

Abstract
The detection, across several genomes, of local conservation of gene content and proximity considerably helps the prediction of features of interest, such as gene fusions or physical and functional interactions. Here, we want to process realistic models of chromosomes, in which genes (or genomic segments of several genes) can be duplicated within a chromosome, or be absent from some other chromosome(s). Our approach adopts the technique of temporarily forgetting genes and working directly with protein “domains” such as those found in Pfam. This allows the detection of strings of domains that are conserved in their content, but not necessarily in their order, which we refer to as domain teams. The prominent feature of the method is that it relaxes the rigidity of the orthology criterion and avoids many of the pitfalls of gene-families identification methods, often hampered by multidomain proteins or low levels of sequence similarity. This approach, that allows both inter- and intrachromosomal comparisons, proves to be more sensitive than the classical methods based on pairwise sequence comparisons, particularly in the simultaneous treatment of many species. The automated and fast detection of domain teams, together with its increased sensitivity at identifying segments of identical (protein-coding) gene contents as well as gene fusions, should prove a useful complement to other existing methods.
Protein structures and sequences can often be split up into “domains.” Databases such as SCOP for the structures (Andreeva et al. 2004) or Pfam for the sequences (Bateman et al. 2004) are devoted to the identification, classification, and storage of protein domains. Recent studies have focused on protein domains as evolutionary units (Patthy 2003; Vogel et al. 2004) or basic elements in protein-protein interactions (Nye et al. 2004). As stated by Koonin et al. (2000) about comparative genomics, the concept of orthology breaks down for genes coding for complex, multidomain proteins and much of the evolutionary process should be thought of and analyzed in terms of domains rather than proteins (genes). In this study, we adopt a novel approach to the search for chromosomal segments with identical or almost identical protein-coding gene content, based on the decomposition of the genes into the domains of the proteins they code for.
Although the term “synteny” originally referred to gene loci on the same chromosome, it is now widely used to refer to gene loci in different organisms, located on a chromosomal region of common evolutionary ancestry (Passarge et al. 1999). Thus, like many others, we shall use the word synteny to mean “local conservation of gene content and proximity across several organisms.” This conservation probably points out, in many cases, to a selection pressure that tends to preserve the very proximity of the genes (Overbeek et al. 1999). As a consequence, the detection, across several genomes, of local conservation of gene content and proximity considerably helps the prediction of features of interest such as the physical interaction of proteins or their participation in common metabolic/regulatory networks (Marcotte et al. 1999a,b; Sali 1999; Galperin and Koonin 2000; Enright and Ouzounis 2001; Suyama and Bork 2001; von Mering et al. 2003; Korbel et al. 2004; Suhre and Claverie 2004). It also enables phylogenetic reconstructions through the identification of some of the numerous rearrangements events that can affect a genome, i.e., transpositions, deletions, insertions, inversions, fusions, and fissions (for review, see Sankoff 2003; Tang and Moret 2003).
Syntenic regions in eucaryotic genomes are generally defined as groups of two or more genes in one species that possess an ortholog on the same chromosome in another species, irrespective of their orientation or order (Pevzner and Tesler 2003; Jaillon et al. 2004). Here, one can speak of macrosynteny. Among prokaryotic genomes, the definition often adds the constraint of gene proximity—not necessarily contiguity—on both of the compared chromosomes (Bergeron et al. 2002; Luc et al. 2003; von Mering et al. 2003). The addition of this constraint results in much shorter conserved regions, in which case, one speaks of microsynteny. In the search for microsyntenies, one can insist on the conservation of gene order (Overbeek et al. 1999), but generally the order, contiguity, and even strandeness of the genes are relaxed to some extent (Fujibuchi et al. 2000; Tamames 2001; Bergeron et al. 2002; Calabrese et al. 2003; Durand and Sankoff 2003; Luc et al. 2003). Such relaxed microsyntenies were formally defined as gene teams by Bergeron et al. (2002).
In this study, we reinvestigate the search for microsyntenies by temporarily forgetting genes and working directly with protein domains, such as those found in Pfam (Bateman et al. 2004). We define chromosomal regions of conserved protein domains as domain teams. This choice has many interesting consequences. First, it allows us to process simultaneously intrachromosomal and interchromosomal comparisons. Indeed, since all of the protein-coding genes are decomposed into the domains of the proteins they code for, the usual step of finding the “bidirectional best hits” (e.g., Overbeek et al. 1999) is avoided, as well as the problem of partitioning sequences into nonoverlapping and biologically coherent clusters when multidomain proteins are present (see, for example, Yona et al. 1999). As a consequence, the rigidity of the orthology criterion is relaxed, and this approach allows us to process more realistic models of chromosomes, in which genes or segments of genes can be duplicated or even be absent from some chromosomes. Moreover, considering genes from the domain point of view enables us to integrate multiple-sequence alignments information; the position-sensitive scoring matrices (Gribskov et al. 1987) or the hidden Markov model profiles (Eddy 1998) that are stored in the Pfam database (Bateman et al. 2004) are known to be more sensitive than pairwise sequence alignments (e.g., Altschul et al. 1997). Finally, this model allows the detection of events such as fusions and duplications that would not be otherwise obvious.
