

Abstract
Transgenerational effects of environmental toxins require either a chromosomal or epigenetic alteration in the germ line. Transient exposure of a gestating female rat during the period of gonadal sex determination to the endocrine disruptors vinclozolin (an antiandrogenic compound) or methoxychlor (an estrogenic compound) induced an adult phenotype in the F1 generation of decreased spermatogenic capacity (cell number and viability) and increased incidence of male infertility. These effects were transferred through the male germ line to nearly all males of all subsequent generations examined (that is, F1 to F4). The effects on reproduction correlate with altered DNA methylation patterns in the germ line. The ability of an environmental factor (for example, endocrine disruptor) to reprogram the germ line and to promote a transgenerational disease state has significant implications for evolutionary biology and disease etiology.
Treatments, such as irradiation and chemotherapy, and compounds, such as environmental toxins, pose a threat to the integrity of the genome. Studies have shown that these agents can result in genetic or developmental defects in the offspring or F1 generation from an exposed gestating mother. The ability of an external agent to induce a transgenerational effect requires stable chromosomal alterations or an epigenetic phenomenon such as DNA methylation (1). In the present study, transgenerational refers to a germline transmission to multiple generations, minimally to the F2 generation. Transgenerational effects of irradiation were the first to be identified through transmission of DNA mutations in the germ line to multiple generations (2), often associated with tumor formation. Chemotherapeutic treatments (3) and environmental toxins such as endocrine disruptors (4) can cause effects in the F1 generation, but they have not been shown to affect the F2 generation. Although no effects have been shown to be transgenerational, the potential impact of such transgenerational effects of endocrine disruptors has been discussed (5).
Epigenetic alterations that could lead to transgenerational transmission of specific genetic traits have recently been identified (1, 6). A transgenerational phenotype or genetic trait requires a permanent reprogramming of the germ line. During mammalian germ cell development the methylation state of the genome is reprogrammed. As primordial germ cells (PGCs) migrate down the genital ridge, a demethylation starts and is complete on colonization in the early gonad (7, 8). Germ cells in the gonad then undergo remethylation in a sex-specific manner during gonadal sex determination (9). Although demethylation may not require the gonadal somatic cells, remethylation of the germ line appears to be dependent on association with the somatic cells in the gonads (7). Gonadal sex determination and testis development occur between embryonic days 12 and 15 (E12 to E15) in the rat (after midgestation in the human) and are initiated by the differentiation of precursor Sertoli cells in response to the testis-determining factor Sry. Aggregation of the precursor Sertoli cells, PGCs, and migrating mesonephros cells (precursor peritubular myoid cells) promotes testis morphogenesis and cord formation (10, 11). During the period of gonadal sex determination, the fetal testis contains steroid receptors and is a target for endocrine agents. The androgen receptor (AR) and estrogen receptor–β (ERβ) are present in Sertoli cells, precursor peritubular myoid cells, and germ cells at the time of cord formation (E14) (12, 13). Although steroids are not produced by the testis at this stage of development, estrogenic and androgenic substances have the ability to influence early testis cellular functions. Therefore, steroidal factors acting inappropriately at the time of gonadal sex determination potentially could reprogram the germ line through an epigenetic mechanism (altered DNA methylation) to cause the transgenerational transmission of an altered phenotype or genetic trait.
