

Abstract
Selection and licensing of mammalian DNA replication origins may be regulated by epigenetic changes in chromatin structure. The Epstein–Barr virus (EBV) origin of plasmid replication (OriP) uses the cellular licensing machinery to regulate replication during latent infection of human cells. We found that the minimal replicator sequence of OriP, referred to as the dyad symmetry (DS), is flanked by nucleosomes. These nucleosomes were subject to cell cycle-dependent chromatin remodeling and histone modifications. Restriction enzyme accessibility assay indicated that the DS-bounded nucleosomes were remodeled in late G1. Remarkably, histone H3 acetylation of DS-bounded nucleosomes decreased during late G1, coinciding with nucleosome remodeling and MCM3 loading, and preceding the onset of DNA replication. The ATP-dependent chromatin-remodeling factor SNF2h was also recruited to DS in late G1, and formed a stable complex with HDAC2 at DS. siRNA depletion of SNF2h reduced G1-specific nucleosome remodeling, histone deacetylation, and MCM3 loading at DS. We conclude that an SNF2h–HDAC1/2 complex coordinates G1-specific chromatin remodeling and histone deacetylation with the DNA replication initiation process at OriP.
Keywords: DNA replication, EBV, histone, OriP, SNF2h
Introduction
Eukaryotic replication origins are highly regulated by cell cycle-dependent activities to ensure they initiate DNA replication once and only once during the S phase of each division cycle (reviewed in Blow and Hodgson, 2002; DePamphilis, 2003; Mendez and Stillman, 2003). The selection, activation, and deactivation of replication origins follow a strict regulatory program that involves an ordered cascade of checkpoints and licensing events. In lower eukaryotes, like budding yeast, DNA replication origins are discrete genetic elements that bind directly to the origin recognition complex (ORC) (reviewed in Bell and Dutta, 2002). Origin selection in higher eukaryotes is less well understood and it is currently not clear whether replication initiates at specific sites or in large initiation zones. Replication origins might be delineated by poorly defined epigenetic factors, like chromatin boundaries and nuclear domains (reviewed in Gilbert, 2001; McNairn and Gilbert, 2003). In human cells, both ORC binding and MCM protein loading are highly regulated by numerous post-translational modifications and accessory proteins, including cdk/cyclins, cdt1, cdc6, and geminin (reviewed in DePamphilis, 2003; Mendez and Stillman, 2003). However, the contribution of chromatin structure and histone modifications to the cell cycle regulation of replication origin function has not been explored in detail.
Viral origins of DNA replication provide valuable models for investigating basic cellular regulatory processes. The Epstein–Barr virus (EBV) is a human lymphotropic herpesvirus that persists as a chromatin-associated episomal genome during latent infections (Kieff, 1996). The EBV origin of plasmid replication (OriP) is licensed to replicate once and only once in latently infected cells that express the viral origin binding protein EBNA1 (Adams, 1987; Yates and Guan, 1991; Hirai and Shirakata, 2001; reviewed in Sugden and Leight, 2001). EBNA1 binds to a minimal replicator sequence, referred to as the dyad symmetry (DS) region, which consists of two pairs of EBNA1 binding sites punctuated by three telomere repeat factor (TRF) binding sites (Yates et al, 2000; Bashaw and Yates, 2001; Koons et al, 2001; Deng et al, 2002). This minimal replicator has been shown to recruit ORC complex and to be genetically dependent upon ORC2 expression for replication function (Chaudhuri et al, 2001; Dhar et al, 2001; Schepers et al, 2001; Ritzi et al, 2003). MCM proteins are recruited to DS in a cell cycle-dependent manner, and ectopic expression of geminin downregulates OriP-dependent replication (Chaudhuri et al, 2001; Dhar et al, 2001). Based on these observations, it is thought that OriP uses the cellular licensing and replication machinery to regulate single-round replication and maintain a stable copy number of viral genomes in proliferating cells latently infected with EBV.
The contribution of chromatin structure and histone modification to nuclear events has become increasingly apparent (Strahl and Allis, 2000; Jenuwein and Allis, 2001). ATP-dependent chromatin-remodeling complexes are thought to be essential for repositioning nucleosomes that preclude the binding of DNA replication and transcription factors (Varga-Weisz, 2001; Becker and Horz, 2002; Bozhenok et al, 2002; Collins et al, 2002; Peterson, 2002; Lusser and Kadonaga, 2003). The SNF2h member of the ISWI family of chromatin-remodeling proteins has been implicated in cellular DNA replication, by facilitating DNA synthesis of higher-ordered heterochromatin in mammalian cells (Bozhenok et al, 2002; Collins et al, 2002; Poot et al, 2004) and facilitating DNA synthesis-associated chromatin assembly in Drosophila (Fyodorov et al, 2004). SNF2h is an ATPase that has been isolated in several multiprotein complexes, including ACF (Ito et al, 1997; LeRoy et al, 2000), CHRAC (Varga-Weisz et al, 1997), WCRF (Bochar et al, 2000; Bozhenok et al, 2002), and a large HDAC1/2–SNF2h–cohesin complex (Hakimi et al, 2002). These distinct complexes suggest that SNF2h performs multiple functions in chromatin regulation.
Post-translational modifications of histone tails have been mechanistically linked to numerous changes in gene regulation, including transcription complex assembly, histone deposition at the DNA replication fork, and response to DNA damage (Carrozza et al, 2003). Typically, histone acetylation correlates well with increased DNA access, while histone deacetylation and histone H3 K9 methylation correlate with the formation of transcriptionally silent chromatin (Strahl and Allis, 2000; Jenuwein and Allis, 2001). Acetylation of histone tails has been mechanistically linked to the recruitment of bromodomain-containing chromatin-remodeling complexes during transcription activation (Agalioti et al, 2000; Hassan et al, 2002; Neely and Workman, 2002). More recently, histone H3 K4 methylation has been linked to recruitment of SNF2h-containing complexes (Santos-Rosa et al, 2003). However, the chromatin events regulating DNA replication initiation have not been extensively investigated.
Cell cycle changes in chromatin remodeling and histone modification at eukaryotic origins may be an important regulatory feature controlling replication and licensing factor access to DNA. In this work, we investigate the nucleosome organization and histone tail modifications at OriP. We found that the DS region of OriP is flanked by nucleosomes that undergo chromatin remodeling and histone deacetylation at the G1/S border of the cell cycle. We also show that an SNF2h–HDAC1/2-dependent chromatin-remodeling activity physically associates with OriP during G1/S and contributes to OriP replication activity.
Results
Nucleosomes border the DS region of OriP
The DS region of OriP consists of four EBNA1 and three TRF binding sites that are occupied throughout most of the cell cycle and are likely to occlude nucleosome binding to the DS sequence itself (Hsieh et al, 1993; Deng et al, 2002; Avolio-Hunter and Frappier, 2003). The nucleosome structure surrounding DS was first examined by micrococcal nuclease (MNase) I digestion patterns of plasmids containing OriP, no OriP (ΔOriP), or no DS (ΔDS) (Figure 1A). Nuclei from transfected cells were digested with MNase I and total cell DNA was then analyzed by Southern blot hybridization with a 200 bp probe specific for vector sequences immediately adjacent to OriP (Figure 1A). We found that plasmids containing OriP established a clear nucleosomal laddering pattern (lanes 1, 3, and 5), while deletion of OriP (lane 2) or DS (lanes 4 and 6) completely abolished nucleosome ladder patterns (Figure 1A). These initial findings suggest that the nucleosome arrays exist in the regions surrounding OriP, and that the DS region of OriP, along with DNA replication competence, contributes to this nucleosome organization.
Figure 1.

