

Abstract
Loss-of-function mutations of the parkin gene are a major cause of early-onset parkinsonism. To explore the mechanism by which loss of parkin function results in neurodegeneration, we are using a genetic approach in Drosophila. Here, we show that Drosophila parkin mutants display degeneration of a subset of dopaminergic (DA) neurons in the brain. The neurodegenerative phenotype of parkin mutants is enhanced by loss-of-function mutations of the glutathione S-transferase S1 (GstS1) gene, which were identified in an unbiased genetic screen for genes that modify parkin phenotypes. Furthermore, overexpression of GstS1 in DA neurons suppresses neurodegeneration in parkin mutants. Given the previous evidence for altered glutathione metabolism and oxidative stress in sporadic Parkinson's disease (PD), these data suggest that the mechanism of DA neuron loss in Drosophila parkin mutants is similar to the mechanisms underlying sporadic PD. Moreover, these findings identify a potential therapeutic approach in treating PD.
Keywords: genetic modifier, neurodegeneration, parkin
Parkinson's disease (PD) is a common neurodegenerative disorder characterized by the loss of dopaminergic (DA) neurons in the substantia nigra and the accumulation of proteinaceous intraneuronal inclusions known as Lewy bodies. The mechanisms responsible for neurodegeneration in PD are largely unknown, although previous work suggests that mitochondrial complex I dysfunction, oxidative stress, and aberrant proteolytic degradation may contribute to pathogenesis (1). The recent identification of genes responsible for rare inherited forms of parkinsonism presents an opportunity to establish neurodegenerative mechanisms that may be relevant to sporadic forms of PD.
Loss-of-function mutations of parkin are a common cause of autosomal recessive juvenile parkinsonism (ARJP), and parkin dysfunction may also contribute to late-onset sporadic PD (2–6). Patients with parkin mutations display many of the typical features of idiopathic PD, including locomotor dysfunction, reduced mitochondrial complex I activity, and degeneration of DA neurons in the substantia nigra. However, most ARJP cases have a significantly earlier age of onset and lack Lewy body pathology. Parkin has been shown to possess ubiquitin-protein ligase activity (7–9), which acts to confer substrate target specificity in the ubiquitin/proteasome protein degradation pathway. This finding has led to the model that toxic accumulation of parkin substrates may be responsible for DA neuron death. A number of putative substrates of parkin have been identified (10). Several of these parkin substrates, including the Lewy body component α-synuclein (11) and the putative G protein-coupled receptor Pael-R (12), have received considerable attention, in part because they implicate specific cellular pathways in parkin-mediated pathogenesis. However, the involvement of many of the identified parkin substrates in the etiology of ARJP remains controversial.
To identify pathways relevant to parkin pathogenesis, we are using a genetic approach to study a Drosophila ortholog of the parkin gene. Although the anatomy of the Drosophila brain differs from that of the vertebrate brain, many features of nervous system development and function are conserved in flies and humans, and this conservation has made Drosophila an excellent system in which to model neurodegeneration (13). In previous work, we showed that loss of Drosophila parkin function results in muscle degeneration and a late developmental defect in the formation of spermatids (14). Mitochondrial pathology is the earliest detectable phenotype associated with muscle degeneration and defective spermatids in Drosophila parkin mutants, suggesting that parkin is required for mitochondrial integrity.
In this study, we have examined DA neuron integrity in Drosophila parkin mutants. We show that parkin is required cell-autonomously to prevent degeneration of a subset of DA neurons in the brain. The neurodegenerative phenotype of parkin mutants is enhanced by a loss-of-function allele of the glutathione S-transferase S1 (GstS1) gene that was identified in a screen for modifiers of a parkin partial lethal phenotype (15). Furthermore, we demonstrate that overexpression of GstS1 prevented DA neuron degeneration in parkin mutants. These observations suggest that a variety of compounds known to induce glutathione S-transferase expression in mammals may provide a promising therapeutic strategy for PD.
Methods
Fly Strains. The park25 null allele, hypomorphic parkZ472 allele, isogenic control parkrvA, and upstream activating sequence (UAS)-parkC2 transgene are described in ref. 14. The parkZ3678 hypomorph, which fails to complement other parkin alleles, has a noncoding point mutation. The tyrosine hydroxylase (TH)-GAL4 driver was a gift from S. Birman (Developmental Biology, Institute of Marseille, Marseille, France) (16). GstS1K9303, GstS1K8805, GstS14227, and GstS1EP2223, 24B-GAL4, and Df(2R)ED1 were obtained from the Bloomington Stock Center (Bloomington, IN). GstS1M26 was isolated by excision of GstS1EP1227 (H.B., unpublished data). UAS-GstS1 was constructed by cloning GstS1 cDNA LD21131 (Berkeley Drosophila Genome Project, Berkeley, CA) into transformation vector pUASP. Germ-line transformants were generated by using standard procedures. Drosophila cultures were kept under standard conditions, and all experiments were performed on flies raised at 25°C.
Analysis of Protein Carbonyl and Glutathione Content. Protein carbonyl content was calculated by using an established procedure (17). Glutathione levels were assayed by using a glutathione detection kit (Chemicon) according to the manufacturer's instructions.
