

Abstract
We introduce a quantum mechanical polarizable force field (QMPFF) fitted solely to QM data at the MP2/aTZ(-hp) level. Atomic charge density is modeled by point-charge nuclei and floating exponentially shaped electron clouds. The functional form of interaction energy parallels quantum mechanics by including electrostatic, exchange, induction, and dispersion terms. Separate fitting of each term to the counterpart calculated from high-quality QM data ensures high transferability of QMPFF parameters to different molecular environments, as well as accurate fit to a broad range of experimental data in both gas and liquid phases. QMPFF, which is much more efficient than ab initio QM, is optimized for the accurate simulation of biomolecular systems and the design of drugs.
Keywords: drug design, quantum mechanics
Accurate simulation of intermolecular interactions is essential in computational studies of chemical and biological systems ranging from multimer spectroscopy in molecular beams, atom-surface interactions, and catalyzed chemical reactions to protein folding and rational drug design. The most reliable and consistent means for such simulations would be to directly use quantum mechanics. However, this is much too computationally demanding, mandating instead the use of a force field, in which the molecular potential surface is approximated by simple analytical formulas. Commonly used force fields including CHARMM, OPLS-AA, MMFF, and AMBER (1–4) originated with Lifson's and Warshel's (5) consistent force field; they all use two basic types of interactions, bonded and nonbonded. The bonded terms are usually modeled formally as functions of stretching, bending, and torsion, whereas the nonbonded components are more physically grounded and involve electrostatic and van der Waals potentials. Electrostatics is described in terms of fixed point charges, and the van der Waals interaction is usually approximated by the classical Leonard–Jones “12–6” potential or its modifications. Empirical parameters that shape the various functional forms are found by fitting to low-level quantum mechanical (QM) and/or experimental data for simple molecules and their interactions in the solid and liquid phases.
Although such force fields have been quite successful in modeling a wide variety of molecular systems, there are significant problems in simulation of liquid-phase solutes (6). These force fields have many possible defects including oversimplified treatment of bonded interactions and approximation of charge distributions by point charges with consequent neglect of charge penetration effects, nonadiabatic motions, and other QM features of intra- and intermolecular interactions. However, the most serious defect is recognized to be the failure to incorporate electronic polarization at a fundamental level, which is especially important in a polar medium such as water. To allow for the effects of polarization, the standard nonpolarizable force fields fit the mean field of the liquid by artificially increased dipole moments, deformed molecular geometry, etc. Doing this decreases both the theoretical grace and practical applicability of such force fields. In addition, the standard force fields have evident methodological restrictions. Fitting the parameters to experiment is limited by insufficient data and generally provides little insight into how the model's inadequacies can be improved. On the other hand, fitting to QM data (if any) generally requires special choice of the QM basis to conform with the mean field approach, so the level of calculations cannot be improved.
Clearly, further progress in force field development requires polarizable models fitted to high-quality QM data, because nonpolarizable pair potentials evidently cannot be further improved (7). The development of polarizable models is motivated by the steadily increasing accuracy, versatility, and completeness of ab initio data, enabling closer agreement to experiment. Polarizable models were first introduced by Applequist and coworkers (8) to describe molecular polarizability and then developed by Warshel and Levitt (9) in the form of a force field. Although there were applications to biomolecular systems (10, 11), the main efforts were concentrated on water and simple compounds (see review in ref. 12 and references therein); more recently (13–15), polarization was introduced into other general-purpose force fields. Two approaches have generally been used to simulate molecular polarizability: (i) inducible multipoles and (ii) fluctuating charges. In the first case, inducible atomic multipoles are formally introduced and their interactions are calculated in a self-consistent iterative way (e.g., see ref. 14). The approach is often confined to a point dipole approximation, although there are extensions using higher multipoles and diffuse distributions (16). In the second case, the partial atomic charges are considered as dynamic variables and the energy of transferring a charge between two sites is usually approximated by a quadratic form (17). In contrast to inducible multipoles, this approach describes polarization to all orders in the charge moments. However, it is often unable to simulate correctly the polarizability tensor, e.g., its normal component for planar molecules. This defect is eliminated in models describing polarization in a universal and intuitively clear way in terms of floating diffuse charges (18–20) tracing back to classical Drude oscillators, and this idea is extended here.
Introducing polarization undoubtedly provides a better physical basis for further progress in simulation of the condensed phase. This is especially important for large organic and biomolecular systems in water solvent, such as proteins or protein–ligand systems, where polarization depends on the local environment and varies widely across the molecular complex. With such a heterogeneous system, the mean-field approximation is intrinsically limited.
