaceuticals, GlaxoSmithKline. J.V.H has a licensing agreement with Spectrum regarding intellectual property for treatment of EGFR and HER2 exon 20 mutations and he has stock ownership interest in Cardinal Spine and Bio-Tree. K.L. has a patent on indel burden and CPI response pending and he received honoraria from Roche tissue diagnostics and research funding from CRUK TDL/Ono/LifeArc alliance, and he reports a consulting role with Monopteros Therapeutics. HK.K. reports funding from Johnson and Johnson. C.S. acknowledges grants from AstraZeneca, Boehringer-Ingelheim, Bristol Myers Squibb, Pfizer, Roche-Ventana, Invitae, Ono Pharmaceutical, and Personalis. He is Chief Investigator for the AZ MeRmaiD 1 and 2 clinical trials and is the Steering Committee Chair. He is also Co-Chief Investigator of the NHS Galleri trial funded by GRAIL and a paid member of GRAIL’s Scientific Advisory Board. He receives consultant fees from Achilles Therapeutics (also a SAB member), Bicycle Therapeutics (also a SAB member), Genentech, Medicxi, China Innovation Centre of Roche (CICoR) formerly Roche Innovation Centre - Shanghai, Metabomed, and the Sarah Cannon Research Institute. C.S has received honoraria from Amgen, AstraZeneca, Bristol Myers Squibb, GlaxoSmithKline, Illumina, MSD, Novartis, Pfizer, and Roche-Ventana. C.S. has previously held stock options in Apogen Biotechnologies and GRAIL, and currently has stock options in Epic Bioscience, Bicycle Therapeutics, and has stock options and is a co-founder of Achilles Therapeutics. C.S declares a patent application (PCT/US2017/028013) for methods for lung cancer); targeting neoantigens (PCT/EP2016/059401); identifying patent response to immune checkpoint blockade (PCT/EP2016/071471), determining HLA LOH (PCT/GB2018/052004); predicting survival rates of patients with cancer (PCT/GB2020/050221), identifying patients who respond to cancer treatment (PCT/GB2018/051912); methods for lung cancer detection (US20190106751A1). C.S. is an inventor on a European patent application (PCT/GB2017/053289) relating to assay technology to detect tumour recurrence. This patent has been licensed to a commercial entity and under their terms of employment, C.S is due a revenue share of any revenue generated from such license(s). The remaining authors declare no competing interests.
Footnotes
Code availability
The R script TCellMap is available at GitHub (https://github.com/Coolgenome/TCM). An open-source implementation of the TESLA algorithm in Python can be downloaded from https://github.com/jianhuupenn/TESLA. The custom script used to overlay the spatial locations of the hypoxia signal and TSTR cells on the same histology image is available at GitHub (https://github.com/Coolgenome/TCM/blob/main/res_largerT.py#L230). Additionally, we have built a user-friendly and interactive online data portal, the Single Cell Research Portal (SCRP, https://singlecell.mdanderson.org/), for visualizing scRNA-seq data. All scRNA-seq data used to build T-cell reference maps in this study can be visualized and queried via SCRP at https://singlecell.mdanderson.org/TCM/.
