

Abstract
With the completion of full genome sequences and advancement in high-throughput technologies, in silico methods have been successfully used to integrate diverse data sources toward unraveling the combinatorial nature of transcriptional regulation. So far, almost all of these studies are restricted to lower eukaryotes such as budding yeast. We describe here a computational search for functional transcription-factor (TF) combinations using phylogenetically conserved sequences and microarray-based expression data. Taking into account both orientational and positional constraints, we investigated the overrepresentation of binding sites in the vicinity of one another and whether these combinations result in more coherent expression profiles. Without any prior biological knowledge, the search led to the discovery of several experimentally established TF associations, as well as some novel ones. In particular, we identified a regulatory module controlling cell cycle-dependent transcription of G2-M genes and expanded its functional generality. We also detected many homotypic combinations, supporting the importance of binding-site density in transcriptional regulation of higher eukaryotes.
Cis-regulation of gene expression by the binding of transcription factors (TFs) is a critical component of cellular physiology. In eukaryotes, a battery of TFs often work together in a combinatorial fashion to enable cells to respond to a wide spectrum of environmental and developmental signals. Integration of genome sequences and/or ChIP-chip data with gene-expression data has facilitated in silico discovery of how the combinatorics and positioning of TF-binding sites underlie gene activation in a variety of cellular processes for relatively simple organisms such as Saccharomyces cerevisiae and Caenorhabditis elegans (Bussemaker et al. 2001; Pilpel et al. 2001; Banerjee and Zhang 2003; Beer and Tavazoie 2004; Kato et al. 2004; Terai and Takagi 2004). Application of these methods to the human genome, however, is complicated by its significantly larger size and substantial repetitive content, as well as the greater complexity of the transcriptional network. Early studies suggest that phylogenetic footprinting (Wasserman and Fickett 1998; Wasserman et al. 2000; Levy et al. 2001; Blanchette and Tompa 2002; Liu et al. 2004) and a focus on motif combinations (Frech et al. 1998; Kel et al. 1999; Krivan and Wasserman 2001; Aerts et al. 2003) may prove essential to computational analyses of human transcription-factor binding sites.
As the functional interactions between TFs often require them to be in physical proximity, their binding sites are likely to be overrepresented in the vicinity of each other. Exploiting such property, we devised a two-step strategy (Fig. 1) to reveal known or novel transcription factors that work in concert. Starting from a TF-binding site of interest, our algorithm first discovers significantly enriched neighboring motif(s) using human-mouse conserved sequences, and then examines the functional significance of their physical proximity through the assessment of similarity in expression profiles. Applying this methodology to human promoter sequences and a cell cycle expression data set, we found a number of motif pairs that not only preferentially co-occur nearby, but also appear to act together in determining gene expression pattern, including a G2-M regulatory module. In addition to heterotypic interactions, we observed a homotypic distribution of transcription-factor binding sites, as many of them are specifically enriched around themselves.
Figure 1.

Strategy used to discover functional combinations for any motif of interest.
Results
Extraction of human promoter sequence and phylogenetic footprinting
We had previously mapped UniGene clusters onto the human genome as well as generated a “mousenized” version of the genome (http://club.med.harvard.edu/hummus/hummus.html). To build a promoter sequences set, we extracted the sequence 1 kb upstream of 11,436 curated RefSeq mRNAs as putative promoter regions. While some regulatory elements can act over very large distances, up to several kilobases from transcriptional start sites (TSS), we focused on sequences in the relative proximity of TSS, as they are most likely to contain regulatory information for evolutionarily conserved biological processes such as the cell cycle.
Accuracy of identifying binding sites by weight matrix
For anchor motifs, we utilized 134 experimentally derived position weight matrices from the TRANSFAC database. They correspond to ∼70 distinct motifs, as estimated by CompareACE (Hughes et al. 2000) with a cut-off of 0.85. Putative binding sites were identified by scanning promoter sequences with the anchor motifs using PATSER (Hertz and Stormo 1999). To estimate the accuracy of our in silico predictions, we compared the list of E2F site (M00516)-containing genes with those identified as E2F4 targets in primary fibroblasts using ChIP-chip technology (Ren et al. 2002). Our promoter set includes 96 of the experimentally determined target genes, and 56 of them were predicted computationally based on 1-kb promoter sequences (P = 1.1 E-11). Phylogenetic footprinting has been demonstrated to be a powerful strategy for filtering out false-positive results of motif discovery algorithms (Loots et al. 2000; Wasserman et al. 2000), as real binding sites are far more likely to be conserved under selection pressure than random sequences. By confining to those hits that are also found in mouse, we refined our in silico predictions from 2759 to 230, 15 of which overlap with the E2F4 target genes by ChIP-chip. The evolutionarily conserved E2F-binding sites have a much sharper peak over the first 100 nucleotides upstream from transcription start site (TSS, as estimated by the start of mRNA sequence; Fig. 2), supporting previous observations regarding the positional distribution of functional E2F sites (Kel et al. 2001). The number of putative sites obtained for each of the 134 anchor motifs before and after incorporating phylogenetic information is available as Supplemental material.
Figure 2.

Distribution of all and human-mouse conserved E2F-binding sites relative to transcription start site (as estimated by the start of mRNA sequence).
Significantly enriched neighbor motifs
To search for enriched neighbor motifs, we extracted both human and corresponding mouse sequences in the vicinity (e.g., 50 and 100 bp) of the conserved anchor motif sites. As the functional interactions between TFs often impose orientational constraints, upstream and downstream sequences were grouped separately. A blind and systematic search was then conducted for shared sequence features with the program AlignACE (Roth et al. 1998; Hughes et al. 2000), which identifies motifs that are overrepresented in a set of unaligned input sequences. AlignACE calculates a statistic called the MAP score, which is an internal metric to determine the statistical significance of an alignment. We only considered those with a MAP score of 10 or higher (Tavazoie et al. 1999), and at least five genes containing the anchor-neighbor combination. This resulted in a total of 6293 neighbor motifs (3227 downstream + 3066 upstream) for the window size of 50 bp, and 9278 (4831 downstream + 4447 upstream) for the window size of 100 bp.