The estrogenic and antiandrogenic endocrine disruptors used in the current study are methoxychlor and vinclozolin, respectively. Vinclozolin is a commonly used fungicide in the wine industry that is metabolized into more active (i.e., higher affinity binding to androgen receptor) compounds (14). Methoxychlor is used as a pesticide to replace DDT and is metabolized into active compounds with the ERα agonist, the ERβ antagonist, and antiandrogenic activity (15–17). Vinclozolin or methoxychlor exposure in the late embryonic or early postnatal period influences sexual differentiation, gonad formation, and reproductive functions in the F1 generation (14, 18, 19). Transient exposure (daily intraperitoneal injection of 100 or 200 mg/kg dose) of a gestating female rat to methoxychlor or vinclozolin between E8 and E15 promotes reduced spermatogenic capacity associated with increased spermatogenic cell apoptosis and decreased sperm number and motility in the adult F1 generation (20, 21). A similar exposure between E15 and E20 had no effect on the F1 generation testis (20, 21). These observations were extended in the present study by treating the gestating mother with vinclozolin. F1 generation male rats were mated with F1 generation females from different litters. Subsequent breeding continued for four generations with sufficient numbers of animals to avoid sibling inbreeding. Adult males from F1, F2, F3, and F4 generations between postnatal days PND60 and PND180 were killed. Testes were isolated for histological examination, and caudal epididymal sperm were collected for sperm counts and motility measurements. Only the original gestating mother (F0) of the F1 generation received a transient endocrine disruptor treatment. Control groups of animals were bred in a similar manner after vehicle treatment (dimethylsulfoxide buffer alone injected) of the F0 gestating mother. Analysis of cellular apoptosis demonstrated a greater than twofold increase in spermatogenic cell apoptosis in the vinclozolin treatment animals for the F1 to F4 generations (Fig. 1A). Sperm numbers were reduced minimally, 20%, and sperm forward motility was reduced about 25 to 35% for vinclozolin generation animals (Fig. 1, B and C). More than 90% of all males analyzed from all generations had the germ cell defect of increased spermatogenic cell apoptosis. Therefore, the frequency of the phenotype was >90% and did not decline between the F1 and F4 generations. A similar experiment was performed with methoxychlor. After transient embryonic methoxychlor exposure (E8 to E15), a similar phenotype was observed in both the F1 and F2 animals (fig. S1). Therefore, both vinclozolin and methoxychlor induced transgenerational defects in spermatogenic capacity and sperm viability.
Fig. 1.
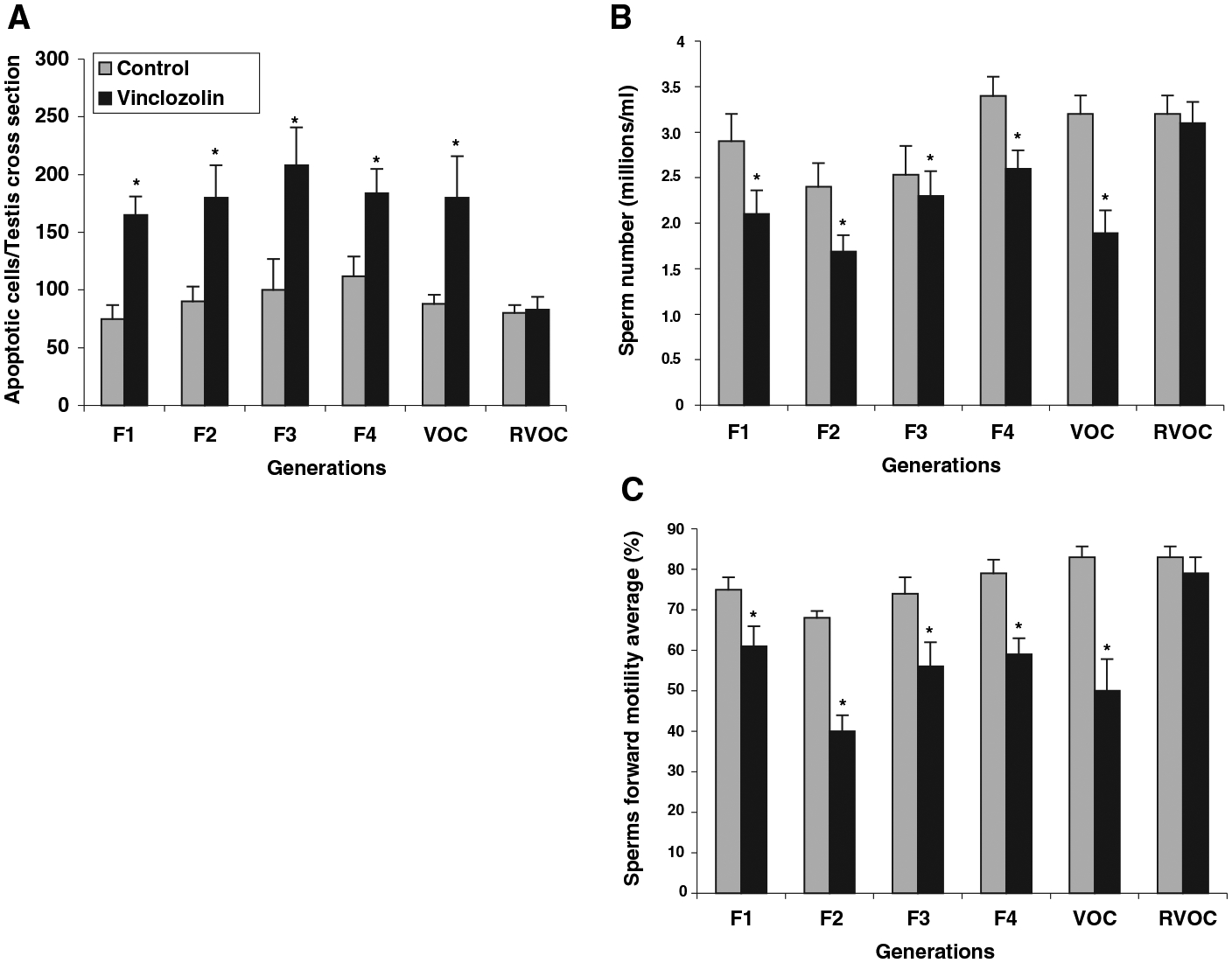