Nucleosomal organization at OriP. (A) Nucleosomal laddering assay of D98/HR1 cells transfected with plasmids N503 (OriP), N530 (ΔOriP), or N564 (ΔDS) as indicated. Plasmid DNA was detected by Southern blot hybridization with a 200 bp probe specific to GFP-vector sequence adjacent to DS. (B) Nucleosome locations at the DS region of OriP were analyzed by indirect end labeling. Purified EBV+ Raji cell DNA (lanes 2 and 3) or nuclei (lane 4) were treated with 0 U/ml (lane 2), 15 U/ml (lane 3), or 75 U/ml (lane 4) MNase I, and subsequently digested with NcoI. The DNA products were then analyzed by Southern blot and probed with a 200 bp fragment covering EBV sequences 8613–8813. Molecular weights of DNA markers (lane 1) are indicated at the left. Asterisks indicate MNase I sites protected in nuclear digest. Deduced positions of nucleosomes (Nuc1 and Nuc2) and EBNA1 binding sites in the DS are indicated by shaded and black ovals in the schematic to the right. EBV coordinates are provided at the bottom and top of the schematic. (C) MNase I-digested mononucleosomal DNA fraction (left lane) and the gel-purified DNA used for primer extension assays (right lane) were visualized with ethidium bromide staining of agarose gels. (D) Primer extension analysis of mononucleosomal DNA isolated from EBV+ Raji cells. Primers a (lane 5), b (lane 6), c (lane 7), and d (lane 8) are indicated above the lane and their position in DS is shown in the schematic below. Sequencing reactions are shown in lanes 1–4. (E) Schematic of DS and primer extension products mapped in (B–D) show Raji nucleosome boundaries (lower thin arrows) and D98/HR1 nucleosome boundaries (upper thick arrows) at the DS region of OriP. EBV genome coordinates are indicated, along with EBNA1 and TRF binding sites. Primer annealing positions a–d are indicated by horizontal arrows.
The nucleosomes surrounding DS were further characterized using indirect end labeling of MNase I-digested nuclei (Figure 1B). The MNase I digestion pattern of EBV-positive Raji nuclei (Figure 1B, lane 4) was compared with the MNase I digestion pattern of naked Raji cell DNA (Figure 1B, lane 3), starting from an NcoI site (EBV coordinates 8613) located 407 bp upstream of DS. We found that the MNase I pattern around the DS region was similar in nuclei and naked, deproteinized DNA. However, we observed that the MNase I cleavage sites at ∼280 and ∼320 bp from the NcoI end-labeled position were present in naked DNA but absent in MNase I digests of chromatin-bound DNA (Figure 1B, compare lanes 3 and 4, noted by asterisks). MNase I cleavage sites at ∼540 bp from the NcoI end label were also protected in nuclear digests. These MNase I-protected sites most likely reflect nucleosome occupancy, and suggest that nucleosomes bind DNA both upstream and downstream of DS. Similar findings were observed when DS was analyzed by an indirect end label placed at the 3′ end of DS (Supplementary Figure 1).
To further verify that nucleosomes flanked the DS, we used a primer extension assay on DNA isolated from gel-purified ∼150 bp mononucleosome fragments (Figure 1C and D) (Lomvardas and Thanos, 2001). Primers were designed for mapping the upstream and downstream nucleosomes relative to DS, designated Nuc1 and Nuc2, respectively. The major primer extension products for Raji-derived nucleosomes were mapped to positions 8850 (EBV genome coordinates) at the 5′ border and 9010 and 9015 at the 3′ border for Nuc1 (Figure 1D, lanes 6 and 7). The major MNase I cleavage sites for Nuc2 were mapped to a 5′ border at 9149 and 9154 and a 3′ border at 9295 (Figure 1D, lanes 7 and 8). This indicates that Nuc1 and Nuc2 occupy the regions immediately adjacent to the DS TRF binding sites but have heterogeneous boundaries (Figure 1E). Similar nucleosome boundaries were found on genomic EBV in latently infected D98/HR1 cells and OriP plasmids transfected in 293 cells (Supplementary Figure 2), suggesting that nucleosomes flank DS independent of viral strain and cell type.
Cell cycle-dependent chromatin remodeling of DS-bound nucleosomes
Nucleosomes surrounding the DS may present a significant barrier to replication factor assembly. Chromatin remodeling provides one potential mechanism to relieve this barrier to replication initiation. Chromatin remodeling in vivo can be detected by an increase in the restriction enzyme accessibility of nucleosome-protected DNA in preparations of isolated nuclei (Almer et al, 1986). Restriction enzyme accessibility assays were used to determine if Nuc1 or Nuc2 were subject to chromatin remodeling at different stages of the cell cycle (Figure 2). Nuclei were isolated from Raji cells arrested in late G1 with mimosine or in M phase with colchicine (Figure 2C). Nuclei were then treated with either EcoRV or MboII, which cut within the boundaries of Nuc1 and Nuc2, respectively, or with HpaI, which cuts between Nuc2 and the 5′ TRF binding site (Figure 2B). Digested DNA was then isolated from the nuclei, linearized with NcoI and KpnI, and analyzed by Southern blot. We found that EcoRV and MboII cleavage of Nuc1 and Nuc2 increased during G1 relative to M-phase-arrested cells (Figure 2A). In contrast, HpaI cleavage outside of the nucleosome boundaries did not change significantly between G1- and M-arrested cells. To verify that restriction enzyme accessibility increased in G1-arrested cells, we compared G1- and M-phase-arrested cells with cells growing asynchronously in logarithmic growth phase (A) and quiescent cells arrested by serum starvation (G0) (Figure 2D and E). We found that restriction enzyme accessibility at the MboII site within Nuc2 was enhanced only in mimosine-treated G1-arrested cells, and not detected in asynchronous or resting cells (Figure 2D).
Figure 2.
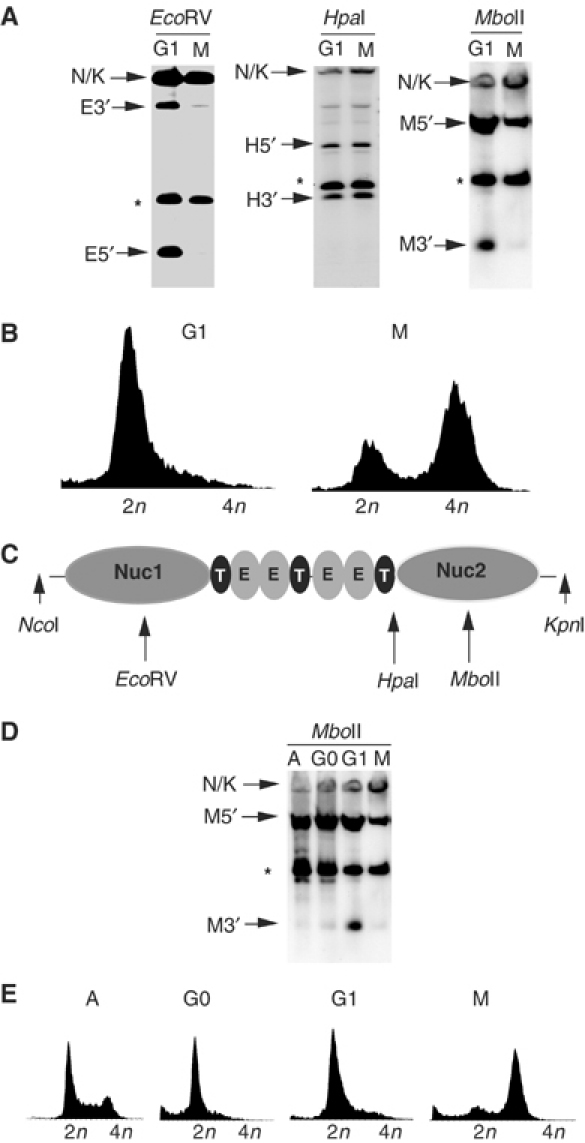
Cell cycle-dependent restriction enzyme accessibility at the DS. (A) Restriction enzyme accessibility assay was performed on nuclei isolated from Raji cells arrested with mimosine (G1) or colchicine (M). Nuclei were digested with EcoRV (left panel), HpaI (middle panel), or MboII (right panel) for 5 min, deproteinized, and then digested with NcoI/KpnI prior to Southern blotting and detection with an OriP-specific probe. The MboII (M), HpaI (H), and EcoRV (E) 5′ and 3′ restriction products are indicated by arrows. Asterisks indicate a control cleavage product outside of nucleosome 1 or 2. (B) Schematic indicating position of MboII, HpaI, and EcoRV relative to DS. EBNA1 (E) and TRFs (T) are indicated by the shaded ovals. (C) FACS profile of cell cycle-arrested Raji cells used for the experiments described above. (D) MboII restriction enzyme accessibility in asynchronous (A), quiescent (G0), mimosine-arrested (G1), or colchicine-arrested (G2) Raji cells. (E) FACS profile of cell cycle for cells used in (D).
