

Abstract
All ligands of the epidermal growth factor (EGF) receptor (EGFR) are synthesized as membrane-anchored precursors. Previous work has suggested that some ligands, such as EGF, must be proteolytically released to be active, whereas others, such as heparin-binding EGF-like growth factor (HB-EGF) can function while still anchored to the membrane (i.e., juxtacrine signaling). To explore the structural basis for these differences in ligand activity, we engineered a series of membrane-anchored ligands in which the core, receptor-binding domain of EGF was combined with different domains of both EGF and HB-EGF. We found that ligands having the N-terminal extension of EGF could not bind to the EGFR, even when released from the membrane. Ligands lacking an N-terminal extension, but possessing the membrane-anchoring domain of EGF, still required proteolytic release for activity, whereas ligands with the membrane-anchoring domain of HB-EGF could elicit full biological activity while still membrane anchored. Ligands containing the HB-EGF membrane anchor, but lacking an N-terminal extension, activated EGFR during their transit through the Golgi apparatus. However, cell-mixing experiments and fluorescence resonance energy transfer studies showed that juxtacrine signaling typically occurred in trans at the cell surface, at points of cell-cell contact. Our data suggest that the membrane-anchoring domain of ligands selectively controls their ability to participate in juxtacrine signaling and thus, only a subclass of EGFR ligands can act in a juxtacrine mode.
INTRODUCTION
Epidermal growth factor (EGF) receptor (EGFR) signaling pathways play important roles in a variety of cellular functions, including proliferation, differentiation, migration, and apoptosis (Wells, 1999). EGFR signaling is initiated by the binding of one of at least seven ligands, including EGF (Carpenter and Cohen, 1990), transforming growth factor-α (TGFα) (Derynck, 1992), heparin-binding EGF-like growth factor (HB-EGF) (Higashiyama et al., 1991), amphiregulin (Shoyab et al., 1989), betacellulin (Shing et al., 1993) epiregulin (Toyoda et al., 1995), and epigen (Strachan et al., 2001). Soluble EGFR ligands are produced through extracellular cleavage of integral membrane precursor proteins, which consist of an EGF motif flanked by an N-terminal extension and a C-terminal membrane-anchoring domain (Massague and Pandiella, 1993; Harris et al., 2003). There is no obvious homology in the predicted cleavage sites among the different EGFR ligands, although many studies have shown that metalloprotease activity is required for their release (Arribas et al., 1996; Brown et al., 1998; Dempsey et al., 1997; Sunnarborg et al., 2002; Hinkle et al., 2004).
TNF alpha converting enzyme (TACE), also known as ADAM 17, is probably responsible for cleaving TGFα, because TACE -/- cells showed a 95% reduction in TGFα release (Black et al., 1997; Peschon et al., 1998). TACE also has been implicated in the cleavage of amphiregulin and HB-EGF (Sunnarborg et al., 2002). ADAM 10 and ADAM 12 also have been implicated in HB-EGF shedding from cells (Asakura et al., 2002; Yan et al., 2002). Substantial evidence indicates that proteolytic release of EGFR ligands is a regulated process, which can be influenced by protein kinase C (PKC) activity (Brown et al., 1998; Pandiella and Massague, 1991a; Raab et al., 1994), calcium influx, and phosphatase activity (Pandiella and Massague, 1991b; Dethlefsen et al., 1998; Vecchi et al., 1998).
Release of EGFR ligands seems to be an important regulatory step in activating the receptor. Mice lacking TACE showed a phenotype similar to that of EGFR -/- mice, probably because of defective processing of TGFα and other EGFR ligands (Peschon et al., 1998). Blocking EGF and TGFα release also inhibits growth and migration of EGFR-dependent cell lines (Dong et al., 1999). In addition, processing of Spitz, a Drosophila TGFα homologue, is a limiting step in the activation of the Drosophila EGFR (Sibilia and Wagner, 1995; Hansen et al., 1997). Transactivation of the EGFR by a variety of different factors, such as insulin-like growth factor 1 and tumor necrosis factor-α, also seem to depend on the proteolytic release of ligands such as amphiregulin, TGFα, and HB-EGF (Gschwind et al., 2003; Chen et al., 2004; El-Shewy et al., 2004). These data suggest that EGFR ligands are only biologically active when they are released. However, a few studies have shown that TGFα, HB-EGF, amphiregulin, and betacellulin can act as juxtacrine factors (Wong et al., 1989; Anklesaria et al., 1990; Higashiyama et al., 1995; Inui et al., 1997; Tada et al., 1999) and can induce tyrosine phosphorylation of EGFR expressed on juxtaposed cells without the release of detectable ligand. The reason for these disparate results is currently unresolved.
There could be numerous reasons why different laboratories have reached divergent opinions regarding the requirement for proteolytic release for ligand activation. One reason could be that the activity of soluble versus juxtacrine ligands differs in their magnitude. Studies comparing the potencies of isolated soluble and membrane-bound forms of HB-EGF have shown that the precursor form has ∼10–25% of the activity of the secreted form in stimulating cell proliferation (Ono et al., 1994). Thus, when low levels of ligand are expressed by cells, only soluble signaling might be observed. The HB-EGF precursor, however, seems to have distinct activities with regard to cell growth and apoptosis (Takemura et al., 1997; Iwamoto et al., 1999; Singh et al., 2004). This suggests that membrane-anchored ligands activate different sets of signaling pathways in cells and argues against there only being a simple qualitative difference between the two signaling modes.
Another possible reason for the different results on the requirement for ligand shedding might be the need for coexpression of accessory proteins, such as CD9. These molecules seem to be crucial for the juxtacrine activity of HB-EGF and TGFα (Goishi et al., 1995; Higashiyama et al., 1995; Nakamura et al., 1995; Miyoshi et al., 1997; Shi et al., 2000). Accessory proteins could be required to remove a steric constraint imposed by another structural feature of the ligand, such as the heparin-binding domain (Takazaki et al., 2004). Alternatively, the accessory protein could be required to “present” the ligand to the receptor by forming part of a multiprotein complex, analogous to the formation of antigen–major histocompatibility complex (MHC) complexes with the T-cell receptor in lymphocytes (Cambier, 1992). If this is the case, then the ability to form a juxtacrine complex could be highly cell type dependent, requiring the expression of a discrete set of proteins in either the ligand-expressing or receptor-bearing cell.
We have previously shown that the biological activity of membrane-anchored EGF constructs has an absolute requirement for proteolytic release (Dong et al., 1999). These constructs consisted of the core receptor binding and the membrane-anchoring domains of EGF, but lacked the amino-terminal extension of the native ligand. The absence of juxtacrine signaling by these engineered ligands could have been due to the absence of an amino-terminal extension that would facilitate formation of a juxtacrine complex. Alternatively, the membrane-anchoring domain of EGF might have prevented juxtacrine signaling. Although the core EGF structure of different EGFR ligands is very homologous and induces almost identical biochemical responses, the membrane-anchoring domain is distinct. Mutational studies with TGFα, HB-EGF and betacellulin suggest that the juxtamembrane structure dictates the cleavage process and that the cytoplasmic tail regulates ligand trafficking (Dempsey and Coffey, 1994; Arribas et al., 1997; Briley et al., 1997; Dethlefsen et al., 1998; Hinkle et al., 2004). It seemed possible that the membrane anchor also could restrict a ligand's ability to act in a juxtacrine manner.
To determine whether specific structural domains of EGFR ligands dictate their ability to act in a juxtacrine mode, we created a series of artificial ligand chimeras and expressed them in EGF-responsive cells. We found that the biological activity of EGF required both the removal of its amino-terminal extension and its proteolytic release from the cell surface. However, when EGF was tethered on the cell surface by the membrane-anchoring domain of HB-EGF, it was able to participate in efficient juxtacrine signaling. This indicates that the membrane-anchoring domain of EGFR ligands controls their ability to act as either soluble or juxtacrine ligands.
MATERIALS AND METHODS
Reagents and Antibodies
Batimastat was kindly provided by British Biotech Pharmaceuticals (Oxford, United Kingdom). All fluorescently labeled antibodies were from Molecular Probes (Eugene, OR). Anti-EGFR 225 monoclonal antibody (mAb) was isolated from a hybridoma cell line obtained from the American Type Culture Collection (Manassas, VA). Anti-EGFR 13A9 mAb was a gift from Genentech (South San Francisco, CA). Anti-phosphotyrosine (RC-20) horseradish peroxidase conjugate, anti-EGFR antibody (C-13), and goat anti-mouse IgG horseradish peroxidase conjugate were from BD Biosciences (San Jose, CA). Sheep antibodies were generated against a phosphopeptide corresponding to the major tyrosine-phosphorylation site of the EGFR (anti-EGFR-1173P) as described previously (Burke et al., 2001). Anti-FLAG mAb M2 was purchased from Sigma-Aldrich (St. Louis, MO). Monoclonal antibodies directed against human EGF (HA, LB, and LC) were isolated from hybridoma cell lines (Yoshitake and Nishikawa, 1988). The HA mAb was directly labeled with Alexa dye 594 according to the manufacturer's instructions (Molecular Probes). Fab fragments of LC and 13A9 were generated using immobilized papain (Pierce Chemical, Rockford. IL) and separated from undigested whole IgG and Fc by using a protein A column. The Fab fragments were labeled with Alexa dye 546 and Alexa dye 647, for LC and 13A9, respectively. The degree of labeling was 1.6–2.0 mol/mol Fab. Endoglycosidase H (Endo H) was purchased from Roche Diagnostics (Indianapolis, IN), and neuraminidase was from Calbiochem-Novabiochem (La Jolla, CA).