We implemented this concept in a software named DomainTeam, freely available on request for academic purposes. The strength and limitations of this approach are discussed in detail in this work.
DomainTeam
For reasons that will be made clear in the Results section, we shall here interest ourselves only in prokaryotic organisms. From a computational point of view, a chromosome can be defined as a collection of genes. Focusing on protein-coding genes, we want to define a chromosome as an ordered sequence of genes, where a unique coding sequence is associated with the nucleic acid sequence of a gene. In addition, we will divide each gene into one or more consecutive domains, each domain having a label. In the present case, the domains will be the Pfam domains of the encoded proteins (Pfam imposes a nonoverlapping rule on domains). In those few cases where a domain is inserted within another one (Bateman et al. 2004), the two domains are considered as adjacent. Overlapping genes (e.g., Fukuda et al. 1999) are similarly noted as contiguous (see Supplemental material, part 1).
The distance between two domains on the same chromosome is the difference between their positions. The position of a domain is defined using the order in which the domains appear on the chromosome (considering both DNA strands). Given a set S of domain labels, and a fixed distance δ, the labels of S divide a set of chromosomes in δ-chains. These are maximal runs of domains whose labels belong to S, such that the distance between two consecutive domains in a run is less than or equal to δ. For example, consider the domains A, B, and C (S ={A, B, C}) and the following set C of chromosomes in which these domains have been underlined:
C = ABD EFBCAGH IJAKBCLM NOPCAQARS
With δ = 2, the set S induces four δ-chains on the chromosomes of C: AB, BCA, AKBC, and CAQA. Note that the domains in different δ-chains can appear in different orders, and are not necessarily contiguous in a given δ-chain.
The content of a δ-chain is the subset of S of the labels that appear in the domains of the run. Each δ-chain that contains all of the labels of a set S is called an occurrence of the set S. A set of labels T is an extension of a set S if S is contained in T, and each occurrence of S is contained in an occurrence of T.
Definition 1
Given δ, a set S of labels is a δ-team of a set of chromosomes C if there is at least one occurrence of the set S in C, and S has no extension.
For example, in the above set C of chromosomes, the set S ={A, B, C} is a δ-team with δ = 2. It has two occurrences: BCA and AKBC. On the other hand, the set {B} is not a δ-team, since the set T ={A, B} is an extension of {B}, which means that each occurrence of label B implies a nearby occurrence of label A (the reverse is not true). Note that for δ = 2, the set T ={A, B} is also a δ-team, even if S contains T because T is not an extension of S. In this case, it has three occurrences: AB, BCA and AKB, which means that teams can be nested. Thus, in a set of n chromosomes, a set {A, B, C} can be a team conserved in m ≤ n chromosomes, but the shorter nested set {A, B} can be conserved in k > m chromosomes. DomainTeam will report both sets. In other words, DomainTeam does not report only those teams conserved in all of the chromosomes. Definition 1 is a direct generalization of the notion of gene teams introduced by Bergeron et al. (2002), which addressed the case of chromosomes containing a unique copy of each gene. He and Goldwasser (2004) also defined an extension of gene teams that allows multiple copies of a gene in a chromosome. However, the number of chromosomes must be restricted to two in order to achieve polynomial time complexity of their algorithm.
Figure 1 shows an example of a domain team found in four different organisms, exhibiting significant rearrangements. The five domains present in Yersima pestis are transposed, reversed, and duplicated in Salmonella typhi, Escherichia coli, and Vibrio cholerae. Another example is shown in the Supplemental material (part 2), depicting a team found in a set of 10 pathogenic bacteria.
Figure 1.

A domain team (δ = 3) of five domains with occurrences in four different organisms, with two occurrences in S. typhi. The first occurrence in S. typhi has the same domain order and content as the occurrence in Y. pestis, except that the whole segment is reversed. In the second occurrence in S. typhi, domain 294 is duplicated in reverse, sandwiching an insertion of a new domain. There is also a transposition of domain 294 and a duplication of domain 359, with respect to the four other occurrences. V. cholerae has a duplication of domain 2379 and E. coli a duplication of domain 294.
The number of teams can be exponential
Without additional constraints, Definition 1 also leads to theoretically exponential algorithms, since the number of domain teams can be exponential in the number of labels. However, as shown in the next sections, real-life examples involving thousands of genes can be computed efficiently or at least in a reasonable time.
In order to show the exponential nature of Definition 1, consider a set L of n labels. Construct n chromosomes, each containing n-1 different labels obtained by removing one different label from L. Then, for δ = n-2, each proper subset of L is a δ-team. For example, with n = 5 and L ={A, B, C, D, E}, one gets the following five chromosomes:
ABCD ABCE ABDE ACDE BCDE
Each proper subset S of L has at least one occurrence, since S is contained in at least one chromosome, and the distance between two labels in a chromosome is always less than δ = n-2. For any domain d not in S, there is an occurrence of S that is not contained in 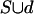 , namely, the chromosome in which d was removed, therefore, S has no extension. Thus, S is a δ-team.
