

Abstract
Ubiquitination is an enzymatic process characterized by the covalent attachment of ubiquitin to target proteins, thereby modulating their degradation, transportation, and signal transduction. By precisely regulating protein quality and quantity, ubiquitination is essential for maintaining protein homeostasis, DNA repair, cell cycle regulation, and immune responses. Nevertheless, the diversity of ubiquitin enzymes and their extensive involvement in numerous biological processes contribute to the complexity and variety of diseases resulting from their dysregulation. The ubiquitination process relies on a sophisticated enzymatic system, ubiquitin domains, and ubiquitin receptors, which collectively impart versatility to the ubiquitination pathway. The widespread presence of ubiquitin highlights its potential to induce pathological conditions. Ubiquitinated proteins are predominantly degraded through the proteasomal system, which also plays a key role in regulating protein localization and transport, as well as involvement in inflammatory pathways. This review systematically delineates the roles of ubiquitination in maintaining protein homeostasis, DNA repair, genomic stability, cell cycle regulation, cellular proliferation, and immune and inflammatory responses. Furthermore, the mechanisms by which ubiquitination is implicated in various pathologies, alongside current modulators of ubiquitination are discussed. Enhancing our comprehension of ubiquitination aims to provide novel insights into diseases involving ubiquitination and to propose innovative therapeutic strategies for clinical conditions.
Keywords: protein degradation, protein homeostasis, ubiquitin, ubiquitination
Ubiquitination regulates protein degradation, trafficking, and signaling through a complex machinery involving ubiquitin and various ligases. It is crucial for protein homeostasis, DNA repair, cell cycle regulation, and immune responses. Dysregulation leads to diseases like cancer and neurodegenerative disorders. Therapeutic strategies targeting ubiquitination show promise, and future research aims to explore its emerging roles and technological advancements.
1. INTRODUCTION
In the early 21st century, the Nobel Prize in Physiology or Medicine was awarded multiple times to scientists investigating endogenous proteins. Notably, in 2004, Aaron Ciechanover, Avram Hershko, and Irwin Rose were honored with the Nobel Prize in Chemistry for their discovery of the ubiquitin (Ub)‐mediated protein degradation mechanism. 1 Although the complexities of protein degradation remained elusive for a long time, research from the mid‐1950s to the late 1970s gradually uncovered the role of nonlysosomal pathways under specific physiological conditions, ultimately confirming the Ub–proteasome pathway. This pathway indicates that Ub tagging can regulate a wide range of cellular processes. 2
Research on ubiquitination has significantly advanced our understanding of cellular physiology and various diseases, contributing to the conceptualization of “protein quality control.” Protein quality control refers to the timely elimination of misfolded or damaged proteins to maintain cellular homeostasis. This process is achieved through targeted protein degradation, often involving molecular chaperones. When molecular chaperones fail to repair a protein, degradation becomes the self‐protective choice of cells. Posttranslational modifications (PTMs) are crucial for controlling protein quality and quantity, with common modifications including phosphorylation, methylation, oxidation, nitration, and ubiquitination, which exponentially expand the proteome. 3 Among these, ubiquitination is the most prevalent PTM involved in protein quality control. The ubiquitination process involves a sequential reaction catalyzed by three enzymes (E1, E2, E3), serving as a regulatory mechanism for numerous cellular activities 4 (Figure 1). By forming conjugates with diverse topologies, ubiquitination can influence the stability, interactions, localization, or activity of thousands of proteins, thereby providing specific signals for broad cellular control. Ub modification of proteins is a critical determinant of cellular fate and function, playing a pivotal role in maintaining human health and cellular homeostasis. Aberrant ubiquitination frequently leads to disease. 5
FIGURE 1.
Overview of the ubiquitination process (by Figdraw). Ubiquitination is an enzyme‐catalyzed reaction that plays a crucial role in various biological processes. Substrates are tagged with ubiquitin and transported to the proteasome for degradation, while ubiquitin molecules are recycled by deubiquitinating enzymes.
Ubiquitination participates in numerous cellular functions, maintaining homeostasis. However, its dysregulation can cause pathological states, including inflammation, or even cell death. Hence, maintaining normal ubiquitination function is crucial for cellular operations, and targeting ubiquitination offers promising avenues for disease treatment. Here, we focus on the pivotal roles of ubiquitination in health and disease. The Ub system comprises multiple components, each collaborating to target a broad array of substrates, regulating diverse cellular processes and functions. While dysregulation of ubiquitination can lead to disease, this very characteristic make each step of ubiquitination process a potential therapeutic target, providing advanced strategies and directions for disease treatment.
2. MECHANISMS OF UBIQUITINATION
2.1. Ub and its structure
Ub was first discovered in bovine thymus during the isolation of thymopoietin, a thymic peptide hormone, and was named for its physiological functions, which were not yet fully understood at the time. Initial amino acid sequencing revealed that Ub is a single polypeptide chain containing 74 amino acids. 6 Subsequently, Wilkinson and Audhya 7 identified a COOH‐terminal sequence of –Arg–Gly–Gly in the active form of Ub, a 76‐amino‐acid protein that is highly conserved across species, from yeast to plants and mammals. Remarkably, only three positions in the Ub structure differ among mammals, yeast, and plants. Structurally, Ub adopts a compact globular fold known as the “ubiquitin fold” or “ubiquitin superfold,” characterized by a helical five‐stranded sheet at the top and an exposed C‐terminal tail that extends to participate in covalent attachment to target proteins. 8 Ub is produced either from the fusion of ribosomal proteins (encoded by UBA52 and RPS27A) or through the action of deubiquitinases on polyubiquitin chains to release free Ub (encoded by UBB and UBC). 9 , 10 As research deepens, the methods currently used for the purification of Ub protein have been refined and generally fall into three categories: epitope tagging, purification through Ub‐binding domains (UBDs), and the use of antibodies. One of the most commonly used antiUb antibodies is the monoclonal antibody FK2. 11
Ub contains seven lysine residues at positions Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63, along with its amino‐terminal, providing eight potential sites for the molecule to form polymeric chains. 12 , 13 Depending on the variability of the lysine linkage sites, Ub chains can be categorized into mono‐ubiquitination or poly‐ubiquitination, with poly‐ubiquitination further subdivided into homogeneous or heterogenous types based on the uniformity of the lysine sites. 14 Protein ubiquitination occurs in two primary forms, each extensively involved in various cellular processes, such as the intricate regulation of inflammatory signaling pathways, 15 modulation of cell death, 16 and control of cell proliferation. 17 Proteins marked by mono‐ubiquitination typically do not undergo degradation; instead, they often regulate protein function and subcellular localization. 18 In contrast, polyubiquitinated proteins are predominantly targeted for degradation via the proteasome, thereby participating in more complex signaling events essential for cellular activities. 19 , 20 Ub chains linked via K48 predominantly target misfolded or aged proteins for degradation through the proteasomal pathway and regulate the turnover of signaling proteins to constrain various immune signaling cascades. 21 In contrast, Ub chains linked via K63, while other Ub chains engage in proteasome‐mediated protein degradation, are predominantly found to participate in cellular signaling processes. 22 Among the various lysine‐linked Ub chains, K33 polyubiquitination is noteworthy for promoting T cell receptor (TCR) ubiquitination and restricting TCR signaling. 23 Lys11‐linked ubiquitination is often found in mixed or branched chains with Lys48 and Lys63 and facilitate proteasomal degradation. For instance, the anaphase‐promoting complex/cyclosome (APC/C) assembles mixed chains containing Lys11, Lys48, and Lys63 bonds through a two‐step mechanism. 24 Lys29 linkage is the most abundant atypical linkage in nonstressed cells and has been predominantly studied in yeast. For example, in the Ub‐fusion degradation (UFD) pathway, Lys29 linkage is formed through the concerted action of homologous to the E6‐associated protein C‐terminal (HECT) ligases Ufd4 and Ubr1, playing a role in cellular protein deposition. 25 , 26
The K27‐linked Ub chain regulates the NF‐κB subunit IKKγ, playing a role in various diseases. For instance, the pathogen Shigella can intercept this Ub chain to suppress immune defense mechanisms of host. The linear Ub chain assembly complex (LUBAC) is a unique structure where one bond is formed through a lysine residue, while the rest of the chain is constructed via the N‐terminal amino group of Ub. LUBAC is composed of SHARPIN, HOIL‐1 (also known as RBCK1), and HOIP. Similarly, linear Ub chains are involved in the activation of the NF‐κB pathway. 27 In the ubiquitination process, in addition to the linkage through lysine residues on Ub itself, Ub can also form covalent bonds via peptide linkage with the N‐terminal α‐amino group, resulting in the formation of linear polyubiquitin chains or N‐terminal Ub fusions. 28 , 29
Ubiquitination is inherently a PTM process; however, Ub itself is a protein, and its seven lysine residues can undergo further modifications, such as phosphorylation, acetylation, and phosphoribosylation. Under the influence of electrostatic and spatial effects, the acetylation of different lysine residues in Ub molecules leads to mono‐acetylated Ub monomers that exert specific impacts on Ub structure, with different acetylated variants connecting to distinct cellular pathways. 30 , 31 The pathogenic Legionella pneumophila effector protein SdeA has been demonstrated to mediate NAD‐dependent and ATP‐independent transfer of Ub to host proteins. Ub phosphoribosylation prevents the activation of conventional ubiquitination cascade enzymes E1 and E2, thereby regulating various cellular activities. 32 This renders the ubiquitination modification process even more intriguing and unpredictable (Figure 2).
FIGURE 2.
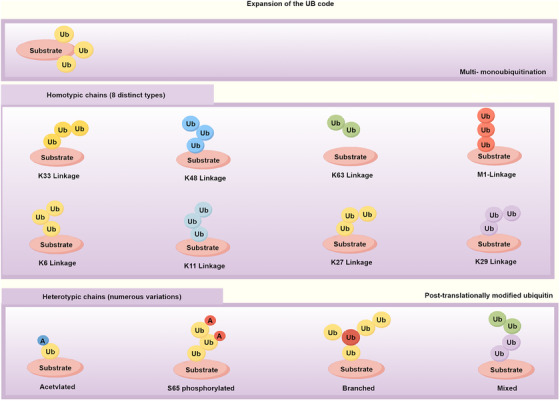
Overview of the ubiquitin chain types (by Figdraw). Different types of ubiquitin chains: monoubiquitination and polyubiquitination. Polyubiquitin chains can be homotypic or heterotypic, indicating the uniformity or diversity in the linkage types within the chain.
2.2. Enzymes involved in ubiquitination: E1, E2, and E3 ligases
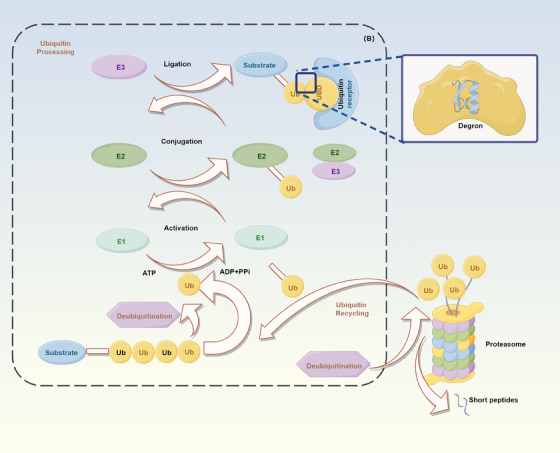
The human genome encodes two E1 enzymes, at least 38 E2 enzymes, and at least 600 E3 enzymes. 33 Generally, enzyme‐mediated ubiquitination can be divided into two processes: the enzymatic cascade and the reversible process. 11 Ub activation involves the formation of a thioester bond between the E1 enzyme and the Ub molecule. The activated Ub is then transferred to an E2 Ub‐conjugating enzyme, which interacts with an E3 Ub ligase to facilitate the transfer of Ub to the lysine residue on the substrate protein. In the presence of ATP, E1 enzyme adenylates the C‐terminus of Ub, then transfers Ub to a conserved Cys residue in E1, resulting in the formation of E1‐Ub thioester, along with the release of AMP and pyrophosphate. 10 The ultimate outcome of the ubiquitination cascade is the linkage of the ε‐amino group of the substrate's lysine side chain to the C‐terminus of Ub. 34
The E1 enzyme is a monomeric protein with a molecular weight of 110−120 kDa, composed of four structural modules: active adenylation domain, first catalytic cysteine half‐domain (FCCH), second catalytic cysteine half‐domain (SCCH), and ubiquitin fold domain (UFD). 35 The catalytic cysteine half‐domains, FCCH and SCCH, include the E1 active site cysteine, which is inserted into each adenylation domain. 36 These domains are crucial for the enzyme's function. Furthermore, the structure includes a four‐helix bundle, which represents a secondary insertion in the inactive adenylation domain immediately following FCCH; and finally, the C‐terminal UFD recruits specific E2, which is essential for subsequent steps in the ubiquitination process. 35 , 37 In all eukaryotes, E1 contains a duplicated sequence of domains derived from bacterial MoeB and ThiF proteins. 38 As a multidomain enzyme, each domain of E1 plays distinct roles in its three catalytic activities: adenylation, thioester bond formation, and thioesterification. The E1 enzyme is regarded as the gatekeeper of the Ub signaling pathway, as it catalyzes the activation of Ub and its transfer to dozens of homologous E2‐conjugating enzymes in a process called E1–E2 thioesterification. 37 During the ubiquitination process, E1 alters the ubiquitination pathway through conformational changes. Ub thioester (Ub(t)) interacts with E1 in an open conformation, while Ub(t) interacts with E2 in a completely different closed conformation, representing two states before and after thiol ester transfer. The SCCH domain can bind two ubiquitins, acting as a molecular switch. During this process, the SCCH domain undergoes a 106° rotation, bringing the catalytic cysteine closer to the Ub C‐terminus. 39
The UBE1‐E2 (Ubc4)/Ub/ATP–Mg complex represents the crystallographic structure generated during the binding of E1 and E2, stabilized by a disulfide bond between their active sites. This structure reveals the combinatorial recognition by the E1 UFD and the Cys domain of E2, where conformational changes in E1 lead to the convergence of the active sites of E1 and E2 during thioester transfer. In this process, E2 binds to the Cys domain of E1 in a fully open configuration within the Ub–E1–E2 complex. 40 The E2 enzyme plays a pivotal role in determining the length and linkage type of the Ub chain. 41 All E2 enzymes possess a core catalytic domain of approximately 150 amino acids, known as the UBC domain, typically comprising four α‐helices and a four‐strand β‐sheet. Some E2s featuring short N‐ and/or C‐terminal extensions that facilitate essential E2 functions. E2s primarily exist in the form of E2–Ub conjugates, being prepared for reaction. In the absence of an E3 ligase, the E2–Ub conjugate exhibits a low rate of Ub transfer, thereby minimizing energy loss. 42 Each E2 enzyme interacts with one E1 enzyme and one or more E3 enzymes. Additionally, E2s may directly bind to target proteins, helping to determine the site and manner of Ub modification on the target protein. 43 Moreover, beyond their role in the ubiquitination cascade, E2 Ub‐conjugating enzymes can regulate the function of other enzymes independently. For example, the E2–Ub conjugate acts as a novel regulator of the OspG effector kinase function in eukaryotic host cells, where OspG is a Shigella effector kinase. 44 , 45 E2‐conjugating enzymes can also enhance the activity of deubiquitinating enzymes (DUBs) like OTUB1 through induced conformational changes. 46
E3 ligases, despite their diversity, are constructed from a small number of basic catalytic cores with various substrate recruitment modules and regulatory elements, allowing them to process different protein substrates and respond to diverse cellular signals. 47 E3 ligases are classified into three main categories: really interesting new gene (RING) ligases, HECT ligases, and RING‐in‐between‐RING (RBR) Ub ligases. RBR ligase contain three tandemly arranged zinc‐binding domains that mediate the direct transfer of Ub from E2 enzymes to target proteins. 48 , 49 Throughout the ubiquitination process, E1 and E2 are not always indispensable. Certain members of the Parabacteroides distasonis family, which interact with multiple Rab small GTPases associated with the endoplasmic reticulum (ER), can catalyze ubiquitination without E1 and E2 enzymes. 50 When E2 and E3 enzymes are linked, E1 is mutually exclusive, meaning that E1 must dissociate from E2 before E2 binding to E3. This is due to the specific residues on the N‐terminal helix of E2 UbcH7, which interact with both the HECT domain‐containing E3 ligase E6AP1 and the RING domain‐containing c‐Cbl2, while also being capable of interacting with the APPBP1–UBA3 of E1. 51 RING E3 ligases contain a RING (or RING‐like) domain responsible for binding to E2 and stimulating Ub transfer, with two conserved Zn2+ cross‐linking the structure. 52 The RING domain binds to the N‐terminal helix of the E2 conjugating enzyme, and it binds even more tightly to the E2–Ub conjugate. In contrast to RING E3 ligases, HECT domain E3 ligases catalyze two distinct reactions: first, a thioesterification reaction where the active site cysteine of E2 is transferred to a cysteine in the HECT domain, followed by a subsequent reaction where the substrate lysine attacks the HECT‐Ub thioester. 53 Additionally, RBR proteins represent a unique family of RING‐HECT hybrid E3s, possessing characteristics of both RING and HECT E3 ligases but catalyzing ubiquitination in a unique manner and autoinhibiting their activity, such as the human homolog ariadne (HHARI) and Parkin, whose RING2 structures contain a catalytic cysteine that mediates ubiquitination in a manner similar to HECT, while RING1 recruits charged E2. 54 The binding of HOIP with HOIL‐1L and SHARPIN in linear Ub chains alleviates this autoinhibition and increases catalytic activity. 55 , 56
Beyond the three types of E3 ligases mentioned above, the recently discovered neuronal‐associated MYCBP2/Phr1 represents a new class of RING‐connected E3s with esterification activity and intrinsic selectivity for threonine over serine. MYCBP2 contains two essential catalytic cysteine residues that transfer Ub to the substrate via thioester intermediates, indicating that higher eukaryotes can also undergo nonlysine ubiquitination. 53 The site where E3 ligases connect with their substrates is referred to as a degron, a specific amino acid sequence of relatively short length (5–20 amino acids) that contains a particular motif for E3 ligases for substrate recruitment. PTMs by kinases and other enzymes on degrons often play a crucial role in determining the timing of E3–substrate interactions and integrating them with upstream events. The presence of degrons ensures a certain degree of precision in the tagging of target proteins. 57 However, degrons are not essential for substrate recognition, as certain proteins can still bind to E3 enzymes through their native structures. 47 , 58
Due to the presence of deubiquitinases (DUBs), ubiquitination is a reversible protein modification. Currently, approximately 100 DUBs have been identified. 59 DUBs can exhibit internal, external, or global‐type activities by cleaving within the chain, cutting from one end of the chain, or removing the entire chain at once. 60 Given the diversity and complexity of Ub chains, recognizing various Ub chains by DUBs is a challenging task. The human genome encodes over 70 DUBs, which can be classified into seven types: USPs (Ub‐specific proteases), UCHs (Ub carboxyl‐terminal hydrolases), MJDs (proteases containing the Machado‐Josephin domain), OTUs (ovarian tumor proteases), MINDYs (motifs interacting with Ub‐like new DUB family), ZUP1 (zinc‐finger Ub peptidase), and JAMMs (JAB1, MPN, MOV34 family). 60 The first six DUB families are cysteine peptidases, while JAMMs are zinc metallopeptidases. 61 Overall, most human DUBs are thiol‐based proteases, typically possessing a catalytic triad that includes a catalytic cysteine involved in nucleophilic attack, a neighboring basic histidine that lowers the pKa of cysteine to enhance its nucleophilicity, and usually a third acidic residue—often aspartate, asparagine, or glutamate—that further polarizes the basic histidine. 60 In essence, most DUB‐catalyzed reactions involve the proteolytic cleavage of the bond between a lysine ε‐amino group and a carboxyl group at the C‐terminus of Ub. DUBs recognize the R72 residue at the C‐terminal tail of Ub to form an acyl intermediate, which is then hydrolyzed by a water molecule to complete the catalytic cycle. 62 , 63 By cleaving Ub attached to the substrates or within the Ub chains, DUBs play a crucial role in regulating fundamental cellular processes, either as switches to remove Ub signals or as rheostats to fine‐tune the amount and type of ubiquitination. 64 Based on this structural foundation, the physiological functions of DUBs are diverse: acquiring free Ub; stabilizing proteins to prevent degradation; and trimming Ub chains to edit the form of Ub modification. 65
In summary, the ubiquitination process begins with the formation of a thioester bond between the Cys residue of the E1 enzyme and the C‐terminus of Ub, a process that requires ATP to release energy. Once E1 is activated, Ub is transferred to the Cys residues on over 40 different E2 Ub‐conjugating enzymes. Last, E3 ligases transfer the Ub molecule to a specific substrate. 66 Additionally, DUBs serve as “recycling stations” during the ubiquitination process.
2.3. Ub‐binding proteins and receptors
UBDs are modular elements within proteins that allow for noncovalent binding to Ub, enabling interactions that facilitate mutual regulation. 67 Due to the reversible nature of ubiquitination, the binding between UBDs and Ub is relatively weak, especially in monoubiquitin chains (with kDa values ranging from 10 to 500 µm). This weak interaction facilitates the assembly and disassembly of Ub and substrates, benefiting various biological processes. 67 Given that the structure of Ub is highly conserved in organisms, the structural diversity and complexity of UBDs are beneficial to multifunctionality of ubiquitination. The existence of different UBDs adds more possibilities to ubiquitination based on various Ub chain structures. According to their structures, UBDs can be classified into several types, including α‐helices, zinc fingers, pleckstrin homology domains, and Ubc domains present in E2 enzymes, among others. 68 These domains reside in different proteins and perform various functions. For instance, UBDs containing α‐helical structures often bind to hydrophobic patches on the β‐sheet of Ub molecules. 69 Similarly, β‐sheets in E2 enzymes, such as the E2 Ub‐conjugating enzyme UBCH5c, can also bind to Ub molecules. 70 The first characterized Ub‐binding site was identified in the proteasomal subunit S5a/RPN10 protein. 71 A sequence motif known as the Ub‐interacting motif (UIM) was identified through hidden Markov modeling and iterative database searches based on the S5a sequence, representing a genuine Ub‐binding motif. 72 Ub‐associated (UBA) domains can directly bind to Ub and are common sequence motifs shared by protein subsets involved in ubiquitination or deubiquitination reactions. 73 The discovery of UIM and UBA motifs marked the beginning of research into UBDs. Proteins typically contain multiple copies of UBD structures; for example, UIMs often occur in tandem, and such tandem arrangements can exhibit different functions. 72 A prominent example is seen in proteins involved in various signaling pathways, where UIM‐containing regions bind to Ub, thereby mediating signal transduction. This is seen in proteins such as Eps15, Eps15R, and epsins, which are induced by active tyrosine kinases. 74 The role of ubiquitination in various signaling pathways will be discussed in detail in the following sections.