Construction of Cell Lines and Ligand Chimeras
The B82L cell line expressing the first plasmid (pUHD 15.1.neo) of the two-plasmid tetracycline-controlled (Tet-off) mammalian expression system has been described previously (Will et al., 1995). They were selected and grown in DMEM containing 10% dialyzed calf serum, 1 μM methotrexate, and 600 μg/ml G418 (Geneticin). Construction and expression of sEGF and EGF-ct in the second plasmid of the Tet-off system (pUHD 13-3) also has been described previously (Will et al., 1995; Wiley et al., 1998).
The full-length EGF precursor in vector pCB6-pEGF was obtained from Barbara Mroczkowski (Agouron Pharmaceutics, San Diego, CA). The pEGF was excised with SmaI and ClaI and subcloned into pBluescript KS cut with HincII and ClaI. The pEGF was excised from pBluescript by cutting with XhoI, endfilling, and cutting with SacII. This fragment was then inserted into pUHD 13-3, which was first digested with BamHI and endfilled before cutting with SacII. The secreted form of the EGF precursor (At-EGF) was made by inserting a stop codon following Arg1023 of the precursor. This corresponds to the carboxy terminus of the mature EGF peptide. The stop codon was generated by PCR mutagenesis. This construct was inserted into pUHD 13-3 as described above for pEGF. Ligands EGF-ctF (EGF with the membrane-anchoring region from EGF and FLAG epitope) and EGF-hcF (EGF with the membrane-anchoring region from HB-EGF and FLAG epitope) were constructed as described previously (Dong and Wiley, 2000). These constructs are shown schematically in Figure 1.
Figure 1.

Natural and artificial EGFR ligands. Shown at the top is native epidermal growth factor (pEGF) followed by the different EGF molecules used in this study. TM includes the transmembrane and juxtamembrane regions, whereas the membrane-anchoring domain includes both the TM region and the cytoplasmic domain. Note that EGF-hcF and EGF-hc contain the membrane-anchoring domain of HB-EGF (shown at the bottom) but have the same receptor-binding core and signal sequence of EGF-ctF.
The human mammary epithelial cell (HMEC) lines HB2 (Berdichevsky et al., 1994) and 184A1 (Stampfer and Yaswen, 1993) were obtained from Joyce Taylor-Papadimitriou (Imperial Cancer Research, London, United Kingdom) and Martha Stampfer (Lawrence Berkley National Laboratory, University of California, Berkeley, Berkeley, CA), respectively. HB2 clones expressing EGF-ctF and EGF-hcF were isolated as described previously (Dong and Wiley, 2000). 184A1 clones expressing EGF-ctF and EGF-hcF were derived in the same way using a retrovirus-based strategy. HB2 clones were cultured DMEM supplemented with 10% cosmic calf serum (Hyclone Laboratories, Logan, UT), 5 μg/ml insulin, and 5 μg/ml dexamethasone. A1 clones were maintained in DFCI-1 medium (Band and Sager, 1989). All experiments were performed using clones before passage 30.
B82L mouse cell lines expressing EGF-ctF and EGF-hcF also were made using the retrovirus-based strategy described previously (Dong and Wiley, 2000). They were maintained in DMEM containing 10% calf serum. The B82L clonal lines expressing EGFR were grown in DMEM containing 10% dialyzed calf serum and 1 μM methotrexate.
Wild-type Chinese hamster ovary (CHO) cells (R-) and cells expressing the EGFR (R+) were a kind gift of Dr. Gordon Gill (University of California, San Diego) and were used to express both EGF-ct and EGF-hc. The cells were grown in α-minimal essential medium: Ham's F-12 containing 5% fetal bovine serum and 5% defined supplemented calf serum (Hyclone Laboratories). The cells were transfected with LipofectAMINE reagent (Invitrogen, Carlsbad, CA) at a rate of 5 μl reagent/2 μg DNA/35-mm plate of cells. The transfection was carried out for 3 h after which time the cells were propagated for 2 d. The cells were split 1:20 and placed into selective medium containing 10 μg/ml puromycin. Individual clones were isolated through limiting dilution and were periodically subjected to fluorescence-activated cell sorting analysis. Cell populations were sorted when the level of expression dropped below 70%. All transfected cells were maintained in selective medium.
EGFR Phosphorylation Analysis
Confluent cells were extracted using RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 7.2, 150 mM NaCl, and 0.02% sodium azide). Equal amounts of protein from each sample were separated on a 7.5% SDS polyacrylamide gel and subsequently transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Phosphotyrosine and EGFR levels were determined on Western blots by using horseradish peroxidase directly labeled anti-phosphotyrosine RC-20, or anti-EGFR mAb C-13 followed by goat anti-mouse IgG horseradish peroxidase conjugate, respectively.
EGF Immunoprecipitations
To identify the different forms of EGF, cells were grown to confluence in 60-mm dishes in the absence of tetracycline to fully induce ligand expression. Cells were changed to cysteine and methionine-free medium for 30 min and then changed to the same medium containing 0.5 mCi/ml Tran35 S-Label (MP Biomedicals, Irvine, CA) for 2 h at 37°C. The cells were rinsed and lysed and scraped from plates in 1 ml of 25 mM Tris, pH 8.0, 0.5% NP-40, 0.5% sodium deoxycholate, and 150 mM NaCl. Debris was removed by centrifugation at 15,000 × g for 10 min. Anti-EGF mAb HA (6 μg) was added to each tube followed by an overnight incubation at 4°C. Immunoprecipitates were isolated using anti-mouse rabbit IgG and protein A-Sepharose. Samples were boiled in SDS and run on 5–15% gradient SDS polyacrylamide gels, transferred to nitrocellulose, and exposed to film.
For the coimmunoprecipitation studies, proteins were isolated in extraction buffer (1% Triton X-100, 50 mM HEPES, pH 7.0, 150 mM NaCl, 10% glycerol, 1 mM EGTA, and 0.02% sodium azide). Equal amounts of protein from each sample were immunoprecipitated with 5 μg/ml M2 anti-FLAG mAb for 0.5 h and then with 10 μg/ml rabbit anti-mouse IgG and 50 μl/ml 50% protein A-Sepharose for 1.5 h. Immunoprecipitates were washed twice with the extraction buffer, separated on a 7.5% SDS polyacrylamide gel, and transferred to a nitrocellulose membrane. The membrane was probed with horseradish peroxidase (HRP)-conjugated anti-phosphotyrosine RC-20 antibody followed by chemiluminescence detection. After incubating with the stripping buffer (2% SDS, 100 mM 2-mercaptoethanol, and 6.25 mM Tris-HCl, pH 6.8) for 30 min at 50°C, the blots were reprobed with anti-EGFR C-13 antibody, followed by HRP-labeled goat anti-mouse IgG. Blots again were developed by the enhanced chemiluminescence reaction.
Immunofluorescence
Cells were plated on fibronectin-coated coverslips for 48 h in media lacking EGF. They were rinsed in ice-cold saline and fixed with 3.6% paraformaldehyde and 0.024% saponin, freshly prepared in phosphate-buffered saline (PBS) as described previously (Wiley et al., 1998). Cells were incubated with 3.5 μg/ml affinity-purified sheep anti-EGFR-1173P antibody for 1 h followed by staining with 1 μg/ml Alexa 594-labeled mAb HA, 5 μg/ml Alexa 488-labeled affinity-purified donkey anti-sheep IgG for 45 min. After rinsing, the coverslips were mounted in 40 μl of Prolong mounting medium (Molecular Probes). Images were captured separately at 520 nm (Alexa 488) and 610 nm (Alexa 594) and corrected for background. Composite images were assembled in Adobe Photoshop. The lack of cross-reactivity of the Alexa 488-labeled affinity-purified donkey anti-sheep IgG for HA mAb was verified experimentally.
For confocal microscopy, cells were grown on coverslips and treated with 10 μM AG 1517 for 1 h to block activity of the EGFR and to prevent receptor desensitization. The cells were then rinsed and incubated with fresh medium at 37°C and incubated a further 15 min before fixation as described above. Activated EGFR were imaged using affinity-purified sheep anti-1173P (see above), and the ligand chimera was visualized using anti-FLAG antibody M2 directly labeled with Alexa 488. We have previously shown that anti-EGF and anti-FLAG antibodies show identical staining patterns in chimera-expressing cells (Dong and Wiley, 2000). Samples were analyzed using a Leica DMIRE2 confocal microscope using a 100× Plan Apo oil immersion objective. Control cells that did not express the ligand chimeras were used to verify that there was no significant staining of either activated EGFR or the FLAG epitope tag.
Fluorescence Resonance Energy Transfer (FRET)
Cells were incubated simultaneously with 20–40 nM labeled Fabs for 10 min at room temperature. Alexa 546 (LC), serving as the donor, and Alexa 647 (13A9), serving as the acceptor of energy, were excited by 532-nm laser (Nd:YAG Verdi V-10, Coherent, Santa Clara, CA), and 632 nm laser (dye laser CR-599; Coherent), respectively, by using a dual-dichroic mirror (Chroma Technology, Brattleboro, VT). Donor and acceptor emissions were sent simultaneously onto two defined areas on a charge-coupled device (CCD) camera, while toggling between the two lasers. The green and red emissions were separated using a dichroic wedge mirror (Chroma Technology). FRET was detected by Alexa 647 indirect excitation with 532 nm, as the energy was transferred from the excited Alexa 546.