Polarization is only one of the components of real QM intermolecular interactions. Providing the correct balance of the different terms of the force field is one of the key requirements for the model to be transferable. In principle, one might accurately fit even a model with oversimplified functional forms and unaccounted effects to a particular training set while ignoring this requirement. When this is done, inaccuracies in various energy components can be made to cancel out. However, such an imbalance will manifest itself in poor model performance beyond the training set, because the relative contributions of different components vary from case to case. To avoid this pitfall, a force field should have a solid physical basis for both the energy decomposition scheme and the functional form, as well as careful individual fitting of all of the components to their QM counterparts. Only in this way can transferability from the training set to other systems be assured.
An additional consideration involves the choice of atom types. In a perfect QM force field, there is only one type of carbon, nitrogen, or oxygen atom. We do not expect to be able to maintain this level of simplicity here but strive for it by choosing components to be mutually well balanced and “as simple as possible, but not simpler.” Because the appropriate levels of simplification are not known a priori, it is reasonable to design the model in a step-by-step manner starting from the simplest components and upgrading only as necessary.
Based on these principles, a general QM polarizable force field (QMPFF) is presented in this article. The model is fitted exclusively to high-level QM data, which provide close agreement with experiment in the gas phase. We find that a relatively small number of atom types for each element (eight for H, three for C, two for O, four for N, and one each for S, F, Cl, and Br) is sufficient to provide strong transferability. QM energies are reproduced to within an average of 0.27 kcal/mol for homo- and heterodimers of small molecules as well as for larger molecules having the same atoms, both inside and outside the training set. The same parameters work well in both gases and liquids, demonstrating good transferability of the model parameters from gas to condensed phase. Together, these results bolster confidence that we have captured the underlying physics in our choice of key energy terms, functional forms, and calibrated parameters. Despite the relatively small number of atom types, our model is able to treat protein molecules and the majority of small-molecule drugs. QMPFF is computationally efficient for evaluation of total energy and forces in large systems such as biomolecules in the condensed phase.
Methods
A molecule is represented as a superposition of interacting atoms. In QMPFF, intramolecular geometry is taken as rigid so only nonbonded interactions contribute to the potential energy (this limitation is being eliminated in subsequent versions of QMPFF that also handle bonded interactions). The potential energy of a molecular complex is decomposed into four components, electrostatic (ES), exchange (EX), induction (IN), and dispersion (DS), as has been done previously in general-purpose polarizable force fields, implying that the model does not describe systems with strong coupling of intra- and intermolecular degrees of freedom, e.g., molecular complexes with large charge transfer and significant overlap of electron densities.
The charge distribution of each atom a is represented as a core atomic point charge, Z̃a, and a diffuse electron density approximated by a negatively charged isotropic electron cloud of exponential form 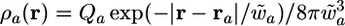 .
.
Positions of the cloud centers ra are written as Ra + ta, where Ra is the position of the nucleus of atom a and ta is the shift of the cloud center from the nucleus. The vectors ta of all of the atoms of the molecular complex under consideration are varied to minimize the total QMPFF potential energy (Fig. 1). These dynamic variables mimic the rapid redistribution of the electron clouds on the nuclear framework.
Fig. 1.

Formulas for potential energy of a molecular complex according to the QMPFF potential. The position of the nucleus of atom a is specified by the vector Ra and the offset of the electron cloud by ta; Ba denotes the set of atoms bonded to atom a. The potential is found by minimization of the sum of ES, EX, and IN terms with respect to vectors ta. Summation of the binary atom–atom ES, EX, and DS interactions is performed over all the atoms in the complex, implying the “1–3 rule” with the terms being dropped for atom pairs separated by one or two chemical bonds (along with the interaction of the clouds with their nuclear cores); for “1–4” interactions, a special renormalization is used (see text). These rules are introduced to comply with the next version of QMPFF, which will allow flexible valence interactions. The unary term 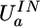 simulates the potential restraining the cloud a to remain close to a reference position. The induction energy can be calculated as the difference between UTOTAL and the energy formally calculated with the clouds fixed at their positions in isolated molecule(s). QMPFF parameters are marked by tilde signs. Parameters for each atom, a: Z̃a, charge;
simulates the potential restraining the cloud a to remain close to a reference position. The induction energy can be calculated as the difference between UTOTAL and the energy formally calculated with the clouds fixed at their positions in isolated molecule(s). QMPFF parameters are marked by tilde signs. Parameters for each atom, a: Z̃a, charge; 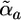 , polarizability; C̃a, exchange strength; w̃a, cloud size; R̃a, dispersion range; Ẽa, dispersion strength. The core charges, Z̃a, are currently fixed at values given in the text; the fixed scale factor R0 is set equal to 1 Å; the parameter t̃max is common for all the atom types. Parameters for each bond, a to b: Q̃ab, bond charge increment; t̃ab, reference cloud shift along bond. (Inset) A schematic representation of a pair of interacting water molecules. (Inset Lower) Atoms drawn as space filling spheres, red for oxygen and gray for hydrogen. (Inset Upper) Nuclei as small spheres with core point charges. The diffuse clouds around (but not necessarily centered on) the nuclei are the electron densities. The arrows illustrate the shifts of the electron cloud centers caused by the external field. It is this movement of diffuse electronic clouds that makes QMPFF polarizable and much more realistic than normal simple point charge force fields. Although the formulae used are much more complicated than for normal force fields, QMPFF is only a factor of 10–20 slower to use; QM would be a million times slower.