Transgenerational phenotype after vinclozolin treatment of F0 gestating mothers. (A) Spermatogenic cell apoptosis, (B) epididymal sperm counts, and (C) epididymal sperm motility in PND60 to 180 control and vinclozolin off-spring Sprague-Dawley rats in the F1, F2, F3, and F4 generations, and vinclozolin F2 generation male outcross (VOC) to wild-type control females, and vinclozolin F2 generation female reverse outcross (RVOC) to wild-type control males. Statistically significant differences between control and vinclozolin treatment generations are indicated by (*) for P G 0.001 with a two-way analysis of variance test. The n value for each bar ranged between 10 and 30 animals. Detailed methods are provided in SOM.
An outcross experiment was performed to determine whether the transgenerational phenotype was transmitted through the male germ line. Vinclozolin F2 generation males (i.e., male progeny from an F0 treated mother) were crossed with wild-type untreated control females, and the offspring were analyzed. The vinclozolin outcross (VOC) male progeny also had an increase in spermatogenic cell apoptosis and a decrease in sperm number and motility (Fig. 1). The reverse vinclozolin outcross (RVOC) with vinclozolin F2 generation females and wild-type control males demonstrated no effect on the spermatogenic cells (Fig. 1). Therefore, the endocrine disruptor–induced transgenerational phenotype appears to be transmitted through the male germ line.
The morphology of the testes from control and treated rats was similar for all animals examined on PND60 in all F1 to F4 vinclozolin generations. Periodically, male rats older than 90 days of age developed complete infertility associated with small testis and severely reduced spermatogenesis, which was not seen in the PND60 animals. This occurred in 4 out of a total of 50 F1, F2, F3, and F4 generation animals. Therefore, 8% of the vinclozolin transgenerational males developed complete infertility. None of the 42 control F1 to F4 generation animals were infertile. The testis histology for a representative infertile vinclozolin F3 generation animal is shown in Fig. 2 and demonstrates a loss of normal spermatogenesis (no germ cells present) and abnormal seminiferous tubule morphology. The control F3 male showed normal morphology with normal spermatogenesis. Although most of the animals older than 90 days of age were fertile, ~20% developed a dramatic decrease in spermatogenic capacity (50% of the tubules having impaired germ cell development) in all the generations examined.The VOC males also had increased infertility in ~20% of the animals over 90 days of age. The treated males that were fertile showed no change in litter size, newborn pup weights, or testis weight per body weight when compared with the control animals of any of the F1 to F4 generations examined. Nearly all the treated male progeny had the minimal phenotype of twofold increase in spermatogenic cell apoptosis, and the majority had a decrease in epididymal sperm number (Fig. 1, A and B).