Analysis of DA and Serotonergic Neurons. DA and serotonergic neurons were analyzed in situ by using anti-TH antiserum Ab152 (1:100, Chemicon) and anti-serotonin antiserum (1:100, Immunostar, Hudson, WI), respectively. Adult heads were dissected in cold PBS, and isolated brains were fixed in 4% paraformaldehyde/PBS for 30 min. Samples were washed in PBS/0.1% Triton X-100 and blocked for 1 h in 0.1 M Tris·Cl, pH 7.5/0.15 M NaCl/0.1% Triton X-100/10% heat-inactivated FBS. Anti-TH was incubated in blocking solution at 4°C overnight. After washing and incubating fluorescent secondary antiserum, samples were again washed and mounted between two glass coverslips by using ProLong antifade medium (Molecular Probes). This method of sample mounting allows visualization of the samples from both sides of the slide to optimize detection of all DA neuron clusters. Confocal microscopy was used to collect optical sections at 1-μm intervals. The number of TH-positive neurons within each of the major DA neuron clusters was determined by visual inspection of individual confocal Z-series images. Additionally, brains from adult flies segregating the TH-GAL4 driver and a UAS-GFP transgene were dissected and fixed as described above. DA neurons were identified by using confocal microscopy to detect intrinsic GFP fluorescence, and the number of GFP-positive neurons was determined as described above. In all studies, DA neurons were counted with the investigator blinded to genotype. The same neuronal phenotypes were observed in completely independent experiments performed by three different investigators (A.J.W., D.A.T., and P.D.W.) at different institutions.
Behavior. Climbing assays were conducted as described in ref. 14. Recombinant chromosomes were generated between 24B-GAL4 and two parkin hypomorphic mutations, parkZ472 and parkZ3678. These recombinants were tested with and without a UAS-GstS1 transgene.
Results
In a previous study, we analyzed DA neuron viability in parkin mutants compared with controls by assessing the number of neurons positive for the DA neuron-specific marker TH in paraffin-imbedded head sections (14). Although no neuronal loss was detected, substantial variance in sample analysis might have obscured a subtle loss of DA neurons. Therefore, we repeated this analysis by using confocal microscopy of whole-mount adult brains stained with antiserum to TH (Fig. 1). This method allowed us to visualize individual DA neurons in the adult Drosophila brain at high resolution and to reproducibly identify all of the previously reported DA neurons (18), thus providing a sensitive approach for a quantitative analysis of DA neurons in parkin mutants.
Fig. 1.

A subset of DA neurons degenerates in parkin mutants. (A) Diagram shows the locations of the major DA neuron clusters in the adult Drosophila brain. PAL, protocerebral anterior lateral; PPM, protocerebral posterior medial; VUM, ventral unpaired medial. The PAL cluster is located in the same position but anterior to the PPL1 cluster, as designated by the arrow. (B) A projected Z-series confocal image from a WT adult brain stained with anti-TH to label DA neurons. DA neurons within the boxed section are shown at higher magnification in C and D. (C and D) Representative images from WT (C) and parkin mutant (D) brains aged 20 days. Fewer DA neurons are detected in the PPL1 cluster in parkin mutants relative to WT. (E and F) The number of DA neurons in each major DA neuron cluster in WT (black bars) and parkin mutants (gray bars) in 1-day-old (E) and 20-day-old (F) adult flies. parkin mutants display a significant decrease in the number of PPL1 neurons at 1 day of age (*, P < 0.05). Neuron loss in parkin mutants is more extensive in the PPL1 cluster at 20 days of age (***, P < 0.0001). Statistical significance was calculated by using Student's t test. n refers to the number of brains used for neuron counts. Animals homozygous for the parkin null allele park25 or bearing the isogenic parkrvA chromosome, which has a WT allele of parkin, were used in these analyses.
Using this method, we compared the number of neurons in each of the DA neuron clusters in parkin mutants and isogenic control animals. Importantly, to control for investigator bias, all experiments were carried out with the experimenter blinded to the sample genotypes throughout the analysis. In agreement with previous work (14, 19), no gross anatomical defects were observed in the cortical or neuropil regions of the adult Drosophila brain, and TUNEL staining experiments did not reveal detectable differences between brains of parkin mutants and controls (data not shown), indicating that the vast majority of neurons are intact and healthy in parkin mutants. Furthermore, no dramatic differences were observed between DA neurons of parkin mutants and control animals (Fig. 1). However, quantification of the different DA neuron clusters of 1-day-old parkin adults relative to age-matched controls revealed a significant reduction in the number of DA neurons in one of the clusters, the protocerebral posterior lateral (PPL) 1 (Fig. 1). In 20-day-old parkin adults, the TH-positive PPL1 neurons showed a further decrease in numbers (Fig. 1). Again, no significant difference was detected in any other DA neuron cluster in 20-day-old parkin mutants. By contrast, the number of TH-positive neurons in the PPL1 cluster during the late pupal stage, in which the adult CNS is already fully formed, was indistinguishable between parkin mutants and controls (data not shown). These data indicate that loss of parkin function in Drosophila results in the progressive degeneration of DA neurons in the PPL1 cluster in the adult brain.
