

Abstract
Extracellular vesicles (EVs) contain cell‐derived lipids, proteins and RNAs; however, determining the tissue‐ and cell‐type‐specific EV abundances in body fluids remains a significant hurdle for our understanding of EV biology. While tissue‐ and cell‐type‐specific EV abundances can be estimated by matching the EV's transcriptome to a tissue's/cell type's expression signature using deconvolutional methods, a comparative assessment of deconvolution methods' performance on EV transcriptome data is currently lacking. We benchmarked 11 deconvolution methods using data from four cell lines and their EVs, in silico mixtures, 118 human plasma and 88 urine EVs. We identified deconvolution methods that estimated cell type‐specific abundances of pure and in silico mixed cell line‐derived EV samples with high accuracy. Using data from two urine EV cohorts with different EV isolation procedures, four deconvolution methods produced highly similar results. The three methods were also concordant in their tissue‐ and cell‐type‐specific plasma EV abundance estimates. We identified driving factors for deconvolution accuracy and highlighted the importance of implementing biological knowledge in creating the tissue/cell type signature. Overall, our analyses demonstrate that the deconvolution algorithms DWLS and CIBERSORTx produce highly similar and accurate estimates of tissue‐ and cell‐type‐specific EV abundances in biological fluids.
Keywords: cell‐conditioned medium, exosome, microvesicle, plasma, single‐cell RNA sequencing, transcriptome, urine
1. INTRODUCTION
Body fluids contain a mosaic of cell type‐specific extracellular vesicles (EVs); however, establishing which tissues and cell types contribute most EVs is challenging and thus limits our understanding of how EVs contribute to health and diseases. Several animal models have been developed to confidently link cell types and their EVs by genetic reporters (Estrada et al., 2022; Gupta et al., 2020; Hegyesi et al., 2022; Luo et al., 2020; Neckles et al., 2019; Nørgård et al., 2022; Rufino‐Ramos et al., 2022); yet, establishing this link in humans is challenging. While traditional methods such as flow cytometry can robustly quantify cell type‐specific EVs, flow cytometry relies on a restricted number of cell type‐specific protein‐based markers that limit its scalability. Nonetheless, advances in EV RNA analyses have provided new approaches to predict the EVs’ cellular source by computational methods termed transcriptome deconvolution. The deconvolution methods can untangle the tissue‐ and cell‐type‐specific EV abundances in body fluids; however, their accuracy has not been thoroughly evaluated, and a comparative assessment of deconvolution methods' performance on EV transcriptome data is currently lacking.
Most transcriptome deconvolution methods have been developed to estimate cell type composition of tissues from bulk RNA sequencing. A general premise for these deconvolution methods is that transcript expression from each cell type is linearly additive, enabling estimation of the tissue's cell type composition from a list of cell‐type‐specific marker transcripts. This reference signature can be established from isolated cells, single‐cell, or single‐nuclei transcriptome data sets. The marker‐based deconvolution methods use this gene list to determine which cell type markers carry the most information for a regression approach to estimate cell type proportions in bulk RNA sequencing data accurately (Jew et al., 2020; Newman et al., 2019; Tsoucas et al., 2019; Wang et al., 2019). In addition to estimating cell type proportions in bulk RNA sequencing data, the deconvolution methods, such as CIBERSORTx (Newman et al., 2019) and Bisque (Jew et al., 2020), have been used to estimate tissue‐ and cell type‐specific EV abundance using plasma and urine EVs (Dwivedi et al., 2023; Shi et al., 2020; Zhu et al., 2021). Moreover, EV‐origin has been created for the deconvolution of EV abundances in plasma (Li et al., 2020; Yu et al., 2020). However, transcriptome deconvolution methods make assumptions about the bulk RNA sequencing data and signature matrix's RNA and cell distribution. For example, deconvolution is most accurate when the bulk and reference transcriptome are from the same cellular compartment, for example, the whole cell (Sutton et al., 2022). Yet, EVs are considered a separate compartment from the cytosol, and RNA transcripts are not always distributed equally between cells and EVs (Almeida et al., 2022; Padilla et al., 2023). Moreover, the EV secretion rate between cell types is highly variable (Auber & Svenningsen 2022; Garcia‐Martin et al., 2022a, 2022b); thus, rare cell types in the signature may contribute a significant number of EV RNA reads in the bulk samples and vice versa. These factors may affect the deconvolution methods’ performance for accurately estimating tissue‐ and cell‐type‐specific EV abundances, and the factors influencing their results remain to be determined.
A significant hurdle for evaluating the deconvolution methods' performance in estimating EV abundances is the lack of samples with known proportions of cell type‐specific EVs. While EVs isolated from cell‐conditioned medium help assess performance in pure systems, biological fluids contain a mixture of cell‐type‐specific EVs. The deconvolution methods’ ability to unmix these samples may, therefore, not be sufficiently assessed using only single‐cell type samples. An essential fact about transcriptome deconvolution methods is that they, rather than proportions of cell types, estimate the proportion of RNA derived from each cell type (Sutton et al., 2022; Zaitsev et al., 2019). Thus, synthetic mixtures with biological heterogeneity in known proportions can be created in silico by sampling and mixing RNA sequencing reads from pure samples (Dong et al., 2023). By leveraging this approach, we used cell and EV transcriptome data from four cell lines to create a dataset containing pure EVs, in silico mixtures of two and three cell‐type‐specific EV samples in different proportions to evaluate 11 deconvolution methods (Table 1) ability to estimate cell type‐specific EV abundance accurately. We also tested the effect of adding cell type‐specific EV missing in the signature matrix. Finally, we evaluate the concordance in EV estimates of transcriptome deconvolution of plasma and urine EVs and highlight the importance of biological knowledge in establishing tissue/cell type signatures.
TABLE 1.
The benchmarked deconvolution methods.
| Algorithm | Regression | Reference |
|---|---|---|
| Bisque | Non‐negative least squares | Jew et al. (2020) |
| CIBERSORTx | Support vector regression | Newman et al. (2019) |
| EV‐origin | Support vector regression | Li et al. (2020) |
| DeconRNASeq | Non‐negative least squares | Gong and Szustakowski (2013) |
| dtangle | Linear mixing model | Hunt et al. (2018) |
| DWLS a | Dampened weighted least squares | Tsoucas et al. (2019) |
| DWLSj a | Dampened weighted least squares b | Tsoucas et al. (2019) |
| DWLS OLS a | Ordinary least squares | Tsoucas et al. (2019) |
| DWLS SVR a | Support vector regression | Tsoucas et al. (2019) |
| MuSiC | Weighted non‐negative least squares | Wang et al. (2019) |
| MuSiC nnls | Non‐negative least squares | Wang et al. (2019) |
The DWLS‐based methods use the same algorithm to identify signature genes for tissue/cell types.
Unlike DWLS, DWLSj does not iterate the proportion estimation to converge on a solution.
2. MATERIALS AND METHODS
2.1. Cell line and cell‐conditioned medium EV RNA sample data
For the cell lines and their EVs, we used RNA sequencing data from two studies that performed paired analyses of cultured cell lines (Almeida et al., 2022; Hinger et al., 2018). Hinger et al. (2018) used colorectal cancer cells (CRC) and their EVs, and Almeida et al. (2022) used prostate cancer cell lines DU145, LNCaP and PC3 and their EVs. To test the deconvolution methods' performance on samples with EVs from cell types missing in the signature matrix, we used EVs RNA sequencing data from co‐cultures of adipocytes and macrophages (Yang et al., 2017). The EV isolation procedures, characterization methods and RNA library preparations are described in Table 2. The paired‐end cell and EV RNA reads were downloaded from the NCBI GEO database (Edgar et al., 2002) and quality‐controlled using FASTQC. Gene expression in cell and EV RNA samples was quantified at the transcript level using Salmon v1.10.2 (Patro et al., 2017) to the human Gencode v44 transcriptome using the following parameters: ‐l A, ‐p 16 and –numBootstraps 100. The transcript quantifications were summarized to gene level with the tximeta package (Love et al., 2020) in R version 4.3.2.
TABLE 2.
Samples used for benchmarking the deconvolution methods.
| Cells/Organisms | EV1 a source | EV isolation b | RNA library kit | Alignment method | EV validation c | GEO/Data source | Reference |
|---|---|---|---|---|---|---|---|
| CRC | CCM | UF/dUC | NEBNext | Salmon |
EM d WB d |
GSE121964 | Hinger et al. (2018) |
|
DU145 LNCaP PC3 |
CCM | dUC | SMARTer | Salmon |
NTA EM WB |
GSE183070 | Almeida et al. (2022) |
|
Adipocytes macrophages |
CCM | dUC | SMART‐Seq v4 | Salmon | WB | GSE94155 | Yang et al. (2017) |
| Human | Plasma | exoRNeasy | SMARTer | HISAT2 e |
EM nanoFCM WB |
exorbase.org | Li et al. (2019), Yu et al. (2020) |
| Human | Urine | exoRNeasy | SMARTer | HISAT2 e | – | exorbase.org | Lai et al. (2021) |
| Human | Urine | dUC | SMART‐Seq | STAR e |
NTA EM WB |
Supplement Table S9 of the paper. | Dwivedi et al. (2023) |
CCM: cell‐conditioned medium.
UF: ultrafiltration; dUC: differential ultracentrifugation, exoRNeasy: a kit that isolates EVs by their affinity to a proprietary membrane.
EM: electron microscopy; NTA: nanotracking analysis; nanoFCM: nano‐flow cytometry; WB: western blot.
Alignment to the human genome was done in the original studies.
2.2. Tissue, single‐cell, plasma EVs and urine EVs RNA data
We used RNA sequencing data from 27 tissues, which was downloaded from the GTEx Portal as reads (GTEx_Analysis_2017‐06‐05_v8_RNASeQCv1.1.9_gene_reads) and transcripts per million (GTEx_Analysis_2017‐06‐05_v8_RNASeQCv1.1.9_gene_tpm). The GTEx consortium used STAR for the alignment of the RNA reads to the human genome. We only included RNA sequencing data and data from patients with a death classification below 4 on the HARDY Scale (i.e., excluding samples from patients who died of long‐term illness). CIBERSORTx has a file size limit of 1 GB, and we, therefore, included a maximum of 200 consecutive samples from each tissue. The number of samples included for each tissue is shown in Table S1. We, moreover, used annotated single‐cell RNA sequencing datasets of human kidneys (McEvoy et al., 2022) and peripheral blood mononuclear cells (PBMCs) (Newman et al., 2019). Both studies used Cell Ranger for alignment to the human genome. For the human kidney single‐cell RNA dataset, we extracted count matrices using Seurat v4 (Hao et al., 2021) for proximal tubules, principal cells, intercalated A cells, intercalated B cells, distal convoluted tubules, cortical thick ascending loop of Henle's, and connecting tubules. Intercalated A and B cells were merged into intercalated cells. For the PBMC dataset, all cell types in the dataset were included. Plasma EV RNA sequencing data was obtained from 118 healthy humans (Table 2). We used urine EV RNA sequencing data from two cohorts that differed in the procedure for EV isolation (Table 2).