, namely, the chromosome in which d was removed, therefore, S has no extension. Thus, S is a δ-team.
Results and Discussion
Sensitivity of DomainTeam as viewed from three closely related genomes
As a way to test the sensitivity of our approach, we compared the results obtained by GeneTeam (Luc et al. 2003) and DomainTeam on a set of three chromosomes from closely related species. Both algorithms implement the same notion of microsynteny, but GeneTeam searches for regions of conserved orthologous protein-coding genes, while DomainTeam looks for regions of conserved protein domains content. The comparison was performed by mapping the chromosome of E. coli according to the syntenic regions it shares with both the S. typhi and Y. pestis chromosomes. In both programs, the δ parameter was set to 3 (allowing gaps of two consecutive genes or domains).
The results are summarized in Figure 2. The first obvious observation is that, for both programs, there are no huge teams that would encompass almost all of the genome. Rather, these three closely related species share a lot of microsyntenic regions (red color in Fig. 2). As expected, the teams obtained by DomainTeam (inner circle) and GeneTeam (outer circle) most often coincide. However, DomainTeam identifies larger and more numerous microsyntenies, as large nonsyntenic regions reported by GeneTeam are broken into several domain teams. The largest teams (green in Fig. 2) contain 31 and 26 genes for DomainTeam and GeneTeam, respectively. On the whole, the domain teams harbor 2207 genes (52% of the E. coli genes) and the gene teams 1662 (40%). This difference can be explained by at least three reasons, i.e., the use of the domain criterion (1) relaxes the need for strict homology, (2) permits various rearrangements of domains such as duplications or fusions, and (3) allows one to take paralogs into account; thus, the identification of duplicated regions. These three points are discussed in the next sections.
Figure 2.

Map of the E. coli chromosome where genes colored red are those genes of E. coli that belong to a team also found in S. typhi and Y. pestis. Genes colored blue do not belong to a microsyntenic region shared by the three species. The inner circle shows the results of DomainTeam (δ = 3). The outer circle shows those of GeneTeam (δ = 3), based on the set of 2106 triplets of orthologous proteins obtained by the bidirectional best hit method. Syntenic regions reported by DomainTeam and GeneTeam coincide, but DomainTeam finds larger syntenic regions and identifies 2207 syntenic genes (52% of the E. coli genes) versus 1662 (40%) for GeneTeam. Green regions indicate the largest teams (31 and 26 genes) for DomainTeam and GeneTeam respectively. Figure 2 was drawn using GenomeViz (Ghai et al. 2004).
The use of domains bypasses the rigidity of pairwise sequence comparisons
As already stated, multiple-sequence alignment profiles make protein sequence comparisons more sensitive than classical pair-wise alignments. Homology inference will inevitably fail in the last case, when sequences diverged too much, while two highly divergent homologous (protein) sequences may well continue to possess a common Pfam domain.
Figure 3 displays a schematic representation of a conserved team between E. coli and S. typhi, in which the proteins share five domains. The proteins encoded by pgtA and pgtB in S. typhi are known to be the members of a two-component regulatory system (Kadner 1996). As shown in the STRING database (von Mering et al. 2003), genes encoding two-component systems are often adjacent. The pairs YfhA/YfhK and Sty2809/Sty2811 are putative proteins that were assigned the same function (two-component regulatory system) by homology with proteins from other bacteria. However, sequence comparisons of PgtB with both YfhK and Sty2811 resulted in high BLAST2 E-values (10 and 0.17, respectively). As a consequence, the teams YfhA/YfhK and Sty2809/Sty2811 are not reported in STRING (they appear, however, in the KEGG database [Kanehisa et al. 2004] which is maintained through considerable manual expertise). Similarly, the probable duplication of pgtA and pgtB in S. typhi would not have been detected by an automated procedure based on pairwise comparisons. Note that the two inserted genes yfhG and sty2810 code for highly similar (hypothetical) proteins, which reinforces the probability that the two teams yfhA/yfhG/yfhK and STY2809/STY2810/STY2811 are genuine orthologous conserved segments whose proteins share the same functions in the two species.
Figure 3.

An example of a team (δ = 3) found in E. coli and S. typhi, corresponding to proteins that belong to the so-called “two-components regulatory system.” The figures near the arrows are the BLAST E-values corresponding to the pairwise alignments of the proteins. It can be seen that the proteins YfhK and PgtB share but little sequence similarity, preventing this team from being detected by automated methods based on sequence comparisons. Similarly, PgtB and STY2811 are poorly similar, but the use of their Pfam labels led to pinpointing the duplication in S. typhi.