Additional experiments were performed to further characterize the neurodegenerative phenotype of parkin mutants. To confirm that loss of TH-positive neurons in parkin mutants was not an artifact of anti-TH immunostaining, we used an alternative method to detect DA neurons. The DA neuron-specific driver TH-GAL4 was used to drive GFP expression in parkin mutants, and GFP fluorescence was used as a marker of DA neurons. Although this GAL4 driver does not express GFP in all of the TH-positive neurons in the PPL1 cluster (ref. 16 and Fig. 2), experiments with this driver revealed significantly decreased numbers of DA neurons in the PPL1 cluster of 20-day-old parkin mutants relative to age-matched control animals (Fig. 2). To verify that the loss of PPL1 neurons derives from loss of parkin function, the TH-GAL4 driver was used to induce expression of a parkin transgene (UAS-park) in an effort to rescue the observed degeneration. Results of this analysis revealed that transgenic expression of parkin in DA neurons significantly attenuated DA neuron loss in the PPL1 cluster (Fig. 2). These results confirm the neurodegenerative phenotype of parkin mutants and demonstrate that parkin is required cell-autonomously for DA neuron integrity. The incomplete rescue of DA neuron loss by transgenic expression of parkin may reflect lack of expression of the TH-GAL4 driver in some of the PPL1 neurons, inefficient expression of transgenic parkin, and/or an additional nonautonomous role of parkin in DA neuron integrity.
Fig. 2.

Neuron loss in parkin mutants is specific to DA neurons and results primarily from loss of a cell-autonomous requirement of parkin.(A–C) Expression of GFP driven by TH-GAL4 (A) and anti-TH (B) staining reveals a high degree of colocalization of signals (C). The boxed area in C marks the PPL1 cluster and is magnified in C′.(D) Analysis of GFP-positive neurons in 20-day-old adult parkin mutants and age-matched WT controls bearing the TH-GAL4 driver and a UAS-GFP transgene confirms the cell loss in the PPL1 DA neuron cluster of parkin mutants (**, P < 0.005, Student's t test). (E) DA neuron loss is significantly reduced by expression of a parkin transgene using the TH-GAL4 driver compared with parkin mutant or parkin mutants bearing the TH-GAL4 driver alone. Expression of GAL4 from the TH-GAL4 driver did not significantly affect DA neuron viability. (**, P < 0.001; Δ, P = 0.3) (F) Diagram shows all major serotonergic neurons in the adult brain. SP, subaesophageal; LP, lateral protocerebral; IP, inferior medial protocerebral. (G) Representative confocal micrograph of a WT adult brain stained with anti-5-hydroxytyrosine to reveal serotonergic neurons. (H) No significant difference in the number of serotonergic neurons is detectable in 20-day-old adult parkin mutants relative to age-matched controls, indicating that neuron loss is specific to a subset of DA neurons. Statistical significance was calculated by using ANOVA and Bonferroni's post hoc test for planned comparisons. Numbers shown in histograms refer to the number of brains used for neuron counts. park25 or parkrvA homozygotes were used in all analyses of neuronal viability.
Neurodegeneration in PD is largely restricted to a subset of DA neurons in the brain. To evaluate the specificity of neuron loss in Drosophila parkin mutants, the integrity of other catecholaminergic neurons was assessed. Specifically, antiserum against serotonin (5-hydroxytyrosine) was used to analyze serotonergic neurons in 20-day-old parkin mutants and age-matched isogenic controls (20). In contrast to our analyses of DA neurons, no significant neuronal loss was observed in any of the serotonergic clusters in parkin mutants (Fig. 2). These results, in conjunction with our previous work and the observation that overall brain volume was not altered in parkin mutants, indicate that neurodegeneration in parkin mutants is selective for a subset of DA neurons in the central nervous system.
In an effort to identify pathways that influence the Drosophila parkin phenotypes, we performed a genetic screen for dominant modifiers of a parkin partial pupal lethal phenotype, which appears to result from muscle dysfunction (15). A loss-of-function allele of GstS1 was the strongest enhancer recovered from this screen. To further assess the involvement of GstS1 in parkin pathogenesis, we explored the effects of altered GstS1 function on other parkin phenotypes. In previous work, we showed that parkin function is required in the musculature for normal geotactic climbing behavior. Thus, we tested whether GstS1 mutations influence the climbing phenotype of parkin mutants. All of the GstS1 alleles tested, including several that were not used in our genetic screen, were also found to enhance a climbing defect of parkin mutants when heterozygous with a WT allele of GstS1 (Fig. 3). By contrast, GstS1 mutations alone had no effect on climbing ability in a background of WT parkin or in parkin heterozygotes (Fig. 3 and data not shown). Moreover, overexpression of a UAS-GstS1 transgene in the musculature, using the mesoderm GAL4 driver, 24B-GAL4, was sufficient to restore the climbing ability of two hypomorphic parkin mutants to the WT (Fig. 3). Considering the extensive muscle degeneration observed in parkin null mutants, hypomorphic mutants were used in this assay to potentiate the detection of small differences in climbing ability.
