

Abstract
MHC–peptide complexes mediate key functions in adaptive immunity. In a classical view, MHC-I molecules present peptides from intracellular source proteins, whereas MHC-II molecules present antigenic peptides from exogenous and membrane proteins. Nevertheless, substantial crosstalk between these two pathways has been observed. We investigated the influence of autophagy on the MHC-II ligandome and demonstrated that peptide presentation is altered considerably upon induction of autophagy. The presentation of peptides from intracellular and lysosomal source proteins was strongly increased on MHC-II in contrast with peptides from membrane and secreted proteins. In addition, autophagy influenced the MHC-II antigen-processing machinery. Our study illustrates a profound influence of autophagy on the class II peptide repertoire and suggests that this finding has implications for the regulation of CD4+ T cell-mediated processes.
Keywords: antigen processing, lysosomal proteases, T helper cells
Peptides of foreign and self proteins are presented on MHC-I and MHC-II molecules at the cell surface and can be recognized by CD8+ and CD4+ T lymphocytes, respectively (1, 2). From a classical point of view, MHC-I molecules present antigenic peptides derived from intracellular proteins, whereas MHC-II molecules do so for exogenous and membrane proteins (3). This phenomenon is reflected in the two major cellular breakdown pathways for proteins: proteasomal degradation, particularly relevant to the generation of MHC-I peptides (4), and degradation by the endosomal/lysosomal system, which is responsible for the processing of MHC-II peptides (5). However, the separation of these distinct pools of source proteins is less stringent than originally believed. It is now well established that MHC-I molecules are able to present peptides derived from exogenous antigens (Ag) by a process known as cross presentation (6). On the other hand, intracellular proteins can be presented by MHC-II molecules (7, 8), even though the underlying processes are less clear. It has been recently shown that peptides from cytosolic model proteins can be presented on MHC-II molecules through autophagy (9–11). Autophagy plays a role in the endosomal/lysosomal degradation pathway and is responsible for feeding intracellular components into this pathway. It is thought to be required for normal turnover of cellular components, particularly in response to starvation (12). Against this background, we hypothesized that autophagy might mediate MHC-II presentation of intracellular Ag, meaning the contents of a cell contained within the plasma membrane, excluding large vacuoles and secretory or ingested material (Gene Ontology classifications), in general. Therefore, we performed a detailed characterization of the MHC-II ligand repertoire (ligandome) presented at the cell surface under normal conditions and after increased autophagy, leading to a comprehensive overall picture of changes in peptide processing and presentation.
Materials and Methods
Cell Culture and Autophagy Induction. Human B-lymphoblastoid Awells cell lines (IHW-No. 9090; HLA-DRB1*0401 and HLA-DRB4*0101) were maintained at 37°C in DMEM. During induction of autophagy, cells were kept in Hank's Balanced Salt Solution (HBSS). For comparative ligand analysis, it was crucial to maintain cells at a density of 0.2 × 106 cells per ml. If cells were kept at higher densities, culture medium was rapidly exhausted, leading to high basal autophagy levels. For autophagy inhibition, cells were kept in DMEM or HBSS supplemented with 10 mM 3-methyladenine. Dead cells were generated by three rounds of freezing in liquid nitrogen and thawing at 37°C. After this procedure, no live cells could be detected by light microscopy.
Analysis of Monodansylcadaverine (MDC)-Labeled Vacuoles. Autophagic vacuoles were labeled with MDC and analyzed by using either fluorescence microscopy (13, 14) or fluorescence spectroscopy in cell lysates (14), essentially as described.
Gene Expression Analysis by High-Density Oligonucleotide Microarrays. Gene expression analysis of six RNA samples (starved and control cells at 6 h and 24 h, 24 h in duplicate) was performed by Affymetrix (Santa Clara, CA) Human Genome U133 Plus 2.0 oligonucleotide microarrays according to the manufacturer's instructions. Microarray data are available from the Gene Expression Omnibus repository (www.ncbi.nih.gov/geo) with the accession no. GSE2435.
Affinity-Labeling of Active Cysteine Proteases and in Vitro Digestions. Crude endocytic fractions were generated by ultracentrifugation of postnuclear supernatants as described in ref. 15. Five micrograms of total endocytic protein were incubated in the presence of DCG-0N, a derivative of DCG-04 that shows the same labeling characteristics (16) for 1 h at room temperature. Samples were resolved by 12.5% SDS/PAGE, then blotted on a poly(vinylidene difluoride)-membrane and visualized by using streptavidine HRP and an enhanced chemiluminescence detection kit (17). Myelin basic protein (MBP)83–99 digestions were performed as described in ref. 18.
Elution and Analysis of MHC-II Bound Peptides. Peptides were isolated from frozen cell pellets (1 × 109 to 5.7 × 1010 cells) as described in refs. 19 and 20 by using the HLA-DR specific mAb L243 (21). Peptides were separated by RP-HPLC, and fractions were analyzed by MALDI-TOF MS and nano-electrospray ionization MS/MS as described in ref. 20.