In addition to UBDs, proteasomes possess receptors that facilitate the recognition of ubiquitinated proteins, guiding them to the proteasome for degradation. In yeast proteasomes, Rpn10, Rpn13, Rad23, Dsk2, and Ddi1 have been identified as key components that assist in the docking of Ub molecules with the proteasome. In mice, RPN10 and RAD23 are essential, suggesting that they may have more complex and unique roles. 75 For instance, the two UIMs of RPN10 in yeast (corresponding to S5a in humans) adopt a helical configuration, capable of binding polyubiquitin chains. Due to the separation of the two UIMs by flexible linker regions, they also possess the ability to independently bind monoubiquitin. 76 , 77 The yeast protein Rad23, which belongs to a family of proteins containing an N‐terminal Ub‐like (UBL) domain, can bind to the proteasome. Experimental studies using model proteins have shown that when the internal unstructured loop of Rad23 is sufficiently long, it can bind to the proteasome. 78 Ubiquitinated proteins can also be degraded through the autophagy pathway, where specific receptors recognize and bind Ub. Autophagy receptors such as p62, NBR1, OPTN, and NDP52 can simultaneously bind Ub and the cargo to be degraded, leading to the initiation of autophagy. 79 For example, p62 contains a UBA domain, and ubiquitination of p62 disrupts the dimerization of the UBA domain, enhancing its ability to selectively recognize polyubiquitinated cargoes for autophagy. 80 The mechanism of p62 ubiquitination involves acetylation of K420 and K435 in the UBA domain, with acetylation of K435 directly increasing the affinity of the UBA–Ub interaction. 81 Similar to p62, other autophagy‐related proteins, such as NBR1, OPTN, and NDR52, also contain UBA domains in their structures, granting them the ability to bind Ub. 82
3. REGULATION OF CELLULAR FUNCTIONS BY UBIQUITINATION
3.1. Protein degradation via the Ub–proteasome system
In the 1930s, isotope labeling techniques confirmed the lysosome as the principal site for cellular protein degradation, mediated by resident acid‐dependent proteases. However, this view was challenged with evidence that the half‐lives of most cellular proteins were insensitive to lysosomal alkalinization. 83 Until the discovery of the Ub–proteasome pathway in 1984, the scientific community was perplexed by the fact that ATP was required for the degradation of proteins. 84 The proteasome pathway primarily degrades short‐lived cytosolic and nuclear proteins as well as misfolded proteins from the ER. Autophagy engulfs large protein aggregates and damaged organelles by forming double‐membraned autophagosomes, which subsequently fuse with lysosomes to degrade the substrates. 85 The UPS was initially identified from an ATP‐dependent protein hydrolysis system in reticulocytes. 86 The proteasome, a multisubunit protease now known as the 26S proteasome, 58 functions as a compartmental protease within the AAA+ (ATPases associated with various cellular activities) protein family. It utilizes ATP hydrolysis to unfold substrates and translocate the denatured polypeptides into an internal degradation chamber for proteolytic cleavage. This capability of unfolding native structures allows the proteasome to regulate the eukaryotic proteome, degrading many regulatory proteins in addition to damaged or misfolded peptides. 87 The autophagic and proteasome pathways are interconnected. Inhibition of autophagy leads to increased levels of proteasomal substrates. For instance, studies have demonstrated that upon autophagy inhibition, p62 (also known as A170/SQSTM1) accumulates and subsequently inhibits the clearance of ubiquitinated proteins by delaying their delivery to the proteasome. 85 Studies have shown that the mechanisms by which misfolded proteins from different cellular compartments are transported to the proteasome for degradation vary. For cytoplasmic proteins, degradation requires tagging with mixed Ub chains linked through K48 and K11, followed by interaction with specific chaperone proteins. In contrast, in the nucleus, the proteasomal degradation of misfolded proteins primarily requires K48‐linked Ub chains and recognition by Ub protein Dsk2 for subsequent entry into the proteasome. 88 The proteasome, formally known as the 26S proteasome, is a complex enzyme composed of a 20S core particle (CP) and 19S regulatory particles (RPs). These two particles work together to regulate protein degradation within the proteasome. The hydrolytic pathway of the proteasome resides within the cavity of the 20S CP, structurally capped at one or both ends by the 19S RPs, thus controlling access to the cavity. 87 Spatially, the active sites of the CP and RPs are separated but interconnected. In yeast, the proteasome‐associated protein Ecm29 maintains the CP–RP complex, with ATP or ADP being indispensable for the stability of this complex. 89 Binding of ubiquitinated proteins to the 26S complex, although of high affinity, is reversible and can be disrupted by competition with other UBDs or high salt concentrations. This binding can occur even at low temperatures, such as 4°C. However, tighter binding requires ATP hydrolysis and the presence of loosely folded regions in the substrate protein, as the energy from ATP hydrolysis is converted into mechanical force. 90 Structural studies, both in vivo and in vitro, have shown that the 26S proteasome exhibits multiple conformations, categorized into the substrate‐free (s1) state and the substrate‐processing (s3‐like) state. The s1 state is the predominant conformation of the ATP‐bound proteasome, whereas the s3‐like state is more conducive to processive degradation. 91 The interaction of substrates with the AAA+ motor pore loops of the 26S proteasome drives this overall conformational switch, a process requiring intricate coordination. Substrates with short or low‐complexity initiation regions rapidly enter the central channel of the proteasome but may fail to stably engage with the AAA+ pore loops, leading to their swift release and a significant increase in substrate KM. This is mainly due to the increased off‐rate of substrates following initial Ub interaction. 91 Recent research has revealed that even uncapped 20S proteasomes are capable of cleaving partially unfolded proteins or proteins with disordered regions. This degradation pathway is strictly regulated by a family of catalytic core regulators. 92 Therefore, structural changes in the proteasome are crucial for its ability to bind target proteins and facilitate their degradation.
3.2. Ub‐mediated protein trafficking and localization
One critical function of ubiquitination is the regulation of protein trafficking and endosomal sorting. One illustrative example of ubiquitination's role in protein trafficking is its involvement in the secretion of secretory proteins. These proteins begin their journey in the ER, where they are folded and assembled into oligomeric complexes. Following a series of modifications, they are packaged into vesicles and transported to the Golgi apparatus. 93 Misfolded polypeptides, however, can be retrotranslocated back to the cytosol and degraded by the UPS, a process known as ER‐associated degradation (ERAD) (Figure 3A). 94 The ER contains a variety of autophagy adaptors, such as transmembrane receptors FAM134B and UBX2, and soluble receptors like p62, which can bind ubiquitinated substrates (e.g., TRIM13) and mediate ER‐phagy. In this process, Hrd1, an E3 Ub ligase, facilitates the translocation of ERAD substrates to the cytosol, reorienting them through the ER membrane via the central pore of the Type II AAA‐ATPase Cdc48 (in yeast) or VCP (also known as p97, in mammals), promoting their extraction into the ERAD pathway. Misfolded proteins are subsequently degraded into short peptides and transported into the cytosol or nucleus with the assistance of cytosolic chaperones and transport factors. 95 , 96 Furthermore, the unfolded protein response and ERAD interact in a coordinated manner with the UPS and autophagy to mitigate protein misfolding or its consequences. 97 Upon leaving the ER, the packaged proteins are transported in vesicles or specialized tubules to the Golgi apparatus. 98 Ubiquitination can increase the size of the export vesicles by approximately fivefold, enabling the accommodation of cargoes, such as collagen, which are 300−400 nm in size, into vesicles with a diameter of only about 60−80 nm. Cul3–Klhl12 is a regulatory factor for COPII coat formation and catalyzes the monoubiquitylation of the COPII‐component SEC31, driving the assembly of large COPII coats 99 (Figure 3B).
FIGURE 3.
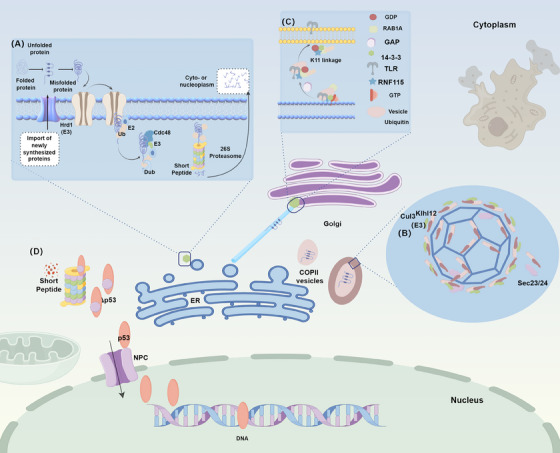
Ubiquitin‐mediated protein trafficking and localization (by Figdraw). (A) Ubiquitin‐mediated protein degradation in the endoplasmic reticulum (ER) involves tagging misfolded proteins for transport to the proteasome. (B) Ubiquitination facilitates the reorganization of COPII vesicles, allowing them to form larger vesicles capable of accommodating substrates. (C) In the Golgi apparatus, ubiquitination mediates the relocation and transport of proteins. (D) Ubiquitin‐mediated transport of proteins within the nucleus ensures proper protein localization and function. Dub, deubiquitinases; NPC, nuclear pore complex; ER, endoplasmic reticulum.
Beyond its established functions, Ub has been identified as a key protein sorting signal, facilitating the transport of damaged and downregulated proteins to the lysosome. This process operates via a unified mechanism in which Ub attachment signals proteins for clathrin‐mediated internalization and endosomal sorting. For instance, studies in mammalian cells have demonstrated that cell surface proteins such as the epidermal growth factor receptor (EGFR) and the epithelial sodium channel directly associate with Ub ligases, supporting the role of Ub as a transport signal within endocytic pathways. Within this model, Ub, as an independent sorting signal, provides a Ub surface for endosomal receptor binding. The underlying mechanisms of Ub‐dependent sorting involve clathrin‐mediated internalization, followed by sorting into multivesicular bodies through the endosomal sorting complex required for transport (ESCRT) system. 100
Secretory proteins undergo further packaging and sorting within the Golgi apparatus. Ubiquitination plays a pivotal role in this process as a sorting signal, facilitating the binding of Ub to receptors on the Golgi membrane and the accumulation of proteins in clathrin‐coated vesicles. In yeast, proteins such as Gga1 and Gga2 (corresponding to human GGA1, 2, 3) regulate membrane protein localization by modulating the ubiquitination process, and they can also cooperate with the ESCRT machinery for protein sorting. 101 The trans‐oligomerization of Golgi proteins GRASP55 and GRASP65 is essential for maintaining Golgi stacking. Ubiquitination of GRASP55 has been found to target it for proteasomal degradation, which rescues phenotypes such as disrupted Golgi structure, reduced protein secretion, and dendritic branching defects. 102 Another compelling example is the post‐ER transport of Toll‐like receptors (TLRs). The subcellular localization and intracellular trafficking of TLRs are crucial for the regulation of TLR‐mediated antimicrobial immunity and autoimmune responses. The E3 Ub ligase ring finger protein (RNF)115 inhibits post‐ER transport of TLRs and TLR‐mediated immune responses by catalyzing the ubiquitination of small GTPases RAB1A and RAB13. The 14‐3‐3 chaperone binds to AKT1‐phosphorylated RNF115, promoting its localization to the ER and Golgi apparatus. This indicates that ubiquitination can alter the trafficking of TLRs post‐ER, thereby influencing cellular activities. 103 Ubiquitination also plays a role when mature proteins leave the Golgi apparatus. Coronin 7 is essential for the budding of transport vesicles derived from the Golgi apparatus, and the Ub ligase Ubr4 impairs the export of secretory proteins from the Golgi by promoting Coronin 7 expression. This function is critical for circadian synchronization and signal processing at the circuit level. 104 In summary, ubiquitination serves to control protein quality and quantity by participating in ERAD degradation, vesicular transport, and the sorting and processing of proteins within the Golgi apparatus (Figure 3C).
Beyond its role in the trafficking of secretory proteins, ubiquitination also regulates the transport of nuclear proteins. Nuclear localization signals (NLSs) are specific topological amino acid sequences within the protein that can be recognized by import proteins, facilitating their transport into the nucleus. This importin‐mediated nuclear transport mechanism is a promising avenue for therapeutic strategies targeting the nucleus. 105 In the Nipah virus matrix protein (NiV‐M), a bipartite NLS has been identified that can undergo mono‐ubiquitination to regulate protein export. Ub overexpression enhances NiV‐M budding. 106
The transcription factor p53 is one of the most commonly mutated tumor suppressors. The mutant form, Δp53, can sequester wild‐type p53, resulting in its retention in the cytoplasm. Δp53 can be ubiquitinated and degraded by mouse double minute 2 homolog (MDM2), indirectly regulating the nuclear localization of p53, thereby affecting tumorigenesis. 107 Research has found that proteasome inhibition, which leads to the accumulation of ubiquitinated TDP‐43 at lysine 95 within its NLS, reduces poly‐GA‐dependent mislocalization of TDP‐43, offering significant therapeutic potential in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia 108 (Figure 3D).
Ubiquitinated proteins can directly interact with nuclear transport proteins that possess specific UBDs, such as importins. This interaction facilitates the translocation of ubiquitinated proteins from the cytoplasm to the nucleus. For instance, in renal clear cell carcinoma cells, the subcellular relocalization of circPPAP2B is dependent on the nondegradative ubiquitination of heterogeneous nuclear ribonucleoprotein C (HNRNPC) and the stabilization of the HNRNPC/vimentin/importin α7 ternary complex, thereby promoting cancer cell metastasis. Key Ub enzymes involved in this process include TRIM and USP10. 109 Ubiquitination also regulates the abundance of plasma membrane receptors and transport proteins, affecting the endosomal degradation of cargoes and the auxin efflux transporter PIN2‐GFP in vivo. In Arabidopsis, OTU 11 and OUT12 are plasma membrane‐localized DUBs. They bind phospholipids through multiple motifs in their OTU domains. The DUB activity of OUT11 and OUT12 on K11‐, K6‐, and K63‐linked Ub is stimulated by association with anionic lipid‐containing membranes. 110
3.3. Ubiquitination and signaling pathways
The nuclear factor κB (NF‐κB) family comprises transcription factors that play a critical role in various cell responses and are regulated by numerous mechanisms to maintain tolerance and cell homeostasis. 111 The activation of NF‐κB is intrinsically linked to ubiquitination. Although NF‐κB is widely expressed, it is typically held inactive in the cytoplasm by members of the IκB inhibitory protein family. Rapid degradation of IκB via the Ub–proteasome pathway permits NF‐κB to translocate into the nucleus. The NF‐κB family consists of five members: p65 (REL‐A), c‐REL, REL‐B, p50, and p52, all of which contain the REL homology domain (RHD) essential for DNA binding, dimerization, nuclear localization, and IκB interaction. Additionally, p65, c‐REL, and REL‐B possess transactivation domains (TAD) necessary for gene activation. The precursor proteins p105 and p100, which contain 5 to 7 ankyrin repeats, undergo proteolytic processing upon activation, leading to the generation of p50 and p52, respectively. 111 When the RHD is masked, NF‐κB cannot translocate to the cytoplasm to exert its functions, with ubiquitination serving as the key to unlocking this inhibition 112 (Figure 4A,B).
FIGURE 4.
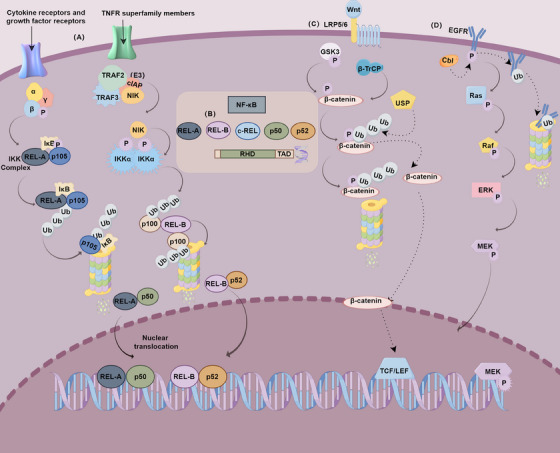
Ubiquitination in key signaling pathways (by Figdraw). (A) Ubiquitination in canonical and noncanonical NF‐κB signaling pathways, regulating the activation and translocation of NF‐κB subunits. (B) Types and structures of NF‐κB, including its various subunits and their functional domains. (C) Ubiquitination in the Wnt‐β‐catenin signaling pathway, influencing the stability and activity of β‐catenin. (D) Ubiquitination in the EGFR signaling pathway, affecting receptor trafficking, degradation, and downstream signaling. TCF, T‐cell factor; LEF, lymphoid enhancer factor; LRP, lipoprotein receptor‐related protein; GSK3, glycogen synthase kinase 3; β‐TrCP, beta‐transducin repeat‐containing protein; RHD, Rel homology domain; TAD, transactivation domains; NIK, NF‐κB‐inducing kinase; cIAP, cellular inhibitor of apoptosis; IKK, IκB kinase; EGFR, epidermal growth factor receptor; P, phosphorylation; Ub, ubiquitin.
The activation of NF‐κB can be divided into canonical and noncanonical pathways. The canonical pathway mediates the activation of NF‐κB1 p50, REL‐A, and c‐Re, while the noncanonical pathway selectively activates NF‐κB members sequestered by p100, primarily NF‐κB2 p52 and REL‐B. Both activation pathways require ubiquitination. In the canonical pathway, IκB kinase (IKK), composed of α and β subunits and a necessary regulatory subunit γ, phosphorylates IκB. The phosphorylated IκB is then recruited by the F‐box protein beta‐transducin repeat‐containing protein (β‐TrcP) to the Ub ligase complex for polyubiquitination and selective degradation, a process independent of the proteasome. The noncanonical pathway selectively responds to a subset of tumor necrosis factor receptor superfamily members. The pathway involves E3 ligases, particularly cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) targeting NF‐κB‐inducing kinase (NIK) for ubiquitination and regulation. NIK phosphorylates and activates IKKα, which subsequently phosphorylates the carboxy‐terminal serine residues of p100, triggering the selective degradation of the C‐terminal IκB‐like structure of p100, leading to the generation of p52 and the nuclear translocation of p52 and REL‐B. 113 , 114 The regulation of NF‐κB activity is also highly dependent on ubiquitination and deubiquitination. For instance, the E3 Ub ligase, PDZ and LIM domain protein 2, inhibits NF‐κB transcriptional activity by removing RelA from DNA‐binding sites and mediating its degradation. 115 Similarly, A20, encoded by Tnfaip3, is a direct NF‐κB target gene that plays a key role in the negative feedback regulation of canonical NF‐κB. A20 contains a DUB domain and a C2–C2 zinc finger E3 Ub ligase domain. The DUB domain of A20 removes K63‐linked Ub chains from RIP1, TRAF6, and NEMO, leading to the disassembly of the IKK complex and downregulation of the inflammatory response. 116
The Wnt/β‐catenin signaling pathway plays a pivotal role in embryogenesis and development and is frequently observed in tumorigenesis, particularly in colorectal cancers. The canonical Wnt signaling pathway is activated when Wnt ligands bind to the Frizzled (Fz) family of seven‐transmembrane receptors and their coreceptors, lipoprotein receptor‐related protein (LRP)5/6, on the cell surface, leading to the recruitment of Dishevelled scaffold proteins to the receptor complex. The regulation of cytosolic β‐catenin through protein degradation pathways is the key to Wnt signaling. 117 The primary E3 Ub ligase responsible for regulating β‐catenin stability is β‐TrCP. Phosphorylation of β‐catenin by glycogen synthase kinase 3 (GSK3) at serines 33 and 37 creates a recognition site for β‐TrCP, which then ubiquitinates β‐catenin, leading to its degradation. Upon translocation into the nucleus, β‐catenin activates T‐cell factor (TCF) and lymphoid enhancer factor (LEF), leading to the transcription of key target genes such as cyclin D1 and c‐Myc. 118 Loss of RNF43 and ZNRF3 induces rapid growth of adenomas, as RNF43 and ZNRF3 target Wnt receptors for degradation by selectively ubiquitinating Fz receptors, thereby attenuating Wnt signaling. 119 Conversely, the DUB USP46 complex acts as a positive regulator of Wnt signaling. Wnt signaling promotes the binding of the USP46 complex to the Wnt coreceptor LRP6 at the cell surface, stabilizing LRP6 on the cell surface. This interaction facilitates LRP6 signalosome assembly by removing obstructive Ub chains, which is essential for Wnt‐dependent intestinal organoid viability 120 (Figure 4C).
Additionally, the Ub ligase Mule inhibits the Wnt pathway by suppressing c‐Myc, thereby controlling unwanted proliferation and stem cell expansion in colorectal cancer. 121 In the regulation of the intestinal ecosystem, the CUL4B–RING Ub ligase (CRL4B) targets immune‐related GTPase family M member 1 (IRGM1) for proteasomal degradation. The absence of Cul4b results in reduced self‐renewal of intestinal stem cells and diminished lineage differentiation towards secretory progenitor cells through downregulation of Wnt signaling. 122 Another example of ubiquitination in regulating the Wnt signaling pathway is JADE‐1, which ubiquitinates both phosphorylated and nonphosphorylated β‐catenin. This modulation of β‐catenin stability during both Wnt‐off and Wnt‐on phases supports the involvement of Jade‐1 and Wnt signaling in renal tumorigenesis. 123 Furthermore, the EGF signaling pathway is deeply involved in ubiquitination processes to maintain protein homeostasis. and itself is also influenced by ubiquitination. In mammalian cells, treatment with EGF leads to rapid modification of ligand‐activated EGFR with K63‐linked Ub chains. 124 LRIG1, a negative regulator of EGF in mammals, is upregulated and upon EGF stimulation, which is accompanied by enhanced EGFR ubiquitination and degradation. c‐Cbl, an E3 Ub ligase, ubiquitinates both EGFR and LRIG1, leading to their degradation 125 (Figure 4D).
4. UBIQUITINATION IN HEALTH
Ubiquitination, due to its widespread distribution, is implicated in a multitude of biological processes. Its prominent role in regulating protein degradation underscores its importance in maintaining protein quality and quantity, thereby supporting protein homeostasis. The intricate architecture of proteins contributes to their diversity and underscores their significance as fundamental components of living systems. Ub, a small molecule ubiquitously present across various cell types, tags and directs specific proteins for degradation in response to cellular signals. This process facilitates the modification of essential proteins implicated in nuclear DNA repair, cell cycle regulation, and inflammation control. Consequently, ubiquitination emerges as a pivotal regulatory mechanism, contributing to the maintenance of protein homeostasis, DNA repair, cell cycle regulation, and the modulation of inflammatory signaling pathways. This, in turn, establishes a stable foundation for the proper functioning of cellular processes.
4.1. Maintenance of protein homeostasis
Protein homeostasis, or proteostasis, is maintained through a complex network of mechanisms that ensure protein quality and support the evolutionary diversity of protein biological functions. 126 Ubiquitination plays a key role in regulating protein quality, quantity, and spatial localization, achieving a state of equilibrium for these proteins. 127 Protein imbalance can lead to abnormal aggregation of normal or aberrant proteins, triggering a range of related diseases and aging. 128 Proteins, with their diverse structures and functions, are distributed throughout the cell and perform different roles by altering their abundance, distribution, and activity. Proteostasis can be categorized into maintenance under normal and stress conditions. 129 , 130 To adapt to various cellular states and external environments, protein homeostasis under normal conditions is reflected by the maturation of proteins through other types of PTMs. Under stress conditions, the UPS mediates the proteolysis of soluble ubiquitinated proteins (in the presence of chaperones) that are misfolded, oxidized, mutated, or otherwise damaged, predominantly participating in the organism's self‐protection mechanism. 131 For instance, Ub inhibits the maturation of amyloid precursor protein (APP) by sequestering it in early secretory pathways, primarily within the Golgi apparatus. This sequestration significantly delays the proteolytic processing of APP by secretases and the proteasome, which is crucial for the onset of late‐onset Alzheimer's disease (AD). 132 Additionally, under acute ER stress, prion proteins are prevented from mislocalizing to the ER and are directed for cytosolic degradation, minimizing prion protein secretion and benefiting the cell. This degradation is attributed to the UPS. 133 The deeper mechanism involves the disposition of such mislocalized proteins depending on the BAG6 complex (such as BAG6, TRC35, and UBL4A). The BAG6 complex recognizes mislocalized proteins, recruits the E2 conjugating enzyme UbcH5 and an unidentified E3 ligase, thus selectively promoting their rapid ubiquitination and proteasomal degradation. 134 Beyond degrading harmful proteins under stress, ubiquitination also degrades excess normal proteins, contributing to cellular homeostasis. Many proteins function within multisubunit complexes requiring proper assembly, and the degradation of unassembled soluble proteins (termed unassembled soluble protein degradation, USPD) necessitates the Ub‐selective chaperone p97, its cofactor nuclear protein localization protein 4 (Npl4), and the proteasome. HUWE1, a protein containing domains homologous to the E6‐AP carboxyl terminus, serves as the Ub ligase for substrates with exposed hydrophobic regions. 135 As previously mentioned, ERAD is another way to control protein homeostasis, capable of degrading functional proteins such as rate‐limiting metabolic enzymes. An example of ERAD controlling ligand‐dependent abundance is the feedback regulation of 3‐hydroxy‐3‐methylglutaryl‐CoA reductase, a key enzyme in the cholesterol biosynthesis pathway. 136 Among the myriad mechanisms maintaining protein homeostasis, modulation through ubiquitination is a relatively late stage of regulation, directly controlling protein levels. Researchers have found that the regulation of epidermal growth factor (EGF) signaling is associated with mechanisms that maintain protein homeostasis. EGF signaling shifts the strategy of maintaining protein homeostasis from a chaperone‐based approach to one involving enhanced UPS activity and polyubiquitination, thereby reducing protein aggregation. 137 Therefore, although multiple strategies are required to maintain protein homeostasis, regulation by the UPS is a more direct method of controlling protein quantity.