Fig. 3.

GstS1 activity modifies locomotor deficits in parkin mutants. (A) Loss-of-function alleles of GstS1 enhance the climbing defect of parkin mutants. Each loss-of-function GstS1 allele, EP2223, k09303, k08805, and 04227, used in this analysis was heterozygous with a WT allele of GstS1. park25 or parkrvA homozygotes were used in all climbing assays. Enhancement of the parkin climbing defect by each of the GstS1 alleles was significant (P < 0.005, Student's t test). (B) Directed expression of a GstS1 transgene (UAS-GstS1) by a muscle GAL4 transgene (24B) alleviates the partial climbing deficits seen in two parkin hypomorphic mutants (parkZ472 and parkZ3678). Suppression of climbing deficits from GstS1 overexpression was significant (**, P < 0.001) by Student's t test.
To extend these findings to the neurodegenerative phenotype of parkin mutants, we analyzed the effect of altered GstS1 activity on DA neuron integrity in parkin mutants. Although GstS1 appears to be predominantly expressed in flight muscle, GstS1 expression is also detected in the adult head (21). To explore the effects of reduced GstS1 dosage on the parkin DA neuron loss phenotype, parkin mutants were crossed to a stock bearing a viable null allele of GstS1 designated GstS1M26. parkin mutants bearing the GstS1M26 mutation in trans to a deletion chromosome that removes the GstS1 gene [Df(2R)ED1] were recovered at low frequency and displayed dramatically shortened lifespan (data not shown). Analysis of DA neuron integrity in 1-day-old parkin mutants bearing this combination of GstS1 alleles revealed significantly enhanced DA neuron loss relative to parkin mutants alone (Fig. 4). Loss of GstS1 function alone had no affect on DA neuron viability in 1-day-old adults (Fig. 4). Conversely, transgenic overexpression of GstS1 only in DA neurons, using the TH-GAL4 driver in conjunction with a UAS-GstS1 transgene, was able to significantly attenuate the loss of DA neurons in 20-day-old parkin mutants (Fig. 4). Moreover, the degree of rescue conferred by GstS1 expression was comparable to that observed with transgenic parkin expression (Fig. 4). These results indicate that GstS1 potently modulates DA neuron viability in parkin mutants.
Fig. 4.
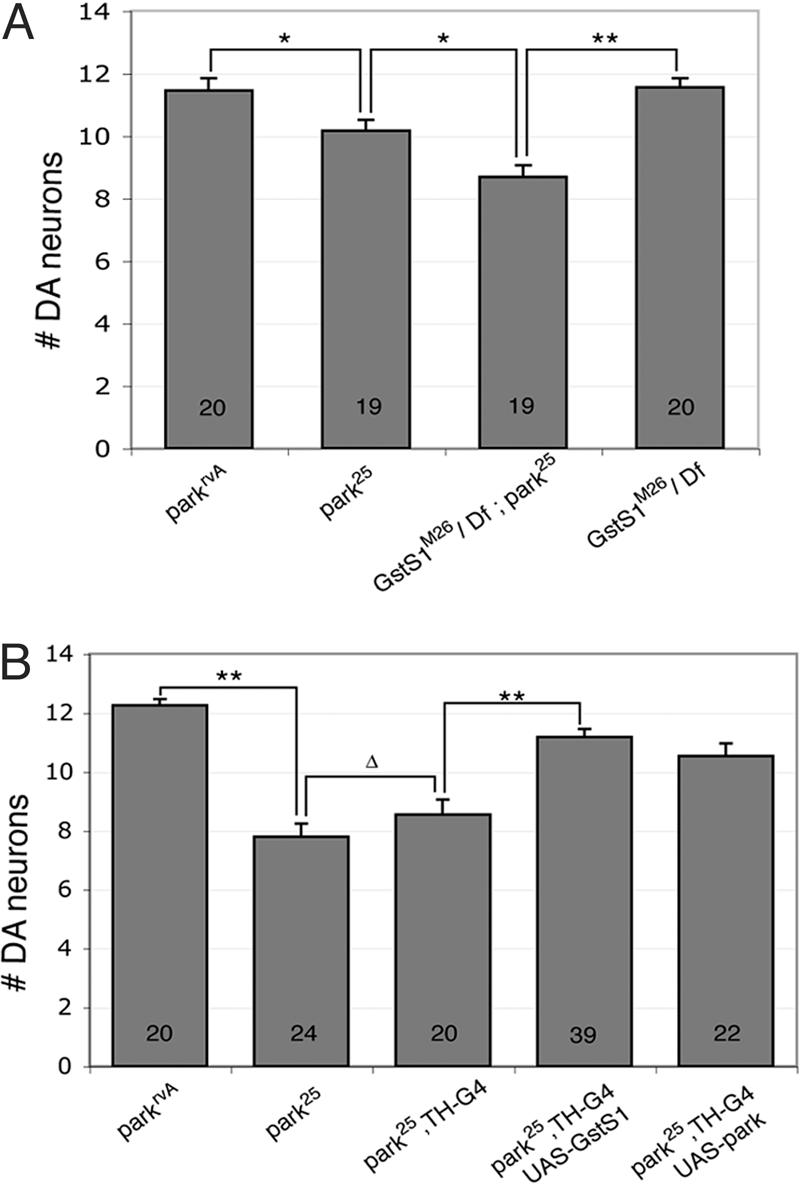
GstS1 activity influences DA neuron viability in parkin mutants. (A)A GstS1 null allele (GstS1M26) in trans to a deletion (Df) of the GstS1 region [Df(2R)ED1] enhances DA neuron loss detected in the PPL1 cluster of 1-day-old parkin mutants. GstS1 null mutants (GstS1M26/Df) alone manifest no DA neuron loss in a WT parkin background. (B) Transgenic overexpression of GstS1 in DA neurons significantly suppresses DA neuron loss in 20-day-old parkin mutants (park25,TH-G4; UAS-GstS1). The degree of rescue conferred by GstS1 is comparable to that achieved by transgenic expression of parkin in DA neurons (park25,TH-G4; UAS-park). Statistical significance was calculated by using ANOVA and Bonferroni's post hoc test for planned comparisons. (*, P <0.05; **, P < 0.001; Δ, P = 0.3). Numbers shown in bars refer to the number of brains analyzed for neuron counts. park25 or parkrvA homozygotes were used in all analyses of neuronal viability.