For comparative peptide analysis between peptides eluted from 1 × 109 to 3 × 109 control cells and 1 × 109 to 2 × 109 cells undergoing autophagy, peptides were analyzed by nano-RP-HPLC directly coupled to the electrospray ionization source. In MS/MS experiments, sequence information was obtained by interpretation of fragment spectra by using computer-assisted database (NCBInr, nonredundant protein database) searching tools (mascot, Matrix Science, London) (22). To differentially quantify the identified peptides, peptide signals in mass chromatograms from serial LC-MS runs (runs performed directly one after the other by using the same settings) were summed, and quantification was done from relative peak heights in the corresponding mass spectra.
Supporting Information. For a more detailed version of materials and methods, see Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Results
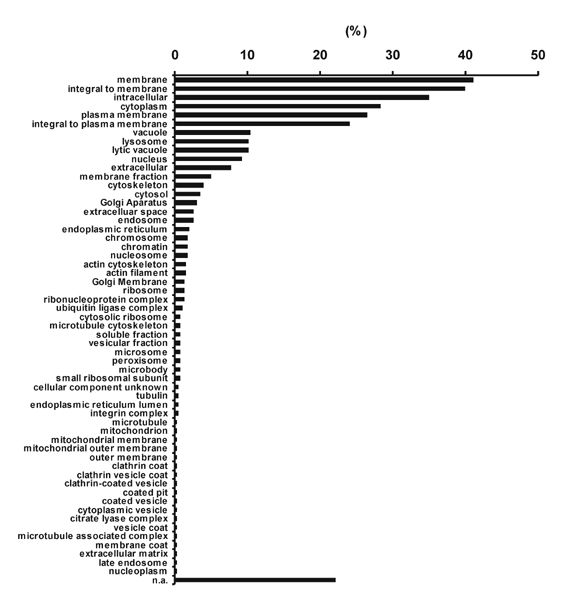
Proteomic Analysis of the Constitutive MHC-II Ligandome: Source Proteins of HLA-DR Presented Peptides Are Allocated Throughout the Cell. We analyzed the constitutive HLA-DR peptide repertoire of human Awells cells by MS and were able to identify 404 peptides from 173 different core sequences (Table 3, which is published as supporting information on the PNAS web site), some of them posttranslationally modified (Table 4, which is published as supporting information on the PNAS web site). This result is the largest number of MHC-presented peptides ever reported from a single experiment. To classify the source proteins according to their cellular localization (Fig. 5, which is published as supporting information on the PNAS web site) and function (Fig. 6, which is published as supporting information on the PNAS web site), we used the david program (23) and the Gene Ontology (go) classifications (24). In contrast to the situation observed for MHC-I (25), the largest fraction of MHC-II source proteins, namely 41.1%, belonged to membrane proteins, which is in concordance with conventional MHC-II antigen processing through the endosomal/lysosomal pathway. However, a rather large proportion of source proteins (34.9%) is localized intracellularly, meaning the contents of a cell contained within the plasma membrane, excluding large vacuoles and secretory or ingested material (go classifications), the site where MHC-I peptide processing is expected to take place. Furthermore, we could identify peptides from proteins localized in virtually every cell compartment: 10.1% lysosome, 9.2% nucleus, 4.0% cytoskeleton, 3.0% Golgi apparatus, 2.0% ER, 1.2% ribosome, 0.7% peroxisome, and 0.2% mitochondrion.
Induction of Autophagy by Starvation. Next, we induced macroautophagy in Awells cells by deprivation of serum and amino acids (14, 26) to perform a comparative, quantitative HLA-DR ligandome analysis between cells undergoing autophagy and control cells by MS. After 6 and 24 h of starvation, an increase in size and the total number of autophagic vacuoles became evident when compared with nonstarved control cells (Fig. 1 A–C). After 6 h of starvation, the formation of autophagic vacuoles, assessed by the overall incorporation of MDC, had already reached the maximum and could not be increased any further by 24 h of starvation (Fig. 1D). We were able to inhibit MDC incorporation by 3-methyladenine, a specific inhibitor of autophagy (data not shown). We would like to point out that we detected basal levels of autophagy in cells even if they were not kept in starvation medium. This finding indicates that Awells cells already display a constitutive level of autophagy, which can be considerably enhanced by starvation. This conclusion has already been demonstrated for other cell lines (14, 26).
Fig. 1.

Starvation enhances the level of autophagic vacuoles. Autophagic vacuoles were stained with the specific dye MDC (13) and analyzed by fluorescence microscopy or fluorescence spectroscopy. Awells cells were incubated for 24 h in DMEM (control cells) (A), 6 h HBSS (B), or 24 h HBSS (starved cells) (C), subsequently, for 10 min with MDC, washed, and immediately analyzed by fluorescence microscopy. Autophagic vacuoles are marked with an arrow. (D) Intracellular MDC measurement by fluorescence spectroscopy; unstained cells were used as negative control.
Comparative gene expression analysis by using oligonucleotide microarrays further supported these observations by revealing in starved cells a distinct transcriptional signature that reflected several hallmarks of autophagy as a process to ensure cell survival in a nutritionally deprived environment (Tables 5 and 6 and Supporting Results, which are published as supporting information on the PNAS web site).