4.2. Role in DNA repair and genome stability
DNA double‐strand breaks (DSBs) are one of the most severe types of DNA damage. In response, mammals have evolved complex cellular pathways to facilitate DNA repair and maintain genome stability: the homologous recombination (HR) and nonhomologous DNA end joining pathways. 138 During DSBs, the breast cancer type 1 (BRCA1)–BRCA1‐associated RING domain protein 1 (BARD1) complex, which possesses E3 ligase activity, aids in DSB repair through HR by promoting nucleolytic resection at DNA ends. 139 The BRCA1–BARD1 complex localizes to damaged chromatin post‐DNA replication and catalyzes the ubiquitination of histone H2A and other cellular targets. The RING domains within BRCA1–BARD1 orient the E2 Ub‐conjugating enzyme atop nucleosomes in a dynamic conformation, preparing for the transfer of Ub to the flexible C‐terminal tails of H2A and its variant H2AX. Concurrently, the nuclear E3 Ub ligase RNF168 rapidly ubiquitinates histone deacetylase 6 (HDAC6) at lysine 116, targeting it for degradation. Under the modification by RNF168, ubiquitinated lysine 15 on H2A binds to the DNA repair protein 53BP1, thus inhibiting 53BP1 and promoting HR repair of DSBs. 140 , 141 , 142 , 143 Moreover, the nucleolytic resection induced by BRCA1–BARD1 also recruits another tumor suppressor complex, BRCA2–PALB2, and the recombinase RAD51 to single‐strand DNA templates, further facilitating DSB repair. 139 HR repair of DSBs is also mediated by MDC1. RNF8 and RNF168 assist DNA repair proteins in reaching sites of DNA damage, and these two E3 ligases are connected by Lethal (3) malignant brain tumor‐like protein 2 (L3MBTL2). MDC1 recruits this crucial factor, which is subsequently ubiquitinated by RNF8, and the ubiquitinated L3MBTL2 then recruits RNF168. 144 Ubiquitination‐associated DNA repair through recombination is also linked to the cell cycle. CDK2 can phosphorylate RNF4 at T26 and T112, enhancing RNF4 E3 ligase activity, which is important for MDC1 degradation during the S phase and proper HR repair. 145
Fanconi anemia (FA) is a prototypical disease associated with DNA crosslink damage. The excision of “toxic replication” intermediates is the key to the pathogenesis of FA, which is also pivotal for the repair of DNA interstrand crosslinks. 146 Within the FA network, FANCD2 and FANCI function together to facilitate the repair of DNA crosslinks. These proteins form a DNA‐binding heterodimeric ID2 complex and undergo mono‐ubiquitination when cells are exposed to DNA crosslinking agents. 147 Studies have shown that Ub is positioned at the interface of FANCD2 and FANCI, acting as a molecular pin to capture the closed conformation of ubD2‐I (a critical factor in DNA crosslink repair) clamped onto the DNA. Thus, the mono‐ubiquitination of the FANCD2–FANCI heterodimer is a critical step in the FA DNA crosslink repair pathway. 148 The interaction between FANCD2 and FANCI within the heterodimer protects monoubiquitinated FANCD2 from polyubiquitination and subsequent proteasomal degradation. 149 RAD18, an E3 ligase, binds to FANCD2 and is essential for the effective mono‐ubiquitination and chromatin localization of both FANCD2 and FANCI. RAD18 facilitates the recruitment of FANCI and FANCD2 to chromatin and their mono‐ubiquitination during the S phase. 150
In addition to its role in repairing DNA DSBs, ubiquitination is also involved in the repair of other types of DNA damage, such as transcription‐coupled nucleotide excision repair (TC‐NER). TC‐NER is initiated when elongating RNA polymerase II (RNAPIIo) stalls at sites of DNA damage. In the UV‐sensitive syndrome (UVSS), the UVSSA protein interacts with the TC‐NER mechanism and stabilizes the ERCC6 complex (associated with Cockayne syndrome genes). UVSSA also promotes the ubiquitination of RNAPIIo stalled at DNA damage sites, thereby initiating the TC‐NER process. 151 A single DNA damage‐induced ubiquitination site has been identified on RNAPIIo at RPB1‐K1268, which plays a critical role in regulating transcription recovery and DNA damage resistance. The ubiquitination of RPB1‐K1268 is crucial for the repair of DNA damage and resolving transcriptional stalling. Mice with a knock‐in mutation at RPB1‐K1268R exhibit shortened lifespans, premature aging, and neurodegeneration. 152 NER is another significant pathway for DNA damage repair that influenced by ubiquitination. DNA–protein crosslinks (DPCs) are large cytotoxic DNA lesions formed following exposure to chemotherapeutic agents and environmental chemicals. In cells proficient in NER, DPCs undergo K48‐linked polyubiquitination and are removed via a proteasome‐dependent mechanism, whereas in NER‐deficient cells, DNA‐conjugated proteins undergo K63‐linked ubiquitination. 153 Furthermore, recent studies have indicated that members of the SWI/SNF and INO80 families, along with poly (ADP‐ribose) polymerase 1 (PARP1), are involved in NER. The H2A‐Ub‐binding protein ZRF1 and the endonuclease DICER influence chromatin conformation through PARP1. Overall, Ub signaling cascades are closely associated with chromatin functions. 154 Besides Ub cascades, deubiquitination also plays a role in regulating genomic stability. For example, the deubiquitinase USP15 interacts with and deubiquitinates PARP1, promoting its stability and thereby stimulating DNA repair, genomic stability, and the proliferation of triple‐negative breast cancer cells. 155 In the context of mismatch repair (MMR), the proteasomal degradation of cyclin‐dependent kinase (CDK) inhibitor p21, which competes with MMR proteins for binding to proliferating cell nuclear antigen, facilitate MMR. This mechanism primarily operates during the G1/S transition, where the timely cullin–RING Ub ligase (CRL)‐dependent degradation of cyclin D and p21 allows for effective MMR activity to correct DNA replication errors 156 (Figure 5).
FIGURE 5.
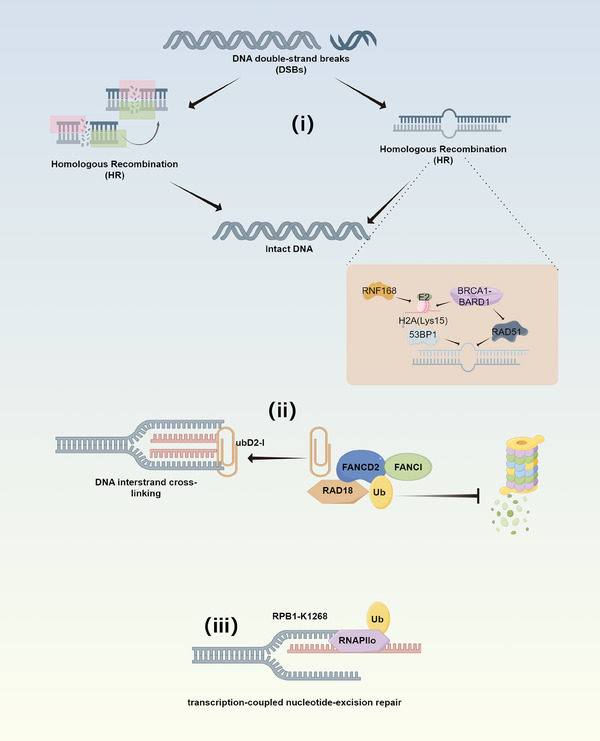
Ubiquitination in DNA repair (by Figdraw). (i) Involvement in DNA double‐strand break repair; (ii) role in DNA crosslink repair; (iii) participation in transcription‐coupled nucleotide excision repair. RNF, ring finger protein; BRCA1, breast cancer type 1; BARD1, BRCA1‐associated RING domain protein 1; Ub, ubiquitin.
4.3. Regulation of cell cycle progression and proliferation
The primary drivers of cell cycle progression are the sequential activations of CDKs, which are regulated, in part, through the Ub‐mediated proteolysis of their cyclin partners and kinase inhibitors (CKIs). Studies have shown that in eukaryotic cells, the TCR/cyclosome (APC/C) is responsible for the ubiquitination and subsequent proteasomal degradation of many CDK regulatory factors, ensuring that the cell cycle proceeds in a timely and precisely regulated manner. 157
In eukaryotes, the APC/C is a multisubunit E3 Ub ligase that regulates the Ub‐independent proteolysis of specific cyclins at different stages of mitosis to coordinate chromosome segregation and transition into G1. During mitosis, CDKs and polo kinase control the activation of APC/C mediated by cell‐division cycle protein 20 homologue (Cdc20) and E‐cadherin gene (Cdh1). 158 , 159 Inhibition of Cdc20 phosphorylation leads to premature activation of APC/C‐Cdc20, causing instability in several substrates, including cyclin B1 and A2, resulting in an extended G2 phase and delayed entry into mitosis. 160 Studies have identified K11‐linked Ub chains as important signaling entities in cell cycle control. Their efficient formation depends on specific interaction between E2 Ub enzyme UbcH10 and APC/C. The APC/C targets cell cycle proteins for degradation by the 26S proteasome. During the G1 phase, APC/C inactivation involves the degradation of its specific E2 UbcH10, which assembles K11‐linked chains. 55 Upon entry into the M phase, the silencing of the spindle assembly checkpoint (SAC) activates the Ub ligase APC/C, leading to the proteasomal destruction of the separase inhibitor securin and cyclin B. This destruction releases separase, which cleaves cohesion, thereby facilitating chromosome segregation. 161 The SAC prevents the APC/C from recognizing cyclin B and securin by incorporating of the APC/C coactivator CDC20 into a complex known as the mitotic checkpoint complex. The SAC generates a diffusible “wait anaphase” signal through unattached kinetochores. 162 In securin‐deficient cells, researchers have identified human shugoshin 2 (Sgo2), which forms a complex with mitotic‐arrest deficient‐1 (Mad2), substituting the role of securin in these cells. 163 The interplay between APC/C, the SAC, and early mitotic inhibitor 1 (Emi1) creates a system of mutual checks that determines whether a cell proceeds to the next cell cycle. 164 Additionally, the CDK inhibitor p27Kip1 is exported from the nucleus to the cytoplasm during G0–G1 transition and is degraded via the Ub–proteasome pathway at this stage. 165
In addition to the large E3 Ub complex APC/C, other Ub‐related molecules also play crucial roles in regulating the cell cycle. Efficient transitions in the cell cycle are crucial for embryonic development and tissue homeostasis, particularly at the G1/S and G2/M checkpoints. The G1/S checkpoint ensures that the cell's condition and nutritional status are optimal before entering the S phase. The PARK2 E3 Ub ligase, a tumor suppressor, coordinately control the stability of cyclin D and cyclin E, acting as a primary regulator of G1/S cyclin stability. 166 In MCF‐7 cells, HDAC inhibitors such as trichostatin A enhance cyclin D1 degradation through a GSK3β/CRM1‐dependent nuclear export and 26S proteasome degradation pathway. 167 Ubiquitination also indirectly regulates the cell cycle by modulating cell cycle inhibitors like p21. Endogenous DNA damage occurring in the S phase leads to p53‐dependent accumulation of p21 during the subsequent G2 phase of the mother cell and G1 phase of the daughter cell, regulating the proliferation‐quiescence decision of daughter cells through CDK2 inhibition. Sub‐threshold accumulation of p21 does not affect the G1/S transition. The Ub ligases CRL4Cdt2 and SCFSkp2 couple the degradation of sub‐threshold p21 prior to the G1/S transition, thereby ensuring an irreversible G1/S transition. 168 Moreover, the transcription factor nuclear casein and cyclin‐dependent kinase substrate 1 (NUCKS1) is recruited to chromatin to activate the expression of S‐phase kinase‐associated protein 2 (SKP2), the F‐box component of the SCFSKP2 Ub ligase, leading to the degradation of p21 and p27 and promoting progression to the S phase. 169
There is also a checkpoint between the G2 and M phases to ensure that all necessary conditions are met before the cell enters mitosis, with ubiquitination playing a crucial role in controlling the progression of mitosis. During centriole replication, USP33 deubiquitinates CP110, leading to CP110 instability, thereby inhibiting centrosome amplification and preventing mitotic defects. This mechanism primarily operates during the S and G2/M phases, the periods of centriole replication and elongation. 170 Furthermore, the human single‐stranded DNA binding protein SSB1 (hSSB1) is a novel DNA damage‐associated protein that can interact with p53 and protect p53 from Ub‐mediated degradation. Consequently, the inactivation of hSSB1 leads to G2/M checkpoint failure. 149
The Golgi apparatus disassembles and disperses its stacks at the onset of mitosis, followed by further vesiculation. The Ub ligase activity of HACE1 during Golgi disassembly in mitosis is essential for subsequent Golgi membrane fusion postmitosis. Depleting HACE1 using small interfering RNA or the expressing inactive HACE1 mutant proteins in cells impairs Golgi membrane fusion after mitosis. 171 The disassembly of integrin‐containing focal adhesions is crucial for cell rounding, the formation of mitotic retraction fibers, bipolar spindle positioning, and chromosome segregation. The underlying mechanism involves the phosphorylation of the integrin activator kindlin by the CDK1–cyclin B1 complex, which subsequently leads to the recruitment of the Cullin 9–FBXL10 Ub ligase complex that mediates kindlin ubiquitination and degradation. 172
In summary, the checkpoints at various stages of the cell cycle are critical for cell cycle regulation. Based on this, Sakaue‐Sawano et al. 173 developed Fucci (fluorescent ubiquitination‐based cell cycle indicator), a genetically encoded optical sensor for monitoring interphase in the cell cycle. Its principle relies on S‐phase‐specific CUL4Ddb1‐mediated ubiquitination to precisely distinguish major cell cycle transitions and phases, particularly G1, S, and G2 (Figure 6).
FIGURE 6.
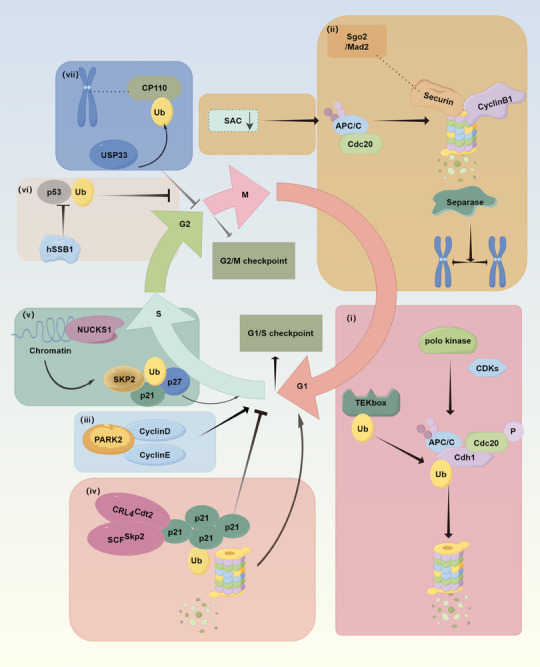
Ubiquitination in cell cycle regulation and genome stability (by Figdraw). (i and ii) APC/C in cell cycle regulation; (iii–v) ubiquitination in the regulation of G1/S transition; (vi and vii) ubiquitination in the regulation of G2/M transition. SAC, spindle assembly checkpoint; Sgo2, shugoshin 2; Mad2, mitotic‐arrest deficient‐1; hSSB1, human single‐stranded DNA binding protein SSB1; NUCKS1, nuclear casein and cyclin‐dependent kinase substrate 1; SKP2, S‐phase kinase‐associated protein 2; APC/C, anaphase‐promoting complex/cyclosome; CDKs, cyclin‐dependent kinases; Cdc20, cell‐division cycle protein 20 homologue; Cdh1, E‐cadherin gene; P, phosphorylation; Ub, ubiquitin.
4.4. Importance in immune response and inflammation
During the initiation of immune responses, ubiquitination mediates various immune processes through interactions with multiple receptors. For instance, Nrdp1, a RING‐type E3 ligase, mediates K33‐linked polyubiquitination of the signaling kinase Zap70 while promoting dephosphorylation of Zap70 by Sts1 and Sts2, thereby blocking TCR signaling in CD8+ T cells. 174 The Ub E3 ligases Itch and WW domain‐containing protein 2 (WWP2) functionally collaborate in regulating CD4+ TCR signal strength. They form a complex and synergistically enhance proximal TCR signal strength by catalyzing the conjugation of atypical Ub chains to the phosphatase SH2 domain‐containing protein tyrosine phosphatase 1 (Shp‐1), reducing SHP‐1′s association with the tyrosine kinase Lck. 175 SHARPIN, as part of the LUBAC, does not directly regulate TCR signal strength; However, SHARPIN deficiency leads to a reduction in the number and function of regulatory T cells, independent of NF‐κB signaling. 176 UBR2, an E3 Ub ligase, acts as a positive regulator of T cell activation. It induces K63‐linked ubiquitination at Lys99 and Lys276 of kinase Lck, which subsequently activates Lck through Tyr394 phosphorylation, thereby promoting T cell activation. 177
T cell homeostasis is a crucial factor in maintaining immune balance. A typical example of its dysregulation involves the deubiquitinase USP8, which has been identified as a new component of the TCR signalosome, interacting with the adaptor molecule GADS and 14‐3‐3 β. Mice with a knockout of the USP8 gene exhibit lethal colitis. 178 In the pathogenesis of allergic asthma, TCR signaling can upregulate the levels of the deubiquitinase USP38, which, in turn, stabilizes the TH2 development factor JunB, ultimately promoting asthma. 179 Ubiquitination processes also influence T cell differentiation. Ndfip1, an activator of the Nedd4 family E3 ligases, regulates cytokine signaling by mediating the degradation of Jak1, thereby limiting the expansion and function of CD4+ effector T cells. 180
In the activation and differentiation of B cells, the ubiquitination process plays a crucial role in regulating the function of E3 ligases Cbl and Cbl‐b (collectively known as Cbls). Ablation of these ligases impairs the clonal expansion of high‐affinity germinal center (GC) B cells, primarily due to an early exit from the GC cycle. Cbls are highly expressed in the light zone of the GC and impede plasma cell differentiation by promoting Irf4 ubiquitination. 181 Smurf2 mediates the ubiquitination and degradation of Yin Yang 1 (YY1), a critical GC transcription factor. Smurf2 deficiency enhances YY1‐mediated transactivation of c‐Myc and B cell proliferation, which can also lead to lymphoma in proliferating B cells. 182 The E3 Ub ligase Itch functions within B cells to limit the numbers of naive and, to a greater extent, GC B cells. 183 The surface levels and degradation of MHCII on GC B cells are dynamically regulated, with fluctuations in surface MHCII levels dependent on ubiquitination and the E3 ligase March1. 184
In inflammatory responses, activator protein‐1 (AP‐1) is a key regulator that can both promote inflammation and cell damage, as well as enhance cellular antioxidant capacity to protect cells from damage. Ubiquitination of AP‐1 is a classical pathway in the regulation of inflammatory mechanisms. 185 As an E3 Ub ligase, TRIM5 enhances innate immune signaling through interaction with retroviral capsid lattices, promoting UBC13–UEV1A‐dependent E3 activity, activating the TAK1 kinase complex, and stimulating AP‐1 and NF‐κB signaling. Additionally, TRIM5 also acts as a pattern recognition receptor, specifically recognizing retroviral capsid lattices and promoting the transcription of AP‐1 and NF‐κB‐dependent factors. 186 Another member of the TRIM family, TRIM7, mediates c‐Jun/AP‐1 activation through Ras signaling. The specific mechanism involves the stabilization of the AP‐1 coactivator RACO‐1 via K63‐linked ubiquitination, thereby promoting AP‐1‐dependent gene expression. 187 Furthermore, DR5 inhibits signaling by promoting sphingosine‐1‐phosphate‐dependent polyubiquitination of TRAF2, which activates the JNK/AP‐1 signaling pathway. 188 These mechanisms illustrate the multifaceted roles of ubiquitination in regulating AP‐1 activity, involving not only direct signal transduction activation but also the stabilization of coactivators and the regulation of downstream signaling pathways, collectively forming a complex regulatory network of ubiquitination in inflammatory responses. Additionally, canonical NF‐κB is rapidly activated in innate and adaptive immune cells through various signals, and its regulation of inflammatory responses has been detailed in previous pages.
Programmed cell death can influence the release of inflammatory factors. There are multiple forms of programmed death, including apoptosis, pyroptosis, and necroptosis, each accompanied by the release of inflammatory factors. 189 For instance, pyroptosis is a form of programmed cell death triggered by inflammasome activation, leading to the formation of pores in cell membranes by gasdermin D (GSDMD), ultimately resulting in the release of cellular contents and the release of inflammatory factors. 190 Ubiquitination plays a critical role in modifying multiple molecules involved in cell death. In the canonical pyroptosis pathway, ubiquitination is involved at every stage, from the initial extracellular stimuli acting on the NLRP family to the ultimate release of inflammatory factors. The NLRP3 inflammasome, the most common initiator of pyroptosis, consists of NLRP3, ASC, and activated caspase‐1. 191 Ubiquitination of the NLRP3 inflammasome generally inhibits its assembly and activation, while deubiquitination often has the opposite effect. 192 , 193 GSDMD, the executor of pyroptosis, when ubiquitinated, often promotes the occurrence of pyroptosis. 194 Even inflammatory factors themselves, such as the IL‐1β, can be ubiquitinated, undergoing various types of ubiquitination. 195 Overall, ubiquitination indirectly but commonly plays a regulatory role in inflammatory responses by modifying molecules involved in cell death (Figure 7).
FIGURE 7.
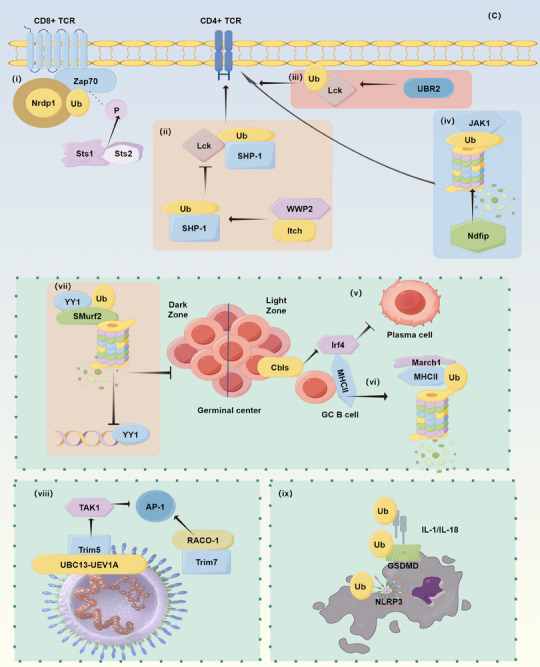
Ubiquitination in inflammation regulation (by Figdraw). (i–iv) Ubiquitination and TCR signaling; (v–vii) ubiquitination in B cell differentiation; (viii) ubiquitination in conjunction with AP‐1 in inflammation regulation; (ix) ubiquitination in cell death (pyroptosis). TCR, T cell receptor; Shp‐1, SH2 domain‐containing protein tyrosine phosphatase 1; WWP2, WW domain‐containing protein 2; YY1, Yin Yang 1; AP‐1, activator protein‐1; GSDMD, gasdermin D; P, phosphorylation; Ub, ubiquitin.