Glutathione S-transferases catalyze the conjugation of reduced glutathione to a variety of substrates, including the products of reactive oxygen species (22), and substantial evidence suggests that oxidative stress is a major contributing factor in sporadic PD (23). Our results demonstrating GstS1 modification of the parkin loss-of-function phenotypes suggest that parkin may act to limit oxidative stress-induced cellular damage. To begin to investigate this hypothesis, we tested whether parkin mutants display other evidence of oxidative stress. One consequence of oxidative stress is the production of protein carbonyls. Examination of the level of protein carbonyl content revealed significantly elevated levels of protein carbonyl content in 1-day-old adult parkin mutants (10.4 nmol of carbonyls per mg of protein) relative to age-matched isogenic controls (2.6 nmol of carbonyls per mg of protein). These results suggest that parkin confers protection from damage by reactive oxygen species. However, we did not detect significant alterations in the levels of glutathione in 1-day-old parkin mutants, indicating that oxidative damage in parkin mutants is not simply a consequence of decreased glutathione levels.
Discussion
Substantial evidence suggests that oxidative stress contributes to DA neuron loss in sporadic PD (24). In particular, increased levels of lipid peroxidation, oxidatively damaged proteins and DNA, and decreased levels of glutathione have been documented in the substantia nigra of PD patients (23). One potential source of this oxidative stress is increased free radical formation resulting from partial inhibition of mitochondrial complex I. Many PD patients display decreased complex I activity in the brain and peripheral tissues, and several complex I inhibitors cause a PD-like syndrome in humans and animal models (25). Our studies of Drosophila parkin mutants reveal many features in common with sporadic PD. We have previously reported that mutations in Drosophila parkin result in profound mitochondrial defects in multiple tissue types. In this report, we demonstrate that fly parkin mutants also display progressive degeneration of a subset of DA neurons and that this degeneration can be rescued by overexpression of a glutathione S-transferase, a factor that has been implicated in the cellular response to oxidative stress (26). These observations demonstrate that fly parkin mutants recapitulate key features of ARJP and suggest that the mechanisms responsible for DA neuron loss in ARJP are similar to the mechanisms responsible for sporadic PD.
Three previous analyses of neuronal integrity in Drosophila parkin mutants, including a study from our laboratory, failed to detect DA neuron loss (14, 19, 27). However, the specific DA neuron cluster that degenerates in parkin mutants (PPL1) was not analyzed in two of these studies (19, 27). Although the PPL1 neuron cluster was assessed in our previous study of Drosophila parkin mutants, the substantial variation in DA neuron counts observed in this study would not have allowed detection of the reduction of DA neurons reported in our current study. A major motivation of the present study was to apply unbiased quantitative methods to analyze all of the DA neuron clusters in the adult Drosophila brain in an effort to detect subtle alterations in neuron structure and integrity. Results described here indicate that confocal microscopy is a powerful method to facilitate detection of subtle neuron loss in Drosophila.
To understand how mutations in parkin result in DA neuron death in the human disease, substantial effort has been invested in the identification of targets of the parkin ubiquitin-protein ligase activity. A number of parkin substrates have been identified (10). Two of these substrates, α-synuclein and Pael-R, have received considerable attention, in part because in vitro studies have shown that these components are cytotoxic when overexpressed, and both of these proteins appear to accumulate in the brains of ARJP individuals (11, 12). Furthermore, overexpression of parkin has been shown to attenuate the toxicity of these proteins in vitro and in vivo (11, 12, 27–31). These findings have led to a model whereby accumulation of these proteins causes DA neuron death in ARJP individuals. Although most of the previously identified parkin substrates appear to have Drosophila orthologs, blast searches of the Drosophila genome sequence have failed to detect orthologs of α-synuclein or Pael-R. Thus, our results demonstrating DA neuron degeneration in Drosophila parkin mutants suggest that α-synuclein and Pael-R are not obligate in DA neurodegeneration. Although our work challenges the absolute requirement of α-synuclein and Pael-R in the etiology of ARJP, these factors may contribute to the severity and/or magnitude of cell loss in ARJP individuals. However, the fact that DA neuron degeneration can occur in the absence of α-synuclein and Pael-R indicates that additional factors are clearly involved in ARJP pathogenesis. The identification of these factors is paramount to our understanding of the molecular etiology of this disorder.