We selected the most statistically significant neighbor motifs using a measure called neighbor specificity (NS) score. It quantifies how specific a neighbor motif targets the neighboring region of the anchor motif, given its rate of occurrence in all promoters. To reduce the bias in assessing the degree of specificity, the calculation was performed based on statistics over the entire 1-kb human sequences, including conserved as well as nonconserved regions. We corrected for multiple testing by calculating NS score cutoffs corresponding to an FDR (Storey and Tibshirani 2003) of 0.05.
Homotypic distribution of TF-binding sites
We noticed that the vicinity of anchor motifs are often specifically enriched with themselves (i.e., anchor = neighbor; Table 1), e.g., 20 instances for 50 bp, and 14 for 100 bp. This observation is in agreement with findings in S. cerevisiae (Wagner 1999) and Drosophila (Lifanov et al. 2003), where statistically significant homotypic clusters of transcription-factor binding sites have been reported. The presence of multiple copies of the same cis-regulatory motifs has been documented in biological literature (Arnone and Davidson 1997). While some represent sites for transcription factors that act as oligomers (e.g., p53, NF-κB, GAGA), others activate morphogen TFs in response to their low local concentration (Gurdon and Bourillot 2001). Meanwhile, they could also contribute to the robustness of regulatory elements (Simpson 2002), or play a role in differentiating real sites from spurious ones by increasing the binding affinity of the former, a task increasingly important in larger genomes, such as that of human.
Table 1.
Significantly overrepresented homotypic motif pairs (anchor = neighbora)
| Anchor motif | Binding factor | Window size, bp | NSb | PECc |
|---|---|---|---|---|
| M00017 | ATF | 50 | 4.5E-5 | Nd |
| M00039 | CREB | 50 | 2.2E-4 | Nd |
| M00119 | Max | 50 | 3.4E-5 | Nd |
| M00121 | USF | 50 | 2.6E-13 | 2.5E-6 |
| M00123 | c-Myc/Max | 50 | 1.6E-3 | 1.3E-6 |
| M00178 | CREB | 50 | 9.7E-8 | Nd |
| M00185 | NF-Y | 50 | 3.0E-59 | 9.3E-7 |
| M00209 | NF-Y | 50 | 3.1E-24 | 1.0E-6 |
| M00210 | Oct-x | 50 | 1.6E-6 | 1.3E-6 |
| M00220 | SREBP-1 | 50 | 6.6E-13 | 6.9E-6 |
| M00236 | Arnt | 50 | 3.1E-7 | Nd |
| M00287 | NF-Y | 50 | 2.7E-12 | 9.8E-7 |
| M00338 | ATF | 50 | 3.9E-5 | Nd |
| M00341 | GABP | 50 | 1.6E-12 | 9.1E-7 |
| M00342 | Oct-1 | 50 | 1.6E-8 | Nd |
| M00491 | MAZR | 50 | 4.6E-42 | Nd |
| M00615 | c-Myc/Max | 50 | 1.4E-10 | Nd |
| M00652 | Nrf-1 | 50 | 1.3E-12 | Nd |
| M00678 | Tel-2 | 50 | 1.6E-12 | 9.6E-7 |
| M00687 | α-CP1 | 50 | 8.4E-13 | 1.0E-6 |
| M00121 | USF | 100 | 3.0E-8 | 1.1E-6 |
| M00185 | NF-Y | 100 | 3.5E-50 | 9.9E-7 |
| M00209 | NF-Y | 100 | 2.2E-45 | 1.0E-6 |
| M00220 | SREBP-1 | 100 | 3.7E-9 | 1.0E-6 |
| M00236 | Arnt | 100 | 1.5E-4 | Nd |
| M00287 | NF-Y | 100 | 3.0E-12 | 9.8E-7 |
| M00341 | GABP | 100 | 7.6E-13 | 1.1E-6 |
| M00342 | Oct-1 | 100 | 2.9E-8 | Nd |
| M00491 | MAZR | 100 | 4.3E-5 | Nd |
| M00539 | Arnt | 100 | 5.3E-8 | Nd |
| M00615 | c-Myc/Max | 100 | 5.8E-9 | 1.5E-6 |
| M00652 | Nrf-1 | 100 | 1.3E-12 | 1.0E-6 |
| M00678 | Tel-2 | 100 | 2.4E-11 | 1.0E-6 |
| M00687 | α-CP1 | 100 | 1.2E-12 | 1.0E-6 |
CompareACE > 0.85.
Neighbor specificity score.
P-value on the hypothesis that the genes with motif combination are equally or less correlated in expression than either anchor or neighborcontaining genes without the combination.
Not statistically significant after taking into account multiple testing (see text for details).
Heterotypic neighbor motifs
It is conceivable that some neighbor motifs may happen to be enriched in the vicinity of anchor motifs simply because they both prefer the same location relative to TSS, but have nothing to do with each other. To filter out such potential positionally biased scenarios, for each statistically overrepresented hetero-neighbor motif (i.e., anchor ≠ neighbor), we randomly selected the same number of promoters as those with its parent anchor motif and extracted segments of same window size from the same distance upstream of TSS as those containing anchor motif, followed by identical motif search procedures. The random sampling process was repeated 100 times, and we rejected the neighbor motif if it was “rediscovered” by any of these runs. After applying such a positional bias filter, we ended up with 636 (852) significant hetero-neighbor motifs from a 50-bp (100-bp) window, 40 (37) of which could be mapped to known TRANSFAC matrices (CompareACE > 0.85).