The intensities in the acceptor and FRET channels were used to define a cut-off value, and signals that overlapped in the two channels were further pursued for analysis. The bleed through of the donor (10%) and the acceptor (7%) into the FRET (red) channel was determined empirically and was subtracted pixel by pixel, to obtain the true FRET signal (Fc):
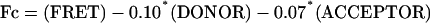 |
1 |
where FRET, DONOR, and ACCEPTOR are the fluorescence intensities in the corresponding channels. FRET efficiency (E) was then calculated pixel by pixel according to equation 2, where γ is the acceptor to donor ratio of the quantum yields of the fluorophores, the objective, and the CCD camera, and equals 0.82.
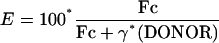 |
2 |
Proliferation Assays
Cells were split at 1:10 into 12-well plates. The next day, cells were changed to control growth medium or medium containing 10 μg/ml 225, 10 μM batimastat, 2.7 μM LB mAb, or 4.7 μM LC mAb. Medium was changed every other day. Cells were counted using a Coulter counter.
Glycosidase Treatments
EGF-ctF and EGF-hcF cells were lysed in RIPA buffer, and the EGFR was immunoprecipitated from the cell lysates with anti-EGFR mAb 225, rabbit anti-mouse IgG, and protein A-Sepharose. Immunoprecipitates were suspended in 50 μl of Endo H buffer (3.6 mM Na2HPO4/NaH2PO4 buffer, pH 5.5, 0.05% SDS, 4 μg/ml phenylmethylsulfonyl fluoride, and 70 mM β-mercaptoethanol) and boiled for 5 min. After centrifugation at 3000 rpm for 3 min, the supernatant was incubated with 5 mU Endo H for 4 h at 37°C. For neuraminidase treatment, the immunoprecipitates were incubated with 50 μl of neuraminidase (1 U/ml) for 30 min at 37°C.
Juxtacrine Activity Assay
B82 cells transfected with or without the gene for human EGFR were transduced either with or without retrovirus containing the EGF-hcF gene. This yielded cells expressing EGFR alone (R+), ligand alone (L+), or both ligand and receptor (R+L+). Before the experiment, cells were first evaluated for receptor number and rate of ligand production. Pure populations of cells (R+ or R+L+) or mixtures of cells (R+/L+) were plated into 100-mm plates such that the number of EGFR (cells × receptors per cell) on each plate was equal. The numbers of ligand producing cells in the mixed cell population were adjusted to equal ligand production by the R+L+ cells. Thus, each plate contained equal numbers of EGFR and ligand production despite varying cell number. However, cell number was always within a factor of 2. Two hours after plating, the medium was replaced with medium containing no additives (control), 10 μg/ml 225 mAb, 50 ng/ml EGF, or 10 μM batimastat; incubated for an additional 16 h; and washed with PBS. Cells were then extracted with 1 ml of RIPA buffer containing 10 mM NaF, 1 mM sodium orthovanadate, and 1 μg/ml each of pepstatin, chymostatin, aprotinin, and leupeptin. After centrifugation, the EGFR was immunoprecipitated from the clarified supernatant as described above, and the relative amount of activated receptor was determined by Western blot analysis.
When using mixtures of CHO cells, equal numbers of receptor-positive and chimera-expressing cells were plated onto 60-mm dishes and allowed to attach for 4 h. The medium was then changed to medium containing 3 nM AG 1517 (Calbiochem-Novabiochem) and either 10 μM batimastat, 10 μg/ml 225 antibody, or 20 μg/ml LC antibody. The cells were incubated overnight and briefly rinsed before the addition of the identical media lacking AG1517. Samples were collected after 2 h by lysing the cells in 1% NP-40 buffer containing 1 mM sodium orthovanadate and then placing samples containing equal amounts of protein on 4–12% gradient bis-Tris polyacrylamide gels. After electrophoresis and Western blotting, the nitrocellulose membranes were probed with anti-phospho 1173 EGFR antibody and then stripped and probed for total receptor mass by using anti-EGFR ab SC-03 (Santa Cruz Biotechnology, Santa Cruz, CA).
RESULTS
The N-terminal Domain of EGF Inhibits Its Activity
We have previously shown that membrane-anchored EGF-ct must be proteolytically released to have biological activity (Dong et al., 1999). Additional studies have suggested that other EGFR ligands also must be released to be active (Peschon et al., 1998; Prenzel et al., 1999). Studies by some investigators, however, have indicated that ligands such as HB-EGF can be biologically active while still membrane associated (Goishi et al., 1995; Inui et al., 1997; Takemura et al., 1997, 1999). A major difference between our studies and those of other investigators was that we were using ligands that lack the domain N terminal of the core receptor binding domain. To determine whether the N-terminal domain of EGF could facilitate its ability to act in a juxtacrine mode, we expressed the full-length EGF precursor (pEGF) as well as three different “preprocessed'' forms of EGF in B82 cells under control of the tetracycline-repressible expression system. As shown in Figure 1, At-EGF (Amino-terminus EGF) lacks the membrane-anchoring domain and corresponds to the high-molecular-weight form of EGF isolated from human breast milk and urine (Mroczkowski and Reich, 1993); EGF-ct (EGF with carboxy terminus) lacks the entire prepro region of At-EGF, as does the mature EGF peptide, but it retains the membrane-anchoring domain; sEGF (secreted EGF) corresponds to the mature EGF peptide with the addition of a signal peptide to allow export from cells. All four forms of EGF were expressed in B82 cells that also were stably transfected with human EGFR so that the activity of these constructs could be evaluated.
EGF was immunoprecipitated from cells expressing the different constructs after induction and metabolic labeling. As shown in Figure 2A, the At-EGF and pEGF forms expressed by cells were ∼160–180 kDa, indicating that they were fully glycosylated. Only a single, high-molecular-weight product was observed in cells expressing pEGF. Immunoprecipitation of the conditioned medium showed that cells expressing sEGF and EGF-ct produced a very similar 7-kDa product, whereas cells producing either pEGF or At-EGF secreted the same 160-kDa product (our unpublished data). Thus, B82 cells are very efficient in cleaving EGF at the juxtamembrane site, but they seem to be unable to process the N-terminal domain of the molecule.
Figure 2.

Expression and activity of different forms of epidermal growth factor. (A) B82 cells expressing the indicated constructs were incubated with Tran35 S label for 2 h and rinsed and lysed as described in Materials and Methods. After immunoprecipitation, the EGFs were separated on a 5–15% gradient denaturing gel. After transfer of the proteins to nitrocellulose, they were visualized by autoradiography. The sample in each lane was normalized to the amount of protein in the original cell extract. (B) B82 cells expressing both the human EGFR and the indicated construct in a Tet-off vector were induced by withdrawal of tetracycline overnight. The parental cells (lacking a ligand vector) were incubated with 25 nM EGF overnight. Cells were extracted and the relative amount of phosphotyrosine associated with the EGFR was determined by a ratiometric ELISA as described in Materials and Methods. All samples were done in triplicate and diluted to keep the ELISA readings within the linear range. Expression of the ligand constructs was verified by nuclease protection assays of parallel plates.
To determine the biological activity of the different EGF constructs, we evaluated EGFR activation after induction of ligand expression by using a ratiometric tyrosine phosphorylation assay (Schooler and Wiley, 2000). As shown in Figure 2B, addition of EGF to the parental (ligand negative) cells resulted in a large increase in the Tyr(P):EGFR ratio. A similar increase was observed after induction of sEGF or EGF-ct expression. The lower absolute level of tyrosine phosphorylation in cells expressing sEGF or EGF-ct is probably due to chronic stimulation of the cell caused by a low level of ligand production even in the Tet-off state (Will et al., 1995). Significantly, we failed to observe any EGFR activation after induction of either At-EGF or pEGF expression. This was not due to low ligand expression in the induced state because the amount of pEGF and At-EGF in the medium was 240 and 500 ng/ml, respectively, as determined by enzyme-linked immunosorbent assay (ELISA). This corresponds to 8 and 16 ng/ml mature EGF. Although this is somewhat less than the amount produced by cells expressing sEGF or EGF-ct, it is ∼10-fold higher than the Kd of the EGF receptor. We also examined four separate clones of cells expressing At-EGF. We were unable to detect any significant change in either EGFR phosphorylation or down-regulation after At-EGF induction (our unpublished data). This indicates that rather than facilitating the ability of EGF to interact with its receptor, the N-terminal domain inhibits receptor binding.
Role of the Membrane-anchoring Domain in Regulating Ligand–Receptor Interactions
The inability of membrane-anchored pEGF to stimulate EGFR activation is very similar to the lack of EGF-ct activity observed when its cleavage is blocked by batimastat (Dong et al., 1999). Thus, in contrast to HB-EGF, it seems that all membrane-anchored forms of EGF are incapable of juxtacrine signaling. One possible explanation for the lack of observable juxtacrine activity for EGF-ct is that its membrane-anchoring domain restricts its ability to engage in this signaling mode. The membrane-anchoring domains of EGF-like ligands are structurally diverse and have already been shown to control the rate of regulated proteolysis (Hinkle et al., 2004). Thus, it seemed reasonable that this domain also could dictate the interactions of membrane-anchored ligands with the EGFR. To explore this idea, we created an EGF chimera that was tethered to the cell surface by the membrane-anchoring domain of HB-EGF, a ligand that has been shown to engage in juxtacrine signaling (Nakamura, 1995). Cells were engineered to express either of two chimeric ligands: EGF-ctF or EGF-hcF. These ligands have the receptor-binding domain of EGF and the membrane-anchoring domains from EGF and HB-EGF, respectively. They also have a FLAG epitope fused to the carboxy terminus of the ligand (Figure 1). We have previously characterized these ligands and found that their distribution and shedding rates mimicked the ligand parent that contributed the membrane-anchoring domain (Dong and Wiley, 2000).