Data availability
A detailed description of data availability including data sources and accession numbers of the scRNA-seq datasets included in the original data collection was provided in Supplementary Tables 1 and 2. In this study, we utilized 10 newly generated scRNA-seq datasets (labeled as “in-house” Supplementary Tables 1, column I). Specifically, the AML dataset31 can be downloaded from the European Genome-Phenome Archive (EGA) database under the accession number EGAD00001007672. The lung cancer (LC_1) dataset can be downloaded from EGA under the accession number EGAS00001005021. The lymphoma dataset (LN_2)23 can be downloaded from EGA under the accession number EGAS00001006052. The scRNA-seq data generated on PBMC samples from healthy donors (PBMC_3) can be downloaded from EGA under the accession number EGAD00001006994. The GBM datasets can be downloaded from the Gene Expression Omnibus (GEO) database under the accession number GSE222522. The breast cancer (BRCA_2) dataset, the lung cancer (LC_5) dataset, the ovarian cancer (OV) dataset, and the STAD dataset can be downloaded from GEO under the accession number GSE222859. The scRNA-seq data generated on reactive lymph nodes from healthy donors (LN_1)23 can be downloaded from GEO under the accession number GSE203610. For the six scRNA-seq datasets generated from patients who received ICB therapy, their data accession numbers, references, and detailed clinical information are provided in Supplementary Table 15. Specifically, the dataset generated by Yost et al.18 can be downloaded from GEO under the accession number GSE123814. The dataset generated by Liu et al.44 can be download from GEO under the accession number GSE179994. The dataset generated by Caushi et al.63 can be downloaded from GEO under the accession number GSE173351. The dataset generated by Zhang et al. can be downloaded from GEO under the accession number GSE169246. The dataset generated by Bi et al.62 can be downloaded from the single cell portal of Broad Institute under the accession number SCP1288. The dataset generated by Bassez et al.48 can be downloaded from EGA under the accession number EGAS00001004809. For the four scRNA-seq datasets used as a demonstration of TCellMap, their data accession numbers and references are provided in Supplementary Table 16. The CosMx SMI datasets generated on NSCLC60 and HCC samples can be downloaded from https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/. For Visium spatial transcriptomics datasets used in this study, the expression count matrices for the BRCA study79 can be downloaded from https://github.com/almaan/her2st. The melanoma Visium data81 can be downloaded from https://www.spatialresearch.org/resources-published-datasets/doi-10-1158-0008-5472-can-18-0747/. The CSCC Visium data can be obtained from GEO under the accession number GSE144240. The ccRCC Visium data82 can be obtained from GEO under the accession number GSE175540. The LUAD Visium data68 can be obtained from EGA under the accession numbers EGAS00001005021. Further information and requests should be directed to and will be fulfilled by the corresponding author Dr. Wang (LWang22@mdanderson.org). All requests for data and materials will be promptly reviewed by The University of Texas MD Anderson Cancer Center to verify if the request is subject to any intellectual property or confidentiality obligations. Any data and materials that can be shared will be released via a Material Transfer Agreement.
References
- 1.Zhang Y & Zhang Z The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cellular & molecular immunology 17, 807–821 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostroumov D, Fekete-Drimusz N, Saborowski M, Kühnel F & Woller N CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cellular and Molecular Life Sciences 75, 689–713 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell JH & Ley TJ Lymphocyte-mediated cytotoxicity. Annual review of immunology 20, 323–370 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Fridman WH, Pagès F, Sautès-Fridman C & Galon J The immune contexture in human tumours: impact on clinical outcome. Nature Reviews Cancer 12, 298–306 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Janssen EM, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 434, 88–93 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Tran E, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreiter S, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 564, 268–272 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Zheng C, et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 169, 1342–1356.e1316 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Papalexi E & Satija R Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol 18, 35–45 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Guo X, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 24, 978–985 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Stubbington MJT, Rozenblatt-Rosen O, Regev A & Teichmann SA Single-cell transcriptomics to explore the immune system in health and disease. Science 358, 58–63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azizi E, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 174, 1293–1308.e1236 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng Q et al. Characteristics of anti-CD19 CAR T cell infusion products associated with eficacy and toxicity in patients with large B cell lymphomas. Nat. Med 26, 1878–1887 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176, 775–789 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sade-Feldman M et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 175, 998–1013 (2018). 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tirosh I et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yost KE et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med 25, 1251–1259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng L et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Aran D et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol 20, 163–172 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pliner HA, Shendure J & Trapnell C Supervised classification enables rapid annotation of cell atlases. Nat. Methods 16, 983–986 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abbas HA et al. Single cell T cell landscape and T cell receptor repertoire profiling of AML in context of PD-1 blockade therapy. Nat. Commun 12, 6071 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han G et al. Follicular lymphoma microenvironment characteristics associated with tumor cell mutations and MHC class II expression. Blood Cancer Discov. 