Functional anchor-neighbor motif combinations
Given the specific enrichment of neighbor motifs, we next asked whether some of them may functionally interact with their corresponding anchors by analyzing a human cell cycle expression data set (Whitfield et al. 2002), which recorded genome-wide expression levels of synchronized HeLa S3 cells using spotted microarray. For any anchor-neighbor motif combination (within a specified window size), we generated three groups of genes, one with the combination, one with anchor motif but not the combination and one with neighbor motif, but not the combination. An anchor-neighbor motif combination was considered “functional” if the expression profiles of the genes from the first set are significantly more highly correlated than both the second and third sets. We used a multivariate hypergeometric model (Banerjee and Zhang 2003) to calculate the probability of obtaining the observed or higher fraction of correlated gene pairs in the set with the combination, given the fractions of correlated gene pairs in the sets without the combination. After accounting for multiple testing with Bonferroni correction, we obtained 167 and 225 significant combinations for 50 and 100 bp, respectively (corrected P-value < 0.05). Among those containing neighbor motifs that could be mapped to known TRANSFAC matrices, 10 (10) homotypic and 8 (10) heterotypic pairs are represented (Tables 1, 2) with a window size of 50 bp (100 bp). A complete list of significant combinations can be found at http://genetics.med.harvard.edu/~zzhu/combination.html.
Table 2.
Functional heterotypic motif pairs (anchor ≠ neighbor) where the neighbor motif may be mapped to known TRANSFAC matrices
| Anchor motif | Neighbor motifa | Window size, bp | Orientationb | NSc | PECd |
|---|---|---|---|---|---|
| M00071 (E47) | M00271 (AML-1a) | 50 | Down | 1.6E-7 | 1.0E-6 |
| M00135 (Oct-1) | M00287 (NF-Y) | 50 | Up | 1.5E-3 | 9.8E-7 |
| M00178 (CREB) | M00216 (TATA) | 50 | Down | 1.5E-6 | 1.9E-6 |
| M00287 (NF-Y) | M00195 (Oct-1) | 50 | Up | 5.2E-7 | 1.3E-6 |
| M00425 (E2F) | M00287 (NF-Y) | 50 | Down | 4.0E-7 | 1.5E-6 |
| M00466 (HIF-1) | M00025 (Elk-1) | 50 | Up | 2.0E-3 | 1.0E-6 |
| M00615 (c-Myc/Max) | M00069 (YY1) | 50 | Down | 4.4E-3 | 1.2E-6 |
| M00651 (NF-muE1) | M00179 (CRE-BP1) | 50 | Up | 9.1E-6 | 1.0E-6 |
| M00136 (Oct-1) | M00622 (C/EBP) | 100 | Down | 2.2E-11 | 1.0E-6 |
| M00177 (CREB) | M00069 (YY1) | 100 | Down | 2.2E-3 | 2.0E-6 |
| M00289 (HFH-3) | M00045 (E4BP4) | 100 | Down | 7.3E-8 | 1.2E-6 |
| M00341 (GABP) | M00281 (RFX1) | 100 | Up | 3.5E-4 | 7.7E-6 |
| M00425 (E2F) | M00287 (NF-Y) | 100 | Down | 6.8E-8 | 1.0E-6 |
| M00464 (POU3F2) | M00216 (TATA) | 100 | Down | 1.4E-13 | 9.8E-7 |
| M00466 (HIF-1) | M00069 (YY1) | 100 | Down | 2.2E-5 | 9.4E-7 |
| M00476 (FOXO4) | M00422 (FOXJ2) | 100 | Down | 5.1E-3 | 9.8E-7 |
| M00615 (c-Myc/Max) | M00651 (NF-muE1) | 100 | Down | 1.8E-3 | 3.7E-6 |
| M00678 (Tel-2) | M00069 (YY1) | 100 | Down | 7.3E-4 | 1.0E-6 |
The best TRANSFAC matrix match (CompareACE > 0.85).
The orientation of neighbor motif with respect to anchor (i.e., down = neighbor motif was derived from sequences downstream of anchor; up = neighbor motif was derived from sequences upstream of anchor).
Neighbor specificity score.
P-value on the hypothesis that the genes with motif combination are equally or less correlated in expression than either anchor or neighbor-containing genes without the combination.
Our approach identified several experimentally established associations between TFs. For instance, the cooperation between E2F and NF-Y, two main regulators of cell cycle, has been well documented (van Ginkel et al. 1997; Caretti et al. 2003), and we uncovered their connection using both 50- and 100-bp window sizes. We found RFX1 to be significantly enriched within 100 bp upstream of GABP, and the combination shows a functional effect on expression, in agreement with previous observations that GABP and RFX-1 act synergistically in boosting activity at ribosomal protein L30 promoter, with a RFX-1 site at -128 and a GABP site at -56 (Safrany and Perry 1995). The cAMP responsiveness via CREB has been demonstrated to require a proximal TATA box (Conkright et al. 2003). We indeed observed TATA overrepresented within 50 bp downstream of CREB, and they appear to functionally interact with each other. In addition, YY1-cMyc, Oct1-C/EBP, CREB-YY1, Oct1-NF-Y, ELK1-HIF1, and POU-TBP, known to either form a physical complex or act in concert at some promoters, were also linked in our analysis (Shrivastava et al. 1993; Zhou et al. 1995; Hatada et al. 2000; Bertolino and Singh 2002; Chang et al. 2003; Hirose et al. 2003).
