

Abstract
[18F]-Flortaucipir PET is considered a good biomarker of Alzheimer’s disease. However, it is unknown how flortaucipir is associated with the distribution of tau across brain regions and how these associations are influenced by β-amyloid. It is also unclear whether flortaucipir can detect tau in definite primary age-related tauopathy (PART). We identified 248 individuals at Mayo Clinic that had undergone [18F]-flortaucipir PET during life, had died, and undergone an autopsy, 239 cases of which also had β-amyloid PET. We assessed nonlinear relationships between flortaucipir uptake in nine medial temporal and cortical regions, Braak tau stage and Thal β-amyloid phase using generalized additive models. We found that flortaucipir uptake was greater with increasing tau stage in all regions. Increased uptake at low tau stages in medial temporal regions was only observed in cases with high β-amyloid phase. Flortaucipir uptake linearly increased with β-amyloid phase in medial temporal and cortical regions. The highest flortaucipir uptake occurred with high Alzheimer’s disease neuropathologic change (ADNC) scores, followed by low-intermediate ADNC scores, then PART, with entorhinal cortex providing the best differentiation between groups. Flortaucipir PET had limited ability to detect PART and imaging defined PART did not correspond with pathologically defined PART. In summary, spatial patterns of flortaucipir mirrored histopathological tau distribution, were influenced by β-amyloid phase, and were useful for distinguishing different ADNC scores and PART.
One sentence summary:
Flortaucipir PET uptake is influenced by both tau distribution and β-amyloid, but cannot detect tau deposition in primary age related tauopathy.
INTRODUCTION
Hyperphosphorylated paired helical filament tau observed in neurofibrillary tangles (NFTs) is one of the key pathological characteristics of Alzheimer’s disease (AD). The progressive spread of tau immunoreactive NFTs through the brain is defined by the Braak NFT staging scheme, whereby NFTs first appear in the transentorhinal cortex (Braak stage I & II), before spreading to the hippocampus and other limbic regions (III & IV), and then to the neocortex (V and VI)(1). Studies show that tau deposition and Braak stage are strongly associated with neurodegeneration of the brain(2–4) and cognitive decline(5, 6). Tau PET ligands, such as the commonly used [18F]-flortaucipir (also known as T807, AV-1451 or Tauvid) (7), show sensitive binding to the tau proteins observed in AD in autoradiographics studies(8–10) and hence allow the possibility of in vivo prediction of tau deposition or even Braak stage. Indeed, studies have observed patterns of flortaucipir uptake in cognitively normal and clinically defined Alzheimer’s dementia populations that appear to mirror Braak stage (11–13). A handful of autopsy studies have found correlations between histologically quantified tau burden and flortaucipir PET uptake in medial temporal and cortical regions (14, 15), although elevated uptake across regions was only consistently observed in Braak stages V and VI (15–17), with little evidence for elevated uptake in Braak stages of IV or lower(14, 15, 17). Hence, flortaucipir PET appears to be best suited for detecting advanced or high AD neuropathologic changes (ADNC).
Tau is not the only important protein present in the brains of patients with AD. Abnormal deposition of another protein, β-amyloid, in the form of senile plaques is also a characteristic and diagnostic feature of AD. The regional spread of β-amyloid through the brain is characterized by the Thal phase, with β-amyloid deposits first appearing in neocortex (phase 1), followed by allocortex (phase 2), diencephalic nuclei, striatum, and the basal forebrain (phase 3), brainstem nuclei (phase 4) and lastly cerebellum (phase 5)(18). The National Institute on Aging-Alzheimer’s Association (NIA-AA) guidelines for the neuropathological assessment of AD stage cases as either not, low, intermediate, or high ADNC considering both β-amyloid plaques and Braak stages(19, 20). Hence, high Braak stage alone is not sufficient to characterize the severity of AD pathology, with high stages of both proteins needed for a diagnosis of high ADNC. Patients with low Braak stages (I-IV) and absent Thal phase (0) are referred to as definite primary age related tauopathy (PART)(21). Tau burden at a given Braak stage may vary, and this variation may depend on the presence and distribution of β-amyloid and, hence, flortaucipir PET uptake may also vary depending on β-amyloid pathology. Previous studies have tried to understand the relationship between tau and β-amyloid using PET(13, 22, 23), demonstrating that β-amyloid is required for high tau PET uptake (23) and that β-amyloid is more strongly associated with tau in cortical regions compared to medial temporal regions(13). These studies, although important, were limited by the lack of autopsy data and the fact that β-amyloid PET ligands have limited sensitivity to low Thal stages(14, 24).
In this PET-autopsy study our first aim was to model the relationships between Braak stage and flortaucipir PET in medial temporal and cortical regions in a large cohort of participants, and to determine whether this relationship is influenced by β-amyloid, as measured histopathologically by Thal phase(18).. Our second aim was to determine the utility of flortaucipir PET to diagnose the degree of underlying pathology by assessing optimum flortaucipir cut points to differentiate between different stages of ADNC and PART. Our third aim was to evaluate how imaging defined PART using β-amyloid PET and flortaucipir PET relates to pathologically defined PART to aid in the interpretation of flortaucipir PET. This is pertinent since studies have suggested that molecular PET can be used to diagnose PART antemortem(25, 26).
RESULTS
Cohort characteristics
We included 248 participants recruited from Mayo Clinic, Rochester, MN, who had flortaucipir PET imaging during life, had died, and had undergone an autopsy evaluation. All patients were enrolled into one of three NIH funded cohort studies including the Mayo Clinic Alzheimer’s Disease Research Center (PI: Petersen), The Mayo Clinic Study of Aging (PI: Petersen) and the Neurodegenerative Research Group (PIs: Josephs/Whitwell). The demographic, pathological and clinical features of the cohort are shown in Table 1. About 60% of the participants in the cohort were male. The median time from flortaucipir scan to death was approximately 2 years (range 1 day-7.6 years). The cohort consisted predominantly of patients who had cognitive impairment/dementia at their last study visit (72%) which included patients with clinically diagnosed amnestic mild cognitive impairment/Alzheimer’s dementia, frontotemporal dementia, and Dementia with Lewy bodies. All 248 cases were assigned a Braak NFT stage (1) and a Thal β-amyloid phase (18) based on published criteria and were classified into not ADNC, PART(21), low/intermediate ADNC, and high ADNC (20). All Braak and Thal phases were represented in the cohort, although there were some combinations of Braak stage and Thal phase with few or no cases, such as low Braak stage with high Thal phase, or High Braak stage with low Thal phase (Fig. 1). This was not a bias but due to the fact that certain combinations of tau and β-amyloid are uncommon (3). The not ADNC cases had clinical diagnoses of cognitively normal (n=3), mild cognitive impairment (n=1), frontotemporal dementia (n=7), parkinsonian disorder (n=1) and atypical AD (n=1), with frontotemporal lobar degeneration (FTLD) pathology observed in 9 (68%) not ADNC cases (Table S1). The distribution of co-pathologies across PART/ADNC scores is shown in Table S1.
Table 1:
Characteristics of the cohort of 248 participants
| Median (IQR) or N (%) | |
|---|---|
|
| |
| Female, n (%) | 96 (39%) |
| APOE ε4 carrier, n (%) | 101 (43%) |
| Race, n (%) | |
| White | 243 (98%) |
| African American | 1 (0.4%) |
| Asian Chinese | 1 (0.4%) |
| Other/Unknown | 3 (1%) |
| Ethnicity, n (%) | |
| Not Hispanic or Latino | 237 (96%) |
| Hispanic or Latino | 1 (0.4%) |
| Other/Unknown | 10 (4%) |
| Education, yr | 15 (12, 16) |
| Age at last tau, yr | 75 (66, 83) |
| Age at death, yr | 76 (68, 85) |
| Time from scan to death, yr | 2.2 (1.3, 3.4) |
| Braak NFT stage | 4.0 (2.0, 6.0) |
| Thal phase | 3.0 (0.0, 5.0) |
| CERAD score | 2.0 (0.0, 3.0) |
| MMSE | 22 (16, 27) |
| CDR-SB | 5.0 (0.5, 10.0) |
| Last known clinical diagnosis, n (%) | |
| Normal | 43 (17%) |
| MCI | 26 (10%) |
| Dementia | 179 (72%) |
| Alzheimer’s dementia | 74 (41%) |
| Typical | 21 (28%) |
| Atypical | 53 (72%) |
| Frontotemporal dementiaβ | 34 (19%) |
| Dementia with Lewy bodies | 21 (12%) |
| Parkinsonian disordersδ | 39 (22%) |
| Other | 11 (6%) |
| Dementia | 1 (9%) |
| Dementia-hard to classify | 9 (82%) |
| Probable vascular dementia | 1 (9%) |
| ADNC pathological diagnosis, n (%) | |
| Not ADNC | 13 (5%) |
| PART | 50 (20%) |
| Low/Intermediate | 95 (38%) |
| High | 90 (36%) |
| Lewy body pathology | 80 (32%) |
| Braak NFT stage of Lewy body cases | 5.0 (3.0, 6.0) |
| TDP-43 pathology | 84 (34%) |
| Non-FTLD | 61 (73%) |
| FTLD | 23 (27%) |
| FTLD pathology | 84 (34%) |
| PiB positive, n (%) | 150 (63%) |
ADNC = Alzheimer’s Disease Neuropathologic Changes; NFT = neurofibrillary tangle; MMSE = Mini-Mental State Examination; CDR-SB = Clinical dementia Rating Scale -sum of boxes; CERAD = Consortium to Establish a Registry for Alzheimer’s Disease; AD = Alzheimer’s Dementia; MCI = Mild Cognitive Impairment; FTD = Frontotemporal Dementia; FTLD = Frontotemporal lobar degeneration; DLB = Dementia with Lewy bodies; PART = primary age related tauopathy; PET = Positron Emission Tomography; PiB = Pittsburgh Compound B; TDP-43 = TAR DNA binding protein of 43 kDa
Parkinsonian disorders included corticobasal syndrome and progressive supranuclear palsy.
Frontotemporal dementia included behavioral variant FTD, semantic dementia, primary progressive apraxia of speech, agrammatic variant of primary progressive aphasia.
Figure 1: Scatterplot illustrating the distribution of participants by Braak stage and Thal phase.