Using domains instead of genes as an atomic unit allows us to detect domain rearrangements such as fusions
The detection of gene fusion events can be used to predict functional associations of proteins, such as functional interaction or complex formation (Enright et al. 1999; Marcotte et al. 1999b; Enright and Ouzounis 2001; Yanai et al. 2001). Fusions can be considered as extreme cases of conservation of gene proximity. Indeed, “evolution of gene fusion often involves an intermediate stage, during which the future fusion components exist as juxtaposed and coregulated, but still distinct genes within operons” (Yanai et al. 2002). In such a context of proximity, DomainTeam can easily detect fusion events, since a two-domains fused protein and the one-domain adjacent unfused proteins will result in the same team.
An example is given in Figure 4, which results from the search for conserved teams across five bacteria. This team is part of the tryptophan operon. While trpC is a stand-alone gene in Bacteroides thetaiotaomicron and Anabaena, it is fused with trpF in E. coli, S. typhi, and Y. pestis. As to trpG, it is fused with trpD in E. coli and S. typhi, but with trpE in Anabaena. These fusions are also detected by other methods based on sequence comparisons and are reported in FusionDB (Suhre and Claverie 2004) and AllFuse (Enright and Ouzounis 2001). However, the simultaneous comparison of several chromosomes by DomainTeam enables an immediate synthetic view of all the domain rearrangements.
Figure 4.

Part of the tryptophan operon as identified in five bacteria (δ = 3), exhibiting rearrangements and fusions of domains. Genes are labeled with their “ordered locus name” and, for E.coli and B. thetaiotaomicron, by their names.
Since DomainTeam detects only the fusions between adjacent genes, it will not replace other methods that rely basically on sequence comparisons, irrespective of the distance between the fusion components. However, the increased sensitivity afforded by the Pfam domains enables us to find otherwise undetected fusions. We examined the fusions concerning adjacent genes in the pairs E. coli/Haemophilus influenzae and E. coli/Helicobacter pylori reported by FusionDB, AllFuse, and DomainTeam. A total of 39 such (predicted) fusions was found, only two of them being reported by the three methods, eight by two methods, and 29 by one method, among which five were predicted by DomainTeam only. As shown in Table 1, in all of these last five cases, one of the fusion (protein) components did not match sufficiently the fused protein to be detected by a similarity search. Conversely, eight fusions predicted by FusionDB or AllFuse were not detected by DomainTeam, because one of their components did not possess a Pfam label. It is therefore clear that while DomainTeam cannot by itself replace other published methods, it can be used usefully as a complementary tool to detect otherwise unpredicted fusions.
Table 1.
Some otherwise undetected composite genes reported by Domain Teams
| N-terminal gene | C-terminal gene | Composite gene |
|---|---|---|
| Hl1549 (lolD) ABC_tran | Hl1548 (lolE) FtsX E> 100 | b0879 (macB) ABC_tran/FtsX |
| Lipoprotein releasing system ATP-binding protein lolD | Lipoprotein releasing system transmembrane protein lolC | Macrolide-specific ABC-type efflux carrier |
| H. influenzae | H. influenzae | E. coli |
| Hl0769 (ftsE) ABC_tran | Hl0770 (ftsX) FtsX E> 100 | b0879 (macB) ABC_tran/FtsX |
| Cell division ATP-binding protein ftsE* | Cell division protein ftsX homolog* | Macrolide-specific ABC-type efflux carrier |
| H. influenzae | H. influenzae | E. coli |
| Hl0291 HMA E=2.10−4 | Hl0290 HMA/E1-E2ATPase/Hydrolase | b0484 (copA) HMA/HMA/E1-E2ATPase/Hydrolase |
| Hypothetical protein | Probable cation-transporting ATPase | Copper-transporting P-type ATPase |
| H. influenzae | H. influenzae | E. coli |
| Hl0988 (leu2) Aconitase | Hl0989 (leuD) Aconitase_C E=0.83 | b1276 (acnA) Aconitase/Aconitase_C |
| 3-isopropylmalate dehydratase large subunit* | 3-isopropylmalate dehydratase small subunit* | Aconitate hydratase 1 |
| H. influenzae | H. influenzae | E. coli |
| b3577 DctQ E=2.4 | b3578 DctM/DedA | Hl0147 DctQ/DctM/DedA |
| Hypothetical protein* | Hypothetical protein* | Hypothetical protein |
| E. coli | E. coli | H. influenzae |
| b2678 (proW) BPD_transp_1 | b2679 (proX) OpuAC E> 100 | HP0818 BPD_transp_1/OpuAC |
| Glycine betaine/L-proline transport system permease* | Glycine betaine-binding periplasmic protein precursor* | Osmoprotection protein (prowx) |
| E. coli | E. coli | H. pylori |
Probable gene fusions between adjacent genes detected by Domain Teams after the comparison of the chromosomes of E. coli, H. influenzae, and H. pylori. Here are listed only the composite genes not reported in AllFuse and FusionDB. Note, however, that the fusion between the two components can be reported in FusionDB or AllFuse, based on evidence from other genomes (*). Each gene is identified by its ordered locus name, followed by its name (if any), followed by the Pfam domain(s) found in the protein they code for. The BLAST2 E-value between one of the components and the composite protein is also reported.