5. UBIQUITINATION IN DISEASE
Ubiquitination plays a crucial role in a wide range of biological processes, throughout the human body. Proteins are essential components of human physiology, with different proteins performing specific functions, intricately linked in a sequential manner to regulate diverse biological pathways. When the ubiquitination process of a critical protein is dysregulated, it can severely impact the multitude of cellular processes. Such dysregulation can escalate to a point where disease is triggered. Currently, extensive research has focused on diseases arising from the aberrant regulation of ubiquitination, including various cancers, neurodegenerative diseases, and metabolic disorders. The following sections will systematically elaborate the role of ubiquitination dysregulation in various disease contexts.
5.1. Dysregulation of ubiquitination in cancer
Aberrant activation or dysregulation of ubiquitination can lead to abnormal pathway activation, improper assembly of protein complexes, and the accumulation of misfolded proteins, all of which can disrupt normal physiological functions and contribute to disease pathogenesis. The complexity and diversity of the ubiquitination process determine its varying roles in different physiological and pathological contexts, playing a critical role in maintaining cellular functions, growth, differentiation, and immune defense. 196 , 197 , 198 Recent studies have indicated that ubiquitination is also extensively involved in the processes of cell death and inflammatory responses. In fact, the scope of the Ub system is still under investigation, as the reactions and signaling processes it influences are likely far more extensive than currently understood. There are approximately 100 specialized proteases, known as DUB, within the human body that can counteract the intricate ubiquitination process. In this context, the balance between E3 ligases and DUBs is a crucial for ensuring the proper functioning of signaling pathways and cellular functions. 199 This delicate interplay between ubiquitination and deubiquitination is likely pivotal in the progression of diseases, including cancer, and could represent a breakthrough in the development of therapeutic interventions.
Compared to normal cells, cancer cells exhibit accelerated proliferation and require significantly higher metabolic activity to support their malignant growth. Dysregulation of metabolic activities is a significant contributor to tumorigenesis. Among the numerous PTMs closely related to metabolic activities, protein ubiquitination stands out as a common and crucial cellular mechanism. Dysregulation of ubiquitination and deubiquitination has been observed in various types of cancer. 200 , 201 , 202 Moreover, the UPS is regulated by transcriptional, translational, and PTMs, thereby playing various roles in both the promotion and inhibition of oncogenesis. In recent years, owing to the extensive involvement of the UPS in the initiation and progression of cancer, it has garnered widespread attention from researchers as a novel therapeutic target for cancer treatment. 14
The UPS is extensively involved in the regulation of various cancer‐related signaling pathways, transcription factors, and metabolic enzymes. Within this broad physiological context, the binding processes of E3 ligases and DUBs with their substrates are notably complex. This complexity arises because a single E3 Ub ligase or DUB can target multiple substrates, while an individual substrate protein can be regulated by multiple E3 ligases or DUBs. 202 For example, FBXW7, an E3 ligase for the crucial oncogene c‐Myc, can function as a tumor suppressor by targeting mTOR, HIF‐1α, c‐Myc, and SREBP1 for degradation. 203 , 204 , 205 Additionally, in response to DNA damage, FBXW7 can mediate the proteasomal degradation of p53, leading to radioresistance. 206 Moreover, the processes of ubiquitination and deubiquitination in cancer are highly context dependent. For instance, in certain environments, DUBs may exhibit pro‐tumorigenic effects, while in other distinct environments, their tumor‐inhibitory roles become more pronounced. This variability and disparity underscore the significance of targeting specific DUBs for cancer therapy. 199
The UPS is extensively involved in various biological processes in tumor cells, such as proliferation, invasion, and apoptosis. For instance, homologous to the E6‐associated protein carboxyl terminus domain containing 3, acting as a prosurvival protein, can promote the stabilization of MALT1 via K63‐linked polyubiquitination, ultimately leading to cancer cell proliferation and invasion. 207 , 208 Furthermore, FBXW7, identified as a tumor suppressor, interacts with Mcl‐1, prompting the ubiquitination and degradation of this Bcl‐2 family member, thereby inducing apoptosis in cancer cells. 209 From a cancer‐type perspective, dysregulation of the UPS also influences the growth, proliferation, and metastasis of various cancers. For example, studies have shown that ATXN3 can promote breast cancer metastasis by deubiquitinating KLF4. 210 Moreover, the lncRNA LNC473 can enhance the proliferation and invasion of liver cancer cells by interacting with USP9X to inhibit the ubiquitination of survivin. 211 Additionally, in renal cell carcinoma, an E3 ligase encoded by the TRC8 gene interacts with and ubiquitinates heme oxygenase‐1, which in turn enhances the tumorigenic and invasive capabilities of renal cancer cells. In prostate and gastric cancers, RBX1 targets CYCLIN E1, promoting tumor cell proliferation. 212 , 213 These examples illustrate the complex and extensive roles of the UPS in cancer. The UPS not only regulates protein degradation but also influences the interaction of proteins with other molecules, thus modulating intracellular signaling networks. In cancer, aberrant expression or mutations of certain E3 ligases can alter cellular responses to specific signaling pathways, leading to uncontrolled cell cycles and enhanced antiapoptotic capabilities. For instance, NEDD4‐1, an E3 Ub ligase, negatively regulates the PI3K/AKT signaling pathway by ubiquitinating and promoting the degradation of PTEN, which facilitates tumor cell proliferation in some cancers, such as lung cancer. 214 Furthermore, APC/C, an important E3 ligase, can activate Cdc20 to regulate β‐catenin, promoting the activation of the WNT/β‐catenin pathway associated with prostate cancer progression. 215 These examples further demonstrate the complexity and diversity of ubiquitination pathways in different cancer types, providing new insights and methods for cancer therapy.
Deubiquitination also exerts considerable influence in cancer biology (Table 1). DUBs can regulate the E3 ligase complexes or modulate the signaling cascades that induce protein degradation, thereby impacting the overall balance between ubiquitination and deubiquitination. This action is not limited to a single target but can affect a multitude of substrate proteins. Normally, these processes are precisely controlled to ensure the proper functioning of physiological processes. However, during cancer progression, this delicate balance can be disrupted. The complexity of deubiquitination in cancer stems from its involvement in nearly every aspect of oncogenesis. DUBs target a wide array of proteins involved in numerous cancer‐related signaling pathways, and a single DUB can also exhibit different roles depending on the tumor type and context—roles that may be either oncogenic or tumor‐suppressive. 199 Like the ubiquitination process, DUBs are also involved in cancer metastasis and proliferation, with different DUBs exerting varying effects on the disease. The research by Han et al. provides a detailed exposition of the roles of different DUBs in promoting and inhibiting cancer. 216 Nevertheless, targeting the key enzymes in ubiquitination and deubiquitination processes may represent a novel and critical approach to cancer therapy. In summary, dysregulation of the ubiquitination pathway in cancer affects not only the stability of tumor‐associated proteins but can also profoundly influence the disease by altering cell signaling and cell cycle regulation. Future research should continue to elucidate the specific mechanisms of the UPS in different types of cancer and develop targeted therapeutic strategies, providing new directions and methods for cancer treatment.
TABLE 1.
Inhibitor drugs targeting UPS and DUBs for cancer treatment.
| UPS and DUBs | Anticancer drugs/inhibitor | References |
|---|---|---|
| Proteasome | Bortezomib, curcumin, b‐AP15 | 217 , 218 , 219 , 220 , 221 , 222 , 223 |
| Ubiquitin proteasome system | Piperlongumine, curcusone D | 220 , 223 |
| USP1 | ML323, SJB2‐043, SJB3‐019A, GW7647, pimozide, rottlerin | 224 , 225 , 226 |
| USP2 | ML364, LCAHA, 6TG | 227 , 228 |
| USP4 | Vialinin A | 229 , 230 |
| USP5 | WP1130, vialinin A | 229 , 230 , 231 , 232 |
| USP7 | FT671, FT827, GNE‐6640, GNE‐6776, compound 1, compound 4, P5091, P22077, HBX41108, HBX19818, Cpd1, Cpd2, Cpd8 | 231 , 233 , 234 , 235 , 236 , 237 , 238 , 239 |
| USP8 | DUBs‐IN‐2, MB7295 | 240 |
| USP9X | WP1130, EOAI3402143 | 233 , 241 |
| USP11 | MIX | 242 |
| USP14 | IU‐1/analogues, b‐AP15, VLX1570, WP1130, auranofin | 233 , 243 , 244 , 245 , 246 |
| USP15 | MIX | 242 |
| USP17 | WP1130 | 233 |
| USP20 | GSK2643943A | 247 |
| USP24 | EOAI3402143 | 241 |
| USP25 | AZ1/2/3/4 | 248 |
| USP28 | AZ1/2/3/4 | 248 |
| USP30 | FT385, MF094, MF095 | 249 |
| USP47 | Compound 1 | 239 |
| POH1 | 8‐Thioquinoline, capzimin, phen | 247 , 250 , 251 |
| UCHL1 | LDN‐57444, GK13S, 6RK73 | 252 , 253 |
| UCHL5 | IU‐1/analogues, b‐AP15, VLX1570, WP1130, auranofin | 231 , 243 , 244 , 245 , 246 |
5.2. Neurodegenerative diseases and protein aggregation
The UPS is responsible for degrading the majority of proteins in the human body and influences biological activities such as cell fate, differentiation, and migration, all of which are crucial for the nervous system. Neurodegenerative diseases, including AD, Parkinson's disease (PD), Huntington's disease (HD), and ALS, are characterized by the accumulation of abnormal proteins and the formation of inclusion bodies. 254 In these diseases, dysfunction of the UPS is a critical factor leading to the accumulation and aggregation of aberrant proteins, such as β‐amyloid (Aβ) and α‐synuclein. Under normal conditions, the UPS is highly selective, maintaining the stability of the nervous system by degrading short‐lived, abnormal, or misfolded proteins. However, genetic factors, aging, or lifestyle changes can disrupt this process, leading to the accumulation of toxic protein aggregates and the emergence of pathological features associated with neurodegenerative diseases. 255 When the UPS malfunctions, the misfolding and aggregation of proteins not only impair neuronal function but also trigger intracellular stress responses, ultimately leading to cell death. 256 Studies have shown that protein aggregates in the brains of many patients with neurodegenerative diseases are Ub positive. 257 For example, the accumulation of Aβ plaques and hyperphosphorylated tau are prominent pathological features of AD. Research indicates that the proteasome extensively regulates the production of these proteins, and inhibiting proteasome function leads to their abnormal accumulation. 258 Additionally, the aggregation of huntingtin protein is a significant pathological marker of HD. These aggregates, which should be marked and degraded by ubiquitination, cannot be digested by the proteasome, resulting in their accumulation. 259 Recent studies in PD have found that the UPS also accelerates the pathological progression of PD by regulating α‐synuclein misfolding and aggregation, mitophagy, neuroinflammation, and oxidative stress. 260 Furthermore, the UPS influences protein quality control and the maintenance of the intracellular environment, participating in the regulation of mitochondrial function. Mitochondrial homeostasis is closely related to the UPS, as the UPS regulates organelle dynamics, mitochondrial proteome, and mitophagy. 261 Numerous studies have confirmed that UPS abnormalities may contribute to the progression of PD through mitochondrial dysfunction. 262 , 263 , 264 ALS is a progressive fatal disease, and its rare early‐onset familial form can be caused by mutations in the superoxide dismutase (SOD1) gene. However, researchers have found that the pathological changes are not due to altered SOD1 activity but rather the accumulation of ubiquitinated aggregates, indicating that UPS dysfunction is involved in the progression of ALS. The UPS is a critical component of the protein quality control system, and targeting the UPS may represent a promising approach for treating neurodegenerative diseases.
Although deubiquitination is the opposite of ubiquitination, DUBs are one of the various checkpoints that ensure the correct ubiquitination of substrates. By removing Ub, DUBs are involved in multiple physiological processes of the nervous system, including neuronal fate determination, axon guidance, synaptic communication, and plasticity. 265 Certain DUBs, such as ataxin‐3, can enhance the degradation of specific proteins by editing the Ub chains attached to substrates. DUBs like USP7 and USP9X can stabilize proteasome substrates by counteracting the activity of certain E3 ligases. Other DUBs, such as USP14, participate in controlling the UPS through various mechanisms, some of which appear to be mutually antagonistic. USP14 can recycle Ub for reuse when the proteasome interacts with substrates, enhancing proteasome function, and can also preemptively ubiquitinate substrates to prevent their degradation when they are not bound to the proteasome. This highlights the significant role of DUBs in stabilizing the UPS and the overall function of the nervous system. 10 We emphasize the balancing role of DUBs in the nervous system. Additionally, due to the specificity of cells and substrates, DUBs may also be potential therapeutic targets for certain neuroinflammatory or degenerative conditions, which is promising for the treatment of neurodegenerative diseases.
5.3. Ubiquitination defects in metabolic disorders
Metabolic syndrome, maybe defined differently across various studies, generally refers to excessive accumulation of lipids in visceral fat tissue, accompanied by hypertension, hyperglycemia, or hyperlipidemia. 266 , 267 Metabolic syndrome is currently estimated to affect over a billion people worldwide, significantly increasing the risks of cardiovascular disease and stroke, two of the leading causes of mortality. 268 Factors such as inflammation, excessive oxidative stress, and more specifically, various key cytokines, protein kinases, and modifications of critical proteins are considered major disruptors of metabolic activities. 268 , 269 , 270 Consequently, the UPS, a key player in protein quality control, is recognized as having a pivotal influence on metabolic diseases. The impact of the UPS on metabolic disorders is multifaceted, affecting various cellular and organismal functions. On a microscopic level, the UPS regulates fundamental protein degradation processes, thereby influencing signal transduction, transcriptional regulation, protein interactions, and even DNA damage repair—all of which are intimately related to metabolic disturbances, including glucose and lipid metabolism. On a macroscopic level, due to its significant impact on basic cellular physiological functions, UPS‐induced disruptions in molecular metabolic pathways can lead to various acute and chronic metabolic diseases, including diabetes, obesity, and related complications. 271 , 272 The regulatory role of the UPS on proteins is undeniable; however, it also modulates glucose and lipid metabolism by controlling the levels and activities of key enzymes or proteins. For instance, in obesity and diabetes, adipocyte differentiation is a crucial process in lipid metabolism involving several ubiquitination‐related proteins. C/EBPβ and PPARγ are key regulatory factors whose ubiquitination status directly affects adipocyte differentiation. Excessive ubiquitination leads to their degradation, severely impacting fat formation. Activating transcription factor 4 (ATF4) is also essential for promoting lipogenesis, and the overexpression of USP7 can restore ATF4 deficiency‐induced lipogenesis impairment and participate in ATF4‐induced adipocyte differentiation. 273 Additionally, DUBs such as USP2, USP7, USP15, USP19, and USP20 have been proven to be involved in lipid metabolism and fat formation, while USP53 may have beneficial effects on obesity control. 272 In summary, the regulation of key enzymes and proteins associated with the UPS is crucial for controlling lipid metabolism and obesity, making the UPS is a significant therapeutic target for obesity and lipid metabolic disorders. Another focus of metabolic diseases is diabetes, a condition involving carbohydrate metabolism disorders, typically classified into Type 1 diabetes mellitus (T1DM) and Type 2 diabetes mellitus (T2DM). 274 T1DM primarily results from autoimmune destruction of pancreatic β‐cells, leading to decreased circulating insulin levels. Although T1DM is more akin to a metabolism disorder caused by autoimmune disease, studies suggest that UPS‐associated key enzymes and proteins might be potential therapeutic targets. For instance, USP22 275 , 276 functions by inhibiting autoimmunity, while USP18 277 may alleviate T1DM stimuli by inhibiting type I interferon signaling or countering certain viral actions. T2DM demonstrates a stronger association with the UPS, as disturbances in blood glucose, inflammatory factors, and various lipid metabolites can lead to β‐cell dysfunction and insulin resistance. 278 Additionally, insulin signaling is a critical factor affecting T2DM, involving several ubiquitination‐related proteins. In T2DM patients, the ubiquitination status of insulin receptor substrates (IRSs) directly affects signaling efficiency. E3 ligases such as WWP1 promote IRS degradation via ubiquitination, weakening insulin signaling and raising blood glucose levels. 279 Furthermore, many DUBs have been shown to participate in insulin signaling across various tissues, including USP1 (in pancreatic B‐cells), USP2 (in the liver), USP19 (in white adipose tissue), and USP20 (in skeletal muscle). 272 In conclusion, given the pivotal role of ubiquitination in metabolic diseases, developing targeted therapeutic strategies is of significant clinical importance. Future research should delve deeper into the specific mechanisms of ubiquitination in metabolic diseases and focus on developing targeted drugs to achieve effective treatment and prevention of metabolic diseases.
5.4. Implications in infectious diseases
The UPS plays a crucial role in the host immune response to pathogens. However, certain pathogens, such as viruses and bacteria, exploit this system for their own advantage during host invasion. The UPS is involved in nearly every aspect of cellular physiology, and during viral infections, viruses manipulate the host's ubiquitination system to facilitate their own replication and spread. Additionally, viral proteins themselves can be ubiquitinated, with some viruses even encoding their own ubiquitination machinery and DUBs. Normally, host cells utilize their UPS to activate immune response pathways and inhibit viral protein synthesis, thereby combating viral infections. 280 However, viruses can evade host defenses by interfering with the ubiquitination and degradation of host surface receptors, thus preventing T‐cell recognition. 281 The exploitation of the UPS by viruses extends beyond these mechanisms. Some viruses, such as human papillomavirus, utilize E3 Ub ligase complexes to degrade cell cycle regulatory proteins. Moreover, viruses can interfere with the host immune response by using the UPS to avoid detection by the immune system while enhancing their own replication. Overall, dysregulation of the ubiquitination pathway can severely impair the host's ability to defend against viral infections. Similar to viral infections, the host has specific signaling pathways to respond to bacterial infections. However, bacteria can interfere with or hijack these pathways, inhibiting the activation of immune responses. To successfully invade the host, these pathogenic microorganisms often disrupt relevant signaling pathways to evade immune detection, relying heavily on their manipulation of the host's UPS. Bacterial pathogens have evolved to mimic host ubiquitination enzymes, including various E3 ligases such as RING, HECT, and novel E3 ligases. 282 Furthermore, due to the reversible nature of ubiquitination, bacteria often produce DUBs that mimic host enzymes, thereby protecting substrate proteins from degradation. 283 In summary, pathogenic infections demonstrate how microorganisms exploit the UPS to manipulate and evade host immune responses, thereby supporting their replication and growth. In infectious diseases, the UPS is extensively involved in pathogen replication, spread, and host immune responses. The interaction between pathogens and the host, as well as the disruption and regulation of immune responses, may provide significant insights for future research in anti‐infective therapies. Therefore, greater emphasis should be placed on the role of the UPS in infectious disease control, as it may present opportunities for novel anti‐infective treatments or vaccine development.
6. THERAPEUTIC TARGETING OF UBIQUITINATION
Ubiquitination functions as a critical regulatory mechanism for maintaining protein quality and quantity across diverse cell types, offering potential avenues for developing specific modulators to either augment or inhibit this process. Precise control of ubiquitination, minimizing detrimental side effects, could signify a transformative breakthrough in influencing human health outcomes. Nonetheless, the intrinsic complexity of proteins and the Ub system presents substantial challenges, rendering this objective largely unattained. At present, the identification of inhibitors targeting ubiquitination has been limited to small molecules, with notable examples including bortezomib and carfilzomib, which are already utilized in clinical oncology. In the context of neurodegenerative diseases, an alternative therapeutic strategy involves exploiting the reversible nature of ubiquitination through the application of DUBs to restore cellular homeostasis. Additionally, a burgeoning area of research in this domain is the development of proteolysis‐targeting chimeras (PROTACs), which facilitate the targeted degradation of specific proteins via recruiting them to E3 ligases. Nevertheless, this system is still under development and investigation, and effective regulation of ubiquitination remains a work in progress. Here, we summarize the drugs and enzymes targeting ubiquitination across various diseases, as outlined in Table 2.
TABLE 2.
The role and function of DUBs in related diseases.
| Disease name | DUBs | The role and function of DUBs | References |
|---|---|---|---|
| Cancer | USP3, USP4, USP5, USP7, USP11, USP14, USP17, USP27X, USP29, USP36, USP37, USP42, USP46, USP47, A20, CSN5, OTUB1, OTUD4, UCHL1 | Upregulation of epithelial–mesenchymal transition in cancer | 199 , 284 |
|
USP2A, USP3, USP4, USP6, USP13, USP15, USP16, USP17, USP21, USP22, USP27X, USP28, USP38, USP39, USP42, USP43, USP49, USP51, CSN5, POH1, TRABID, UCHL1, UCHL3, BRCC36, COPS6, COPS5, ATXN3L, ATXN3, OTUD7B, OTUD2 |
Upregulation of tumor invasion in cancer | 284 , 285 , 286 , 287 , 288 , 289 , 290 , 291 , 292 , 293 , 294 , 295 , 296 , 297 , 298 , 299 | |
| USP9X, USP53, OTUD3 | Downregulation of tumor invasion in cancer | 199 , 284 , 300 , 301 | |
|
USP3, USP4, USP8, USP9X, USP17, USP22, USP27X, USP29, USP37, USP45, UCHL1 |
Upregulation of tumor migration in cancer | 199 , 284 | |
| USP11, USP53 | Downregulation of tumor migration in cancer | 199 , 284 | |
|
USP1, USP4, USP7, USP10, USP11, USP20, USP21, USP24, USP26, USP33, A20, ataxin 3, CSN5, OTUB2, UCHL1, AMSH, COPS6, COPS5, UCHL5 |
Upregulation of metastatic dissemination in cancer | 199 , 217 , 284 , 298 , 299 , 300 , 301 , 302 , 303 , 304 , 305 , 306 , 307 | |
| OTUD1, TRABID, OTUD7A, OTUD6B, CYLD | Downregulation of metastatic dissemination in cancer | 199 , 284 , 308 , 309 , 310 , 311 , 312 , 313 , 314 , 315 , 316 | |
| Neurodegenerative diseases and other nervous system disorders | ATXN3, USP7, USP8, USP14, USP15, USP30, BAP1, CYLD, PSMD14, TNFAIP3, UCH‐L1, UCHL‐5, USP1, USP4, USP10, USP11, USP12, USP13, USP18, USP22, USP33, USP36, USPL1, USP9X, YOD1 | Involved in the upregulation of neurodegeneration formation | 61 , 243 , 317 , 318 , 319 , 320 , 321 , 322 , 323 , 324 , 325 , 326 , 327 , 328 , 329 , 330 , 331 , 332 , 333 , 334 , 335 , 336 , 337 , 338 , 339 , 340 |
| USP16, A20, CYLD, OTULIN, USP18, USP25, UCHL1, UCHL3, UCHL5/UCH37, USP2, USP5, OTUD4, PSMD14, EIF3H, AMSH | Involved in the progression of other nervous system disorders | 10 , 341 , 342 , 343 , 344 , 345 , 346 , 347 , 348 , 349 | |
| Obesity | USP2, USP7, USP15, USP19, USP20 | Involved in the progression of worsening obesity | 271 , 350 , 351 , 352 |
| USP53 | Involved in the progression of reducing obesity | 271 , 353 | |
| T1DM | USP18, USP22 | Involved in the progression of worsening T1DM | 275 , 277 , 350 |
| T2DM | USP1, USP19, USP21, USP2, USP14, USP20 | Involved in the progression of worsening T2DM | 354 , 355 , 356 , 357 , 358 |
| USP22, USP2, USP9X, USP20, USP33, USP4, USP7, USP10, USP18 | Involved in the progression of reducing T2DM | 356 , 359 , 360 , 361 , 362 , 363 , 364 , 365 , 366 |
6.1. Small molecule inhibitors and activators of ubiquitination enzymes
The ubiquitination pathway plays a crucial role in intracellular protein degradation and signal transduction, with its dysregulation closely linked to the onset and progression of various diseases, particularly cancer and neurodegenerative disorders. Therefore, developing small molecule drugs targeting specific enzymes within the ubiquitination pathway, such as E1, E2, and E3 enzymes, as well as proteasome inhibitors or activators, holds significant therapeutic potential. 14 In the field of cancer, recent research has focused on small molecule inhibitors targeting E3 ligases. These inhibitors can specifically prevent the ubiquitination and degradation of tumor‐associated proteins, thereby affecting tumor growth and spread. 47 Additionally, PROTACs represent an emerging therapeutic strategy. PROTACs work by linking E3 ligases to target proteins (proteins of interest, POIs), inducing the ubiquitination and subsequent degradation of the POIs. Due to their high selectivity and specificity, PROTACs offer a novel direction for cancer treatment. 367 , 368 Furthermore, activating Ub ligases is also a therapeutic strategy for addressing abnormal protein accumulation in neurodegenerative and metabolic diseases. For example, NRBP1‐containing CRL2/CRL4A complexes can target and promote the degradation of Aβ protein in AD, enhancing related ubiquitination pathways and slowing disease progression. 369 While the development of agonists for Ub ligases is still in its early stages, research on small molecule inhibitors targeting DUBs is more advanced and extensive, with significant therapeutic potential. DUBs can remove Ub tags from proteins, affecting their stability and function and playing a role in protein quality control. In certain disease states, such as cancer and neurodegenerative diseases, overactivation of DUBs may lead to the accumulation of abnormal proteins. 256 Developing small molecule inhibitors for DUBs can inhibit their activity, enhance the ubiquitination process, and promote the degradation of abnormally accumulated proteins, thereby effectively inhibiting disease progression. For example, small molecule inhibitors targeting USP7 can stabilize p53 and inhibit cancer progression. 369 Additionally, USP7 inhibitors can potentially target neurological diseases by inhibiting neuroinflammation. 370 Other examples include inhibitors targeting USP14, which have been widely studied for the treatment of neurodegenerative diseases. 243 In summary, developing small molecule drugs targeting specific enzymes within the UPS, such as inhibitors or activators of E1, E2, or E3 enzymes, as well as DUB inhibitors, holds significant potential for disease treatment. These drugs can influence the ubiquitination process of specific substrates, aiding in disease control and providing new strategies for treating various diseases. Future research should focus more on the specific mechanisms of ubiquitination in related diseases, especially the key molecular mechanisms underlying abnormal proteins or disease progression, to achieve effective treatment and prevention. Next, we will discuss the potential of targeting the UPS in cancer and neurodegenerative disease therapies.