Autophagy Promotes the Presentation of Peptides from Intracellular and Lysosomal Source Proteins on MHC-II Molecules. To determine whether autophagy contributes to the endogenous presentation of intracellular antigens on HLA class II in general or if this process represents a minor event followed only by some model antigens (9–11), the presentation levels of peptides from different inherent source proteins were quantified and compared between starved cells (6 and 24 h) undergoing autophagy and nonstarved control cells by MS. For the quantitation experiments, smaller cell numbers were used, resulting in a smaller subset of MHC-II ligands that could be analyzed. To exclude possible influences caused by an altered MHC surface expression, we measured MHC-I and -II levels by flow cytometry and observed no substantial changes upon autophagy induction (data not shown).
Fifty-four HLA-DR-bound peptides from 31 different source proteins were sequenced, quantified, and divided into two groups: peptides from membrane and secreted proteins, which should be preferentially presented on MHC-II molecules, and peptides from intracellular, especially nuclear, proteins, which should be preferentially presented on MHC-I molecules (Table 1). Additionally, we analyzed peptides from lysosomal proteins, because lysosomes take part in the autophagic turnover of the cell. After 6 h of starvation, the presentation of peptides from intracellular and lysosomal proteins rose on average by 27%, and after 24 h of starvation by 56% (Fig. 2), compared with peptides from membrane and secreted proteins. Upon application of unpaired two-tail Student's t tests to the two groups of quantified ligands, the means turned out to be significantly different (P < 0.001) with nonoverlapping 99% confidence intervals. Enhancement of presentation appeared to be selective for the cellular localization of peptide source proteins. From the four source proteins that showed the highest presentation levels of peptides after 24 h of starvation, three are localized in the nucleus and one in lysosomes (Table 1 and Fig. 2). In the case of these four peptides, presentation levels were raised on average by 131% after 24 h of autophagy. These changes represent relative differences in presentation levels normalized to the abundant peptide LSSWTAADTAAQITQR (Table 1). The absolute changes in presentation levels could not be assessed by our assay.
Table 1. Differential presentation of peptides on HLA-DR molecules and corresponding mRNA data.
| 6 h of starvation
|
24 h of starvation
|
|||||
|---|---|---|---|---|---|---|
| Source protein | Entrez gene ID | Peptide sequence | Peptide ratio | mRNA ratio | Peptide ratio | mRNA ratio |
| Membrane proteins | ||||||
| HLA-A*0201 | 3105 | FVRFDSDAASQR | 0.64 | NC | 1.53 | NC |
| 3105 | FVRFDSDAASQRME | 0.68 | NC | 1.30 | NC | |
| 3105 | DTQFVRFDSDAASQRME | 1.58 | NC | 0.36 | NC | |
| 3105 | VDDTQFVRFDSDAASQR | 1.15 | NC | 0.82 | NC | |
| 3105 | KHKWEAAHVAEQLR | 1.09 | NC | 1.23 | NC | |
| 3105 | DDTQFVRFDSDAASQRME | 1.18 | NC | 0.95 | NC | |
| HLA-B*4402 | 3106 | EDLSSWTAADTAAQITQRKWE | 1.18 | NC | 0.42 | NC |
| 3106 | LSSWTAADTAAQITQR | 1.11 | NC | 1.07 | NC | |
| HLA-Cw*0501 | 3107 | VDDTQFVQFDSDAASPRGEPR | 1.20 | NC | 0.50 | NC |
| 3107 | KDYIALNEDLRSWTA | 1.11 | NC | — | NC | |
| 3107 | DGKDYIALNEDLRSWTA | 1.01 | NC | 0.61 | NC | |
| 3107 | FVQFDSDAASPRGEPR | 0.76 | NC | 1.12 | NC | |
| HLA-E | 3133 | DLRSWTAVDTAAQISEQ | 0.97 | 1.87 | 0.70 | 2.46 |
| HLA-DQB1*0301 | 3119 | DVEVYRAVTPLGPPD | 1.25 | NC | — | NC |
| Lymphocyte antigen Ly-6E | 4061 | KPTICSDQDNYCVT | 1.18 | NC | — | 0.54 |
| 4061 | LKPTICCSDQDNYCVT | 1.40 | NC | 0.97 | 0.54 | |
| Immunoglobulin heavy chain | 3492 | YLQMNSLKTEDT | 0.75 | — | 1.33 | |