We first examined the expression and rate of release of EGF-ctF and EGF-hcF in HMECs. This cell type was used because of its robust biological response to EGFR activation (Stampfer et al., 1993). As shown in Figure 3A, the constitutive release of EGF from cells expressing EGF-ctF was significantly higher than from cells expressing EGF-hcF. The metalloprotease inhibitor batimastat inhibited the release of both ligands. These data are consistent with previously published reports (Dempsey et al., 1997; Dong and Wiley, 2000).
Figure 3.

Expression and activity of EGF-hcF and EGF-ctF in mammary epithelial cells. (A) Cells expressing the indicated construct were solubilized, and EGF levels were determined by ELISA. To measure released EGF, cells were incubated with mAb 225 (133 nM) in combination with or without 10 μM batimastat (Bat) for 24 h before medium was harvested and assayed. Error bars represent SD of ELISA data. (B) Cells were split 1:10 into 12-well dishes. Eighteen hours later, cells were changed to medium containing 67 mM mAb 225 or 10 μM batimastat. Medium was changed every 2 d, and cells were counted on the indicated days. Data are average of duplicate plates of cells. The data are representative of those obtained from at least three separate clonal cell lines expressing the ligand chimeras.
As shown in Figure 3B, growth of cells expressing EGF-ctF was strongly inhibited by either blocking the EGFR with the antagonistic antibody 225 or by inhibiting ligand shedding with batimastat. This shows that proliferation of HMECs is dependent on the activation of the EGFR and shedding of either EGF-ctF or their endogenous ligands. In cells expressing the EGF-hcF chimera (Figure 3C), blocking the EGFR with 225 antibody also blocked cell proliferation, showing that these cells retained their dependence on EGFR activation. However, batimastat treatment did not block proliferation of these cells. Its minor effect was consistent with its previously demonstrated effect on EGF-induced HMEC growth (Dong et al., 1999). Because batimastat totally blocked release of EGF-hcF (Figure 3A), these data suggest that EGF-hcF could activate the EGFR while still anchored to the membrane (i.e., through a juxtacrine mechanism).
To directly demonstrate the ability of membrane-anchored EGF-hcF to activate the EGFR, we examined EGFR phosphorylation by using Western blots. In addition, we used two distinct HMEC strains (184A1L5 and HB2) expressing EGF-hcF and EGF-ctF to make sure that the results were independent of cell type. Cells were treated with 225 antibody or batimastat for 24 h, and the EGFR was immunoprecipitated and then analyzed on Western blots for phosphotyrosine content. As shown in Figure 4A, treating cells expressing either EGF-hcF or EGF-ctF with 225 antibody efficiently blocked EGFR phosphorylation. Significantly, batimastat strongly inhibited EGFR phosphorylation only in cells expressing EGF-ctF, but it had little effect on cells expressing EGF-hcF, demonstrating that proteolytic release of EGF-hcF is not required for its activity.
Figure 4.

Differential inhibition of EGFR phosphorylation in EGF-hcF and EGF-ctF cells by batimastat. (A) Both 184A1 and HB2 clones expressing the indicated construct were analyzed. Cells were treated with 67 nM mAb 225 or 10 μM batimastat for 22 h. EGFR was immunoprecipitated and analyzed by Western blots by using an anti-phosphotyrosine HRP conjugate (RC20-HRP) or anti-EGFR antibody (C-13), followed by HRP-labeled goat anti-mouse IgG. (B) 184A1 cells expressing full-length HB-EGF or EGF-hcF were treated as described in A. The separating gel was a 4–12% gradient.
Although EGF-hcF contains the membrane-anchoring domain of HB-EGF, it lacks its N-terminal glycosylaminoglycan binding domains. To determine whether the juxtacrine activity of EGF-hcF was similar to native HB-EGF, we expressed full-length HB-EGF in HMEC 184 A1L5 cells by using retroviral transduction. Phosphorylation of the EGFR was evaluated in the presence or absence of either 225 antibody or batimastat. As shown in Figure 4B, 225 antibody blocked EGFR phosphorylation in cells expressing either HB-EGF or EGF-hc. Batimastat had a similar, minor effect on EGFR phosphorylation in cells expressing either ligand. This result suggests that native HB-EGF and the chimera have similar potential for juxtacrine activity in HMEC and thus the N-terminal extension of HB-EGF is not a requirement.
If membrane-anchored EGF-hcF was directly binding to the EGFR, we should be able to coprecipitate the EGFR with anti-ligand antibodies. Because both EGF-ctF and EGF-hcF have a FLAG epitope at their carboxy terminus, we used anti-FLAG antibodies in an attempt to isolate ligand–receptor complexes. Extracts of cells expressing either EGF-hcF or EGF-ctF were incubated with anti-FLAG antibody M2 or an anti-EGFR mAb. The immunoprecipitates were separated on a 7.5% SDS polyacrylamide gel and immunoblotted for phosphotyrosine and EGFR. As shown in Figure 5, phosphorylated EGFR was effectively coimmunoprecipitated with anti-FLAG antibody only in the case of cells expressing EGF-hcF, although a faint band was sometimes observed in the case of EGF-ctF.
Figure 5.

Activated EGFR can be coimmunoprecipitated with full-length EGF-hcF. HB2 parental cells and cells expressing EGF-hcF or EGF-ctF were grown in 100-mm dishes to confluence and extracted in 1% Triton X-100 lysis buffer. Cell extracts were immunoprecipitated with anti-FLAG M2 (right three lanes) or anti-EGFR 13A9 mAb (left three lanes). The immunoprecipitates were analyzed on Western blots for phosphotyrosine (top) and then stripped and reprobed for the EGFR (middle). Alternately, the cells were first treated with the cross-linking agent disuccinimidyl suberate before extraction (bottom). In this case, only one-third of the anti-EGFR 13A9 mAb sample was loaded to facilitate band visualization.
The low efficiency of the coprecipitation indicated that the ligand–receptor complexes were rare or unstable. Alternately, it seemed possible that the complexes between the receptor and the ligands formed after cell solubilization. To explore these different hypotheses, we incubated cells with a cross-linking agent before they were solubilized. Chemical cross-linkers only work efficiently on preformed protein complexes (Sorkin and Carpenter, 1991). As shown in Figure 5 (bottom), the cross-linker substantially increased the amount of coprecipitated EGFR only in the case of EGF-hcF, demonstrating that this chimera formed a complex with the EGFR in intact cells. Because the amount of complexes between EGF-ctF and the EGFR were not enhanced by a cross-linker, they probably formed after cell solubilization.
Intracellular Binding and Activation of EGFR by EGF-hcF
During our studies of EGFR phosphorylation, we noticed that the molecular weight of EGFR in some cells expressing EGF-hcF was significantly lower than that observed either in the parental cells or in cells expressing EGF-ctF (Figure 5). This lower molecular weight receptor was especially evident in the anti-Tyr(P) blots by using single-percentage gels (i.e., 7.5% acrylamide) and was independent of cell type (Figure 4A). In an attempt to determine the reason for the lower molecular weight form, we probed blots with antibodies against individual phosphorylation sites of the EGFR. No differences were seen between receptors activated by exogenous EGF, EGF-ctF, or EGF-hcF (our unpublished data). Likewise, anti-phosphoserine and anti-phosphothreonine antibodies did not reveal any differences (our unpublished data). The distribution of EGFR between the cell surface and intracellular compartments also was indistinguishable between cells expressing the different ligands. However, EGFR cells expressing EGF-hcF displayed sensitivity to endoglycosidase H treatment (Figure 6A), indicating that they were incompletely glycosylated.
Figure 6.

Differential glycosylation of EGFR in cells expressing EGF-ctF versus EGF-hcF. (A) The EGFR was immunoprecipitated from lysates of cells expressing either EGF-hcF (left three lanes) or EGF-ctF (right three lanes). The immunoprecipitates were treated with either endoglycosidase H or neuraminidase as described in Materials and Methods and analyzed by Western blots by using polyclonal anti-EGFR antibody SC-003. (B) Cells expressing the indicated ligand constructs were grown on coverslips, fixed, permeabilized, and stained with anti-EGF mAb (left) or affinity-purified antibodies against the major phosphorylation site in the EGFR (right) as described in Materials and Methods. Arrows indicate colocalization.
Terminal glycosylation of the EGFR takes place in the Golgi apparatus. The ability of the EGFR to bind to ligands, however, is gained before processing of the core oligosaccharides (Slieker and Lane, 1985). This suggested to us the possibility that EGF -hcF was binding to the immature EGFR in the Golgi apparatus, thus interfering with the synthesis of complex carbohydrate chains. If this were the case, then we would expect to see activated EGFR colocalized in the perinuclear region with EGF-hcF.