Duplications are detected by intrachromosomal comparisons
The classical step of finding orthologous genes before searching for syntenies prevents the detection of intrachromosomal duplications. We have already shown in Figure 3 that the use of domains and intrachromosomal comparisons not only enables one to find duplications, but also to detect duplications where the sequence similarities are weak. Another example containing a duplication of a whole syntenic region will be found in the Supplemental material (part 2), showing a team found in a set of 10 pathogenic bacteria.
Sensitivity of DomainTeam in massive comparisons
The simultaneous detection of a local conservation of orthologous genes in a number of chromosomes is a difficult task, since the sequence similarities can be weak in distant species. As a way to explore the sensitivity of DomainTeam across many genomes, we took as a test case the collection of E. coli operons stored in the RegulonDB database (Salgado et al. 2004; J. Collado-Vides, pers. comm.) and searched for their being conserved in a set of 14 other Gram-negative bacteria. From the set of 309 E. coli operons, 245 (79%) were fully recovered by at least one domain team. The conserved regions, hence, the teams, were always larger than the operons per se. In some cases, one or more genes within a team encompassing an operon were considered as insertions as they corresponded to proteins that had no Pfam label (an example is given in Fig. 5). The fifty operons that could not be entirely recovered as a single domain team were operons that contained too many consecutive Pfam unlabeled genes. They were thus broken into several partial segments. Fourteen operons in E. coli have no counterpart in any of the 14 other bacteria.
Figure 5.

An example of a team (δ = 3) found in four bacteria. This team corresponds to the “superoperon” yjeFE-amiB-mutL-miaA-hfq-hflXKC in the RegulonDB database, from b4167 to b4175 in E. coli. The conserved team thus extends beyond this operon. Some proteins do not contain a Pfam label (arrowheads). However, DomainTeams could retrieve the entire operon (and more) because these proteins are considered as insertions. The proteins are labeled by the “ordered locus name” of their genes.
Each fully recovered operon was classified according to the number of chromosomes the team was found in, from two to 16 (the set of 15 Gram-negative bacteria comprised 16 chromosomes, since the genome of V. cholerae consists of two chromosomes; see Methods). Each class was then divided into three groups in the following way: (1) group 1, containing the teams found only in two or more of the eight gammaproteobacteria chromosomes; (2) group 2, containing the teams found in both gammaproteobacteria and other proteobacteria (comprising two epsilon-proteo-bacteria and one alphaproteobacterium); (3) group 3, containing the teams found simultaneously in gammaproteobacteria, other proteobacteria, and more distant taxons (the set included one cyanobacterium, one bacteroidete, one spirochete, one chlamydiae, and one thermotogae). Figure 6 illustrates the phylogenetic distribution of the 245 fully recovered operons. While 14 operons are specific to E. coli, 96 operons were recovered only within the gammaproteobacteria (group 1), and 33 extra operons were also found in other proteobacteria (group 2). Surprisingly enough, the 116 remaining operons were also fully recovered within at least one of the more distant species (group 3). See Supplemental material, part 3, for the list of operons and their phylogenetic distribution.
Figure 6.

Diagram of the phylogenetic distribution of 245 E. coli operons (of 309) fully recovered by at least one domain team in the set of 15 Gram-negative bacteria. The figure shows the distribution of the operons as a function of the number of chromosomes in which the operons were identified as syntenic. Each class has been divided into three categories, depending on the species where the teams were found, i.e., only in gammaproteobacteria or only in proteobacteria, or also in other taxons. Thus 96 operons (gray) were recovered only within close species (gammaproteobacteria), but the diagram shows that 149 other operons are conserved in more distant bacteria. Fourteen operons (class 1) were found only in E. coli.
Limitations of domain teams identification
However sensitive the method is, DomainTeam may report false negatives in those cases where adjacent protein-coding genes are not labeled with a Pfam domain. Conversely, DomainTeam may result in false positives due to “promiscuous domains” of broad specificity (Marcotte et al. 1999b; see also, Harlow et al. 2004) that link otherwise unrelated proteins. An empirical score aimed at ranking the observed sets of teams has been designed to reduce the number of false positives.
The DomainTeam algorithm relies on pre-existing Pfam annotations of proteomes. As of December 2004, the Pfam library covers 74% of the proteins in SWISS-PROT/TrEMBL. This means that, on average, one protein in four is not (so far) labeled with a Pfam domain. As shown in Table 2, the Pfam coverage of complete proteomes is heterogeneous and varies from 96% for Buchnera aphidicola (a symbiotic bacterium endowed with a small genome) down to 40% for the archaebacterium Aeropyrum pernix. Obviously, DomainTeam will inevitably miss these unlabeled proteins and their corresponding genes. Most of the time, however, they will simply be considered as insertions within the teams (a false negative will be obtained when n consecutive genes are unlabeled, with n ≥ δ). In order to apply DomainTeam to a newly sequenced genome, one would have first to annotate the proteins with the HMMER series of programs (http://hmmer.wustl.edu/), which may not be trivial. Since the aim of DomainTeam is not to supercede other tools dedicated to the search of microsyntenies, but to allow a more sensitive approach, we would rather advise using GeneTeam (Luc et al. 2003) as a first global approach for the study of a genome devoid of Pfam annotations.