| 3492 | TLYLQMNSLKTEDT | 1.38 | — | — | ||
| Immunoglobulin λ chain | 3537 | SHKSYSCQVTHEGSTVE | 1.02 | — | 1.45 | |
| B-lymphocyte antigen CD 20 | 931 | INIYNCEPANPSEK | 1.16 | NC | 1.53 | NC |
| Class I cytokine receptor | 9466 | VGVPYRITVTAVSASG | 1.20 | NC | — | NC |
| Transferrin receptor protein 1 | 7037 | FTYINLDKAVLGTSN | 1.18 | NC | 0.85 | NC |
| Carboxypeptidase D | 1362 | VPGTYKITASARGYNPV | 1.27 | 1.23 | 1.37 | 1.52 |
| 1362 | VPGTYKITASARGYN | 1.13 | 1.23 | — | 1.52 | |
| Extracellular proteins | ||||||
| Serotransferrin (bovine) | FVKDQTVIQNTD | 0.66 | — | 1.37 | — | |
| DVAFVKDQTVIQNTD | 1.13 | — | — | — | ||
| DVAFVKDQTVIQ | 1.24 | — | — | — | ||
| Serum albumin (bovine) | SPDLPKLKPDPNTLCDEF | 1.24 | — | 1.01 | — | |
| Apolipoprot B-100 (bovine) | SASYKADTVAKVQGT | 1.08 | — | 1.02 | — | |
| SASYKADTVAKVQGTE | 0.98 | — | 0.44 | — | ||
| Intracellular proteins | ||||||
| Heat shock 70-kDa protein 1 | 3303 | NVLRIINEPTAAAIAYG | 1.50 | 3.48 | 1.48 | NC |
| 3303 | VLRIINEPTAAAIAY | 1.03 | 3.48 | 1.24 | NC | |
| 3303 | RIINEPTAAAIA | 1.49 | 3.48 | 2.25 | NC | |
| 3303 | VLRIINEPTAAAIAYG | 1.12 | 3.48 | 1.30 | NC | |
| Heat shock cognate 71-kDa protein | 3312 | GILNVSAVDKSTGKE | 1.67 | NC | 1.51 | NC |
| 3312 | ERAMTKDNNLLGKFE | 1.19 | NC | 1.50 | NC | |
| 3312 | GERAMTKDNNLLGKFE | 1.48 | NC | 1.30 | NC | |
| Elongation factor 1-α 1 | 1917 | IEKFEKEAAEMGKGSF | 1.49 | NC | 2.87 | NC |
| TNF, α-induced protein 3 | 7128 | EIIHKALIDRNIQ | 1.32 | 2.14 | — | 2.64 |
| RAD23 homolog B | 5887 | LLQQISQHQEHF | 1.88 | NC | 1.79 | NC |
| Actin, cytoplasmic 2 | 71 | TDYLMKILTERGYS | 1.30 | NC | 1.09 | NC |
| NEDD4La | 23327 | DGRTFYIDHNSKITQ | 1.26 | NC | 1.51 | NC |
| T complex protein 1, β subunit | 10576 | SLMVTNDGATILKN | 1.15 | NC | — | NC |
| Ubiquitin | 7311 | SDYNIQKESTLHLV | 1.05 | — | 1.42 | — |
| α enolase | 2023 | VPLYRHIADLAGNSEV | 1.50 | NC | 1.14 | NC |
| syntaxin 6 | 10228 | NPRKFNLDATELSIRK | 1.60 | NC | — | NC |
| tubulin β-5 chain | 10382 | EPYNATLSVHQL | 1.50 | NC | 1.23 | NC |
| Lysosomal proteins | ||||||
| Cathepsin C | 1075 | YDHNFVKAINAIQKSWT | 1.31 | NC | 1.28 | NC |
| 1075 | YDHNFVKAINAIQKSW | 1.28 | NC | 1.27 | NC | |
| 1075 | YDHNFVKAINAIQKS | 1.56 | NC | 1.40 | NC | |
| Cathepsin D | 1509 | LSRDPDAQPGGE | 0.83 | NC | 2.30 | NC |
| Cathepsin S | 1520 | TTAFQYIIDNKGIDSD | 1.61 | 2.30 | — | 4.92 |
| 1520 | TTAFQYIIDNKGID | 1.90 | 2.30 | 1.56 | 4.92 | |
| Lysosomal α-mannosidase | 4125 | VDYFLNVATAQGRYY | 1.64 | NC | — | NC |
The given peptide and mRNA ratios refer to the comparison of cells grown under starvation with control cells. For peptides, ratios were calculated from the signal intensities in LC-MS experiments. mRNA ratios were calculated from the signal log ratios given by the microarray analysis. No change (NC) is displayed if no significant change in the expression level was observed according to the change algorithm.
Fig. 2.

Altered peptide presentation on HLA-DR under starvation. Displayed are the relative intensity ratios of HLA-DR-eluted peptides from starved cells (6 and 24 h) and control cells as assessed by LC-MS. Peptides were quantified by their relative peak heights in mass spectra and grouped according to the cellular localization of their source proteins: membrane plus secreted proteins and intracellular plus lysosomal proteins. Data of serial LC-MS runs were normalized to the abundant peptide LSSWTAADTAAQITQR, which showed only marginal differences in presentation levels (Table 1). Horizontal bars indicate the mean intensity ratios for each group. Marked in a box are the four peptides that showed the highest presentation levels after 24 h of starvation. Their source proteins are localized in the nucleus and in lysosomes.