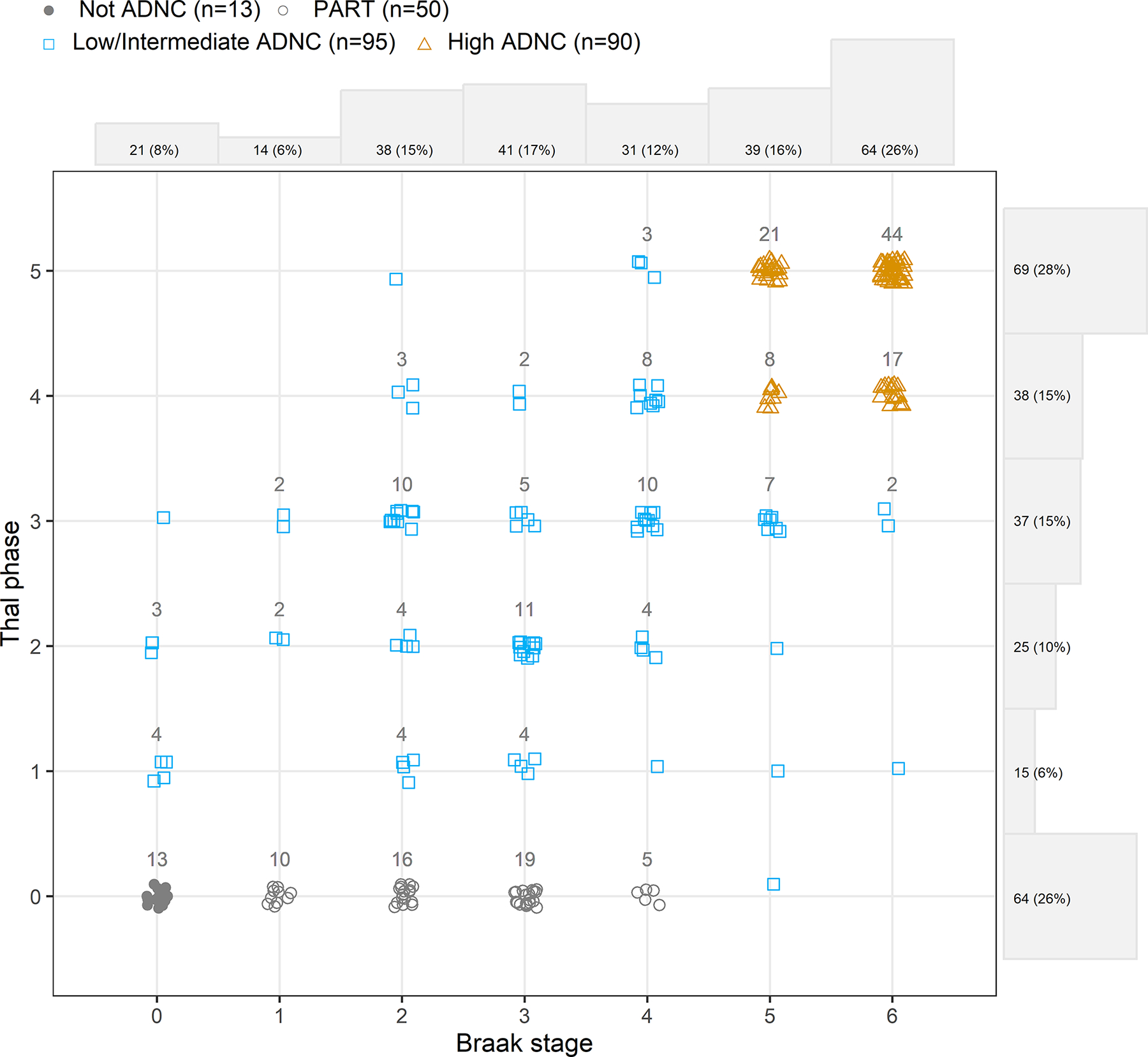
PART, not ADNC, low/intermediate ADNC and high ADNC cases are illustrated with different colors and symbols with the numbers of participants shown for each group. Density plots on the axes illustrate the number of participants in each row/column.
Relationship between flortaucipir, Braak stage and Thal phase
Flortaucipir standardized uptake value ratios (SUVRs) were calculated for the following nine regions that were selected to capture the different Braak stages: entorhinal cortex (Braak stages I-II), anterior hippocampus (Braak stage II), fusiform cortex (Braak stage III), inferior temporal cortex (Braak stage IV), superior frontal cortex, precuneus and superior parietal cortex (Braak stage V), calcarine cortex and precentral cortex (primary motor cortex) (Braak stage VI). We opted to assess the anterior portion of the hippocampus instead of the whole hippocampus to reduce the problem of bleed-in from off-target signal in the choroid plexus, given that the choroid plexus is close in proximity to the posterior hippocampus. For each region, we fit a generalized additive model to capture nonlinear relationships between combinations of Braak stage and Thal phase (predictors) and flortaucipir SUVR (outcome), accounting for age at the time of PET scan and time from PET scan to death.
Flortaucipir relationships with Braak stage
Plots illustrating the relationship between regional flortaucipir SUVR and Braak NFT stage, and how this relationship is influenced by Thal β-amyloid phase, are shown in Fig. 2A. The percentage changes in flortaucipir SUVR with each increment in Braak stage for each Thal phase are shown in fig. S1 and Table S2. with fig. S1 illustrating whether increases in uptake by Braak stage differed, represented by the confidence interval not crossing the 0 line. In all regions, flortaucipir uptake showed a non-linear relationship with Braak stage, with SUVR increase per Braak unit generally greater at higher Braak stages. For example, in the entorhinal cortex, flortaucipir uptake increased by 1% between Braak 0 and I, 6% between Braak II and III, and 11% between Braak V and VI, in Thal 5 cases (Table S2). All regions showed greater uptake at higher Braak stages (IV-VI) compared to lower Braak stages (Braak 0-III). The Braak stage at which elevated flortaucipir uptake was observed was influenced by Thal phase. In the medial temporal regions (entorhinal cortex and anterior hippocampus), flortaucipir uptake increased between Braak stage I and II in Thal 5 cases, which was significant for the anterior hippocampus (p=0.03), and between Braak stage II and III in Thal 3–4 cases (Fig. 2A and fig. S1). However, flortaucipir did not increase until between Braak stage III and IV in Thal 1–2 cases and after Braak stage IV in Thal 0 cases. Increases in flortaucipir uptake between Braak stages were generally observed at higher Braak stages in the cortical regions, with increases in uptake only observed after Braak stage III in those with Thal 3–5, and after Braak stage IV in those with lower Thal (Fig. 2A and fig. S1). Overall, elevated uptake at low Braak stages was only observed in medial temporal regions with high Thal phase. In cases with definite PART (Thal 0, Braak I-IV), there was little evidence for elevated flortaucipir uptake (Fig. S1). Identical models were repeated using partial volume corrected (PVC) PET data (Fig. S2 and Table S3). The PVC model results were very similar to the non-PVC models.
Figure 2: Model estimated flortaucipir standardized uptake value ratios (SUVRs) by Braak stage and Thal phase in each brain region.
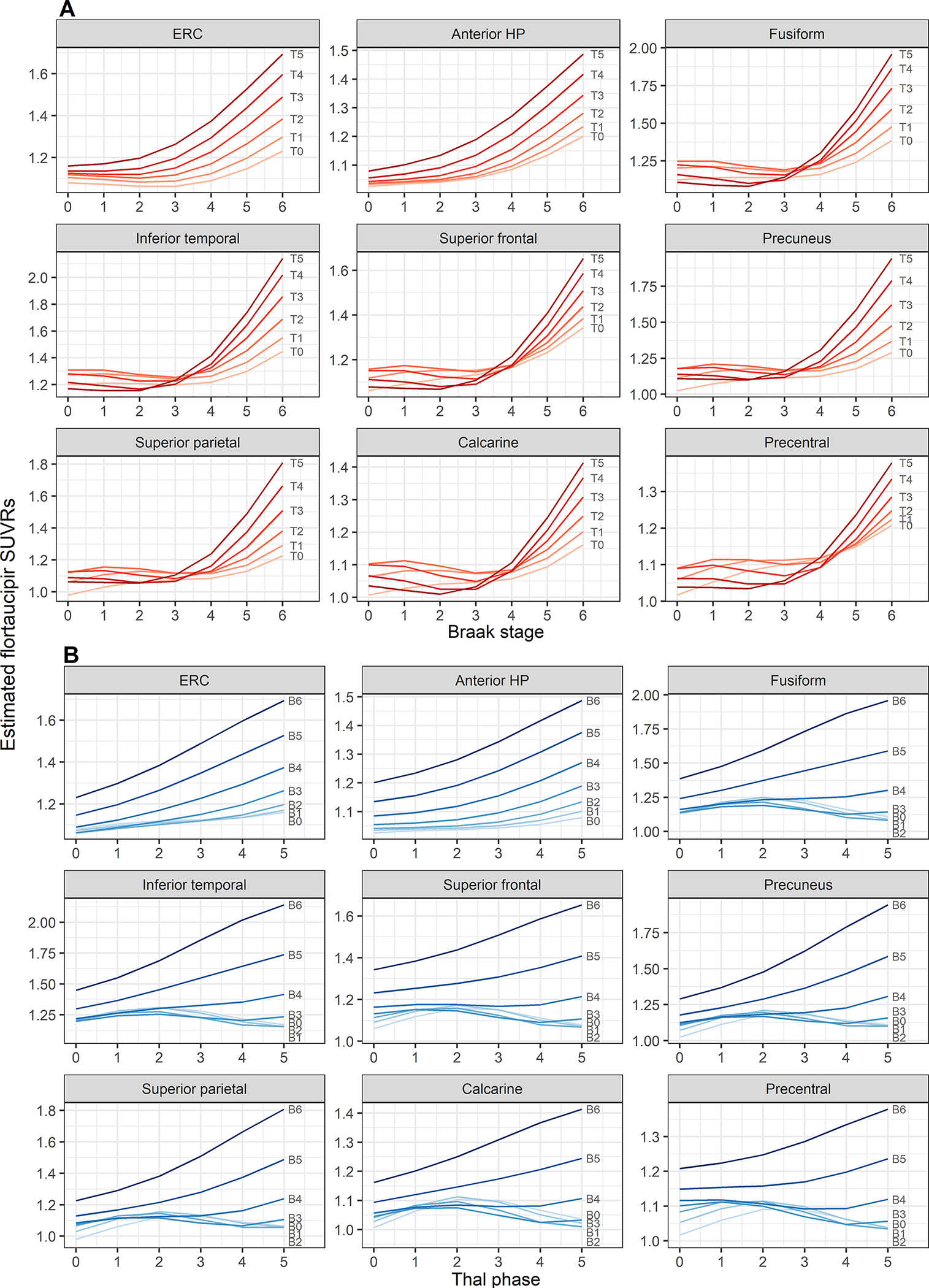
A) Estimated SUVR by Braak stage with separate curves for each Thal phase (T0 – T5). B) Estimated SUVR by Thal phase with separate curves for each Braak stage (B0 – B6). ERC=entorhinal cortex; HP=hippocampus
Flortaucipir relationships with Thal phase
The model estimates plotted to illustrate the relationship between regional flortaucipir SUVR and Thal phase plotted for different Braak stages are shown in Fig. 2B. The percentage changes in flortaucipir SUVR with each increment in Thal phase for each Braak stage are shown in Fig. S3 and Table S4, with Fig. S3 illustrating whether increases in uptake by Thal phase differed, represented by the confidence interval not crossing the 0 line.
In the medial temporal regions, flortaucipir uptake increased with Thal phase across all Braak stages, although increases in flortaucipir uptake with increasing Thal phase were greater in those with higher Braak stage. For example, in the entorhinal cortex, flortaucipir uptake increased significantly by 5–8% for each progressive increment in Thal phase in cases with Braak stage VI (p<0.001 for all contrasts) and increased by only 2–4% per Thal phase increment in cases with Braak stage II and was only significant for an increase between Thal 4–5 (p=0.02) (Fig. S3 and Table S4). In the cortical regions, flortaucipir uptake increased with Thal phase in cases with higher Braak stages (V-VI). In cases with lower Braak stage, flortaucipir uptake in the cortical regions was not strongly influenced by Thal phase, except for a relatively low burden with high Thal phase in cases with Braak stage 0-III. The PVC model results were very similar to the non-PVC models (Fig. S4 and Table S5).