Previous work on GstS1 function in Drosophila suggests that this factor may play a role in detoxifying products of oxidative damage (26). Our finding that altered GstS1 activity influences the parkin mutant phenotypes raises the possibility that parkin may also offer protection from the effects of oxidative stress. Further support for this hypothesis comes from the findings that oxidative damage in flies, mice, and cell lines and sensitivity to oxidative stress agents in flies and cell lines correlates inversely with parkin activity (19, 32–34). Our recent results from transcriptional profiling of parkin mutants and a genetic screen for parkin modifiers also demonstrate that oxidative stress response elements are up-regulated and that mutations in oxidative stress response components enhance the parkin mutant phenotypes (15).
There are potentially many ways that parkin might confer protection from the effects of oxidative stress. For example, parkin may recognize substrates bearing a specific oxidative modification and target these damaged proteins for degradation. Support for this model is provided by the finding that the HOIL-1 ubiquitin-protein ligase, a protein with a similar domain structure to parkin, has recently been shown to recognize and ubiquitinate an oxidatively modified form of its substrate IRP2 (35). Alternatively, the findings that altered parkin function results in mitochondrial defects in cell lines, flies, mice, and humans raises the possibility that parkin may directly influence mitochondrial integrity (14, 32, 36, 37). Studies in yeast have identified a ubiquitin ligase involved in mitochondria fission (38), and ubiquitination is also important for the insertion of proteins into the outer mitochondrial membrane (39, 40). An intriguing possibility is that parkin may label oxidatively damaged mitochondrial proteins and target the entire mitochondrion for destruction by autophagy. Indeed, the ultrastructural morphology of mitochondria in parkin mutant flies is strikingly similar to the appearance of mitochondria that are engulfed during autophagy (41). Furthermore, previous work has shown that ubiquitination of mitochondrial proteins and the formation of other ubiquitin-like conjugates can target mitochondria for autophagic degradation (42, 43).
Our findings suggest a potential therapeutic approach for ARJP and possibly sporadic PD. There are a number of compounds that are known to induce glutathione S-transferase activity in vertebrates (44). Although these compounds have been studied primarily because of their protective effects against cancer, our current results suggest that these compounds might be useful in the treatment of ARJP. There are several reasons to believe that this potential treatment strategy could extend beyond ARJP. First, as mentioned above, significant evidence suggests that oxidative stress is involved in the etiology of sporadic PD (24). Furthermore, alleles of the Gst omega-1 gene have been found to influence the age of onset of PD in humans (45), and loss-of-function mutations of the yeast GstS1 homolog, gtt1, enhance the toxicity of α-synuclein in this organism (46). Because mutations that increase α-synuclein expression result in heritable forms of PD and α-synuclein is a component of the Lewy body inclusions associated with sporadic PD, the finding that reduced gtt1 activity in yeast enhances α-synuclein toxicity suggests that altered glutathione S-transferase activity may be relevant to sporadic PD pathogenesis. A number of excellent models of α-synucleinopathy currently exist (46–49) that can be used to test this hypothesis.
In summary, our findings that DA neuron loss occurs in Drosophila parkin mutants and that DA neuron loss can be modified by a putative oxidative damage detoxification factor suggest that the mechanisms responsible for DA neuron death in Drosophila parkin mutants and sporadic PD are conserved. Furthermore, the observation that a modifier of the reduced viability and climbing phenotypes of Drosophila parkin mutants also modifies the DA neuron loss phenotype suggests an underlying similarity in the etiology of the muscle and neuron phenotypes in Drosophila parkin mutants. The ease of detecting modifiers of the Drosophila parkin viability and behavioral phenotypes and our current demonstration of the relevance of one of these factors to DA neuron integrity illustrate the potential of this system to identify genetic pathways and therapeutic factors relevant to an understanding of PD pathogenesis and treatment.
Acknowledgments
We thank the Berkeley Drosophila Genome Project and the Bloomington Stock Center for providing fly stocks; L. Andrews, A. Khan, and A. Peterson for help with technical aspects of this work; and P. Muchowski, A. La Spada, F. Perez, and members of L.J.P.'s laboratory for scientific discussion and critical comments on the manuscript. This work was supported by National Institutes of Health Grant 1RO1NS41780-01 (to L.J.P.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PD, Parkinson's disease; DA, dopaminergic; GstS1, glutathione S-transferase S1; ARJP, autosomal recessive juvenile parkinsonism; TH, tyrosine hydroxylase; PPL, protocerebral posterior lateral; UAS, upstream activating sequence.