Cells expressing either EGF-ctF or EGF-hcF were fixed, and the distribution of ligands was determined using an anti-EGF antibody. The distribution of activated EGFR was determined using an affinity-purified sheep antibody against the major phosphorylation site of the EGFR (Burke et al., 2001). As shown in Figure 6B, EGF-ctF was diffusely distributed on the cell surface and in small vesicles. Activated EGFR displayed a weak, diffuse pattern in those cells. In contrast, EGF-hcF was found in intracellular vesicles, as we have described previously (Dong and Wiley, 2000). Activated EGFRs also were found concentrated in small vesicles in the perinuclear region of the cell (Figure 6B, arrows). Most of the intracellular, ligand-containing vesicles, however, did not contain activated EGFR. These data indicate that EGF-hcF is able to form direct complexes with the receptor in some, but not all, intracellular vesicles. These data are also consistent with EGF-hcF binding to the EGFR before its exit from the Golgi apparatus.
The images shown in Figure 6B were taken at a focal plane through the midpoint of the cell, to better image ligand–receptor complexes in the perinuclear region. When the focal plane was changed to visualize the area of cell-cell contact, we noticed that most activated receptors were not intracellular, but instead could be found at the plasma membrane at areas of contact (Figure 7, top). This was specific for the cells expressing EGF-hcF, but it did not correspond to an area of high receptor density. Confocal microscopy of cells expressing EGF-hcF confirmed that areas of cell-cell contact generally displayed increased levels of phosphorylated EGFR (Figure 7, bottom), suggesting that much of the juxtacrine signaling at the cell surface occurred between opposing cells. In the case of cells expressing EGF-ctF, no significant anti-phosphotyrosine staining was observed at points of cell-cell contact (our unpublished data).
Figure 7.

Activated EGFR in cells expressing EGF-hcF are found at points of cell-cell contact. (A) Cells expressing EGF-hcF were fixed, permeabilized, and stained for both EGF (top) and tyrosine-phosphorylated EGFR as described in the legend for Figure 6B. The focal plane was adjusted to optimally visualize the area of cell-cell contact. (B) Cells were treated as described in A, but the immunofluorescence was by confocal microscopy. Shown is a group of four cells imaged through their midplane.
Juxtacrine Signaling Primarily Operates in Trans
The observed concentration of activated EGFR at areas of cell-cell contact suggests that juxtacrine signaling operates as a mechanism to signal between cells in trans. The ability of EGF-hcF to form complexes within the Golgi apparatus suggests that juxtacrine signaling could potentially operate within a cell in cis. It is important to determine whether juxtacrine signaling normally operates in cis or trans because it defines the flow of information within the system (either between cells or within a single cell). To investigate this issue, we expressed either EGF-ct or EGF-hc in Chinese hamster ovary (CHO) cells, which lack EGFR. We then mixed these with CHO cells in which the EGFR was expressed as a transgene. As shown in Figure 8A, the EGFR was activated in the mixed cell population and blocking the EGFR with 225 antibody inhibited this activation. Interestingly, adding the anti-EGF antibody LC had little effect on receptor activation, indicating that this antibody does not interfere with receptor binding. Batimastat blocked signaling mediated by EGF-ct, but not EGF-hc, indicating that EGF-hc participated in efficient juxtacrine signaling in this mixed cell system. Because the receptors and ligands were on different cells, juxtacrine signaling seems to operate in trans.
Figure 8.

Juxtacrine activation primarily operates in trans. (A) CHO cells expressing either EGF-ct (left) or the EGF-hc chimera (right) were mixed with an equal number of cells expressing the EGFR and allowed to plate for 2 h. The cells were then changed to fresh medium (Con) or together with 10 μM batimastat (Bat), 10 μg/ml 225 mAb (225), or 10 μg/ml LC mAb (LC). After 18 h, the cells were extracted, and the EGFR was immunoprecipitated and analyzed for phosphotyrosine (top) or EGFR content (bottom) as described in Materials and Methods. (B) Mouse B82L cells expressing either human EGFR alone (R+) or both receptor and EGF-hcF (R+L+) were plated alone. Alternately, R+ cells were mixed together with cells expressing EGF-hcF alone (R+/L+). The number of cells on each plate was adjusted so that the net receptor number and ligand production was the same. Two hours after plating, the medium was replaced with that containing 10 μg/ml 225 mAb or 10 μM batimastat. After 18 h, the cells were processed as described in A. Numbers under the Tyr(P) bands are their relative intensities (in arbitrary densitometric units). This blot is representative of three replicated experiments.
We confirmed these results by using B82 cells transfected either with or without the human EGFR gene, yielding R+ and R-cells, respectively. These cells were then transduced with retrovirus containing the ligand genes. As shown in Figure 8B, receptor activation in cells expressing both receptors and ligands (R+L+) was essentially the same as observed in the mixed cell population (R+/L+). This is consistent with trans-juxtacrine signaling. Blocking the EGFR with mAb 225 inhibited ligand-induced receptor activation in all cases. Adding batimastat increased receptor activation, suggesting that the membrane-anchored growth factor was the active species. These results suggest that although EGF-hc can form intracellular cis-juxtacrine complexes, the majority of receptor signaling is due to its action as a trans-juxtacrine growth factor.
The purpose of the experiment shown in Figure 8B was to compare cells expressing both ligand and receptors (R+L+) to those expressing either ligands or receptors (R+/L+). To accomplish this, we adjusted the number of ligand-expressing cells in the R+/L+ group to be equivalent to the R+L+ group. An intrinsic consequence of this experimental design was nonequivalency in cell density because the receptors and ligands were now on different cells. However, we found that varying cell density in the R+/L+ group by a factor of five had little effect on the level of observed juxtacrine signaling. To explore this puzzling observation, we visualized the interactions of the two cell types to determine whether cell-cell contacts were proportional to cell density. Mixtures of live R+ and L+ cells were incubated with low levels of fluorescently labeled mAb 13A9 and mAb LC to identify the R+ and L+ cells, respectively. We found that the presence of L+ cells resulted in R+ cells forming extensive protrusions and contacts with the L+ cells (Figure 9). Extensions from the R+ cells seemed to be attached to the surface of L+ cells, particularly in the space between the cells and the culture dish (Figure 9F). In contrast, cells expressing both EGF-hcF and the EGFR (R+L+ cells) were more rounded and uniform in appearance (our unpublished data; also see Figures 6B and 7). Thus, juxtacrine signaling does not seem to be a passive process simply dependent on cell density but is instead a facilitated process in which R+ cells make numerous contacts with the ligand-producing cells.
Figure 9.

Cells involved in juxtacrine signaling display extensive intercellular contacts. B82 cells expressing either the EGFR or EGF-hcF were mixed and incubated overnight in a Bioptechs coverslip dish for live cell imaging. Cells were incubated for 1 h at 37°C with 1 μg/ml anti-EGFR mAb 13A9 labeled with Alexa 488 (B, green, C and D) or anti-EGF mAb LC labeled with Alexa 594 (A, red, C and D). Arrows show areas of contact between R+ and L+ cells. Shown in E is a differential interference contrast image of D. (F) Outlined composite image of D.
The extensive contacts we observed between cells expressing EGF-hcF and EGFR suggest that juxtacrine complexes form between opposing cell surfaces. To directly test this idea, we used fluorescence resonance energy transfer (FRET). By labeling mAb LC with Alexa 546 and the non-antagonistic anti-EGFR antibody 13A9 with Alexa 647, complexes between membrane-anchored EGF and the EGFR should be indicated by a FRET signal. Neither LC nor 13A9 interferes with juxtacrine signaling (our unpublished observations; also see Figure 8A). We used Fab fragments of both antibodies to prevent antibody-induced aggregation and to reduce the potential of steric hindrance by bulky, full-sized antibodies. Cells expressing either EGF-ct or EGF-hc were used. As shown in Figure 10, we did not observe any significant FRET signal in cells expressing EGF-ct. In contrast, cells expressing EGF-hc displayed a robust FRET signal at points of cell-cell contact. These results demonstrate that the membrane-anchoring domain of HB-EGF, but not EGF, allows the formation of juxtacrine complexes between cells in trans.
Figure 10.

FRET between ligands and the EGFR in juxtacrine complexes. Cells expressing EGF-hcF chimeras show substantial FRET signals along cell-cell contacts, indicating molecular interactions between the chimera and EGFR. Typical images of donor, acceptor and FRET efficiency, taken from cells expressing EGF-ctF (top row) and EGF-hcF (bottom row) chimeras, are shown. The right column shows images of the chimeras, tagged with labeled antibodies against the extracellular domain of EGF. The center column shows images of EGFR, tagged with the antibody against the receptor. The left column shows images of FRET efficiency, calculated pixel by pixel (see Materials and Methods).
DISCUSSION
One of the most important aspects of receptor regulation is control of receptor activation. Most studies have focused on downstream signaling events, but few have focused on the control of ligand availability, which is rate limiting for EGFR activation. This has been demonstrated by studies in which inhibition of ligand binding blocks most of the endogenous activity of EGFR (Stampfer et al., 1993; Dong et al., 1999). Although ligand-independent mechanisms of EGFR activation have been reported (reviewed in Gschwind et al., 2001; Yan et al., 2002; Bill et al., 2004), the magnitude of their effect seems minor, at least in mammary epithelium at steady state.