A module controlling transcription of G2-M genes
One of the most significant motif combinations uncovered in our analysis (P = 9.97 E-7; Fig. 3A) involves a potentially novel neighbor motif derived from sequences downstream of NF-Y binding sites, as it does not match any known motif matrix in TRANSFAC. Expression coherence (EC) score is a measure devised to quantify the similarity of the expression among a set of genes (Pilpel et al. 2001). Genes with the combination have an EC score of 0.23, while those containing anchor (NF-Y) or neighbor motif, but without it, only have a score of 0.08 and 0.07, respectively (Fig. 3B-D). After some literature searches, we noticed that the neighbor motif derived in this case is rather similar to the tandem repressor element experimentally identified from the promoters of a number of G2-M-specific genes, cell cycle-dependent element (CDE) and cell cycle genes homology region (CHR) (Tanaka et al. 2002). A comprehensive structure-function analysis of the CDC25C promoter (Lucibello et al. 1995) showed that repression via the CDE-CHR occurs only in the presence of an upstream activating sequence (UAS), and that the functionally crucial elements in the CDC25C UAS include recognition sites for NF-Y. Here, without any prior biological knowledge, our algorithm not only identified this regulatory module in silico, but also substantially expanded its functional generality. CDE-CHR was discovered from neighboring sequences downstream of NF-Y only, supporting the proposed molecular mechanism underlying its action, in which it is assumed to interfere with the activation function of upstream activators, which are present throughout the cell cycle (Zwicker et al. 1995).
Figure 3.


A functional combination discovered from our analysis involving NF-Y binding site and a significantly enriched neighbor motif within 50 bp downstream. (A) Sequence logo of the neighbor motif, produced by the World Wide Web service at http://www.bio.cam.ac.uk/cgi-bin/seqlogo/logo.cgi. The height of each letter is proportional to its frequency of occurrence in the binding-site matrix, times the information content at each position. (B) The expression profiles of genes containing the NF-Y neighbor-motif combination. The blue lines represent expression profiles of individual genes, and the red line represents mean expression profile of the group. (C) The expression profiles of genes containing NF-Y motif, but without neighbor motif within 50 bp downstream. (D) The expression profile of genes containing the neighbor motif, but without NF-Y within 50 bp upstream.
Most of the genes containing the NF-Y-CDE-CHR combination are indeed cell cycle periodic and peak in G2-M phases. We identified 20 genes with the module in their 1-kb promoter sequences, 17 of which are included in the cell cycle expression data set. Based on their microarray data set, Whitfield et al. (2002) reported 872 cell cycle periodic genes by Fourier Transform analysis. Each of them was assigned to a cell cycle phase by their peak correlation to an idealized expression profile from well-studied genes. Among our 17 putative targets, 14 were characterized as cell cycle periodic (Table 3), with all but one peaking in G2 or G2/M phases (P = 1.18 E-12).
Table 3.
Genes containing the NF-Y-CDE-CHR combination in their 1-kb promoter sequences
| UniGene ID | Gene name | Gene description | Phase assignmenta |
|---|---|---|---|
| Hs.656 | CDC25C | Cell division cycle 25C | G2 |
| Hs.334562 | CDC2 | Cell division cycle 2, G1 to S and G2 to M | G2/M |
| Hs.85137 | CCNA2 | Cyclin A2 | G2 |
| Hs.77597 | PLK | Polo-like kinase (Drosophila) | G2/M |
| Hs.180655 | STK12 | Serine/threonine kinase 12 | G2 |
| Hs.120996 | STK17B | Serine/threonine kinase 17b (apoptosis-inducing) | G2 |
| Hs.14870 | MGC14386 | Similar to cyclin-E binding protein 1 (H. sapiens) | G2 |
| Hs.348669 | CKS1 | CDC28 protein kinase 1 | G2/M |
| Hs.83758 | CKS2 | CDC28 protein kinase 2 | G2/M |
| Hs.77204 | CENPF | Centromere protein F (350/400 kD, mitosin) | G2/M |
| Hs.3104 | KIAA0042 | KIAA0042 gene product | G2/M |
| Hs.86211 | FLJ10156 | Hypothetical protein | G2/M |
| Hs.30114 | MGC2577 | Hypothetical protein MGC2577 | G2/M |
| Hs.37035 | HLXB9 | Homeo box HB9 | G1/S |
| Hs.179526 | TXNIP | Thioredoxin interacting protein | |
| Hs.144505 | DKFZP566F0546 | DKFZP566F0546 protein | |
| Hs.174795 | RA-GEF-2 | Rap guanine nucleotide exchange factor |
The cell cycle phase assignment (if applicable) were obtained from Whitfield et al. (2002).
Combinations enriched in other expression clusters
We also looked for TF modules that may regulate genes of other phases of the cell cycle. E2F has a well-established role in controlling G1/S transition. We found two E2F combinations with GC-rich motifs within 100 bp downstream of E2F that are overrepresented among G1/S genes (P = 1.40 E-5 and 8.12 E-5, respectively). Another enriched combination involves E2F and a neighbor motif strongly resembling the binding site of NF-Y (CompareACE score = 0.98). While genes with this combination have a clear preference for peaking in G1/S and S phases (P = 4.39 E-4), it should also be noted that a small number of them belong to G2 and G2/M clusters instead, including CDC2 and CYCLIN B1, whose promoter elements have recently been characterized to contain functional E2F and NF-Y sites (Zhu et al. 2004). Our in silico observations are in line with a newly emerged role for E2F beyond G1/S transition as uncovered from microarray and ChIP-chip studies (Ishida et al. 2001; Ren et al. 2002). We also noticed several combinations enriched with S-genes. But a close inspection of their putative targets revealed that all are histones. Given the cross-hybridization of histone genes on the microarray and their manual assignments to S phase (Whitfield et al. 2002), this result has to be interpreted with caution. None of our combinations appears to be predictive of M/G1.
Discussion
Understanding the regulation of the human cell-division cycle is central to the study of many diseases. A recent genome-wide in silico study identified TF binding sites that are overrepresented in the promoters of cell cycle periodic genes (Elkon et al. 2003). Here, in an effort to explore the combinatorial aspect of the transcriptional control in the human cell cycle, we developed a computational algorithm to identify transcription factors that preferentially act together. Based on de novo motif finding, our method is not limited to interactions involving known TF target sites, but rather has the potential of discovering novel ones. Considering the small number of binding motifs that have been well characterized in mammals so far, we believe such capability is imperative to a thorough understanding of transcriptional regulatory networks.