6.2. Targeting ubiquitination for cancer therapy
Approximately 80−90% of intracellular proteins are removed through the UPS. Tumor cells often exploit the UPS to reduce apoptosis or increase proliferation. Consequently, targeting the ubiquitination or proteasome system with various drugs has become a viable strategy for cancer treatment. Increasing recognition of the UPS as a target for cancer therapy has stimulated the development of small molecules, peptides, and proteasome inhibitors that can restore cellular balance and disrupt tumor growth. 371 These targeted therapies can be classified based on their targets: E1 enzymes (e.g., MLN7243 and MLN4924), E2 enzymes (e.g., leucettamol A and CC0651), E3 ligases (e.g., nutlin and MI‐219), proteasome inhibitors (e.g., bortezomib, carfilzomib, oprozomib, and ixazomib), and DUB inhibitors (e.g., G5 and F6). 14 Many of these targeted drugs have achieved tangible success in cancer treatment, offering new hope. For instance, proteasome inhibitors like Bortezomib or MG132 block the degradation of proteins entirely. However, while effective, such drugs may lack specificity and selectivity, suggesting that drugs targeting specific E3 ligases might offer better selectivity and lower toxicity, fulfilling clinical needs more effectively. 47 Among various targeted small molecule inhibitors, those targeting E3 ligases are the most extensively researched. 215
E3 ligases play a pivotal role in tumorigenesis by promoting protein ubiquitination and degradation, thereby influencing various physiological processes. 372 , 373 A notable example is the RING‐type E3 ligase MDM2, which is overexpressed in many human cancers. 374 MDM2 directly interacts with p53, a critical gene involved in cell cycle arrest, DNA repair, and apoptosis regulation, making it a significant cancer therapy target. 375 MDM2 induces the ubiquitination and proteasomal degradation of p53, reducing its tumor suppressor functions. Consequently, numerous small molecule inhibitors targeting MDM2 have been designed. 376 Nutlin, a cis‐imidazoline analog, was among the first inhibitors identified to bind MDM2, preventing p53 degradation. Similarly, MI‐219 disrupts the MDM2–p53 interaction. 377 Unlike the aforementioned inhibitors, RITA (Reactivation of p53 and Induction of Tumor cell Apoptosis) directly binds to p53, blocking its interaction with MDM2 and potentially other similar proteins, thereby promoting p53 activation in tumors. 377 , 378 The second‐generation MDM2 inhibitor, RG7388 (idasanutlin), has demonstrated reduced toxicity and increased efficacy compared to early Nutlins. 379 Another promising E3 ligase target within the SCF (Skp1–cullin–F‐box) complex is the F‐box protein Skp2, which is overexpressed in various cancers and involved in tumor regulation. 380 Skp2 inhibitors like SKPin C1 and SMIP004 have shown tumor‐inhibitory effects. 381
Compared to traditional small molecule inhibitors, PROTACs offer a practical approach to protein degradation. By recruiting E3 ligases to POIs, PROTACs induce the ubiquitination and proteasomal degradation of the POIs. Commonly targeted E3 ligases for PROTACs include MDM2, IAPs, cereblon, and VHL. 382 Several PROTAC‐based inhibitors, such as ARV‐471 and ARV‐110, have entered clinical trials. 367 Numerous PROTACs have been successfully developed, with many undergoing clinical validation for cancer treatment. 368 Overall, drugs targeting E3 ligases represent an effective strategy among UPS‐targeted therapies, with PROTAC technology offering superior selectivity and potency. While other UPS targets, such as E1 and E2 enzymes, are also being actively researched and tested, each has its own advantages and limitations. Finding and developing more efficient targets and drugs remain crucial for advancing cancer treatment.
6.3. Potential for drug development in neurodegenerative diseases
Proteasome dysfunction is observed in AD, PD, HD, and ALS, a common feature across nearly all neurodegenerative diseases. These conditions often involve the accumulation of abnormal proteins, suggesting an insufficient proteasomal clearance capability. Thus, enhancing ubiquitination or inhibiting deubiquitination processes through targeting the UPS may be an optimal approach for treating neurodegenerative diseases. One strategy is to promote the degradation of abnormal proteins. For instance, in AD, the aggregation of Aβ is a key factor in disease progression. Drugs can be designed to activate E3 ligases, such as NRBP1‐containing CRL2/CRL4A complexes, to accelerate Aβ degradation by targeting regulators like BRI2 and BRI3. 369 In PD, developing drugs that activate E3 ligases like CHIP could promote the ubiquitination and degradation of α‐synuclein. 383 Another strategy involves inhibiting DUB, making them potential therapeutic targets for neurodegeneration. 265 While the structure and characteristics of many DUBs have been studied, their precise physiological functions remain largely unknown. However, growing research highlights their potential role and core involvement in treating neurodegenerative diseases. 265 The integrity of neural development is closely linked to specific DUBs and their associated ubiquitination pathways, with a balance between ubiquitination and deubiquitination essential for maintaining neural stability. Neurodegenerative diseases frequently involve neuronal death, mitochondrial dysfunction, ER stress, and inflammation, with ubiquitination and deubiquitination playing significant roles in these processes. Additionally, UPS regulation can affect synaptic function, with synaptic loss being a critical cause of cognitive and motor deficits in many neurodegenerative diseases. Ubiquitination is crucial for the renewal and maintenance of synaptic proteins. Thus, developing drugs to modulate the ubiquitination state of synaptic‐related proteins may help restore synaptic function and improve cognitive and motor abilities. Specific DUBs known to function at synapses include UCHL1 (involved in maintaining synaptic function and structure), USP14 (enhancing proteasome function by recycling Ub in the synaptic proteasome), Fat Facets (Faf), and USP9X (both involved in cell differentiation and synaptic function). 265 Beyond promoting abnormal protein degradation, protecting neuronal survival and function, and improving synaptic function, DUB inhibitors might offer significant advantages due to their substrate specificity, making them potential therapeutic targets in the Ub–proteasome pathway. Similar to cancer treatment, drugs directly targeting the proteasome have shown success but often cause unintended side effects due to a lack of specificity. High‐throughput screening has identified several small molecule inhibitors, including those targeting USP7 and UCHL1. 384
7. FUTURE PERSPECTIVES AND CHALLENGES
7.1. Emerging roles of ubiquitination in biology and medicine
The discovery of canonical ubiquitination dates back several decades, but its significance extends far beyond the classical understanding of ubiquitination process. Initially, ubiquitination was thought to be confined to proteins. However, recent research has revealed the diversity of ubiquitination substrates, encompassing nucleotides, sugars, and lipids. These substrates perform various functions postubiquitination, such as glycogen clearance and bacterial elimination. Additionally, the chemical bonds between the Ub complex and its substrates include ester and amide bonds, in addition to the conventional peptide and isopeptide bonds 385 , 386 , 387 Consequently, the concept of noncanonical ubiquitination has been proposed. One aspect of noncanonical ubiquitination involves the variation in Ub linkage types. Studies indicate that, besides lysine, amino acids like serine (S), threonine (T), and cysteine (C) can serve as alternative Ub linkage sites. Notably, S and T residues form ester bonds during ubiquitination, although these bonds are less stable than peptide bonds. Despite its potential, the abundance and kinetics of nonpeptide‐linked ubiquitination remain underexplored, marking a promising avenue for future research. 21 , 388
In addition to variations in canonical ubiquitination processes, researchers have identified Ub‐like molecules that perform functions analogous to ubiquitination, yet with distinct cellular roles. One of the extensively studied examples is the small Ub‐like modifier (SUMO). The SUMO system, while simpler in composition than the ubiquitination system, modifies a broader range of substrates. Similar to ubiquitination, SUMOylation can alter protein localization, activity, and stability. 389 At the microscopic level, the SUMO system participates in various cellular functions and processes. SUMO modifies functionally related protein groups to primarily regulate nuclear processes, including gene expression, DNA damage response, RNA processing, cell cycle progression, and protein deposition. Current research has expanded SUMO's role to include immunoregulation, cell migration, and pathophysiological processes. 390 , 391 On a broader scale, SUMOylation is implicated in the pathogenesis of multiple diseases. For example, due to its role in stress response, SUMO is upregulated in many types of cancer cells, serving as a mechanism to protect these cells. 392 The relationship between SUMOylation and ubiquitination extends beyond mere similarity. SUMO can integrate within the Ub modification system, forming Ub–SUMO hybrid polymers. The ubiquitination of SUMO can induce proteasomal degradation of substrates. 24 Analogous to SUMO in its function, ISG15 is a 15‐kDa Ub‐like protein composed of two Ub‐like domains connected by a short linker. The corresponding process, known as ISGylation, involves a three‐enzyme cascade. Similar to ubiquitination, ISGylation is reversible. Linked with interferon responses, ISG15 plays a crucial role in antiviral defenses. 393 Additionally, another well‐known Ub‐like modifier is NEDD8, which has become a significant target in the development of anticancer therapeutics. 394
7.2. Technological advancements and tools for studying ubiquitination
The tools for investigating ubiquitination have become increasingly diverse. For example, super‐resolution microscopy enables the observation of subcellular structural details of ubiquitination events, such as the subcellular localization of the 19S and 20S proteasome complexes. This technique has also revealed the nuclear localization of the E3 Ub ligase RAD18 during its participation in membrane‐associated DNA repair. 395 Furthermore, live‐cell imaging allows for real‐time tracking of the ubiquitination pathways within cells. It has been used to observe the recruitment of OPTN to PARK2‐mediated ubiquitinated mitochondria. 396 Technologies such as live‐cell imaging and mass spectrometry are highly convenient for mechanistic studies of ubiquitination. 397
Molecular dynamics simulations can further elucidate the molecular mechanisms involved in ubiquitination and assist in studying the structure‐function relationships of ubiquitination regulatory factors. For instance, it has been validated that ubiquitination within the reticulon homology domain of the ER‐phagy receptor FAM134B promotes receptor aggregation and its binding with lipidated LC3B, thus stimulating ER autophagy. 398
With the evolution of artificial intelligence (AI), its application in ubiquitination research has become increasingly significant. Current machine learning models can predict binding behaviors among Ub, E3 ligases, and substrate ligands. 399 Moreover, a network system named DEGRONOPEDIA has been developed; it searches for degrons, maps them onto adjacent residues that can undergo ubiquitination and disordered regions, and assesses N‐/C‐terminal stability. 285 The application of AI in ubiquitination research is not limited to classical ubiquitination. For instance, GPS‐SUMO is a predictor constructed using three computational algorithms (deep neural networks, penalized logistic regression, and transformers), allowing users to accurately predict SUMO sites within a cell by inputting one or multiple protein sequences or identifiers, and it has been made freely available by its developers. 286 Furthermore, the PROTAC system for targeted protein degradation has also been integrated with a deep neural network model, resulting in the development of a system called DeepPROTACs. 287 AI is still in its infancy regarding the precise localization of protein molecules and the functionality optimization of targeted degradation, offering substantial room for research and advancement.
7.3. Challenges in targeting ubiquitination for therapeutic interventions
Modulating the ubiquitination process to regulate disease progression remains a significant challenge in current research. Although several ubiquitination inhibitors have received United States Food and Drug Administration (US FDA) approval, no ubiquitination agonists have been approved to date. For instance, ixazomib, an oral proteasome inhibitor, has been used in the treatment of multiple myeloma. 288 Earlier US FDA‐approved ubiquitination inhibitors include Bortezomib and Carfilzomib, which are also used for treating multiple myeloma. 289 , 290 These targeted drugs are part of the broader strategy of targeted protein degradation. However, due to the diversity of Ub enzymes and substrates, and the specificity required for targeted therapies, developing universally applicable targeted drugs is quite challenging. In this context, drug research has also focused on improving drug delivery systems, such as enhancing the efficacy of Bortezomib in treating hepatocellular carcinoma through nanosystem‐mediated delivery. 291 Despite these advances, more efficient and safer technologies are still under development. Additionally, the reversible nature of the ubiquitination process presents complex challenges for therapeutic interventions. Understanding the dynamics of E3 ligases and DUBs is crucial for studying the broader dynamics of ubiquitination. In summary, research on ubiquitination is still in its early stages. Addressing issues of specificity and universality, integrating advanced drug delivery technologies, and undergoing extensive basic and clinical trials are essential steps for the maturation of ubiquitination research.
8. CONCLUSION
8.1. Summary of key findings
The discovery of ubiquitination has expanded our understanding of protein quality control mechanisms beyond the lysosomal pathway. Subsequent findings revealed that ubiquitinated proteins can be targeted to both the proteasome and the autophagosome, thereby adding more possibilities to the process of autophagy. Ubiquitination is a three‐enzyme cascade reaction involving E1, E2, and E3 ligases. The wide variety of these enzymes contributes to the specificity and diversity of the ubiquitination process. DUBs, acting as Ub recyclers, render ubiquitination reversible. Originally, it was believed that ubiquitination was exclusive to proteins. However, it has been proven that sugars and lipids can also be ubiquitinated. The sites on substrates where Ub attaches are known as UBDs. Based on the discovery of UIM and UBA, UBDs exist in various topologies within substrates, guiding Ub attachment and subsequent action. Ub receptors present in the proteasome recognize ubiquitinated proteins, which are then degraded into short peptides after Ub binds to these receptors. Ub is a ubiquitous molecule in living organisms, and its widespread distribution underpins its complex and diverse functions. The most well‐known function of ubiquitination is protein degradation. Misfolded proteins from various cellular locations are directed to the proteasome for degradation, facilitated by conformational changes in the proteasome. In protein localization and transport, especially for secretory proteins, ubiquitination plays a critical role in sorting and signaling across different organelles. For instance, in ERAD pathway, misfolded proteins are recognized, relocalized, and subsequently degraded by the proteasome into short peptides, which are then transported to the cytoplasm or nucleus as needed. Ubiquitination can also influence vesicle size and assembly. In the downstream processes of protein secretion, ubiquitination acts as a signal for proteins to enter and exit the Golgi apparatus. Nuclear import of proteins is also regulated by ubiquitination, underscoring its importance in altering protein localization and various cellular functions. In numerous signaling pathways, ubiquitination targets specific key proteins, thereby altering the direction and outcome of these pathways. The structural and functional attributes of ubiquitination make it crucial for maintaining biological homeostasis. When functioning properly, ubiquitination helps maintain protein homeostasis, nucleic acid stability, cellular proliferation, and immune balance. Conversely, its dysregulation can lead to a range of diseases, including tumors, neurodegenerative disorders, metabolic conditions, and infectious diseases. Understanding the mechanisms of these diseases and targeting the ubiquitination process—either by inhibiting or activating it—presents a promising therapeutic approach.
8.2. Implications for understanding disease mechanisms and therapeutic development
Ubiquitination is a complex process that involves the attachment of Ub molecules to target proteins via various Ub enzymes, followed by the interaction between these enzymes and substrate molecules, and ultimately the transport of these substrates to the proteasome for degradation. Each step in this process presents potential therapeutic targets for disease treatment. Currently identified therapeutic targets within the ubiquitination pathway include: (1) the reversible nature of the UPS; (2) targeting various enzymes involved in ubiquitination to block the process; (3) development of modulators specific to E3 ligases; (4) she emerging PROTACs system. Although various molecules have been identified in existing studies, the progress in treating specific diseases remains unclear. Continued investigation into the molecular basis of diseases caused by UPS dysregulation and the development of targeted therapeutics is crucial for advancing disease treatment and prevention. With the rapid development of other disciplines, the integration of AI technology and materials science with ubiquitination‐targeted drugs or strategies can potentially enhance the efficiency of diagnostic and therapeutic methods.
8.3. Future directions for research in ubiquitination biology
Ubiquitination is an “ancient” molecular process, earning its name “ubiquitin” due to its ubiquitous presence across cellular life. It is indispensable in cellular processes, with its dysregulation leading to various pathological conditions and diseases. However, the number of drugs targeting ubiquitination, especially those approved by the US FDA, remains scarce. Notably, there are currently no activators of the ubiquitination process available, posing a challenge for treating diseases caused by suppressed ubiquitination. Given the advancement of emerging technologies, researchers are now exploring the integration of ubiquitination with nanotechnology, AI, and other cutting‐edge technologies, aiming to develop more precise and intelligent diagnostic and therapeutic techniques targeting ubiquitination. Nonetheless, continued research in the field of ubiquitination is essential. The molecular diversity of ubiquitination and its various forms inherently pose challenges in achieving specificity. Furthermore, given the widespread distribution of Ub and its substrates (proteins, sugars, lipids), there remains substantial scope for research. The ongoing decryption of ubiquitination processes should persist to unlock its full potential and therapeutic applications.
AUTHOR CONTRIBUTIONS
Conceptualization and writing—original draft: Yan Liao. Writing—review: Wangzheqi Zhang. Supervision: Yang Liu. Conceptualization and writing—review: Chenglong Zhu. Supervision and funding acquisition: Zui Zou. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
This work was supported by Science and Technology Commission of Shanghai Municipality (20XD1434400), Talent Development Fund of Shanghai (2020075), and Shanghai Industrial Collaborative Innovation Project (HCXBCY‐2023‐041).
Liao Y, Zhang W, Liu Y, Zhu C, Zou Z. The role of ubiquitination in health and disease. MedComm. 2024;5:e736. 10.1002/mco2.736
Contributor Information
Chenglong Zhu, Email: smmuzcl1997@163.com.
Zui Zou, Email: zuizou@smmu.edu.cn.
DATA AVAILABILITY STATEMENT
All data are freely available from the corresponding author upon request.