What regulates ligand availability? A series of recent reports suggests that proteolytic processing is the primary regulator for a number of EGFR ligands (Peschon et al., 1998; Dong et al., 1999; Prenzel et al., 1999; Sunnarborg et al., 2002; Hinkle et al., 2004). However, there is strong evidence that HB-EGF does not require proteolytic release for activity. Instead, its ability to form a complex with accessory proteins, such as CD9, seems to be crucial for its activity (Goishi et al., 1995; Higashiyama et al., 1995; Nakamura et al., 1995; Miyoshi et al., 1997). Accessory proteins could be required to remove a steric constraint imposed by another structural feature of the ligand, such as the heparin-binding domain. Alternatively, the accessory protein could be required to “present” the ligand to the receptor by forming part of a multiprotein complex, analogous to the formation of antigen–MHC complexes with the T-cell receptor in lymphocytes (Cambier, 1992).
By expressing EGF constructs that contained or lacked the prepro extension in an inducible expression system, we found that removal of this domain was required for ligand activity. The lack of activity of 160-kDa EGF contradicts previous work by Parries et al. (1995), but is consistent with results published by Dempsey et al. (1997). The difference between these studies is that the Parries study used purified 160-kDa EGF, whereas the Dempsey study (and the current study) examined native 160-kDa EGF produced by cultured cells. It seems possible that the conditions used to purify EGF from large quantities of urine could have resulted in partial denaturation of the precursor, leading to its ability to bind to the EGFR. We could find no evidence that the 160-kDa EGF produced by cultured cells could bind or activate EGFR, and so removal of the prepro extension is probably necessary for its biological activity in situ.
It has previously been shown that membrane-anchored EGF or TGFα also must be proteolytically released to have biological activity (Dong et al., 1999; Borrell-Pages et al., 2003). Our current studies confirm that EGF must be released to be active because membrane-anchored EGF could neither bind nor activate the EGFR. Our results are in contrast with previous studies with EGF and TGFα (Brachmann et al., 1989; Mroczkowski et al., 1989). However, the endogenous metalloprotease activity of cells was not inhibited in those studies. Instead, they used ligand mutants that were believed to be noncleavable (Brachmann et al., 1989; Wong et al., 1989). More recent evidence indicates that the cleavage of these mutants proceeds at a low but significant rate and that assays to detect ligand release in the presence of EGFR-bearing cells are inaccurate unless the EGFR is blocked (Dempsey and Coffey, 1994; Dong et al., 1999). Thus, the observed EGFR activation could have arisen from a small amount of released ligand.
In contrast to the results obtained with EGF or TGFα, we found that a chimera between the membrane-anchoring domain of HB-EGF and the receptor-binding domain of EGF resulted in a molecule that was fully active as a juxtacrine factor. Protease inhibitors could no longer inhibit EGFR activation. Our ability to isolate a complex of EGFR and EGF-hcF directly demonstrates the formation of juxtacrine complexes by the chimera. Imaging studies using fluorescently labeled Fab fragments against EGF and the receptor demonstrated FRET at points of cell-cell contact only in the case of the HB-EGF chimera. Although full-length HB-EGF expressed in our cells also displayed juxtacrine activity, the activity of the chimera in the presence of metalloprotease inhibitors was similar. Because the chimera lacks the domains of HB-EGF required for binding to accessory proteins such as CD9, the membrane-anchoring domain seems to be the critical juxtacrine determinant. Recent reports suggest that the heparin-binding domain of HB-EGF functions as a negative regulator of its activity (Takazaki et al., 2004). Our results cannot address this point directly because the cells used to express native HB-EGF contain high levels of CD9 and other cell surface proteins that have been reported to relieve the inhibitory activity of that domain (our unpublished observations).
It is not clear how the membrane-anchoring region of EGFR ligands restricts receptor access. The “stem” (juxtamembrane) domain between the last critical leucine in EGF (L47) and the membrane-spanning domain of the ligand consist of only 18 and 15 hydrophilic residues for EGF-ctF and EGF-hcF, respectively. Both stems are predicted to assume bend configurations, but display no other obvious structural motifs. However, the anchoring domain could position the core EGF domain in an appropriate orientation for binding in the cleft between EGFR domains I and III or could directly associate with the receptor itself (Garrett et al., 2002). The membrane-spanning domains also could affect the conformation of the stem. Alternately, ligand trafficking and localization could be determined by their membrane-anchoring domain (Dempsey and Coffey, 1994; Brown et al., 2001), and the chimeric ligand may be localized to a compartment or domain of the cell surface where the EGFR is accessible (Dong and Wiley, 2000). Finally, there may be accessory molecules associating with the two different membrane-anchoring domains that stabilize the association of EGFR with EGF-hcF and/or prevent the association of EGFR with the membrane-bound EGF-ctF. Further studies will be required to discriminate between these possibilities.
We noticed that when the EGFR was expressed in the same cell as the EGF-hcF chimera, it migrated at a lower molecular weight on denaturing gels, apparently because of incomplete glycosylation. The simplest explanation for this observation is that EGF-hcF ligand binds to the EGFR during its transit through the Golgi apparatus, thereby interfering with processing of its complex carbohydrates. Consistent with this explanation, we observed that EGF-hcF and activated EGFR were colocalized in the perinuclear region of cells. The incomplete glycosylation indicates that most if not all of the EGFR is interacting with EGF-hcF before it exits from the Golgi. What is not clear is the amount of signaling that arises from this interaction and its stability. The number and distribution of EGFR found in cells expressing EGF-hcF is not substantially different from that found in cells expressing EGF-ctF, suggesting that the “intracrine” interactions do not result in down-regulation of the receptor. In addition, we only see significant numbers of juxtacrine complexes at points of cell-cell contact. The simplest explanation for these observations is that juxtacrine interactions require the receptor and membrane-anchored ligand be juxtaposed on facing membrane surfaces. Such a situation exists at both points of cell-cell contact and in the stacks of membranes found in the Golgi apparatus. When the receptor and ligand enter vesicles for transport to the cell surface, the juxtacrine complex could dissemble because of steric considerations. After delivery to cell surface, new juxtacrine complexes could only form at points of cell-cell contact.
The “intracrine signaling” we observe due to EGF-hcF expression is probably due to the lack of a prepro domain in the ligand chimera. Indeed, we have never seen underglycosylation of the EGFR in cells expressing high levels of native HB-EGF (our unpublished observations). Under normal circumstances, prepro domains of ligands are removed, probably at the cell surface. Significantly, it has been shown that removing the prepro region of amphiregulin prevents its transport to the cell surface (Thorne and Plowman, 1994). In addition, the normal removal of the prepro domain of HB-EGF has been shown to significantly enhance its juxtacrine activity (Nakagawa et al., 1996). We therefore propose that an important role of the prepro domain of EGFR ligands is to prevent binding of the ligands to receptors during their transport to the cell surface. The regulated removal of the prepro domain at the cell surface could restrict receptor activation to that compartment.
It seems that juxtacrine complexes can function as points of cell adhesion. When we mixed cells expressing only EGFR with cells expressing only EGF-hcF, we observed the formation of numerous contacts between the two cell types. The presence of cells expressing EGF-hcF induced the formation of extensive protrusions and ruffling in cells expressing EGFR (Figure 9), probably due to the constitutive release of small amounts of EGF from the cells expressing EGF-hcF (Dong and Wiley, 2000). The contacts between the two cell types are probably due to the formation of stable juxtacrine complexes because we were able to detect FRET between ligand and receptors at these points (Figure 10). It has previously been shown that HB-EGF can act as a cell adhesion factor (Singh et al., 2004), but it is has been unclear whether this was mediated by the heparin-binding or receptor-binding domains of the molecule. Our results indicate that the receptor binding domain itself can mediate cell adhesions.
Our data are consistent with a model in which there are two distinct steps that regulate ligand access to the EGFR. The first step involves the maintenance of the prepro domain during ligand transport to the cell surface. The retention of this structural element is probably necessary to prevent intracrine signaling and unregulated cell proliferation (Wiley et al., 1998). The second step involves either the proteolytic release of the membrane-anchored ligand or the formation of a juxtacrine complex with neighboring cells. The ability of any given ligand to form a juxtacrine complex would require the appropriate membrane-anchoring domain and the presence of auxiliary proteins, such as the tetraspanin CD9. In this model, ligand activation is a highly regulated process that depends on the involvement of a number of distinct molecules and processing events. The multiple steps required for activation probably serve as important control points of regulation for cell proliferation and differentiation. Further investigation into the coordination of these processes should reveal important new insights into how the activity of the EGFR system is regulated.
Acknowledgments
We thank Margaret Woolf for excellent technical assistance, Birgit Will for help with the metabolic labeling studies, Serdar Ozcelik and Haluk Resat for assistance with the FRET analysis, and Yoshino Yoshitake and Katsuzo Nishikawa for the anti-EGF hybridomas. This research was in part sponsored by Laboratory Directed Research and Development funds from the U.S. Department of Energy and by National Institutes of Health Grant GM-62575.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-11-0994) on April 13, 2005.