Table 2.
Coverage of the Pfam database
| Number of genomes | Mean coverage (%) | Highest coverage (%) | Lowest coverage (%) | |
|---|---|---|---|---|
| Eukaryota | 17 | 64 | 75 Arabidopsis thaliana | 49 Plasmodium falciparum |
| Bacteria | 157 | 76 | 96 Buchnera apidicola | 44 Rhodospirellula baltica |
| Archae | 19 | 66 | 79 Pyrococcus abyssi | 40 Aeropyrum pernix |
The coverage of a complete proteome is the number of its proteins (in percent) that contain one or more Pfam domain(s). The data have been extracted from the Pfam Web site (December 2004).
Although microsyntenic regions can be found across eukaryotic genomes (e.g., Oh et al. 2002; Jaillon et al. 2004), the situation here is so complicated by the presence of promiscuous domains, tandemly duplicated genes, and alternative splicing, that DomainTeam does not seem to perform better than other existing tools for higher eukaryotic species.
Some “promiscuous domains,” such as DNA-binding domains, increase the number of small uninteresting teams. We addressed this problem through the use of a simple and empirical score, aimed at ranking the observed sets of teams as a function of the number of different domains they contain and the number of different chromosomes they belong to. For one set of a given δ-team, let np be the number of proteins in the team (not counting those proteins having one or more orphan Pfam label[s]), nd the number of different domains, no the number of occurrences of the team, and m the weighted mean of the frequencies of the domains in the set (m = ∑i ni * fi with ni the number of times the domain i appears in the team and fi the frequency of the domain i in the set). The score S is defined as
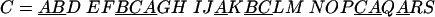 |
The best ranks are for those teams having a high number of proteins per chromosome (np/no) with a high number of different domains (nd) and a low number of promiscuous domains (1/m). It is our experience that teams with S > 90 are potentially interesting. See Supplemental material, part 4, as an example of the average number of proteins per occurrence in those teams having a score ≥ 90.
Practical computing considerations
The computation time required to compare a set of chromosomes is a function of the number of chromosomes, the number of proteins in the set, the value of δ, and the degree of conservation between the organisms under study. We tested the efficiency of DomainTeam on a 1 Ghz Sun ultrasparc III+ processor. The comparison with δ = 3 was performed in 5 min for the set of 16 Archaebacteria, 320 min for the set of 15 Gram-negative bacteria (containing very close species), and 29 min for the set of 13 Gram-positive bacteria. Thus, DomainTeam can compare a large number of chromosomes in a reasonable time. See Supplemental material, part 5, for more information about computing considerations.
Conclusions
Most of the methods aimed at detecting chromosomal regions of conserved gene content are based on the sequence similarities between the encoded proteins. We have shown that labeling the genes with the Pfam domain(s) of the proteins they code for, coupled with the notion of teams, adds an extra sensitivity to the process and makes it possible to compare simultaneously more than 10 chromosomes in a reasonable time. In addition, the program DomainTeam performs both inter- and intrachromosomal comparisons at the same time. It should prove a useful complement to other existing methods.
Methods
Chromosome tables and Pfam annotations
The chromosomal ordered lists (chromosome tables) of the bacterial genes and their products (together with their UniProt IDs) were downloaded from the EBI “proteome” site (http://www.ebi.ac.uk/integr8/EBI-Integr8-HomePage.do). The Pfam annotations pertaining to the above-mentioned proteomes were downloaded from ftp://ftp.sanger.ac.uk/pub/databases/Pfam/database-files.
Bacterial sets
The bacterial sets used in this study were as follows:
Set of 15 Gram-negative bacteria: Anabaena sp, Bacteroides thetaiotaomicron, Borrelia burgdorferi, Campylobacter jejuni NCTC 11168, Chlamydia muridarum, Escherichia coli K12, Haemophilus influenzae, Helicobacter pylori ATCC 700392, Pseudomonas aeruginosa, Rhizobium loti, Salmonella typhi, Thermotoga maritima, Vibrio cholerae, Xylella fastidiosa, Yersinia pestis CO-92.
Set of 13 Gram-positive bacteria: Bacillus subtilis, Bifidobacterium longum, Clostridium perfringens, Corynebacterium efficiens, Deinococcus radiodurans, Enterococcus faecalis, Lactococcus lactis, Lactobacillus plantarum, Listeria monocytogenes, Mycobacterium leprae, Oceanobacillus iheyensis, Staphylococcus aureus N315, Streptococcus agalactiae serotype V.