REFERENCES
- 1. Wilkinson KD. Ubiquitin: a nobel protein. Cell. 2004;119(6):741‐745. [DOI] [PubMed] [Google Scholar]
- 2. Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005;6(1):79‐87. [DOI] [PubMed] [Google Scholar]
- 3. Wang X, Pattison JS, Su H. Posttranslational modification and quality control. Circ Res. 2013;112(2):367‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dikic I, Schulman BA. An expanded lexicon for the ubiquitin code. Nat Rev Mol Cell Biol. 2023;24(4):273‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ikeda F, Crosetto N, Dikic I. What determines the specificity and outcomes of ubiquitin signaling? Cell. 2010;143(5):677‐681. [DOI] [PubMed] [Google Scholar]
- 6. Schlesinger DH, Goldstein G. Molecular conservation of 74 amino acid sequence of ubiquitin between cattle and man. Nature. 1975;255(5507):423‐424. [DOI] [PubMed] [Google Scholar]
- 7. Wilkinson KD, Audhya TK. Stimulation of ATP‐dependent proteolysis requires ubiquitin with the COOH‐terminal sequence Arg‐Gly‐Gly. J Biol Chem. 1981;256(17):9235‐9241. [PubMed] [Google Scholar]
- 8. Kiel C, Serrano L. The ubiquitin domain superfold: structure‐based sequence alignments and characterization of binding epitopes. J Mol Biol. 2006;355(4):821‐844. [DOI] [PubMed] [Google Scholar]
- 9. Kumar D, Sharma A, Sharma L. A comprehensive review of Alzheimer's association with related proteins: pathological role and therapeutic significance. Curr Neuropharmacol. 2020;18(8):674‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbé S. Deubiquitylases from genes to organism. Physiol Rev. 2013;93(3):1289‐1315. [DOI] [PubMed] [Google Scholar]
- 11. Vertegaal ACO. Uncovering ubiquitin and ubiquitin‐like signaling networks. Chem Rev. 2011;111(12):7923‐7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yau R, Rape M. The increasing complexity of the ubiquitin code. Nat Cell Biol. 2016;18(6):579‐586. [DOI] [PubMed] [Google Scholar]
- 13. Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203‐229. [DOI] [PubMed] [Google Scholar]
- 14. Deng L, Meng T, Chen L, Wei W, Wang P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct Target Ther. 2020;5(1):11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang T, Wang J. K63‐linked polyubiquitination of IRF1: an essential step in the IL‐1 signaling cascade. Cell Mol Immunol. 2014;11(5):407‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hrdinka M, Gyrd‐Hansen M. The Met1‐linked ubiquitin machinery: emerging themes of (de)regulation. Mol Cell. 2017;68(2):265‐280. [DOI] [PubMed] [Google Scholar]
- 17. Zhang H, Jin X, Huang H. Deregulation of SPOP in cancer. Cancer Res. 2023;83(4):489‐499. [DOI] [PubMed] [Google Scholar]
- 18. Engelender S. Ubiquitination of alpha‐synuclein and autophagy in Parkinson's disease. Autophagy. 2008;4(3):372‐374. [DOI] [PubMed] [Google Scholar]
- 19. Srivastava A, McGrath B, Bielas SL. Histone H2A monoubiquitination in neurodevelopmental disorders. Trends Genet. 2017;33(8):566‐578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Humphreys LM, Smith P, Chen Z, Fouad S, D'Angiolella V. The role of E3 ubiquitin ligases in the development and progression of glioblastoma. Cell Death Differ. 2021;28(2):522‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rahman S, Wolberger C. Breaking the K48‐chain: linking ubiquitin beyond protein degradation. Nat Struct Mol Biol. 2024;31(2):216‐218. [DOI] [PubMed] [Google Scholar]
- 22. Cohen P, Strickson S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017;24(7):1153‐1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buneeva O, Medvedev A. Atypical ubiquitination and Parkinson's disease. Int J Mol Sci. 2022;23(7):3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon YT, Ciechanover A. The ubiquitin code in the ubiquitin‐proteasome system and autophagy. Trends Biochem Sci. 2017;42(11):873‐886. [DOI] [PubMed] [Google Scholar]
- 25. Kulathu Y, Komander D. Atypical ubiquitylation—the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13(8):508‐523. [DOI] [PubMed] [Google Scholar]
- 26. Lindsten K, de Vrij FMS, Verhoef LGGC, et al. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J Cell Biol. 2002;157(3):417‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tokunaga F, Nakagawa T, Nakahara M, et al. SHARPIN is a component of the NF‐κB‐activating linear ubiquitin chain assembly complex. Nature. 2011;471(7340):633‐636. [DOI] [PubMed] [Google Scholar]
- 28. Scaglione KM, Basrur V, Ashraf NS, et al. The ubiquitin‐conjugating enzyme (E2) Ube2w ubiquitinates the N terminus of substrates. J Biol Chem. 2013;288(26):18784‐18788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tokunaga F, Sichi S, Saeki Y, et al. Involvement of linear polyubiquitylation of NEMO in NF‐κB activation. Nat Cell Biol. 2009;11(2):123‐132. [DOI] [PubMed] [Google Scholar]
- 30. Kienle SM, Schneider T, Stuber K, et al. Electrostatic and steric effects underlie acetylation‐induced changes in ubiquitin structure and function. Nat Commun. 2022;13(1):5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Swatek KN, Komander D. Ubiquitin modifications. Cell Res. 2016;26(4):399‐422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhogaraju S, Kalayil S, Liu Y, et al. Phosphoribosylation of ubiquitin promotes serine ubiquitination and impairs conventional ubiquitination. Cell. 2016;167(6):1636‐1649. e13. [DOI] [PubMed] [Google Scholar]
- 33. Schulman BA, Harper JW. Ubiquitin‐like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10(5):319‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Budhidarmo R, Nakatani Y, Day CL. RINGs hold the key to ubiquitin transfer. Trends Biochem Sci. 2012;37(2):58‐65. [DOI] [PubMed] [Google Scholar]
- 35. Lee I, Schindelin H. Structural insights into E1‐catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell. 2008;134(2):268‐278. [DOI] [PubMed] [Google Scholar]
- 36. Jaremko M, Jaremko Ł, Nowakowski M, et al. NMR structural studies of the first catalytic half‐domain of ubiquitin activating enzyme. J Struct Biol. 2014;185(1):69‐78. [DOI] [PubMed] [Google Scholar]
- 37. Yuan L, Lv Z, Adams MJ, Olsen SK. Crystal structures of an E1‐E2‐ubiquitin thioester mimetic reveal molecular mechanisms of transthioesterification. Nat Commun. 2021;12(1):2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Johnson ES, Schwienhorst I, Dohmen RJ, Blobel G. The ubiquitin‐like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 1997;16(18):5509‐5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lv Z, Yuan L, Atkison JH, Aldana‐Masangkay G, Chen Y, Olsen SK. Domain alternation and active site remodeling are conserved structural features of ubiquitin E1. J Biol Chem. 2017;292(29):12089‐12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Olsen SK, Lima CD. Structure of a ubiquitin E1‐E2 complex: insights to E1‐E2 thioester transfer. Mol Cell. 2013;49(5):884‐896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ye Y, Rape M. Building ubiquitin chains: e2 enzymes at work. Nat Rev Mol Cell Biol. 2009;10(11):755‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stewart MD, Ritterhoff T, Klevit RE, Brzovic PS. E2 enzymes: more than just middle men. Cell Res. 2016;26(4):423‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McGinty RK, Henrici RC, Tan S. Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome. Nature. 2014;514(7524):591‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pruneda JN, Smith FD, Daurie A, et al. E2∼Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J. 2014;33(5):437‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Olsen SK, Capili AD, Lu X, Tan DS, Lima CD. Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature. 2010;463(7283):906‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wiener R, DiBello AT, Lombardi PM, et al. E2 ubiquitin‐conjugating enzymes regulate the deubiquitinating activity of OTUB1. Nat Struct Mol Biol. 2013;20(9):1033‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86:129‐157. [DOI] [PubMed] [Google Scholar]
- 48. Malynn BA, Ma A. Ubiquitin makes its mark on immune regulation. Immunity. 2010;33(6):843‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuan L, Lv Z, Atkison JH, Olsen SK. Structural insights into the mechanism and E2 specificity of the RBR E3 ubiquitin ligase HHARI. Nat Commun. 2017;8(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qiu J, Sheedlo MJ, Yu K, et al. Ubiquitination independent of E1 and E2 enzymes by bacterial effectors. Nature. 2016;533(7601):120‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang R, Shi S. The role of NEDD4 related HECT‐type E3 ubiquitin ligases in defective autophagy in cancer cells: molecular mechanisms and therapeutic perspectives. Mol Med. 2023;29:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang XS, Cotton TR, Trevelyan SJ, et al. The unifying catalytic mechanism of the RING‐between‐RING E3 ubiquitin ligase family. Nat Commun. 2023;14(1):168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pao KC, Wood NT, Knebel A, et al. Activity‐based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature. 2018;556(7701):381‐385. [DOI] [PubMed] [Google Scholar]
- 54. Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474(7349):105‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin‐chain formation by the human anaphase‐promoting complex. Cell. 2008;133(4):653‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Berndsen CE, Wolberger C. New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol. 2014;21(4):301‐307. [DOI] [PubMed] [Google Scholar]
- 57. Sherpa D, Chrustowicz J, Schulman BA. How the ends signal the end: regulation by E3 ubiquitin ligases recognizing protein termini. Mol Cell. 2022;82(8):1424‐1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Varshavsky A. The ubiquitin system, autophagy, and regulated protein degradation. Annu Rev Biochem. 2017;86:123‐128. [DOI] [PubMed] [Google Scholar]
- 59. Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33(3):275‐286. [DOI] [PubMed] [Google Scholar]
- 60. Lange SM, Armstrong LA, Kulathu Y. Deubiquitinases: from mechanisms to their inhibition by small molecules. Mol Cell. 2022;82(1):15‐29. [DOI] [PubMed] [Google Scholar]
- 61. Harrigan JA, Jacq X, Martin NM, Jackson SP. Deubiquitylating enzymes and drug discovery: emerging opportunities. Nat Rev Drug Discov. 2018;17(1):57‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu F, Zhuang W, Song B, et al. MAVS‐loaded unanchored Lys63‐linked polyubiquitin chains activate the RIG‐I‐MAVS signaling cascade. Cell Mol Immunol. 2023;20(10):1186‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang Y, Shi M, Feng H, et al. Structural insights into non‐canonical ubiquitination catalyzed by SidE. Cell. 2018;173(5):1231‐1243. e16. [DOI] [PubMed] [Google Scholar]
- 64. Clague MJ, Urbé S, Komander D. Breaking the chains: deubiquitylating enzyme specificity begets function. Nat Rev Mol Cell Biol. 2019;20(6):338‐352. [DOI] [PubMed] [Google Scholar]
- 65. Komander D, Clague MJ, Urbé S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009;10(8):550‐563. [DOI] [PubMed] [Google Scholar]
- 66. Rape M. Ubiquitylation at the crossroads of development and disease. Nat Rev Mol Cell Biol. 2018;19(1):59‐70. [DOI] [PubMed] [Google Scholar]
- 67. Hicke L, Schubert HL, Hill CP. Ubiquitin‐binding domains. Nat Rev Mol Cell Biol. 2005;6(8):610‐621. [DOI] [PubMed] [Google Scholar]
- 68. Husnjak K, Dikic I. Ubiquitin‐binding proteins: decoders of ubiquitin‐mediated cellular functions. Annu Rev Biochem. 2012;81:291‐322. [DOI] [PubMed] [Google Scholar]
- 69. Dikic I, Wakatsuki S, Walters KJ. Ubiquitin binding domains — from structures to functions. Nat Rev Mol Cell Biol. 2009;10(10):659‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu K, DeVita RJ, Pan ZQ. Monoubiquitination empowers ubiquitin chain elongation. J Biol Chem. 2024;300(3):105753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Crosas B, Hanna J, Kirkpatrick DS, et al. Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell. 2006;127(7):1401‐1413. [DOI] [PubMed] [Google Scholar]
- 72. Harper JW, Schulman BA. Structural complexity in ubiquitin recognition. Cell. 2006;124(6):1133‐1136. [DOI] [PubMed] [Google Scholar]
- 73. Raasi S, Varadan R, Fushman D, Pickart CM. Diverse polyubiquitin interaction properties of ubiquitin‐associated domains. Nat Struct Mol Biol. 2005;12(8):708‐714. [DOI] [PubMed] [Google Scholar]
- 74. Di Fiore PP, Polo S, Hofmann K. When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol. 2003;4(6):491‐497. [DOI] [PubMed] [Google Scholar]
- 75. Liang RY, Chen L, Ko BT, et al. Rad23 interaction with the proteasome is regulated by phosphorylation of its ubiquitin‐like (UbL) domain. J Mol Biol. 2014;426(24):4049‐4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Isasa M, Katz EJ, Kim W, et al. Monoubiquitination of RPN10 regulates substrate recruitment to the proteasome. Mol Cell. 2010;38(5):733‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kim HT, Kim KP, Uchiki T, Gygi SP, Goldberg AL. S5a promotes protein degradation by blocking synthesis of nondegradable forked ubiquitin chains. EMBO J. 2009;28(13):1867‐1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fishbain S, Prakash S, Herrig A, Elsasser S, Matouschek A. Rad23 escapes degradation because it lacks a proteasome initiation region. Nat Commun. 2011;2:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24(3):167‐185. [DOI] [PubMed] [Google Scholar]
- 80. Peng H, Yang J, Li G, et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017;27(5):657‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. You Z, Jiang WX, Qin LY, et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat Commun. 2019;10(1):5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Turco E, Savova A, Gere F, et al. Reconstitution defines the roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation and autophagy initiation. Nat Commun. 2021;12(1):5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Clague MJ, Urbé S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143(5):682‐685. [DOI] [PubMed] [Google Scholar]
- 84. Pickart CM. Back to the future with ubiquitin. Cell. 2004;116(2):181‐190. [DOI] [PubMed] [Google Scholar]
- 85. Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin‐proteasome pathway substrates. Mol Cell. 2009;33(4):517‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hershko A, Ciechanover A, Rose IA. Resolution of the ATP‐dependent proteolytic system from reticulocytes: a component that interacts with ATP. Proc Natl Acad Sci USA. 1979;76(7):3107‐3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A. Structure and function of the 26S proteasome. Annu Rev Biochem. 2018;87:697‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Samant RS, Livingston CM, Sontag EM, Frydman J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature. 2018;563(7731):407‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kleijnen MF, Roelofs J, Park S, et al. Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat Struct Mol Biol. 2007;14(12):1180‐1188. [DOI] [PubMed] [Google Scholar]
- 90. Collins GA, Goldberg AL. The logic of the 26S proteasome. Cell. 2017;169(5):792‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bard JAM, Bashore C, Dong KC, Martin A. The 26S proteasome utilizes a kinetic gateway to prioritize substrate degradation. Cell. 2019;177(2):286‐298. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Deshmukh FK, Ben‐Nissan G, Olshina MA, et al. Allosteric regulation of the 20S proteasome by the catalytic core regulators (CCRs) family. Nat Commun. 2023;14(1):3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lu A, Pfeffer SR. A CULLINary ride across the secretory pathway: more than just secretion. Trends Cell Biol. 2014;24(7):389‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Betegon M, Brodsky JL. Unlocking the door for ERAD. Nat Cell Biol. 2020;22(3):263‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Chino H, Mizushima N. ER‐Phagy: quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 2020;30(5):384‐398. [DOI] [PubMed] [Google Scholar]
- 96. Ji CH, Kim HY, Heo AJ, et al. The N‐Degron pathway mediates ER‐phagy. Mol Cell. 2019;75(5):1058‐1072. e9. [DOI] [PubMed] [Google Scholar]
- 97. Cybulsky AV. The intersecting roles of endoplasmic reticulum stress, ubiquitin‐ proteasome system, and autophagy in the pathogenesis of proteinuric kidney disease. Kidney Int. 2013;84(1):25‐33. [DOI] [PubMed] [Google Scholar]
- 98. Chung KP, Zeng Y, Jiang L. COPII paralogs in plants: functional redundancy or diversity? Trends Plant Sci. 2016;21(9):758‐769. [DOI] [PubMed] [Google Scholar]
- 99. Jin L, Pahuja KB, Wickliffe KE, et al. Ubiquitin‐dependent regulation of COPII coat size and function. Nature. 2012;482(7386):495‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Piper RC, Lehner PJ. Endosomal transport via ubiquitination. Trends Cell Biol. 2011;21(11):647‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Lauwers E, Erpapazoglou Z, Haguenauer‐Tsapis R, André B. The ubiquitin code of yeast permease trafficking. Trends Cell Biol. 2010;20(4):196‐204. [DOI] [PubMed] [Google Scholar]
- 102. Litterman N, Ikeuchi Y, Gallardo G, et al. An OBSL1‐Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 2011;9(5):e1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhang ZD, Li HX, Gan H, et al. RNF115 inhibits the post‐ER trafficking of TLRs and TLRs‐mediated immune responses by catalyzing K11‐linked ubiquitination of RAB1A and RAB13. Adv Sci (Weinh). 2022;9(16):e2105391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hegazi S, Cheng AH, Krupp JJ, et al. UBR4/POE facilitates secretory trafficking to maintain circadian clock synchrony. Nat Commun. 2022;13(1):1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Goswami R, Gupta A, Bednova O, et al. Nuclear localization signal‐tagged systems: relevant nuclear import principles in the context of current therapeutic design. Chem Soc Rev. 2024;53(1):204‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wang YE, Park A, Lake M, et al. Ubiquitin‐regulated nuclear‐cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010;6(11):e1001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chan WM, Poon RYC. The p53 Isoform Deltap53 lacks intrinsic transcriptional activity and reveals the critical role of nuclear import in dominant‐negative activity. Cancer Res. 2007;67(5):1959‐1969. [DOI] [PubMed] [Google Scholar]
- 108. Khosravi B, LaClair KD, Riemenschneider H, et al. Cell‐to‐cell transmission of C9orf72 poly‐(Gly‐Ala) triggers key features of ALS/FTD. EMBO J. 2020;39(8):e102811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zheng Z, Zeng X, Zhu Y, et al. CircPPAP2B controls metastasis of clear cell renal cell carcinoma via HNRNPC‐dependent alternative splicing and targeting the miR‐182‐5p/CYP1B1 axis. Mol Cancer. 2024;23(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Vogel K, Bläske T, Nagel MK, et al. Lipid‐mediated activation of plasma membrane‐localized deubiquitylating enzymes modulate endosomal trafficking. Nat Commun. 2022;13(1):6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang L, Xiao X, Arnold PR, Li XC. Transcriptional and epigenetic regulation of immune tolerance: roles of the NF‐κB family members. Cell Mol Immunol. 2019;16(4):315‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature. 2009;458(7237):430‐437. [DOI] [PubMed] [Google Scholar]
- 113. Sun SC. The non‐canonical NF‐κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu Z, Mar KB, Hanners NW, et al. A NIK–SIX signalling axis controls inflammation by targeted silencing of non‐canonical NF‐κB. Nature. 2019;568(7751):249‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Tanaka T, Grusby MJ, Kaisho T. PDLIM2‐mediated termination of transcription factor NF‐kappaB activation by intranuclear sequestration and degradation of the p65 subunit. Nat Immunol. 2007;8(6):584‐591. [DOI] [PubMed] [Google Scholar]
- 116. Ruland J. Return to homeostasis: downregulation of NF‐κB responses. Nat Immunol. 2011;12(8):709‐714. [DOI] [PubMed] [Google Scholar]
- 117. Kumari N, Jaynes PW, Saei A, Iyengar PV, Richard JLC, Eichhorn PJA. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim Biophys Acta Rev Cancer. 2017;1868(2):456‐483. [DOI] [PubMed] [Google Scholar]
- 118. Xu X, Zheng L, Yuan Q, et al. Transforming growth factor‐β in stem cells and tissue homeostasis. Bone Res. 2018;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Koo BK, Spit M, Jordens I, et al. Tumour suppressor RNF43 is a stem‐cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012;488(7413):665‐669. [DOI] [PubMed] [Google Scholar]
- 120. Ng VH, Spencer Z, Neitzel LR, et al. The USP46 complex deubiquitylates LRP6 to promote Wnt/β‐catenin signaling. Nat Commun. 2023;14(1):6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dominguez‐Brauer C, Hao Z, Elia AJ, et al. Mule regulates the intestinal stem cell niche via the Wnt pathway and targets EphB3 for proteasomal and lysosomal degradation. Cell Stem Cell. 2016;19(2):205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Fan Y, Huo X, Guo B, et al. Cullin 4b‐RING ubiquitin ligase targets IRGM1 to regulate Wnt signaling and intestinal homeostasis. Cell Death Differ. 2022;29(9):1673‐1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Chitalia VC, Foy RL, Bachschmid MM, et al. Jade‐1 inhibits Wnt signalling by ubiquitylating β‐catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. 2008;10(10):1208‐1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tsuchiya H, Burana D, Ohtake F, et al. Ub‐ProT reveals global length and composition of protein ubiquitylation in cells. Nat Commun. 2018;9(1):524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Gur G, Rubin C, Katz M, et al. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. EMBO J. 2004;23(16):3270‐3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Inagi R, Ishimoto Y, Nangaku M. Proteostasis in endoplasmic reticulum–new mechanisms in kidney disease. Nat Rev Nephrol. 2014;10(7):369‐378. [DOI] [PubMed] [Google Scholar]
- 127. You K, Wang L, Chou CH, et al. QRICH1 dictates the outcome of ER stress through transcriptional control of proteostasis. Science. 2021;371(6524):eabb6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tawo R, Pokrzywa W, Kevei É, et al. The ubiquitin ligase CHIP integrates proteostasis and aging by regulation of insulin receptor turnover. Cell. 2017;169(3):470‐482. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Chiosis G, Digwal CS, Trepel JB, Neckers L. Structural and functional complexity of HSP90 in cellular homeostasis and disease. Nat Rev Mol Cell Biol. 2023;24(11):797‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Pessa JC, Joutsen J, Sistonen L. Transcriptional reprogramming at the intersection of the heat shock response and proteostasis. Mol Cell. 2024;84(1):80‐93. [DOI] [PubMed] [Google Scholar]
- 131. Henning RH, Brundel BJJM. Proteostasis in cardiac health and disease. Nat Rev Cardiol. 2017;14(11):637‐653. [DOI] [PubMed] [Google Scholar]
- 132. El Ayadi A, Stieren ES, Barral JM, Boehning D. Ubiquilin‐1 regulates amyloid precursor protein maturation and degradation by stimulating K63‐linked polyubiquitination of lysine 688. Proc Natl Acad Sci USA. 2012;109(33):13416‐13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Rane NS, Kang SW, Chakrabarti O, Feigenbaum L, Hegde RS. Reduced translocation of nascent prion protein during ER stress contributes to neurodegeneration. Dev Cell. 2008;15(3):359‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Leznicki P, High S. SGTA antagonizes BAG6‐mediated protein triage. Proc Natl Acad Sci USA. 2012;109(47):19214‐19219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Xu Y, Anderson DE, Ye Y. The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell Discov. 2016;2:16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Avci D, Lemberg MK. Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol. 2015;25(10):611‐622. [DOI] [PubMed] [Google Scholar]
- 137. Liu G, Rogers J, Murphy CT, Rongo C. EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J. 2011;30(15):2990‐3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ceccaldi R, Rondinelli B, D'Andrea AD. Repair pathway choices and consequences at the double‐strand break. Trends Cell Biol. 2016;26(1):52‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Zhao W, Steinfeld JB, Liang F, et al. BRCA1‐BARD1 promotes RAD51‐mediated homologous DNA pairing. Nature. 2017;550(7676):360‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hu Q, Botuyan MV, Zhao D, Cui G, Mer E, Mer G. Mechanisms of BRCA1‐BARD1 nucleosome recognition and ubiquitylation. Nature. 2021;596(7872):438‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Tarsounas M, Sung P. The antitumorigenic roles of BRCA1‐BARD1 in DNA repair and replication. Nat Rev Mol Cell Biol. 2020;21(5):284‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Becker JR, Clifford G, Bonnet C, Groth A, Wilson MD, Chapman JR. BARD1 reads H2A lysine 15 ubiquitination to direct homologous recombination. Nature. 2021;596(7872):433‐437. [DOI] [PubMed] [Google Scholar]