Enriched histone H3 K4 methylation at DS-bound nucleosomes
Histone modifications may also regulate chromatin dynamics at OriP. We next used the chromatin immunoprecipitation (ChIP) assay to characterize DS-associated proteins and histone post-translational modifications in Raji cells (Figure 3). As expected, EBNA1 was highly enriched at DS relative to the inactive viral lytic origin OriLyt located over 30 kb from OriP (Figure 3A). ORC2 and MCM3 proteins were modestly enriched at DS, consistent with published findings (Chaudhuri et al, 2001; Dhar et al, 2001; Schepers et al, 2001; Ritzi et al, 2003). Most significantly, ChIP with antibodies to several histone tail modifications revealed that DS-associated nucleosomes were highly enriched for histone H3 dimethyl-K4 (H3mK4) relative to OriLyt (Figure 3A). Acetylated histone H3 (AcH3) was also enriched at DS, although to a lesser extent than that seen for H3mK4. In contrast, acetylated histone H4 (AcH4) was slightly more enriched at OriLyt relative to DS.
Figure 3.
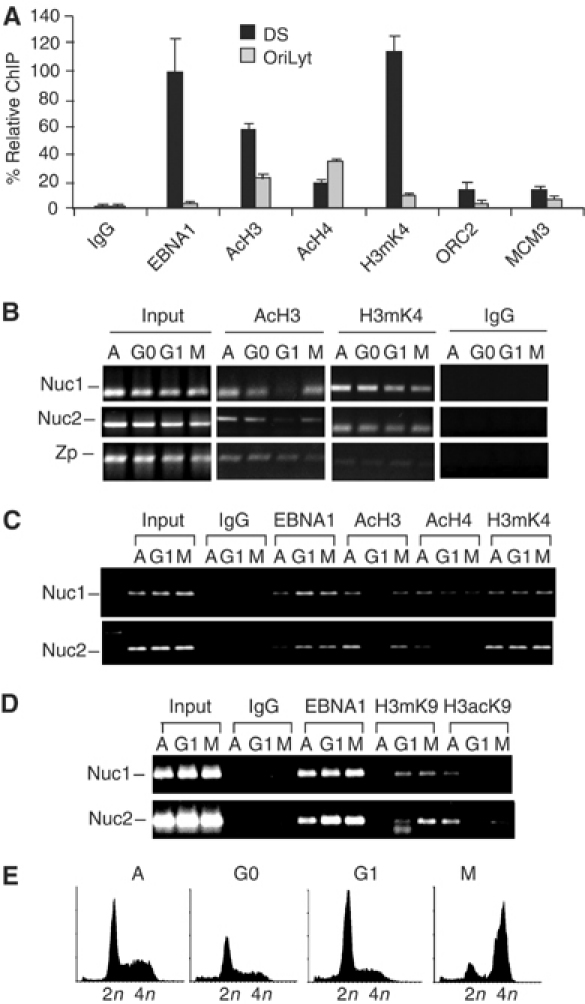
Cell cycle-associated changes in histone modifications at OriP. (A) Protein interactions at DS were determined by ChIP assay with either control IgG or antibodies specific to EBNA1, AcH3, AcH4, H3mK4, ORC2, and MCM3. Chromatin immunoprecipitated DNA was analyzed by real-time PCR with primers specific for DS or OriLyt. (B) ChIP assay of MNase I-treated Raji cell nuclei derived from asynchronous (A), serum-starved (G0), mimosine-arrested (G1), or colchicine-arrested (M) cells. Immunoprecipitation with antibodies specific for acetylated H3 or H3mK4 was analyzed by PCR with primers specific for DS-associated leftward nucleosome (Nuc1), the DS-associated rightward nucleosome (Nuc2), or the BZLF1 promoter region (Zp). (C) Same as (B), but with antibodies specific for EBNA1, AcH3, AcH4, H3mK4, and control IgG. (D) Same as above, but with IgG, EBNA1, H3mK9, and histone H3 acetyl K9 (H3acK9). (E) FACS profile of propidium iodide-treated cells after treatments for cell cycle arrests shown above.
G1-specific histone deacetylation at DS-bound nucleosomes
Histone modifications at OriP were examined for changes at various stages of the cell cycle (Figure 3B). To measure more accurately the histone modifications at Nuc1 and Nuc2, we enriched for mononucleosomes by digesting DNA with MNase I and performed ChIP with primer sets located within the boundary of each nucleosome. Remarkably, we found that AcH3 was significantly reduced at Nuc1 and Nuc2 in mimosine-treated G1-arrested cells (Figure 3B). This loss of AcH3 modification in G1 was specific for DS nucleosomes since we did not observe a similar loss at control region Zp, located ∼40 kb from DS. In contrast to AcH3, H3mK4 was not reduced in mimosine-arrested cells, suggesting that histones still occupy the DS region during G1. A more extensive analysis of Nuc1 and Nuc2 shows that the association with EBNA1, AcH4, and H3mK4 did not change significantly at various stages of the cell cycle, while AcH3 was consistently reduced at both nucleosome positions (Figure 3C). Modification of the K9 position of H3 has been associated with changes in gene expression and heterochromatin formation (Jenuwein and Allis, 2001). Consistent with the results found for the hyperacetylated H3 antibody, ChIP analysis revealed a strong reduction in H3acK9 at Nuc1 and Nuc2 in G1 as well as in M (Figure 3D). Conversely, H3mK9 showed a modest increase at Nuc2 in M-phase-arrested cells. No significant change in EBNA1 binding could be detected, suggesting that MNase I digestion was incomplete or that Nuc1 and Nuc2 interact with neighboring EBNA1 or DS-associated proteins throughout the cell cycle.
Cell cycle association of SNF2h with DS
Since DS-associated nucleosomes were subject to chromatin remodeling during G1 (Figure 2), we reasoned that an ATP-dependent chromatin-remodeling complex is likely to be responsible for this activity. In mammalian cells, the predominant chromatin-remodeling complexes belong to the SNF2h, BRG1, or Mi-2/NuRD families (Varga-Weisz, 2001; Peterson, 2002; Lusser and Kadonaga, 2003; Bowen et al, 2004). Antibodies specific to SNF2h, BRG1, or the BRG1-associated protein Ini1 were compared for their ability to precipitate DS DNA in a ChIP assay (Figure 4A). We found that SNF2h specifically associated with DS relative to GAPDH DNA, and to a greater extent than BRG1 or Ini1 (Figure 4A). We next asked whether SNF2h associated with OriP in a cell cycle-dependent manner using real-time PCR analysis of ChIP DNA (Figure 4B and C). Raji cells grown asynchronously (A) or cell cycle arrested with serum starvation (G0), mimosine (G1), hydroxyurea (S), or colchicine (M) were assayed with antibodies specific for AcH3, SNF2h, MCM3, EBNA1, or control IgG (Figure 4B). First, we confirmed our previous observations that AcH3 levels were reduced at DS in G1 (∼4-fold relative to M-phase levels). Next, we found that SNF2h was significantly enriched at DS in G1 stage (∼5-fold relative to M-phase levels). SNF2h was not similarly enriched at the control DNA site of OriLyt. We also demonstrate that MCM3 was enriched at DS in G1 (∼8-fold relative to M-phase levels). EBNA1 remained bound to DS through all stages of the cell cycle examined and IgG did not precipitate significant levels of DS or OriLyt in any condition. These results indicate that SNF2h bound DS in G1-arrested cells where nucleosome remodeling was detected (Figure 2), AcH3 levels decreased (Figures 3 and 4B), and MCM3 binding increased (Figure 4B) at DS.
Figure 4.