Functional interactions between TFs not only require their co-occurrence on the same promoter (enhancer), but often with positional (Makeev et al. 2003) and orientational (Terai and Takagi 2004) constraints as well. By grouping together neighboring sequences upstream and downstream of anchor motifs separately, our algorithm provided an additional layer of insights regarding whether there is a preference in a relative location of the two motifs with respect to the genes they regulate. Some of our findings are consistent with experimental observations (see Results). Furthermore, we incorporated a distance parameter into the algorithm, as the statistical overrepresentation of two potentially cooperating motifs in the vicinity of each other is more likely to be biologically relevant. In this study, we experimented with window sizes of 50 and 100 bp, respectively.
The success of our approach clearly relies on the correct identification of true TF-binding sites from sequences. To estimate the reliability of in silico predictions, we compared the list of E2F site-containing genes with those in vivo targets determined using ChIP-chip technology. While there is a significant overlap between the two, it is worth noting that some ChIP-chip targets are “missed” by sequence-based search. Such apparent discrepancy has been reported before (Iyer et al. 2001; Ren et al. 2002; Weinmann et al. 2002; Cawley et al. 2004; Euskirchen et al. 2004), and may be contributed to by a number of factors. For instance, binding could occur indirectly through association with other proteins; there may exist unknown sequence variants or elements beyond primary sequence recognized by the transcription factor. In addition, as a relatively new experimental technique, ChIP-chip is prone to noise itself, and additional strategies have been utilized to filter out false positives (Garten et al. 2005).
The findings of many significant motif pairs, where neighbor seems to be the same as anchor, underscores the importance of homotypic interactions in transcriptional regulation. Two recent bioinformatics studies have based their search for cis- regulatory modules (CRM) in Drosophila upon the clustering of a single motif (Markstein et al. 2002; Papatsenko et al. 2002). What we observed here using human sequence and expression data supports a functional role of binding-site densities, suggesting an analogous search strategy may also be applied to the genome of higher eukaryotes.
Several TF combinations uncovered in our analysis appear to control specific phases of the cell cycle. For example, we found the NF-Y-CDE-CHR module predictive of G2 and G2/M genes. E2F-NF-Y, on the other hand, is preferably associated with those peaking in G1/S and S. NF-Y binding has been reported in many cell cycle promoters (Bolognese et al. 1999; Farina et al. 1999; Yun et al. 1999; Caretti et al. 2003), and its activity can be regulated through nuclear localization, splicing, or post-transcriptional modification (Mantovani 1999). Our results further suggest that as a master transcriptional regulator of cell cycle progression, it may achieve phase specificity by coupling with different functional partners. In addition to combinations of cell cycle-related regulators, we also identified a number of experimentally established regulatory modules involved in other biological processes.
Deciphering transcription regulatory networks from genomic sequence is an exciting but challenging task, especially given the enormous size and complexity of the human genome. In this study, we attempted to uncover the signals that may direct gene expression by searching for evolutionarily conserved and overrepresented TF-binding site combinations associated with more coherent mRNA patterns. While the current analysis was performed with an expression data set obtained from synchronized HeLa cells, it can be readily extended to probe different cellular conditions and types. We anticipate such approaches will be useful for understanding how gene regulation is encoded in the genomic instruction book of life.
Methods
Promoter sequences
Sequence and human-mouse (HUMMUS) alignment information were obtained from a previous study (Shendure and Church 2002). For each of the 11436 curated human mRNA RefSeq in the data set, we extracted the sequence 1 kb upstream of the mRNA as putative promoter region. A total of 8141 have at least portions of their promoter regions conserved in mouse.
Anchor motifs
Position weight matrices (PWM) for transcription-factor binding sites were obtained from the TRANSFAC database (Wingender 2004) (release 6.1). There are 281 matrices annotated as bound by human factors (based on BF field). In consideration of computational time, we limited our analysis to 134 of those with no more than 7000 gene targets and 2000 human-mouse conserved sites in our human promoter set.
Expression data
We utilized the cell cycle expression data from Whitfield et al. (2002). In their study, in order to obtain better resolution at various cell cycle phases, HeLa S3 cells were synchronized with three different methods, double thymidine block, thymidine-nocodazole block, and mitotic shake off. For our analysis, we used the time series from experiments Thy_Thy3, Thy_Noc, and mitotic shake-off, each of which covers one to two cell cycles at 1- to 2-h intervals.
Genome-wide scanning for anchor motifs
PATSER (Hertz and Stormo 1999) (v. 3e) was used to scan our promoter set for matches to the PWM. It was run with the following command line options “-c -li -s -u2.” An “alphabet” file was used to provide the following background frequencies: A/T = 0.48 and C/G = 0.52. These frequencies were determined from our 1-kb human promoter set.
Search for neighbor motifs
AlignACE was used to search for enriched neighbor motifs. It was run with default parameters and a GC background frequency of 0.54, which was calculated using the human-mouse conserved regions of our promoter set.
Statistics of overrepresented neighbor motifs
To determine whether the enrichment of neighbor motif around anchor motif is statistically significant, we devised a measure called neighbor specificity (NS) score based on the binomial distribution. Consider constructing a set of sample genes (promoters) with all those containing anchor motifs. “Success” is scored if the sampled promoter contains a neighbor motif within a specified window around the anchor, or “failure” otherwise. The probability of a random success for each sampled promoter, p, is approximated by:
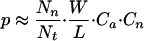 |
where Nn is the number of promoters with neighbor motif, Nt is the total number of promoters (11436), W is the specified window size (50 or 100 bp), L is the promoter length (1000 bp), Ca is the average copy number of anchor motifs per anchor-containing promoter, and Cn is the average copy number of neighbor motifs per neighbor-containing promoter. The W/L term is included, because a success is scored only if the neighbor motif falls within a particular window around the anchor; the Ca and Cn terms take into account scenarios where there may be more than one copy of anchor or neighbor motif on a promoter, which essentially leads to a larger window size or multiple tests for success, respectively. The probability of getting at least the observed number of (anchor-containing) promoters with neighbor motifs within a specified window by chance follows as:
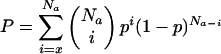 |
where Na is the number of promoters with anchor motif, and x is the number of promoters with anchor-neighbor motif combination.