- 143. Qiu L, Xu W, Lu X, et al. The HDAC6‐RNF168 axis regulates H2A/H2A.X ubiquitination to enable double‐strand break repair. Nucleic Acids Res. 2023;51(17):9166‐9182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Nowsheen S, Aziz K, Aziz A, et al. L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nat Cell Biol. 2018;20(4):455‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Luo K, Deng M, Li Y, et al. CDK‐mediated RNF4 phosphorylation regulates homologous recombination in S‐phase. Nucleic Acids Res. 2015;43(11):5465‐5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Jeong E, Lee SG, Kim HS, et al. Structural basis of the fanconi anemia‐associated mutations within the FANCA and FANCG complex. Nucleic Acids Res. 2020;48(6):3328‐3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Longerich S, Kwon Y, Tsai MS, Hlaing AS, Kupfer GM, Sung P. Regulation of FANCD2 and FANCI monoubiquitination by their interaction and by DNA. Nucleic Acids Res. 2014;42(9):5657‐5670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Alcón P, Shakeel S, Chen ZA, Rappsilber J, Patel KJ, Passmore LA. FANCD2‐FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat Struct Mol Biol. 2020;27(3):240‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Rego MA, Kolling FW, Vuono EA, Mauro M, Howlett NG. Regulation of the Fanconi anemia pathway by a CUE ubiquitin‐binding domain in the FANCD2 protein. Blood. 2012;120(10):2109‐2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Williams SA, Longerich S, Sung P, Vaziri C, Kupfer GM. The E3 ubiquitin ligase RAD18 regulates ubiquitylation and chromatin loading of FANCD2 and FANCI. Blood. 2011;117(19):5078‐5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Nakazawa Y, Sasaki K, Mitsutake N, et al. Mutations in UVSSA cause UV‐sensitive syndrome and impair RNA polymerase IIo processing in transcription‐coupled nucleotide‐excision repair. Nat Genet. 2012;44(5):586‐592. [DOI] [PubMed] [Google Scholar]
- 152. Nakazawa Y, Hara Y, Oka Y, et al. Ubiquitination of DNA damage‐stalled RNAPII promotes transcription‐coupled repair. Cell. 2020;180(6):1228‐1244. e24. [DOI] [PubMed] [Google Scholar]
- 153. Essawy M, Chesner L, Alshareef D, Ji S, Tretyakova N, Campbell C. Ubiquitin signaling and the proteasome drive human DNA‐protein crosslink repair. Nucleic Acids Research. 2023;51(22):12174‐12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Chitale S, Richly H. DICER and ZRF1 contribute to chromatin decondensation during nucleotide excision repair. Nucleic Acids Res. 2017;45(10):5901‐5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sun X, Tang H, Chen Y, et al. Loss of the receptors ER, PR and HER2 promotes USP15‐dependent stabilization of PARP1 in triple‐negative breast cancer. Nat Cancer. 2023;4(5):716‐733. [DOI] [PubMed] [Google Scholar]
- 156. Rona G, Miwatani‐Minter B, Zhang Q, et al. CDK‐independent role of D‐type cyclins in regulating DNA mismatch repair. Mol Cell. 2024;84(7):1224‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Dang F, Nie L, Wei W. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ. 2021;28(2):427‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Zhang S, Chang L, Alfieri C, et al. Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature. 2016;533(7602):260‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Fujimitsu K, Grimaldi M, Yamano H. Cyclin‐dependent kinase 1‐dependent activation of APC/C ubiquitin ligase. Science. 2016;352(6289):1121‐1124. [DOI] [PubMed] [Google Scholar]
- 160. Hein JB, Nilsson J. Interphase APC/C‐Cdc20 inhibition by cyclin A2‐Cdk2 ensures efficient mitotic entry. Nat Commun. 2016;7:10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Zhou CJ, Wang XY, Dong YH, et al. CENP‐F‐dependent DRP1 function regulates APC/C activity during oocyte meiosis I. Nat Commun. 2022;13(1):7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Izawa D, Pines J. The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature. 2015;517(7536):631‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Hellmuth S, Gómez‐H L, Pendás AM, Stemmann O. Securin‐independent regulation of separase by checkpoint‐induced shugoshin‐MAD2. Nature. 2020;580(7804):536‐541. [DOI] [PubMed] [Google Scholar]
- 164. Tischer T, Hörmanseder E, Mayer TU. The APC/C inhibitor XErp1/Emi2 is essential for Xenopus early embryonic divisions. Science. 2012;338(6106):520‐524. [DOI] [PubMed] [Google Scholar]
- 165. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF‐κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5(1):209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Gong Y, Zack TI, Morris LGT, et al. Pan‐cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet. 2014;46(6):588‐594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Alao JP, Stavropoulou AV, Lam EWF, Coombes RC, Vigushin DM. Histone deacetylase inhibitor, trichostatin A induces ubiquitin‐dependent cyclin D1 degradation in MCF‐7 breast cancer cells. Mol Cancer. 2006;5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Barr AR, Cooper S, Heldt FS, et al. DNA damage during S‐phase mediates the proliferation‐quiescence decision in the subsequent G1 via p21 expression. Nat Commun. 2017;8:14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Hume S, Grou CP, Lascaux P, et al. The NUCKS1‐SKP2‐p21/p27 axis controls S phase entry. Nat Commun. 2021;12(1):6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Li J, D'Angiolella V, Seeley ES, et al. USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature. 2013;495(7440):255‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Tang D, Xiang Y, De Renzis S, et al. The ubiquitin ligase HACE1 regulates Golgi membrane dynamics during the cell cycle. Nat Commun. 2011;2:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Chen NP, Aretz J, Fässler R. CDK1‐cyclin‐B1‐induced kindlin degradation drives focal adhesion disassembly at mitotic entry. Nat Cell Biol. 2022;24(5):723‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Sakaue‐Sawano A, Yo M, Komatsu N, et al. Genetically encoded tools for optical dissection of the mammalian cell cycle. Mol Cell. 2017;68(3):626‐640. [DOI] [PubMed] [Google Scholar]
- 174. Yang M, Chen T, Li X, et al. K33‐linked polyubiquitination of Zap70 by Nrdp1 controls CD8(+) T cell activation. Nat Immunol. 2015;16(12):1253‐1262. [DOI] [PubMed] [Google Scholar]
- 175. Aki D, Li H, Zhang W, et al. The E3 ligases Itch and WWP2 cooperate to limit TH2 differentiation by enhancing signaling through the TCR. Nat Immunol. 2018;19(7):766‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Park Y, Jin HS, Lopez J, et al. SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat Immunol. 2016;17(3):286‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Shih YC, Chen HF, Wu CY, et al. The phosphatase DUSP22 inhibits UBR2‐mediated K63‐ubiquitination and activation of Lck downstream of TCR signalling. Nat Commun. 2024;15(1):532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Dufner A, Kisser A, Niendorf S, et al. The ubiquitin‐specific protease USP8 is critical for the development and homeostasis of T cells. Nat Immunol. 2015;16(9):950‐960. [DOI] [PubMed] [Google Scholar]
- 179. Chen S, Yun F, Yao Y, et al. USP38 critically promotes asthmatic pathogenesis by stabilizing JunB protein. J Exp Med. 2018;215(11):2850‐2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. O'Leary CE, Riling CR, Spruce LA, et al. Ndfip‐mediated degradation of Jak1 tunes cytokine signalling to limit expansion of CD4+ effector T cells. Nat Commun. 2016;7:11226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Li X, Gadzinsky A, Gong L, et al. Cbl ubiquitin ligases control B cell exit from the germinal‐center reaction. Immunity. 2018;48(3):530‐541. e6. [DOI] [PubMed] [Google Scholar]
- 182. Ramkumar C, Cui H, Kong Y, Jones SN, Gerstein RM, Zhang H. Smurf2 suppresses B‐cell proliferation and lymphomagenesis by mediating ubiquitination and degradation of YY1. Nat Commun. 2013;4:2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Moser EK, Roof J, Dybas JM, et al. The E3 ubiquitin ligase Itch restricts antigen‐driven B cell responses. J Exp Med. 2019;216(9):2170‐2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Bannard O, McGowan SJ, Ersching J, et al. Ubiquitin‐mediated fluctuations in MHC class II facilitate efficient germinal center B cell responses. J Exp Med. 2016;213(6):993‐1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Karin M, Gallagher E. TNFR signaling: ubiquitin‐conjugated TRAFfic signals control stop‐and‐go for MAPK signaling complexes. Immunol Rev. 2009;228(1):225‐240. [DOI] [PubMed] [Google Scholar]
- 186. Pertel T, Hausmann S, Morger D, et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature. 2011;472(7343):361‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Chakraborty A, Diefenbacher ME, Mylona A, Kassel O, Behrens A. The E3 ubiquitin ligase Trim7 mediates c‐Jun/AP‐1 activation by Ras signalling. Nat Commun. 2015;6:6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Oh YT, Yue P, Sun SY. DR5 suppression induces sphingosine‐1‐phosphate‐dependent TRAF2 polyubiquitination, leading to activation of JNK/AP‐1 and promotion of cancer cell invasion. Cell Commun Signal. 2017;15(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2024;25(5):379‐395. [DOI] [PubMed] [Google Scholar]
- 190. Elias EE, Lyons B, Muruve DA. Gasdermins and pyroptosis in the kidney. Nat Rev Nephrol. 2023;19(5):337‐350. [DOI] [PubMed] [Google Scholar]
- 191. Liu X, Zhang Z, Ruan J, et al. Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535(7610):153‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Song H, Zhao C, Yu Z, et al. UAF1 deubiquitinase complexes facilitate NLRP3 inflammasome activation by promoting NLRP3 expression. Nat Commun. 2020;11(1):6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Zhao W, Shi CS, Harrison K, et al. AKT regulates NLRP3 inflammasome activation by phosphorylating NLRP3 serine 5. J Immunol. 2020;205(8):2255‐2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhu Y, Zhang J, Yao X, et al. Ubiquitinated gasdermin D mediates arsenic‐induced pyroptosis and hepatic insulin resistance in rat liver. Food Chem Toxicol. 2022;160:112771. [DOI] [PubMed] [Google Scholar]
- 195. Vijayaraj SL, Feltham R, Rashidi M, et al. The ubiquitylation of IL‐1β limits its cleavage by caspase‐1 and targets it for proteasomal degradation. Nat Commun. 2021;12(1):2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Schwartz AL, Ciechanover A. Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol. 2009;49:73‐96. [DOI] [PubMed] [Google Scholar]
- 197. Hoeller D, Dikic I. Targeting the ubiquitin system in cancer therapy. Nature. 2009;458(7237):438‐444. [DOI] [PubMed] [Google Scholar]
- 198. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20(11):1242‐1253. [DOI] [PubMed] [Google Scholar]
- 199. Dewson G, Eichhorn PJA, Komander D. Deubiquitinases in cancer. Nat Rev Cancer. 2023;23(12):842‐862. [DOI] [PubMed] [Google Scholar]
- 200. Antao AM, Tyagi A, Kim KS, Ramakrishna S. Advances in deubiquitinating enzyme inhibition and applications in cancer therapeutics. Cancers (Basel). 2020;12(6):1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Park HB, Kim JW, Baek KH. Regulation of Wnt signaling through ubiquitination and deubiquitination in cancers. Int J Mol Sci. 2020;21(11):3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Sun T, Liu Z, Yang Q. The role of ubiquitination and deubiquitination in cancer metabolism. Mol Cancer. 2020;19(1):146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Flügel D, Görlach A, Kietzmann T. GSK‐3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28‐dependent degradation of HIF‐1α. Blood. 2012;119(5):1292‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Gonzalez‐Pecchi V, Kwan AK, Doyle S, Ivanov AA, Du Y, Fu H. NSD3S stabilizes MYC through hindering its interaction with FBXW7. J Mol Cell Biol. 2020;12(6):438‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Mao JH, Kim IJ, Wu D, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008;321(5895):1499‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Cui D, Xiong X, Shu J, Dai X, Sun Y, Zhao Y. FBXW7 confers radiation survival by targeting p53 for degradation. Cell Rep. 2020;30(2):497‐509. e4. [DOI] [PubMed] [Google Scholar]
- 207. Ekambaram P, Lee JYL, Hubel NE, et al. The CARMA3‐Bcl10‐MALT1 signalosome drives NFκB activation and promotes aggressiveness in angiotensin II receptor‐positive breast cancer. Cancer Res. 2018;78(5):1225‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Jiang Q, Li F, Cheng Z, Kong Y, Chen C. The role of E3 ubiquitin ligase HECTD3 in cancer and beyond. Cell Mol Life Sci. 2020;77(8):1483‐1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Koo J, Yue P, Deng X, Khuri FR, Sun SY. mTOR complex 2 stabilizes Mcl‐1 protein by suppressing its glycogen synthase kinase 3‐dependent and SCF‐FBXW7‐mediated degradation. Mol Cell Biol. 2015;35(13):2344‐2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Zou H, Chen H, Zhou Z, Wan Y, Liu Z. ATXN3 promotes breast cancer metastasis by deubiquitinating KLF4. Cancer Lett. 2019;467:19‐28. [DOI] [PubMed] [Google Scholar]
- 211. Mai H, Zhou B, Liu L, et al. Molecular pattern of lncRNAs in hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38(1):198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Ju LG, Zhu Y, Long QY, et al. SPOP suppresses prostate cancer through regulation of CYCLIN E1 stability. Cell Death Differ. 2019;26(6):1156‐1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Migita K, Takayama T, Matsumoto S, et al. Prognostic impact of RING box protein‐1 (RBX1) expression in gastric cancer. Gastric Cancer. 2014;17(4):601‐609. [DOI] [PubMed] [Google Scholar]
- 214. Bernassola F, Chillemi G, Melino G. HECT‐type E3 ubiquitin ligases in cancer. Trends Biochem Sci. 2019;44(12):1057‐1075. [DOI] [PubMed] [Google Scholar]
- 215. Zhang Q, Huang H, Liu A, et al. Cell division cycle 20 (CDC20) drives prostate cancer progression via stabilization of β‐catenin in cancer stem‐like cells. EBioMedicine. 2019;42:397‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Han S, Wang R, Zhang Y, et al. The role of ubiquitination and deubiquitination in tumor invasion and metastasis. Int J Biol Sci. 2022;18(6):2292‐2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Lim SO, Li CW, Xia W, et al. Deubiquitination and stabilization of PD‐L1 by CSN5. Cancer Cell. 2016;30(6):925‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Larasati YA, Yoneda‐Kato N, Nakamae I, Yokoyama T, Meiyanto E, Kato JY. Curcumin targets multiple enzymes involved in the ROS metabolic pathway to suppress tumor cell growth. Sci Rep. 2018;8(1):2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Rajkumar SV, Kumar S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020;10(9):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Berenson JR. Hematology: bortezomib in newly diagnosed multiple myeloma. Nat Rev Clin Oncol. 2009;6(5):255‐256. [DOI] [PubMed] [Google Scholar]
- 221. Jana NR, Dikshit P, Goswami A, Nukina N. Inhibition of proteasomal function by curcumin induces apoptosis through mitochondrial pathway. J Biol Chem. 2004;279(12):11680‐11685. [DOI] [PubMed] [Google Scholar]
- 222. Ma YS, Wang XF, Yu F, et al. Inhibition of USP14 and UCH37 deubiquitinating activity by b‐AP15 as a potential therapy for tumors with p53 deficiency. Signal Transduct Target Ther. 2020;5(1):30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Tyagi A, Haq S, Ramakrishna S. Redox regulation of DUBs and its therapeutic implications in cancer. Redox Biol. 2021;48:102194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Das DS, Das A, Ray A, et al. Blockade of deubiquitylating enzyme USP1 inhibits DNA repair and triggers apoptosis in multiple myeloma cells. Clin Cancer Res. 2017;23(15):4280‐4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Mistry H, Hsieh G, Buhrlage SJ, et al. Small‐molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol Cancer Ther. 2013;12(12):2651‐2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Liang Q, Dexheimer TS, Zhang P, et al. A selective USP1‐UAF1 inhibitor links deubiquitination to DNA damage responses. Nat Chem Biol. 2014;10(4):298‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Davis MI, Pragani R, Fox JT, et al. Small molecule inhibition of the ubiquitin‐specific protease USP2 accelerates cyclin D1 degradation and leads to cell cycle arrest in colorectal cancer and mantle cell lymphoma models. J Biol Chem. 2016;291(47):24628‐24640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Magiera K, Tomala M, Kubica K, et al. Lithocholic acid hydroxyamide destabilizes cyclin D1 and induces G0/G1 arrest by inhibiting deubiquitinase USP2a. Cell Chem Biol. 2017;24(4):458‐470. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Yang J, Xu P, Han L, et al. Cutting edge: ubiquitin‐specific protease 4 promotes Th17 cell function under inflammation by deubiquitinating and stabilizing RORγt. J Immunol. 2015;194(9):4094‐4097. [DOI] [PubMed] [Google Scholar]
- 230. Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ. Deubiquitinase inhibition by small‐molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70(22):9265‐9276. [DOI] [PubMed] [Google Scholar]
- 231. Okada K, Ye YQ, Taniguchi K, et al. Vialinin A is a ubiquitin‐specific peptidase inhibitor. Bioorg Med Chem Lett. 2013;23(15):4328‐4331. [DOI] [PubMed] [Google Scholar]
- 232. Wang S, Juan J, Zhang Z, et al. Inhibition of the deubiquitinase USP5 leads to c‐Maf protein degradation and myeloma cell apoptosis. Cell Death Dis. 2017;8(9):e3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Colland F, Formstecher E, Jacq X, et al. Small‐molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol Cancer Ther. 2009;8(8):2286‐2295. [DOI] [PubMed] [Google Scholar]
- 234. Gavory G, O'Dowd CR, Helm MD, et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat Chem Biol. 2018;14(2):118‐125. [DOI] [PubMed] [Google Scholar]
- 235. Di Lello P, Pastor R, Murray JM, et al. Discovery of small‐molecule inhibitors of ubiquitin specific protease 7 (USP7) using integrated NMR and in silico techniques. J Med Chem. 2017;60(24):10056‐10070. [DOI] [PubMed] [Google Scholar]
- 236. Jing B, Liu M, Yang L, et al. Characterization of naturally occurring pentacyclic triterpenes as novel inhibitors of deubiquitinating protease USP7 with anticancer activity in vitro. Acta Pharmacol Sin. 2018;39(3):492‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Kategaya L, Di Lello P, Rougé L, et al. USP7 small‐molecule inhibitors interfere with ubiquitin binding. Nature. 2017;550(7677):534‐538. [DOI] [PubMed] [Google Scholar]
- 238. Turnbull AP, Ioannidis S, Krajewski WW, et al. Molecular basis of USP7 inhibition by selective small‐molecule inhibitors. Nature. 2017;550(7677):481‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Weinstock J, Wu J, Cao P, et al. Selective dual inhibitors of the cancer‐related deubiquitylating proteases USP7 and USP47. ACS Med Chem Lett. 2012;3(10):789‐792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Byun S, Lee SY, Lee J, et al. USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin Cancer Res. 2013;19(14):3894‐3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Peterson LF, Sun H, Liu Y, et al. Targeting deubiquitinase activity with a novel small‐molecule inhibitor as therapy for B‐cell malignancies. Blood. 2015;125(23):3588‐3597. [DOI] [PubMed] [Google Scholar]
- 242. Burkhart RA, Peng Y, Norris ZA, et al. Mitoxantrone targets human ubiquitin‐specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol Cancer Res. 2013;11(8):901‐911. [DOI] [PubMed] [Google Scholar]
- 243. Lee BH, Lee MJ, Park S, et al. Enhancement of proteasome activity by a small‐molecule inhibitor of USP14. Nature. 2010;467(7312):179‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Jiang L, Sun Y, Wang J, et al. Proteasomal cysteine deubiquitinase inhibitor b‐AP15 suppresses migration and induces apoptosis in diffuse large B cell lymphoma. J Exp Clin Cancer Res. 2019;38(1):453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Boselli M, Lee BH, Robert J, et al. An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J Biol Chem. 2017;292(47):19209‐19225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Moghadami AA, Aboutalebi Vand Beilankouhi E, Kalantary‐Charvadeh A, et al. Inhibition of USP14 induces ER stress‐mediated autophagy without apoptosis in lung cancer cell line A549. Cell Stress Chaperones. 2020;25(6):909‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Perez C, Li J, Parlati F, et al. Discovery of an inhibitor of the proteasome subunit Rpn11. J Med Chem. 2017;60(4):1343‐1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Wrigley JD, Gavory G, Simpson I, et al. Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem Biol. 2017;12(12):3113‐3125. [DOI] [PubMed] [Google Scholar]
- 249. Rusilowicz‐Jones EV, Jardine J, Kallinos A, et al. USP30 sets a trigger threshold for PINK1‐PARKIN amplification of mitochondrial ubiquitylation. Life Sci Alliance. 2020;3(8):e202000768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Li J, Yakushi T, Parlati F, et al. Capzimin is a potent and specific inhibitor of proteasome isopeptidase Rpn11. Nat Chem Biol. 2017;13(5):486‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Song Y, Li S, Ray A, et al. Blockade of deubiquitylating enzyme Rpn11 triggers apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Oncogene. 2017;36(40):5631‐5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Grethe C, Schmidt M, Kipka GM, et al. Structural basis for specific inhibition of the deubiquitinase UCHL1. Nat Commun. 2022;13(1):5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Liu S, González‐Prieto R, Zhang M, et al. Deubiquitinase activity profiling identifies UCHL1 as a candidate oncoprotein that promotes TGFβ‐induced breast cancer metastasis. Clin Cancer Res. 2020;26(6):1460‐1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10‐S17. Suppl. [DOI] [PubMed] [Google Scholar]
- 255. Galves M, Rathi R, Prag G, Ashkenazi A. Ubiquitin signaling and degradation of aggregate‐prone proteins. Trends Biochem Sci. 2019;44(10):872‐884. [DOI] [PubMed] [Google Scholar]
- 256. Ristic G, Tsou WL, Todi SV. An optimal ubiquitin‐proteasome pathway in the nervous system: the role of deubiquitinating enzymes. Front Mol Neurosci. 2014;7:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Dugger BN, Dickson DW. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol. 2017;9(7):a028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Tseng BP, Green KN, Chan JL, Blurton‐Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29(11):1607‐1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Venkatraman P, Wetzel R, Tanaka M, Nukina N, Goldberg AL. Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine‐containing proteins. Mol Cell. 2004;14(1):95‐104. [DOI] [PubMed] [Google Scholar]
- 260. Liang Y, Zhong G, Ren M, et al. The role of ubiquitin‐proteasome system and mitophagy in the pathogenesis of Parkinson's disease. Neuromolecular Med. 2023;25(4):471‐488. [DOI] [PubMed] [Google Scholar]
- 261. Ross JM, Olson L, Coppotelli G. Mitochondrial and ubiquitin proteasome system dysfunction in ageing and disease: two sides of the same coin?. Int J Mol Sci. 2015;16(8):19458‐19476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Hauser DN, Hastings TG. Mitochondrial dysfunction and oxidative stress in Parkinson's disease and monogenic parkinsonism. Neurobiol Dis. 2013;51:35‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Martin I, Dawson VL, Dawson TM. Recent advances in the genetics of Parkinson's disease. Annu Rev Genomics Hum Genet. 2011;12:301‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal. 2011;14(10):1929‐1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Todi SV, Paulson HL. Balancing act: deubiquitinating enzymes in the nervous system. Trends Neurosci. 2011;34(7):370‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Huang PL. A comprehensive definition for metabolic syndrome. Dis Model Mech. 2009;2(5‐6):231‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365(9468):1415‐1428. [DOI] [PubMed] [Google Scholar]
- 268. Saklayen MG. The global epidemic of the metabolic syndrome. Curr Hypertens Rep. 2018;20(2):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. 2017;113(4):389‐398. [DOI] [PubMed] [Google Scholar]
- 270. Nosalski R, Guzik TJ. Perivascular adipose tissue inflammation in vascular disease. Br J Pharmacol. 2017;174(20):3496‐3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Kitamura H. Ubiquitin‐specific proteases (USPs) and metabolic disorders. Int J Mol Sci. 2023;24(4):3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Yang YH, Wen R, Yang N, Zhang TN, Liu CF. Roles of protein post‐translational modifications in glucose and lipid metabolism: mechanisms and perspectives. Mol Med. 2023;29(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Yu K, Mo D, Wu M, et al. Activating transcription factor 4 regulates adipocyte differentiation via altering the coordinate expression of CCATT/enhancer binding protein β and peroxisome proliferator‐activated receptor γ. FEBS J. 2014;281(10):2399‐2409. [DOI] [PubMed] [Google Scholar]
- 274. Petersmann A, Müller‐Wieland D, Müller UA, et al. Definition, classification and diagnosis of diabetes mellitus. Exp Clin Endocrinol Diabetes. 2019;127(S 01):S1‐S7. [DOI] [PubMed] [Google Scholar]