, polarizability; C̃a, exchange strength; w̃a, cloud size; R̃a, dispersion range; Ẽa, dispersion strength. The core charges, Z̃a, are currently fixed at values given in the text; the fixed scale factor R0 is set equal to 1 Å; the parameter t̃max is common for all the atom types. Parameters for each bond, a to b: Q̃ab, bond charge increment; t̃ab, reference cloud shift along bond. (Inset) A schematic representation of a pair of interacting water molecules. (Inset Lower) Atoms drawn as space filling spheres, red for oxygen and gray for hydrogen. (Inset Upper) Nuclei as small spheres with core point charges. The diffuse clouds around (but not necessarily centered on) the nuclei are the electron densities. The arrows illustrate the shifts of the electron cloud centers caused by the external field. It is this movement of diffuse electronic clouds that makes QMPFF polarizable and much more realistic than normal simple point charge force fields. Although the formulae used are much more complicated than for normal force fields, QMPFF is only a factor of 10–20 slower to use; QM would be a million times slower.
Approximating the molecular charge density by a diffuse distribution is natural from the physical point of view: It allows adequate description of the electrostatic interaction and takes into account the penetration effect, which contributes to the total energy at small and medium distances.
Atom types are assigned by a simple scheme according to which heavy atoms are classified by atomic number, multiplicity of the chemical bond, and aromatic attribute. Hydrogen atoms are classified by the heavy atom type to which the hydrogen is bonded. The bond types are determined by the pair of atom types, the bond multiplicity, and the bond aromatic attribute.
The QM data for parameterization of QMPFF were calculated with the gamess package (www.msg.ameslab.gov/GAMESS/GAMESS.html) at QM level MP2/aTZ(-hp), which is the Dunning basis MP2/aug-cc-pVTZ (21) with the highest orbital momentum functions removed. Preliminary tests of four basis sets at the MP2 level, 6–311G**, TZ(-hp), aTZ(-hp), and aTZ, showed that the fit to experiment of dipole moments and molecular polarizabilities of H2O, NH3, CH4, and CH3OH was best for the last two sets. On the other hand, the energies of dimers of these molecules calculated with aTZ(-hp) and aTZ were close to each other. Our choice of the basis set is the most complete that in practice allows the treatment with gamess of the largest dimers in the training set. Details of QM calculations are given in Supporting Text, which is published as supporting information on the PNAS web site.
The training set includes the data on molecular properties (components of polarizability tensor, dipole, and quadrupole moments) for all representative molecules as well as components of intermolecular energy of representative dimers. For each dimer in the training set, we choose two types of conformations: (i) the optimal conformations and (ii) “random” conformations covering the most important intermolecular distances and orientations.
Functional Form. The electrostatic interaction between two atoms, a and b, each with a nucleus and electron cloud, includes four terms: nucleus–nucleus, cloud–cloud, cloud–nucleus, and nucleus–cloud. Each term uses the function ϕ(r; u, v) to calculate the electrostatic potential between two exponentially shaped clouds with width parameters u and v separated by distance r. For analytical expressions, see ref. 22. Each atom has an adjustable parameter for the cloud size, w̃a, and for the fixed charge, Z̃a, which is set equal to 1.0 for hydrogen, 4.0 for C, 5.0 for N, 6.0 for O, 7.0 for F, 6.0 for S, 7.0 for Cl, and 7.0 for Br in the current version of QMPFF.
The cloud charges Qa are defined as Qa = –Z̃a + ΣbQ̃ba, where the bond charge transfers, Q̃ab, are QMPFF parameters, and the summation is over all atoms bonded to atom a. For 1–4 interactions, the sum over bonded atoms is dropped for pairs 1–2 and 3–4.
The exchange repulsion is known to decay almost exponentially with the distance; the preexponential factor was found by trial and error. Given that electron cloud sizes are generally similar to each other, the decay rate of the exchange interaction of electron clouds a and b can be taken as (w̃a + w̃b)/2 to give the EX term in Fig. 1. The strength parameters, C, are adjustable.