Fig. 2.
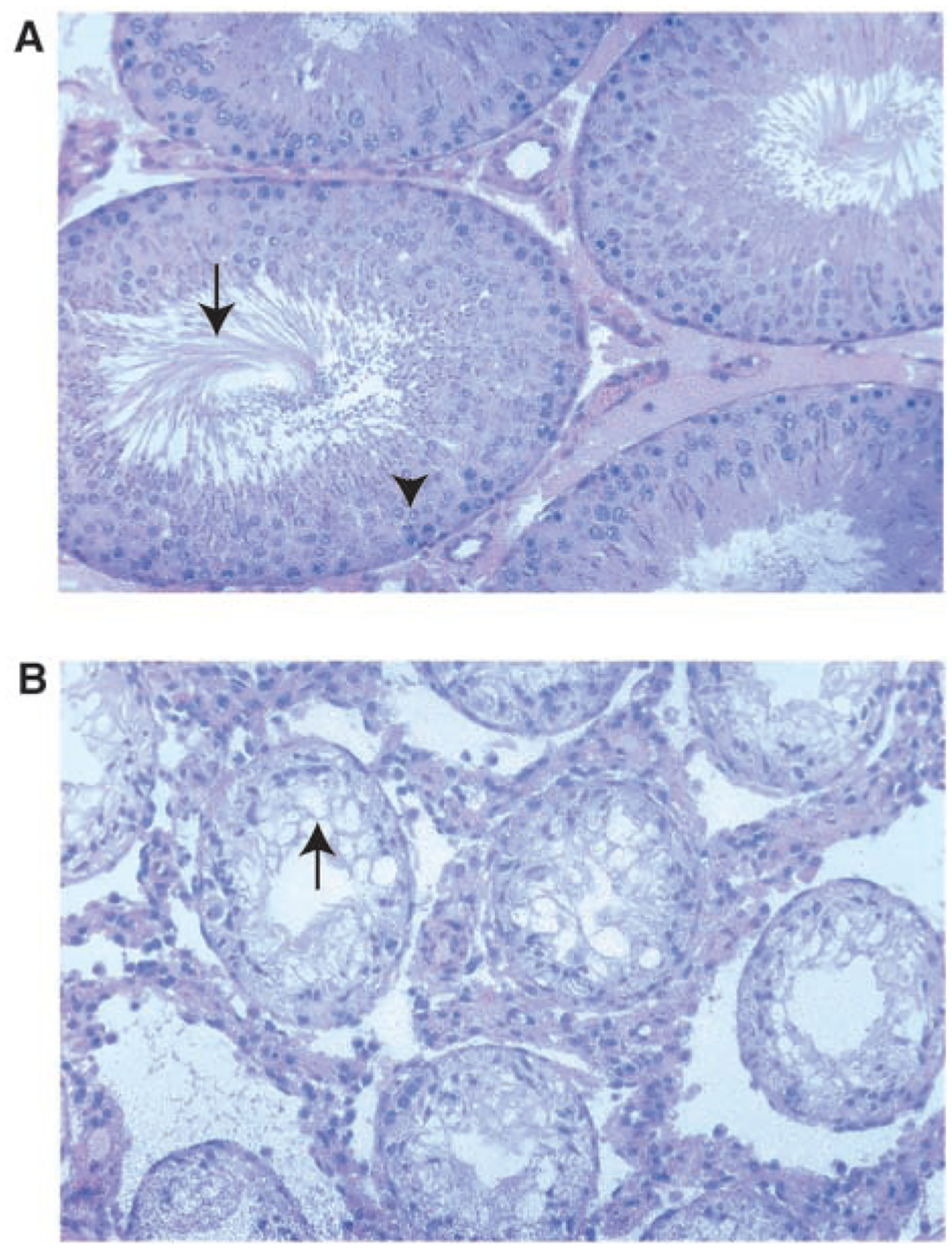
Testis histology from control (A) and vinclozolin treatment (B) 100-day-old F3 generation animals, ×200 magnification. The vinclozolin F3 generation male is a representative infertile male. Arrow in (A) identifies the tails of elongate spermatozoa in the seminiferous tubule lumen; arrowhead labels spermatocytes in the tubule epithelial layer. Arrow in (B) identifies the lack of germ cells in the seminiferous tubule. Methods are provided in SOM.