2.3. In silico EV mixtures
To create EV samples in silico with precise control of mixing ratios and equal library sizes, we adapted a method for simulating RNA sequencing data from mixtures of cell types described and verified by Dong et al. (2023). Briefly, this method enables the creation of in silico mixtures by combining randomly selected reads from pure RNA sequencing samples. To verify the approach, we re‐analysed RNA sequencing data from pure and mixed samples of NCI‐H1975 and HCC827 cells (Holik et al., 2017, data accessible at NCBI GEO database, accession GSE64098) similar to Dong et al. (2023). The samples consist of RNA sequencing data from H1975 and HCC827 cells mixed physically at five different ratios: 0:100, 25:75, 50:50, 75:25 and 100:0. In silico mixtures were created by randomly extracting 7.5, 15 and 22.5 million reads from each pure H1975 and HCC827 sample using seqtk (https://github.com/lh3/seqtk) with a fixed random seed number to keep the pairing of the paired‐end reads. The downsampled H1975 sequencing files were merged with downsampled HCC827 sequencing files to create an in silico mix with 30 million reads in ratios of 75:25, 50:50 and 25:75 in three replicates. Moreover, we also made mixtures of pure H1975 and HCC827 cells in silico by combining two 15 million read subsamples for each cell type. To make in silico mixed EV samples, the DU145 and LNCaP‐derived EV RNA sequencing files were downsampled and mixed to create in silico mixed files with 30 million reads containing DU145 and LNCaP EVs in 75:25, 50:50, 25:75, 10:90, 7.5:92.5, 5:95, 2.5:97.5, 1:99 and 0.5:99.5 ratios in three replicates. In silico mixtures of pure DU145 and LNCaP EV samples were also created in triplicate by combining two downsampled 15 million read files. We also created more complex mixtures of different combinations of 0%, 12.5%, 25%, 37.5%, 50% of mapped DU145, LNCaP and PC3‐derived EV reads. To test the impact on the deconvolution of EVs from cell types not present in the cell matrix, we in silico mixed pure DU145 EV samples with the EV RNA from co‐cultured adipocytes and macrophages. Count matrices were produced as described above.
2.4. Transcriptional deconvolution methods
We used 11 deconvolution methods (Table 1) that use different algorithms to identify signature genes and the subsequent regression to estimate cell type proportions. The data were used as raw counts or counts per million (CPM) normalized as specified below.
Bisque (Jew et al., 2020) was run using the Bisque Bioconductor R package with default setting and tissue/cell and EV RNA as count matrices.
CIBERSORTx (Newman et al., 2019) was run on the website https://cibersortx.stanford.edu in absolute mode with B‐mode batch correction and without quantile normalization. Tissue/cell and EV RNA expression data were CPM‐normalized.
DeconRNASeq (Gong & Szustakowski 2013) was run using the DeconRNASeq v1.26 Bioconductor R package with default parameters. Tissue/cell and EV RNA expression data were CPM‐normalized.
Dtangle (Hunt et al., 2018) was run in R using the default setting. Tissue/cell and EV RNA expression data were CPM‐normalized.
EV‐origin (Li et al., 2020) was run using the R script downloaded from https://github.com/HuangLab‐Fudan/EV‐origin/blob/master/EV‐origin.new.txt. The EV‐origin script was run with default settings. Cell and EV RNA expression data were CPM‐normalized. In contrast to Li et al. (2020), who used a limited list of genes with high tissue/cell type‐specificity, we included the 10.000 and 20.000 genes with the highest expression variability between GTEX tissue and cells, respectively, to better compare the performance of EV‐origin to the other deconvolution methods.
DWLS (Tsoucas et al., 2019) was run using the default setting in R. Tissue/cell RNA data were used as count matrices, and EV data as CPM normalized data. The function solveDampenedWLS() was used to produce the DWLS estimates. The DWLSj estimates were made by the functions findDampeningConstant() and then solveDampenedWLSj(). The DWLS OLS and DWLS SVR estimates were produced with the functions solveOLS() and solveSVR().
MuSiC v0.1.1 (Wang et al., 2019) was run using the music_prop() function from the R package available at https://github.com/xuranw/MuSiC. The estimates of the MuSiC and MuSiC nnls algorithms were retrieved from the music_prop() function output data frames Est.prop.weighted and Est.prop.allgene. Raw count data was used as input for both signatures and mixtures.
2.5. Benchmarking scores
The deconvolution methods’ estimates were converted to relative proportions, and the total sum within one sample is always 1. Thus, a change in the abundance of one tissue's/cell type's EV affects the estimate of other tissue's/cell type's EV abundance. To assess the overall sample‐to‐sample performance by the deconvolution methods, we used the Bray‐Curtis dissimilarity metric. This metric uses the deconvolution methods proportion estimates as a multidimensional data point, for example, a sample consisting of EVs from the four cell lines DU145, LNCaP, PC3 and CRC is a four‐dimensional data point. The accuracy of the deconvolution methods is how similar the estimates of multidimensional data points are to the true data points. The Bray–Curtis dissimilarity was calculated as:
where C estimate/true is the sum of the lesser values for each cell type EVs in estimates and true samples, S estimate and S true are the total number of EVs in the estimate and true samples. For visualization purposes, we used the Bray–Curtis index, defined as 1‐ Bray–Curtis dissimilarity. Thus, a Bray–Curtis dissimilarity value and Bray–Curtis index of 0 and 1, respectively, indicate that the proportions in the estimate and true sample are similar. Moreover, the root mean squared error (RMSE) was used to measure the standard deviation of the estimation error, and Pearson's correlation coefficient R was used to measure the correlation between estimated and true cell type EV abundances.
2.6. Data presentation
Sample numbers, definitions of values and error bars are described in the figure legends. The analyses and data visualizations were done with R version 4.3.2.
3. RESULTS
3.1. Deconvolution of EVs from cultured cells
For our benchmarking strategy, we used RNA sequencing data from four different cell lines and their EVs isolated from cell‐conditioned medium (Figure 1a). A multidimensional scaling plot of the cells and EVs showed cell type‐specific clustering (Figure 1b), and we employed the 11 deconvolution methods to test their ability to estimate cell type‐specific EV abundances in these pure samples (Figure 1a). Compared to the truth (Figure 1c, top left, Figure S1a), the four deconvolution methods, that is, DWLS, DWLSj, CIBERSORTx and DWLS OLS, created Bray–Curtis index scores > 0.75 with the DWLS method estimates closest to the true samples (Bray–Curtis index 0.92, Figure 1c,d).
FIGURE 1.
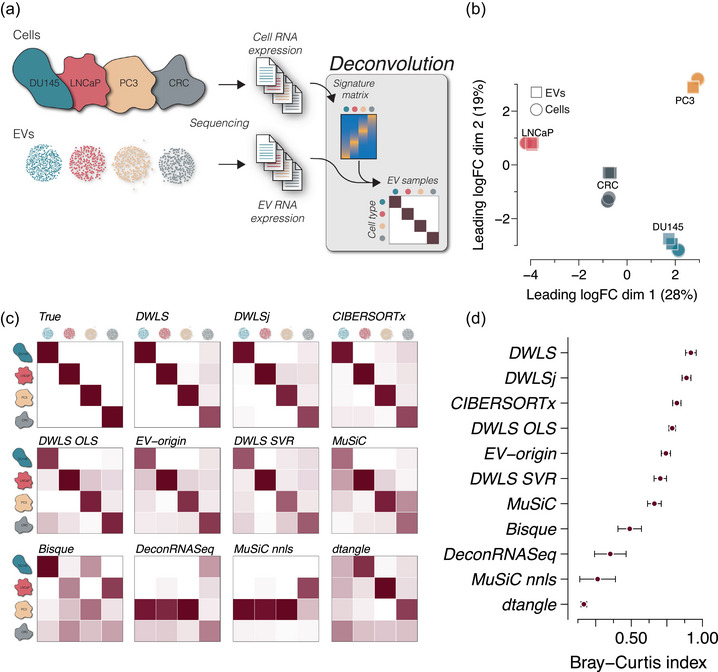
Deconvolution accuracy across methods in pure EV samples from cell cultures. (a) Cell and EV RNA sequencing data from three prostate cancer cell lines DU145, LNCaP and PC3 (GSE183070) and a colorectal cancer (CRC, GSE121964) cell line (n = 3 cell and EV sample for each cell type). The cell RNA data was used to create a signature matrix for deconvolution of the cell‐derived EV RNA data. (b) Multidimensional scaling plot of the cell and EV samples showing cell type‐specific clustering. Circles indicate cells, and squares indicate EVs (n = 3 for each sample type). (c) Heatmaps of deconvolution results of the EV samples. The estimated EV abundance is the average of triplicates and is scaled between 0 and 1. The estimate for each sample is shown in Figure S1. The numbers are represented by the darkness of the colour, that is, 0 is white, and 1 is dark red. The upper left heatmap shows the true distribution. (d) Similarity to the true samples (upper left heatmap) is calculated as the Bray–Curtis index for all 12 samples, that is, four cell types and three replicates. The points indicate the mean Bray–Curtis index values and the error bars indicate SEM.