Induction (IN) is simulated in QMPFF by floating electron clouds. Each cloud a moves in the external field and in the nonharmonic unary restraint potential 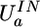 . It constrains the cloud shift, ta, to remain close to the reference position
. It constrains the cloud shift, ta, to remain close to the reference position 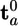 , depending on the atoms b bonded to atom a and defined as Σb∈Bat̃abnab, where nab ≡ Rab/Rab is the unit vector directed from atom a to b. t̃ab is a parameter depending on the bond type a–b; it characterizes the tendency to shift the cloud from atom a toward atom b (note that, in general, t̃ab ≠ t̃ba). Thus, the electron cloud is attached to the nucleus by a spring whose stiffness increases with the extent of the stretching. The stiffness becomes infinite as the argument
, depending on the atoms b bonded to atom a and defined as Σb∈Bat̃abnab, where nab ≡ Rab/Rab is the unit vector directed from atom a to b. t̃ab is a parameter depending on the bond type a–b; it characterizes the tendency to shift the cloud from atom a toward atom b (note that, in general, t̃ab ≠ t̃ba). Thus, the electron cloud is attached to the nucleus by a spring whose stiffness increases with the extent of the stretching. The stiffness becomes infinite as the argument  approaches the parameter value t̃max (Fig. 1). Therefore, the restraint potential prevents the polarization catastrophe in nonuniform fields, whereas at small values of the argument, it is close to a harmonic potential.
approaches the parameter value t̃max (Fig. 1). Therefore, the restraint potential prevents the polarization catastrophe in nonuniform fields, whereas at small values of the argument, it is close to a harmonic potential.
The dispersion term is expected from QM considerations to be finite at r = 0 and vary like r–6 at large distances, so in QMPFF, it was chosen as a simple buffered r–6 (see Fig. 1). Because the positions of cloud centers are close to their corresponding nuclei, the DS term is taken as between nuclei rather than clouds; this simplifies minimization of the total potential energy with respect to cloud positions. In the QMPFF parameterization procedure, the DS term is fitted to the difference between the total QM intermolecular energy and the sum of QM electrostatic, exchange, and induction terms. Adjustable parameters are the buffering range, R̃a, and strength, Ea, for each atom, whereas R0 is a common scaling factor.
Parameterization Procedure. The full list of current QMPFF adjustable parameters is given in Fig. 1, and the list of atom types is given in Table 1. The determination of parameter values was done in a step-wise fashion starting with atom type group 1 (tetravalent carbon and its associated hydrogen atom), for which we used methane and ethane as representative molecules. For each representative dimer conformation, the electrostatics and exchange terms of the QMPFF model were fitted to those of the QM calculation. The induction term of the model was fitted to QM molecular polarizability tensors, and the dispersion term of the model was fitted to the differences between the total QM intermolecular energy and the other terms. In addition, data on dipole and quadrupole moments of representative molecules were used to choose the best parameter set. Next, parameters were found for group 2 (divalent oxygen and its hydrogen atom) by using data on water and methanol and their dimers as well as their mixed dimers with methane (parameters already determined for group 1 were not changed). The procedure was repeated for each new group without recalculation of the previously determined parameters, radically reducing computational effort while allowing a more thorough search of the high-dimensional parameter space.
Table 1. Atom types used for QMPFF.
| Group | Atoms | Example | Aromaticity | Representative molecules |
|---|---|---|---|---|
| 1C, H | >C< | CH4, C–CH2–C>CH– | F | Methane, Ethane |
| 2O, H | –O– | OH2, C–OH, C–O–C | F | Water, Methanol Methyl ether |
| 3N, H | >N– | NH3, C–NH2 C–NH–C | F | Ammonia, Methylamine |
| 4C, H |
|
|
F | Ethylene, Propylene |
| 5O | O |
C–CO–C, >C |
F | Acetone, MethylFormate Formic Acid, Formamide |
| 6C, H |
|
>CH | T | Benzene, Phenol, Aniline |
| 7N |
|
C |
T | Pyridine |
| 8N, H | >N– | >NH | T | Pyrrole |
| 9S, H | –S– | SH2, C–S–C | F | Hydrogen Sulfide Methylethiole |
| 10F | F– | C–F | F | Methyl Fluoride |
| 11Cl | Cl– | C–Cl | F | Methyl Chloride |
| 12Br | Br– | C–Br | F | Methyl Bromide |
| 13N |
|
|
F | Methylimine, Guanidine |
> and < both designate two single bonds. There are a total of 21 atom types (13 heavy atoms and 8 hydrogen atoms), each with five energy parameters (W̃a, 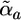 C̃a, R̃a, and Ẽa), not including the fixed charge parameters Za. There are also a total of 43 bond types, each with three energy parameters (Q̃ab, t̃ab, and t̃ba). Together, this gives a total of 105 + 106 adjustable parameters (23 bond parameters are eliminated by symmetry) plus the universal parameter t̃max.