3, 428–443 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hay SB, Ferchen K, Chetal K, Grimes HL & Salomonis N The Human Cell Atlas bone marrow single-cell interactive web portal. Exp. Hematol 68, 51–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jerby-Arnon L et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell 175, 984–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lambrechts D et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med 24, 1277–1289 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Laughney AM et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat. Med 26, 259–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; Hay SB, Ferchen K, Chetal K, Grimes HL & Salomonis N The Human Cell Atlas bone marrow single-cell interactive web portal. Exp Hematol 68, 51–61 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma L, et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 36, 418–430 e416 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng J, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 29, 725–738 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Puram SV, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 171, 1611–1624 e1624 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sinjab A, et al. Resolving the Spatial and Cellular Architecture of Lung Adenocarcinoma by Multiregion Single-Cell Sequencing. Cancer Discov 11, 2506–2523 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L, et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 181, 442–459 e429 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Zhang S et al. Longitudinal single-cell profiling reveals molecular heterogeneity and tumor-immune evolution in refractory mantle cell lymphoma. Nat. Commun 12, 2877 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zilionis R, et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334 e1310 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerlach C, et al. The Chemokine Receptor CX3CR1 Defines Three Antigen-Experienced CD8 T Cell Subsets with Distinct Roles in Immune Surveillance and Homeostasis. Immunity 45, 1270–1284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Naluyima P, et al. Terminal Effector CD8 T Cells Defined by an IKZF2(+)IL-7R(−) Transcriptional Signature Express FcγRIIIA, Expand in HIV Infection, and Mediate Potent HIV-Specific Antibody-Dependent Cellular Cytotoxicity. J Immunol 203, 2210–2221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meister M, et al. Dickkopf-3, a tissue-derived modulator of local T-cell responses. Front Immunol 6, 78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu KH, et al. Dickkopf-3 Contributes to the Regulation of Anti-Tumor Immune Responses by Mesenchymal Stem Cells. Front Immunol 6, 645 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang X, Bogomolovas J, Trexler C & Chen J The BAG3-dependent and -independent roles of cardiac small heat shock proteins. JCI Insight 4(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiebel C, et al. BAG3 Proteomic Signature under Proteostasis Stress. Cells 9(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stürner E & Behl C The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front Mol Neurosci 10, 177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mercurio F & Manning AM NF-kappaB as a primary regulator of the stress response. Oncogene 18, 6163–6171 (1999). [DOI] [PubMed] [Google Scholar]
- 43.ElTanbouly MA & Noelle RJ Rethinking peripheral T cell tolerance: checkpoints across a T cell’s journey. Nat Rev Immunol 21, 257–267 (2021). [DOI] [PubMed] [Google Scholar]
- 44.Liu B, et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat Cancer 3, 108–121 (2022). [DOI] [PubMed] [Google Scholar]
- 45.Trapnell C, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiu X, et al. Reversed graph embedding resolves complex single-cell trajectories. Nature Methods 14, 979–982 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cao J, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bassez A, et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat Med 27, 820–832 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Tai YT, et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: therapeutic implications. Leukemia 33, 426–438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran DQ, et al. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 106, 13445–13450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fergusson JR, et al. CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell Rep 9, 1075–1088 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ling GS, et al. C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science 360, 558–563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Subramanian Vignesh K & Deepe GS Jr. Metallothioneins: Emerging Modulators in Immunity and Infection. Int J Mol Sci 18(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghorani E, et al. The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nat Cancer 1, 546–561 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trucco LD, et al. Ultraviolet radiation-induced DNA damage is prognostic for outcome in melanoma. Nat Med 25, 221–224 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Litchfield K, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 184, 596–614 e514 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mariathasan S, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McDermott DF, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 24, 749–757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Flanagan CH, et al. Dissociation of solid tumor tissues with cold active protease for single-cell RNA-seq minimizes conserved collagenase-associated stress responses. Genome Biol 20, 210 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He SS, et al. High-plex imaging of RNA and proteins at subcellular resolution in fixed tissue by spatial molecular imaging. Nature Biotechnology 40, 1794–1806 (2022). [DOI] [PubMed] [Google Scholar]