While some of the deconvolution methods estimate cell‐specific EV abundances of pure samples with high accuracy, EVs isolated from body fluids are a mixture derived from various cell types. To evaluate the deconvolution methods’ performance in estimating cell type‐specific EV proportions in mixed samples, we mixed the pure DU145 and LNCaP EV samples in silico (Figure 2a). The in silico mixing strategy was previously described and validated by Dong et al. (2023). Briefly, RNA sequencing reads were randomly extracted from pure samples, and the downsampled reads were mixed to control mixing ratios precisely and maintain equal library size (Figure 2a). This strategy was verified by re‐analysing physically mixed NCI‐H1975 and HCC827 cells, demonstrating highly similar multidimensional scaling plot clustering and gene expression profiles to the in silico mixtures made from pure samples (Figure S2). The in silico mixing strategy was used on pure DU145 and LNCaP EV samples, which were downsampled to 22.5, 15 and 7.5 million reads. The downsampling of the pure EV samples did not affect the cell type‐specific clustering, gene expression profile (Figure S3) or the estimates of cell type‐specific EV abundances by the 11 deconvolution methods (Figure S4). The downsampled DU145 and LNCaP EV samples were randomly combined in ratios of 100:0, 75:25, 50:50, 25:75 and 0:100 in triplicate with 30 million reads per sample. A multidimensional scaling plot demonstrated that pure DU145 and LNCaP EV samples were closely clustered with in silico mixed DU145 and LNCaP EV samples created by combining two downsampled EV samples from the same cell type, and the in silico DU145/ LNCaP EV mixtures clustering was dependent on the mixing ratio (Figure 2b). The 15 in silico DU145/LNCaP mixtures, that is, five mixing ratios in triplicate, were used to evaluate the deconvolution methods’ performances. DWLS estimated the EV proportions close to the expected ratio (Bray–Curtis index = 0.97), and the correlation between estimated and in silico mixed DU145 and LNCaP was high (RDU145 = 0.99, RLNCaP = 0.99, Figure 2c). Similarly, DWLSj, CIBERSORTx and DWLS OLS estimated DU145 and LNCaP EV proportions close to the expected ratios (RDU145 > 0.9 and RLNCaP > 0.9, Figure S5a). We, then, tested the deconvolution methods’ ability to detect lower fractions of cell type‐specific EVs, and created DU145/LNCaP EV mixed ratios of 0:100, 0.5:99.5, 1:99, 2.5:97.5, 5:95, 7.5:92.5, 10:90, 90:10, 92.5:7.5, 95:5, 97.5:2.5, 99:1, 99.5:0.5 and 100:0. The DWLS method had the most accurate estimates of cell type‐specific EV abundance (Bray–Curtis index = 0.98) with high correlation between the estimated and expected proportion in the in silico mixed samples (RDU145 = 0.98, RLNCaP = 0.95, Figure 2d). DWLSj and CIBERSORTx also estimated the cell type‐specific EV abundances close to the expected mixing ratio (Bray–Curtis index > 0.9); however, the DU145 abundance estimates in the mixed samples with DU145 EV abundance ≤10% were more overestimated by CIBERSORTx (Figure S5b). Overall, the DWLS, DWLSj and CIBERSORTx deconvolution estimates were similar and close to the expected mixing ratios (Figure 2e).
FIGURE 2.
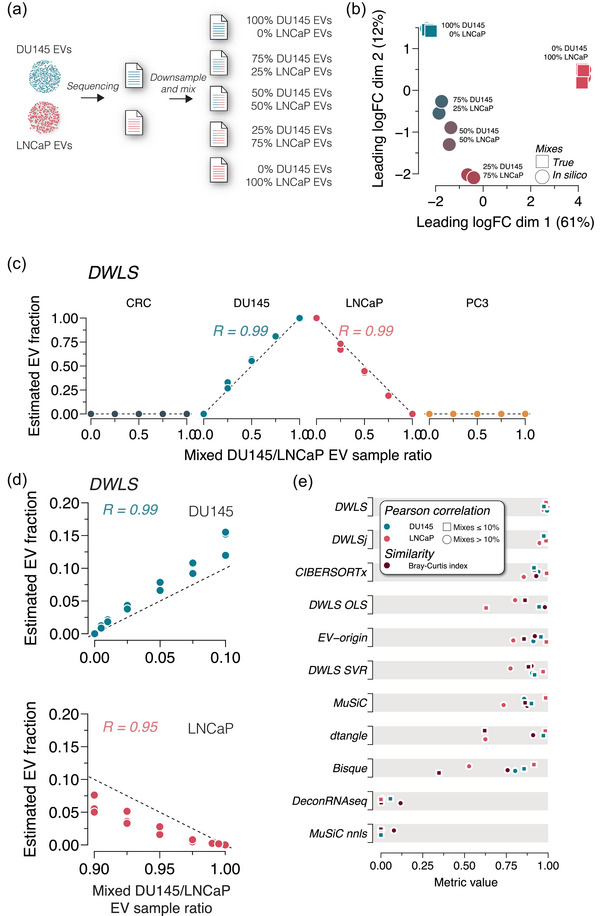
Deconvolution accuracy across methods in mixed DU145 and LNCaP EV samples. (a) Raw RNA sequencing reads from DU145 and LNCaP EV samples were downsampled and mixed in ratios of 100:0, 75:25, 50:50, 25:75 and 0:100. All mixed files contained 30 million reads. (b) Multidimensional scaling plots of true (square) and in silico mixed (circle) RNA sequencing reads show ratio‐specific clustering (n = 3 for each ratio). (c) DWLS deconvolution of DU145 and LNCaP EV samples estimates the in silico mixing ratio accurately. R indicates the Pearson correlation coefficient. The deconvolution estimates with the remaining 10 methods are shown in Figure S5a. (d) Raw RNA sequencing reads from DU145 and LNCaP EV samples were downsampled and mixed in ratios of 0:100, 0.5:99.5, 1:99, 2.5:97.5, 5:95, 7.5:92.5, 10:90, 90:10, 92.5:7.5, 95:5, 97.5:2.5, 99:1, 99.5:0.5 and 100:0. DWLS convolution was most accurate in estimating the low abundance in silico mixing ratios of DU145 and LNCaP EVs. R indicates the Pearson correlation coefficient. The deconvolution estimates of the remaining 10 methods are shown in Figure S5b. (e) Summary of Pearson correlation coefficients and Bray–Curtis index for deconvolution of DU145 and LNCaP in silico mixed EV samples for the 11 transcriptome deconvolution methods. The metrics are colour‐coded, as indicated in the legend.
To assess the 11 deconvolution methods’ performance on more complex mixtures, we created 12 different samples with 0%, 12.5%, 25%, 37.5% and 50% DU145, LNCaP and PC3 EVs (Figure 3a). CRC EV samples were not included because, for example, the library build differed from the three other cell types. The DWLS‐based methods and CIBERSORTx correctly estimated the absence of CRC EVs in the 12 mixtures, and DWLS, DWLSj and CIBERSORTx estimated the cell type‐specific EV abundances close to the true mixing proportions (Bray–Curtis index > 0.9, Figures 3b and S6). Thus, transcriptome deconvolution can accurately predict cell type‐specific EV abundances in mixed samples.
FIGURE 3.
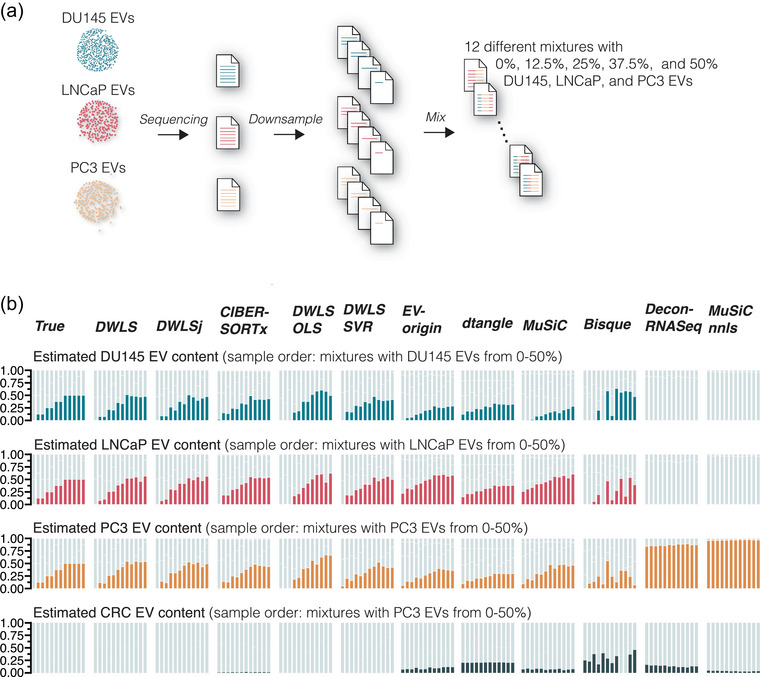
Deconvolution accuracy of complex mixtures of DU145, LNCaP and PC3 EV samples. (a) Raw sequencing reads from DU145, LNCaP and PC3 EV samples were downsampled and mixed to yield 12 different samples with cell type‐specific EV content of 0%, 12.5%, 25%, 37.5% and 50%. (b) Compared to the true samples, DWLS, DWLSj, CIBERSORTx, DWLS OLS and DWLS SVR deconvolution methods accurately estimated the cell type‐specific EV content. The estimated EV abundance is the average of three in silico mixes. For visualization, the samples are ordered to show mixtures with an expected increase in the cell type‐specific EV content. Graphs with unsorted samples are shown in Figure S6.
Next, we evaluated whether EVs from cell types missing in the signature matrix interfered with the deconvolution. To this end, we made in silico mixed DU145 EVs with adipocyte and macrophage EV RNA (GSE94155, Yang et al. 2017) in ratios of 100:0, 75:25, 50:50 and 25:75 (Figure 4a). While samples with >50% adipocyte and macrophage EV affected the deconvolution by Bisque, DeconRNASeq and MuSiC nnls, the EV proportions estimated by DWLS, DWLSj, CIBERSORTx, DWLS OLS, EV‐origin, DWLS SVR, dtangle and MuSiC deconvolution methods were only minimally affected by the presence of adipocyte and macrophage EV RNA even at high proportions (Figure 4b,c).
FIGURE 4.