C̃a, R̃a, and Ẽa), not including the fixed charge parameters Za. There are also a total of 43 bond types, each with three energy parameters (Q̃ab, t̃ab, and t̃ba). Together, this gives a total of 105 + 106 adjustable parameters (23 bond parameters are eliminated by symmetry) plus the universal parameter t̃max.
Results
The parameterization of QMPFF presented here includes the most common atom types in biomolecules. With a total of just 13 atom groups (21 atom types and 43 bond types; see Table 1), all 20 standard amino acids and about two-thirds of known drugs presented in the Dobashi Drug Data Base (www.ps.toyaku.ac.jp/dobashi) can be parameterized. A total of 132 molecules were used in the training set, with 44 of these molecules giving rise to 68 representative dimers. For these dimers, there were 81 optimal conformations and 5,093 random dimer conformations. The representative molecules and their dimers are listed in Tables 4–6, which are published as supporting information on the PNAS web site.
Quality of QMPFF Parameterization. The quality of the QMPFF parameterization is demonstrated by the error distribution (Fig. 2) and distance dependence of dimer energies (Fig. 3). Table 2 presents statistics characterizing the quality of the fit of QMPFF to QM for the training set. In addition, corresponding results calculated with MMFF94 are given. Although a direct comparison of QMPFF with a nonpolarizable force field is not strictly correct, because the latter is oriented mainly to simulations in the liquid phase, it nevertheless provides some reference point of accuracy for gas-phase properties. As seen, the fit of QMPFF to QM is quite satisfactory, with the most accurate fit being for the polarizability values. In all cases, the percentage errors between QMPFF and QM are smaller than those between MMFF94 and QM.
Fig. 2.

Error distributions for dimer energies. (a) Distribution of energy differences between QMPFF results and the QM results at the MP2/6–311G** level for the interaction of 53 dimers in random orientations. The distributions for the training set (red, 1,881 conformations) and test set (blue, 4,220 conformations) are essentially identical, with rms energy errors of 0.39 and 0.42 kcal/mol, respectively. (b) The distribution of errors with QMPFF fitted to better quantum calculations at the MP2/aTZ(-hp) level are even closer to the QM results (0.27 kcal/mol rms error). The errors between the Merck force field MMFF94 and the QM results are larger (blue) with rms error of 0.51 kcal/mol. In b, the number of random dimer conformations is 5,093.
Fig. 3.

Distance dependence of dimer energies. (a–c) QMPFF with energy parameters for carbon and oxygen is able to reproduce the interaction energy of homodimers (a and b) and heterodimers (c) of water and methane. The QMPFF energy (red line) is much closer to the QM values (shaded plus signs) calculated at the MP2/aTZ(-hp) level than are the results from the Merck force field MMFF94 (blue line). (d–f) QMPFF fits the QM results for molecules outside the training set for the interaction of water–cyclohexane (d), ethanediol–ethanediol (e), and glycine–glycine (f). The QMPFF energy functions achieve this fit while using a comparatively small number of atom types (eight in all, with two carbon, two oxygen, and one nitrogen types and three associated hydrogen atom types). The QMPFF energy (red line) is much closer to the MP2/aTZ(-hp) QM values (plus signs) than are the results from the best potentials like the Merck force field MMFF94 (blue line).
Table 2. Fit of QMPFF to QM for various types of data in training set.
| Number of cases
|
rms error
|
Percent error
|
||
|---|---|---|---|---|
| Property fitted | QMPFF | MMFF | ||
| Polarizability, Å3 | 132 | 0.44 | 2.7 | — |
| Dipole moment, Debye | 110 | 0.29 | 12.5 | 30.0 |
| Quadrupole moment, Buckingham | 128 | 2.20 | 11.9 | 30.0 |
| Random dimer energies, kcal/mol | 5,093 | 0.27 | 22.0 | 44.0 |
| Optimized dimer energies, kcal/mol | 81 | 0.95 | 13.7 | 18.7 |
| Optimized dimer geometry, Å | 81 | 0.10 | 3.3 | 5.0 |
Detailed data on molecular properties of dimers are given in Tables 5-7.