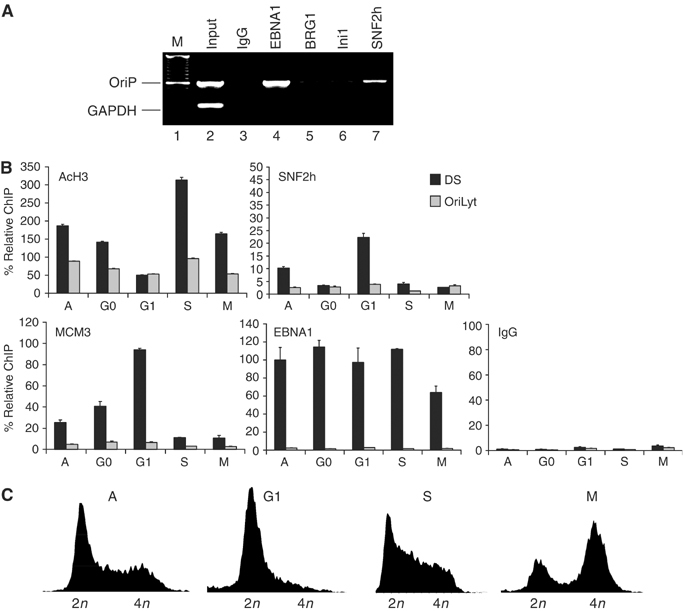
SNF2h is recruited to OriP in a cell cycle-dependent manner and is required for DNA replication. (A) ChIP assays with antibodies specific for EBNA1, BRG1, Ini1, SNF2h, or control IgG were analyzed for DNA from OriP or GAPDH. (B) Chromatin dynamics were analyzed in asynchronous (A), serum-starved (G0), mimosine-treated (G1), hydroxyurea-treated (S), or colchicine-treated (M) Raji cells. Real-time PCR analyses of DS (black box) or OriLyt (gray box) DNA precipitated by ChIP assay with antibodies specific for AcH3, SNF2h, MCM3, EBNA1, or control IgG are indicated. (C) FACS analysis of propidium iodide-stained cells used in the experiments shown in (B).
Cell cycle arrest experiments also revealed that histone AcH3 levels oscillate through the cell cycle (Figure 4B). Remarkably, we found that AcH3 levels decreased four-fold in G1/S, followed by a rapid increase in S (∼7-fold relative to G1-phase levels), and then a return to median levels in M-phase-arrested cells. This oscillation in AcH3 levels was not observed at control OriLyt DNA (Figure 4B), or at the Zp region of EBV (Figure 3B). Similar patterns of oscillation were not observed for SNF2h, MCM3, EBNA1, or IgG. These observations suggest that DS-associated histone H3 acetylation has a markedly distinct cell cycle pattern.
HDAC2 colocalizes with SNF2h at DS during G1
SNF2h interacts with DS in G1-arrested cells, which coincides with the loss of AcH3 (Figures 3 and 4). SNF2h has been isolated as a multiprotein complex with HDAC1 and 2 (Hakimi et al, 2002), and we sought to determine whether HDAC1 and 2 may also interact with DS in a cell cycle- and SNF2h-associated manner. We first determined whether SNF2h and HDAC2 could be isolated as a stable complex in EBV-positive Raji cells (Figure 5A). Raji cell nuclear extracts were subject to immunoprecipitation with SNF2h or HDAC2, followed by immunoblotting with the reciprocal antibodies. We found that SNF2h was detected in HDAC2 immunoprecipitates, and HDAC2 was detected in SNF2h immunoprecipitates. Thus, SNF2h and HDAC2 can be isolated as a stable complex in EBV-positive Raji cells. To determine if SNF2h-associated HDACs interact with OriP in a cell cycle-dependent manner similar to SNF2h, we used the ChIP assay (Figure 5B). Antibodies specific for HDAC1, HDAC2, or EBNA1 were assayed for their ability to chromatin immunoprecipitate OriP DNA in various stages of the cell cycle. Significantly, we found that HDAC1 and HDAC2 were enriched at OriP in mimosine-arrested G1 cells relative to asynchronous (A), serum-starved (G0), or colchicine-arrested (M) cells (Figure 5B). HDAC antibodies did not precipitate DNA from the cellular GAPDH promoter region, indicating that the interactions at OriP were specific. These results suggest that HDAC1 and HDAC2 associate with DS at the same time as SNF2h in G1-arrested cells.
Figure 5.
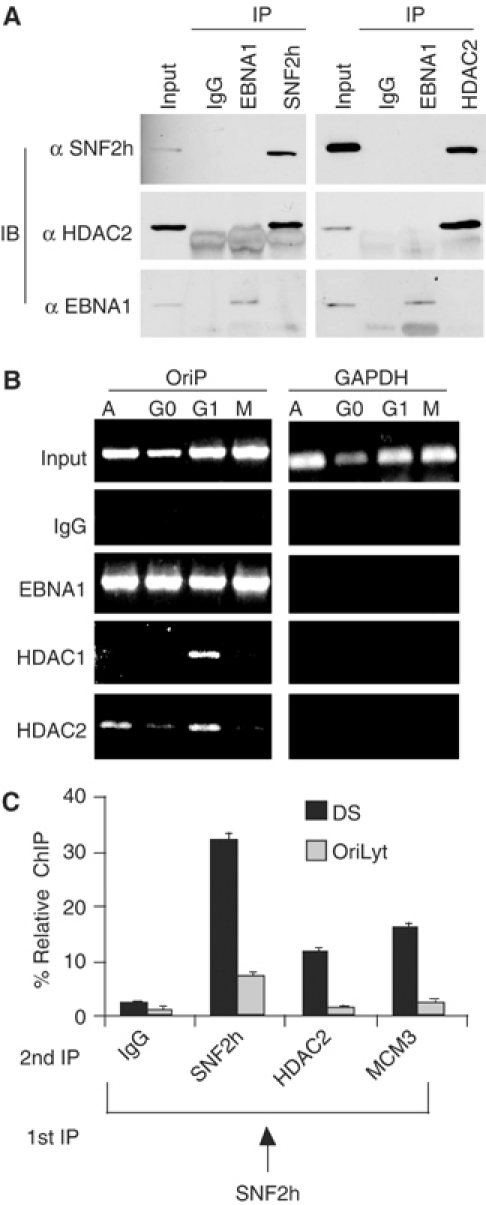
HDACs, SNF2h, and MCMs colocalize at OriP. (A) Immunoprecipitates (IP) of EBNA1, SNF2h, HDAC2, or IgG were analyzed by immunoblotting (IB) with antibodies specific for SNF2h, HDAC2, or EBNA1 as indicated. (B) ChIP assays with antibodies specific for EBNA1, HDAC1, or HDAC2 were used with extracts from asynchronous (A), serum-starved (G0), mimosine-arrested (G1), or colchicine-arrested (M) Raji cells and assayed for OriP (left panel) or cellular GAPDH (right panel) DNA. (C) G1-arrested Raji cells were subject to double ChIP assay with first-round SNF2h, followed by second-round ChIP with antibodies to SNF2h, HDAC2, MCM3, or control IgG.
Since EBV genomes are multicopy in latently infected cells, it is essential to demonstrate that SNF2h bound to the same genomes that were bound by HDAC and MCM components. To demonstrate this, we performed a double ChIP assay in which the SNF2h was used for the first-round immunoprecipitation from mimosine-arrested Raji cells (Figure 5C). Eluted complexes were then subject to a second round of immunoprecipitation with HDAC2, MCM3, or SNF2h as positive control. We found that HDAC2 and MCM3 were specifically associated with SNF2h-precipitated DS DNA relative to other regions of the viral genome and were not detected in control ChIPs with IgG antibody (Figure 5C). These data strongly suggest that SNF2h and HDAC2 function as a coordinated complex at DS during the G1 phase of the cell cycle.
Cell cycle-regulated SNF2h–HDAC2 binding at DS of mini-EBV
Several reports have raised concern that replication initiation may occur in zones outside of the DS at a relatively high frequency in the Raji strain (Norio et al, 2000; Norio and Schildkraut, 2001, 2004). Thus, chromatin modifications at the Raji DS may not reflect a highly active origin of DNA replication. In contrast to Raji, replication initiation at DS occurs at a much higher frequency in the ∼80 kB EBV derivative referred to as mini-EBV (Ritzi et al, 2003; Norio and Schildkraut, 2004). To determine if the cell cycle-associated chromatin modifications that we have observed in Raji DS correlate with a high-frequency replicon, we assayed the protein–DNA interactions and histone modifications at DS in a mini-EBV-transformed cell line using the ChIP assay (Figure 6). Mini-EBV cells were sorted into different stages of the cell cycle by centrifugal elutriation (Figure 6A). MCM3 binding to DS was highly enriched in G1/S (55 ml/min), as expected for a functional replication origin. Significantly, we found that SNF2h was enriched at DS in G1/S, similar to that observed in Raji. AcH3 levels were notably lower relative to EBNA1 levels in mini-EBV lymphoblastoid cell lines (LCLs) compared to Raji. Nevertheless, AcH3 levels decreased in G1/S (55 ml/min) and increased in S (65 ml/min), similar to that observed in Raji. HDAC2 binding also increased in G1/S. EBNA1 and H3mK4 levels remained high at DS throughout the cell cycle. Cell cycle changes did not occur at control OriLyt region of the mini-EBV genome, and control IgG levels remained low at DS throughout the cell cycle. These ChIP profiles were nearly identical to those observed with Raji cells (Figures 4 and 5), suggesting that SNF2h–HDAC association at DS correlates with MCM loading and replication origin competence. Similar observations, including G1-specific chromatin remodeling, were observed at DS in MutuI cells, which were shown to have a relatively high rate of replication initiation at DS (Supplementary Figure 4). Together, these results indicate that the cell cycle changes in SNF2h binding and histone deacetylation are a general property of the DS replication origin in various strains and cell backgrounds, and independent of replication initiation frequency.