Apart from an increased uptake into autophagic vacuoles, several other processes might contribute to an enhanced presentation of peptides derived from intracellular proteins under starvation. To examine whether a higher mRNA expression for specific proteins upon autophagy induction led to an increased peptide presentation, gene expression for all 31 source proteins was assessed by oligonucleotide microarrays (Table 1). On average, mRNA levels of most genes remained stable under starvation. Among the membrane proteins, only HLA-E and carboxypeptidase D displayed an increased expression. For intracellular and lysosomal proteins, the same could be observed for TNF α-induced protein 3, heat shock 70-kDa protein 1, and cathepsin S. Peptides from the corresponding source proteins were also presented in higher amounts at the cell surface after induction of autophagy. We therefore cannot exclude the possibility that overexpression of these particular proteins during autophagy was the reason for elevated presentation levels of the corresponding peptides at the cell surface. However, only intracellular source proteins from 7 of 24 analyzed peptides showed elevated mRNA expression levels during autophagy. It is therefore highly unlikely that altered source gene expression was a major contributor to the observed changes in presentation levels.
In addition, an enhanced presentation of intracellular peptides on MHC-II molecules on cells undergoing autophagy might be due to an enhanced uptake of cellular debris by live cells, although this reaction should affect intracellular and membrane proteins similarly. To exclude this possibility we incubated control cells and cells undergoing autophagy with the corresponding amounts of dead cells (three freeze–thaw rounds) and analyzed the MHC-II ligands as described. We observed no enhanced presentation of intracellular peptides if dead cells were present (data not shown). Therefore, an enhanced uptake of dead cells in the starved samples does not contribute to the observed changes in MHC-II peptide presentation levels.
Autophagy Leads to a Time-Dependent Decrease of Lysosomal Proteases and Altered Antigen Processing. Interestingly, presentation levels of peptides derived from the same source protein were differentially affected by starvation. This result applied both to proteins processed by the classical MHC-II pathway, for example HLA-A*0201, and to intracellular proteins, such as heat shock 70-kDa protein 1 (Table 1). These data led us to hypothesize that activation of the autophagic pathway might concomitantly affect the MHC-II processing machinery by altering the activity levels of lysosomal proteases. Therefore, we assessed the activity of the major cathepsins during autophagy by affinity labeling (Fig. 3A). Active cathepsins Z, B, H, S, and C could be detected in control cells by using this method, largely in agreement with previous studies in other cells (17). Starvation of cells led to a time-dependent decrease of the activity signals for all cathepsins without a clear preference for any individual cathepsin. The same pattern of down-regulation was observed when control cells and cells undergoing autophagy were probed for cathepsin polypeptides by Western blot (Fig. 3B). This effect was not due to nonselective breakdown of total cellular protein or lysosomal protein in general, because the amounts of β-actin and the lysosome-resident protein LAMP-1 remained unaffected by autophagy.
Fig. 3.

Changes of lysosomal protease composition during autophagy. (A) Affinity labeling of active cathepsins. Endocytic extracts were generated from control cells, cells after 6 h and 24 h of starvation, and human peripheral blood monoyctes, respectively, by differential centrifugation as reported in refs. 15 and 27. Five-microgram total endocytic protein (1.5 μg in monocytes) were either directly incubated with the active site-restricted biotinylated affinity label DCG-0N as described (lanes: 2, control cells; 3, 6 h of starvation; 4, 24 h of starvation; and 9, monocytes) or were subjected to 95°C as negative control (lane 1). In addition, control cells were incubated with the CatS-inhibitor LHVS (25 nM), the CatB-inhibitor Ca074 (1 μM), the pan-cysteine protease inhibitors leupeptin (1 mM), or E64 (25 μM) (lanes 5–8) for 45 min at 37°C before labeling as further controls. Active cathepsins were visualized after resolution by SDS/PAGE by streptavidin-HRP blot: CatZ, CatB, CatH, and CatS at 36, 33, 30, and 28 kDa, respectively. (B) Cathepsin polypeptides probed by Western blot. Identical amounts of total cellular protein from control cells (lane 1) and cells undergoing autophagy (6 and 24 h of starvation, respectively; lane 2 and lane 3) were probed for CatS, CatC, CatD, CatH, β-actin, and LAMP-1 by Western blot.
To assess whether cathepsin down-regulation had an effect on the generation of antigenic peptides, we incubated MBP peptide ENPVVHFFKNIVTPRTP (MBP83–99), a well characterized model peptide for MHC-II processing (28), with active lysosomal extracts from control cells and cells undergoing autophagy and analyzed the degradation products by RP-HPLC, MALDI-MS, and Edman microsequencing. Fig. 4A shows the detected cleavage points that are largely in agreement with ref. 18. A quantitative analysis of the degradation products (Fig. 4B) revealed that the down-regulation of lysosomal proteases by autophagy affected the breakdown products to a different extent. As expected, the amount of undigested peptide was higher in autophagic cells, which exhibited lower cathepsin levels than control cells, and, in agreement with this finding, most of the breakdown products were more abundant in control cells than in autophagic cells (Table 2). However, the ratios of down-regulation of breakdown products differed markedly corresponding to the involved proteases. Whereas asparagine endoproteinase appeared to be quite sensitive to autophagy, the corresponding peptides dropped at an average of 33.2%, cathepsin D seemed to be more resistant, which is in concordance with the Western blot results (Fig. 3B). Thus, an overall down-regulation of active lysosomal proteases with only subtle differences between the key enzymes can nevertheless markedly influence the generation of different MHC-II ligands corresponding to the involved proteases. These data could explain the differences in MHC-II presentation levels of different peptides from the same source proteins that were observed in cells undergoing autophagy and control cells (Table 1).