The transmission of this endocrine disruptor–induced testis phenotype in a transgenerational manner suggests an epigenetic alteration of the male germ line. The frequency observed for the phenotype >90% of all males in all generations) cannot be explained through genetic DNA sequence mutational mechanisms. A high-frequency genetic (DNA sequence) mutation hotspot event would be an order of magnitude less than that observed (2, 22, 23). Subsequent segregation of this mutation would likely result in a reduction of the phenotype frequency with each generation. In addition, the random nature of genetic (DNA sequence) mutations is generally more variable in the phenotype observed (2, 22, 23). In contrast, an epigenetic mechanism involving reprogramming of the germ line could result in the high frequency observed. In addition, the developmental period used for the endocrine disruptor exposure was during the remethylation programming of the germ line. Although we cannot exclude the possibility of a novel genetic (DNA sequence) mutational event, available information suggests an epigenetic mechanism is involved.
The only epigenetic mechanism currently known to influence germline transmission involves the methylation pattern of imprinted genes. This study does not investigate specific known imprinted genes, but focuses on the effects of the endocrine disruptor on the total genome. Fisher rats were used because of reduced polymorphisms, which made methylation studies more consistent and reproducible. PND6 testes were collected from male F1 progeny of vinclozolin-treated and control rats. A methylation-sensitive restriction enzyme digestion analysis involving a polymerase chain reaction (PCR)–based procedure was used to assess changes in DNA methylation patterns (Methods in SOM). Animals (n = 4) from different litters of control and treated animals were analyzed. The vinclozolin-induced methylation patterns were similar in the replicate animals. About 25 different PCR products were identified that had altered DNA methylation patterns after the endocrine disruptor treatment. A representative change in methylation pattern is shown in Fig. 3A and fig. S2. These methylation experiments were extended by isolating and cloning several of the DNA fragments with apparent altered methylation. Two of the DNA fragments were sequenced and mapped to CpG-rich regions on chromosomes 6q32 and 8q32 (Fig. 3B). Clone 7 mapped to 6q32 and was within the lysophospholipase (LPLase) gene (accession NM144750). LPLase is a critical enzyme in the synthesis of important bioactive lipids and associated signaling (24). Clone 17 mapped to 8q32 and is within 1 kb of the start site of an uncharacterized protein termed cytokineinducible SH2 protein (accession AJ243907). Whether the genes identified are causal factors or simply markers (i.e., downstream) of the transgenerational epigenetic phenotype remains to be determined.