Correcting for multiple hypothesis testing
Correction for multiple testing was conducted with a Q-value package (http://faculty.washington.edu/~jstorey/qvalue/), which uses an FDR method. FDR, or false discovery rate, is the rate that significant features are truly null. It has increased power over the Bonferroni-type approach. We determined NS score thresholds corresponding to a FDR of 0.05: 5.75 E-3 and 5.09 E-3 for 50 and 100 bp, respectively.
Identifying functional anchor-neighbor motif combinations
We quantified the similarity of expression profiles within a given set of genes using the expression-coherence score (Pilpel et al. 2001), which is defined as the fraction of gene pairs whose expression profiles are closely correlated (i.e., correlation coefficient falls within the top fifth percentile of all gene pairs in the genome). To determine whether the expression-coherence score of genes with the motif combination is significantly higher than both anchor- and neighbor-containing genes without the combination, we adopted a model based on multivariate hypergeometric distribution as previously described (Banerjee and Zhang 2003). The probability of observing at least mcomb out of ncomb gene pairs closely correlated was calculated as following:
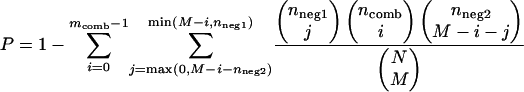 |
where n = the number of gene pairs in a set, m = the number of closely correlated gene pairs in a set, comb = the set of genes with anchor-neighbor motif combination, neg1 = the set of genes with anchor motif but without neighbor in the vicinity, neg2 = the set of genes with neighbor motif but without anchor in the vicinity, N =nneg1+ncomb+nneg2, and M =mneg1+mcomb+mneg2. We reported the combinations with P-values <7.5 E-5 and 5.7 E-5 for 50 and 100 bp, respectively, as the implied significance level for these cut-offs is 0.05 when applying Bonferroni correction for multiple testing.
Search for genes containing the NF-Y-CDE-CHR module
Based on a few experimentally characterized instances of CDE-CHR repressor, we expanded our search for genes containing the NF-Y-CDE-CHR module by allowing a flexible link of up to 10 bp between CDE ([G/C]GCG[G/C]) and CHR ([G/A][T/C]TTGAA). We then scanned 100 bp upstream of CDE-CHR for NF-Y motif (M00287). Hypergeometric distribution was used to estimate the chance probability of obtaining at least the observed number of NF-Y-CDE-CHR module containing genes peaking in G2 and G2/M phases. More specifically, it is calculated as following:
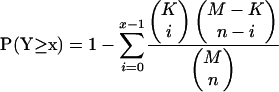 |
where x is the number of genes with potential NF-Y-CDE-CHR regulatory module and peak in G2 and G2/M phases, M is the total number of genes that we have both expression data and sequence data for (8162), n is the number of genes with potential NF-Y-CDE-CHR regulatory module, and K is the total number of G2 and G2/M genes (230).
Acknowledgments
We are grateful to John Aach, Patrik D'haeseleer, Philippe Marc, Allegra Petti, and Fritz Roth for inspiring discussions and valuable comments. We also thank the anonymous reviewers for their helpful feedback. Z.Z. was a Howard Hughes Medical Institute predoctoral fellow. This work was supported by CEGS, DOE-GTL, DARPA, and the Lipper Foundation.
Footnotes
[Supplemental material is available online at www.genome.org and http://genetics.med.harvard.edu/~zhu/combination.html.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3394405.
References
- Aerts, S., Van Loo, P., Thijs, G., Moreau, Y., and De Moor, B. 2003. Computational detection of cis-regulatory modules. Bioinformatics 19: II5-II14. [DOI] [PubMed] [Google Scholar]
- Arnone, M.I. and Davidson, E.H. 1997. The hardwiring of development: Organization and function of genomic regulatory systems. Development 124: 1851-1864. [DOI] [PubMed] [Google Scholar]
- Banerjee, N. and Zhang, M.Q. 2003. Identifying cooperativity among transcription factors controlling the cell cycle in yeast. Nucleic Acids Res. 31: 7024-7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer, M.A. and Tavazoie, S. 2004. Predicting gene expression from sequence. Cell 117: 185-198. [DOI] [PubMed] [Google Scholar]
- Bertolino, E. and Singh, H. 2002. POU/TBP cooperativity: A mechanism for enhancer action from a distance. Mol. Cell 10: 397-407. [DOI] [PubMed] [Google Scholar]
- Blanchette, M. and Tompa, M. 2002. Discovery of regulatory elements by a computational method for phylogenetic footprinting. Genome Res. 12: 739-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognese, F., Wasner, M., Dohna, C.L., Gurtner, A., Ronchi, A., Muller, H., Manni, I., Mossner, J., Piaggio, G., Mantovani, R., et al. 1999. The cyclin B2 promoter depends on NF-Y, a trimer whose CCAAT-binding activity is cell-cycle regulated. Oncogene 18: 1845-1853. [DOI] [PubMed] [Google Scholar]
- Bussemaker, H.J., Li, H., and Siggia, E.D. 2001. Regulatory element detection using correlation with expression. Nat. Genet. 27: 167-171. [DOI] [PubMed] [Google Scholar]
- Caretti, G., Salsi, V., Vecchi, C., Imbriano, C., and Mantovani, R. 2003. Dynamic recruitment of NF-Y and histone acetyltransferases on cell-cycle promoters. J. Biol. Chem. 278: 30435-30440. [DOI] [PubMed] [Google Scholar]
- Cawley, S., Bekiranov, S., Ng, H.H., Kapranov, P., Sekinger, E.A., Kampa, D., Piccolboni, A., Sementchenko, V., Cheng, J., Williams, A.J., et al. 2004. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116: 499-509. [DOI] [PubMed] [Google Scholar]