For the separate components in the energy decomposition scheme (Fig. 1), the rms errors were 0.21, 0.17, and 0.27 kcal/mol for ES, EX, and DS, respectively. The correlation coefficients between the QMPFF and QM data were 0.97, 0.98, and 0.87 for ES, EX, and DS, respectively. The agreement for ES and EX was reduced by strained conformations with very large energy terms. The most problematic component is the DS term. This is expected because in QMPFF (and many other force fields) the DS term accounts not only for the dispersion interaction but also for all other unaccounted effects.
The results presented here demonstrate the highly satisfactory quality of QMPFF simulation of QM data. In all cases the systematic shifts of QMPFF results with respect to QM values were negligible in comparison with random deviations. The accuracy was ≈3% for molecular polarizability and ≈12% for multipoles (see Table 2). The accuracy of optimized dimer energies was 0.95 kcal/mol; however, the weighted rms deviation for the random dimer conformations was only ≈0.3 kcal/mol, which is comparable with or better than the accuracy of the QM data itself. The reason for this difference in accuracy is primarily that the energies of random conformations are much less in absolute value than for optimal conformations, so the rms deviation is correspondingly smaller. In addition, a significant contribution to the rms deviation of the random conformations is from polar dimers, which are accurately described by QMPFF due to accurate simulation of the dipole moments. On the other hand, in optimal conformations the other components, e.g., DS, contribute more. Overall, the data demonstrate that QMPFF conforms well with QM calculations at the basis-set level MP2/aTZ(-hp).
Transferability. Preparation of MP2/aTZ(-hp) QM data is an expensive procedure. Thus, it was not feasible to statistically validate the force field at this level with a large number of molecules outside the training set. Because the QMPFF approach is only weakly dependent on the level of the QM calculations, transferability can be analyzed by fitting QMPFF to less accurate QM data that can be generated rapidly enough to permit sufficient statistics. The distribution of the dimer energy differences for a QMPFF model fitted to QM at basis set level MP2/6–311G** is shown in Fig. 2a for random dimer conformations involving atom groups 1–5. The number of conformations in the training and test sets was 1,881 from 17 dimers and 4,220 from 36 dimers, respectively, providing good statistics. The distributions of errors for the training and test sets are indistinguishable, with rms errors of 0.39 and 0.42 kcal/mol, respectively showing good fitting quality and almost perfect transferability. This use of the MP2/6–311G** basis is done solely for testing purposes.
Error distributions in the training set were also determined for the real QMPFF force field, i.e., fitted to high-level QM calculations (Fig. 2b). These errors are very similar to those found for the test and training sets by using MP2/6–311G**, with an rms error of 0.27 kcal/mol. The rms error for the MMFF94 potential is much larger, at 0.51 kcal/mol, which can be partially explained by systematic shifts of aTZ(-hp) QM data relative to the 6–31G* QM data basically used for MMFF94 parameterization.
A good illustration of model transferability comes from the variation of dimer energy with distance between the monomers. These calculations, which were carried out at our usual basis-set level, MP2/aTZ(-hp), start from the optimal conformation of the dimer and then move one of the monomers along the direction connecting two predefined reference atoms (usually the central heavy atoms). Fig. 3 a–c shows how the parameters fitted to the training set, which includes methane–methane and water–water homodimers, reproduce the test water–methane heterodimer well. Fig. 3 d–f shows the dimer interaction between larger molecules that were not used in the training set. In all cases, the QMPFF potential results in better agreement with QM data than the MMFF94 potential.
Validation. Our QMPFF potential is designed to reproduce the properties of molecules in any phase by virtue of its physically meaningful parameterization that captures essential QM effects. Fig. 4 shows that the model is indeed able to fit both gaseous and condensed phases well. Dimerization Gibbs energy data closely related to the second virial coefficient are shown in Table 7, which is published as supporting information on the PNAS web site, for homogeneous vapors and mixtures.
Fig. 4.

Comparison of QMPFF and MMFF94 with experimental data for methane and methanol in gas and condensed phases. The second virial coefficient characterizes the gas phase and is calculated as Bij = (1/2)∫〈1–e–Uij/kT〉ΩdR, where 〈...〉Ω denotes the averaging over molecular rotations; Uij is the interaction energy between the two molecules i, j; T is the temperature; and the integration is performed over the translational degrees of freedom. (a) For methane, the fit of B calculated for QMPFF (red line) to experiment (shaded plus signs) is no better than that of MMFF94 (blue line); these errors are partly due to insufficient accuracy of the QM calculations (dotted black line). (b) For methanol, QMPFF fits experiment much better than MMFF. (c) The radial distribution function, which characterizes the liquid phase, shows that both QMPFF and MMFF fit experiment well for methane. (d) For methanol, the QMPFF fit to experiment is clearly better than for MMFF. Liquids are simulated by using a box of 219 CH4 or 96 CH3OH molecules for 100 ps. For QMPFF, these simulations were slower by a factor of 15 than for MMFF.