Model robustness and sensitivity analyses.
Model estimated flortaucipir SUVRs with 95% confidence intervals are shown compared to raw mean flortaucipir uptake for all Braak stage and Thal phase combinations in Table S6. The estimated flortaucipir SUVRs for the models were very similar in our sensitivity analyses accounting for sex, co-pathologies and FTLD, and when limiting the cohort to cases with PET within two years of death (Fig. S5). Furthermore, there was no evidence that the presence of FTLD influenced raw flortaucipir SUVRs in the different PART/ADNC groups (Fig. S6).
Relative regional flortaucipir uptake by Braak stage and Thal phase
Since the dynamic range of flortaucipir uptake in each region differed, the model-based estimated mean flortaucipir SUVR in each region was scaled relative to the modelled mean minimum (set as 0) and maximum (set as 1) SUVR across combinations of Braak stage and Thal phase. Relative mean uptake within each ROI was then plotted on the heatmap in Fig. 3 to illustrate three-dimensional relationships with Braak stage and Thal phase. In cases with high Thal phase (4 and 5), higher relative uptake was observed in the medial temporal lobe at low-intermediate Braak stages (Braak 0-V), with medial temporal and cortical regions showing equivalent uptake at Braak VI. In cases with lower Thal phase (0–3), little relative uptake was observed in most regions until Braak stage V and VI.
Figure 3: Heatmap visualizing predicted flortaucipir standardized uptake value ratios (SUVRs) within and among regions by Braak stage and Thal phase.
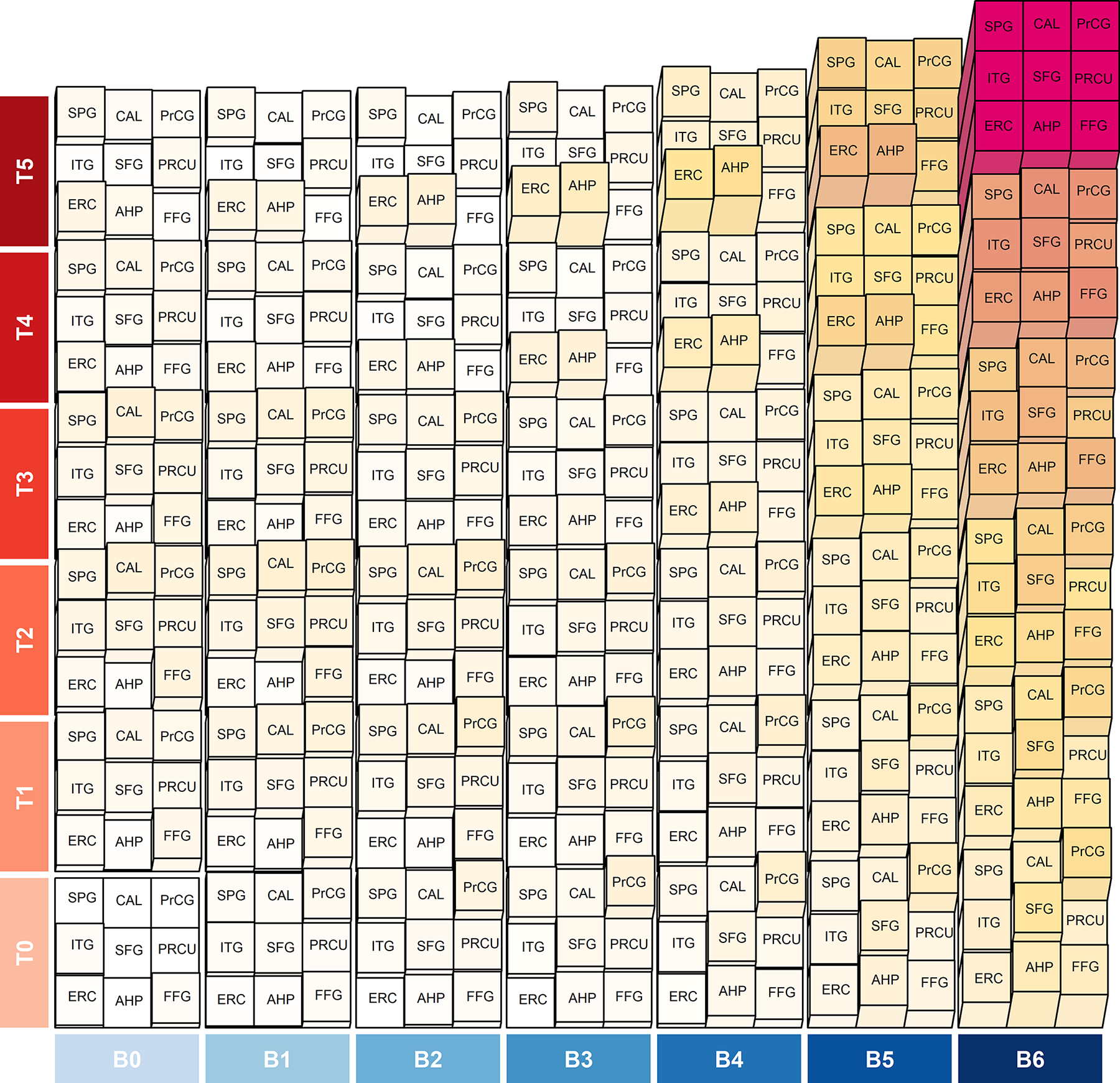
SUVRs normalized to a scale of 0 to 1. There are 9 blocks in each combination of Braak stage (B) and Thal phase (T) representing the nine brain regions. The normalized values are signified by colors (white=0, red=1) and height (flat=0, full height=1) of the blocks. The figure illustrates how the entorhinal cortex and amygdala show uptake close to their capacity at earlier Braak stages than the cortical regions. ERC=entorhinal cortex; AHP=anterior hippocampus; FFG=fusiform gyrus; ITG=inferior temporal gyrus; SFG=superior frontal gyrus; PRCU=precuneus; SPG=superior parietal gyrus; CAL=calcarine; PrCG=precentral gyrus.
Flortaucipir relationship to PART and ADNC
Flortaucipir uptake in all regions was greater in the high ADNC cases compared to both the definite PART and low/intermediate ADNC cases (Table 2). Flortaucipir uptake was also greater in low/intermediate ADNC compared to PART in the entorhinal cortex, fusiform and inferior temporal cortex. The entorhinal cortex provided excellent differentiation of high ADNC from both low/intermediate ADNC and PART, with area under the receiver operator characteristic curves (AUROCs) of 0.92 and 0.98 respectively (Table 3). These AUROCs were greater than the AUROCs for all other regions in the comparison of high ADNC and PART, and greater than all regions, except the inferior temporal and fusiform gyri, in the comparison of high ADNC and low/intermediate ADNC (Table 3). The entorhinal cortex could also differentiate low/intermediate ADNC from PART with an AUROC of 0.70 and provided better differentiation than all other regions (Table 3). Similar findings were observed in the PVC analysis (Table S7 and S8). As a secondary analysis we compared the low and intermediate ADNC cases (Table S9). The entorhinal cortex could differentiate these groups with an AUROC of 0.69 and provided better differentiation compared to all other regions, except the hippocampus (Table S10). Flortaucipir uptake did not differ between low ADNC and PART (Table S9 and S10) or between PART and the not ADNC cases, for any of the regions.
Table 2:
Regional flortaucipir uptake comparisons across PART and ADNC groups
| Region | High ADNC | Low/Intermediate ADNC | PART | High ADNC vs Low/Intermediate ADNC | High ADNC vs PART | Low/Intermediate ADNC vs PART |
|---|---|---|---|---|---|---|
|
| ||||||
| Entorhinal cortex | 1.61 (1.46, 1.84) | 1.16 (1.07, 1.25) | 1.09 (1.02, 1.11) | <0.001 | <0.001 | <0.001 |
| Anterior HP | 1.46 (1.27, 1.68) | 1.07 (1.02, 1.17) | 1.08 (0.98, 1.12) | <0.001 | <0.001 | 0.16 |
| Fusiform | 1.76 (1.46, 2.24) | 1.20 (1.14, 1.28) | 1.16 (1.12, 1.22) | <0.001 | <0.001 | 0.009 |
| Inferior temporal | 1.95 (1.56, 2.43) | 1.28 (1.19, 1.36) | 1.23 (1.16, 1.29) | <0.001 | <0.001 | 0.02 |
| Superior frontal | 1.39 (1.21, 1.97) | 1.16 (1.06, 1.24) | 1.13 (1.09, 1.22) | <0.001 | <0.001 | 0.63 |
| Precuneus | 1.52 (1.26, 2.48) | 1.17 (1.09, 1.25) | 1.13 (1.07, 1.20) | <0.001 | <0.001 | 0.06 |
| Superior parietal | 1.47 (1.21, 2.28) | 1.11 (1.05, 1.20) | 1.08 (1.04, 1.17) | <0.001 | <0.001 | 0.26 |
| Calcarine | 1.26 (1.12, 1.51) | 1.07 (1.02, 1.14) | 1.05 (1.01, 1.12) | <0.001 | <0.001 | 0.20 |
| Precentral | 1.22 (1.09, 1.44) | 1.10 (1.03, 1.15) | 1.10 (1.06, 1.16) | <0.001 | <0.001 | 0.60 |
Median (IQR) flortaucipir standardized uptake value ratios (SUVRs) are shown by pathological group. Pairwise comparison p-values are from Wilcoxon Rank Sum test. HP = hippocampus.