- 275. Cortez JT, Montauti E, Shifrut E, et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature. 2020;582(7812):416‐420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Lee GR. The balance of Th17 versus Treg cells in autoimmunity. Int J Mol Sci. 2018;19(3):730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Santin I, Moore F, Grieco FA, Marchetti P, Brancolini C, Eizirik DL. USP18 is a key regulator of the interferon‐driven gene network modulating pancreatic beta cell inflammation and apoptosis. Cell Death Dis. 2012;3(11):e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Wu H, Ballantyne CM. Metabolic inflammation and insulin resistance in obesity. Circ Res. 2020;126(11):1549‐1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Hirata Y, Nomura K, Senga Y, et al. Hyperglycemia induces skeletal muscle atrophy via a WWP1/KLF15 axis. JCI Insight. 2019;4(4):e124952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Kumar R, Mehta D, Mishra N, Nayak D, Sunil S. Role of host‐mediated post‐translational modifications (PTMs) in RNA virus pathogenesis. Int J Mol Sci. 2020;22(1):323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Isaacson MK, Ploegh HL. Ubiquitination, ubiquitin‐like modifiers, and deubiquitination in viral infection. Cell Host Microbe. 2009;5(6):559‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Rytkönen A, Holden DW. Bacterial interference of ubiquitination and deubiquitination. Cell Host Microbe. 2007;1(1):13‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Sheedlo MJ, Qiu J, Tan Y, Paul LN, Luo ZQ, Das C. Structural basis of substrate recognition by a bacterial deubiquitinase important for dynamics of phagosome ubiquitination. Proc Natl Acad Sci USA. 2015;112(49):15090‐15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Ren J, Yu P, Liu S, et al. Deubiquitylating enzymes in cancer and immunity. Adv Sci (Weinh). 2023;10(36):e2303807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Szulc NA, Stefaniak F, Piechota M, et al. DEGRONOPEDIA: a web server for proteome‐wide inspection of degrons. Nucleic Acids Res. 2024;52(W1):W221‐W232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Gou Y, Liu D, Chen M, et al. GPS‐SUMO 2.0: an updated online service for the prediction of SUMOylation sites and SUMO‐interacting motifs. Nucleic Acids Res. 2024;2(W1):W238‐W247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Li F, Hu Q, Zhang X, et al. DeepPROTACs is a deep learning‐based targeted degradation predictor for PROTACs. Nat Commun. 2022;13(1):7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Guan X, Wang Y, Yu W, et al. Blocking ubiquitin‐specific protease 7 induces ferroptosis in gastric cancer via targeting stearoyl‐CoA desaturase. Adv Sci (Weinh). 2024;11(18):e2307899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Landgren O, Hultcrantz M, Diamond B, et al. Safety and effectiveness of weekly carfilzomib, lenalidomide, dexamethasone, and daratumumab combination therapy for patients with newly diagnosed multiple myeloma: the MANHATTAN nonrandomized clinical trial. JAMA Oncol. 2021;7(6):862‐868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Wang Q, Lin Z, Wang Z, et al. RARγ activation sensitizes human myeloma cells to carfilzomib treatment through the OAS‐RNase L innate immune pathway. Blood. 2022;139(1):59‐72. [DOI] [PubMed] [Google Scholar]
- 291. Mostafaei F, Hemmati S, Valizadeh H, et al. Enhanced intracellular accumulation and cytotoxicity of bortezomib against liver cancer cells using N‐stearyl lactobionamide surface modified solid lipid nanoparticles. Int J Pharm. 2024;649:123635. [DOI] [PubMed] [Google Scholar]
- 292. Kim Y, Kim W, Song Y, et al. Deubiquitinase YOD1 potentiates YAP/TAZ activities through enhancing ITCH stability. Proc Natl Acad Sci USA. 2017;114(18):4691‐4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Spel L, Nieuwenhuis J, Haarsma R, et al. Nedd4‐binding protein 1 and TNFAIP3‐interacting protein 1 control MHC‐1 display in neuroblastoma. Cancer Res. 2018;78(23):6621‐6631. [DOI] [PubMed] [Google Scholar]
- 294. Bonacci T, Suzuki A, Grant GD, et al. Cezanne/OTUD7B is a cell cycle‐regulated deubiquitinase that antagonizes the degradation of APC/C substrates. EMBO J. 2018;37(16):e98701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Pareja F, Ferraro DA, Rubin C, et al. Deubiquitination of EGFR by Cezanne‐1 contributes to cancer progression. Oncogene. 2012;31(43):4599‐4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Wang B, Jie Z, Joo D, et al. TRAF2 and OTUD7B govern a ubiquitin‐dependent switch that regulates mTORC2 signalling. Nature. 2017;545(7654):365‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Ge F, Chen W, Qin J, et al. Ataxin‐3 like (ATXN3L), a member of the Josephin family of deubiquitinating enzymes, promotes breast cancer proliferation by deubiquitinating Krüppel‐like factor 5 (KLF5). Oncotarget. 2015;6(25):21369‐21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Zhang S, Hong Z, Chai Y, et al. CSN5 promotes renal cell carcinoma metastasis and EMT by inhibiting ZEB1 degradation. Biochem Biophys Res Commun. 2017;488(1):101‐108. [DOI] [PubMed] [Google Scholar]
- 299. Huang M, Xiong H, Luo D, Xu B, Liu H. CSN5 upregulates glycolysis to promote hepatocellular carcinoma metastasis via stabilizing the HK2 protein. Exp Cell Res. 2020;388(2):111876. [DOI] [PubMed] [Google Scholar]
- 300. Yuan L, Lv Y, Li H, et al. Deubiquitylase OTUD3 regulates PTEN stability and suppresses tumorigenesis. Nat Cell Biol. 2015;17(9):1169‐1181. [DOI] [PubMed] [Google Scholar]
- 301. Pu Q, Lv YR, Dong K, Geng WW, Gao HD. Tumor suppressor OTUD3 induces growth inhibition and apoptosis by directly deubiquitinating and stabilizing p53 in invasive breast carcinoma cells. BMC Cancer. 2020;20(1):583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Zhou F, Pan Y, Wei Y, et al. Jab1/Csn5‐thioredoxin signaling in relapsed acute monocytic leukemia under oxidative stress. Clin Cancer Res. 2017;23(15):4450‐4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Li J, Li Y, Wang B, Ma Y, Chen P. CSN5/Jab1 facilitates non‐small cell lung cancer cell growth through stabilizing survivin. Biochem Biophys Res Commun. 2018;500(2):132‐138. [DOI] [PubMed] [Google Scholar]
- 304. Hou J, Deng Q, Zhou J, et al. CSN6 controls the proliferation and metastasis of glioblastoma by CHIP‐mediated degradation of EGFR. Oncogene. 2017;36(8):1134‐1144. [DOI] [PubMed] [Google Scholar]
- 305. Su L, Guo W, Lou L, et al. EGFR‐ERK pathway regulates CSN6 to contribute to PD‐L1 expression in glioblastoma. Mol Carcinog. 2020;59(5):520‐532. [DOI] [PubMed] [Google Scholar]
- 306. Wicks SJ, Haros K, Maillard M, et al. The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF‐beta signalling. Oncogene. 2005;24(54):8080‐8084. [DOI] [PubMed] [Google Scholar]
- 307. Iwakami Y, Yokoyama S, Watanabe K, Hayakawa Y. STAM‐binding protein regulates melanoma metastasis through SLUG stabilization. Biochem Biophys Res Commun. 2018;507(1‐4):484‐488. [DOI] [PubMed] [Google Scholar]
- 308. Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fässler R. Cyld inhibits tumor cell proliferation by blocking Bcl‐3‐dependent NF‐kappaB signaling. Cell. 2006;125(4):665‐677. [DOI] [PubMed] [Google Scholar]
- 309. Brummelkamp TR, Nijman SMB, Dirac AMG. Bernards R. Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF‐kappaB. Nature. 2003;424(6950):797‐801. [DOI] [PubMed] [Google Scholar]
- 310. Fernández‐Majada V, Welz PS, Ermolaeva MA, et al. The tumour suppressor CYLD regulates the p53 DNA damage response. Nat Commun. 2016;7:12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Tauriello DVF, Haegebarth A, Kuper I, et al. Loss of the tumor suppressor CYLD enhances Wnt/beta‐catenin signaling through K63‐linked ubiquitination of Dvl. Mol Cell. 2010;37(5):607‐619. [DOI] [PubMed] [Google Scholar]
- 312. Massoumi R, Kuphal S, Hellerbrand C, et al. Down‐regulation of CYLD expression by Snail promotes tumor progression in malignant melanoma. J Exp Med. 2009;206(1):221‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Miliani de Marval P, Lutfeali S, Jin JY, Leshin B, Selim MA, Zhang JY. CYLD inhibits tumorigenesis and metastasis by blocking JNK/AP1 signaling at multiple levels. Cancer Prev Res (Phila). 2011;4(6):851‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. de Jel MM, Schott M, Lamm S, Neuhuber W, Kuphal S, Bosserhoff AK. Loss of CYLD accelerates melanoma development and progression in the Tg(Grm1) melanoma mouse model. Oncogenesis. 2019;8(10):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Liu X, Zhang X, Peng Z, et al. Deubiquitylase OTUD6B governs pVHL stability in an enzyme‐independent manner and suppresses hepatocellular carcinoma metastasis. Adv Sci (Weinh). 2020;7(8):1902040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Xu Z, Pei L, Wang L, Zhang F, Hu X, Gui Y. Snail1‐dependent transcriptional repression of Cezanne2 in hepatocellular carcinoma. Oncogene. 2014;33(22):2836‐2845. [DOI] [PubMed] [Google Scholar]
- 317. Anta B, Martín‐Rodríguez C, Gomis‐Perez C, et al. Ubiquitin‐specific protease 36 (USP36) controls neuronal precursor cell‐expressed developmentally down‐regulated 4‐2 (Nedd4‐2) actions over the neurotrophin receptor TrkA and potassium voltage‐gated channels 7.2/3 (Kv7.2/3). J Biol Chem. 2016;291(36):19132‐19145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Bai P, Virág L. Role of poly(ADP‐ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012;586(21):3771‐3777. [DOI] [PubMed] [Google Scholar]
- 319. Basic M, Hertel A, Bajdzienko J, et al. The deubiquitinase USP11 is a versatile and conserved regulator of autophagy. J Biol Chem. 2021;297(5):101263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Ceriani M, Amigoni L, D'Aloia A, Berruti G, Martegani E. The deubiquitinating enzyme UBPy/USP8 interacts with TrkA and inhibits neuronal differentiation in PC12 cells. Exp Cell Res. 2015;333(1):49‐59. [DOI] [PubMed] [Google Scholar]
- 321. Fang X, Zhou W, Wu Q, et al. Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14‐mediated Myc ubiquitination. J Exp Med. 2017;214(1):245‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Huang TT, Nijman SMB, Mirchandani KD, et al. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8(4):339‐347. [DOI] [PubMed] [Google Scholar]
- 323. Kon N, Zhong J, Kobayashi Y, et al. Roles of HAUSP‐mediated p53 regulation in central nervous system development. Cell Death Differ. 2011;18(8):1366‐1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Liu X, Hebron M, Shi W, Lonskaya I, Moussa CEH. Ubiquitin specific protease‐13 independently regulates parkin ubiquitination and alpha‐synuclein clearance in alpha‐synucleinopathies. Hum Mol Genet. 2019;28(4):548‐560. [DOI] [PubMed] [Google Scholar]
- 325. Vaden JH, Bhattacharyya BJ, Chen PC, et al. Ubiquitin‐specific protease 14 regulates c‐Jun N‐terminal kinase signaling at the neuromuscular junction. Mol Neurodegener. 2015;10:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Yeates EFA, Tesco G. The endosome‐associated deubiquitinating enzyme USP8 regulates BACE1 enzyme ubiquitination and degradation. J Biol Chem. 2016;291(30):15753‐15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Yuasa‐Kawada J, Kinoshita‐Kawada M, Wu G, Rao Y, Wu JY. Midline crossing and slit responsiveness of commissural axons require USP33. Nat Neurosci. 2009;12(9):1087‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Abbracchio MP, Cattabeni F. Brain adenosine receptors as targets for therapeutic intervention in neurodegenerative diseases. Ann N Y Acad Sci. 1999;890:79‐92. [DOI] [PubMed] [Google Scholar]
- 329. Alexopoulou Z, Lang J, Perrett RM, et al. Deubiquitinase Usp8 regulates α‐synuclein clearance and modifies its toxicity in Lewy body disease. Proc Natl Acad Sci USA. 2016;113(32):E4688‐4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Bello AI, Goswami R, Brown SL, et al. Deubiquitinases in neurodegeneration. Cells. 2022;11(3):556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Bernardi KM, Williams JM, Inoue T, Schultz A, Tsai B. A deubiquitinase negatively regulates retro‐translocation of nonubiquitinated substrates. Mol Biol Cell. 2013;24(22):3545‐3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Bingol B, Tea JS, Phu L, et al. The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature. 2014;510(7505):370‐375. [DOI] [PubMed] [Google Scholar]
- 333. Dupont S, Mamidi A, Cordenonsi M, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136(1):123‐135. [DOI] [PubMed] [Google Scholar]
- 334. Gong B, Cao Z, Zheng P, et al. Ubiquitin hydrolase Uch‐L1 rescues beta‐amyloid‐induced decreases in synaptic function and contextual memory. Cell. 2006;126(4):775‐788. [DOI] [PubMed] [Google Scholar]
- 335. Hong S, Kim SJ, Ka S, Choi I, Kang S. USP7, a ubiquitin‐specific protease, interacts with ataxin‐1, the SCA1 gene product. Mol Cell Neurosci. 2002;20(2):298‐306. [DOI] [PubMed] [Google Scholar]
- 336. Matsumoto M, Yada M, Hatakeyama S, et al. Molecular clearance of ataxin‐3 is regulated by a mammalian E4. EMBO J. 2004;23(3):659‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Schimmack G, Schorpp K, Kutzner K, et al. YOD1/TRAF6 association balances p62‐dependent IL‐1 signaling to NF‐κB. Elife. 2017;6:e22416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Schulz S, Chachami G, Kozaczkiewicz L, et al. Ubiquitin‐specific protease‐like 1 (USPL1) is a SUMO isopeptidase with essential, non‐catalytic functions. EMBO Rep. 2012;13(10):930‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Tanji K, Mori F, Miki Y, et al. YOD1 attenuates neurogenic proteotoxicity through its deubiquitinating activity. Neurobiol Dis. 2018;112:14‐23. [DOI] [PubMed] [Google Scholar]
- 340. Wilson SM, Bhattacharyya B, Rachel RA, et al. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin‐specific protease. Nat Genet. 2002;32(3):420‐425. [DOI] [PubMed] [Google Scholar]
- 341. Margolin DH, Kousi M, Chan YM, et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N Engl J Med. 2013;368(21):1992‐2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Sano Y, Furuta A, Setsuie R, et al. Photoreceptor cell apoptosis in the retinal degeneration of Uchl3‐deficient mice. Am J Pathol. 2006;169(1):132‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Suzuki S, Tamai K, Watanabe M, et al. AMSH is required to degrade ubiquitinated proteins in the central nervous system. Biochem Biophys Res Commun. 2011;408(4):582‐588. [DOI] [PubMed] [Google Scholar]
- 344. Tao BB, He H, Hua SX, et al. Up‐regulation of USP2a and FASN in gliomas correlates strongly with glioma grade. J Clin Neurosci. 2013;20(5):717‐720. [DOI] [PubMed] [Google Scholar]
- 345. Tsou WL, Burr AA, Ouyang M, Blount JR, Scaglione KM, Todi SV. Ubiquitination regulates the neuroprotective function of the deubiquitinase ataxin‐3 in vivo. J Biol Chem. 2013;288(48):34460‐34469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Verma R, Aravind L, Oania R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298(5593):611‐615. [DOI] [PubMed] [Google Scholar]
- 347. Wang Q, Li L, Ye Y. Regulation of retrotranslocation by p97‐associated deubiquitinating enzyme ataxin‐3. J Cell Biol. 2006;174(7):963‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419(6905):403‐407. [DOI] [PubMed] [Google Scholar]
- 349. Yao T, Song L, Xu W, et al. Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat Cell Biol. 2006;8(9):994‐1002. [DOI] [PubMed] [Google Scholar]
- 350. Bolton J, Montastier E, Carayol J, et al. Molecular biomarkers for weight control in obese individuals subjected to a multiphase dietary intervention. J Clin Endocrinol Metab. 2017;102(8):2751‐2761. [DOI] [PubMed] [Google Scholar]
- 351. Coyne ES, Bédard N, Gong YJ, Faraj M, Tchernof A, Wing SS. The deubiquitinating enzyme USP19 modulates adipogenesis and potentiates high‐fat‐diet‐induced obesity and glucose intolerance in mice. Diabetologia. 2019;62(1):136‐146. [DOI] [PubMed] [Google Scholar]
- 352. Lu XY, Shi XJ, Hu A, et al. Feeding induces cholesterol biosynthesis via the mTORC1‐USP20‐HMGCR axis. Nature. 2020;588(7838):479‐484. [DOI] [PubMed] [Google Scholar]
- 353. Yang H, Seo SG, Shin SH, et al. 3,3’‐Diindolylmethane suppresses high‐fat diet‐induced obesity through inhibiting adipogenesis of pre‐adipocytes by targeting USP2 activity. Mol Nutr Food Res. 2017;61(10). [DOI] [PubMed] [Google Scholar]
- 354. Liu B, Jiang S, Li M, et al. Proteome‐wide analysis of USP14 substrates revealed its role in hepatosteatosis via stabilization of FASN. Nat Commun. 2018;9(1):4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Molusky MM, Li S, Ma D, Yu L, Lin JD. Ubiquitin‐specific protease 2 regulates hepatic gluconeogenesis and diurnal glucose metabolism through 11β‐hydroxysteroid dehydrogenase 1. Diabetes. 2012;61(5):1025‐1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. An S, Zhao LP, Shen LJ, et al. USP18 protects against hepatic steatosis and insulin resistance through its deubiquitinating activity. Hepatology. 2017;66(6):1866‐1884. [DOI] [PubMed] [Google Scholar]
- 357. Gorrepati KDD, Lupse B, Annamalai K, Yuan T, Maedler K, Ardestani A. Loss of deubiquitinase USP1 blocks pancreatic β‐cell apoptosis by inhibiting DNA damage response. iScience. 2018;1:72‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Kim A, Koo JH, Jin X, et al. Ablation of USP21 in skeletal muscle promotes oxidative fibre phenotype, inhibiting obesity and type 2 diabetes. J Cachexia Sarcopenia Muscle. 2021;12(6):1669‐1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Berthouze M, Venkataramanan V, Li Y, Shenoy SK. The deubiquitinases USP33 and USP20 coordinate beta2 adrenergic receptor recycling and resensitization. EMBO J. 2009;28(12):1684‐1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Elumalai S, Karunakaran U, Moon JS, Won KC. High glucose‐induced PRDX3 acetylation contributes to glucotoxicity in pancreatic β‐cells: prevention by Teneligliptin. Free Radic Biol Med. 2020;160:618‐629. [DOI] [PubMed] [Google Scholar]
- 361. Forand A, Koumakis E, Rousseau A, et al. Disruption of the phosphate transporter pit1 in hepatocytes improves glucose metabolism and insulin signaling by modulating the USP7/IRS1 interaction. Cell Rep. 2016;16(10):2736‐2748. [DOI] [PubMed] [Google Scholar]
- 362. Hashimoto M, Fujimoto M, Konno K, et al. Ubiquitin‐specific protease 2 in the ventromedial hypothalamus modifies blood glucose levels by controlling sympathetic nervous activation. J Neurosci. 2022;42(23):4607‐4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Kitamura H, Kimura S, Shimamoto Y, et al. Ubiquitin‐specific protease 2–69 in macrophages potentially modulates metainflammation. FASEB J. 2013;27(12):4940‐4953. [DOI] [PubMed] [Google Scholar]
- 364. Niu Y, Jiang H, Yin H, et al. Hepatokine ERAP1 disturbs skeletal muscle insulin sensitivity via inhibiting USP33‐Mediated ADRB2 deubiquitination. Diabetes. 2022;71(5):921‐933. [DOI] [PubMed] [Google Scholar]
- 365. Wang P, Zhang RY, Song J, et al. Loss of AMP‐activated protein kinase‐α2 impairs the insulin‐sensitizing effect of calorie restriction in skeletal muscle. Diabetes. 2012;61(5):1051‐1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Zhao Y, Wang F, Gao L, et al. Ubiquitin‐specific protease 4 is an endogenous negative regulator of metabolic dysfunctions in nonalcoholic fatty liver disease in mice. Hepatology. 2018;68(3):897‐917. [DOI] [PubMed] [Google Scholar]
- 367. Lin X, Xiang H, Luo G. Targeting estrogen receptor α for degradation with PROTACs: a promising approach to overcome endocrine resistance. Eur J Med Chem. 2020;206:112689. [DOI] [PubMed] [Google Scholar]
- 368. Li X, Song Y. Proteolysis‐targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J Hematol Oncol. 2020;13(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Yasukawa T, Tsutsui A, Tomomori‐Sato C, et al. NRBP1‐containing CRL2/CRL4A regulates amyloid β production by targeting BRI2 and BRI3 for degradation. Cell Rep. 2020;30(10):3478‐3491. e6. [DOI] [PubMed] [Google Scholar]
- 370. Zhang XW, Feng N, Liu YC, et al. Neuroinflammation inhibition by small‐molecule targeting USP7 noncatalytic domain for neurodegenerative disease therapy. Sci Adv. 2022;8(32):eabo0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Dagar G, Kumar R, Yadav KK, Singh M, Pandita TK. Ubiquitination and deubiquitination: implications on cancer therapy. Biochim Biophys Acta Gene Regul Mech. 2023;1866(4):194979. [DOI] [PubMed] [Google Scholar]
- 372. Chen YJ, Wu H, Shen XZ. The ubiquitin‐proteasome system and its potential application in hepatocellular carcinoma therapy. Cancer Lett. 2016;379(2):245‐252. [DOI] [PubMed] [Google Scholar]
- 373. Huang X, Dixit VM. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016;26(4):484‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Lee JT, Gu W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010;17(1):86‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Tuval A, Strandgren C, Heldin A, Palomar‐Siles M, Wiman KG. Pharmacological reactivation of p53 in the era of precision anticancer medicine. Nat Rev Clin Oncol. 2024;21(2):106‐120. [DOI] [PubMed] [Google Scholar]
- 376. Cheok CF, Verma CS, Baselga J, Lane DP. Translating p53 into the clinic. Nat Rev Clin Oncol. 2011;8(1):25‐37. [DOI] [PubMed] [Google Scholar]
- 377. Shangary S, Qin D, McEachern D, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci USA. 2008;105(10):3933‐3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Issaeva N, Bozko P, Enge M, et al. Small molecule RITA binds to p53, blocks p53‐HDM‐2 interaction and activates p53 function in tumors. Nat Med. 2004;10(12):1321‐1328. [DOI] [PubMed] [Google Scholar]
- 379. Ding Q, Zhang Z, Liu JJ, et al. Discovery of RG7388, a potent and selective p53‐MDM2 inhibitor in clinical development. J Med Chem. 2013;56(14):5979‐5983. [DOI] [PubMed] [Google Scholar]
- 380. Hao Z, Huang S. E3 ubiquitin ligase Skp2 as an attractive target in cancer therapy. Front Biosci (Landmark Ed). 2015;20(3):474‐490. [DOI] [PubMed] [Google Scholar]
- 381. Li C, Du L, Ren Y, et al. SKP2 promotes breast cancer tumorigenesis and radiation tolerance through PDCD4 ubiquitination. J Exp Clin Cancer Res. 2019;38(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Békés M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022;21(3):181‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Tiwari S, Singh A, Gupta P, Singh S. UBA52 is crucial in HSP90 ubiquitylation and neurodegenerative signaling during early phase of Parkinson's disease. Cells. 2022;11(23):3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Colland F. The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem Soc Trans. 2010;38:137‐143. Pt 1. [DOI] [PubMed] [Google Scholar]
- 385. Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus . Science. 2004;306(5698):1037‐1040. [DOI] [PubMed] [Google Scholar]
- 386. Kelsall IR, McCrory EH, Xu Y, et al. HOIL‐1 ubiquitin ligase activity targets unbranched glucosaccharides and is required to prevent polyglucosan accumulation. EMBO J. 2022;41(8):e109700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Sakamaki JI, Mizushima N. Ubiquitination of non‐protein substrates. Trends Cell Biol. 2023;33(11):991‐1003. [DOI] [PubMed] [Google Scholar]
- 388. Akizuki Y, Kaypee S, Ohtake F, Ikeda F. The emerging roles of non‐canonical ubiquitination in proteostasis and beyond. J Cell Biol. 2024;223(5):e202311171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Chang HM, Yeh ETH. SUMO: from bench to bedside. Physiol Rev. 2020;100(4):1599‐1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Vertegaal ACO. Signalling mechanisms and cellular functions of SUMO. Nat Rev Mol Cell Biol. 2022;23(11):715‐731. [DOI] [PubMed] [Google Scholar]
- 391. Li C, McManus FP, Plutoni C. Quantitative SUMO proteomics identifies PIAS1 substrates involved in cell migration and motility. Nat Commun. 2020;11(1):834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Seeler JS, Dejean A. SUMO and the robustness of cancer. Nat Rev Cancer. 2017;17(3):184‐197. [DOI] [PubMed] [Google Scholar]
- 393. Skaug B, Chen ZJ. Emerging role of ISG15 in antiviral immunity. Cell. 2010;143(2):187‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Fu DJ, Wang T. Targeting NEDD8‐activating enzyme for cancer therapy: developments, clinical trials, challenges and future research directions. J Hematol Oncol. 2023;16(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Palek M, Palkova N, consortium CZECANCA , Kleiblova P, Kleibl Z, Macurek L, consortium CZECANCA . RAD18 directs DNA double‐strand break repair by homologous recombination to post‐replicative chromatin. Nucleic Acids Res. 2024;52(13):7687‐7703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Wong YC, Holzbaur ELF. Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy. 2015;11(2):422‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Liao YC, Pang S, Li WP, et al. COPII with ALG2 and ESCRTs control lysosome‐dependent microautophagy of ER exit sites. Dev Cell. 2024;59(11):1410‐1424. e4. [DOI] [PubMed] [Google Scholar]
- 398. González A, Covarrubias‐Pinto A, Bhaskara RM, et al. Ubiquitination regulates ER‐phagy and remodelling of endoplasmic reticulum. Nature. 2023;618(7964):394‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Offensperger F, Tin G, Duran‐Frigola M, et al. Large‐scale chemoproteomics expedites ligand discovery and predicts ligand behavior in cells. Science. 2024;384(6694):eadk5864. [DOI] [PubMed] [Google Scholar]
- 400. Xu S, Wu Y, Chen Q, et al. hSSB1 regulates both the stability and the transcriptional activity of p53. Cell Res. 2013;23(3):423‐435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Mao Z, Sang MM, Chen C, Zhu WT, Gong YS, Pei DS. CSN6 promotes the migration and invasion of cervical cancer cells by inhibiting autophagic degradation of cathepsin L. Int J Biol Sci. 2019;15(6):1310‐1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Yan K, Li L, Wang X, et al. The deubiquitinating enzyme complex BRISC is required for proper mitotic spindle assembly in mammalian cells. J Cell Biol. 2015;210(2):209‐224. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are freely available from the corresponding author upon request.