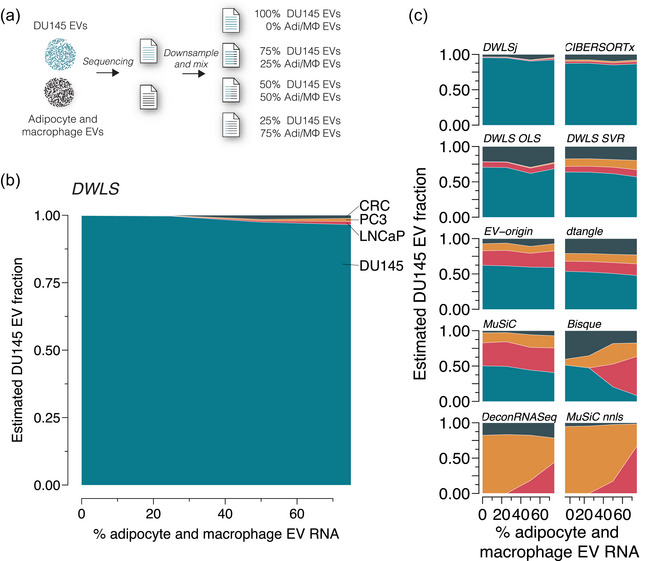
Effect of missing cell types in the signature matrix. (a) In silico mixed EV samples were created by downsampling and mixing RNA reads from DU145 and physically mixed adipocyte and macrophage EV samples (GSE94155) in ratios of 100:0, 75:25, 50:50, 25:75 (n = 3 for each ratio). The samples were deconvoluted using the transcript signatures for DU145, LNCaP, PC3 and CRC cells. (b) The adipocyte and macrophage EV RNA did not significantly affect the cell type‐specific EV fraction estimated by DWLS. (c) Similarly, the cell‐specific EV estimated of the mixed samples were not significantly affected by adipocyte and macrophage EV RNA for DWLSj, CIBERSORTx, DWLS OLS, DWLS SVR, EV‐origin, dtangle and MuSiC deconvolution methods; however, the presence of 50% or more adipocyte and macrophage EV RNA changed cell type‐specific EV estimates for Bisque, DeconRNASeq and MuSiC nnls deconvolution methods.
To evaluate the deconvolution methods' performance overall, we combined their scores (RMSE, Bray–Curtis dissimilarity and correlation coefficients) from deconvoluting pure, downsampled and mixed samples (Figure 5). For visualization purposes, we used 1 ‐ Pearson correlation coefficient (PCC) values in which 0 is a perfect correlation. DWLS created deconvolution results closest to the expected (Figure 5a), followed by DWLSj and CIBERSORTx (Figure 5b). Together, our deconvolution analyses of the cell line‐derived EVs demonstrate that current methods for transcriptome deconvolution enable cell type‐specific EV abundance estimation with high accuracy.
FIGURE 5.
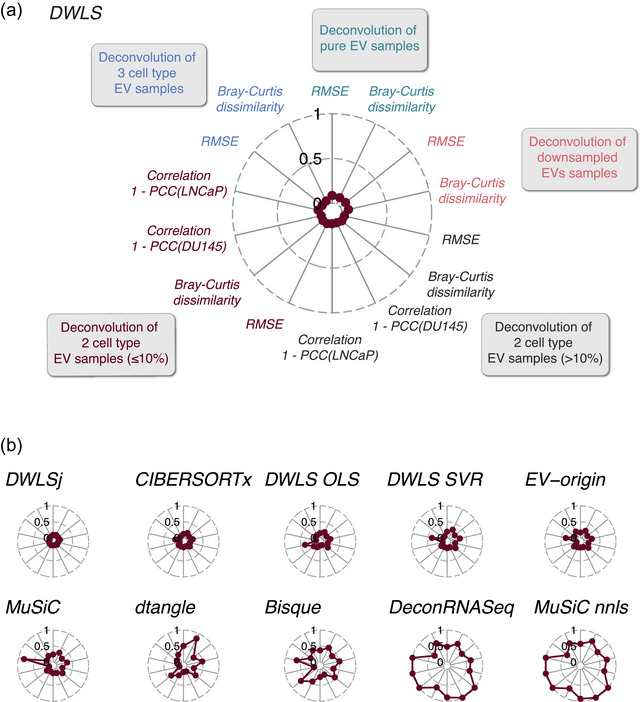
Benchmarking the deconvolution accuracy for cell line‐derived EV samples across methods. (a) Overall benchmarking scores of DWLS deconvolution of the pure, the downsampled, and the in silico mixed EV samples with expected cell types‐specific EV content > or ≤10%, respectively, and complex mixtures. RMSE indicates root mean square error. Correlations for the deconvolution of the in silico mixed DU145 and LNCaP EV samples are shown as 1‐PCC, where PCC indicates the Pearson Correlation Coefficient. (b) Overall benchmarking of the remaining 10 transcriptome deconvolution methods.
3.2. Deconvolution of urine EVs
To evaluate the deconvolution methods’ ability to predict the tissue‐specific EV abundances in urine samples, we used urine EV RNA sequencing data from two independent cohorts, which we will refer to as exoRbase (n = 16 samples, Lai et al., 2021) and Dwivedi (n = 72 samples, Dwivedi et al., 2023) cohorts. Notably, the urine EV isolation methods for the two cohorts were different: the exoRbase cohort's urine EVs were isolated by exoRNAeasy, and the Dwivedi cohort's urine EVs were isolated by differential ultracentrifugation.
First, we created a signature matrix with tissue‐specific gene expression from 27 human tissues. For the exoRbase and Dwivedi cohorts, the deconvolution methods’ tissue‐specific EV abundance estimates were highly variable (Figures S7 and S8), and the correlation coefficients between the methods’ estimates were low (Figure S9). We noticed that epithelial‐rich organs, such as the pancreas and stomach, were predicted to contribute a significant fraction of EVs to the urine. This contrasts with other lines of experimental evidence suggesting that urine EVs are predominantly derived from epithelial cells in the genitourinary system (Blijdorp et al., 2022; Nørgård et al., 2022; Svenningsen et al., 2020). We, thus, restricted the tissue signature matrix to nine tissues of the genitourinary system. Using the genitourinary‐restricted tissue signature matrix for deconvolution of the exoRbase cohort samples demonstrated that the DWLS, DWLSj, CIBERSORTx and DWLS OLS deconvolution methods estimated the kidneys to be dominant contributors of urine EVs (80% ± 6%, Figure 6a). The correlations between the DWLS, DWLSj, CIBERSORTx and DWLS OLS deconvolution methods were high (R > 0.95, Figure 6b). Urine EV samples from the Dwivedi cohort were estimated by DWLS, DWLSj, CIBERSORTx and DWLS OLS deconvolution to contain EVs derived from kidneys (49% ± 5%) and bladder (39% ± 2%), accounting for 88% ± 6% of the urine EVs (Figure 6c) and with high correlation between the four deconvolution methods estimates R > 0.95 (Figure 6d). We then compared the deconvolution methods’ estimates for the two cohorts. The DWLS method's deconvolution of the two cohorts was highly correlated (R = 0.95, Figure S10a). CIBERSORTx demonstrated a lower correlation (R = 0.56), but DWLSj and DWLS OLS showed high correlations between the two cohorts’ estimates (RDWLSj = 0.85 and RDWLS OLS = 0.85, Figure S10b). To assess the cell type‐specific contribution of urine EV from the kidney, we used single‐cell RNA data from human living‐donor kidneys (McEvoy et al., 2022). For the urine EV data from the exoRobase and Dwivedi cohorts, there was a high correlation between the estimates of DWLS, DWLSj, CIBERSORTx, DWLS SVR, MuSiC and MuSiC nnls (Figure S11a,b). Moreover, these deconvolution methods’ estimates were highly correlated between the cohorts (Figures 6f and S11c), indicating that, among the tested kidney epithelial cells, proximal tubular cells (PT), principal cells (PC) and intercalated cells (IC) contribute with the highest proportion of urine EVs. Proximal tubular cells are the most abundant kidney epithelial cell type; however, when normalizing the estimated cell type‐specific urine EV abundances to the number of each cell type in human kidneys (Hatton et al., 2023), intercalated cell‐derived urine EVs were estimated to be present at a ∼5‐fold higher EV/cell ratio than proximal tubule and principal cells. This could indicate that, compared to the other kidney epithelial cell types, intercalated cells secrete EVs at a higher rate. In conclusion, the tissue/cell type gene expression matrix is crucial for obtaining plausible results, and signatures matrices created from bulk and single‐cell RNA sequencing data yield a high agreement between the results created by DWLS, DWLSj and CIBERSORTx and a strong positive correlation between these deconvolution methods’ estimates between cohorts.
FIGURE 6.
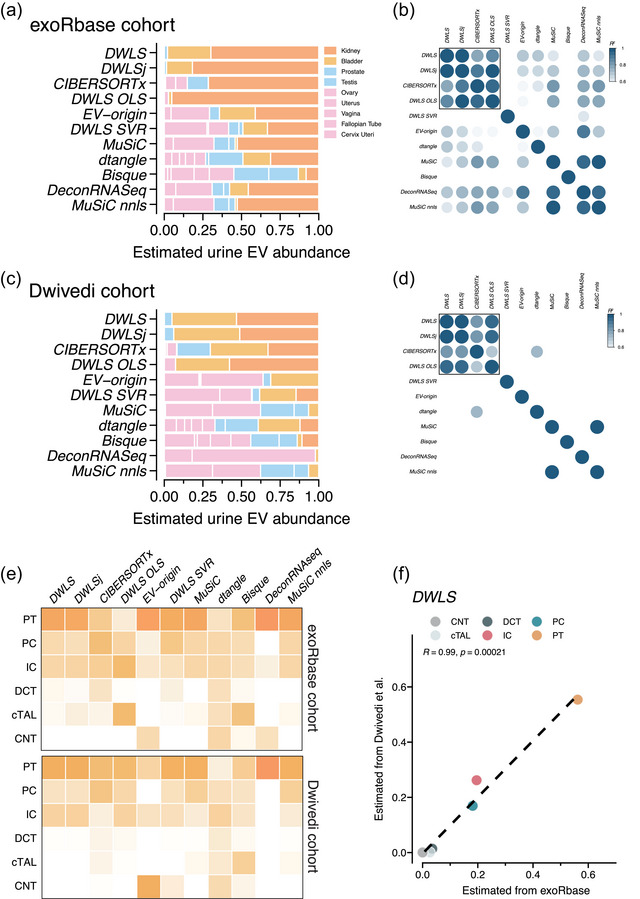
Deconvolution of tissue‐ and kidney‐cell type‐specific EV abundances in urine across methods. (a) Deconvolution of urine EV samples from the exoRbase cohort (n = 16 samples) by the 11 deconvolution methods. The deconvolution methods’ estimated tissue/organ source is colour‐coded according to the legend. (b) Pearson correlation coefficients (R2 indicated by colour) between the 11 deconvolution methods’ estimates of tissue‐specific urine EV abundance. (c) Estimated tissue‐specific EV abundance and (d) Pearson correlation coefficients (R2 indicated by colour) between the 11 deconvolution methods’ estimates of the urine EVs from the Dwivedi et al. (2023) cohort (n = 72). (e) Heatmaps showing the estimated abundances of kidney epithelial cell type‐specific urine EV abundances in the exoRbase (top) and Dwivedi (bottom) cohorts by the 11 deconvolution methods. The estimated EV abundance is the average of all samples in the respective cohorts and is scaled between 0 and 1. The numbers are represented by the darkness of the colour, that is, 0 is white and 1 is dark orange. (f) Correlation between the DWLS method's deconvolution of urine samples from the exoRbase and Dwivedi cohorts. R indicates the Pearson correlation coefficient. The correlations for the remaining 10 deconvolution methods are shown in Figure S11. PT, proximal tubule; PC, principal cell; IC, intercalated cell; DCT, distal convoluted tubule; cTAL, cortical thick ascending limb of the loop of Henle's; CNT, connecting tubule.