Table 3:
Area under the receiver operator characteristic curve analysis to differentiate PART and ADNC groups
| Region | High ADNC vs Low/Intermediate ADNC | High ADNC vs PART | Low/Intermediate ADNC vs PART | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||
| AUROC (95% CI) | Cut-off | Sens (%) | Spec (%) | AUROC (95% CI) | Cut-off | Sens (%) | Spec (%) | AUROC (95% CI) | Cut-off | Sens (%) | Spec (%) | |
|
| ||||||||||||
| Entorhinal cortex | 0.92 (0.88, 0.96) | 1.39 | 92 | 81 | 0.98 (0.96, 1.00) | 1.26 | 98 | 92 | 0.70 (0.62, 0.79) | 1.13 | 80 | 60 |
| Anterior HP | 0.89 (0.84, 0.94) | 1.23 | 87 | 82 | 0.94 (0.90, 0.98)** | 1.19 | 90 | 88 | 0.57 (0.47, 0.67)*** | 1.11 | 74 | 43 |
| Fusiform | 0.90 (0.85, 0.95) | 1.40 | 94 | 80 | 0.95 (0.92, 0.99)* | 1.32 | 100 | 84 | 0.63 (0.54, 0.72)* | 1.17 | 62 | 62 |
| Inferior temporal | 0.91 (0.86, 0.95) | 1.46 | 91 | 82 | 0.95 (0.92, 0.99)* | 1.48 | 100 | 82 | 0.62 (0.53, 0.71)** | 1.25 | 62 | 62 |
| Superior frontal | 0.79 (0.73, 0.86)*** | 1.30 | 94 | 58 | 0.81 (0.75, 0.88)*** | 1.23 | 84 | 72 | 0.52 (0.43, 0.62)*** | 1.15 | 62 | 54 |
| Precuneus | 0.83 (0.77, 0.89)*** | 1.34 | 93 | 70 | 0.88 (0.82, 0.93)*** | 1.29 | 96 | 72 | 0.59 (0.50, 0.69)* | 1.20 | 78 | 43 |
| Superior parietal | 0.83 (0.77, 0.89)** | 1.22 | 84 | 74 | 0.85 (0.79, 0.92)*** | 1.21 | 88 | 74 | 0.56 (0.46, 0.66)** | 1.08 | 48 | 66 |
| Calcarine | 0.77 (0.70, 0.84)*** | 1.18 | 89 | 59 | 0.81 (0.74, 0.88)*** | 1.25 | 100 | 51 | 0.57 (0.47, 0.66)** | 1.07 | 62 | 53 |
| Precentral | 0.72 (0.64, 0.80)*** | 1.25 | 95 | 49 | 0.70 (0.62, 0.79)*** | 1.30 | 94 | 44 | 0.47 (0.37, 0.57)*** | 1.12 | 62 | 44 |
Area under the receiver operator curve (AUROC), Sensitivity, and Specificity (based on the Youden criteria maximizing the sum of sensitivity and specificity) are shown for each region. HP = hippocampus. P values compare AUROCs to entorhinal AUROC
p ≤ 0.05
p ≤ 0.01
p ≤ 0.001
Investigating flortaucipir and β-amyloid PET in PART
To investigate how imaging defined PART relates to pathologically defined PART, we plotted both flortaucipir PET SUVR and β-amyloid PET findings (+/−) against Braak stage, Thal phase and PART/ADNC groups in our cases in Fig. 4A. Fig. S7 shows the same plot with participant numbers provided. 239 cases had undergone β-amyloid PET using Pittsburgh Compound B (PiB) during life. We focused on entorhinal flortaucipir SUVR since this region provided optimum differentiation of PART from ADNC, and tau deposition is observed in this region in both PART and early Braak stage. Our analyses utilized Thal phase as the primary measure of β-amyloid, although previous studies have utilized Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuritic plaque scores and hence, we also plotted CERAD scores for all 248 cases. Entorhinal flortaucipir SUVRs are also plotted for each PART/ADNC group, with β-amyloid PET status shown, in Fig. 4B.
Figure 4: Relationship of CERAD scores and PiB status with Thal phase, Braak stage and entorhinal flortaucipir SUVR.
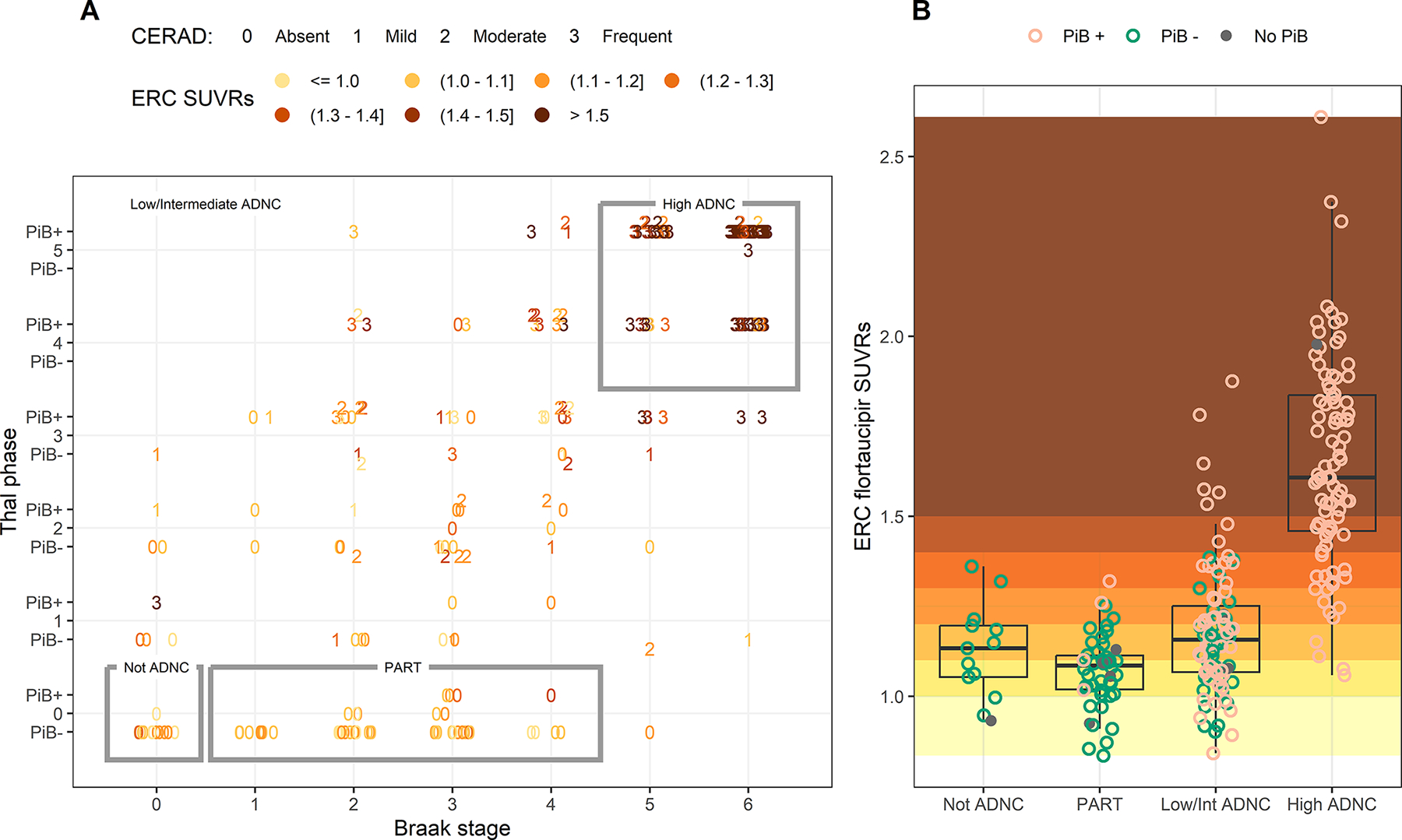
A) The distribution of participants by Braak stage and Thal phase, with CERAD score shown by numbers (0=absent, 1=mild, 2=moderate, 3=frequent), PiB status (PiB+/PiB-) shown by the horizontal position of the data point (above the line indicates PiB+, below the line indicates PiB- and on the line indicates missing PiB) and entorhinal (ERC) SUVR shown by shaded color. Not ADNC, definite PART and high ADNC cases are outlined, with the remaining cases classified as low/intermediate ADNC. B) Entorhinal SUVR for not ADNC, PART, low/intermediate ADNC and high ADNC, with individual points colored by PiB status. The background has been shaded by entorhinal SUVR with values below 1.0 in pale yellow, values above 1.5 in dark orange, and values in between colored by 0.1 increments. Boxes represent 25th (lower hinge) 50th, and 75th (upper hinge) percentiles. The upper whisker extends from the hinge to the highest value that is within 1.5*IQR of the hinge. The lower whisker extends from the hinge to the lowest value within 1.5*IQR of the hinge.
Flortaucipir SUVR in the entorhinal cortex was elevated in some of the PART cases with SUVRs overlapping with the range observed in high ADNC cases (approximately 1.1–1.4 SUVR). However, elevated SUVRs in this range were also observed in some of the not ADNC cases where NFTs were absent. As expected, absent CERAD was observed in all Thal 0 cases and a CERAD score of frequent was most common in cases with Thal phases 3–5. Absent CERAD was, however, observed in cases with Thal phases 1–4. Hence, many of the low/intermediate ADNC cases would be mis-classified as definite PART based on CERAD alone and many of these cases show elevated flortaucipir SUVR. A high proportion of Thal 1–2 cases (27/38, 71%) were PiB-negative, and several PiB-negative cases were Thal 3, with many of these having moderate or frequent CERAD. Hence, many PiB-negative participants that show elevated entorhinal flortaucipir uptake have low/intermediate ADNC with diffuse and even neuritic plaques. Flortaucipir scans from PiB-negative cases with the greatest entorhinal or inferior temporal uptake and most likely to be given an imaging diagnosis of PART are shown in Fig. 5; all these cases had low/intermediate ADNC or not ADNC.
Figure 5: Flortaucipir PET scans from PiB-PET negative cases with the highest entorhinal cortex and inferior temporal flortaucipir uptake.
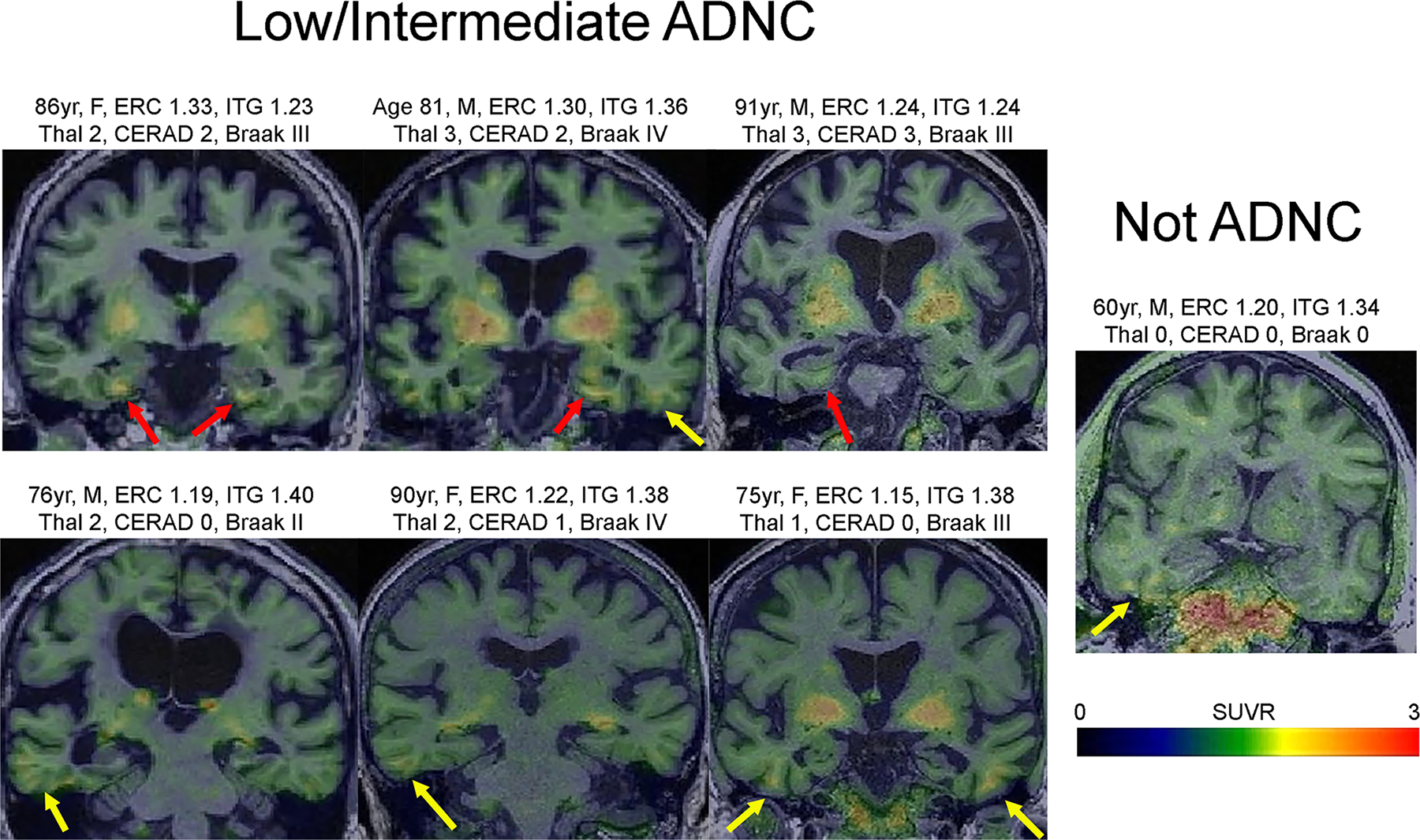
Representative Flortaucipir PET scans of PiB-negative cases. The greatest flortaucipir uptake was observed in the entorhinal cortex (all >1.20 SUVR; red arrows) or the inferior temporal gyrus (all >1.34 SUVR; yellow arrows). Patients with frontotemporal dementia were excluded. Clinical diagnoses before death included two cognitively normal (top left, bottom right), one with Alzheimer’s dementia (bottom left) and four with mild cognitive impairment (all the rest). The patient with Alzheimer’s dementia had advanced leukoaraiosis. Age=age at PET; M=male; F=female ERC=entorhinal cortex; ITG=inferior temporal gyrus.