Set of 16 archaebacteria: Aeropyrum pernix, Archaeoglobus fulgidus, Halobacterium sp, Methanobacterium thermoautotrophicum, Methanococcus jannaschii, Methanopyrus kandleri, Methanosarcina acetivorans, Methanosarcina mazei, Pyrococcus abyssi, Pyrobaculum aerophilum, Pyrococcus furiosus, Pyrococcus horikoshii, Sulfolobus solfataricus, Sulfolobus tokodaii, Thermoplasma acidophilum, Thermoplasma volcanium.
DomainTeam
The program DomainTeam is written in standard ANSI C and was run under both the Linux kernel 2.4.21 (Intel Pentium III at 1.3 GHz) and Sun Solaris 9 (Ultrasparc III+ at 1 Ghz) operating systems. The full results of DomainTeam for the Gram-negative and Gram-positive and archaebacteria can be viewed and queried by gene name from http://lgi.infobiogen.fr/DomainTeams. The DomainTeam program is freely available on request for academic purposes. Binary codes and scripts to display graphical outputs can be obtained from the same URL (Downloads). See also the link `Overview of the software' for an explanation of the text output format of DomainTeam.
Acknowledgments
We thank the Infobiogen team for their patience and understanding during very long runs and M. Marshall from the Pfam team for her help in retrieving the proper annotation files.
Footnotes
[Supplemental material is available online at www.genome.org.]
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3638405. Article published online before print in May 2005.
References
- Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva, A., Howorth, D., Brenner, S.E., Hubbard, T.J., Chothia, C., and Murzin, A.G. 2004. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 32: D226-D229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32: D138-D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron, A., Corteel, S., and Raffinot, M. 2002. The algorithmic of gene teams. Lecture Notes Comput. Sci. 2452: 464-476. [Google Scholar]
- Calabrese, P.P., Chakravarty, S., and Vision, T.J. 2003. Fast identification and statistical evaluation of segmental homologies in comparative maps. Bioinformatics 19: i74-i80. [DOI] [PubMed] [Google Scholar]
- Durand, D. and Sankoff, D. 2003. Tests for gene clustering. J. Comput. Biol. 10: 453-482. [DOI] [PubMed] [Google Scholar]
- Eddy, S.R. 1998. Profile hidden Markov models. Bioinformatics 14: 755-763. [DOI] [PubMed] [Google Scholar]
- Enright, A.J. and Ouzounis, C.A. 2001. Functional associations of proteins in entire genomes by means of exhaustive detection of gene fusions. Genome Biol. 2: research0034.1-0034.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright, A.J., Iliopoulos, I., Kyrpides, N.C., and Ouzounis, C.A. 1999. Protein interaction maps for complete genomes based on gene fusion events. Nature 402: 86-90. [DOI] [PubMed] [Google Scholar]
- Fujibuchi, W., Ogata, H., Matsuda, H., and Kanehisa, M. 2000. A heuristic graph comparison algorithm and its application to detect functionally related enzyme clusters. Nucleic Acids Res. 28: 4021-4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, Y., Washio, T. and Tomita, M. 1999. Comparative study of overlapping genes in the genomes of Mycoplasma genitalium and Mycoplasma pneumoniae. Nucleic Acids Res. 27: 1847-1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin, M.Y. and Koonin, E.V. 2000. Who's your neighbor? New computational approaches for functional genomics. Nat. Biotech. 18: 609-613. [DOI] [PubMed] [Google Scholar]
- Ghai, R., Torsten Hain, T. and Chakraborty, T. 2004. GenomeViz: Visualizing microbial genomes. BMC Bioinformatics 5: 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribskov, M., McLachlan, A.D., and Eisenberg, D. 1987. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. 84: 4355-4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow, T.J., Gogarten, J.P., and Ragan, M.A. 2004. A hybrid clustering approach to recognition of protein families in 114 microbial genomes. BMC Bioinformatics 5: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, X. and Goldwasser, M. 2004. Identifying conserved gene clusters in the presence of orthologous groups. In Proceedings of the Eighth Annual International Conference on Computational Molecular Biology (RECOMB) 2004 (eds. P.E. Bourne and D. Gusfield), pp. 272-280. ACM, New York.
- Jaillon, O., Aury, J-M., Brunet, F., Petit, J-L., Stange-Thomann, N., Mauceli, E., Bouneau, L., Fischer, C., Ozouf-Costaz, C., Bernot, A., et al. 2004. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431: 946-957. [DOI] [PubMed] [Google Scholar]
- Kadner, R.J. 1996. Cytoplasmic membrane. In Escherichia coli and Salmonella typhimurium, cellular and molecular biology (eds. F.C. Neidhardt et al.), pp. 58-87. ASM Press, Washington, DC.
- Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. 2004. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32: D277-D280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin, E.V., Arawind, L., and Kondrashov, A.S. 2000. The impact of comparative genomics on our understanding of evolution. Cell 101: 573-576. [DOI] [PubMed] [Google Scholar]
- Korbel, J.O., Jensen, L.J., von Mering, C., and Bork, P. 2004. Analysis of genomic context: Prediction of functional associations from conserved bidirectionally transcribed gene pairs. Nat. Biotech. 22: 911-917. [DOI] [PubMed] [Google Scholar]
- Luc, N., Risler, J-L., Bergeron, A., and Raffinot, M. 2003. Gene teams: A new formalization of gene clusters for comparative genomics. Comput. Biol. Chem. 27: 59-67. [DOI] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Thompson, M.J., Yeates, T.O., and Eisenberg, D. 1999a. A combined algorithm for genome-wide prediction of protein function. Nature 402: 83-86. [DOI] [PubMed] [Google Scholar]
- Marcotte, E.M., Pellegrini, M., Ho-Leung, N., Rice, D.W., Yeates, T.O., and Eisenberg, D. 1999b. Detecting protein function and protein-protein interactions from genome sequences. Science 30: 751-753. [DOI] [PubMed] [Google Scholar]
- Nye, T.M., Berzuini, C., Gilks, W.R., Babu, M.M., and Teichmann, S.A. 2004. Statistical analysis of domains in interacting protein pairs. Bioinformatics 21: 993-1001. [DOI] [PubMed] [Google Scholar]
- Oh, K.C., Hardeman, C., Ivanchenko, M.G., Ellard-Ivet, M., Nebenfür, A., White, T.J., and Lomax, T.L. 2002. Fine mapping in tomato using microsynteny with the Arabidopsis genome: The Diageotropica (Dgt) locus. Genome Biol. 3: research0049.1-0049.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek, R., Fonstein, M., D'Souza, M., Pusch, G.D., and Maltsev, N. 1999. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. 96: 2896-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passarge, E., Horsthemke, B., and Farber, R.A. 1999. Incorrect use of the term synteny. Nat. Genet. 23: 387. [DOI] [PubMed] [Google Scholar]
- Patthy, L. 2003. Modular assembly of genes and the evolution of new functions. Genetica 118: 217-231. [PubMed] [Google Scholar]
- Pevzner, P. and Tesler, G. 2003. Genome rearrangements in mammalian evolution: Lessons from human and mouse genomes. Genome Res. 13: 37-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado, H., Gama-Castro, S., Martinez-Antonio, A., Diaz-Peredo, E., Sanchez-Solano, F., Peralta-Gil, M., Garcia-Alonso, D., Jimenez-Jacinto, V., Santos-Zavaleta, A., Bonavides-Martinez, C., et al. 2004. RegulonDB (version 4.0): Transcriptional regulation, operon organization and growth conditions in Escherichia coli K-12. Nucleic Acids Res. 32: D303-D306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali, A. 1999. Functional links between proteins. Nature 402: 23-26. [DOI] [PubMed] [Google Scholar]
- Sankoff, D. 2003. Rearrangements and genome evolution. Curr. Opin. Gen. Dev. 13: 583-587. [DOI] [PubMed] [Google Scholar]
- Suhre, K. and Claverie, J-M. 2004. FusionDB: A database for in-depth analysis of prokaryotic gene fusion events. Nucleic Acids Res. 32: D273-D276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama, M. and Bork, P. 2001. Evolution of prokaryotic gene order: Genome rearrangements in closely related species. Trends Genet. 17: 10-13. [DOI] [PubMed] [Google Scholar]
- Tamames, J. 2001. Evolution of gene order conservation in prokaryotes. Genome Biol. 2: 0020.1-0020.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, J. and Moret, B.M. 2003. Scaling up accurate phylogenetic reconstruction from gene-order data. Bioinformatics 19: i305-i312. [DOI] [PubMed] [Google Scholar]
- Vogel, C., Bashton, M., Kerrison, N.D., Chothia, C., and Teichmann, S.A. 2004. Structure, function and evolution of multidomains proteins. Curr. Opin. Struct. Biol. 14: 208-216. [DOI] [PubMed] [Google Scholar]
- von Mering, C., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., and Snel, B. 2003. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 31: 258-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai, I., Derti, A., and DeLisi, C. 2001. Genes linked by fusion events are generally of the same functional category: A systematic analysis of 30 microbial genomes. Proc. Natl. Acad. Sci. 98: 7940-7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai, I., Wolf, Y.I., and Koonin, E.V. 2002. Evolution of gene fusions: Horizontal transfer versus independent events. Genome Biol. 3: research0024.1-0024.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yona, G., Linial, N., and Linial, M. 1999. Protomap: Automatic classification of protein sequences, a hierarchy of protein families, and local maps of the protein space. Proteins 37: 360-378. [PubMed] [Google Scholar]
WEB SITE REFERENCES
- ftp://ftp.sanger.ac.uk/pub/databases/Pfam/database-files; The directory of the Pfam ftp server that contains the Pfam annotations of the proteins in UniProt.
- http://hmmer.wustl.edu/; HMMER series of programs.
- http://www.ebi.ac.uk/integr8/EBI-Integr8-HomePage.do; The proteome Home Page at EBI.
- http://lgi.infobiogen.fr/DomainTeams; DomainTeams full results and code downloads.