References
- 1.Dawson, T. M. & Dawson, V. L. (2003) Science 302, 819–822. [DOI] [PubMed] [Google Scholar]
- 2.West, A. B. & Maidment, N. T. (2004) Hum. Genet. 114, 327–336. [DOI] [PubMed] [Google Scholar]
- 3.Yao, D., Gu, Z., Nakamura, T., Shi, Z. Q., Ma, Y., Gaston, B., Palmer, L. A., Rockenstein, E. M., Zhang, Z., Masliah, E., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 10810–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung, K. K., Thomas, B., Li, X., Pletnikova, O., Troncoso, J. C., Marsh, L., Dawson, V. L. & Dawson, T. M. (2004) Science 304, 1328–1331. [DOI] [PubMed] [Google Scholar]
- 5.Pawlyk, A. C., Giasson, B. I., Sampathu, D. M., Perez, F. A., Lim, K. L., Dawson, V. L., Dawson, T. M., Palmiter, R. D., Trojanowski, J. Q. & Lee, V. M. (2003) J. Biol. Chem. 278, 48120–48128. [DOI] [PubMed] [Google Scholar]
- 6.Kalia, S. K., Lee, S., Smith, P. D., Liu, L., Crocker, S. J., Thorarinsdottir, T. E., Glover, J. R., Fon, E. A., Park, D. S. & Lozano, A. M. (2004) Neuron 44, 931–945. [DOI] [PubMed] [Google Scholar]
- 7.Shimura, H., Hattori, N., Kubo, S., Mizuno, Y., Asakawa, S., Minoshima, S., Shimizu, N., Iwai, K., Chiba, T., Tanaka, K. & Suzuki, T. (2000) Nat. Genet. 25, 302–305. [DOI] [PubMed] [Google Scholar]
- 8.Imai, Y., Soda, M. & Takahashi, R. (2000) J. Biol. Chem. 275, 35661–35664. [DOI] [PubMed] [Google Scholar]
- 9.Zhang, Y., Gao, J., Chung, K. K., Huang, H., Dawson, V. L. & Dawson, T. M. (2000) Proc. Natl. Acad. Sci. USA 97, 13354–13359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Coelln, R., Dawson, V. L. & Dawson, T. M. (2004) Cell Tissue Res. 318, 175–184. [DOI] [PubMed] [Google Scholar]
- 11.Shimura, H., Schlossmacher, M. G., Hattori, N., Frosch, M. P., Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K. S. & Selkoe, D. J. (2001) Science 293, 263–269. [DOI] [PubMed] [Google Scholar]
- 12.Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y. & Takahashi, R. (2001) Cell 105, 891–902. [DOI] [PubMed] [Google Scholar]
- 13.Marsh, J. L. & Thompson, L. M. (2004) BioEssays 26, 485–496. [DOI] [PubMed] [Google Scholar]
- 14.Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B. & Pallanck, L. J. (2003) Proc. Natl. Acad. Sci. USA 100, 4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greene, J. C., Whitworth, A. J., Andrews, L. A., Parker, T. J. & Pallanck, L. J. (2005) Hum. Mol. Genet. 14, 799–811. [DOI] [PubMed] [Google Scholar]
- 16.Friggi-Grelin, F., Coulom, H., Meller, M., Gomez, D., Hirsh, J. & Birman, S. (2003) J. Neurobiol. 54, 618–627. [DOI] [PubMed] [Google Scholar]
- 17.Reznick, A. Z. & Packer, L. (1994) Methods Enzymol. 233, 357–363. [DOI] [PubMed] [Google Scholar]
- 18.Nassel, D. R. & Elekes, K. (1992) Cell Tissue Res. 267, 147–167. [DOI] [PubMed] [Google Scholar]
- 19.Pesah, Y., Pham, T., Burgess, H., Middlebrooks, B., Verstreken, P., Zhou, Y., Harding, M., Bellen, H. & Mardon, G. (2004) Development (Cambridge, U.K.) 131, 2183–2194. [DOI] [PubMed] [Google Scholar]
- 20.Valles, A. M. & White, K. (1988) J. Comp. Neurol. 268, 414–428. [DOI] [PubMed] [Google Scholar]
- 21.Clayton, J. D., Cripps, R. M., Sparrow, J. C. & Bullard, B. (1998) J. Muscle Res. Cell Motil. 19, 117–127. [DOI] [PubMed] [Google Scholar]
- 22.Hayes, J. D., Flanagan, J. U. & Jowsey, I. R. (2005) Annu. Rev. Pharmacol. Toxicol. 45, 51–88. [DOI] [PubMed] [Google Scholar]
- 23.Bharath, S., Hsu, M., Kaur, D., Rajagopalan, S. & Andersen, J. K. (2002) Biochem. Pharmacol. 64, 1037–1048. [DOI] [PubMed] [Google Scholar]
- 24.Dauer, W. & Przedborski, S. (2003) Neuron 39, 889–909. [DOI] [PubMed] [Google Scholar]
- 25.Sherer, T. B., Betarbet, R. & Greenamyre, J. T. (2002) Neuroscientist 8, 192–197. [DOI] [PubMed] [Google Scholar]