We also examined how continuous PiB-PET uptake (shown as centiloids) was related to Thal and CERAD (Fig. S8). PiB-PET uptake was increased in cases with Thal phase of 3 or higher, without increases at Thal phase 1 and 2. PiB-PET uptake increased between all CERAD stages. PiB-PET was able to differentiate PART from low/intermediate ADNC with a good AUROC of 0.83 which was significantly higher (p=0.006) than the AUROC generated using entorhinal flortaucipir SUVR (Table S11). PiB-PET was also able to differentiate PART from low ADNC with a fair AUROC of 0.71, although specificity was low and there was no evidence that PiB-PET performed better than entorhinal cortex flortaucipir SUVR (Table S12).
DISCUSSION
In this study we used flortaucipir PET as an in vivo marker of regional tau burden combined with autopsy confirmed Braak NFT stage and Thal phase to better understand how the relationship between tau burden and distribution is influenced by β-amyloid, in aging and AD. We observed a non-linear relationship between flortaucipir uptake and Braak stage across medial temporal and cortical regions and observed that β-amyloid has an influence on flortaucipir uptake across regions. Cases with high ADNC showed dramatically higher uptake than cases with low/intermediate ADNC and cases with definite PART across the brain, and were well differentiated using in vivo tau PET. However, flortaucipir PET had limited ability to detect definite PART and a molecular PET definition of PART did not correspond well with pathologically defined definite PART.
As expected, flortaucipir uptake was greater at high Braak stages (V/VI) compared to low Braak stages across the brain, concurring with previous studies(15, 17, 27). However, we found that the relationship between flortaucipir uptake and Braak stage was non-linear. In cases with low Braak stage, prior to tau extending into limbic regions, flortaucipir uptake was relatively low, even in medial temporal lobe regions. Mean flortaucipir uptake then increased with increasing Braak stage, with this pattern starting at lower Braak stage in medial temporal regions than in cortical regions. Once tau extended into neocortical regions (Braak V/VI), flortaucipir uptake was high across all brain regions. This suggests that as Braak stage increases in aging and ADNC, there is a greater flortaucipir uptake throughout the brain, including in regions affected earlier in the disease.
Somewhat surprisingly, we found that flortaucipir uptake, and hence tau burden, was greater with higher β-amyloid Thal phase when holding Braak stage constant. Although one study did find some evidence of flortaucipir binding to β-amyloid since binding was observed in cored plaques which do not have tau components(28), most autoradiographic studies have found that flortaucipir does not bind to β-amyloid(8–10). Flortaucipir does, however, show inconsistent binding to tau in neuritic plaques(8) which may explain some of the β-amyloid-associated increased flortaucipir uptake, although flortaucipir binding to NFTs has been suggested to be stronger based on immunohistochemistry profiles in one autoradiographic study(8). The suggestion that flortaucipir PET may be binding to tau in neuritic plaques is critical for the interpretation of flortaucipir PET in AD studies and as an outcome measure in clinical treatment trials. For example, lowering of tau PET signal in association with β-amyloid antibody treatment may be related to neuritic plaque clearance, as opposed to changes in NFT pathology. Alternatively, our findings may reflect disease interactions between tau and β-amyloid(29). Hence, for any Braak stage, increased flortaucipir uptake within a given region is linked to higher β-amyloid burden. Previous studies have found some evidence suggesting that higher β-amyloid burden is associated with higher tau PET uptake in aging and ADNC(13, 30, 31). One study showed that flortaucipir uptake in medial temporal regions was related to aging, whereas uptake in cortical regions was related to β-amyloid, as measured by PET(13). Using pathological data, we found evidence that a greater spread of β-amyloid from cortical regions to subcortical regions and cerebellum is also associated with greater flortaucipir uptake. This relationship was observed both in the cortex and medial temporal regions, accounting for age. Two potential explanations for the differing findings in the medial temporal lobe are that we assessed β-amyloid at autopsy rather than on PET and that we assessed β-amyloid distribution rather than burden. Although Thal phase correlates well with β-amyloid burden measured on PET(24), our study shows that β-amyloid PET does not detect the vast majority of Thal 1 and 2 cases. Differences may also be cohort driven since we modeled relationships across all participants, many of whom had MCI or dementia and the full range of Thal phase, whereas the previous study assessed relationships in cognitively healthy older adults which had relatively low β-amyloid PET uptake(24).
We found that β-amyloid influenced flortaucipir uptake at all Braak stages in the entorhinal cortex and hippocampus, but only in Braak stages of IV and greater in the cortical regions. These findings in the cortical regions would be expected given that they do not have appreciable tau deposition at the lower Braak stages. The models suggest that in the medial temporal regions, flortaucipir uptake only increases in the low Braak stages when β-amyloid Thal phase is high. Hence, flortaucipir PET will not be able to detect tau in cases with PART. This has been alluded to in previous smaller studies(14, 15). Pathological studies have shown that the burden of NFTs is less in the entorhinal cortex, Cornu Ammonis 1 (CA1), CA3, CA4 and dentate gyrus of the hippocampus in PART compared to ADNC cases(32) which may explain why it cannot be detected by flortaucipir PET. We did observe elevated flortaucipir uptake in some of the definite PART cases, although similar degrees of uptake were observed in the not ADNC cases that have no NFTs, suggesting that uptake in PART may represent off-target binding. These findings influence the interpretation of flortaucipir PET. For example, previous PET studies detected medial temporal tau uptake in the absence of β-amyloid on PET and hypothesized that these cases represented PART(25, 26). Data from PiB-PET in our cohort would suggest that such cases are likely to have had a low/intermediate ADNC and could even have had moderate or frequent CERAD with neuritic plaques, and hence PiB-PET is not specific to definite PART. We found that PiB-PET uptake was clearly increased in Thal phase 3 or higher but was less sensitive to Thal phase 1 and 2, which concurs with previous studies(33, 34). Furthermore, the PiB-negative cases in our cohort that had the highest temporal lobe flortaucipir uptake, and hence would meet imaging criteria for PART, all had low/intermediate ADNC or not ADNC at autopsy, and none of them had definite PART. The cut-points used to determine positivity in PET studies will be critical to determine the proportion of cases identified as β-amyloid negative and tau PET positive. In previous studies, the proportion of such cases has varied between 2% and 51% (22, 35, 36). Our findings suggest that previously utilized β-amyloid PET cut-points may be too high leading to many cases with β-amyloid in their brain being labelled at β-amyloid negative on PET. However, lower cut-points still do not appear to be able to separate Thal 0 from Thal1/Thal2 cases. Regarding flortaucipir PET, the overlap in uptake between no ADNC and definitive PART, irrespective of FTLD co-pathology, suggests that flortaucipir PET will not be able to differentiate these groups, regardless of cut-point.
We observed slightly different relationships between flortaucipir uptake and Thal phase in the medial temporal versus cortical regions in cases with low Braak stage. In the medial temporal regions flortaucipir uptake increased with Thal phase in cases with low Braak stage, but that was not the case in cortical regions. In cortical regions, we observed slightly lower flortaucipir uptake in cases with low Braak stage but high β-amyloid Thal phase (4/5). Some investigators consider such cases to have age-related β-amyloid deposition, or pathological aging(37), and may be unique, with some not having a neurodegenerative disease. In fact, cases with pathological aging have widespread diffuse β-amyloid senile plaques but not neuritic plaques(37). A relatively small amount of tau in the form of threads and pretangles can be found in cortical regions, even in cases with Braak stage III or less(25), which may account for the cases with lower Thal phase having slightly higher cortical uptake. It is also possible that the model estimated flortaucipir values are inaccurate given that there was very scant data for patients with low Braak stage and high Thal phase.
One of the biological unknowns in AD that cannot be answered with pathological analyses alone is how tau accumulates to maximum capacity in one region relative to accumulation to maximum capacity in other regions. A larger SUVR would imply more average tau accumulation across all the voxels in that region. Hence, the inferior temporal gyrus has the capacity to accumulate more tau than the entorhinal cortex and hippocampus given the larger maximum mean estimated SUVR of that region, as others have found(38). To control for this biological unknown, we set each regional maximum mean accumulation at 1 and each regional minimum mean accumulation at 0, based on flortaucipir SUVR. Medial temporal regions had relatively higher uptake and, hence, were closer to maximum capacity than other regions up to Braak stage V for cases with Thal phase 4 and 5, with all regions reaching capacity at Braak VI. Hence, even when the cortical regions start to accumulate tau, the entorhinal cortex and hippocampus are still closer to reaching their maximum capacity until end stage disease. This pattern of medial temporal prominence was not observed in cases with lower Thal phase, again supporting the strong influence of β-amyloid deposition on flortaucipir uptake in the medial temporal regions. It also suggests different mechanisms of tau accumulation in patients with high β-amyloid versus those with low β-amyloid. It should be acknowledged, however, that the maximum model estimated mean which is set to “1” will be lower than the maximum patient-level raw SUVR. It is possible that many patients in our cohort may not have reached maximum flortaucipir SUVR in these regions, particularly in cortical regions. This does not imply that we did not have cases with relatively high cortical uptake because our cohort did include patients with atypical AD which would have high flortaucipir uptake in cortical regions(39).
Approximately one third of our cohort had FTLD pathology, with most of these cases having PART or low/intermediate ADNC. Although mild uptake on flortaucipir PET can be observed in FTLD(14, 17), we did not see any evidence that uptake was elevated in FTLD at any of the ADNC or PART scores. The models were also rerun including FTLD as a covariate and the model estimated flortaucipir SUVRs were similar to the original models, suggesting our findings would generalize to autopsy cohorts that either do or do not contain FTLD. There is no reason to exclude cases with FTLD since PART and low/intermediate ADNC are what they are and are no less important if they occur in the presence of FTLD. The models were also robust to the presence of Lewy bodies and co-existent TDP-43 pathology.