Figure 6.
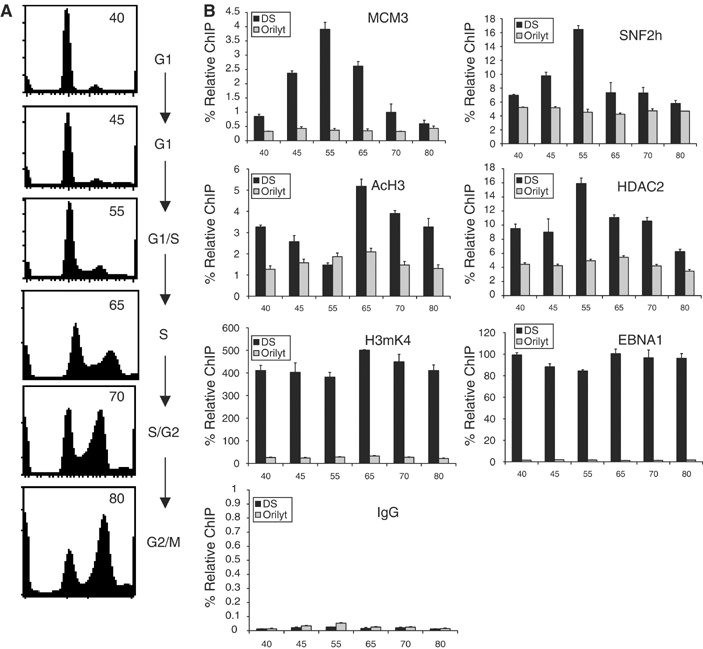
G1/S-specific SNF2h–HDAC association at the primary origin (OriP) of mini-EBV. (A) Cell cycle FACS profiles of mini-EBV LCLs sorted by centrifugal elutriation at 40, 45, 55, 65, 70, and 80 ml/min. (B) Real-time PCR analysis of mini-EBV ChIP assays with antibodies specific for MCM3, SNF2h, AcH3, HDAC2, H3mK4, EBNA1, and control IgG at DS (black) or OriLyt (gray) in the various cell cycle stages as indicated.
SNF2h and HDAC2 are required for OriP-dependent DNA replication
The functional importance of SNF2h in OriP replication was demonstrated using siRNA depletion. OriP-containing plasmid was cotransfected with siRNA directed against SNF2h, BRG1, or control luciferase gene and then assayed for transient replication activity. Both siRNAs were shown to reduce their targeted proteins to less than 10% of control levels, without affecting PCNA or EBNA1 expression levels (Figure 7A). We found that SNF2h-directed siRNA reduced replication to 23% of control levels, while BRG1 siRNA reduced replication to 76% of the control (Figure 7B). SNF2h and BRG1 siRNA produced a slight accumulation of cells in G1, consistent with the known function of these proteins in regulating cellular gene expression and replication through heterochromatin.
Figure 7.
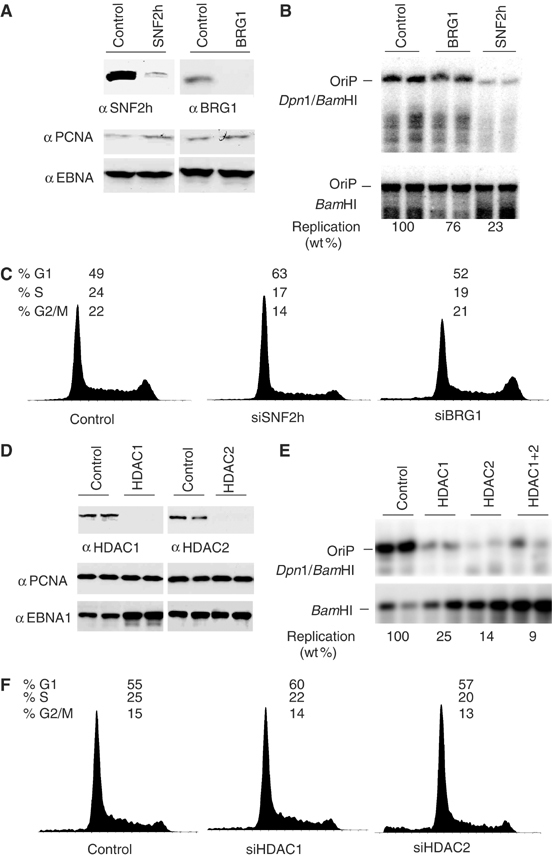
SNF2h and HDACs promote replication and maintenance of OriP. (A) Western blots of cells transfected with siRNA specific for SNF2h (left) or BRG1 (right) were analyzed with antibodies to SNF2h, BRG1, or against control proteins EBNA1 and PCNA (as indicated). (B) Replication assays of OriP in cells cotransfected with control, BRG1, or SNF2h siRNAs. Transfected DNA was analyzed by Dpn1/BamHI or BamHI resistance followed by Southern blot. (C) FACS analysis of propidium iodide-stained cells transfected with siRNAs for SNF2h or BRG1 as indicated. (D) Western blots of extracts derived from D98/HR1 cells transfected with HDAC1 or HDAC2 siRNAs (upper panels), or with control antibodies specific to EBNA1 or cellular protein PCNA (lower panels). (E) OriP-dependent DNA replication was assayed in D98/HR1 cells transfected with OriP plasmid and siRNA specific for HDAC1, HDAC2, or HDAC1+2 as indicated above each lane. (F) FACS analysis of propidium iodide-stained cells transfected with siRNA specific for HDAC1 or HDAC2 as indicated.
The functional significance of HDAC association with OriP was also demonstrated with siRNA directed against HDAC1 and HDAC2 (Figure 6D–F). Western blots for HDAC1 and HDAC2 showed that the efficiency of siRNA depletion was greater than 90% (Figure 7D). Transient knockdown of HDACs did not significantly alter PCNA or EBNA1 levels (Figure 7D, lower panels). We found that siRNA depletion of HDAC1 and 2 inhibited OriP replication to 25 and 14% of control levels, respectively. Combining HDAC1 and 2 siRNA further reduced OriP replication to 9% of the control siRNA, suggesting that these proteins are not redundant. Transient reduction of HDAC1 and HDAC2 did not have a gross effect on cell cycle distribution, as determined by FACS analysis (Figure 7F). We also found that the pharmacological inhibitors of HDACs, sodium butyrate and trichostatin A, potently blocked OriP-dependent DNA replication and plasmid maintenance (Supplementary Figure 5). Together, these results indicate that HDAC1 and 2 proteins and HDAC enzymatic activity contribute to OriP replication function.
SNF2h functions in chromatin remodeling, histone modification, and MCM loading at DS
We have shown that SNF2h binds DS in the G1 phase of the cell cycle (Figures 4, 5 and 6) and siRNA depletion of SNF2h inhibits OriP replication (Figure 7). We next determined whether SNF2h depletion disrupted chromatin dynamics at DS (Figure 8). EBV-positive D98/HR1 cells were transfected with control or SNF2h-specific siRNA, arrested in G1 with mimosine, and then assayed for restriction enzyme accessibility at the Nuc1 site using EcoRV (Figure 8A and B). We found that cells depleted for SNF2h had significantly less EcoRV restriction enzyme accessibility at Nuc1 relative to cells transfected with control siRNA (Figure 8B). We next asked whether SNF2h depletion led to a loss of MCM3 protein association at DS in G1-arrested cells using the ChIP assay (Figure 8C). We found that SNF2h depletion led to an ∼3-fold decrease in MCM3 binding to DS. We also observed that SNF2h depletion resulted in a corresponding increase in histone H3 acetylation at DS, but not at control OriLyt region of the EBV genome (Figure 8D). Surprisingly, we also observed that SNF2h depletion led to an ∼2-fold decrease in histone H3mK4 at DS, with no significant change at OriLyt. Taken together, these data indicate that SNF2h contributes to chromatin remodeling and preinitiation complex assembly at DS.