Fig. 4.

MBP83–99 digestion with lysosomal extracts from control cells and cells undergoing autophagy. (A) Preferential detected cleavage sites. MBP83–99 was incubated at pH 5.4 with lysosomal extracts from control cells and cells undergoing 24 h autophagy for 3 h. Breakdown products were subsequently separated by RP-HLPC and analyzed by MALDI-MS and Edman microsequencing. (B) RP-HLPC chromatogram of control cell MBP breakdown products at 214 nm. The annotated peaks correspond to the major breakdown products of MBP83–99 as identified by MALDI MS and Edman microsequencing. The corresponding chromatogram of autophagic cells is not shown.
Table 2. Quantitative distribution of MBP83–99 breakdown products.
| Peak | Peptide sequence | Protease | Control cells, % | Autophagic cells, % |
|---|---|---|---|---|
| 1 | IVTPRTP | AEP | 21.5 | 12.6 |
| 2 | NIVTPRTP | Cat S/L | 3.3 | 2.2 |
| 3 | FKNIVTPRTP | Cat D | 2.2 | 3.2 |
| 4 | ENPVVHF | Cat D | 7.0 | 5.0 |
| 5 | NPVVHFFKN | — | 2.5 | 0.5 |
| 6 | FFKNIVTPRTP | Cat S/L | 1.4 | 1.2 |
| 7 | ENPVVHFFKN | AEP | 17.2 | 12.9 |
| 8 | ENPVVHFFK | Cat S/L | 3.6 | 1.5 |
| 9 | VVHFFKNIVTPRTP | — | 1.3 | 1.2 |
| PVVHFFKNIVTPRT | ||||
| 10 | NPVVHFFKNIVTPRTP | — | 3.2 | 1.1 |
| 11 | ENPVVHFFKNIVTPRTP | Undigested | 36.6 | 58.7 |
Breakdown products were quantified by their peak heights in the RP-HPLC chromatogram. The total amount of identified peptides was set to 100%. Shown are the differences between control cells and cells undergoing 24 h of autophagy and the lysosomal proteases, which are known to be responsible for the generation of the corresponding fragments (18).
Discussion
To assess the impact of autophagy on the HLA class II ligandome, we started by performing a detailed characterization of the MHC-II self peptide repertoire of a cell line grown under normal conditions. This analysis revealed that peptides from source proteins that are localized in almost all cell compartments and take part in general cellular processes are presented on MHC-II molecules. Some examples of peptides from intracellular proteins on MHC-II have been described in refs. 29 and 30. However, in our case, the number of such source proteins was surprisingly high. This result possibly reflects the detected basal level of autophagy that might be responsible for a constant shuttling of intracellular source proteins into the endosomal/lysosomal compartment. Thus, peptides from intracellular antigens are likely to have a larger impact on CD4+ helper T cell regulation than was originally believed. It has already been reported that CD4+ T cells are able to recognize peptides from intracellular melanoma antigens (31, 32) and the viral antigen EBNA1 (33), and that under inflammatory conditions, peptides from intracellular antigens are presented on HLA class II molecules on epithelial cells that are target cells in autoimmunity (34).
This study demonstrates that autophagy constitutes a general pathway promoting the processing of intracellular proteins by lysosomes and presentation of the resulting peptides on MHC-II molecules. Autophagy is a constitutive process responsible for the turnover of intracellular proteins (35). Basal levels have been observed in most tissues (36) and can be particularly enhanced by starvation. In addition, autophagy is involved in tumor development (36, 37). Starvation-induced autophagy has been observed, for example, in lymphocytes isolated from patients with chronic lymphocytic leukemia (38). This finding might indicate an important role of this process in tumor survival under nutrient-limiting conditions. In contrast, autophagy as a form of programmed cell death may accelerate tumor development if it is decreased (39). Some anticancer drugs potentially act through triggering autophagy (40) and, by doing so, could cause an enhanced presentation of intracellular CD4+ T cell epitopes in MHC-II-expressing tumor cells. Autophagy has also been described as a constitutive process under nutrient-rich conditions for several tissues in vivo, including thymic epithelial cells (41). For this reason, it might play an important role in the presentation of intracellular self-antigens to CD4+ T cells during negative selection.
Our results indicate a profound impact of enhanced autophagy on MHC-II antigen processing caused by a decrease of active cathepsins in the endocytic compartment. Decreased cathepsin levels might favor the generation of MHC-II peptides due to a less efficient lysosomal protein digestion. This hypothesis has been suggested as a mechanism to explain the superiority of dendritic cells over macrophages as antigen-presenting cells (42). Similarly, autophagy might subject the cell to an enhanced immune surveillance by CD4+ T cells under potentially dangerous stress conditions.