Given the large sample size and different combinations of Braak stage and Thal phase, we were able to assess for differences between high ADNC, low/intermediate ADNC and definite PART. We showed that regional flortaucipir SUVR differs between these three groups. In fact, for all nine regions there was a gradient of flortaucipir SUVR that was always highest in the patients with high ADNC, followed by the low/intermediate group with the PART cases having the lowest SUVR. This difference in flortaucipir uptake allowed us to identify cut-points to differentiate between groups with excellent sensitivities and high specificities. In our sample the entorhinal cortex had the highest AUROC to differentiate between groups and could differentiate high ADNC from low/intermediate ADNC with sensitivity of 89% and specificity of 81% and differentiate high ADNC from PART with sensitivity of 100% and specificity of 91%. The entorhinal cortex performed better than all other regions to differentiate high ADNC from PART, although the hippocampus and fusiform gyrus performed similarly to the entorhinal cortex in differentiating low/intermediate ADNC from high ADNC. Hence, our data suggests that the entorhinal cortex would be the optimal brain region to select for imaging studies aiming to identify high ADNC, although the hippocampus and fusiform would also be appropriate choices if the goal was to differentiate high ADNC from low/intermediate ADNC. The entorhinal cortex also performed the best to differentiate low/intermediate ADNC from PART, although with only a fair AUROC and relatively low specificity. Within the low/intermediate ADNC cases, entorhinal SUVR was higher in the intermediate cases and entorhinal SUVR did not differ between low ADNC and PART. Given that PART is defined by the absence of β-amyloid, it would be reasonable to assume that PiB-PET would provide good differentiation of PART from low/intermediate ADNC cases. Indeed, PiB-PET did provide better differentiation of these groups compared to entorhinal flortaucipir SUVR. PiB-PET also differed between the low ADNC cases and PART, but the AUROC was only fair, specificity was low, and there was no evidence that this differentiation was better than entorhinal flortaucipir SUVR. The relatively poor differentiation of PART and low ADNC reflects the fact that many of the low ADNC cases are incorrectly classified as PiB-PET negative. The differentiation of PART from low/intermediate ADNC is particularly important given the availability of anti-amyloid treatment and these findings suggest that PiB PET may be the best option as a biomarker to select appropriate candidates for treatment, with the caveat that cases with low ADNC and low Thal phase will be missed. This is the first study to provide flortaucipir cut-points and sensitivity/specificity to differentiate between high, low/intermediate ADNC and definite PART. One previous study of 64 cases found that visual reads of cortical uptake had a sensitivity of 94.7% and specificity of 80.8% to differentiate high ADNC cases from other cases(16). Our previous study of 26 cases found that a flortaucipir temporal meta-ROI could differentiate AD/pathological aging from other cases (mixture of definite PART, low/intermediate ADNC and not ADNC), with sensitivity of 87% and specificity of 82%(15); the much larger cohort in this present study allowed a more fine-grained analysis of how flortaucipir can differentiate specific ADNC scores and definite PART.
There are many strengths of our study including the large sample size of 248 autopsy confirmed cases and the range of combinations of Braak stages and Thal phases that allowed us to model how flortaucipir uptake is related to tau, and how such relationships are influenced by β-amyloid. Our cohort also included a range of individuals with different clinical and pathological diagnoses, and hence our results may generalize to other diverse pathological cohorts. Our findings did not change when a two-compartment PVC were utilized.
Limitations of the study included the small number of cases with certain combinations of Braak stage and Thal phase. A consequence of not having all combinations is that some of the conclusions made were predominantly based on statistical modelling in the presence of a limited number of data points. The generalized additive model we used was chosen because it provides a smoothed, or de-noised, estimated mean across combinations of Braak stage and Thal phase and facilitates a degree of extrapolation. The missing combinations are not specific to this study but underscore the fact that there are some combinations of Braak stage and Thal phase that are rare(3, 40). The fact that our models are based on pathological scales that measure the spatial spread of proteins (Braak stage and Thal phase) rather than quantitative burden measures limits our ability to determine direct relationships between flortaucipir and the burden of tau and β-amyloid. Previous studies have, however, shown positive relationships between NFT counts and Braak stage(41) and senile plaque counts and Thal phase (33) suggesting these staging schemes provide indirect measures of protein burden. The average two-year temporal gap between flortaucipir PET and death in our patients may have added noise into our models given that increased spread and burden of pathology may have occurred between PET and death. However, we reran the models using only the cases with PET within two years of autopsy and the model estimates were very similar. The flortaucipir PET thresholds generated from our AUROC analysis may be dependent upon our specific imaging pipeline which limits their generalizability. Standardized quantification of tau PET in ongoing work such as the CenTauR project(42) should allow better translation of results between different academic sites by creating a standard reference scheme for tau PET SUVR values. Lastly, a limitation of the study was that the cohort consisted predominantly of white non-Hispanic participants and hence the findings may not generalize to more racially and ethnically diverse cohorts. Our study should be cross-validated using additional, more racially and ethnically diverse cohorts.
In conclusion, the presented findings help to understand how tau spreads throughout the brain with aging and in AD, and how β-amyloid plays a role in determining this spread. The study furthermore has implications for the interpretation and use of molecular PET biomarkers. Our data support the use of flortaucipir PET to help predict ADNC scores during life and highlight the fact that uptake will reflect underlying tau pathology but will also be influenced by β-amyloid in both medial temporal and cortical regions. Importantly, caution is warranted in the use of molecular PET to predict the presence of PART and cut points should be carefully considered in this regard.
MATERIALS AND METHODS
Study design
This is a retrospective case-control study that aimed to model the relationships between Braak stage and flortaucipir PET, and to determine whether this relationship is influenced by β-amyloid, as measured histopathologically by Thal phase. Two secondary aims were to determine optimum flortaucipir cut points to differentiate between different ADNC scores and PART and evaluate how imaging defined PART using β-amyloid PET and flortaucipir PET relates to pathologically defined PART. We identified all individuals at Mayo Clinic that had undergone flortaucipir PET during life, had died between 4/8/2016 and 11/18/2023, and had undergone an autopsy evaluation (n=248). Participants had been enrolled and followed during life in either the Mayo Clinic ADRC, the Mayo Clinic Study of Aging/Patient Registry(43), or the Neurodegenerative Research Group (NRG). The study was approved by the Mayo Clinic IRB. All participants provided written informed consent to participate in this study.
Clinical evaluations
All participants had undergone detailed neurological and neuropsychological evaluations(43), including the Mini-Mental Status Examination (MMSE)(44) and the Clinical Dementia Rating Scale (CDR)(45). All had been evaluated by a Behavioral Neurologist and diagnosed as cognitively unimpaired, mild cognitive impairment (MCI)(46) or dementia(47) according to published diagnostic criteria. Participants were classified as cognitively unimpaired if they were judged to have no cognitive impairment according to published criteria(45). Participants who met criteria for dementia were given specific syndromic diagnoses. Apolipoprotein E testing was performed as previously described(48).
Neuropathological evaluations
All participants had died and underwent standard neuropathological examination performed by a board-certified neuropathologist (DWD, RRR or AN) as previously described in detail(14). Each case had been assigned a Braak neurofibrillary tangle (NFT) stage(1) using anti-tau antibodies (clone AT8, 1:1,000 dilution; Endogen, Woburn, MA)(1), a Thal phase(18) using thioflavin-S or antibodies to β-amyloid (clone 6F/3D, 1:10 dilution; Novocastra Vector Labs, Burlingame, CA). All patients were given a Braak NFT stage based on published criteria as follows: Braak stage 0 = no neurofibrillary tangles found throughout the entire brain; Braak stage I = NFTs are present and confined to the transentorhinal cortex; Braak stage II = NFTs extends into the entorhinal cortex; Braak stage III = NFTs extends beyond entorhinal into fusiform cortex; Braak stage IV = NFTs extend beyond fusiform cortex into inferior and medial temporal cortex; Braak stage V = NFTs extend beyond lateral temporal cortex into occipital and frontal association cortices and Braak stage VI = NFTs are present in peristriate and striate areas of the occipital cortex. All patients had NFT deposition that followed Braak staging and allowed us to generate Braak NFT stage. All patients were given a Thal β-amyloid phase based on published criteria as follows: Thal phase 0 = no β-amyloid deposition is present throughout the entire brain; Thal phase 1 = β-amyloid positive senile plaques are present and confined to cortical regions; Thal phase 2 = β-amyloid senile plaques are identified in neocortex as well as in the cornu ammonis region 1 or subiculum of the hippocampus; Thal phase 3 = β-amyloid senile plaques are found in the basal ganglia; Thal phase 4 = β-amyloid senile plaques are found in the cornu ammonis region 4 or midbrain and Thal phase 5 = β-amyloid deposition extends into the cerebellar molecular layer. A CERAD(49) score for neuritic plaques was also assigned using modified Bielschowsky silver stain or thioflavin-S, as follows: CERAD absent = 0; mild = 1; moderate = 2 and frequent = 3. Cases were classified as definite PART if the Thal phase was 0, CERAD score was 0 and the Braak stage was I-IV(21) (Fig. 1). Using the National Institute of Aging-Alzheimer’s Association recommendations(19, 20), we classified cases into not ADNC (Thal 0, Braak 0), low/intermediate ADNC (Thal 0 and Braak stage V-VI, Thal 1–3 and Braak stage 0-VI, or Thal 4–5 and Braak stage 0-IV) or high-ADNC (Thal 4–5 and Braak V-VI) (Fig. 1).
Frontotemporal lobar degeneration was diagnosed if there were neuronal loss and gliosis affecting frontal or temporal lobe (50) and included FTLD disorders characterized by 4R tau, 3R tau, or TAR DNA binding protein of 43 kDa (TDP-43). All FTLD cases had screened negative for FTLD-associated mutations. Lewy body disease was diagnosed based on the presence of Lewy bodies observed on sections immunostained for α-synuclein (clone LB509, 1:200; Zymed, San Francisco, CA). In non-FTLD cases, the amygdala and hippocampus were screened for the presence of TDP-43 immunoreactivity using antibody (pS409/410, 1:5,000, Cosmo Bio, Tokyo, Japan).
PET analysis
All 248 participants underwent a structural head MRI and flortaucipir PET using consistent protocols. If more than one flortaucipir PET scan was available, the last scan closest to death was selected for analysis. Flortaucipir PET scans were acquired using a PET/CT scanner. Approximately 370MBq (range 333–407 MBq) of [18F]-flortaucipir was administered, followed by a 20-minute PET acquisition performed 80 minutes after injection. The scans consisted of four 5-minute dynamic frames following a low dose CT transmission scan. We applied standard corrections and data were reconstructed into a 256×256 matrix with a 30-cm field of view (in-plane pixel size=1.17mm). Dynamic PET frames were coregistered and averaged. All participants also underwent a 3T head MRI protocol using GE scanners that included a magnetization prepared rapid gradient echo (MPRAGE) sequence (TR/TE/TI, 2300/3/900 ms; flip angle 8°, 26-cm field of view (FOV); 256×256 in-plane matrix; phase FOV of 0.94; slice thickness of 1.2 mm). The MRI scans were performed a median of one day from the flortaucipir PET scans.