- Chang, H., Shyu, K.G., Lin, S., Tsai, S.C., Wang, B.W., Liu, Y.C., Sung, Y.L., and Lee, C.C. 2003. The plasminogen activator inhibitor-1 gene is induced by cell adhesion through the MEK/ERK pathway. J. Biomed. Sci. 10: 738-745. [DOI] [PubMed] [Google Scholar]
- Conkright, M.D., Guzman, E., Flechner, L., Su, A.I., Hogenesch, J.B., and Montminy, M. 2003. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol. Cell 11: 1101-1108. [DOI] [PubMed] [Google Scholar]
- Elkon, R., Linhart, C., Sharan, R., Shamir, R., and Shiloh, Y. 2003. Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res. 13: 773-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euskirchen, G., Royce, T.E., Bertone, P., Martone, R., Rinn, J.L., Nelson, F.K., Sayward, F., Luscombe, N.M., Miller, P., Gerstein, M., et al. 2004. CREB binds to multiple loci on human chromosome 22. Mol. Cell. Biol. 24: 3804-3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farina, A., Manni, I., Fontemaggi, G., Tiainen, M., Cenciarelli, C., Bellorini, M., Mantovani, R., Sacchi, A., and Piaggio, G. 1999. Down-regulation of cyclin B1 gene transcription in terminally differentiated skeletal muscle cells is associated with loss of functional CCAAT-binding NF-Y complex. Oncogene 18: 2818-2827. [DOI] [PubMed] [Google Scholar]
- Frech, K., Quandt, K., and Werner, T. 1998. Muscle actin genes: A first step towards computational classification of tissue specific promoters. In Silico Biol. 1: 29-38. [PubMed] [Google Scholar]
- Garten, Y., Kaplan, S., and Pilpel, Y. 2005. Extraction of transcription regulatory signals from genome-wide DNA-protein interaction data. Nucleic Acids Res. 33: 605-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon, J.B. and Bourillot, P.Y. 2001. Morphogen gradient interpretation. Nature 413: 797-803. [DOI] [PubMed] [Google Scholar]
- Hatada, E.N., Chen-Kiang, S., and Scheidereit, C. 2000. Interaction and functional interference of C/EBPβ with octamer factors in immunoglobulin gene transcription. Eur. J. Immunol. 30: 174-184. [DOI] [PubMed] [Google Scholar]
- Hertz, G.Z. and Stormo, G.D. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563-577. [DOI] [PubMed] [Google Scholar]
- Hirose, T., Sowa, Y., Takahashi, S., Saito, S., Yasuda, C., Shindo, N., Furuichi, K., and Sakai, T. 2003. p53-independent induction of Gadd45 by histone deacetylase inhibitor: Coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 22: 7762-7773. [DOI] [PubMed] [Google Scholar]
- Hughes, J.D., Estep, P.W., Tavazoie, S., and Church, G.M. 2000. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J. Mol. Biol. 296: 1205-1214. [DOI] [PubMed] [Google Scholar]
- Ishida, S., Huang, E., Zuzan, H., Spang, R., Leone, G., West, M., and Nevins, J.R. 2001. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol. Cell. Biol. 21: 4684-4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, V.R., Horak, C.E., Scafe, C.S., Botstein, D., Snyder, M., and Brown, P.O. 2001. Genomic binding sites of the yeast cell-cycle transcription factors SBF and MBF. Nature 409: 533-538. [DOI] [PubMed] [Google Scholar]
- Kato, M., Hata, N., Banerjee, N., Futcher, B., and Zhang, M.Q. 2004. Identifying combinatorial regulation of transcription factors and binding motifs. Genome Biol. 5: R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kel, A., Kel-Margoulis, O., Babenko, V., and Wingender, E. 1999. Recognition of NFATp/AP-1 composite elements within genes induced upon the activation of immune cells. J. Mol. Biol. 288: 353-376. [DOI] [PubMed] [Google Scholar]
- Kel, A.E., Kel-Margoulis, O.V., Farnham, P.J., Bartley, S.M., Wingender, E., and Zhang, M.Q. 2001. Computer-assisted identification of cell cycle-related genes: New targets for E2F transcription factors. J. Mol. Biol. 309: 99-120. [DOI] [PubMed] [Google Scholar]
- Krivan, W. and Wasserman, W.W. 2001. A predictive model for regulatory sequences directing liver-specific transcription. Genome Res. 11: 1559-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy, S., Hannenhalli, S., and Workman, C. 2001. Enrichment of regulatory signals in conserved non-coding genomic sequence. Bioinformatics 17: 871-877. [DOI] [PubMed] [Google Scholar]
- Lifanov, A.P., Makeev, V.J., Nazina, A.G., and Papatsenko, D.A. 2003. Homotypic regulatory clusters in Drosophila. Genome Res. 13: 579-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., Liu, X.S., Wei, L., Altman, R.B., and Batzoglou, S. 2004. Eukaryotic regulatory element conservation analysis and identification using comparative genomics. Genome Res. 14: 451-458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots, G.G., Locksley, R.M., Blankespoor, C.M., Wang, Z.E., Miller, W., Rubin, E.M., and Frazer, K.A. 2000. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288: 136-140. [DOI] [PubMed] [Google Scholar]
- Lucibello, F.C., Truss, M., Zwicker, J., Ehlert, F., Beato, M., and Muller, R. 1995. Periodic cdc25C transcription is mediated by a novel cell cycle-regulated repressor element (CDE). EMBO J. 14: 132-142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeev, V.J., Lifanov, A.P., Nazina, A.G., and Papatsenko, D.A. 2003. Distance preferences in the arrangement of binding motifs and hierarchical levels in organization of transcription regulatory information. Nucleic Acids Res. 31: 6016-6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani, R. 1999. The molecular biology of the CCAAT-binding factor NF-Y. Gene 239: 15-27. [DOI] [PubMed] [Google Scholar]