3.3. Deconvolution of plasma EVs
For plasma EVs, we used the 27‐tissue signature to estimate the tissue‐specific EV abundances for plasma samples from 118 healthy persons. While DWLS, DWLSj and CIBERSORTx estimated that blood and spleen contributed 95% ± 2% of the plasma EVs (Figure 7a), they estimated different proportions of these two tissues. CIBERSORTx estimated that 50% of the plasma EVs were derived from the spleen, but DWLS and DWLSj estimated that the spleen‐derived EVs only contributed with <5% of plasma EVs (Figure 7a). Consistent with this, the correlation between the tissue‐specific EV abundances estimated by the different deconvolution methods was low (Figure S12). The spleen is a reservoir for blood cells, and we therefore, combined the estimates for blood and spleen‐derived plasma EVs. This improved the correlations between DWLS, DWLSj, CIBERSORTx and DWLS OLS algorithms (R > 0.98, Figure S12b) and produced highly similar estimates for each of the 118 samples (Figure 7b). Although several tissue types contribute to the plasma EV pool, the deconvolution results indicate that plasma EVs are primarily derived from blood cells, for example, red blood cells, platelets and circulating immune cells. The intravascular blood cell types are dominated by red blood cells and platelets, which outnumber other blood cell types by >1000‐fold (Hatton et al., 2023). While the red blood cell‐ and platelet‐derived EVs are expected to be present in large amounts in plasma samples, we used a single‐cell RNA sequencing dataset of human peripheral blood mononuclear cells (PBMCs) (Newman et al., 2019) to estimate the cell type‐specific plasma EV abundances of monocytes, B cells, NK cells, NKT cells and CD4 and CD8 T cells. Except for dtangle, the deconvolution methods estimated that monocytes were the PBMC cell type that contributed the most plasma EVs (Figure 7c). Additionally, DWLS, DWLSj, CIBERSORTx and DWLS OLS created highly correlated estimates of the cell type‐specific source of the plasma EVs (R ≥ 0.9, Figure S13). Interestingly, the estimated cell type‐specific EV abundances did not reflect the cell type's fraction of PBMCs; NK cells, CD4 and CD8 T cells are more abundant than B cells and monocytes. This could indicate differences in cell type‐specific EV secretion rates and/or plasma half‐lives. In sum, the DWLS‐based and CIBERSORTx deconvolution methods had highly concordant estimates of tissue‐ and cell‐specific EV abundances in plasma samples.
FIGURE 7.
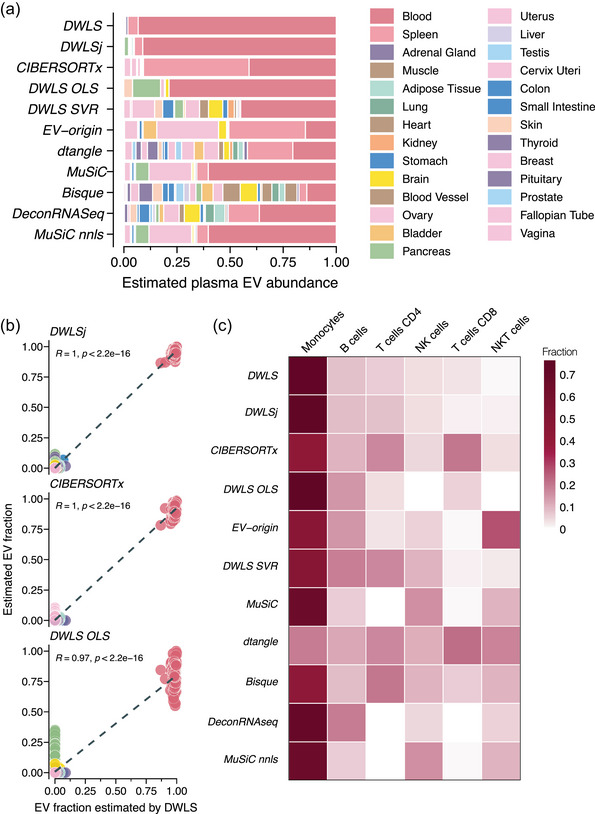
Deconvolution of tissue‐specific EV abundances in plasma across methods. (a) Estimated tissue‐specific plasma EV abundance of plasma EV samples (n = 118, GEO accession numbers GSE100206 and GSE133684) by the 11 deconvolution methods. The tissues/organs are colour‐coded according to the legend. (b) Correlation between the individual person's tissue‐specific plasma EV abundance estimated by DLWS and (top panel) DWLSj, (middle panel) CIBERSORTx and (bottom panel) DWLS OLS. R indicates the Pearson correlation coefficient. (c) Heatmap showing the estimated abundances of peripheral blood mononuclear cells‐derived plasma EVs by the 11 deconvolution methods. The estimated EV abundance is the average of all samples in the cohorts and is scaled between 0 and 1. The numbers are represented by the darkness of the colour, that is, 0 is white and 1 is dark red. The correlation between the 11 deconvolution methods estimates is shown in Figure S13.
4. DISCUSSION
By meticulous analyses of transcriptomic deconvolution methods on EVs from cultured cells, plasma, and urine samples, we here provide comprehensive benchmarking data that can help address the question of tissue‐ and cell‐type‐specificity of EVs isolated from biological fluids. We analysed 11 deconvolution methods. By comparing their performances on several sample types, we identify the DWLS and CIBERSORTx methods with highly concordant results in all the benchmarking tests. Importantly, our analyses highlighted the tissue/cell type signature matrix's essential role in the EV abundance estimates. Overall, the benchmarking suggests that the best performing transcriptomic deconvolution methods, that is, DWLS and CIBERSORTx, can convert EV RNA sequencing data into reliable tissue‐ and cell‐type‐specific EV abundance estimates in biofluids.
The deconvolution methods differ in the algorithms used to identify the most informative signature genes, how these genes are weighted, and the final regression analyses. Three deconvolution methods, CIBERSORTx, EV‐origin and DWLS SVR, are based on support vector regression (SVR). Although the SVR‐based methods’ estimates for cell line‐derived EV were similar, they demonstrated low correlations in urine and plasma EV samples. CIBERSORTx's estimates were most consistent with the best‐performing deconvolution methods, DWLS, DWLSj and DWLS OLS. While these three methods use the same algorithm to establish the tissue/cell type‐specific genes by differential expression analyses, they treat this information differently. DWLS OLS uses ordinary least square regression (OLS); however, this algorithm tends to ignore low abundant transcripts, such as genes only expressed in rare cells or genes only expressed at low levels (Tsoucas et al., 2019). DWLS and DWLSj are also based on OLS regression, but they reduce this bias by a weighted least square approach to include the contribution for each gene (Tsoucas et al., 2019). Importantly, DWLSj estimates the weights based on the initial OLS regression, but DWLS uses an iterative approach that updates the weighted least square solution until the convergence of its estimates (Tsoucas et al., 2019). Consistent with these additional optimization steps, DWLS makes the most accurate estimates on the cell line‐derived EVs, even in samples with a low abundance of cell type‐specific EVs, and demonstrates the importance of identifying the most informative transcripts for deconvolution. Moreover, the processes involved in acquiring RNA sequencing data from tissues, cells and EVs differ. This technical variation may also affect the deconvolution method's performance. CIBERSORTx minimizes this technical variation through a batch correction step (Newman et al., 2019), and DWLS uses an iterative to optimize its estimates (Tsoucas et al., 2019). Although DWLS's iterative approach is not exactly similar to CIBERSORTx's batch correction, the high agreement between the DWLS and CIBERSORTx estimates indicates that approaches to minimize the technical variation between EV samples and the tissue/cell reference matrix improve the estimates.
A significant challenge for the deconvolution methods is cell types with similar gene expression. Consistent with previous results using EV‐origin and a carefully constructed tissue signature matrix (Li et al., 2020), the estimates of the four deconvolution methods showed that blood and spleen cells contribute significantly to the plasma EV pool; however, the same 27‐tissue signature estimated a high abundance of urine EVs derived from epithelial‐rich tissue, such as the pancreas and stomach. Yet, by restricting the number of tissues to only include tissues of the genitourinary systems in the signature matrix, the four best‐performing deconvolution methods produced highly concordant estimates for the two independent urine EV cohorts that, among others, differed in their EV isolation procedure. Consistent with experimental evidence from mice and humans (Blijdorp et al., 2022; Nørgård et al., 2022; Svenningsen et al., 2020), the four best‐performing deconvolution methods indicated that the bladder and kidneys are the dominant sources of urine EVs. Using CIBERSORTx, Zhu et al. (2021) have previously reported that bladder‐ and lung‐derived EVs were abundantly present in urine, but kidney‐derived EVs were rarer. The discrepancy between Zhu et al. (2021) and our findings is multifactorial, but choices of which tissues and genes to include in the signature are likely significant drivers. Nonetheless, data from transgenic EV reporter mice suggest that the kidneys do not freely filter the plasma EVs (Nørgård et al., 2022), and proteome data of human EVs sampled from a nephrostomy drain or normally voided urine support a substantial role of the kidneys as a source for urine EVs (Blijdorp et al., 2022). Thus, including biological knowledge of which tissues/cell types are likely to contribute EVs, can improve the deconvolution methods’ estimates.
The use of in silico mixes provides a well‐controlled approach to benchmarking the EV deconvolution methods; however, in complex biological EV samples, such as from plasma and urine, RNA extraction, library builds and sequencing may introduce technical biases in the amplification of rare versus abundant EV RNAs. Thus, the sensitivity to detect low abundant EVs of the in silico mixtures cannot be directly extrapolated to the deconvolution of the plasma and urine samples. Moreover, EVs are highly heterogeneous, and the choice of EV isolation method may introduce a technical variation. Although comparative studies are needed to determine the impact of EV isolation methods, library preparation and sequencing instruments on the estimates, our benchmarking still demonstrated a high correlation between the best‐performing deconvolution methods estimates of tissue‐ and cell‐type‐specific EV abundance in urine and plasma samples.