Each PET image was rigidly registered to its corresponding MPRAGE using SPM12 (Welcome Trust Centre for Neuroimaging, London, UK). Using ANTs, the Mayo Clinic Adult Lifespan Template (MCALT) (https://www.nitrc.org/projects/mcalt/) atlases were propagated to the native MPRAGE space and used to calculate regional flortaucipir PET values in the grey and white matter. Tissue probabilities were determined for each MPRAGE using Unified Segmentation in SPM12, with MCALT tissue priors and settings. Standard uptake value ratios (SUVRs) were created normalizing uptake in each region of interest (ROI) to the cerebellar crus grey matter(51). We analyzed the following nine regions-of-interest (ROIs) that were selected to capture different Braak stages; entorhinal cortex (Braak stages I-II), anterior portion of the hippocampus (Braak stage II), fusiform gyrus (Braak stage III), inferior temporal gyrus (Braak stage IV), superior frontal, precuneus and superior parietal (Braak stage V), calcarine cortex and precentral (motor) (Braak stage VI). All analyses were performed without PVC and then the analyses were repeated using a two-compartment PVC(52, 53).
Of the 248 participants, 239 had also undergone a PiB PET to assess β-amyloid deposition. The acquisition protocol consisted of administering 628MBq (385–723 MBq) of PiB, followed by a 20-minute PET acquisition performed 40 minutes after injection. Acquisition, corrections, and data reconstruction were identical to flortaucipir PET. A global PiB SUVR was calculated by creating a weighted average of uptake in prefrontal, orbitofrontal, parietal, temporal, anterior and posterior cingulate and precuneus normalized to uptake in the cerebellar crus grey matter(54), and a cut-off of 1.48 was used to defined PiB-positivity(55). Global PiB SUVR values were converted to the centiloid scale as previously described(56) (SUVR cut-off of 1.48 is equivalent to centiloid of 22).
Statistical analysis
We used generalized additive models (GAMs) to model how mean log-transformed tau PET SUVR varies with numeric Thal phase and numeric Braak stage. We fit a separate model for each of nine regions of interest and included age at PET scan and time from PET scan to death as covariates. The models were of the form log(SUVR) = β0 + β1 XAge + β2 XTime + s(XThal, XBraak) + ε where s() is a two-dimensional, semiparametric spline-based smoother and can be thought of as representing a smooth interaction between Thal phase and Braak phase which is more flexible than including a product of these two terms in the model. Holding age and time from scan to death constant, the model provides an estimate and confidence intervals for the mean for all combinations of Thal phase and Braak stage. We used the fitted models to summarize the estimated mean difference and corresponding 95% CI for a one-phase difference in Thal at each Braak stage, and for a one-stage difference in Braak at each Thal phase. We also used the fitted models to provide a comprehensive summary of the relationship between Thal phase, Braak stage, and mean tau PET uptake across regions in a 3D figure that uses shading and bar height (normalized to the unit interval [0,1] based on the minimum and maximum mean values for a given region). We note that this was a rescaling of the estimated mean values from the GAM models and done to facilitate comparisons across region, Thal phase, and Braak stage. We also performed five separate sensitivity analyses to further evaluate the GAM models: (i) limiting the analysis to individuals who died within two years of FTP imaging; (ii) adjusting for sex in the models; (iii) adjusting for TDP-43 status (0/1); (iv) adjusting for Lewy body disease status (0/1); and (v) adjusting for FTLD status (0/1). For sensitivity analyses ii–iii we report exponentiated mean values after setting the indicator variable to the sample mean.
Nonparametric area under the receiver operating characteristic curve (AUROC) analyses, which have p-values equivalent to Mann-Whitney U/Wilcoxon rank sum test p-values, were used to summarize the difference between PART/ADNC groups and Youden cut-offs for discriminating between groups are reported. Dependent AUROC estimates were compared directly(57).
Analyses were performed using R Statistical Software version 4.1.3 and GAM modeling used the mgcv package version 1.8–42.
Supplementary Material
Acknowledgements:
We thank AVID Radiopharmaceuticals for their support in supplying the AV-1451 precursor, chemistry production advice and oversight, and US Food and Drug Administration regulatory cross-filing permission and documentation needed for this work.
Funding:
This study was funded by NIH grants RF1-NS112153 to K.A.J. and J.L.W., R01-AG50603 to J.L.W., R01-AG37491 to K.A.J., R01-DC12519 to J.L.W., R01-DC14942 to K.A.J., R01-NS89757 to J.L.W. and K.A.J., R37-AG11378 to C.R.J., P30 AG062677 to R.C.P., U01-AG 006786 to R.C.P., the Elsie and Marvin Dekelboum Family Foundation, and the Oxley Foundation.
Footnotes
Competing interests: Dr. Knopman serves on a Data Safety Monitoring Board for the DIAN study, serves on a Data Safety Monitoring Board for Biogen but receives no personal compensation, is an investigator in clinical trials sponsored by Biogen, Lilly Pharmaceuticals, and the University of Southern California, and serves as a consultant for Roche, Samus Therapeutics, Third Rock, and Alzeca Biosciences but receives no personal compensation. Dr. Petersen serves as a consultant for Roche Inc., Merck Inc., and Biogen, Inc., serves on the Data Safety Monitoring Board for Genentech, Inc., and receives royalty from Oxford University Press and UpToDate. Dr. Lowe consults for Bayer Schering Pharma, Piramal Life Sciences, Life Molecular Imaging, Eisai Inc., AVID Radiopharmaceuticals, and Merck Research and receives research support from GE Health care, Siemens Molecular Imaging, and AVID Radiopharmaceuticals. The other authors report no disclosures.
Data and materials availability:
All data associated with this study are present in the paper and the Supplementary Materials and a Dryad repository: https://doi.org/10.5061/dryad.v6wwpzh48.
References and Notes
- 1.Braak H, Braak E, Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Frigerio I, Boon BDC, Lin CP, Galis-de Graaf Y, Bol J, Preziosa P, Twisk J, Barkhof F, Hoozemans JJM, Bouwman FH, Rozemuller AJM, van de Berg WDJ, Jonkman LE, Amyloid-beta, p-tau and reactive microglia are pathological correlates of MRI cortical atrophy in Alzheimer’s disease. Brain Commun 3, fcab281 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Josephs KA, Martin PR, Weigand SD, Tosakulwong N, Buciuc M, Murray ME, Petrucelli L, Senjem ML, Spychalla AJ, Knopman DS, Boeve BF, Petersen RC, Parisi JE, Dickson DW, Jack CR, Whitwell JL, Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 143, 3463–3476 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD, Vemuri P, Senjem ML, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Dickson DW, Jack CR Jr., MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology 71, 743–749 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Josephs KA, Murray ME, Tosakulwong N, Whitwell JL, Knopman DS, Machulda MM, Weigand SD, Boeve BF, Kantarci K, Petrucelli L, Lowe VJ, Jack CR Jr., Petersen RC, Parisi JE, Dickson DW, Tau aggregation influences cognition and hippocampal atrophy in the absence of beta-amyloid: a clinico-imaging-pathological study of primary age-related tauopathy (PART). Acta Neuropathol 133, 705–715 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinson AC, Davidson YS, Horan MA, Pendleton N, Mann DMA, Pathological Correlates of Cognitive Impairment in The University of Manchester Longitudinal Study of Cognition in Normal Healthy Old Age. J Alzheimers Dis 64, 483–496 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, Mu F, Sinha A, Su H, Szardenings AK, Walsh JC, Wang E, Yu C, Zhang W, Zhao T, Kolb HC, [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 9, 666–676 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Lowe VJ, Curran G, Fang P, Liesinger AM, Josephs KA, Parisi JE, Kantarci K, Boeve BF, Pandey MK, Bruinsma T, Knopman DS, Jones DT, Petrucelli L, Cook CN, Graff-Radford NR, Dickson DW, Petersen RC, Jack CR Jr., Murray ME, An autoradiographic evaluation of AV-1451 Tau PET in dementia. Acta Neuropathol Commun 4, 58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marquie M, Normandin MD, Vanderburg CR, Costantino IM, Bien EA, Rycyna LG, Klunk WE, Mathis CA, Ikonomovic MD, Debnath ML, Vasdev N, Dickerson BC, Gomperts SN, Growdon JH, Johnson KA, Frosch MP, Hyman BT, Gomez-Isla T, Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol 78, 787–800 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sander K, Lashley T, Gami P, Gendron T, Lythgoe MF, Rohrer JD, Schott JM, Revesz T, Fox NC, Arstad E, Characterization of tau positron emission tomography tracer [18F]AV-1451 binding to postmortem tissue in Alzheimer’s disease, primary tauopathies, and other dementias. Alzheimers Dement 12, 1116–1124 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Schwarz AJ, Yu P, Miller BB, Shcherbinin S, Dickson J, Navitsky M, Joshi AD, Devous MD Sr., Mintun MS, Regional profiles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 139, 1539–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Das SR, Xie L, Wisse LEM, Vergnet N, Ittyerah R, Cui S, Yushkevich PA, Wolk DA, I. Alzheimer’s Disease Neuroimaging, In vivo measures of tau burden are associated with atrophy in early Braak stage medial temporal lobe regions in amyloid-negative individuals. Alzheimers Dement 15, 1286–1295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scholl M, Lockhart SN, Schonhaut DR, O’Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J, Schwimmer HD, Rabinovici GD, Jagust WJ, PET Imaging of Tau Deposition in the Aging Human Brain. Neuron 89, 971–982 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghirelli A, Tosakulwong N, Weigand SD, Clark HM, Ali F, Botha H, Duffy JR, Utianski RL, Buciuc M, Murray ME, Labuzan SA, Spychalla AJ, Pham NTT, Schwarz CG, Senjem ML, Machulda MM, Baker M, Rademakers R, Filippi M, Jack CR Jr., Lowe VJ, Parisi JE, Dickson DW, Josephs KA, Whitwell JL, Sensitivity-Specificity of Tau and Amyloid β Positron Emission Tomography in Frontotemporal Lobar Degeneration. Annals of neurology 88, 1009–1022 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lowe VJ, Lundt ES, Albertson SM, Min HK, Fang P, Przybelski SA, Senjem ML, Schwarz CG, Kantarci K, Boeve B, Jones DT, Reichard RR, Tranovich JF, Hanna Al-Shaikh FS, Knopman DS, Jack CR Jr., Dickson DW, Petersen RC, Murray ME, Tau-positron emission tomography correlates with neuropathology findings. Alzheimers Dement 16, 561–571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleisher AS, Pontecorvo MJ, Devous MD Sr., Lu M, Arora AK, Truocchio SP, Aldea P, Flitter M, Locascio T, Devine M, Siderowf A, Beach TG, Montine TJ, Serrano GE, Curtis C, Perrin A, Salloway S, Daniel M, Wellman C, Joshi AD, Irwin DJ, Lowe VJ, Seeley WW, Ikonomovic MD, Masdeu JC, Kennedy I, Harris T, Navitsky M, Southekal S, Mintun MA, Investigators AS, Positron Emission Tomography Imaging With [18F]flortaucipir and Postmortem Assessment of Alzheimer Disease Neuropathologic Changes. JAMA Neurol 77, 829–839 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soleimani-Meigooni DN, Iaccarino L, La Joie R, Baker