Fig. 3.
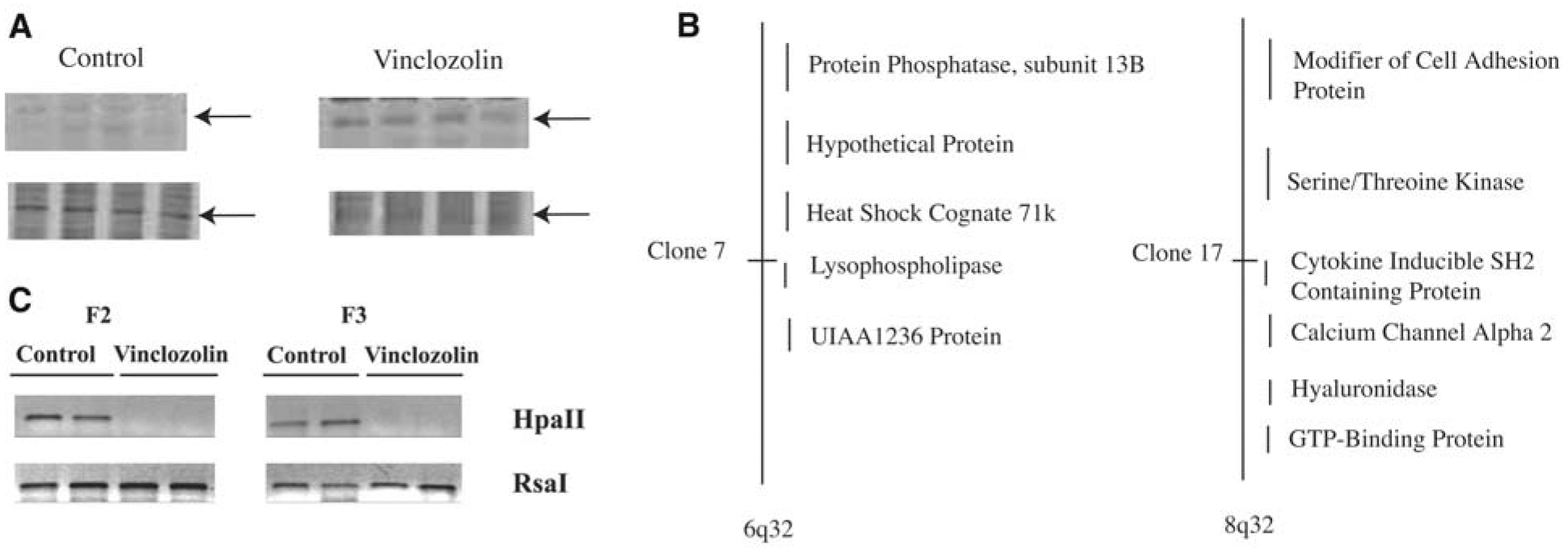
DNA methylation analysis from control and vinclozolin offspring testis. (A) Representative gel images of the PCR-based methylation-sensitive Hpa II restriction enzyme digest analysis with representative band (arrow) affected in PND6 testis from control and vinclozolin treatment animals. Each lane represents a different individual animal (n = 4). (B) Location of selected sequences on specific chromosomes for two representative DNA sequences with altered DNA methylation patterns termed clone 7 and 17. (C) Methylation-sensitive restriction enzyme PCR analysis of the methylation state of clone 17 (i.e., cytokine-inducible SH2 protein) gene in epididymal sperm from F2 and F3 generations from control and vinclozolin-treated animals. The bands presented are representative of sperm DNA collected from different animals from different litters and are consistent in four out of eight F2 animals and two out of five F3 animals analyzed. Methods are provided in SOM.
The epigenetic transgenerational transmission of the altered methylation pattern through the male germ line was investigated. PCR primers were designed for the flanking regions of the clone 17 gene, cytokine-inducible SH2 protein and were used to investigate altered methylation with the methylation-sensitive restriction enzyme digest procedure. Epididymal sperm were isolated from vinclozolin treatment F2 and F3 generation animals. As shown in Fig. 3C, the control animals had the PCR product, whereas the vinclozolin treatment F2 and F3 animals did not. The control Rsa I digest had a PCR product in all samples. Therefore, the vinclozolin treatment F2 and F3 generation sperm samples appeared to have altered DNA methylation in this clone 17 gene compared with control animal sperm. An alternate bisulfite DNA sequence analysis was used to confirm the methylation changes. Bisulfite analysis confirmed the altered methylations of the LPLase gene within the CpG island identified. This bisulfite altered sequence (fig. S3) was observed in ~25% of the vinclozolin treatment sperm DNA samples analyzed. A single-gene methylation event alone is not likely sufficient to promote the phenotype, but could cause alterations in a subset of genes. For example, 25 DNA sequences with altered methylation were identified, and if a random subset of genes with altered methylation promotes the phenotype, a 25% frequency for altered methylation of a single gene would be significant. Observations indicate that the endocrine disruptors can induce an epigenetic transgenerational change in the DNA methylation pattern of the male germ line. The epigenetic alterations observed involve both hypermethylation and hypomethylation events.