Figure 8.
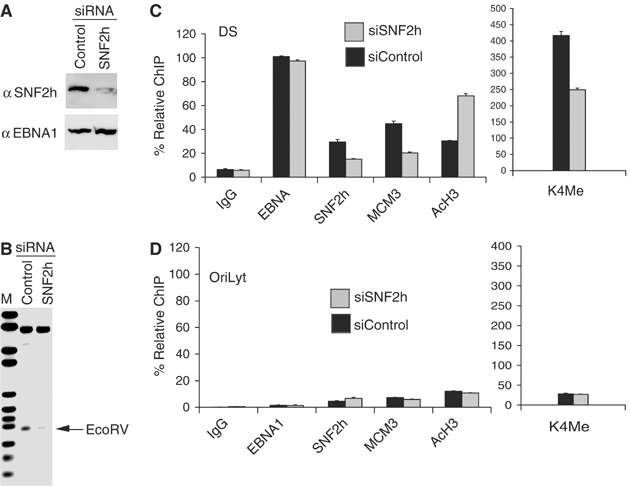
SNF2h depletion reduces chromatin remodeling, histone deacetylation, and MCM3 binding at DS. (A) Western blot of D98/HR1 cells transfected with SNF2h-specific or control siRNA and assayed for SNF2h (top) or EBNA1 (bottom) protein levels. (B) Southern blot of EcoRV restriction enzyme accessibility assay in G1-arrested D98/HR1 cells transfected with control or SNF2h-specific siRNA. (C) Real-time PCR analysis of ChIP assay from D98/HR1 cells transfected with SNF2h or control siRNA assayed at DS (C) or at control region OriLyt (D).
Discussion
Cell cycle regulation of DNA replication is subject to numerous levels of control that alter the composition, stability, post-translational modification, and activity of the prereplication complex (Blow and Hodgson, 2002; Nasheuer et al, 2002; Hyrien et al, 2003). Epigenetic influences, like chromatin structure and modification, are also thought to contribute to prereplication complex assembly and origin selection, but the precise mechanisms operating at mammalian origins have not been well characterized (Gilbert, 2001; McNairn and Gilbert, 2003). In this study, we have examined the cell cycle changes in chromatin organization and histone tail modifications in the nucleosomes surrounding the EBV origin of plasmid replication, OriP. We found that the DS region of OriP was flanked by nucleosomes located 5′ and 3′ of the outermost TRF sites of DS (Figures 1 and 2). These nucleosomes were subject to cell cycle-dependent chromatin remodeling and histone H3 deacetylation. These chromatin changes correlated with MCM3 binding in the G1/S phase of the cell cycle, suggesting that cell cycle changes in chromatin are coordinated with replication licensing at OriP.
Nucleosome occupancy is known to influence replication preinitiation complex assembly and origin activity (Simpson, 1990; Lipford and Bell, 2001). Our data suggest that the nucleosomes flanking the DS may function as a cell cycle-dependent barrier to replication factor assembly at OriP. We found that the nucleosomal DNA adjacent to DS was subject to a G1/S-specific increase in restriction enzyme accessibility (Figure 2). We interpreted these data as an indication that DS-flanked nucleosomes are remodeled in G1/S. Consistent with this interpretation, we found that the chromatin-remodeling factor SNF2h was enriched at DS in G1/S-arrested cells (Figures 4, 5 and 6). Depletion of SNF2h inhibited OriP replication (Figure 7) and decreased G1/S-associated binding of MCM3 (Figure 8). Our data are consistent with a role of SNF2h in the remodeling of nucleosomes contemporaneously with the loading of the MCM complex. The remodeled nucleosomes at DS remained in close association with DNA, since we observed no obvious change in nucleosome boundaries by indirect end-labeling (data not shown) or primer extension assay (Supplementary Figure 3), and no loss of histone H3mK4 binding in ChIP assays (Figures 3 and 6). The precise DNA contacts of the remodeled nucleosome are not clear, but must be altered sufficiently to allow MCM recruitment. MCM complex is thought to be the replicative helicase and its loading correlates with the licensing and activation of a replication origin (Blow and Hodgson, 2002; McNairn and Gilbert, 2003). Thus, we consider it likely that SNF2h-associated chromatin remodeling potentiates replication preinitiation complex assembly by increasing DNA accessibility at DS-neighboring sites (Figure 9).
Figure 9.

Model of cell cycle coordinated histone tail modification and chromatin remodeling at OriP. An SNF2h/HDAC2 complex can be colocalized to DS during G1, simultaneously with histone H3 deacetylation, chromatin remodeling, and MCM loading.
Nucleosomes flanking DS were also examined for cell cycle changes in histone tail modifications. In G1/S-arrested cells, DS-associated AcH3 was substantially reduced relative to other regions of the viral and cellular genome, with no corresponding change in H3mK4 (Figures 3, 4 and 6). We interpreted these observations as evidence for G1/S-specific histone H3 deacetylation. This interpretation was further supported by finding G1/S-specific enrichment of HDAC1 and HDAC2 at DS (Figures 5 and 6). We also observed that histone H3 acetylation levels increased at DS in S-phase-arrested cells, and then returned to intermediate levels in M-phase-arrested cells. While the oscillation of histone acetylation and deacetylation across the cell cycle is not unexpected (Verreault, 2003), we were surprised to find histone deacetylation at DS in G1/S, when prereplication factors are assembling. Histone deacetylation is typically associated with repressive chromatin, and histone acetylation has been linked to activation of transcription and DNA replication in other systems (Sterner and Berger, 2000; Aggarwal and Calvi, 2004; Stedman et al, 2004). In Drosophila follicle cells, histone acetylation enhanced, while deacetylation repressed replication initiation frequency (Aggarwal and Calvi, 2004). Similarly, constitutively high levels of histone H3 acetylation were found at the origin of plasmid replication in the related viral genome of HHV8/KSHV (Stedman et al, 2004). Paradoxically, chromatin remodeling and replication factor assembly at EBV OriP correlated with histone deacetylation and not histone hyperacetylation.
The atypical pattern of histone modification at DS may be linked to the complex epigenetic behavior of OriP. Replication initiation at OriP occurs at relatively low frequency in Raji cells, but at high frequency in mini-EBV-transformed LCLs (Norio and Schildkraut, 2001, 2004). Despite the difference in replication initiation frequency at OriP, we found no significant difference in the G1/S-specific histone H3 deacetylation, or SNF2h, HDAC1/2, or MCM3 binding (Figure 6). It has been reported that OriP-dependent DNA replication initiates in late S phase (Carroll et al, 1991). Histone deacetylation at OriP may delay replication firing until the burst of histone acetylation observed at DS in later S phase (Figures 4 and 6). This would suggest that late S-phase firing origins load MCM, but remain inactive due to histone deacetylation. Histone deacetylation and late S-phase firing at OriP may also explain the reduced replication initiation frequency observed in Raji cells, where alternative replication zones may fire in early S phase. Future experiments will be required to determine whether these and other epigenetic factors regulate the replication timing and initiation frequency at OriP in the Raji and min-EBV genomes.
Histone modifications may also create a protein-recognition code that regulates replication activity (Strahl and Allis, 2000; Jenuwein and Allis, 2001). We found that DS nucleosomes were enriched for H3mK4 relative to other regions of the EBV genome at all stages of the cell cycle examined. H3mK4 has been shown to serve as a recognition site for SNF2h-containing complexes (Santos-Rosa et al, 2003). However, the constitutively elevated levels of H3mK4 cannot account for the G1-specific recruitment of SNF2h to DS. Other histone modifications may also modulate SNF2h binding, as was shown for the inhibition of Drosophila ISWI to acetylated histone H4 K16 (Corona et al, 2002). Thus, it is possible that histone H3 deacetylation may be required in combination with H3 K4 methylation to provide the necessary specificity for the cell cycle-dependent recruitment of SNF2h to DS.