- 61.Nakayama K & Kataoka N Regulation of Gene Expression under Hypoxic Conditions. Int J Mol Sci 20(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bi K, et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell 39, 649–661 e645 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Caushi JX, et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature 596, 126–132 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang Y, et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593 e1578 (2021). [DOI] [PubMed] [Google Scholar]
- 65.Mathewson ND, et al. Inhibitory CD161 receptor identified in glioma-infiltrating T cells by single-cell analysis. Cell 184, 1281–1298 e1226 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sautes-Fridman C, Petitprez F, Calderaro J & Fridman WH Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 19, 307–325 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Anderson AC, et al. Spatial transcriptomics. Cancer Cell 40, 895–900 (2022). [DOI] [PubMed] [Google Scholar]
- 68.Hao D et al. The single-cell immunogenomic landscape of B and plasma cells in early-stage lung adenocarcinoma. Cancer Discov. 12, 2626–2645 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Butler A, Hofman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across diferent conditions, technologies, and species. Nat. Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Becht E et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol 37, 38–44 (2019). [DOI] [PubMed] [Google Scholar]
- 71.Korsunsky I et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hao Y et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lovmar L, Ahlford A, Jonsson M & Syvanen AC Silhouette scores for assessment of SNP genotype clusters. BMC Genomics 6, 35 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tran HTN et al. A benchmark of batch-efect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu B et al. An entropy-based metric for assessing the purity of single cell populations. Nat. Commun 11, 3155 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu J et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Thorsson V et al. The immune landscape of cancer. Immunity 48, 812–830 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Becht E et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17, 218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andersson A et al. Spatial deconvolution of HER2-positive breast cancer delineates tumor-associated cell type interactions. Nat. Commun 12, 6012 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thrane K, Eriksson H, Maaskola J, Hansson J & Lundeberg J Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res. 78, 5970–5979 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Ji AL et al. Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182, 497–514 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meylan M et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity 55, 527–541 (2022). [DOI] [PubMed] [Google Scholar]
- 83.Liu B, Zhang Y, Wang D, Hu X & Zhang Z Single-cell meta-analyses reveal responses of tumor-reactive CXCL13(+) T cells to immune-checkpoint blockade. Nat. Cancer 3, 1123–1136 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed description of data availability including data sources and accession numbers of the scRNA-seq datasets included in the original data collection was provided in Supplementary Tables 1 and 2. In this study, we utilized 10 newly generated scRNA-seq datasets (labeled as “in-house” Supplementary Tables 1, column I). Specifically, the AML dataset31 can be downloaded from the European Genome-Phenome Archive (EGA) database under the accession number EGAD00001007672. The lung cancer (LC_1) dataset can be downloaded from EGA under the accession number EGAS00001005021. The lymphoma dataset (LN_2)23 can be downloaded from EGA under the accession number EGAS00001006052. The scRNA-seq data generated on PBMC samples from healthy donors (PBMC_3) can be downloaded from EGA under the accession number EGAD00001006994. The GBM datasets can be downloaded from the Gene Expression Omnibus (GEO) database under the accession number GSE222522. The breast cancer (BRCA_2) dataset, the lung cancer (LC_5) dataset, the ovarian cancer (OV) dataset, and the STAD dataset can be downloaded from GEO under the accession number GSE222859. The scRNA-seq data generated on reactive lymph nodes from healthy donors (LN_1)23 can be downloaded from GEO under the accession number GSE203610. For the six scRNA-seq datasets generated from patients who received ICB therapy, their data accession numbers, references, and detailed clinical information are provided in Supplementary Table 15. Specifically, the dataset generated by Yost et al.18 can be downloaded from GEO under the accession number GSE123814. The dataset generated by Liu et al.44 can be download from GEO under the accession number GSE179994. The dataset generated by Caushi et al.63 can be downloaded from GEO under the accession number GSE173351. The dataset generated by Zhang et al. can be downloaded from GEO under the accession number GSE169246. The dataset generated by Bi et al.62 can be downloaded from the single cell portal of Broad Institute under the accession number SCP1288. The dataset generated by Bassez et al.48 can be downloaded from EGA under the accession number EGAS00001004809. For the four scRNA-seq datasets used as a demonstration of TCellMap, their data accession numbers and references are provided in Supplementary Table 16. The CosMx SMI datasets generated on NSCLC60 and HCC samples can be downloaded from https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/. For Visium spatial transcriptomics datasets used in this study, the expression count matrices for the BRCA study79 can be downloaded from https://github.com/almaan/her2st. The melanoma Visium data81 can be downloaded from https://www.spatialresearch.org/resources-published-datasets/doi-10-1158-0008-5472-can-18-0747/. The CSCC Visium data can be obtained from GEO under the accession number GSE144240. The ccRCC Visium data82 can be obtained from GEO under the accession number GSE175540. The LUAD Visium data68 can be obtained from EGA under the accession numbers EGAS00001005021. Further information and requests should be directed to and will be fulfilled by the corresponding author Dr. Wang (LWang22@mdanderson.org). All requests for data and materials will be promptly reviewed by The University of Texas MD Anderson Cancer Center to verify if the request is subject to any intellectual property or confidentiality obligations. Any data and materials that can be shared will be released via a Material Transfer Agreement.