Recently, it has been reported that peptides from cytosolic antigens (9, 10) and from a cytosolic viral antigen (11) can be presented through autophagy on MHC-II molecules. It was not clear, however, if this represents a minor event or if autophagy contributes to the endogenous presentation of intracellular antigens on HLA class II in general. This study sheds more light on this issue by demonstrating that autophagy affects MHC-II presentation of peptides from intracellular proteins in general and by providing clear evidence for altered lysosomal processing. Thus, apart from its various known implications in stress responses and cell death, autophagy might play an important role in the regulation of CD4+ T cell-mediated processes.
Supplementary Material
Acknowledgments
We thank L. Yakes for carefully reading the manuscript and A. Nordheim and the Proteom Centrum Tübingen for the use of mass spectrometers. This work was supported by the Deutsche Krebshilfe (10-2189-St 2), the Deutsche Forschungsgemeinschaft (Graduiertenkolleg 794), the Studienstiftung des Deutschen Volkes, the Pinguin-Stiftung, and the German Federal Ministry of Education and Research (Fö. 01KS9602), in association with Interdisciplinary Center of Clinical Research Tübingen Project S.04.00088.
Author contributions: J.D., O.S., H.K., R.B., C.D., H.-G.R., and S.S. designed research; J.D., O.S., R.F., M.R., M.K., M.M., K.K., F.A., and J.B. performed research; J.D. and O.S. analyzed data; and J.D., O.S., H.-G.R., and S.S. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: MDC, monodansylcadaverine.
Data deposition: The microarray data reported in this paper have been deposited in the Gene Expression Omnibus database (accession no. GSE2435).
See Commentary on page 7779.
References
- 1.Rammensee, H. G., Falk, K. & Rotzschke, O. (1993) Curr. Opin. Immunol. 5, 35–44. [DOI] [PubMed] [Google Scholar]
- 2.Villadangos, J. A. (2001) Mol. Immunol. 38, 329–346. [DOI] [PubMed] [Google Scholar]
- 3.Rammensee, H. G., Bachmann, J. & Stevanovic, S. (1997) MHC Ligands and Peptide Motifs (Springer-Verlag, Heidelberg, Germany).
- 4.Princiotta, M. F., Finzi, D., Qian, S. B., Gibbs, J., Schuchmann, S., Buttgereit, F., Bennink, J. R. & Yewdell, J. W. (2003) Immunity 18, 343–354. [DOI] [PubMed] [Google Scholar]
- 5.Neefjes, J. (1999) Eur. J. Immunol. 29, 1421–1425. [DOI] [PubMed] [Google Scholar]
- 6.Moron, G., Dadaglio, G. & Leclerc, C. (2004) Trends Immunol. 25, 92–97. [DOI] [PubMed] [Google Scholar]
- 7.Chicz, R. M., Urban, R. G., Gorga, J. C., Vignali, D. A., Lane, W. S. & Strominger, J. L. (1993) J. Exp. Med. 178, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lechler, R., Aichinger, G. & Lightstone, L. (1996) Immunol. Rev. 151, 51–79. [DOI] [PubMed] [Google Scholar]
- 9.Nimmerjahn, F., Milosevic, S., Behrends, U., Jaffee, E. M., Pardoll, D. M., Bornkamm, G. W. & Mautner, J. (2003) Eur. J. Immunol. 33, 1250–1259. [DOI] [PubMed] [Google Scholar]
- 10.Dorfel, D., Appel, S., Grunebach, F., Weck, M. M., Muller, M. R., Heine, A. & Brossart, P. (2005) Blood 105, 3199–3205. [DOI] [PubMed] [Google Scholar]
- 11.Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T. & Munz, C. (2005) Science 307, 593–596. [DOI] [PubMed] [Google Scholar]
- 12.Klionsky, D. J. & Emr, S. D. (2000) Science 290, 1717–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biederbick, A., Kern, H. F. & Elsasser, H. P. (1995) Eur. J. Cell Biol. 66, 3–14. [PubMed] [Google Scholar]
- 14.Munafo, D. B. & Colombo, M. I. (2001) J. Cell Sci. 114, 3619–3629. [DOI] [PubMed] [Google Scholar]
- 15.Schroter, C. J., Braun, M., Englert, J., Beck, H., Schmid, H. & Kalbacher, H. (1999) J. Immunol. Methods 227, 161–168. [DOI] [PubMed] [Google Scholar]
- 16.Greenbaum, D., Medzihradszky, K. F., Burlingame, A. & Bogyo, M. (2000) Chem. Biol. 7, 569–581. [DOI] [PubMed] [Google Scholar]