It is important to highlight that plasma and urine EVs are not only derived from a few sources but that many tissues/cell types contribute. Identifying these tissues/cell types is challenging with the marked‐based deconvolution methods. As discussed above, too many tissues/cell types with similar gene expression may confound the results; however, the opposite is also true. Our test with the addition of EV RNA from adipocytes and macrophages, which was not present in the signature matrix, did not affect the deconvolution estimates of best‐performing methods. While marker‐based deconvolution methods cannot classify EVs from the “unknown” cell types, complete deconvolution methods can unmix samples without knowledge about cell types (Zaitsev et al., 2019). Nonetheless, our analyses showed that the relative abundances of the known cell type‐specific EVs were maintained, demonstrating that the deconvolution estimates are highly sensitive to the composition of the signature matrix.
Our benchmarking indicates that the transcriptome deconvolution of EV RNA data from biofluid is an attractive means to unmix tissue/cell type‐specific EV abundances and thereby provide an additional method to aid the interpretation of bulk analyses of EV cargo such as proteins, lipids and RNAs. EV cargo abundance is a product of two main factors: the level of cargo within each EV and the abundance of cell‐type‐specific EVs in the sample. The relative contribution of these two factors is dynamically regulated by cellular gene expression level and cell type‐specific EV release rate. While isolation or enrichment for cell type‐specific EVs could unmix these two factors, only a restricted number of affinity reagents are currently available. This constraint does not limit the deconvolution methods, which can effectively quantify the influence of the tissue/cell type‐specific EV abundance. Indeed, Shi et al. (2020) used CIBERSORTx to infer the cell populations represented in plasma EVs from patients with metastatic melanoma and used it to develop a tool to monitor tumour/non‐tumour ratios in response to treatment. Thus, the transcriptome deconvolution of EVs to identify the parental cell types creates new opportunities to understand human health and diseases.
Our overall recommendations for transcriptome deconvolution of EV samples are that (i) DWLS (Tsoucas et al., 2019) and CIBERSORTx (Newman et al., 2019) are good method choices, (ii) cellular subtypes or tissues with highly similar cell type composition can significantly bias the EV estimates and (iii) the tissue/cell type signature matrix should match biological knowledge and cover the tissue/cell types expected to contribute to the biofluid's EV pool. Although additional optimization on the genes included in the signature matrix may improve the estimates of the other methods, DWLS and CIBERSORTx made accurate estimates using their default settings for the construction of the signature matrix. Moreover, their highly similar and robust estimates of the DWLS and CIBERSORTx methods suggest that these two existing transcriptome deconvolution methods offer tools to determine tissue‐ and cell‐type‐specific EV abundances. Combined with expression analyses and, for example, proteomics data, this can provide new insights into the role of EVs in health and diseases.
AUTHOR CONTRIBUTIONS
Jannik Hjortshøj Larsen: Data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); validation (equal); visualization (equal); writing—review and editing (equal). Iben Skov Jensen: Data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); validation (equal); visualization (equal); writing—review and editing (equal). Per Svenningsen: Conceptualization (lead); data curation (lead); formal analysis (lead); funding acquisition (lead); investigation (lead); methodology (lead); project administration (lead); resources (lead); software (lead); supervision (lead); validation (lead); visualization (lead); writing—original draft (lead); writing—review and editing (lead).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Supplementary Figure S1: Deconvolution of pure cell line‐derived EV samples. Heatmaps of deconvolution results of the individual EV samples scaled between 0 and 1. The upper left heatmap shows the true distribution.
Supplementary Figure S2: Comparison of in silico and physical mixtures of cell samples. (a) Multidimensional plot showing RNA sampled mixed physically and in silico. (b‐f) Scatter plots of the log‐CPM values of transcript abundances from physically and in silico mixed samples for mix (b) 100:0, (c) 75:25, (d) 50:50, (e) 25:75, and (f) 0:100. R indicates the Pearson correlation coefficient for each plot. Data is from GSE64098 (Holik et al., 2017)
Supplementary Figure S3: Comparison of downsampled DU145, LNCaP, PC3, and CRC EV samples. (a) Multidimensional plot showing original and downsampled DU145, LNCaP, PC3, and CRC EV samples. The original samples are indicated by a triangle, and the samples downsampled to 22.5 million, 15 million, and 7.5 million reads are shown as squares, circles, and diamonds. (b‐e) Scatter plots of log‐CPM values of the transcript abundances from the original and downsampled EV samples for (b) DU145, (c) LNCaP, (d) PC3, and (e) CRC cells. R indicates Pearson correlation for each plot.
Supplementary Figure S4: Deconvolution of downsampled DU145 and LNCaP EV samples. (a) Color coding for cell types. The effect of downsampling the DU145 and LNCaP EV samples on deconvolution by (b) DWLS, (c) DWLSj, (d) CIBERSORTx, (e) DWLS OLS, (f) EV‐origin, (g) DWLS SVR, (h) MuSiC, (i) dtangle, (j) Bisque, (k) DeconRNASeq, and (l) MuSiC nnls.
Supplementary Figure S5: Deconvolution of mixed DU145 and LNCaP EV samples. (a) Estimated cell‐specific EV abundance in mixed DU145 and LNCaP EV samples at different ratios > 10% by (from top to bottom): DWLSj, CIBERSORTx, DWLS OLS, EV‐origin, DWLS SVR, MuSiC, dtangle, Bisque, DeconRNASeq, and MuSiC nnls. (b) Similar to a, but with samples ≤10%.
Supplementary Figure S6: Deconvolution accuracy of complex mixtures of DU145, LNCaP, and PC3 EV samples. The cell type‐specific EV estimates for the 12 different DU145, LNCaP, and PC3 EV mixtures by the 11 deconvolution methods. The estimated EV abundance is the average of three in silico mixes.
Supplementary Figure S7: Deconvolution of exoRbase urine EV samples using a 27‐tissue signature matrix. Plots showing deconvolution of the 16 urine EV samples from the exoRbase cohort using a signature matrix created from RNA sequencing data from GTEX of 27 tissues by (a) DWLS, (b) DWLSj, (c) CIBERSORTx, (d) DWLS OLS, (e) DWLS SVR, (f) EV‐origin, (g) dtangle, (h) MuSiC, (i) Bisque, (j) DeconRNASeq, and (k) MuSiC nnls.
Supplementary Figure S8: Deconvolution of Dwivedi cohort's urine EV samples using a 27‐tissue signature. Plots showing deconvolution of the 72 urine EV samples from Dwivedi et al. (2023) cohort using a signature matrix created from RNA sequencing data from GTEX of 27 tissues by (a) DWLS, (b) DWLSj, (c) CIBERSORTx, (d) DWLS OLS, (e) DWLS SVR, (f) EV‐origin, (g) dtangle, (h) MuSiC, (i) Bisque, (j) DeconRNASeq, and (k) MuSiC nnls.
Supplementary Figure S9: Correlation matrix of deconvolution of exoRbase and Dwivedi cohorts’ urine EV samples with the 27‐tissue signature matrix. Correlation coefficients (R indicated by color) between the 11 deconvolution methods’ estimates of the tissue‐specific EV abundance in (a) exoRbase and (b) Dwivedi urine samples.
Supplementary Figure S10: Correlation between tissue‐specific urine EV abundance estimates between exoRbase and Dwivedi cohorts. (a) Correlation between the tissue‐specific urine EV abundance estimates between the two cohorts by DWLS, and (b) the remaining ten transcriptome deconvolution methods. R indicates the Pearson correlation coefficient.
Supplementary Figure S11: Correlation between kidney epithelial cell type‐specific urine EV abundance estimates between exoRbase and Dwivedi cohorts. Correlation matrix showing correlation coefficients between kidney epithelial cell‐derived urine EV abundances estimated by the deconvolution methods from the (a) exoRbase and (b) Dwivedi cohorts. (c) Correlation between the kidney epithelial cell‐specific urine EV abundance estimates between the two cohorts by the remaining 10 different deconvolution methods. R indicates the Pearson correlation coefficient. Abbreviations: PT, Proximal tubule; PC, Principal cell; IC, Intercalated cell; DCT, Distal convoluted tubule; cTAL, cortical Thick ascending limb of the loop of Henle's; CNT, Connecting tubule.
Supplementary Figure S12: Correlation matrix of the deconvolution methods’ estimates of tissue‐specific abundances of plasma EVs without merging blood and spleen. (a) Correlation matrix showing correlation coefficients between tissue‐specific plasma EV abundances estimated by the deconvolution methods without combining blood and spleen estimates. (b) Pearson correlation coefficients (R2 indicated by color) between the 11 deconvolution methods’ estimates of the tissue‐specific EV abundance in plasma samples with combined blood and spleen estimates.
Supplementary Figure S13: Correlation matrix of the deconvolution methods’ estimates of cell type‐specific plasma EV. Correlation matrix showing correlation coefficients between peripheral blood mononuclear cells‐derived plasma EV abundances estimated by the deconvolution methods.
Supplementary Tabe S1: Number of tissue samples included from the GTEX portal
ACKNOWLEDGEMENTS
This work was supported by the Independent Research Fund Denmark under Grant #10.46540/3103‐00263B, and the Augustinus Fonden under Grant #22‐2015. The research in the PS lab is supported by CARE‐IN‐HEALTH funded by the European Union's Horizon Europe Research and Innovation Framework program for Health under grant agreement 101095413. Views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Health and Digital Executive Agency. Alfred Gandrup Svenningsen is thanked for helping to download the data from the GEO database and transcript quantification.
Larsen, J. H. , Jensen, I. S. , & Svenningsen, P. (2024). Benchmarking transcriptome deconvolution methods for estimating tissue‐ and cell‐type‐specific extracellular vesicle abundances. Journal of Extracellular Vesicles, 13, e12511. 10.1002/jev2.12511
Jannik Hjortshøj Larsen, Iben Skov Jensen and Per Svenningsen contributed equally to this work.