Two different endocrine disruptors, vinclozolin and methoxychlor, after a transient embryonic exposure at a critical time during gonadal sex determination (E8 to E15 in the rat), promoted an adult testis phenotype of decreased spermatogenic capacity and male infertility. No gross abnormality was observed in any other tissues examined, and serum testosterone levels were found to be normal in all the animals examined. This phenotype was found to be transgenerational and appears associated with altered DNA methylation of the male germ line. The phenotype was observed in nearly all males from all vinclozolin generations, such that a genetic mutation event (e.g., alteration in DNA sequence) is not likely to be a major factor. The frequency of a genetic mutation would be orders of magnitude less than the transmission frequency observed in the current study (2, 22, 23). Preliminary data demonstrate no major effect on the female, but a number of abnormal pregnancy outcomes were observed in pregnant females from offspring of vinclozolin-treated animals, but not controls. The abnormal phenotype has similarities to preeclampsia that include death, severe anemia, and blood cell defects. This study shows that environmental factors can induce an epigenetic transgenerational phenotype through an apparent reprogramming of the male germ line. It should be noted that the exposure levels used in these studies are higher than anticipated for environmental exposure; hence, future toxicology studies would be needed to ascertain the possible impact on animal populations.
Supplementary Material
Acknowledgments
We acknowledge the technical contributions of I. Sadler-Riggleman, S. Rekow, and B. Johnston and the assistance of H. Suzuki with the methylation PCR procedure. This research was supported in part by a grant to M.K.S. from the U.S. Environmental Protection Agency’s Science to Achieve Results (STAR) program involving endocrine disruptors.
Footnotes
Supporting Online Material
References and Notes
- 1.Rakyan V, Whitelaw E, Curr. Biol 13, R6 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Barber R, Plumb MA, Boulton E, Roux I, Dubrova YE, Proc. Natl. Acad. Sci. U.S.A 99, 6877 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morris ID, Int. J. Androl 25, 255 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Foran CM, Peterson BN, Benson WH, Toxicol. Sci 68, 389 (2002). [DOI] [PubMed] [Google Scholar]
- 5.DeRosa C, Richter P, Pohl H, Jones DE, J. Toxicol. Environ. Health B Crit. Rev 1, 3 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Rakyan VK et al. , Proc. Natl. Acad. Sci. U.S.A 100, 2538 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hajkova P et al. , Mech. Dev 117, 15 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Durcova-Hills G, Ainscough J, McLaren A, Differentiation 68, 220 (2001). [DOI] [PubMed] [Google Scholar]
- 9.Reik W, Walter J, Nat. Rev. Genet 2, 21 (2001). [DOI] [PubMed] [Google Scholar]
- 10.Jost A, Magre S, Agelopoulou R, Hum. Genet 58, 59 (1981). [DOI] [PubMed] [Google Scholar]
- 11.Buehr M, Gu S, McLaren A, Development 117, 273 (1993). [DOI] [PubMed] [Google Scholar]
- 12.Majdic G, Millar MR, Saunders PT, J. Endocrinol 147, 285 (1995). [DOI] [PubMed] [Google Scholar]
- 13.Goyal HO et al. , Anat. Rec 249, 54 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Kelce WR, Monosson E, Gamcsik MP, Laws SC, Gray LE Jr., Toxicol. Appl. Pharmacol 126, 276 (1994). [DOI] [PubMed] [Google Scholar]
- 15.Cummings AM, Crit. Rev. Toxicol 27, 367 (1997). [DOI] [PubMed] [Google Scholar]
- 16.Gaido KW et al. , Endocrinology 140, 5746 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Kelce WR, Lambright CR, Gray LE Jr., Roberts KP, Toxicol. Appl. Pharmacol 142, 192 (1997). [DOI] [PubMed] [Google Scholar]
- 18.Fisher JS, Reproduction 127, 305 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Chapin RE et al. , Fundam. Appl. Toxicol 40, 138 (1997). [DOI] [PubMed] [Google Scholar]
- 20.Cupp AS et al. , J. Androl 24, 736 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Uzumcu M, Suzuki H, Skinner MK, Reprod. Toxicol 18, 765 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Shi BS, Cai ZN, Yang J, Yu YN, Mutat. Res 556, 1 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Dong H et al. , Biochemistry 43, 15922 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Tokumura A, J. Cell. Biochem 92, 869 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.