S, Bourakova V, Boxer AL, Edwards L, Eser R, Gorno-Tempini ML, Jagust WJ, Janabi M, Kramer JH, Lesman-Segev OH, Mellinger T, Miller BL, Pham J, Rosen HJ, Spina S, Seeley WW, Strom A, Grinberg LT, Rabinovici GD, 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain 143, 3477–3494 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thal DR, Rub U, Orantes M, Braak H, Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ, National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, National Institute on A, Alzheimer’s A, National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol 123, 1–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL 3rd, Wisniewski T, Woltjer RL, Yamada M, Nelson PT, Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128, 755–766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costoya-Sanchez A, Moscoso A, Silva-Rodriguez J, Pontecorvo MJ, Devous MD Sr., Aguiar P, Scholl M, Grothe MJ, Alzheimer’s Disease Neuroimaging I, the S Harvard Aging Brain, Increased Medial Temporal Tau Positron Emission Tomography Uptake in the Absence of Amyloid-beta Positivity. JAMA Neurol, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack CR, Wiste HJ, Botha H, Weigand SD, Therneau TM, Knopman DS, Graff-Radford J, Jones DT, Ferman TJ, Boeve BF, Kantarci K, Lowe VJ, Vemuri P, Mielke MM, Fields JA, Machulda MM, Schwarz CG, Senjem ML, Gunter JL, Petersen RC, The bivariate distribution of amyloid-beta and tau: relationship with established neurocognitive clinical syndromes. Brain 142, 3230–3242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowe VJ, Lundt ES, Albertson SM, Przybelski SA, Senjem ML, Parisi JE, Kantarci K, Boeve B, Jones DT, Knopman D, Jack CR Jr., Dickson DW, Petersen RC, Murray ME, Neuroimaging correlates with neuropathologic schemes in neurodegenerative disease. Alzheimers Dement 15, 927–939 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wuestefeld A, Pichet Binette A, Berron D, Spotorno N, van Westen D, Stomrud E, Mattsson-Carlgren N, Strandberg O, Smith R, Palmqvist S, Glenn T, Moes S, Honer M, Arfanakis K, Barnes LL, Bennett DA, Schneider JA, Wisse LEM, Hansson O, Age-related and amyloid-beta-independent tau deposition and its downstream effects. Brain 146, 3192–3205 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoon B, Guo T, Provost K, Korman D, Ward TJ, Landau SM, Jagust WJ, Alzheimer’s Disease Neuroimaging I, Abnormal tau in amyloid PET negative individuals. Neurobiol Aging 109, 125–134 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kotari V, Southekal S, Navitsky M, Kennedy IA, Lu M, Morris A, Zimmer JA, Fleisher AS, Mintun MA, Devous MD Sr., Pontecorvo MJ, Early tau detection in flortaucipir images: validation in autopsy-confirmed data and implications for disease progression. Alzheimers Res Ther 15, 41 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ono M, Sahara N, Kumata K, Ji B, Ni R, Koga S, Dickson DW, Trojanowski JQ, Lee VM, Yoshida M, Hozumi I, Yoshiyama Y, van Swieten JC, Nordberg A, Suhara T, Zhang MR, Higuchi M, Distinct binding of PET ligands PBB3 and AV-1451 to tau fibril strains in neurodegenerative tauopathies. Brain 140, 764–780 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Busche MA, Hyman BT, Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat Neurosci 23, 1183–1193 (2020). [DOI] [PubMed] [Google Scholar]
- 30.Rabin JS, Nichols E, La Joie R, Casaletto KB, Palta P, Dams-O’Connor K, Kumar RG, George KM, Satizabal CL, Schneider JA, Pa J, Brickman AM, Cerebral amyloid angiopathy interacts with neuritic amyloid plaques to promote tau and cognitive decline. Brain 145, 2823–2833 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Price JL, Morris JC, Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45, 358–368 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Walker JM, Fudym Y, Farrell K, Iida MA, Bieniek KF, Seshadri S, White CL, Crary JF, Richardson TE, Asymmetry of Hippocampal Tau Pathology in Primary Age-Related Tauopathy and Alzheimer Disease. J Neuropathol Exp Neurol 80, 436–445 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murray ME, Lowe VJ, Graff-Radford NR, Liesinger AM, Cannon A, Przybelski SA, Rawal B, Parisi JE, Petersen RC, Kantarci K, Ross OA, Duara R, Knopman DS, Jack CR Jr., Dickson DW, Clinicopathologic and 11C-Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer’s disease spectrum. Brain 138, 1370–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.La Joie R, Ayakta N, Seeley WW, Borys E, Boxer AL, DeCarli C, Dore V, Grinberg LT, Huang E, Hwang JH, Ikonomovic MD, Jack C Jr., Jagust WJ, Jin LW, Klunk WE, Kofler J, Lesman-Segev OH, Lockhart SN, Lowe VJ, Masters CL, Mathis CA, McLean CL, Miller BL, Mungas D, O’Neil JP, Olichney JM, Parisi JE, Petersen RC, Rosen HJ, Rowe CC, Spina S, Vemuri P, Villemagne VL, Murray ME, Rabinovici GD, Multisite study of the relationships between antemortem [(11)C]PIB-PET Centiloid values and postmortem measures of Alzheimer’s disease neuropathology. Alzheimers Dement 15, 205–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ossenkoppele R, Pichet Binette A, Groot C, Smith R, Strandberg O, Palmqvist S, Stomrud E, Tideman P, Ohlsson T, Jogi J, Johnson K, Sperling R, Dore V, Masters CL, Rowe C, Visser D, van Berckel BNM, van der Flier WM, Baker S, Jagust WJ, Wiste HJ, Petersen RC, Jack CR Jr., Hansson O, Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat Med 28, 2381–2387 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jack CR Jr., Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, Vemuri P, Mielke MM, Roberts RO, Machulda MM, Senjem ML, Gunter JL, Rocca WA, Petersen RC, Age-specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol 16, 435–444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK, Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 13, 179–189 (1992). [DOI] [PubMed] [Google Scholar]
- 38.Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, Rentz D, Mormino E, Chhatwal J, Amariglio R, Papp K, Marshall G, Albers M, Mauro S, Pepin L, Alverio J, Judge K, Philiossaint M, Shoup T, Yokell D, Dickerson B, Gomez-Isla T, Hyman B, Vasdev N, Sperling R, Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79, 110–119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tetzloff KA, Graff-Radford J, Martin PR, Tosakulwong N, Machulda MM, Duffy JR, Clark HM, Senjem ML, Schwarz CG, Spychalla AJ, Drubach DA, Jack CR, Lowe VJ, Josephs KA, Whitwell JL, Regional Distribution, Asymmetry, and Clinical Correlates of Tau Uptake on [18F]AV-1451 PET in Atypical Alzheimer’s Disease. J Alzheimers Dis 62, 1713–1724 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braak H, Braak E, Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18, 351–357 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Gertz HJ, Xuereb J, Huppert F, Brayne C, McGee MA, Paykel E, Harrington C, Mukaetova-Ladinska E, Arendt T, Wischik CM, Examination of the validity of the hierarchical model of neuropathological staging in normal aging and Alzheimer’s disease. Acta Neuropathol 95, 154–158 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Villemagne VL, Leuzy A, Bohorquez SS, Bullich S, Shimada H, Rowe CC, Bourgeat P, Lopresti B, Huang K, Krishnadas N, Fripp J, Takado Y, Gogola A, Minhas D, Weimer R, Higuchi M, Stephens A, Hansson O, Dore V, I. Alzheimer’s Disease Neuroimaging, A. r. g. the, CenTauR: Toward a universal scale and masks for standardizing tau imaging studies. Alzheimers Dement (Amst) 15, e12454 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts RO, Geda YE, Knopman DS, Cha RH, Pankratz VS, Boeve BF, Ivnik RJ, Tangalos EG, Petersen RC, Rocca WA, The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 30, 58–69 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Folstein MF, Folstein SE, McHugh PR, “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12, 189–198 (1975). [DOI] [PubMed] [Google Scholar]
- 45.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL, A new clinical scale for the staging of dementia. Br J Psychiatry 140, 566–572 (1982). [DOI] [PubMed] [Google Scholar]
- 46.Petersen RC, Mild cognitive impairment as a diagnostic entity. J Intern Med 256, 183–194 (2004). [DOI] [PubMed] [Google Scholar]
- 47.D. American Psychiatric Association, A. P. Association, Diagnostic and statistical manual of mental disorders: DSM-5. (American Psychiatric Association, Washington, 2013). [Google Scholar]
- 48.Crook R, Hardy J, Duff K, Single-day apolipoprotein E genotyping. J Neurosci Methods 53, 125–127 (1994). [DOI] [PubMed] [Google Scholar]
- 49.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L, The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41, 479–486 (1991). [DOI] [PubMed] [Google Scholar]
- 50.Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, White CL 3rd, Schneider JA, Grinberg LT, Halliday G, Duyckaerts C, Lowe JS, Holm IE, Tolnay M, Okamoto K, Yokoo H, Murayama S, Woulfe J, Munoz DG, Dickson DW, Ince PG, Trojanowski JQ, Mann DM, D. Consortium for Frontotemporal Lobar, Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114, 5–22 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lowe VJ, Wiste HJ, Senjem ML, Weigand SD, Therneau TM, Boeve BF, Josephs KA, Fang P, Pandey MK, Murray ME, Kantarci K, Jones DT, Vemuri P, Graff-Radford J, Schwarz CG, Machulda MM, Mielke MM, Roberts RO, Knopman DS, Petersen RC, Jack CR Jr., Widespread brain tau and its association with ageing, Braak stage and Alzheimer’s dementia. Brain 141, 271–287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meltzer CC, Zubieta JK, Links JM, Brakeman P, Stumpf MJ, Frost JJ, MR-based correction of brain PET measurements for heterogeneous gray matter radioactivity distribution. J Cereb Blood Flow Metab 16, 650–658 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Schwarz CG, Therneau TM, Weigand SD, Gunter JL, Lowe VJ, Przybelski SA, Senjem ML, Botha H, Vemuri P, Kantarci K, Boeve BF, Whitwell JL, Josephs KA, Petersen RC, Knopman DS, Jack CR Jr., Selecting software pipelines for change in flortaucipir SUVR: Balancing repeatability and group separation. Neuroimage 238, 118259 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jack CR Jr., Wiste HJ, Weigand SD, Therneau TM, Lowe VJ, Knopman DS, Gunter JL, Senjem ML, Jones DT, Kantarci K, Machulda MM, Mielke MM, Roberts RO, Vemuri P, Reyes DA, Petersen RC, Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement 13, 205–216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jack CR Jr., Therneau TM, Weigand SD, Wiste HJ, Knopman DS, Vemuri P, Lowe VJ, Mielke MM, Roberts RO, Machulda MM, Graff-Radford J, Jones DT, Schwarz CG, Gunter JL, Senjem ML, Rocca WA, Petersen RC, Prevalence of Biologically vs Clinically Defined Alzheimer Spectrum Entities Using the National Institute on Aging-Alzheimer’s Association Research Framework. JAMA Neurol 76, 1174–1183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klunk WE, Koeppe RA, Price JC, Benzinger TL, Devous MD Sr., Jagust WJ, Johnson KA, Mathis CA, Minhas D, Pontecorvo MJ, Rowe CC, Skovronsky DM, Mintun MA, The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 11, 1–15 e11–14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeLong ER, DeLong DM, Clarke-Pearson DL, Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845 (1988). [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper and the Supplementary Materials and a Dryad repository: https://doi.org/10.5061/dryad.v6wwpzh48.