- 26.Singh, S. P., Coronella, J. A., Beneš, H., Cochrane, B. J. & Zimniak, P. (2001) Eur. J. Biochem. 268, 2912–2923. [DOI] [PubMed] [Google Scholar]
- 27.Yang, Y., Nishimura, I., Imai, Y., Takahashi, R. & Lu, B. (2003) Neuron 37, 911–924. [DOI] [PubMed] [Google Scholar]
- 28.Lo Bianco, C., Schneider, B. L., Bauer, M., Sajadi, A., Brice, A., Iwatsubo, T. & Aebischer, P. (2004) Proc. Natl. Acad. Sci. USA 101, 17510–17515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haywood, A. F. & Staveley, B. E. (2004) BMC Neurosci. 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrucelli, L., O'Farrell, C., Lockhart, P. J., Baptista, M., Kehoe, K., Vink, L., Choi, P., Wolozin, B., Farrer, M., Hardy, J. & Cookson, M. R. (2002) Neuron 36, 1007–1019. [DOI] [PubMed] [Google Scholar]
- 31.Oluwatosin-Chigbu, Y., Robbins, A., Scott, C. W., Arriza, J. L., Reid, J. D. & Zysk, J. R. (2003) Biochem. Biophys. Res. Commun. 309, 679–684. [DOI] [PubMed] [Google Scholar]
- 32.Palacino, J. J., Sagi, D., Goldberg, M. S., Krauss, S., Motz, C., Wacker, M., Klose, J. & Shen, J. (2004) J. Biol. Chem. 279, 18614–18622. [DOI] [PubMed] [Google Scholar]
- 33.Hyun, D. H., Lee, M., Hattori, N., Kubo, S., Mizuno, Y., Halliwell, B. & Jenner, P. (2002) J. Biol. Chem. 277, 28572–28577. [DOI] [PubMed] [Google Scholar]
- 34.Jiang, H., Ren, Y., Zhao, J. & Feng, J. (2004) Hum. Mol. Genet. 13, 1745–1754. [DOI] [PubMed] [Google Scholar]
- 35.Yamanaka, K., Ishikawa, H., Megumi, Y., Tokunaga, F., Kanie, M., Rouault, T. A., Morishima, I., Minato, N., Ishimori, K. & Iwai, K. (2003) Nat. Cell Biol. 5, 336–340. [DOI] [PubMed] [Google Scholar]
- 36.Darios, F., Corti, O., Lucking, C. B., Hampe, C., Muriel, M. P., Abbas, N., Gu, W. J., Hirsch, E. C., Rooney, T., Ruberg, M. & Brice, A. (2003) Hum. Mol. Genet. 12, 517–526. [DOI] [PubMed] [Google Scholar]
- 37.Muftuoglu, M., Elibol, B., Dalmizrak, O., Ercan, A., Kulaksiz, G., Ogus, H., Dalkara, T. & Ozer, N. (2004) Movement Disorders 19, 544–548. [DOI] [PubMed] [Google Scholar]
- 38.Fritz, S., Weinbach, N. & Westermann, B. (2003) Mol. Biol. Cell 14, 2303–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhaung, Z. P. & McCauley, R. (1989) J. Biol. Chem. 264, 14594–14596. [PubMed] [Google Scholar]
- 40.Zhuang, Z. P., Marks, B. & McCauley, R. B. (1992) J. Biol. Chem. 267, 591–596. [PubMed] [Google Scholar]
- 41.Brunk, U. T. & Terman, A. (2002) Eur. J. Biochem. 269, 1996–2002. [DOI] [PubMed] [Google Scholar]
- 42.Thompson, W. E., Ramalho-Santos, J. & Sutovsky, P. (2003) Biol. Reprod. 69, 254–260. [DOI] [PubMed] [Google Scholar]
- 43.Ohsumi, Y. & Mizushima, N. (2004) Semin. Cell Dev. Biol. 15, 231–236. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen, T., Sherratt, P. J. & Pickett, C. B. (2003) Annu. Rev. Pharmacol. Toxicol. 43, 233–260. [DOI] [PubMed] [Google Scholar]
- 45.Li, Y. J., Oliveira, S. A., Xu, P., Martin, E. R., Stenger, J. E., Scherzer, C. R., Hauser, M. A., Scott, W. K., Small, G. W., Nance, M. A., et al. (2003) Hum. Mol. Genet. 12, 3259–3267. [DOI] [PubMed] [Google Scholar]
- 46.Willingham, S., Outeiro, T. F., DeVit, M. J., Lindquist, S. L. & Muchowski, P. J. (2003) Science 302, 1769–1772. [DOI] [PubMed] [Google Scholar]
- 47.Masliah, E., Rockenstein, E., Veinbergs, I., Mallory, M., Hashimoto, M., Takeda, A., Sagara, Y., Sisk, A. & Mucke, L. (2000) Science 287, 1265–1269. [DOI] [PubMed] [Google Scholar]
- 48.Feany, M. B. & Bender, W. W. (2000) Nature 404, 394–398. [DOI] [PubMed] [Google Scholar]
- 49.Giasson, B. I., Duda, J. E., Quinn, S. M., Zhang, B., Trojanowski, J. Q. & Lee, V. M. (2002) Neuron 34, 521–533. [DOI] [PubMed] [Google Scholar]