References
- Anklesaria, P., Teixido, J., Laiho, M., Pierce, J. H., Greenberger, J. S., and Massague, J. (1990). Cell-cell adhesion mediated by binding of membrane-anchored transforming growth factor α to epidermal growth factor receptors promotes cell proliferation. Proc. Natl. Acad. Sci. USA 87, 3289-3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribas, J., Coodly, L., Vollmer, P., Kishimoto, T. K., Rose-John, S., and Massague, J. (1996). Diverse cell surface protein ectodomains are shed by a system sensitive to metalloprotease inhibitors. J. Biol. Chem. 271, 11376-11382. [DOI] [PubMed] [Google Scholar]
- Arribas, J., Lopez-Casillas, F., and Massague, J. (1997). Role of the juxtamembrane domains of the transforming growth factor-α precursor and the beta-amyloid precursor protein in regulated ectodomain shedding. J. Biol. Chem. 272, 17160-17165. [DOI] [PubMed] [Google Scholar]
- Asakura, M., et al. (2002). Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat. Med. 8, 35-40. [DOI] [PubMed] [Google Scholar]
- Band, V., and Sager, R. (1989). Distinctive traits of normal and tumor-derived human mammary epithelial cells expressed in a medium that supports long-term growth of both cell types. Proc. Natl. Acad. Sci. USA 86, 1249-1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdichevsky, F., Alford, D., D'Souza, B., and Taylor-Papadimitriou, J. (1994). Branching morphogenesis of human mammary epithelial cells in collagen gels. J. Cell Sci. 107, 3557-3568. [DOI] [PubMed] [Google Scholar]
- Bill, H. M., Knudsen, B., Moores, S. L., Muthuswamy, S. K., Rao, V. R., Brugge, J. S., and Miranti, C. K. (2004). Epidermal growth factor receptor-dependent regulation of integrin-mediated signaling and cell cycle entry in epithelial cells. Mol. Cell. Biol. 24, 8586-8599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, R. A., et al. (1997). A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 385, 729-733. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages, M., Rojo, F., Albanell, J., Baselga, J., and Arribas, J. (2003). TACE is required for the activation of the EGFR by TGF-α in tumors. EMBO J. 22, 1114-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann, R., Lindquist, P. B., Nagashima, M., Kohr, W., Lipari, T., Napier, M., and Derynck, R. (1989). Transmembrane TGF-α precursors activate EGF/TGF-α receptors. Cell 56, 691-700. [DOI] [PubMed] [Google Scholar]
- Briley, G. P., Hissong, M. A., Chiu, M. L., and Lee, D. C. (1997). The carboxyl-terminal valine residues of proTGF α are required for its efficient maturation and intracellular routing. Mol. Biol. Cell 8, 1619-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C. L., Meise, K. S., Plowman, G. D., Coffey, R. J., and Dempsey, P. J. (1998). Cell surface ectodomain cleavage of human amphiregulin precursor is sensitive to a metalloprotease inhibitor. Release of a predominant N-glycosylated 43-kDa soluble form. J. Biol. Chem. 273, 17258-17268. [DOI] [PubMed] [Google Scholar]
- Brown, C. L., Coffey, R. J., and Dempsey, P. J. (2001). The proamphiregulin cytoplasmic domain is required for basolateral sorting, but is not essential for constitutive or stimulus-induced processing in polarized Madin-Darby canine kidney cells. J. Biol. Chem. 276, 29538-29549. [DOI] [PubMed] [Google Scholar]
- Burke, P. M., Schooler, K., and Wiley, H. S. (2001). Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol. Biol. Cell 12, 1897-1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier, J. C. (1992). Signal transduction by T-and B-cell antigen receptors: converging structures and concepts. Curr. Opin. Immunol. 4, 257-264. [DOI] [PubMed] [Google Scholar]
- Carpenter, G., and Cohen, S. (1990). Epidermal growth factor. J. Biol. Chem. 265, 7709-7712. [PubMed] [Google Scholar]
- Chen, W. N., Woodbury, R. L., Kathmann, L. E., Opresko, L. K., Zangar, R. C., Wiley, H. S., and Thrall, B. D. (2004). Induced autocrine signaling through the epidermal growth factor receptor contributes to the response of mammary epithelial cells to tumor necrosis factor α. J. Biol. Chem. 279, 18488-18496. [DOI] [PubMed] [Google Scholar]
- Dempsey, P. J., and Coffey, R. J. (1994). Basolateral targeting and efficient consumption of transforming growth factor-α when expressed in Madin-Darby canine kidney cells. J. Biol. Chem. 269, 16878-16889. [PubMed] [Google Scholar]
- Dempsey, P. J., Meise, K. S., Yoshitake, Y., Nishikawa, K., and Coffey, R. J. (1997). Apical enrichment of human EGF precursor in Madin-Darby canine kidney cells involves preferential basolateral ectodomain cleavage sensitive to a metalloprotease inhibitor. J. Cell Biol. 138, 747-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck, R. (1992). The physiology of transforming growth factor-α. Adv. Cancer Res. 58, 27-52. [DOI] [PubMed] [Google Scholar]
- Dethlefsen, S. M., Raab, G., Moses, M. A., Adam, R. M., Klagsbrun, M., and Freeman, M. R. (1998). Extracellular calcium influx stimulates metalloproteinase cleavage and secretion of heparin-binding EGF-like growth factor independently of PKC. J. Cell. Biochem. 69, 143-153. [DOI] [PubMed] [Google Scholar]
- Dong, J., Opresko, L. K., Dempsey, P. J., Lauffenburger, D. A., Coffey, R. J., and Wiley, H. S. (1999). Metalloprotease-mediated ligand release regulates autocrine signaling through the epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 96, 6235-6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, J., and Wiley, H. S. (2000). Trafficking and proteolytic release of epidermal growth factor receptor ligands are modulated by their membrane-anchoring domains. J. Biol. Chem. 275, 557-564. [DOI] [PubMed] [Google Scholar]
- El-Shewy, H. M., Kelly, F. L., Barki-Harrington, L., and Luttrell, L. M. (2004). Ectodomain shedding-dependent transactivation of epidermal growth factor receptors in response to insulin-like growth factor type I. Mol. Endocrinol. 18, 2727-2739. [DOI] [PubMed] [Google Scholar]
- Garrett, T. P., et al. (2002). Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor α. Cell 110, 763-773. [DOI] [PubMed] [Google Scholar]
- Goishi, K., Higashiyama, S., Klagsbrun, M., Nakano, N., Umata, T., Ishikawa, M., Mekada, E., and Taniguchi, N. (1995). Phorbol ester induces the rapid processing of cell surface heparin-binding EGF-like growth factor: conversion from juxtacrine to paracrine growth factor activity. Mol. Biol. Cell 6, 967-980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschwind, A., Zwick, E., Prenzel, N., Leserer, M., and Ullrich, A. (2001). Cell communication networks: EGF receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 20, 1594-1600. [DOI] [PubMed] [Google Scholar]
- Gschwind, A., Hart, S., Fischer, O. M., and Ullrich, A. (2003). TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. EMBO J. 22, 2411-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, L. A., Alexander, N., Hogan, M. E., Sundberg, J. P., Dlugosz, A., Threadgill, D. W., Magnuson, T., and Yuspa, S. H. (1997). Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am. J. Pathol. 150, 1959-1975. [PMC free article] [PubMed] [Google Scholar]
- Harris, R. C., Chung, E., and Coffey, R. J. (2003). Epidermal growth factor receptor ligands. Exp. Cell Res. 284, 2-13. [DOI] [PubMed] [Google Scholar]
- Higashiyama, S., Abraham, J. A., Miller, J., Fiddes, J. C., and Klagsbrun, M. (1991). A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science 251, 936-939. [DOI] [PubMed] [Google Scholar]
- Higashiyama, S., Iwamoto, R., Goishi, K., Raab, G., Taniguchi, N., Klagsbrun, M., and Mekada, E. (1995). The membrane protein CD9/DRAP 27 potentiates the juxtacrine growth factor activity of the membrane-anchored heparin-binding EGF-like growth factor. J. Cell Biol. 128, 929-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle, C. L., Sunnarborg, S. W., Loiselle, D., Parker, C. E., Stevenson, M., Russell, W. E., and Lee, D. C. (2004). Selective roles for tumor necrosis factor α-converting enzyme/ADAM17 in the shedding of the EGF receptor ligand family: the juxtamembrane stalk determines cleavage efficiency. J. Biol. Chem. 279, 24179-24188. [DOI] [PubMed] [Google Scholar]
- Inui, S., Higashiyama, S., Hashimoto, K., Higashiyama, M., Yoshikawa, K., and Taniguchi, N. (1997). Possible role of coexpression of CD9 with membrane-anchored heparin-binding EGF-like growth factor and amphiregulin in cultured human keratinocyte growth. J. Cell. Physiol. 171, 291-298. [DOI] [PubMed] [Google Scholar]
- Iwamoto, R., Handa, K., and Mekada, E. (1999).. Contact-dependent growth inhibition and apoptosis of epidermal growth factor (EGF) Receptor-expressing cells by the membrane-anchored form of heparin-binding EGF-like growth factor. J. Biol. Chem. 274, 25906-25912. [DOI] [PubMed] [Google Scholar]
- Massague, J., and Pandiella, A. (1993). Membrane-anchored growth factors. Annu. Rev. Biochem. 62, 515-541. [DOI] [PubMed] [Google Scholar]