REFERENCES
- Almeida, A. , Gabriel, M. , Firlej, V. , Martin‐Jaular, L. , Lejars, M. , Cipolla, R. , Petit, F. , Vogt, N. , San‐Roman, M. , Dingli, F. , Loew, D. , Destouches, D. , Vacherot, F. , de la Taille, A. , Théry, C. , & Morillon, A. (2022). Urinary extracellular vesicles contain mature transcriptome enriched in circular and long noncoding RNAs with functional significance in prostate cancer. Journal of Extracellular Vesicles, 11(5), e12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auber, M. , & Svenningsen, P. (2022). An estimate of extracellular vesicle secretion rates of human blood cells. Journal of Extracellular Biology, 1(6), e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckler, M. D. , Higginbotham, J. N. , Franklin, J. L. , Ham, A. J. , Halvey, P. J. , Imasuen, I. E. , Whitwell, C. , Li, M. , Liebler, D. C. , & Coffey, R. J. (2013). Proteomic analysis of exosomes from mutant KRAS colon cancer cells identifies intercellular transfer of mutant KRAS. Molecular & Cellular Proteomics: MCP, 12(2), 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blijdorp, C. J. , Hartjes, T. A. , Wei, K. Y. , van Heugten, M. H. , Bovée, D. M. , Budde, R. P. J. , van de Wetering, J. , Hoenderop, J. G. J. , van Royen, M. E. , Zietse, R. , Severs, D. , & Hoorn, E. J. (2022). Nephron mass determines the excretion rate of urinary extracellular vesicles. Journal of Extracellular Vesicles, 11(1), e12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, X. , Du, M. R. M. , Gouil, Q. , Tian, L. , Jabbari, J. S. , Bowden, R. , Baldoni, P. L. , Chen, Y. , Smyth, G. K. , Amarasinghe, S. L. , Law, C. W. , & Ritchie, M. E. (2023). Benchmarking long‐read RNA‐sequencing analysis tools using in silico mixtures. Nature Methods, 20(11), 1810–1821. [DOI] [PubMed] [Google Scholar]
- Dwivedi, O. P. , Barreiro, K. , Käräjämäki, A. , Valo, E. , Giri, A. K. , Prasad, R. B. , Roy, R. D. , Thorn, L. M. , Rannikko, A. , Holthöfer, H. , Gooding, K. M. , Sourbron, S. , Delic, D. , Gomez, M. F. , iBEAt . Groop, P. H. , Tuomi, T. , Forsblom, C. , Groop, L. , & Puhka, M. . (2023). Genome‐wide mRNA profiling in urinary extracellular vesicles reveals stress gene signature for diabetic kidney disease. iScience, 26(5), 106686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. , Domrachev, M. , & Lash, A. E. (2002). Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research, 30(1), 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada, A. L. , Valenti, Z. J. , Hehn, G. , Amorese, A. J. , Williams, N. S. , Balestrieri, N. P. , Deighan, C. , Allen, C. P. , Spangenburg, E. E. , Kruh‐Garcia, N. A. , & Lark, D. S. (2022). Extracellular vesicle secretion is tissue‐dependent ex vivo and skeletal muscle myofiber extracellular vesicles reach the circulation in vivo. American Journal of Physiology Cell Physiology, 322(2), C246–C259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Martin, R. , Brandao, B. B. , Thomou, T. , Altindis, E. , & Kahn, C. R. (2022a). Tissue differences in the exosomal/small extracellular vesicle proteome and their potential as indicators of altered tissue metabolism. Cell Reports, 38(3), 110277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Martin, R. , Wang, G. , Brandão, B. B. , Zanotto, T. M. , Shah, S. , Kumar Patel, S. , Schilling, B. , & Kahn, C. R. (2022b). MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature, 601, 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, T. , & Szustakowski, J. D. (2013). DeconRNASeq: A statistical framework for deconvolution of heterogeneous tissue samples based on mRNA‐Seq data. Bioinformatics (Oxford, England), 29(8), 1083–1085. [DOI] [PubMed] [Google Scholar]
- Gupta, D. , Liang, X. , Pavlova, S. , Wiklander, O. P. B. , Corso, G. , Zhao, Y. , Saher, O. , Bost, J. , Zickler, A. M. , Piffko, A. , Maire, C. L. , Ricklefs, F. L. , Gustafsson, O. , Llorente, V. C. , Gustafsson, M. O. , Bostancioglu, R. B. , Mamand, D. R. , Hagey, D. W. , Görgens, A. , … El Andaloussi, S. (2020). Quantification of extracellular vesicles in vitro and in vivo using sensitive bioluminescence imaging. Journal of Extracellular Vesicles, 9(1), 1800222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, Y. , Hao, S. , Andersen‐Nissen, E. , Mauck, W. M. 3rd , Zheng, S. , Butler, A. , Lee, M. J. , Wilk, A. J. , Darby, C. , Zager, M. , Hoffman, P. , Stoeckius, M. , Papalexi, E. , Mimitou, E. P. , Jain, J. , Srivastava, A. , Stuart, T. , Fleming, L. M. , Yeung, B. , … Satija, R. (2021). Integrated analysis of multimodal single‐cell data. Cell, 184(13), 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatton, I. A. , Galbraith, E. D. , Merleau, N. S. C. , Miettinen, T. P. , Smith, B. M. , & Shander, J. A. (2023). The human cell count and size distribution. Proceedings of the National Academy of Sciences of the United States of America, 120(39), e2303077120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyesi, H. , Pallinger, É. , Mecsei, S. , Hornyák, B. , Kovácsházi, C. , Brenner, G. B. , Giricz, Z. , Pálóczi, K. , Kittel, Á. , Tóvári, J. , Turiak, L. , Khamari, D. , Ferdinandy, P. , & Buzás, E. I. (2022). Circulating cardiomyocyte‐derived extracellular vesicles reflect cardiac injury during systemic inflammatory response syndrome in mice. Cellular and Molecular Life Sciences: CMLS, 79(2), 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham, J. N. , Demory Beckler, M. , Gephart, J. D. , Franklin, J. L. , Bogatcheva, G. , Kremers, G. J. , Piston, D. W. , Ayers, G. D. , McConnell, R. E. , Tyska, M. J. , & Coffey, R. J. (2011). Amphiregulin exosomes increase cancer cell invasion. Current Biology: CB, 21(9), 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinger, S. A. , Cha, D. J. , Franklin, J. L. , Higginbotham, J. N. , Dou, Y. , Ping, J. , Shu, L. , Prasad, N. , Levy, S. , Zhang, B. , Liu, Q. , Weaver, A. M. , Coffey, R. J. , & Patton, J. G. (2018). Diverse long RNAs are differentially sorted into extracellular vesicles secreted by colorectal cancer cells. Cell Reports, 25(3), 715–725.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holik, A. Z. , Law, C. W. , Liu, R. , Wang, Z. , Wang, W. , Ahn, J. , Asselin‐Labat, M. L. , Smyth, G. K. , & Ritchie, M. E. (2017). RNA‐seq mixology: Designing realistic control experiments to compare protocols and analysis methods. Nucleic Acids Research, 45(5), e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt, G. J. , Freytag, S. , Bahlo, M. , & Gagnon‐Bartsch, J. A. (2018). dtangle: Accurate and robust cell type deconvolution. Bioinformatics (Oxford, England), 35(12), 2093–2099. [DOI] [PubMed] [Google Scholar]
- Jew, B. , Alvarez, M. , Rahmani, E. , Miao, Z. , Ko, A. , Garske, K. M. , Sul, J. H. , Pietiläinen, K. H. , Pajukanta, P. , & Halperin, E. (2020). Accurate estimation of cell composition in bulk expression through robust integration of single‐cell information. Nature Communications, 11(1), 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, H. , Li, Y. , Zhang, H. , Hu, J. , Liao, J. , Su, Y. , Li, Q. , Chen, B. , Li, C. , Wang, Z. , Li, Y. , Wang, J. , Meng, Z. , Huang, Z. , & Huang, S. (2021). exoRBase 2.0: An atlas of mRNA, lncRNA and circRNA in extracellular vesicles from human biofluids. Nucleic Acids Research, 50(D1), D118–D128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , He, X. , Li, Q. , Lai, H. , Zhang, H. , Hu, Z. , Li, Y. , & Huang, S. (2020). EV‐origin: Enumerating the tissue‐cellular origin of circulating extracellular vesicles using exLR profile. Computational and Structural Biotechnology Journal, 18, 2851–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Zhao, J. , Yu, S. , Wang, Z. , He, X. , Su, Y. , Guo, T. , Sheng, H. , Chen, J. , Zheng, Q. , Li, Y. , Guo, W. , Cai, X. , Shi, G. , Wu, J. , Wang, L. , Wang, P. , He, X. , & Huang, S. (2019). Extracellular vesicles long RNA sequencing reveals abundant mRNA, circRNA, and lncRNA in human blood as potential biomarkers for cancer diagnosis. Clinical Chemistry, 65(6), 798–808. [DOI] [PubMed] [Google Scholar]
- Love, M. I. , Soneson, C. , Hickey, P. F. , Johnson, L. K. , Pierce, N. T. , Shepherd, L. , Morgan, M. , & Patro, R. (2020). Tximeta: Reference sequence checksums for provenance identification in RNA‐seq. PLoS Computational Biology, 16(2), e1007664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, W. , Dai, Y. , Chen, Z. , Yue, X. , Andrade‐Powell, K. C. , & Chang, J. (2020). Spatial and temporal tracking of cardiac exosomes in mouse using a nano‐luciferase‐CD63 fusion protein. Communications Biology, 3(1), 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy, C. M. , Murphy, J. M. , Zhang, L. , Clotet‐Freixas, S. , Mathews, J. A. , An, J. , Karimzadeh, M. , Pouyabahar, D. , Su, S. , Zaslaver, O. , Röst, H. , Arambewela, R. , Liu, L. Y. , Zhang, S. , Lawson, K. A. , Finelli, A. , Wang, B. , MacParland, S. A. , Bader, G. D. , … Crome, S. Q. (2022). Single‐cell profiling of healthy human kidney reveals features of sex‐based transcriptional programs and tissue‐specific immunity. Nature Communications, 13(1), 7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckles, V. N. , Morton, M. C. , Holmberg, J. C. , Sokolov, A. M. , Nottoli, T. , Liu, D. , & Feliciano, D. M. (2019). A transgenic inducible GFP extracellular‐vesicle reporter (TIGER) mouse illuminates neonatal cortical astrocytes as a source of immunomodulatory extracellular vesicles. Scientific Reports, 9(1), 3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, A. M. , Steen, C. B. , Liu, C. L. , Gentles, A. J. , Chaudhuri, A. A. , Scherer, F. , Khodadoust, M. S. , Esfahani, M. S. , Luca, B. A. , Steiner, D. , Diehn, M. , & Alizadeh, A. A. (2019). Determining cell type abundance and expression from bulk tissues