SNF2h can be isolated in several distinct multiprotein complexes (Varga-Weisz, 2001). SNF2h can be recruited to replication foci through a physical interaction with WSTF and PCNA (Poot et al, 2004). SNF2h can also be isolated in a multiprotein complex containing HDAC1 and 2 (Hakimi et al, 2002). We found that SNF2h and HDAC2 co-immunoprecipitated and were colocalized to DS in double ChIP assays (Figure 5). Histone deacetylation and nucleosome remodeling colocalized spatially and temporally with MCM3 binding to DS, indicating that these changes correlate with preinitiation complex assembly at DS. The physical association of SNF2h with HDACs at DS suggests that histone deacetylation is functionally linked to chromatin remodeling. Chromatin remodeling during G1/S provides a mechanism for MCM loading and replication factor assembly at DNA sites occluded by DS-flanked nucleosomes. However, the precise function of G1/S-specific histone H3 deacetylation at DS remains unclear. It is possible that G1/S-specific histone deacetylation, in combination with high histone H3 K4 methylation, provides a unique epigenetic mark on the origin-associated nucleosomes that modulates SNF2h remodeling, or replication preinitiation complex assembly. It is also possible that histone deacetylation of the remodeled nucleosomes at OriP restricts replication initiation to late S phase or to once per cell cycle. It will be important to further characterize these cell cycle-dependent changes in chromatin structure at OriP and to determine if these modifications are common features of other viral and cellular replication origins, and how these chromatin dynamics correlate with replication timing and initiation frequency.
Materials and methods
Plasmids and cell culture
OriP plasmid (N503) has been described previously and consists of OriP sequences, EBNA1, eGFP, and hygromycin genes as a pREP10 (Invitrogen) derivative (Deng et al, 2002). pΔOriP (N530) and pΔDS (N564) are replication-incompetent derivatives of N503 lacking sequences of OriP and the DS region, respectively. Raji and MutuI cells are EBV+ suspension cells that were maintained in RPMI supplemented with 10% FBS, glutamine, penicillin, and streptomycin sulfate (Cellgro). D98/HR1 (EBV+ adherent cells) and 293 cells were maintained in DMEM with 10% FBS, glutamine, penicillin, and streptomycin sulfate (Cellgro). Adherent cells were transfected with Lipofectamine 2000, according to the manufacturer's recommendations (Invitrogen). The A39 mini-EBV LCL was cultured as described previously (Schepers et al, 2001; Ritzi et al, 2003).
Cell cycle arrests
Asynchronous Raji or D98/HR1 cells were arrested in G0 by growth in 0.5% FBS for 48 h, in G1/S by treatment with 400 μM mimosine (Sigma) for 24 h, in S phase by treatment with 100 μM hydroxyurea for 18 h, or in G2/M by treatment with 1 μM colchicine (Sigma) for 24 h. Mini-EBV LCL was fractionated according to cell cycle stage by centrifugal elutriation using the same parameters as described (Ritzi et al, 2003).
Primer extension analysis of mononucleosomes
Isolation of nuclei and primer extension assay were performed as described (Lomvardas and Thanos, 2001) with some modifications (see Supplementary Methods).
Indirect end-labeling assay
Nucleosome positions were analyzed by indirect end-labeling method described previously (Hager and Fragoso, 1999; Ryan et al, 1999).
Restriction enzyme accessibility assay
Raji, MutuI, and siRNA transfected D98/HR1 cell nuclei were prepared as described. Nuclei (107) were resuspended in 50 μl restriction digestion buffer for either MboII, HpaI, or EcoRV as specified by the manufacturer (NEB). Restriction enzyme digestion was performed at 37°C for 10 min and stopped by the addition of Stop Buffer. After incubation at 50°C for 2 h with proteinase K, DNA was phenol/chloroform extracted, purified by ethanol precipitation, cut with KpnI and NcoI, and analyzed by Southern blotting. DNA was detected using the DIG detection kit (Roche) with a 150 bp PCR-generated probe to DS region.
Chromatin immunoprecipitation assays
The ChIP assay was a modification of the protocol provided by Upstate Biotechnology Inc., and has been described in detail elsewhere (Deng et al, 2002, 2003). Real-time PCR analysis of ChIP DNA was the average of three independent experiments, quantified using the standard curve method on ABI 7000 thermocycler, and normalized to EBNA1 bound to DS in asynchronous cells (set at 100%).
OriP replication assay
DNA replication and plasmid maintenance assays have been described previously (Yates et al, 2000; Deng et al, 2002). For transient replication assays with siRNA, all cells were cotransfected with OriP plasmid and siRNA.
siRNA
siRNAs were synthesized as duplex RNA (Dharmacon Inc.) with the following target sequences for Luciferase control (cgtacgcggaatacttcga) and SNF2h (aagaggaggaugaagagcuau) as described previously (Collins et al, 2002). siRNAs for HDAC1, 2, and 3 were commercially available as Smartpool products (Dharmacon Inc.). siRNA to BRG1 was generated by the Hanon method (Paddison et al, 2002) with the following oligonucleotide primer (aaaaaagacgttaacgctgtcacagacgctaccg caagcttccagtagcatctgtaacagcattaactg tcggtgtttcgtcctttccacaa). Plasmid or siRNA controls were used in parallel for each siRNA experiment.
Additional details of methods can be found in Supplementary Methods.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Methods
Acknowledgments
We thank Latasha Day for technical support and the Wistar Cancer Center Core Facilities for their assistance. This work was funded by NCI CA93606 and DOD BC022095 to PML and NIH (GM61204) to RS. Charles Chau was funded by the Wistar NCI postdoctoral training grant, and Zhong Deng is a fellow of the Leukemia-Lymphoma Society.
References
- Adams A (1987) Replication of latent Epstein–Barr virus genomes in Raji cells. J Virol 61: 1743–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 103: 667–678 [DOI] [PubMed] [Google Scholar]
- Aggarwal BD, Calvi BR (2004) Chromatin regulates origin activity in Drosophila follicle cells. Nature 430: 372–376 [DOI] [PubMed] [Google Scholar]
- Almer A, Rudolph H, Hinnen A, Horz W (1986) Removal of positioned nucleosomes from the yeast PHO5 promoter upon PHO5 induction releases additional upstream activating DNA elements. EMBO J 5: 2689–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avolio-Hunter TM, Frappier L (2003) EBNA1 efficiently assembles on chromatin containing the Epstein–Barr virus latent origin of replication. Virology 315: 398–408 [DOI] [PubMed] [Google Scholar]
- Bashaw JM, Yates JL (2001) Replication from oriP of Epstein–Barr virus requires exact spacing of two bound dimers of EBNA1 which bend DNA. J Virol 75: 10603–10611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker PB, Horz W (2002) ATP-dependent nucleosome remodeling. Annu Rev Biochem 71: 247–273 [DOI] [PubMed] [Google Scholar]
- Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Blow JJ, Hodgson B (2002) Replication licensing—defining the proliferative state? Trends Cell Biol 12: 72–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochar DA, Savard J, Wang W, Lafleur DW, Moore P, Cote J, Shiekhattar R (2000) A family of chromatin remodeling factors related to Williams syndrome transcription factor. Proc Natl Acad Sci USA 97: 1038–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen NJ, Fujita N, Kajita M, Wade PA (2004) Mi-2/NuRD: multiple complexes for many purposes. Biochim Biophys Acta 1677: 52–57 [DOI] [PubMed] [Google Scholar]
- Bozhenok L, Wade PA, Varga-Weisz P (2002) WSTF–ISWI chromatin remodeling complex targets heterochromatic replication foci. EMBO J 21: 2231–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SM, Trotter J, Wahl GM (1991) Replication timing control can be maintained in extrachromosomally amplified genes. Mol Cell Biol 11: 4779–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Utley RT, Workman JL, Cote J (2003) The diverse functions of histone acetyltransferase complexes. Trends Genet 19: 321–329 [DOI] [PubMed] [Google Scholar]