- Miyoshi, E., Higashiyama, S., Nakagawa, T., Hayashi, N., and Taniguchi, N. (1997). Membrane-anchored heparin-binding epidermal growth factor-like growth factor acts as a tumor survival factor in a hepatoma cell line. J. Biol. Chem. 272, 14349-14355. [DOI] [PubMed] [Google Scholar]
- Mroczkowski, B., and Reich, M. (1993). Identification of biologically active epidermal growth factor precursor in human fluids and secretions. Endocrinology 132, 417-425. [DOI] [PubMed] [Google Scholar]
- Mroczkowski, B., Reich, M., Chen, K., Bell, G. I., and Cohen, S. (1989). Recombinant human epidermal growth factor precursor is a glycosylated membrane protein with biological activity. Mol. Cell. Biol. 9, 2771-2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, T., Higashiyama, S., Mitamura, T., Mekada, E., and Taniguchi, N. (1996). Amino-terminal processing of cell surface heparin-binding epidermal growth factor-like growth factor up-regulates its juxtacrine but not its paracrine growth factor activity. J. Biol. Chem. 271, 30858-30863. [DOI] [PubMed] [Google Scholar]
- Nakamura, K., Iwamoto, R., and Mekada, E. (1995). Membrane-anchored heparin-binding EGF-like growth factor (HB-EGF) and diphtheria toxin receptor-associated protein (DRAP27)/CD9 form a complex with integrin α3β 1 at cell-cell contact sites. J. Cell Biol. 129, 1691-1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono, M., Raab, G., Lau, K., Abraham, J. A., and Klagsbrun, M. (1994). Purification and characterization of transmembrane forms of heparin-binding EGF-like growth factor. J. Biol. Chem. 269, 31315-31321. [PubMed] [Google Scholar]
- Pandiella, A., and Massague, J. (1991a). Cleavage of the membrane precursor for transforming growth factor α is a regulated process. Proc. Natl. Acad. Sci. USA 88, 1726-1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandiella, A., and Massague, J. (1991b). Multiple signals activate cleavage of the membrane transforming growth factor-α precursor. J. Biol. Chem. 266, 5769-5773. [PubMed] [Google Scholar]
- Parries, G., Chen, K., Misono, K. S., and Cohen, S. (1995). The human urinary epidermal growth factor (EGF) precursor. Isolation of a biologically active 160-kilodalton heparin-binding pro-EGF with a truncated carboxyl terminus. J. Biol. Chem. 270, 27954-27960. [DOI] [PubMed] [Google Scholar]
- Peschon, J. J., et al. (1998). An essential role for ectodomain shedding in mammalian development. Science 282, 1281-1284. [DOI] [PubMed] [Google Scholar]
- Prenzel, N., Zwick, E., Daub, H., Leserer, M., Abraham, R., Wallasch, C., and Ullrich, A. (1999). EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402, 884-888. [DOI] [PubMed] [Google Scholar]
- Raab, G., Higashiyama, S., Hetelekidis, S., Abraham, J. A., Damm, D., Ono, M., and Klagsbrun, M. (1994). Biosynthesis and processing by phorbol ester of the cells surface-associated precursor form of heparin-binding EGF-like growth factor. Biochem. Biophys. Res. Commun. 204, 592-597. [DOI] [PubMed] [Google Scholar]
- Schooler, K., and Wiley, H. S. (2000). Ratiometric assay of epidermal growth factor receptor tyrosine kinase activation. Anal. Biochem. 277, 135-142. [DOI] [PubMed] [Google Scholar]
- Shi, W., Fan, H., Shum, L., and Derynck, R. (2000). The tetraspanin CD9 associates with transmembrane TGF-α and regulates TGF-α-induced EGF receptor activation and cell proliferation. J. Cell Biol. 148, 591-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shing, Y., Christofori, G., Hanahan, D., Ono, Y., Sasada, R., Igarashi, K., and Folkman, J. (1993). Betacellulin: a mitogen from pancreatic beta cell tumors. Science 259, 1604-1607. [DOI] [PubMed] [Google Scholar]
- Shoyab, M., Plowman, G. D., McDonald, V. L., Bradley, J. G., and Todaro, G. J. (1989). Structure and function of human amphiregulin: a member of the epidermal growth factor family. Science 243, 1074-1076. [DOI] [PubMed] [Google Scholar]
- Sibilia, M., and Wagner, E. F. (1995). Strain-dependent epithelial defects in mice lacking the EGF receptor [correction published in Science (1995). 269, 909]. Science 269, 234-238. [DOI] [PubMed] [Google Scholar]
- Singh, A. B., Tsukada, T., Zent, R., and Harris, R. C. (2004). Membrane-associated HB-EGF modulates HGF-induced cellular responses in MDCK cells. J. Cell Sci. 117, 1365-1379. [DOI] [PubMed] [Google Scholar]
- Slieker, L. J., and Lane, M. D. (1985). Post-translational processing of the epidermal growth factor receptor. Glycosylation-dependent acquisition of ligand-binding capacity, J. Biol. Chem. 260, 687-690. [PubMed] [Google Scholar]
- Sorkin, A., and Carpenter, G. (1991). Dimerization of internalized epidermal growth factor receptors. J. Biol. Chem. 266, 23453-23460. [PubMed] [Google Scholar]
- Stampfer, M. R., Pan, C. H., Hosoda, J., Bartholomew, J., Mendelsohn, J., and Yaswen, P. (1993). Blockage of EGF receptor signal transduction causes reversible arrest of normal and immortal human mammary epithelial cells with synchronous reentry into the cell cycle. Exp. Cell Res. 208, 175-188. [DOI] [PubMed] [Google Scholar]
- Strachan, L., Murison, J. G., Prestidge, R. L., Sleeman, M. A., Watson, J. D., and Kumble, K. D. (2001). Cloning and biological activity of epigen, a novel member of the epidermal growth factor superfamily. J. Biol. Chem. 276, 18265-18271. [DOI] [PubMed] [Google Scholar]
- Stampfer, M. R., and Yaswen, P. (1993). Culture systems for study of human mammary epithelial cell proliferation, differentiation and transformation. Cancer Surv. 18, 7-34. [PubMed] [Google Scholar]
- Sunnarborg, S. W., et al. (2002). Tumor Necrosis Factor-a converting enzyme (TACE) regulates epidermal growth factor receptor ligand availability. J. Biol. Chem. 277, 12838-12845. [DOI] [PubMed] [Google Scholar]
- Tada, H., Sasada, R., Kawaguchi, Y., Kojima, I., Gullick, W. J., Salomon, D. S., Igarashi, K., Seno, M., and Yamada, H. (1999). Processing and juxtacrine activity of membrane-anchored betacellulin. J. Cell. Biochem. 72, 423-434. [PubMed] [Google Scholar]
- Takazaki, R., Shishido, Y., Iwamoto, R., and Mekada, E. (2004). Suppression of the biological activities of the EGF-like domain by the heparin-binding domain of heparin-binding EGF-like growth factor. J. Biol. Chem. 279, 47335-47343. [DOI] [PubMed] [Google Scholar]
- Takemura, T., Hino, S., Murata, Y., Yanagida, H., Okada, M., Yoshioka, K., and Harris, R. C. (1999). Coexpression of CD9 augments the ability of membrane-bound heparin-binding EGF-like growth factor (proHB-EGF) to preserve renal epithelial cell viability. Kidney Int. 55, 71-81. [DOI] [PubMed] [Google Scholar]
- Takemura, T., Kondo, S., Homma, T., Sakai, M., and Harris, R. C. (1997). The membrane-bound form of heparin-binding epidermal growth factor-like growth factor promotes survival of cultured renal epithelial cells. J. Biol. Chem. 272, 31036-31042. [DOI] [PubMed] [Google Scholar]
- Thorne, B. A., and Plowman, G. D. (1994). The heparin-binding domain of amphiregulin necessitates the precursor pro-region for growth factor secretion. Mol. Cell. Biol. 14, 1635-1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda, H., Komurasaki, T., Uchida, D., Takayama, Y., Isobe, T., Okuyama, T., and Hanada, K. (1995). Epiregulin. A novel epidermal growth factor with mitogenic activity for rat primary hepatocytes. J. Biol. Chem. 270, 7495-7500. [DOI] [PubMed] [Google Scholar]
- Vecchi, M., Rudolph-Owen, L. A., Brown, C. L., Dempsey, P. J., and Carpenter, G. (1998). Tyrosine phosphorylation and proteolysis. Pervanadate-induced, metalloprotease-dependent cleavage of the ErbB-4 receptor and amphiregulin. J. Biol. Chem. 273, 20589-20595. [DOI] [PubMed] [Google Scholar]
- Wells, A. (1999). EGF receptor. Int. J. Biochem. Cell Biol. 31, 637-643. [DOI] [PubMed] [Google Scholar]
- Wiley, H. S., Woolf, M. F., Opresko, L. K., Burke, P. M., Will, B., Morgan, J. R., and Lauffenburger, D. A. (1998). Removal of the membrane-anchoring domain of epidermal growth factor leads to intracrine signaling and disruption of mammary epithelial cell organization. J. Cell Biol. 143, 1317-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will, B. H., Lauffenburger, D. A., and Wiley, H. S. (1995). Studies on engineered autocrine systems: requirements for ligand release from cells producing an artificial growth factor. Tissue Eng. 1, 83-96. [DOI] [PubMed] [Google Scholar]
- Wong, S. T., Winchell, L. F., McCune, B. K., Earp, H. S., Teixido, J., Massague, J., Herman, B., and Lee, D. C. (1989). The TGF-α precursor expressed on the cell surface binds to the EGF receptor on adjacent cells, leading to signal transduction. Cell 56, 495-506. [DOI] [PubMed] [Google Scholar]
- Yan, Y., Shirakabe, K., and Werb, Z. (2002). The metalloprotease Kuzbanian (ADAM10) mediates the transactivation of EGF receptor by G protein-coupled receptors. J. Cell Biol. 158, 221-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitake, Y., and Nishikawa, K. (1988). Production of monoclonal antibodies with specificity for different epitopes on the human epidermal growth factor molecule. Arch. Biochem. Biophys. 263, 437-446. [DOI] [PubMed] [Google Scholar]