with digital cytometry. Nature Biotechnology, 37(7), 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørgård, M. Ø. , Steffensen, L. B. , Hansen, D. R. , Füchtbauer, E. M. , Engelund, M. B. , Dimke, H. , Andersen, D. C. , & Svenningsen, P. (2022). A new transgene mouse model using an extravesicular EGFP tag enables affinity isolation of cell‐specific extracellular vesicles. Scientific Reports, 12(1), 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla, J. A. , Barutcu, S. , Malet, L. , Deschamps‐Francoeur, G. , Calderon, V. , Kwon, E. , & Lécuyer, E. (2023). Profiling the polyadenylated transcriptome of extracellular vesicles with long‐read nanopore sequencing. BMC Genomics, 24(1), 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro, R. , Duggal, G. , Love, M. I. , Irizarry, R. A. , & Kingsford, C. (2017). Salmon provides fast and bias‐aware quantification of transcript expression. Nature Methods, 14(4), 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufino‐Ramos, D. , Lule, S. , Mahjoum, S. , Ughetto, S. , Cristopher Bragg, D. , Pereira de Almeida, L. , Breakefield, X. O. , & Breyne, K. (2022). Using genetically modified extracellular vesicles as a non‐invasive strategy to evaluate brain‐specific cargo. Biomaterials, 281, 121366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, A. , Kasumova, G. G. , Michaud, W. A. , Cintolo‐Gonzalez, J. , Díaz‐Martínez, M. , Ohmura, J. , Mehta, A. , Chien, I. , Frederick, D. T. , Cohen, S. , Plana, D. , Johnson, D. , Flaherty, K. T. , Sullivan, R. J. , Kellis, M. , & Boland, G. M. (2020). Plasma‐derived extracellular vesicle analysis and deconvolution enable prediction and tracking of melanoma checkpoint blockade outcome. Science Advances, 6(46), eabb3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, G. J. , Poppe, D. , Simmons, R. K. , Walsh, K. , Nawaz, U. , Lister, R. , Gagnon‐Bartsch, J. A. , & Voineagu, I. (2022). Comprehensive evaluation of deconvolution methods for human brain gene expression. Nature Communications, 13(1), 1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsen, P. , Sabaratnam, R. , & Jensen, B. L. (2020). Urinary extracellular vesicles: Origin, role as intercellular messengers and biomarkers; efficient sorting and potential treatment options. Acta physiologica (Oxford, England)[Internet], 228(1), e13346. https://onlinelibrary.wiley.com/doi/abs/10.1111/apha.13346 [DOI] [PubMed] [Google Scholar]
- Tsoucas, D. , Dong, R. , Chen, H. , Zhu, Q. , Guo, G. , & Yuan, G. C. (2019). Accurate estimation of cell‐type composition from gene expression data. Nature Communications, 10(1), 2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Park, J. , Susztak, K. , Zhang, N. R. , & Li, M. (2019). Bulk tissue cell type deconvolution with multi‐subject single‐cell expression reference. Nature Communications, 10(1), 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Hagen, J. , Guntur, K. V. , Allette, K. , Schuyler, S. , Ranjan, J. , Petralia, F. , Gesta, S. , Sebra, R. , Mahajan, M. , Zhang, B. , Zhu, J. , Houten, S. , Kasarskis, A. , Vishnudas, V. K. , Akmaev, V. R. , Sarangarajan, R. , Narain, N. R. , Schadt, E. E. , … Tu, Z. (2017). A next generation sequencing based approach to identify extracellular vesicle mediated mRNA transfers between cells. BMC Genomics, 18(1), 987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, S. , Li, Y. , Liao, Z. , Wang, Z. , Wang, Z. , Li, Y. , Qian, L. , Zhao, J. , Zong, H. , Kang, B. , Zou, W. B. , Chen, K. , He, X. , Meng, Z. , Chen, Z. , Huang, S. , & Wang, P. (2020). Plasma extracellular vesicle long RNA profiling identifies a diagnostic signature for the detection of pancreatic ductal adenocarcinoma. Gut, 69(3), 540–550. [DOI] [PubMed] [Google Scholar]
- Zaitsev, K. , Bambouskova, M. , Swain, A. , & Artyomov, M. N. (2019). Complete deconvolution of cellular mixtures based on linearity of transcriptional signatures. Nature Communications, 10(1), 2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, Q. , Cheng, L. , Deng, C. , Huang, L. , Li, J. , Wang, Y. , Li, M. , Yang, Q. , Dong, X. , Su, J. , Lee, L. P. , & Liu, F. (2021). The genetic source tracking of human urinary exosomes. Proceedings of the National Academy of Sciences of the United States of America, 118(43), e2108876118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: Deconvolution of pure cell line‐derived EV samples. Heatmaps of deconvolution results of the individual EV samples scaled between 0 and 1. The upper left heatmap shows the true distribution.
Supplementary Figure S2: Comparison of in silico and physical mixtures of cell samples. (a) Multidimensional plot showing RNA sampled mixed physically and in silico. (b‐f) Scatter plots of the log‐CPM values of transcript abundances from physically and in silico mixed samples for mix (b) 100:0, (c) 75:25, (d) 50:50, (e) 25:75, and (f) 0:100. R indicates the Pearson correlation coefficient for each plot. Data is from GSE64098 (Holik et al., 2017)
Supplementary Figure S3: Comparison of downsampled DU145, LNCaP, PC3, and CRC EV samples. (a) Multidimensional plot showing original and downsampled DU145, LNCaP, PC3, and CRC EV samples. The original samples are indicated by a triangle, and the samples downsampled to 22.5 million, 15 million, and 7.5 million reads are shown as squares, circles, and diamonds. (b‐e) Scatter plots of log‐CPM values of the transcript abundances from the original and downsampled EV samples for (b) DU145, (c) LNCaP, (d) PC3, and (e) CRC cells. R indicates Pearson correlation for each plot.
Supplementary Figure S4: Deconvolution of downsampled DU145 and LNCaP EV samples. (a) Color coding for cell types. The effect of downsampling the DU145 and LNCaP EV samples on deconvolution by (b) DWLS, (c) DWLSj, (d) CIBERSORTx, (e) DWLS OLS, (f) EV‐origin, (g) DWLS SVR, (h) MuSiC, (i) dtangle, (j) Bisque, (k) DeconRNASeq, and (l) MuSiC nnls.
Supplementary Figure S5: Deconvolution of mixed DU145 and LNCaP EV samples. (a) Estimated cell‐specific EV abundance in mixed DU145 and LNCaP EV samples at different ratios > 10% by (from top to bottom): DWLSj, CIBERSORTx, DWLS OLS, EV‐origin, DWLS SVR, MuSiC, dtangle, Bisque, DeconRNASeq, and MuSiC nnls. (b) Similar to a, but with samples ≤10%.
Supplementary Figure S6: Deconvolution accuracy of complex mixtures of DU145, LNCaP, and PC3 EV samples. The cell type‐specific EV estimates for the 12 different DU145, LNCaP, and PC3 EV mixtures by the 11 deconvolution methods. The estimated EV abundance is the average of three in silico mixes.
Supplementary Figure S7: Deconvolution of exoRbase urine EV samples using a 27‐tissue signature matrix. Plots showing deconvolution of the 16 urine EV samples from the exoRbase cohort using a signature matrix created from RNA sequencing data from GTEX of 27 tissues by (a) DWLS, (b) DWLSj, (c) CIBERSORTx, (d) DWLS OLS, (e) DWLS SVR, (f) EV‐origin, (g) dtangle, (h) MuSiC, (i) Bisque, (j) DeconRNASeq, and (k) MuSiC nnls.
Supplementary Figure S8: Deconvolution of Dwivedi cohort's urine EV samples using a 27‐tissue signature. Plots showing deconvolution of the 72 urine EV samples from Dwivedi et al. (2023) cohort using a signature matrix created from RNA sequencing data from GTEX of 27 tissues by (a) DWLS, (b) DWLSj, (c) CIBERSORTx, (d) DWLS OLS, (e) DWLS SVR, (f) EV‐origin, (g) dtangle, (h) MuSiC, (i) Bisque, (j) DeconRNASeq, and (k) MuSiC nnls.
Supplementary Figure S9: Correlation matrix of deconvolution of exoRbase and Dwivedi cohorts’ urine EV samples with the 27‐tissue signature matrix. Correlation coefficients (R indicated by color) between the 11 deconvolution methods’ estimates of the tissue‐specific EV abundance in (a) exoRbase and (b) Dwivedi urine samples.
Supplementary Figure S10: Correlation between tissue‐specific urine EV abundance estimates between exoRbase and Dwivedi cohorts. (a) Correlation between the tissue‐specific urine EV abundance estimates between the two cohorts by DWLS, and (b) the remaining ten transcriptome deconvolution methods. R indicates the Pearson correlation coefficient.
Supplementary Figure S11: Correlation between kidney epithelial cell type‐specific urine EV abundance estimates between exoRbase and Dwivedi cohorts. Correlation matrix showing correlation coefficients between kidney epithelial cell‐derived urine EV abundances estimated by the deconvolution methods from the (a) exoRbase and (b) Dwivedi cohorts. (c) Correlation between the kidney epithelial cell‐specific urine EV abundance estimates between the two cohorts by the remaining 10 different deconvolution methods. R indicates the Pearson correlation coefficient. Abbreviations: PT, Proximal tubule; PC, Principal cell; IC, Intercalated cell; DCT, Distal convoluted tubule; cTAL, cortical Thick ascending limb of the loop of Henle's; CNT, Connecting tubule.
Supplementary Figure S12: Correlation matrix of the deconvolution methods’ estimates of tissue‐specific abundances of plasma EVs without merging blood and spleen. (a) Correlation matrix showing correlation coefficients between tissue‐specific plasma EV abundances estimated by the deconvolution methods without combining blood and spleen estimates. (b) Pearson correlation coefficients (R2 indicated by color) between the 11 deconvolution methods’ estimates of the tissue‐specific EV abundance in plasma samples with combined blood and spleen estimates.
Supplementary Figure S13: Correlation matrix of the deconvolution methods’ estimates of cell type‐specific plasma EV. Correlation matrix showing correlation coefficients between peripheral blood mononuclear cells‐derived plasma EV abundances estimated by the deconvolution methods.
Supplementary Tabe S1: Number of tissue samples included from the GTEX portal