Discussion
Nonadditivity Effects. Table 1 presents atom types generally encountered in biomolecules and drug-like molecules. Other atom types can also be parameterized in the framework of the QMPFF approach. As an example, we parameterized the charged sp3-hybridized nitrogen (along with corresponding hydrogen and bonds) by using dimers 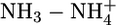 ,
, 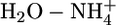 ,
, 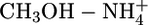 , and
, and 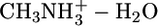 . Because of the strong electric field due to nonzero charge, the nonadditivity polarization effects in interactions of this atom type are expected to be significant. Table 3 compares the QMPFF predictions with QM results for decomposition of the total energy of the pentamer
. Because of the strong electric field due to nonzero charge, the nonadditivity polarization effects in interactions of this atom type are expected to be significant. Table 3 compares the QMPFF predictions with QM results for decomposition of the total energy of the pentamer 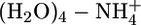 calculated at the optimal multimer conformation.
calculated at the optimal multimer conformation.
Table 3. Nonadditivity effects in 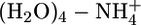 .
.
| Method | E2 | E3 - 3E2 | E4 - 2E3 + 3E2 | E5 - E4 + E3 - E2 | E5 |
|---|---|---|---|---|---|
| MP2/aTZ(-hp) | -74.0 | 10.3 | -0.5 | 0.0 | -64.2 |
| QMPFF | -75.5 | 10.3 | -0.3 | 0.0 | -65.5 |
Values are in kcal/mol.
In Table 3, the Ek values denote the sums of energies of all of the k-mers calculated with the geometry corresponding to the optimized pentamer. The second through fifth columns represent the contribution of two-, three-, four-, and five-particle nonadditivities, respectively; in the last column, the total energy is given. As shown, QMPFF gives many-particle nonadditivities that are in good agreement with MP2 results. For comparison, MMFF94 gave a total energy of –77.7 kcal/mol, satisfactorily simulating two-particle polarization by artificially increasing dipole moments of water molecules; however, all high-order nonadditivity effects are naturally zero in the pairwise additive MMFF94 potential, so E5 is off by ≈13 kcal/mol.
Condensed-Phase Polarization. The most evident advantage of the physically grounded polarizable model over fixed-charge models is transferability from the gas to the condensed phase. Several liquids have been simulated by using molecular dynamics and Monte Carlo techniques and found to be in good agreement with experimental data on thermodynamic and structural properties in all cases, similarly to the results presented in Fig. 4.
Nonharmonic form of the restraint term 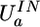 not only prevents the polarization catastrophe but decreases the mean molecular polarizability in the liquid phase. It is also reduced by the exchange repulsion between the electron clouds. This emulates the quantum induction component of intermolecular interaction (23). Such effects are not simulated by other force fields, because the polarization is associated only with the electrostatic field, and electron displacement is treated as a harmonic spring. The effect can be significant in a highly polar liquid like water. For QMPFF, the estimated 18% decrease of polarizability in liquid water relative to vapor compares well with literature estimates ranging from 7% (24) to 29% (19).
not only prevents the polarization catastrophe but decreases the mean molecular polarizability in the liquid phase. It is also reduced by the exchange repulsion between the electron clouds. This emulates the quantum induction component of intermolecular interaction (23). Such effects are not simulated by other force fields, because the polarization is associated only with the electrostatic field, and electron displacement is treated as a harmonic spring. The effect can be significant in a highly polar liquid like water. For QMPFF, the estimated 18% decrease of polarizability in liquid water relative to vapor compares well with literature estimates ranging from 7% (24) to 29% (19).
Limitations Due to QM Accuracy. The basis level of QM calculations used here [MP2/aTZ(-hp)] is generally adequate for polar interactions, where the accuracy is ≈5%. For nonpolar and van der Waals complexes, the accuracy is not as good (see Fig. 4a, comparing ab initio QM and experimental virial coefficients for methane). Errors are even larger for large nonpolar molecules: The calculated energy of the optimal benzene homodimer is –7.3 kcal/mol, more than twice the generally accepted value. The problem is worse for strained conformations, so more advanced QM calculations are needed with emphasis on better treatment of electron correlations. Indeed, because QMPFF, unlike other force fields, has no free parameters that are not fitted to QM data, the only way to improve QMPFF is by using such advanced QM calculations.