- Chaudhuri B, Xu H, Todorov I, Dutta A, Yates JL (2001) Human DNA replication initiation factors, ORC and MCM, associate with oriP of Epstein–Barr virus. Proc Natl Acad Sci USA 98: 10085–10089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins N, Poot RA, Kukimoto I, Garcia-Jimenez C, Dellaire G, Varga-Weisz PD (2002) An ACF1–ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet 32: 627–632 [DOI] [PubMed] [Google Scholar]
- Corona DF, Clapier CR, Becker PB, Tamkun JW (2002) Modulation of ISWI function by site-specific histone acetylation. EMBO Rep 3: 242–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Atanasiu C, Burg JS, Broccoli D, Lieberman PM (2003) Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate replication of Epstein–Barr virus OriP. J Virol 77: 11992–12001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z, Lezina L, Chen CJ, Shtivelband S, So W, Lieberman PM (2002) Telomeric proteins regulate episomal maintenance of Epstein–Barr virus origin of plasmid replication. Mol Cell 9: 493–503 [DOI] [PubMed] [Google Scholar]
- DePamphilis ML (2003) The ‘ORC cycle': a novel pathway for regulating eukaryotic DNA replication. Gene 310: 1–15 [DOI] [PubMed] [Google Scholar]
- Dhar SK, Yoshida K, Machida Y, Khaira P, Chaudhuri B, Wohlschlegel JA, Leffak M, Yates J, Dutta A (2001) Replication from oriP of Epstein–Barr virus requires human ORC and is inhibited by geminin. Cell 106: 287–296 [DOI] [PubMed] [Google Scholar]
- Fyodorov DV, Blower MD, Karpen GH, Kadonaga JT (2004) Acf1 confers unique activities to ACF/CHRAC and promotes the formation rather than disruption of chromatin in vivo. Genes Dev 18: 170–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DM (2001) Making sense of eukaryotic DNA replication origins. Science 294: 96–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, Fragoso G (1999) Analysis of nucleosome positioning in mammalian cells. Methods Enzymol 304: 626–638 [DOI] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Schmiesing JA, Dong Y, Barak OG, Speicher DW, Yokomori K, Shiekhattar R (2002) A chromatin remodelling complex that loads cohesin onto human chromosomes. Nature 418: 994–998 [DOI] [PubMed] [Google Scholar]
- Hassan AH, Prochasson P, Neely KE, Galasinski SC, Chandy M, Carrozza MJ, Workman JL (2002) Function and selectivity of bromodomains in anchoring chromatin-modifying complexes to promoter nucleosomes. Cell 111: 369–379 [DOI] [PubMed] [Google Scholar]
- Hirai K, Shirakata M (2001) Replication licensing of the EBV oriP minichromosome. Curr Top Microbiol Immunol 258: 13–33 [DOI] [PubMed] [Google Scholar]
- Hsieh DJ, Camiolo SM, Yates JL (1993) Constitutive binding of EBNA1 protein to the Epstein–Barr virus replication origin, oriP, with distortion of DNA structure during latent infection. EMBO J 12: 4933–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrien O, Marheineke K, Goldar A (2003) Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. BioEssays 25: 116–125 [DOI] [PubMed] [Google Scholar]
- Ito T, Bulger M, Pazin MJ, Kobayashi R, Kadonaga JT (1997) ACF, an ISWI-containing and ATP-utilizing chromatin assembly and remodeling factor. Cell 90: 145–155 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Kieff E (1996) Epstein–Barr virus and its replication. In Field's Virology, Knipe D, Howley PM (eds) Vol. 2, pp 2343–2396. Philadelphia: Lippincott-Raven Publishers [Google Scholar]
- Koons MD, Scoy SV, Hearing J (2001) The replicator of the Epstein–Barr virus latent cycle origin of DNA replication, oriP, is composed of multiple functional elements. J Virol 75: 10582–10592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeRoy G, Loyola A, Lane WS, Reinberg D (2000) Purification and characterization of a human factor that assembles and remodels chromatin. J Biol Chem 275: 14787–14790 [DOI] [PubMed] [Google Scholar]
- Lipford JR, Bell SP (2001) Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol Cell 7: 21–30 [DOI] [PubMed] [Google Scholar]
- Lomvardas S, Thanos D (2001) Nucleosome sliding via TBP DNA binding in vivo. Cell 106: 685–696 [DOI] [PubMed] [Google Scholar]
- Lusser A, Kadonaga JT (2003) Chromatin remodeling by ATP-dependent molecular machines. BioEssays 25: 1192–1200 [DOI] [PubMed] [Google Scholar]
- McNairn AJ, Gilbert DM (2003) Epigenomic replication: linking epigenetics to DNA replication. BioEssays 25: 647–656 [DOI] [PubMed] [Google Scholar]
- Mendez J, Stillman B (2003) Perpetuating the double helix: molecular machines at eukaryotic DNA replication origins. BioEssays 25: 1158–1167 [DOI] [PubMed] [Google Scholar]
- Nasheuer HP, Smith R, Bauerschmidt C, Grosse F, Weisshart K (2002) Initiation of eukaryotic DNA replication: regulation and mechanisms. Prog Nucleic Acid Res Mol Biol 72: 41–94 [DOI] [PubMed] [Google Scholar]
- Neely KE, Workman JL (2002) Histone acetylation and chromatin remodeling: which comes first? Mol Genet Metab 76: 1–5 [DOI] [PubMed] [Google Scholar]
- Norio P, Schildkraut CL (2001) Visualization of DNA replication on individual Epstein–Barr virus episomes. Science 294: 2361–2364 [DOI] [PubMed] [Google Scholar]
- Norio P, Schildkraut CL (2004) Plasticity of DNA replication initiation in Epstein–Barr virus episomes. PLoS Biol 2: e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norio P, Schildkraut CL, Yates JL (2000) Initiation of DNA replication within oriP is dispensable for stable replication of the latent Epstein–Barr virus chromosome after infection of established cell lines. J Virol 74: 8563–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 16: 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL (2002) Chromatin remodeling enzymes: taming the machines. Third in review series on chromatin dynamics. EMBO Rep 3: 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot RA, Bozhenok L, van den Berg DL, Steffensen S, Ferreira F, Grimaldi M, Gilbert N, Ferreira J, Varga-Weisz PD (2004) The Williams syndrome transcription factor interacts with PCNA to target chromatin remodelling by ISWI to replication foci. Nat Cell Biol 6: 1236–1244 [DOI] [PubMed] [Google Scholar]
- Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, Hammerschmidt W, Schepers A (2003) Complex protein–DNA dynamics at the latent origin of DNA replication of Epstein–Barr virus. J Cell Sci 116: 3971–3984 [DOI] [PubMed] [Google Scholar]
- Ryan MP, Stafford GA, Yu L, Cummings KB, Morse RH (1999) Assays for nucleosome positioning in yeast. Methods Enzymol 304: 376–399 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bernstein BE, Karabetsou N, Morillon A, Weise C, Schreiber SL, Mellor J, Kouzarides T (2003) Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell 12: 1325–1332 [DOI] [PubMed] [Google Scholar]
- Schepers A, Ritzi M, Bousset K, Kremmer E, Yates JL, Harwood J, Diffley JF, Hammerschmidt W (2001) Human origin recognition complex binds to the region of the latent origin of DNA replication of Epstein–Barr virus. EMBO J 20: 4588–4602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson RT (1990) Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature 343: 387–389 [DOI] [PubMed] [Google Scholar]
- Stedman W, Deng Z, Lu F, Lieberman PM (2004) ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J Virol 78: 12566–12575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner DE, Berger SL (2000) Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64: 435–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- Sugden B, Leight ER (2001) Molecular mechanisms of maintenance and disruption of virus latency. In Epstein–Barr Virus and Human Cancer, Takada K (ed) Vol. 258, pp 3–11. Heidelberg: Springer [Google Scholar]
- Varga-Weisz P (2001) ATP-dependent chromatin remodeling factors: nucleosome shufflers with many missions. Oncogene 20: 3076–3085 [DOI] [PubMed] [Google Scholar]
- Varga-Weisz PD, Wilm M, Bonte E, Dumas K, Mann M, Becker PB (1997) Chromatin-remodelling factor CHRAC contains the ATPases ISWI and topoisomerase II. Nature 388: 598–602 [DOI] [PubMed] [Google Scholar]
- Verreault A (2003) Histone deposition at the replication fork: a matter of urgency. Mol Cell 11: 283–284 [DOI] [PubMed] [Google Scholar]
- Yates JL, Camiolo SM, Bashaw JM (2000) The minimal replicator of Epstein–Barr virus oriP. J Virol 74: 4512–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates JL, Guan N (1991) Epstein–Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J Virol 65: 483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Methods