- 17.Lautwein, A., Kraus, M., Reich, M., Burster, T., Brandenburg, J., Overkleeft, H. S., Schwarz, G., Kammer, W., Weber, E., Kalbacher, H., et al. (2004) J. Leukocyte Biol. 75, 844–855. [DOI] [PubMed] [Google Scholar]
- 18.Beck, H., Schwarz, G., Schroter, C. J., Deeg, M., Baier, D., Stevanovic, S., Weber, E., Driessen, C. & Kalbacher, H. (2001) Eur. J. Immunol. 31, 3726–3736. [DOI] [PubMed] [Google Scholar]
- 19.Schirle, M., Keilholz, W., Weber, B., Gouttefangeas, C., Dumrese, T., Becker, H. D., Stevanovic, S. & Rammensee, H. G. (2000) Eur. J. Immunol. 30, 2216–2225. [DOI] [PubMed] [Google Scholar]
- 20.Dengjel, J., Rammensee, H. G. & Stevanovic, S. (2005) J. Mass Spectrom. 40, 100–104. [DOI] [PubMed] [Google Scholar]
- 21.Lampson, L. A. & Levy, R. (1980) J. Immunol. 125, 293–299. [PubMed] [Google Scholar]
- 22.Perkins, D. N., Pappin, D. J., Creasy, D. M. & Cottrell, J. S. (1999) Electrophoresis 20, 3551–3567. [DOI] [PubMed] [Google Scholar]
- 23.Dennis, G., Jr., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C. & Lempicki, R. A. (2003) Genome Biol. 4, 3. [PubMed] [Google Scholar]
- 24.Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., et al. (2000) Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hickman, H. D., Luis, A. D., Buchli, R., Few, S. R., Sathiamurthy, M., VanGundy, R. S., Giberson, C. F. & Hildebrand, W. H. (2004) J. Immunol. 172, 2944–2952. [DOI] [PubMed] [Google Scholar]
- 26.Stromhaug, P. E. & Klionsky, D. J. (2001) Traffic 2, 524–531. [DOI] [PubMed] [Google Scholar]
- 27.Burster, T., Beck, A., Tolosa, E., Marin-Esteban, V., Rotzschke, O., Falk, K., Lautwein, A., Reich, M., Brandenburg, J., Schwarz, G., et al. (2004) J. Immunol. 172, 5495–5503. [DOI] [PubMed] [Google Scholar]
- 28.Ota, K., Matsui, M., Milford, E. L., Mackin, G. A., Weiner, H. L. & Hafler, D. A. (1990) Nature 346, 183–187. [DOI] [PubMed] [Google Scholar]
- 29.Friede, T., Gnau, V., Jung, G., Keilholz, W., Stevanovic, S. & Rammensee, H. G. (1996) Biochim. Biophys. Acta 1316, 85–101. [DOI] [PubMed] [Google Scholar]
- 30.Dongre, A. R., Kovats, S., deRoos, P., McCormack, A. L., Nakagawa, T., Paharkova-Vatchkova, V., Eng, J., Caldwell, H., Yates, J. R., III, & Rudensky, A. Y. (2001) Eur. J. Immunol. 31, 1485–1494. [DOI] [PubMed] [Google Scholar]
- 31.Topalian, S. L., Gonzales, M. I., Parkhurst, M., Li, Y. F., Southwood, S., Sette, A., Rosenberg, S. A. & Robbins, P. F. (1996) J. Exp. Med. 183, 1965–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang, Y., Chaux, P., Stroobant, V., Eggermont, A. M., Corthals, J., Maillere, B., Thielemans, K., Marchand, M., Boon, T. & van der Bruggen, P. (2003) J. Immunol. 171, 219–225. [DOI] [PubMed] [Google Scholar]
- 33.Munz, C. (2004) J. Exp. Med. 199, 1301–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muntasell, A., Carrascal, M., Serradell, L., Veelen, P. P., Verreck, F., Koning, F., Raposo, G., Abian, J. & Jaraquemada, D. (2002) J. Immunol. 169, 5052–5060. [DOI] [PubMed] [Google Scholar]
- 35.Seglen, P. O. & Bohley, P. (1992) Experientia 48, 158–172. [DOI] [PubMed] [Google Scholar]
- 36.Shintani, T. & Klionsky, D. J. (2004) Science 306, 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H. & Levine, B. (1999) Nature 402, 672–676. [DOI] [PubMed] [Google Scholar]
- 38.Seglen, P. O., Munthe-Kaas, A. C. & Dybedal, M. A. (1984) Exp. Cell Res. 155, 121–128. [DOI] [PubMed] [Google Scholar]
- 39.Okada, H. & Mak, T. W. (2004) Nat. Rev. Cancer 4, 592–603. [DOI] [PubMed] [Google Scholar]
- 40.Scarlatti, F., Bauvy, C., Ventruti, A., Sala, G., Cluzeaud, F., Vandewalle, A., Ghidoni, R. & Codogno, P. (2004) J. Biol. Chem. 279, 18384–18391. [DOI] [PubMed] [Google Scholar]
- 41.Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T. & Ohsumi, Y. (2004) Mol. Biol. Cell 15, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Delamarre, L., Pack, M., Chang, H., Mellman, I. & Trombetta, E. S. (2005) Science 307, 1630–1634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}