Conclusions. QMPFF simulates vacuum dimer properties in satisfactory agreement with experimental data. The fit of QMPFF to the QM data is generally comparable to the fit of QM to experiment. Further increase in the accuracy of QMPFF simulations will require not only refinements in the functional form and the model parameterization procedure, but also the use of more accurate QM data for parameterization. Fitting the QMPFF model to data obtained with more advanced QM techniques will let QMPFF fit experimental data even more precisely. QMPFF shows almost perfect transferability in simulations of dimer energies for a wide range of chemical compounds, raising hopes for successful extension to large biomolecules.
Supplementary Material
Acknowledgments
We thank C. Queen for inspiring the QMPFF program and for careful review of the manuscript; M. Levitt for critical discussions; and A. Artemyev, A. Bibikov, I. Bodrenko, N. Galkin, A. Illarionov, and M. Olevanov for assistance with calculations.
Abbreviations: DS, dispersion; ES, electrostatic; EX, exchange; IN, induction; QM, quantum mechanical; QMPFF, QM polarizable force field.
References
- 1.MacKerell, A. D., Jr., Wiorkiewicz-Kuczera, J. & Karplus, M. (1995) J. Am. Chem. Soc. 117, 11946–11975. [Google Scholar]
- 2.Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. (1996) J. Am. Chem. Soc. 118, 11225–11236. [Google Scholar]
- 3.Halgren, T. A. (1996) J. Comput. Chem. 17, 490–519. [Google Scholar]
- 4.Wang, J., Cieplack, P. & Kollman, P. (2000) J. Comput. Chem. 21, 1049–1074. [Google Scholar]
- 5.Lifson, S. & Warshel, A. (1968) J. Chem. Phys. 49, 5116–5129. [Google Scholar]
- 6.Kaminski, G. A. & Jorgensen, W. L. (1996) J. Phys. Chem. 100, 18010–18013. [Google Scholar]
- 7.Guillot, B. & Guissani, Y. (2001) J. Chem. Phys. 114, 6720–6733. [Google Scholar]
- 8.Applequist, J., Carl, J. & Fung, K.-K. (1972). J. Am. Chem. Soc. 94, 2952–2960. [Google Scholar]
- 9.Warshel, A. & Levitt, M. (1976) J. Mol. Biol. 103, 227–249. [DOI] [PubMed] [Google Scholar]
- 10.Lee, F. S., Chu, Z. T. & Warshel, A. (1993) J. Comput. Chem. 14, 161–185. [Google Scholar]
- 11.Muegge, I., Qi, P. X., Wand A. J., Chu Z. T. & Warshel, A. (1997) J. Phys. Chem. 101, 825–836. [Google Scholar]
- 12.Halgren, T. A. & Damm, W. (2001) Curr. Opin. Struct. Biol. 11, 236–242. [DOI] [PubMed] [Google Scholar]
- 13.Cieplak, P., Caldwell, J. & Kollman, P. (2001) J. Comput. Chem. 22, 1048–1057. [Google Scholar]
- 14.Kaminski, G. A., Stern, H. A., Berne, B. J., Friesner, R. A., Cao, Y. X., Murphy, R. B., Zhou, R. & Halgrem, T. A. (2002) J. Comput. Chem. 23, 1515–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel, S., Mackerell, A. D., Jr., & Brooks, C. L., III (2004) J. Comput. Chem. 25, 1–16. [DOI] [PubMed] [Google Scholar]
- 16.Ren, P. & Ponder. J. (2002) J. Comput. Chem. 23, 1497–1506. [DOI] [PubMed] [Google Scholar]
- 17.Stern, H. A., Ritter, F., Berne, B. J. & Friesner, R. A. (2001) J. Chem. Phys. 115, 2237–2251. [Google Scholar]
- 18.Saint-Martin, H., Hernandez-Cobos, J., Bernal-Uruchurtu, M. I., Ortega-Blake, I. & Berendsen, H. J. C. (2000) J. Chem. Phys. 113, 10899–10912. [Google Scholar]
- 19.Lamoreux, G., MacKerell, A. D., Jr., & Roux, B. (2003) J. Chem. Phys. 119, 5185–5197. [Google Scholar]
- 20.Yu, H., Hansson, T. & van Gunsteren, W. F. (2003) J. Chem. Phys. 118, 221–234. [Google Scholar]
- 21.Dunning, T., Jr. (1989) J. Chem. Phys. 90, 1007–1023. [Google Scholar]
- 22.Slater, J. C. (1963) Quantum Theory of Molecules and Solids (McGraw–Hill, New York), Vol. 1.
- 23.Chalasinski, G. & Szczesniak, M. M. (1994) Chem. Rev. 94, 1723–1765. [Google Scholar]
- 24.Morita, A. (2002) J. Comput. Chem. 23, 1466–1471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.