- Markstein, M., Markstein, P., Markstein, V., and Levine, M.S. 2002. Genome-wide analysis of clustered Dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc. Natl. Acad. Sci. 99: 763-768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papatsenko, D.A., Makeev, V.J., Lifanov, A.P., Regnier, M., Nazina, A.G., and Desplan, C. 2002. Extraction of functional binding sites from unique regulatory regions: The Drosophila early developmental enhancers. Genome Res. 12: 470-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilpel, Y., Sudarsanam, P., and Church, G.M. 2001. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat. Genet. 29: 153-159. [DOI] [PubMed] [Google Scholar]
- Ren, B., Cam, H., Takahashi, Y., Volkert, T., Terragni, J., Young, R.A., and Dynlacht, B.D. 2002. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes & Dev. 16: 245-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, F.P., Hughes, J.D., Estep, P.W., and Church, G.M. 1998. Finding DNA regulatory motifs within unaligned noncoding sequences clustered by whole-genome mRNA quantitation. Nat. Biotechnol. 16: 939-945. [DOI] [PubMed] [Google Scholar]
- Safrany, G. and Perry, R.P. 1995. The relative contributions of various transcription factors to the overall promoter strength of the mouse ribosomal protein L30 gene. Eur. J. Biochem. 230: 1066-1072. [DOI] [PubMed] [Google Scholar]
- Shendure, J. and Church, G.M. 2002. Computational discovery of sense-antisense transcription in the human and mouse genomes. Genome Biol. 3: research0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava, A., Saleque, S., Kalpana, G.V., Artandi, S., Goff, S.P., and Calame, K. 1993. Inhibition of transcriptional regulator Yin-Yang-1 by association with c-Myc. Science 262: 1889-1892. [DOI] [PubMed] [Google Scholar]
- Simpson, P. 2002. Evolution of development in closely related species of flies and worms. Nat. Rev. Genet. 3: 907-917. [DOI] [PubMed] [Google Scholar]
- Storey, J.D. and Tibshirani, R. 2003. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. 100: 9440-9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, M., Ueda, A., Kanamori, H., Ideguchi, H., Yang, J., Kitajima, S., and Ishigatsubo, Y. 2002. Cell-cycle-dependent regulation of human aurora A transcription is mediated by periodic repression of E4TF1. J. Biol. Chem. 277: 10719-10726. [DOI] [PubMed] [Google Scholar]
- Tavazoie, S., Hughes, J.D., Campbell, M.J., Cho, R.J., and Church, G.M. 1999. Systematic determination of genetic network architecture. Nat. Genet. 22: 281-285. [DOI] [PubMed] [Google Scholar]
- Terai, G. and Takagi, T. 2004. Predicting rules on organization of cis-regulatory elements, taking the order of elements into account. Bioinformatics 20: 1119-1128. [DOI] [PubMed] [Google Scholar]
- van Ginkel, P.R., Hsiao, K.M., Schjerven, H., and Farnham, P.J. 1997. E2F-mediated growth regulation requires transcription factor cooperation. J. Biol. Chem. 272: 18367-18374. [DOI] [PubMed] [Google Scholar]
- Wagner, A. 1999. Genes regulated cooperatively by one or more transcription factors and their identification in whole eukaryotic genomes. Bioinformatics 15: 776-784. [DOI] [PubMed] [Google Scholar]
- Wasserman, W.W. and Fickett, J.W. 1998. Identification of regulatory regions which confer muscle-specific gene expression. J. Mol. Biol. 278: 167-181. [DOI] [PubMed] [Google Scholar]
- Wasserman, W.W., Palumbo, M., Thompson, W., Fickett, J.W., and Lawrence, C.E. 2000. Human-mouse genome comparisons to locate regulatory sites. Nat. Genet. 26: 225-228. [DOI] [PubMed] [Google Scholar]
- Weinmann, A.S., Yan, P.S., Oberley, M.J., Huang, T.H., and Farnham, P.J. 2002. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes & Dev. 16: 235-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield, M.L., Sherlock, G., Saldanha, A.J., Murray, J.I., Ball, C.A., Alexander, K.E., Matese, J.C., Perou, C.M., Hurt, M.M., Brown, P.O., et al. 2002. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell 13: 1977-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender, E. 2004. TRANSFAC, TRANSPATH and CYTOMER as starting points for an ontology of regulatory networks. In Silico Biol. 4: 55-61. [PubMed] [Google Scholar]
- Yun, J., Chae, H.D., Choy, H.E., Chung, J., Yoo, H.S., Han, M.H., and Shin, D.Y. 1999. p53 negatively regulates cdc2 transcription via the CCAAT-binding NF-Y transcription factor. J. Biol. Chem. 274: 29677-29682. [DOI] [PubMed] [Google Scholar]
- Zhou, Q., Gedrich, R.W., and Engel, D.A. 1995. Transcriptional repression of the c-fos gene by YY1 is mediated by a direct interaction with ATF/CREB. J. Virol. 69: 4323-4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, W., Giangrande, P.H., and Nevins, J.R. 2004. E2Fs link the control of G1/S and G2/M transcription. EMBO J. 23: 4615-4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwicker, J., Lucibello, F.C., Wolfraim, L.A., Gross, C., Truss, M., Engeland, K., and Muller, R. 1995. Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO J. 14: 4514-4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
WEB SITE REFERENCES
- http://club.med.harvard.edu/hummus/hummus.html; Human-mouse sequence conservation.
- http://faculty.washington.edu/~storey/qvalue/; Q-value package for determining false discovery rate (FDR).
- http://www.bio.cam.ac.uk/cgi-bin/seqlogo/logo.cgi; Tools for generating sequence logo of motifs.